

Abstract
Drugs that target the main protease (Mpro) of SARS-CoV-2 are effective therapeutics that have entered clinical use. Wide-scale use of these drugs will apply selection pressure for the evolution of resistance mutations. To understand resistance potential in Mpro, we performed comprehensive surveys of amino acid changes that can cause resistance to nirmatrelvir (Pfizer), and ensitrelvir (Xocova) in a yeast screen. We identified 142 resistance mutations for nirmatrelvir and 177 for ensitrelvir, many of which have not been previously reported. 99 mutations caused apparent resistance to both inhibitors, suggesting likelihood for the evolution of cross-resistance. The mutation with the strongest drug resistance score against nirmatrelvir in our study (E166V) was the most impactful resistance mutation recently reported in multiple viral passaging studies. Many mutations that exhibited inhibitor-specific resistance were consistent with the distinct interactions of each inhibitor in the substrate binding site. In addition, mutations with strong drug resistance scores tended to have reduced function. Our results indicate that strong pressure from nirmatrelvir or ensitrelvir will select for multiple distinct resistant lineages that will include both primary resistance mutations that weaken interactions with drug while decreasing enzyme function and compensatory mutations that increase enzyme activity. The comprehensive identification of resistance mutations enables the design of inhibitors with reduced potential of developing resistance and aids in the surveillance of drug resistance in circulating viral populations.
Keywords: Mpro, resistance, protease, SARS-CoV-2, deep mutational scanning, virology
Graphical Abstract
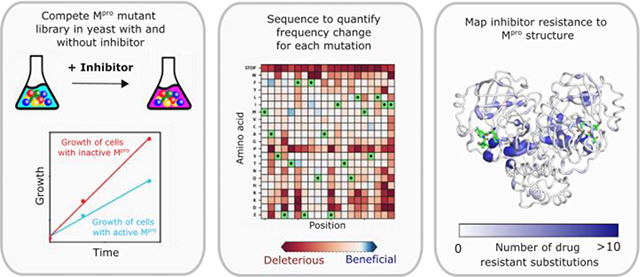
The COVID-19 pandemic continues to have broad impacts on human health, economics, and day to day life. Multiple strategies have been implemented to reduce the spread of the virus and disease progression, including mRNA vaccines 1, monoclonal antibody therapeutics 2, and most recently small molecule direct acting antivirals 3–5. However, SARS-CoV-2, the virus that causes COVID-19, is capable of evolving rapidly to adapt to new selection pressures 6–7. For example, recently evolved omicron variants of SARS-CoV-2 have reduced the protection provided by vaccines as well as the efficacy of monoclonal antibody therapeutics 8. Evasion of therapeutic interventions is a hallmark of rapidly evolving diseases including many cancers and viral infections 9–10. Evaluating the evolutionary potential of therapeutic approaches can provide a valuable guide for genetic surveillance and treatment choices as well as improving the design of new therapeutics with reduced potential for resistance (Figure 1A&B).
Figure 1. Practical motivations for systematic analyses of drug resistance mutations in SARS-CoV-2 Mpro.
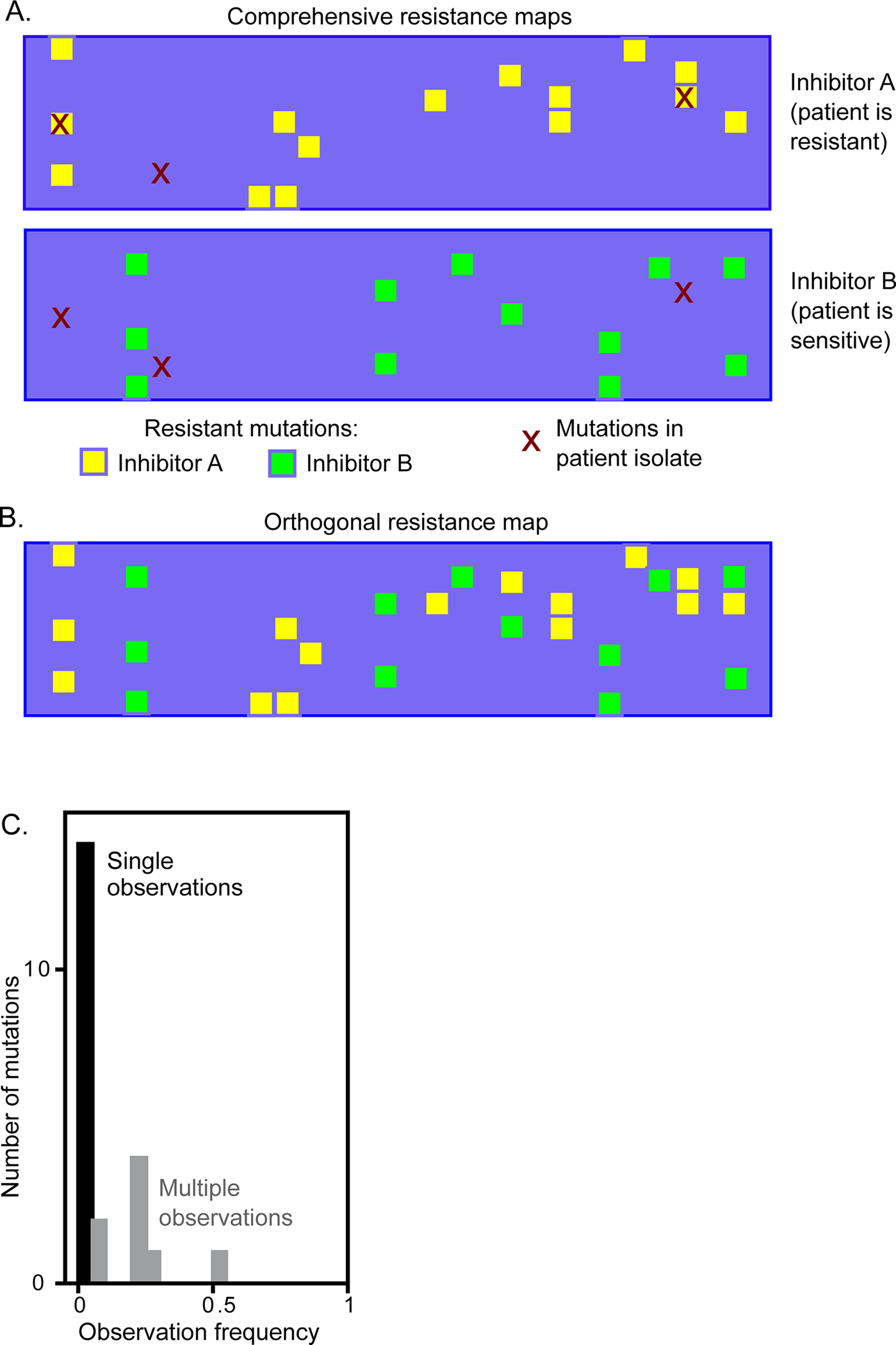
(A) Comprehensive maps of drug resistance can enable a sequence-informed choice of inhibitors for effective treatments. (B) Potential to identify inhibitors with orthogonal mutational profiles that could be utilized in combination to reduce the likelihood of resistance evolution. (C) A viral passaging study 19 of SARS-CoV-2 reports multiple mutations observed once, suggesting that sampling is insufficient to completely reveal all potential resistance mutations. Comprehensive analyses of resistance potential will likely be of use in efforts to survey for evolution of resistance in patient isolates.
The SARS-CoV-2 genome encodes two overlapping polyproteins, pp1a and pp1b which are proteolytically cleaved to generate 16 nonstructural proteins (nsps) and 4 structural proteins 11. The nsp5 gene encodes for main protease (Mpro) which initiates autoproteolysis out of the polyproteins and then subsequently cuts at 11 additional cleavage sites to yield the nonstructural proteins including the replication machinery essential for viral function 12–13. Mpro is a dimeric cysteine protease; each monomer is composed of three domains with the active site comprising of a His41-Cys145 catalytic dyad located in the clefts between domain I and domain II 14.
Mpro is an important target for the development of inhibitors against SARS-CoV-2 and future coronavirus-mediated pandemics due to its essential functional role in the viral life cycle and its high conservation amongst all coronaviruses. Inhibitors targeting Mpro have demonstrated strong clinical effectiveness in treating infected individuals 3, 15. Approximately half a dozen peptidomimetic and small molecule inhibitors have entered clinical trials 16. The drug PAXLOVID™, a combination of the Mpro inhibitor nirmatrelvir (PF −07321332) and the CYP3A4 inhibitor ritonavir, which boosts the serum half-life of many protease inhibitors, was recently approved for emergency use authorization in treating COVID-19 17. Nirmatrelvir is a reversible-covalent, peptidomimetic inhibitor with potent anti-viral effect. Ensitrelvir (S-217622), a non-peptidic, non-covalent Mpro inhibitor developed by Shionogi, is currently being evaluated in late-stage phase III trials and recently received emergency regulatory approval in Japan 18.
The wide-scale clinical usage of drugs that target Mpro to treat patients infected with SARS-CoV-2 will likely drive selection pressure for resistance; in fact, the evolution of resistance to nirmatrelvir is under active investigation. While the clearest understanding of drug resistance will come from examining the natural resistance that occurs following prolonged drug exposure in the clinical population, it is currently early in the pandemic and to date there has been no evidence of resistance to Mpro inhibitors arising in the human population. In lieu of this, it is important to use alternative methods such as viral passaging in cell culture and systematic mutational experiments such as reported in this paper to measure the resistance potential of drugs. In cell culture experiments, several recent reports indicate that multiple mutations in Mpro can provide resistance to nirmatrelvir 19–25. Resistance mutations raised against nirmatrelvir in vitro using cell culture exhibit little overlap between different experimental repeats, with many mutations being observed only one time (Figure 1C), suggesting that the scale of these traditional approaches is insufficient to provide complete experimental coverage of potential resistance mutations. Additionally, as other Mpro inhibitors reach the clinic, it will become important to identify their unique resistance profiles to better guide the development of drugs such that their efficacy is not immediately lost to cross-resistance from prior antivirals (Figure 1A&B).
Analyses of purified Mpro harboring drug resistance mutations identified in viral passaging studies indicate that they fall into two categories 19–20, 24; mutations in the first category weaken binding to drug, while those in the second category increase the proteolytic activity of Mpro. Similar categories of mutations contribute to drug resistance in many other systems including HIV protease 26–27. In HIV protease, a majority of initial mutations arise at the active site, directly affect inhibitor binding, and are the primary cause of resistance to protease inhibitors. These mutations typically also cause reduced enzymatic activity. Subsequently, compensatory mutations arise that rescue the enzyme defects caused by primary mutations; occasionally such mutations appear to also contribute to drug resistance both in HIV and SARS-CoV-2 in cell culture 19, 24, 28–29. In principle, mutations that increase enzymatic activity should provide a small growth advantage in the presence of drugs, and this might be amplified for viral proteases such as Mpro which must cleave themselves out of polyproteins to generate additional active protease molecules 30. The highest level of drug resistance observed in HIV protease and in SARS-CoV-2 Mpro appears to arise from a combination of primary mutations that disrupt drug binding and compensatory mutations that increase enzyme activity.
The potential to evolve resistance to nirmatrelvir provides a strong motivation to comprehensively understand the mutations that can cause resistance. Prior studies in other rapidly evolving diseases have found that identification of drug resistance mutations from clinical samples and/or cell culture passage does not exhaustively identify all resistance mutations 31–34. Here, we describe a comprehensive approach to assess the resistance landscape of Mpro from SARS-CoV-2. We provide a systematic map of primary resistance mutations for two chemically distinct inhibitors, nirmatrelvir and ensitrelvir. We find that these two inhibitors exhibit partially overlapping drug resistance profiles accordant with their chemotypes and interactions in the substrate binding site. We identified both mutations that cause cross-resistance to the two drugs and inhibitor-specific resistance mutations. These comprehensive resistance maps can be used as a guide to evaluate the evolution of drug resistance in circulating SARS-CoV-2 and to aid in the development of drugs with distinct resistance profiles to treat viruses that may evade current drugs.
Results and Discussion
Comparison of the binding modes of Mpro inhibitors.
Mpro is a widely targeted protein in SARS-CoV-2 antiviral development. To date, hundreds of co-crystal structures of Mpro in complex with inhibitors at its active site have been made available on the Protein Databank (PDB), providing extensive insights into inhibitor-protein interactions. We hypothesized that similarities in inhibitor binding interactions may predict similarities in resistance profiles. To investigate this correlation, we compared the interactions of nirmatrelvir and ensitrelvir with Mpro to that of other structurally characterized inhibitors. To assess similarities and distinctions in inhibitor binding profiles, we calculated the van der Waals (vdW) interactions between 134 Mpro-inhibitor crystal structures acquired from the PDB. This measurement describes the packing of active site residues around individual inhibitors and provides an overall contact pattern for inhibitors. Subsequently, we performed principal component analysis (PCA) and k-means clustering to assess the variance in the dataset and group inhibitors in three main clusters (Figure 2A). The three clusters can be differentiated by PC1, which primarily reflects vdW interactions with the inhibitor at the S1 and S2 subsites (where S1 refers to the binding pocket for the substrate amino acid immediately N-terminal to the cut-site, and S2 the binding pocket for the preceding amino acid). Nirmatrelvir and ensitrelvir belong to different clusters (1 and 2 respectively) suggesting that they may exhibit distinct resistance profiles. In future efforts it will be interesting to extensively explore how inhibitor binding classification relates to drug resistance profile.
Figure 2. Principal component analysis of vdW interactions show distinct modes of nirmatrelvir and ensitrelvir binding.

A) Primary component analysis of potent compounds (IC50 < 1 uM) based on strength of van der Waals interactions. PCA data was grouped on three clusters. Nirmatrelvir and ensitrelvir are in clusters 1 and 2 respectfully. B) Heat map based on strength of van der Waals interactions between inhibitor and Mpro residues. C) Strength of van der Waals interactions between Mpro residues and nirmatrelvir (PDB 7RFS) 3 or ensitrelvir (PDB 7vu6) 18 mapped to structure.
Consistent with their classification in distinct PCA clusters, nirmatrelvir and ensitrelvir contact the Mpro active site in distinct modes. Nirmatrelvir spans the S4-S1 portion of the active site and forms an extensive interaction network with the active site involving both vdW interactions with non-prime site residues as well as hydrogen bonding with H163, E166, and Q189 (Figure 2B and C). In contrast to nirmatrelvir and most other Mpro active site inhibitors, ensitrelvir extends into the S1′ pocket and interacts with the threonine cluster (T24-T27). Moreover, its P2 moiety forms a π–π stacking interaction with the catalytic residue H41, displacing it from the active conformation. We chose to initially focus our systematic characterization of Mpro drug resistance on these two inhibitors, as they are both currently in clinical use, have strong anti-viral potency, and exhibit distinct binding modes within the Mpro active site.
Systematic identification of drug resistance mutations
We previously reported multiple yeast-based approaches to systematically quantify the function of all point mutations in Mpro 35. In the current work, we utilized the effect of Mpro expression on yeast growth as a measure of enzymatic activity. Expression of SARS-CoV-2 Mpro reduces the growth rate of yeast cells, but catalytically-inactive variants such as C145A do not35–36, indicating that changes in growth rate are due to cleavage of endogenous yeast proteins. While this assay relies on cleavage of proteins that may not be directly relevant to SARS-CoV-2, the impacts on yeast growth correlate strongly (R2=0.9) with measures from a FRET-based assay that directly measures cleavage of the SARS-CoV-2 Nsp4/5 cut-site 35. In addition, we found that functional scores from yeast growth assays correlated linearly with catalytic rates measured for individual variants (though increased activity variants have not been assessed carefully by alternate approaches). The approach also identified known critical residues (such as catalytic residues), and circulating clinical variants (which must contain active Mpro) overwhelmingly show wild-type-like functional scores 35. Together, these results suggest that our yeast growth assay provides useful estimates of Mpro function for the purpose of assessing SARS-CoV-2 fitness.
To systematically identify potential resistance mutations, we performed deep mutational scanning experiments in the presence of nirmatrelvir and ensitrelvir. Because yeast possess numerous drug efflux pumps and are protected by a cell wall, they often require genetic engineering to make them accessible to drugs 37–39. To increase the druggability of the yeast in our assays, we used a strain called Δ4 with four efflux pumps deleted (Δsnq2 Δpdr5 Δpdr1 Δyap1) and added a low concentration of SDS to increase permeability. Under these conditions, we found that addition of either nirmatrelvir or ensitrelvir restored the yeast growth rate that was retarded due to Mpro expression (Figure S1). In our mutational scanning experiments, we found that addition of either 10 μM ensitrelvir or 20 μM nirmatrelvir to the culture inhibited approximately 50% of the activity of the wild-type Mpro in our library.
We transformed the Δ4 yeast strain with our previously described barcoded Mpro plasmid library 35 that contains both internal positive controls (wild-type Mpro) and negative controls (internal stop codons known to destroy function). The resulting yeast libraries were amplified without induction of Mpro and subsequently treated with either 20 μM nirmatrelvir, 10 μM ensitrelvir, or DMSO, followed by addition of 2 μM β-estradiol to induce expression of Mpro. Cells were collected at 0- and 16-hour time points and subjected to Illumina sequencing to estimate the frequency of each mutant in each sample (Figure 3A). We calculated a functional score for each variant based on its change in frequency before and after induction of Mpro as described in Methods (See Table S1 for all sequencing counts and functional scores). Functional scores were normalized such that a score of 0 represents an inactive mutant that abolishes Mpro activity and thus restores yeast growth. Wild-type Mpro had a functional score of 0.67 in the absence of inhibitors, and we were able to reproducibly measure mutations that both increased and decreased enzyme activity. Experiments were performed in triplicate; functional scores between the three replicates were strongly correlated. Figure 3B shows the correlation between the functional scores of all mutations in two replicates measured in the absence of inhibitor (R2=0.95) or in the presence of nirmatrelvir (R2=0.95) or ensitrelvir (R2=0.94). There was a narrow distribution of functional scores for barcodes associated with wild-type Mpro and for stop codons, excluding amino acids 300–306 that are dispensable for Mpro activity 35 (Figure S2A). Additionally, initial reads per variant were not correlated with functional score indicating minimal selection prior to induction with β-estradiol (Figure S2B).
Figure 3. Systematic analyses of mutations that decrease sensitivity to nirmatrelvir and ensitrelvir.
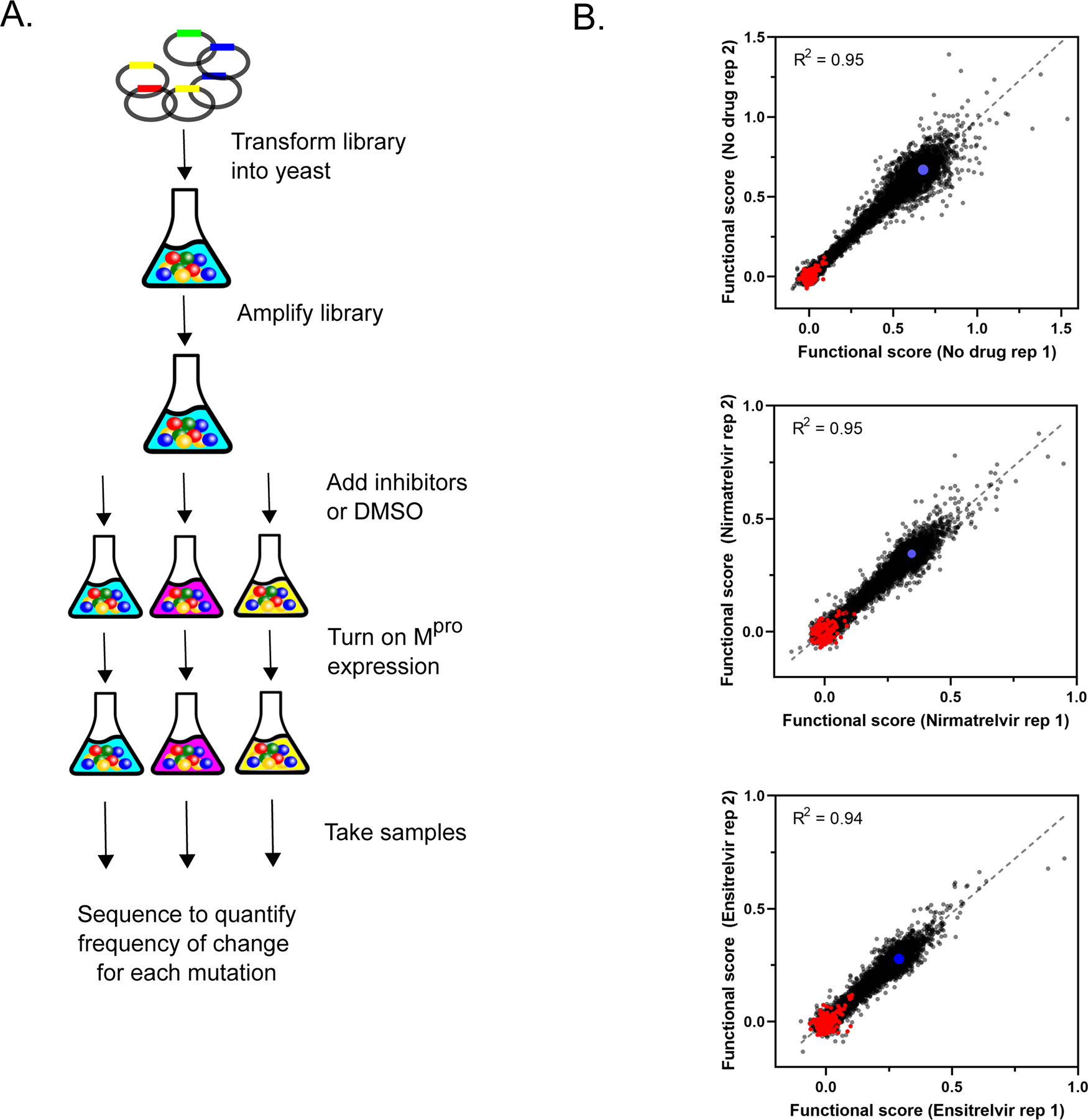
A) Illustration of yeast selection screen B) Correlation between replicates of functional scores of all Mpro variants in the absence of drug or presence of nirmatrelvir or ensitrelvir. Stop codons are shown in red and wild-type Mpro in blue.
The distributions of functional scores of all mutations in the presence or absence of inhibitors indicates that both nirmatrelvir and ensitrelvir were effective in reducing the activity of most Mpro variants (Figure 4A). Wild-type Mpro was inhibited approximately 50% by nirmatrelvir or ensitrelvir, and we could clearly distinguish the functional scores of stop codons (shown in red) from wild-type Mpro in each experiment (Figure 4A and Figure S2A). Similar to wild-type Mpro, both inhibitors reduced the functional scores of most mutants by about 2-fold as well, consistent with the conclusion that most mutations did not affect drug resistance.
Figure 4. Comprehensive analysis of mutations that show drug resistance.
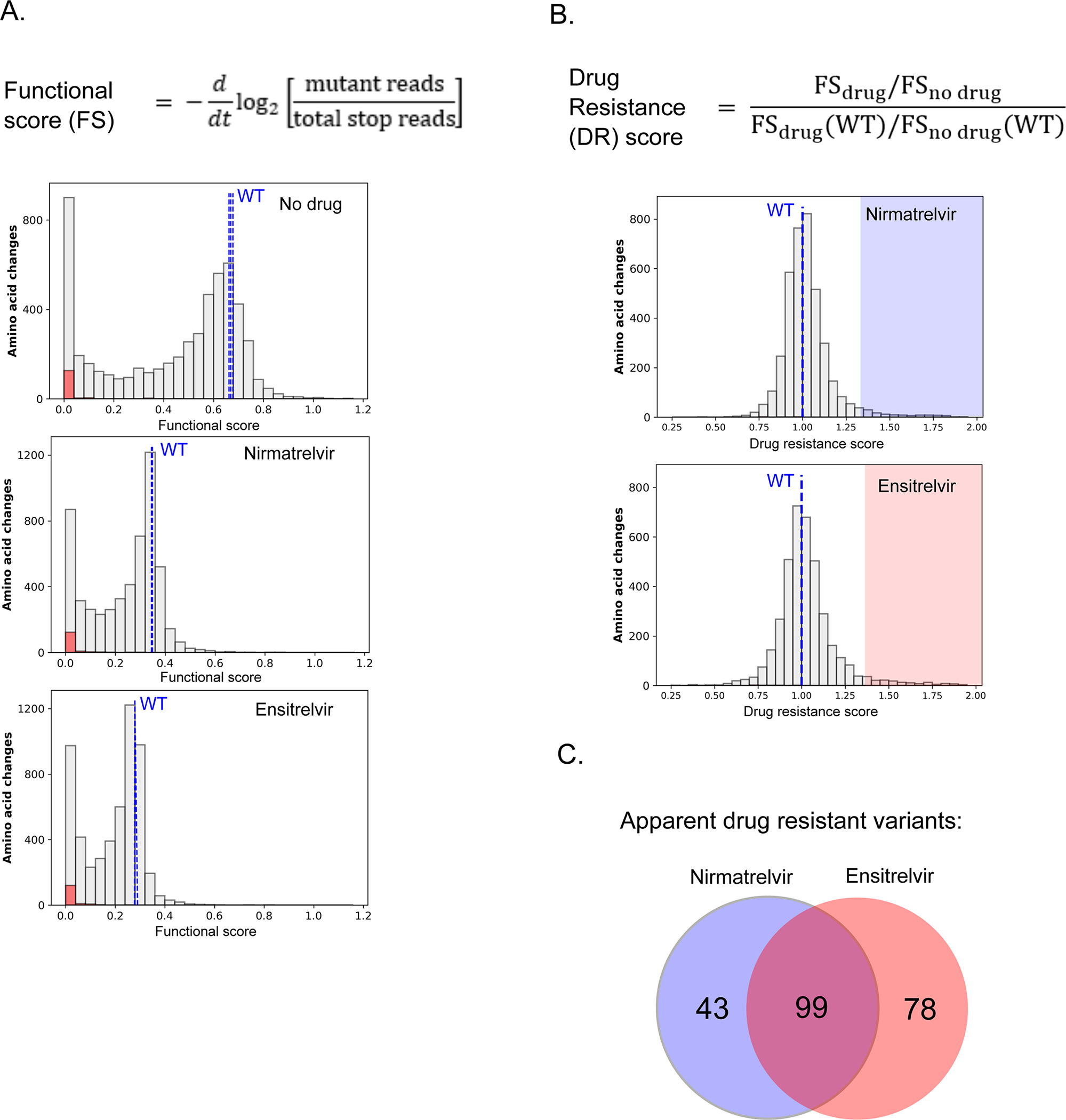
A) Equation to calculate functional scores (top panel). Distribution of all functional scores for all variants (gray), stop codons (red) and wild-type (blue dashed lines) in the absence of drug or presence of nirmatrelvir or ensitrelvir (bottom panel). B) Equation to calculate drug resistance (DR) scores (top). Distribution of all DR scores in the presence of nirmatrelvir or ensitrelvir (bottom panel). Mutants with DR scores above two standard deviations of wild-type are highlighted and are considered apparent drug resistance mutations. C) Venn diagram of nirmatrelvir and ensitrelvir resistance mutations.
However, a subset of mutations had functional scores that were relatively unaffected by the inhibitors (for example, see Figure S3A), suggesting these variants may be relevant for drug resistance. To systematically assess the impacts of mutations on inhibition, we calculated a drug resistance score (DR score) (see Table S1 for all DR scores). We defined the DR score as the ratio of the functional score of each mutant in the absence of the inhibitor to that in the presence of the inhibitor, compared to the same ratio with wild-type Mpro (Figure 4B and Figure S3B). By computing the ratio of functional scores with and without drug, the underlying impacts of mutations on enzyme function are canceled out such that the DR score estimates drug sensitivity. This is similar to IC50 estimates of DR that quantify changes in sensitivity to drug independently from how a variant impacts function. Under the conditions of our experiments, DR scores have a ceiling of about 2 even if the inhibitors have no measurable impact on function, while wild-type Mpro has a DR score of 1. We chose these conditions to make the DR score sensitive to small changes in IC50, but experiments with higher concentrations of inhibitor would be required to resolve changes in IC50 greater 10-fold (Figure S3C). DR scores were only calculated for variants with a functional score at least 1/3 that of wild-type, which approximates the lowest functional score we observed for common, already-circulating Mpro variants discussed in our previous work 35. This threshold was applied to limit the potential for elevated noise derived from ratios of small numbers as well as to account for the requirement of Mpro activity for viral propagation. Our approach was designed to sensitively identify mutations that cause drug resistance and distinguish the impact these mutations are predicted to have on enzymatic activity.
Figure 4B shows the distribution of DR scores calculated for both nirmatrelvir and ensitrelvir. We considered mutants with DR scores above two standard deviations (SD) of wild-type as apparent drug resistance mutations. We identified 142 mutants with apparent resistance to nirmatrelvir and 177 to ensitrelvir (Figure 4C, Table S2). Given the total number of DR measurements made for each inhibitor (3858 variants with function above the 1/3 wild-type threshold), random expectations would lead to 6 variants with apparent resistance to both inhibitors. However, we observed 99 mutations that caused apparent DR to both drugs (p<0.0001, chi2), suggesting a largely shared basis of drug resistance for both inhibitors.
Mutations with the greatest likelihood of contributing to drug resistance in nature
Both the likelihood of mutations occurring and their impacts on fitness will determine the likelihood of contributing to drug resistance in nature. While detailed estimates of evolutionary likelihoods will require extensive modeling that are beyond the scope of this work, we have performed analyses of primary factors including the number of nucleotide changes required for an amino acid change and the magnitude of the DR score. Under the recommended treatment regimen of Paxlovid, the concentration of nirmatrelvir is maintained well above that needed to inhibit wild-type Mpro 40, providing selection pressure for mutations with large impacts on drug binding. Single nucleotide changes are by far the most common mutation observed in SARS-CoV-2 isolates (Figure S4), consistent with reported mutational biases 41. In contrast, amino acid changes that cause apparent drug resistance to either nirmatrelvir or ensitrelvir tend to require multiple nucleotide changes, consistent with the greater number of amino acid changes that can be accessed. Multiple nucleotide changes can evolve, especially if they provide a unique fitness advantage. However, in the case of both nirmatrelvir and ensitrelvir, the strongest DR scores of single nucleotide variants are similar to those accessible by multiple nucleotide mutations (Table S2). These findings indicate that amino acid changes with the highest DR scores accessible by single nucleotide mutations have the most likelihood of contributing to the natural evolution of drug resistance.
The 16 single nucleotide variants with the strongest DR scores for nirmatrelvir and ensitrelvir are listed in Table 1. Viral passaging with these inhibitors has identified some of the same variants, suggesting that our findings are relevant to viral evolution. For example, H172Y is a mutation that was disclosed by Pfizer to cause strong resistance to nirmatrelvir with a greater than 200-fold increase of Ki 42. Our data identify M49L and P52S as apparent DR mutations for ensitrelvir and these mutations have also been reported in viral experiments with ensitrelvir (https://www.japic.or.jp/mail_s/pdf/23-11-1-07.pdf). In addition, E166V evolved in multiple lineages of SARS-CoV-2 under selection with gradual (2-fold) increases in nirmatrelvir 19.
Table 1.
Single nucleotide mutations with largest nirmatrelvir or ensitrelvir resistance scores.*
| Mutation | DR score | References | ||
|---|---|---|---|---|
| Nirmatrelvir | Ensitrelvir | |||
|
| ||||
| E166V | 2.0 | 1.8 | 19, 24 | |
| G2R | 2.0 | 1.9 | ||
| G2C | 1.9 | 1.7 | ||
| S139P | 1.8 | 1.0 | 19 | |
| P168R | 1.8 | 2.2 | ||
| Q299H | 1.8 | 1.9 | ||
| G2S | 1.7 | 1.7 | ||
| N214Y | 1.7 | 1.7 | ||
| G2A | 1.7 | 1.7 | ||
| C300R | 1.6 | 1.7 | ||
| M49L | 1.2 | 2.1 | 21, www.japic.or.jp/mail_s/pdf/23–11-1–07.pdf | |
| H172Y | 1.5 | 1.9 | 19, 21, 42 | |
| E166A | 1.6 | 1.8 | 19–20 | |
| P52L | 1.0 | 1.8 | ||
| S139L | 1.6 | 1.8 | ||
| C44G | 0.9 | 1.8 | ||
References are provided for previously described drug resistance mutations.
The evolved lineage with E166V showed the highest reported drug resistance with an ability to tolerate roughly 1000-fold higher concentrations of nirmatrelvir than wild-type. Interestingly, many lineages evolved with gradual increases in nirmatrelvir concentration did not contain mutations with strong DR scores in our study and instead contained multiple mutations with either small increases in DR score and/or increased functional scores relative to wild-type (Table S3). Because the treatment regimen of Paxlovid maintains the concentration of nirmatrelvir 10-fold above the sensitivity of SARS-CoV-2 with wild-type Mpro, clinical drug pressure may preferentially select for stronger resistance than observed with 2-fold increases in drug concentration in cell culture.
We further examined the validity of our yeast screen by measuring the inhibition of two Mpro variants (T25E and M49L) that showed apparent DR specifically for ensitrelvir. Consistent with our DR scores, both variants exhibited increased Ki for ensitrelvir (40-fold for M49L and 300-fold for T25E) compared to wild-type and minimal to no observed impact on Ki for nirmatrelvir (Figure 5 and Figure S5). Of note, the T25E amino acid change requires three nucleotide changes, reducing the likelihood of its natural evolution.
Figure 5. M49L and T25E Mpro exhibit decreased potency for ensitrelvir.
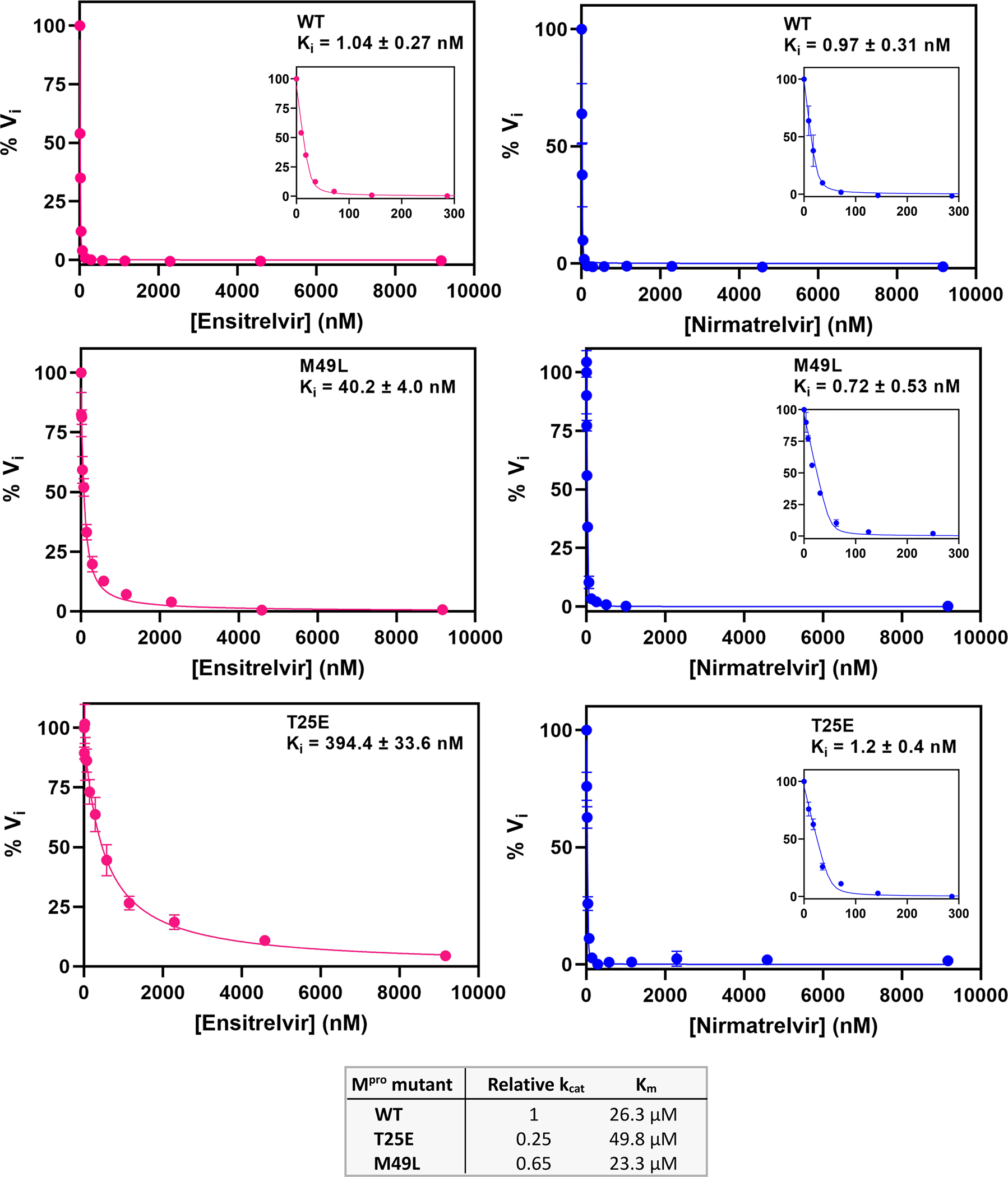
Determination of Ki values for nirmatrelvir or ensitrelvir against Mpro using the FRET peptide assay. Curves were fit using the Morrison equation in Prism software (see Methods). Each measurement was performed in triplicate.
There are four variants accessible by single nucleotide mutations that were in the top ten of DR scores for both nirmatrelvir and ensitrelvir (E166V, G2R, P168R, and Q299H), suggesting these variants may lead to cross resistance and are of particular health concern. The cross-resistant mutations indicate that given enough time nirmatrelvir and ensitrelvir are unlikely to provide a benefit in combination, necessitating the development of additional therapeutic tools to treat SARS-CoV-2 infections.
Evaluation of resistance with regards to the substrate envelope
We explored the structural basis of variants likely to contribute to natural resistance by mapping them to the structure of Mpro and the substrate envelope. The substrate envelope represents the consensus volume occupied by the multiple Mpro cut sites in the viral proteome 43. Previous investigations of drug resistance demonstrate that inhibitors that fit within this consensus volume are less likely to be susceptible to drug resistance mutations. However, an inhibitor that protrudes beyond the substrate envelope provides opportunities for the evolution of resistance 43–45. We compared the substrate envelope with the location of bound inhibitors as well as the strongest apparent drug resistance mutations (Figure 6A). We focused on the strongest apparent drug resistance mutations here because steric clashes where inhibitors protrude from the substrate envelope typically have large impacts on inhibitor binding. Nirmatrelvir and ensitrelvir both protrude from the substrate envelope in the S1 pocket close to E166. Both inhibitors hydrogen bond to E166: nirmatrelvir to the side-chain and ensitrelvir to the main-chain.
Figure 6. Structural distribution of apparent drug resistance mutations.
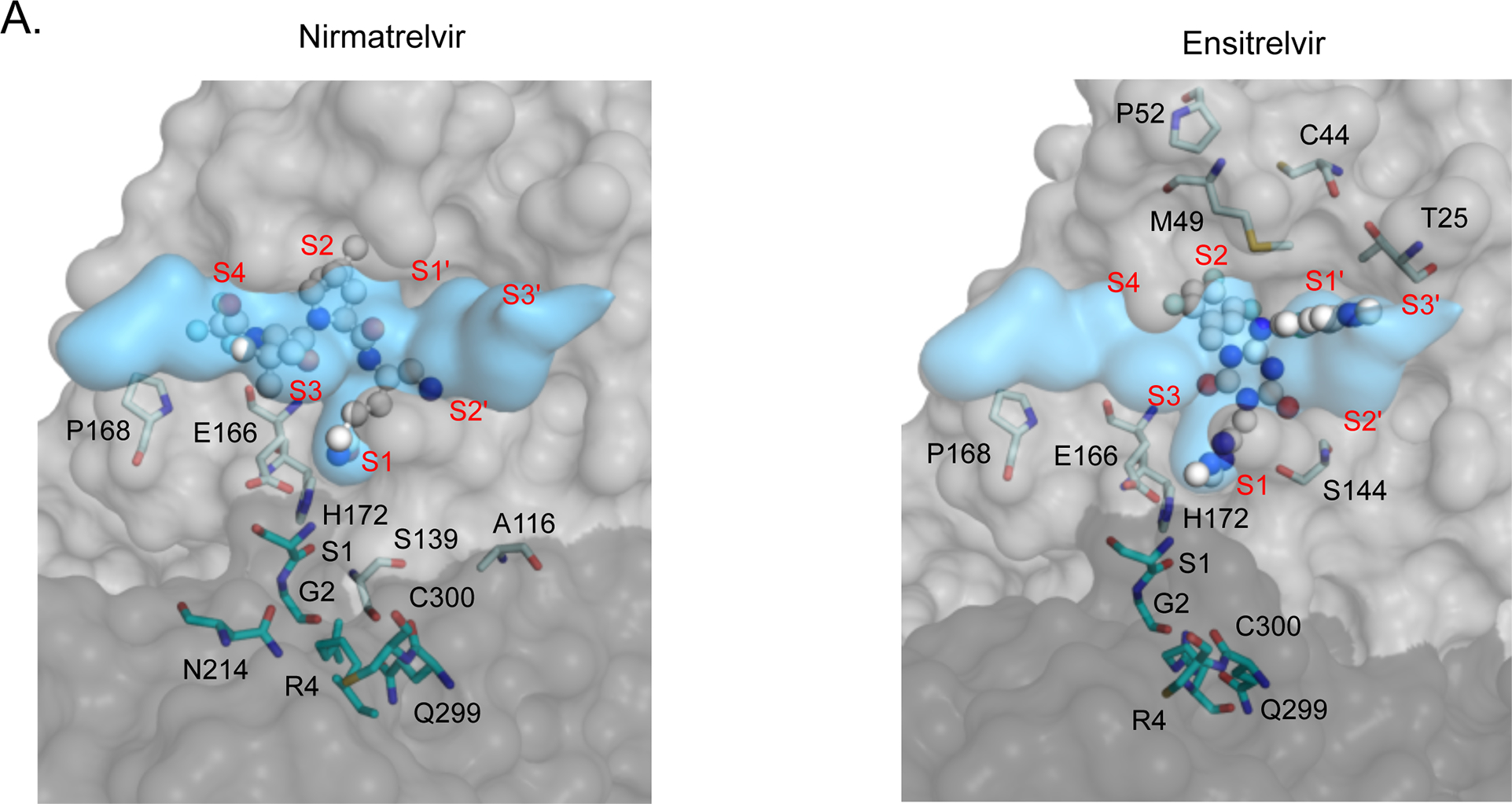
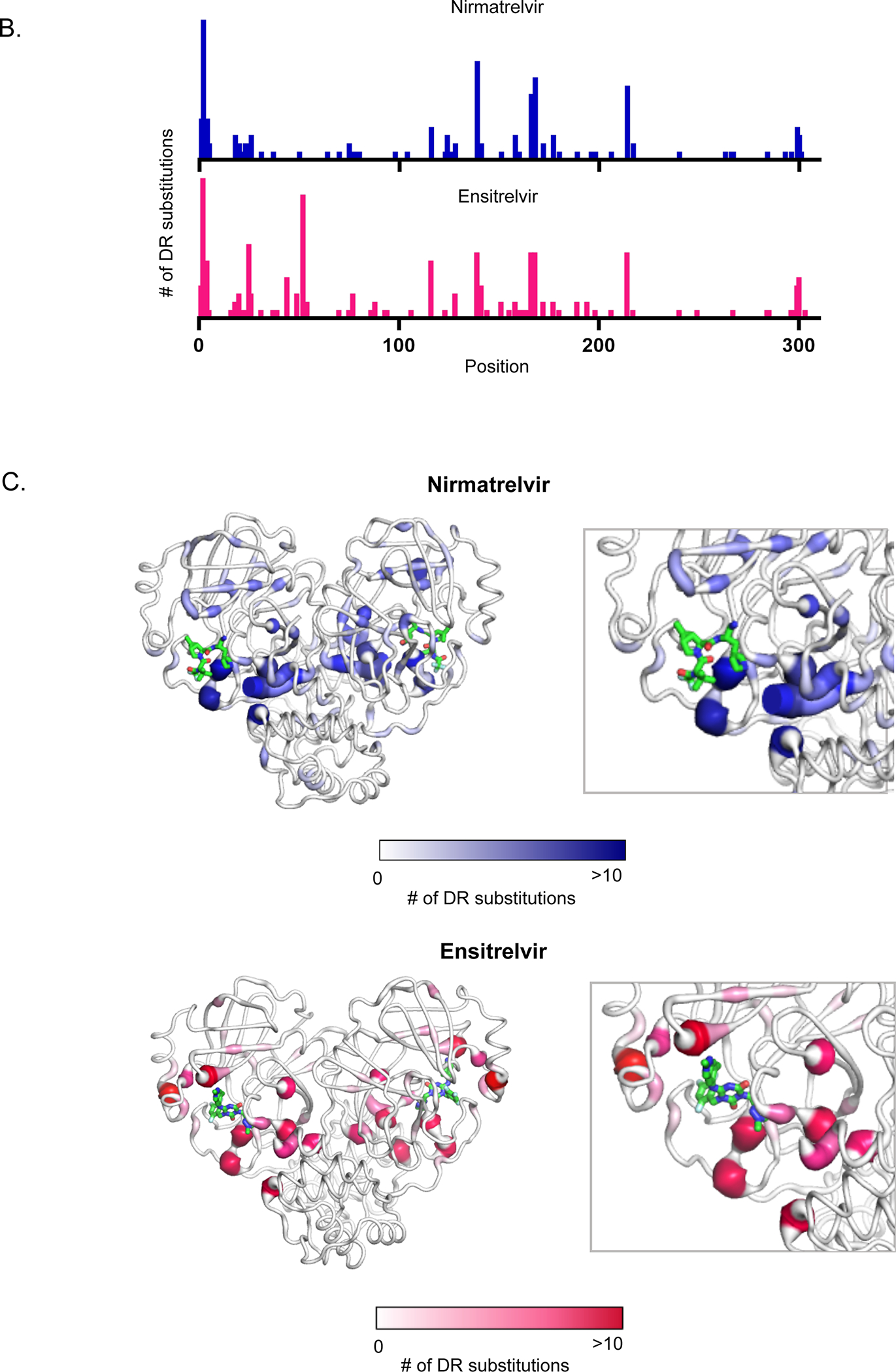
A) The fit of nirmatrelvir (left panel) and ensitrelvir (right panel) within the substrate envelope. The labeled Mpro positions are those that contain the 30 mutations with the highest DR scores. S1, G2, R4, N214, Q299 and C300 are located on monomer B (shown in darker gray) and are represented as cyan sticks. The S4-S3’ binding subsites are labeled in red. B) The number of nirmatrelvir and ensitrelvir resistance mutations at each position of Mpro. C) The number of nirmatrelvir and ensitrelvir resistance mutations at each position of Mpro are mapped to the Mpro-inhibitor structures. Inhibitors are shown in green.
Mutations that cause the strongest cross-resistance to both inhibitors including G2R, E166V, P168R, and Q299H are located at or close to E166 where both nirmatrelvir and ensitrelvir protrude from the substrate envelope (Figure 6A). Mpro forms a dimer with two active sites. The active sites are formed predominantly by residues from one monomer, but the N-terminus of the trans-subunit extends close to the active site and forms a hydrogen bond with the side-chain of E166. G2 and Q299 are in steric contact with each other and mutations at either of these residues likely impact the dynamics and/or structure of the N-terminus of Mpro and the region of the active site near position 166. Similarly, P168 also appears positioned such that mutations are likely to impact the dynamics and/or structure of the active site in this region. P168 is in a beta-turn, where prolines are known to help stabilize or rigidify structure 46–47. Consistent with destabilization of this turn contributing to drug resistance, many other mutations at P168 also cause apparent drug resistance including K, V, and T.
Inhibitor specific resistance mutations
We also observe some mutations that cause strong DR scores to one inhibitor but that did not impact the other inhibitor (Table 2). To explore the structural basis of inhibitor-specific resistance, we chose to investigate all amino acid changes regardless of the number of nucleotide mutations. The strongest inhibitor-specific resistance mutations were S1H and S139P for nirmatrelvir and mutations at C44, M49, T25, P52 and Y54 for ensitrelvir. Both position 1 and 139 are in proximity to E166 where both inhibitors protrude slightly from the substrate envelope. The amino terminus of position 1 hydrogen bonds to the side-chain of E166. S139 is in steric contact with positions 2 and 299 and is close to position 166. Many mutations at both position 1 and 139 (e.g, S1E, S1D, S139L and S139Q) show high DR scores for both inhibitors, consistent with similar protrusions from the substrate envelope closest to these residues. Thus, S1H and S139P may reshape the active site in a highly specific manner to preferentially disrupt interactions with nirmatrelvir. The strongest ensitrelvir-specific apparent resistance mutations including C44G, M49L, and P52L are all located close to unique protrusions of ensitrelvir from the substrate envelope in the S2 and S1’ pockets (Figure 6A). These results indicate that protrusions from the substrate envelope generally lead to resistance potential, though the specific drug resistance mutations themselves are challenging to predict from the substrate envelope alone.
Table 2.
Mutations with largest differences in drug resistance score between nirmatrelvir and ensitrelvir
| Mutation | nirmatrelvir | DR score | difference |
|---|---|---|---|
| ensitrelvir | |||
|
| |||
| C44N | 0.8 | 1.9 | 1.1 |
| M49L | 1.1 | 2.1 | 1.0 |
| T25E | 1.1 | 2.0 | 0.9 |
| C44G | 0.9 | 1.8 | 0.9 |
| P52L | 1.0 | 1.8 | 0.8 |
| P52N | 0.9 | 1.8 | 0.9 |
| S139P | 1.8 | 1.0 | 0.8 |
| Y54W | 0.9 | 1.7 | 0.8 |
| C44M | 0.9 | 1.7 | 0.8 |
| T25H | 1.1 | 1.8 | 0.7 |
| S1H | 1.7 | 1.0 | 0.7 |
| C44I | 1.0 | 1.7 | 0.7 |
Location of all apparent drug resistance mutations provide mechanistic insights
To explore potential mechanisms of drug resistance, we mapped all apparent resistance mutations to structure (Figure 6B and C). Apparent DR mutations were identified at 63 different positions in Mpro (Table S2). However, the majority (190 of 319) of the apparent DR mutations occurred at 14 hot-spot positions (Table 3). The mutations driving the largest effects (e.g., Table 1) are almost all at these hot-spot positions. The main exceptions are H172Y (DR score of 1.9 for ensitrelvir) and M49L (DR score of 2.1 for ensitrelvir). Both of these positions were on the fringe of the cutoff we used for hot-spots (6 apparent DR mutations). At position 172 the only tolerated mutations that result in enzymatic function in our assay are aromatic amino acids, and indeed H172Y and H172F were apparent DR mutations for both inhibitors. At M49, three mutations (L, I, H) caused apparent DR for ensitrelvir. Position 49 is located close to unique protrusions of ensitrelvir from the substrate envelope and there were no apparent DR mutations for nirmatrelvir at this position. Upon examination of the structure of Mpro, all of the hot-spot positions were located at or adjacent (via bridging contacts with one reside) to the active site and/or the dimerization interface (Table 3).
Table 3.
Positions with the most apparent drug resistance mutations
| Position | Apparent DR mutations | Location | |
|---|---|---|---|
| Nirmatrelvir | Ensitrelvir | ||
|
| |||
| G2 | 17 | 17 | Dimer interface adjacent to active site |
| S139 | 12 | 8 | Dimer interface adjacent to active site |
| P168 | 10 | 8 | Active site and dimer interface |
| N214 | 9 | 8 | Adjacent to dimer interface |
| E166 | 8 | 8 | Active site and dimer interface |
| P52 | 0 | 15 | Adjacent to active site |
| R4 | 5 | 7 | Dimer interface |
| A116 | 5 | 7 | Adjacent to active site and dimer interface |
| S1 | 5 | 4 | Dimer interface adjacent to active site |
| T25 | 0 | 9 | Active site |
| Q299 | 4 | 4 | Dimer interface |
| C300 | 3 | 5 | Dimer interface |
| T26 | 3 | 3 | Active site |
| L141 | 2 | 4 | Dimer interface adjacent to active site |
These observations suggest that the dynamics and/or structure of the active site is coupled to the structure of the dimer interface. The majority of the indirect resistance hot-spots for both inhibitors including positions 1, 2, 4, 214, 298 and 299 contact one another and span from one active site across the dimer interface to the second active-site (Figure 6C). In our previous functional scan of Mpro without inhibitors 35 we noted a hypersensitive amino acid communication network spanning between active sites that we proposed modulated the shape and/or dynamics of the active sites. The resistance hot-spot positions provide further evidence that dimerization can strongly influence the physical properties of the active site. Of note, dimerization of Mpro is essential for its catalytic activity 48–49. In addition, mutations at the substrate binding site including E166A 50 and S147A 51 weaken Mpro dimerization, consistent with the shape of the active site being linked to dimerization. Structurally, the two Mpro subunits are oriented perpendicular to each other with an extensive interface formed primarily between domain II of one monomer and the N-terminal residues (N-finger) of the other monomer 52. Direct interaction between the N-finger from one subunit and E166 at the active site in the other subunit have been shown to strongly impact Mpro activity 50. Our comprehensive drug resistance study demonstrates that changes in a network of amino acids that bridge between the active sites can disrupt binding to both nirmatrelvir and ensitrelvir.
Functional cost of apparent drug resistance mutations and implications for evolution
Both Mpro function and affinity for inhibitor may contribute to the evolution of drug resistance in circulating viral populations. Mpro variants with wild-type-like function that strongly disrupt inhibitor binding pose the greatest chance of increasing to high frequency and causing the most potential harm to human health. We examined the relationship between DR score and functional score (Figure 7). As DR scores increase, there is a clear trend towards lower functional scores. Mutations that cause the strongest disruption of drug binding tend to also disrupt the processing of substrates. Indeed, only one amino acid change accessible by a single nucleotide change (P168R for nirmatrelvir) led to a DR score greater than 1.8 and a functional score equal to or greater than wild-type. The rarity of this type of mutation increases the chance of evolving resistance through alternative pathways that require multiple mutations as has been observed in SARS-CoV-2 passaging studies 19. This is similar to what has been observed for the evolution of drug resistance in HIV to protease inhibitors. Primary resistance mutations that disrupt inhibitor binding to HIV protease typically possess reduced enzyme activity. Continued drug pressure results in the emergence of compensatory secondary mutations that rescue these enzymatic defects 28, 53–54.
Figure 7. Functional cost of apparent drug resistance mutations.
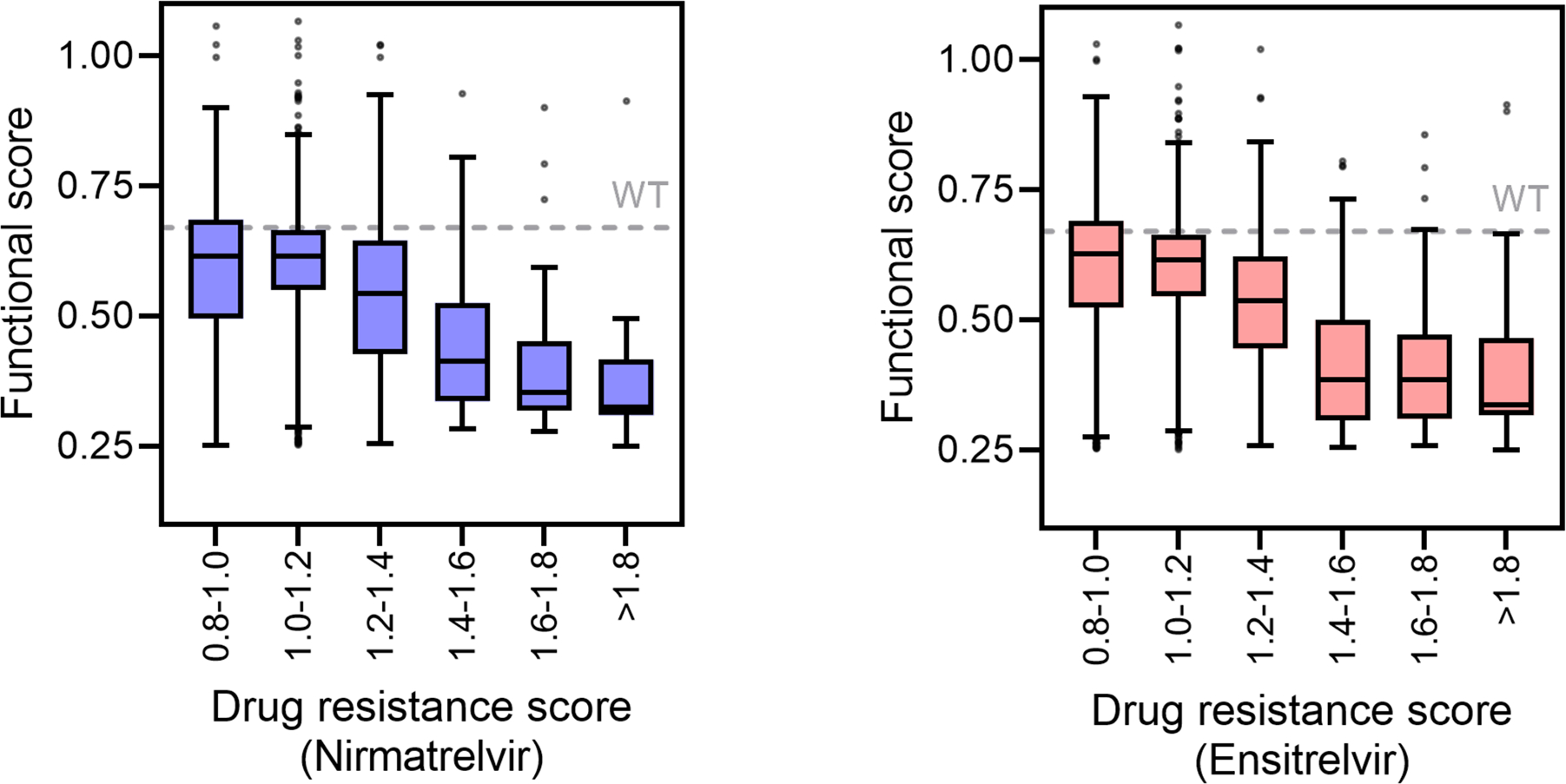
Mutants with increased DR scores for nirmatrelvir (left panel) or ensitrelvir (right panel) tend to have reduced functional scores. The box and whisker plots are plotted using the Tukey method. The box contains the 25th to 75th percentiles of the dataset with the center line denoting the median value.
In our data, we identified a large group of mutations that increase the enzyme activity of Mpro (Table S1). The large decreases in function of variants with the strongest DR scores (Figure 7) should select for compensatory mutations with large impacts on function. For example, T21I and L50F were observed multiple times in viral selection experiments with nirmatrelvir 19–20, 24 and both of these variants exhibit strong increases in functional score (0.92 and 0.89 for T21I and L50F respectively compared to 0.67 for wild-type). Because enzyme function can depend on substrate, we are pursuing analyses of increased activity mutants using substrates directly relevant to viral infections. A thorough analysis of activating mutations is beyond the scope of this work and will be the topic of future efforts.
Survey of drug resistance in natural isolates
Our results as well as those from passaging studies indicate that clinical use of drugs targeting Mpro will select for multiple different mutational pathways. Therefore, surveillance efforts for detecting early steps in drug resistance evolution will be more powerful if they integrate over all likely pathways to resistance. Our comprehensive survey of mutations that can cause apparent drug resistance provides a promising approach to this issue. We surveyed for all variants that we identify as likely to contribute to drug resistance based on DR score and accessibility by single nucleotide mutations. We also assessed the frequency of these mutations in SARS-CoV-2 isolates obtained in the U.S. both before and after approval of Paxlovid (data not shown). These initial analyses indicated that resistance in circulating virus populations is currently low and/or undetectable. The reproducible measurements of drug resistance potential described here provide a valuable tool for future efforts to assess resistance in data from ongoing sequencing efforts. With this motivation, we are currently working towards developing publicly available computational tools to survey the frequency of variants with either strong DR scores and/or high functional scores as a function of geographic location and time relative to clinical use of Mpro inhibitors.
Caveats and sources of uncertainty
Simplified readouts of the impacts of mutations such as those presented in this work are not perfect mimics of impacts in viruses. For example, we analyzed Mpro mutations expressed in yeast, which provides a distinct environment including distinct chaperones compared to human hosts. While some proteins are highly sensitive to chaperones for folding, Mpro can be readily expressed and folded in bacteria 52. In addition, Mpro is active in purified form indicating that it is not highly sensitive to chaperones or other cellular factors for function. We used yeast growth as a readout of Mpro function that we assume is caused by cleavage of endogenous yeast proteins that are not directly related to viral infection. Because our analyses of DR score are determined by changes in function with and without drug, the impact of using non-viral substrates is mitigated as inhibitors should impact all substrates similarly. Indeed, independent measures of IC50’s of Mpro variants in purified form correlate with our DR scores (Figure S6) 21.
While it is clear that Mpro function is required for SARS-CoV-2 infectivity, the quantitative relationship between function and fitness has not been accurately assessed. Thus, we do not know how viral fitness is determined based on the activity of Mpro nor inhibitor concentration and affinity. Because there are numerous viral cut-sites, developing an accurate model of relationships between fitness and Mpro function is complex. In the absence of an accurate quantitative model, we can still make qualitative assessments. For example, there is strong evidence that Mpro mutants with very low functional scores are selected against in viral populations 35 and that mutations that reduce binding affinity for inhibitors increase the ability of virus to expand in the presence of drugs 19. Thus, the evolution of Mpro variants with wild-type-like function and decreased affinity for inhibitors clearly represents an undesirable outcome – a virus that would be both highly drug resistant and highly fit in the absence of drug. For viral maturation, Mpro must cut itself out of the viral polyprotein such that sequences at the N- and C-terminus of Mpro may have special relevance in the context of viral replication that are not captured in our screen. Therefore, the viral relevance of DR scores for Mpro mutations near the termini should be interpreted with caution.
Our approach describes point mutations only and therefore does not provide information on the potential interdependence of mutations at multiple positions in the Mpro protein. Our observations and those from others 19–20, 24 indicate that the natural evolution of high levels of drug resistance is likely to involve multiple mutations including primary mutations that strongly disrupt drug binding (but come with functional costs) and compensatory mutations that rescue function. Our data provides a reliable view of mutations that disrupt drug binding and a first pass at mutations that increase function in the wild-type Mpro genetic background. In considering multiple mutations, independent or near-independent effects of mutations can be a reasonable first approximation 55, but epistasis or mutational dependencies are also a common feature of protein evolution 56. In future work, it will be important to evaluate the contributions of epistasis to Mpro evolution.
Conclusions
Many mutations in Mpro can cause cross-resistance to both nirmatrelvir and ensitrelvir, providing strong motivation for the development of additional therapeutics to combat SARS-CoV-2 infections. Mutations that can cause cross-resistance were predominantly located at or near E166. Both nirmatrelvir and ensitrelvir protrude slightly from the substrate envelope to make hydrogen bonds to either the side-chain or main-chain of E166. The close contact of nirmatrelvir and ensitrelvir with E166 appear to make them susceptible to similar resistance mutations. From the perspective of resistance potential, these findings provide motivation for developing inhibitors that do not hydrogen bond with E166 and bind within the substrate envelope and/or inhibitors that target other sites or other proteins as has been successful in treatments for HIV 57.
Mutations with the largest DR scores tend to reduce the enzymatic activity of Mpro such that drug pressure is likely to select for a combination of primary resistance mutations that reduce function together with compensatory mutations that rescue activity. Because many mutations cause either drug resistance or increased function, there are a wide array of variants likely to evolve in response to drug pressure. The comprehensive identification of resistance mutations of interest provides for sensitive and thorough surveillance in the early stages of evolution of drug resistance in Mpro. Variants with strong drug resistance together with wild-type-like enzymatic function provide the greatest potential to expand to high prevalence with significant impacts to public health. This pattern of resistance evolution is reminiscent of recent observations in influenza A virus 58, where selective pressure against oseltamivir caused the evolution of a primary neuraminidase mutation that disrupted inhibitor binding together with secondary mutations that rescued function. The resulting fit and resistant neuraminidase variant rose in frequency until it was dominant in viral isolates in 2008 59. The frequency of resistant variants of Mpro in SARS-CoV-2 isolates may be used as a barometer for the likelihood of widespread resistance evolution.
Methods
Generation of mutant libraries and barcode association
The SARS-CoV-2 single site variant library was synthesized by Twist Biosciences (twistbioscience.com) with each amino acid position modified to all 19 amino acid positions plus a stop codon using the preferred yeast codon for each substitution. The library was fused to a Ubiquitin gene, combined via Gibson Assembly with a linearized p416LexA destination vector, and barcoded with a randomized 18 bp nucleotide sequence as described 35. The p416 LexA destination vector contains a β-estradiol-activated LexA-ER-B112 transcription factor 60.
Bulk competition experiment of Mpro libraries in presence of inhibitors
The barcoded plasmid library was combined with a plasmid containing wild-type Mpro linked to approximately 150 unique barcodes 35 and was transformed using the large-scale lithium acetate procedure 61 into CMB855 cells (Mat a ura3 leu2 his3 lys2 snq2::kanMX pdr5::kanMX pdr1::nat1 yap1::nat1). Sufficient transformation reactions were performed to attain 2 million independent yeast transformants representing a 20-fold sampling of the average barcode. Following a 6-hour recovery in YPAD, transformed cells were washed three times in SD-U media (synthetic dextrose media lacking uracil to select for the plasmid library) to remove extracellular DNA and grown in 1 L SD-U at 30°C for 40 hours with repeated dilution to maintain the cells in log phase of growth (OD600 = 0.05 – 1) and to expand the library. The library was diluted to early log phase in SD-U, grown for 3 hours, and a 0-hour time point sample of 108 cells was collected by centrifugation and cell pellets were stored at −80°C. Cells were subsequently split into flasks containing 10 mL each, and either 20 μM nirmatrelvir (from a 50 mM stock in 100% DMSO), 10 uM ensitrelvir (from a 50 mM stock in 100% DMSO) or 2 μL of 100% DMSO was added to each flask. Additionally, 0.005% SDS was added to permeabilize the cell membrane in order to increase uptake of the drug. We performed three technical replicates with each inhibitor and the DMSO control. For replicates, we transformed the library into yeast cells in bulk and then separated the culture into three flasks and performed the growth competitions and sequence analyses independently. 2 μM β-estradiol (from a 10 mM stock in 95% ethanol) was then added to activate Mpro expression. Cultures were grown with shaking at 180 rpm for 16 hours with dilution after 8 hours (into media containing the appropriate concentrations of β-estradiol, nirmatrelvir, ensitrelvir, DMSO, and SDS). 16-hour samples were collected by centrifugation and cell pellets were stored at −80°C.
Determination of functional scores
DNA was isolated, amplified and sequenced as described 35 with minor modifications. Barcodes were amplified with 22 cycles of PCR using Phusion polymerase (NEB) with primers that add Illumina adaptor sequences and a 6 bp identifier sequence to distinguish the various drug conditions and time points. PCR products were purified two times over silica columns (Zymo research) and DNA concentration was quantified using the KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems) on a BioRad CFX Machine. PCR samples were combined and sequenced on an Illumina NextSeq instrument using a NextSeq 500/550 High Output Kit v.2.5 (75 cycles). The Illumina barcode reads were calculated using custom scripts that have been deposited on Github (https://github.com/JuliaFlynn/BolonLab). Illumina sequences were filtered for reads with a Phred score >10 and strict matching to the expected template and identifier sequence. Filtered reads were parsed based on their identifier sequences. For each condition/time point each unique N18 barcode read was counted. Using the variant-barcode association table that was generated by PacBio sequencing 35 the frequency of each mutant in each condition/time point was tabulated. Functional scores were determined by linear fits to the change in mutant abundance relative to total number of stop mutation reads using the following equation.
Functional scores were normalized to stop reads because stop mutations abolish Mpro activity and restore normal yeast growth. Stop codon reads for amino acids 300–306 were excluded because Mpro mutants containing stops at these positions remain functional 35. Example data and calculations for wild-type and stop mutants are shown in Table S4. All figures in this paper use the average functional score between the three replicates. Standard deviation was used to represent experimental variation between replicates. DR scores were calculated as the ratio of the average functional score of each mutant in the absence of the inhibitor to that in the presence of the inhibitor, compared to the same ratio of wild-type Mpro. The SDs between the functional scores in the three replicates with and without drug were propagated to assess statistical significance and calculate p-values for the DR scores.
Purification of Mpro
His6-SUMO-SARS-CoV-2 Mpro (WT) was cloned into a pETite vector 43. M49L and T25E mutations were created by site specific mutagenesis. The vectors were transformed into Hi-Control BL21(DE3) E. coli cells and the Mpro proteins were expressed and purified as previously described 43 with minor modifications. An overnight culture was used to inoculate 1 L of Terrific Broth (TB) medium containing 50 μg/ml of kanamycin at 37°C. Protein expression was initiated at OD600 = 1.5 with 0.5 mM IPTG, the temperature was reduced to 19°C, and cells were grown overnight. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris pH 8.0, 400 mM NaCl, 1mM TCEP) prior to lysis by two passes through a cell disrupter at 15,400 psi. The lysed cell suspension was clarified by centrifugation at 19,000 rpm for 30 min at 4°C and the supernatant was incubated for two hours with Co-NTA resin that had been pre-equilibrated with lysis buffer. The following steps were all performed at room temperature due to a proclivity for the protein to precipitate at 4°C. The resin was loaded onto a column and was washed with 20 column volumes of lysis buffer containing 10 mM imidazole. Mpro was eluted with lysis buffer containing 500 mM Imidazole. The SUMO tag was cleaved by addition of Ulp1 to the eluted protein while dialyzing into 2L of lysis buffer in a 10,000 MWCO dialysis cassette overnight. The SUMO protease and tag were subsequently removed by CoNTA resin. Protein was concentrated to 2.5 ml and run over a Sephadex G-25 desalting column (Cytiva) pre-equilibrated with 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP. Proteins were frozen in liquid nitrogen and stored at −80°C.
Determination of Michaels-Menten (Km) constant
To measure the Km of Mpro mutants, 100 nM enzyme was added to a series of 0–125 μM FRET substrate (Dabcyl-KTSAVLQSGFRKME-Edans (LifeTein)) in assay buffer (50 mM Tris pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT and 4% DMSO). The proteolytic reaction was monitored using a Perkin Elmer EnVision plate reader (355 nm excitation, 460 nm emission). Three replicates were performed with each mutant. The absorbance of the FRET substrate at the highest concentration (125 uM) used in these assays was less than 0.1, and so interference due to the inner filter effect was assumed to be minimal. The initial velocity was plotted against FRET substrate concentrations and were fit using GraphPad Prism 9 to the Michaelis-Menten equation.
Enzyme inhibition assays
For Ki measurements, 25 nM mutant Mpro enzyme was pre-incubated at 30°C with increasing concentrations of ensitrelvir or nirmatrelvir for 45 min in assay buffer. The proteolytic reaction was initiated with a final concentration of 10 μM protease FRET substrate (LifeTein) and monitored using a Perkin Elmer Envision plate reader. Three replicates were performed for each mutant and each inhibitor. The reaction was monitored for 1 hour and the initial velocity was calculated by linear regression. The Ki was calculated by plotting the inhibitor concentration versus the initial velocity and fit to the Morrison equation (tight binding, see below) using GraphPad Prism 9 software.
van der Waals calculations
All crystal structures were obtained from the Protein DataBank. Prior to van der Waals calculations, Mpro-inhibitor complexes were prepared using the Schrodinger Protein Preparation Wizard. Hydrogen atoms were added, protonation states determined using PropKa, and the hydrogen bonding network was optimized. A restrained minimization was performed using the OPLS2005 force field within an RMSD of 0.3 Å. All crystallographic waters were retained during structure minimization. Interaction energies between the inhibitor and protease were estimated using a simplified Lennard-Jones potential using a custom Python script, as previously described 62.
PCA analysis
Prior to analysis, all vdW interactions were standardized by calculating the Z score (Equation 1); the sample mean was subtracted from each interaction and divided by the standard deviation. The Z scores were used as input for the scikit-learn implementation of principal components analysis. Principal components accounting for 65% of the variance were used to reproject the vdW data, and we used k-means clustering to separate inhibitors into three clusters.
Structure analysis
The inhibitor co-crystal structure (PDB 7vu6 for ensitrelvir 15 and PDB 7RFS for nirmatrelvir 3 were aligned onto the co-crystal structure of SARS-CoV-2 Mpro with the Nsp4/5 substrate peptide (PDB 7T70) using the carbon alpha atoms of residues 41, 144, 145, 163 and 164 43. This allows for proper visualization of each inhibitor within the substrate envelope. Because 7vu6 lacks the two N-terminal residues S1 and G2, these were modeled in using the conformations seen in PDB 6xhm as a reference 63. Figures were generated using Matplotlib 64, PyMOL, and GraphPad Prism version 9.3.1.
Supplementary Material
Footnotes
Supporting Information
- Additional supplemental figures and tables (PDF)
- Sequencing counts, functional scores and drug resistance scores for all Mpro variants in the absence and presence of inhibitors (Table S1) (XLSX)
- DR scores of all nirmatrelvir and ensitrelvir apparent drug resistance mutations (Table S2) (XLSX)
Data availability
Next generation sequencing data has been deposited to the NCBI short read archive (PRJNA933538).
References
- 1.Polack FP; Thomas SJ; Kitchin N; Absalon J; Gurtman A; Lockhart S; Perez JL; Perez Marc G; Moreira ED; Zerbini C; Bailey R; Swanson KA; Roychoudhury S; Koury K; Li P; Kalina WV; Cooper D; Frenck RW Jr.; Hammitt LL; Tureci O; Nell H; Schaefer A; Unal S; Tresnan DB; Mather S; Dormitzer PR; Sahin U; Jansen KU; Gruber WC; Group CCT, Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 2020, 383 (27), 2603–2615. DOI: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen P; Nirula A; Heller B; Gottlieb RL; Boscia J; Morris J; Huhn G; Cardona J; Mocherla B; Stosor V; Shawa I; Adams AC; Van Naarden J; Custer KL; Shen L; Durante M; Oakley G; Schade AE; Sabo J; Patel DR; Klekotka P; Skovronsky DM; Investigators B −. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med 2021, 384 (3), 229–237. DOI: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owen DR; Allerton CMN; Anderson AS; Aschenbrenner L; Avery M; Berritt S; Boras B; Cardin RD; Carlo A; Coffman KJ; Dantonio A; Di L; Eng H; Ferre R; Gajiwala KS; Gibson SA; Greasley SE; Hurst BL; Kadar EP; Kalgutkar AS; Lee JC; Lee J; Liu W; Mason SW; Noell S; Novak JJ; Obach RS; Ogilvie K; Patel NC; Pettersson M; Rai DK; Reese MR; Sammons MF; Sathish JG; Singh RSP; Steppan CM; Stewart AE; Tuttle JB; Updyke L; Verhoest PR; Wei L; Yang Q; Zhu Y, An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374 (6575), 1586–1593. DOI: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 4.Beigel JH; Tomashek KM; Dodd LE; Mehta AK; Zingman BS; Kalil AC; Hohmann E; Chu HY; Luetkemeyer A; Kline S; Lopez de Castilla D; Finberg RW; Dierberg K; Tapson V; Hsieh L; Patterson TF; Paredes R; Sweeney DA; Short WR; Touloumi G; Lye DC; Ohmagari N; Oh MD; Ruiz-Palacios GM; Benfield T; Fatkenheuer G; Kortepeter MG; Atmar RL; Creech CB; Lundgren J; Babiker AG; Pett S; Neaton JD; Burgess TH; Bonnett T; Green M; Makowski M; Osinusi A; Nayak S; Lane HC; Members A-SG, Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med 2020, 383 (19), 1813–1826. DOI: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jayk Bernal A; Gomes da Silva MM; Musungaie DB; Kovalchuk E; Gonzalez A; Delos Reyes V; Martin-Quiros A; Caraco Y; Williams-Diaz A; Brown ML; Du J; Pedley A; Assaid C; Strizki J; Grobler JA; Shamsuddin HH; Tipping R; Wan H; Paschke A; Butterton JR; Johnson MG; De Anda C; Group MO-OS, Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med 2022, 386 (6), 509–520. DOI: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P; Nair MS; Liu L; Iketani S; Luo Y; Guo Y; Wang M; Yu J; Zhang B; Kwong PD; Graham BS; Mascola JR; Chang JY; Yin MT; Sobieszczyk M; Kyratsous CA; Shapiro L; Sheng Z; Huang Y; Ho DD, Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593 (7857), 130–135. DOI: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- 7.Starr TN; Greaney AJ; Addetia A; Hannon WW; Choudhary MC; Dingens AS; Li JZ; Bloom JD, Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371 (6531), 850–854. DOI: 10.1126/science.abf9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L; Iketani S; Guo Y; Chan JF; Wang M; Liu L; Luo Y; Chu H; Huang Y; Nair MS; Yu J; Chik KK; Yuen TT; Yoon C; To KK; Chen H; Yin MT; Sobieszczyk ME; Huang Y; Wang HH; Sheng Z; Yuen KY; Ho DD, Striking antibody evasion manifested by the Omicron variant of SARS-CoV-2. Nature 2022, 602 (7898), 676–681. DOI: 10.1038/s41586-021-04388-0. [DOI] [PubMed] [Google Scholar]
- 9.Housman G; Byler S; Heerboth S; Lapinska K; Longacre M; Snyder N; Sarkar S, Drug resistance in cancer: an overview. Cancers (Basel) 2014, 6 (3), 1769–92. DOI: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurt Yilmaz N; Schiffer CA, Introduction: Drug Resistance. Chem Rev 2021, 121 (6), 3235–3237. DOI: 10.1021/acs.chemrev.1c00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herold J; Raabe T; Schelle-Prinz B; Siddell SG, Nucleotide sequence of the human coronavirus 229E RNA polymerase locus. Virology 1993, 195 (2), 680–91. DOI: 10.1006/viro.1993.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim KP; Ng LF; Liu DX, Identification of a novel cleavage activity of the first papain-like proteinase domain encoded by open reading frame 1a of the coronavirus Avian infectious bronchitis virus and characterization of the cleavage products. J Virol 2000, 74 (4), 1674–85. DOI: 10.1128/jvi.74.4.1674-1685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziebuhr J; Herold J; Siddell SG, Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J Virol 1995, 69 (7), 4331–8. DOI: 10.1128/JVI.69.7.4331-4338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin Z; Du X; Xu Y; Deng Y; Liu M; Zhao Y; Zhang B; Li X; Zhang L; Peng C; Duan Y; Yu J; Wang L; Yang K; Liu F; Jiang R; Yang X; You T; Liu X; Yang X; Bai F; Liu H; Liu X; Guddat LW; Xu W; Xiao G; Qin C; Shi Z; Jiang H; Rao Z; Yang H, Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582 (7811), 289–293. DOI: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 15.Tyndall JDA, S-217622, a 3CL Protease Inhibitor and Clinical Candidate for SARS-CoV-2. J Med Chem 2022, 65 (9), 6496–6498. DOI: 10.1021/acs.jmedchem.2c00624. [DOI] [PubMed] [Google Scholar]
- 16.Cully M, A tale of two antiviral targets - and the COVID-19 drugs that bind them. Nat Rev Drug Discov 2022, 21 (1), 3–5. DOI: 10.1038/d41573-021-00202-8. [DOI] [PubMed] [Google Scholar]
- 17.Lamb YN, Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82 (5), 585–591. DOI: 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unoh Y; Uehara S; Nakahara K; Nobori H; Yamatsu Y; Yamamoto S; Maruyama Y; Taoda Y; Kasamatsu K; Suto T; Kouki K; Nakahashi A; Kawashima S; Sanaki T; Toba S; Uemura K; Mizutare T; Ando S; Sasaki M; Orba Y; Sawa H; Sato A; Sato T; Kato T; Tachibana Y, Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J Med Chem 2022, 65 (9), 6499–6512. DOI: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iketani S; Mohri H; Culbertson B; Hong SJ; Duan Y; Luck MI; Annavajhala MK; Guo Y; Sheng Z; Uhlemann AC; Goff SP; Sabo Y; Yang H; Chavez A; Ho DD, Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 2023, 613 (7944), 558–564. DOI: 10.1038/s41586-022-05514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jochmans D; Liu C; Donckers K; Stoycheva A; Boland S; Stevens SK; De Vita C; Vanmechelen B; Maes P; Trueb B; Ebert N; Thiel V; De Jonghe S; Vangeel L; Bardiot D; Jekle A; Blatt LM; Beigelman L; Symons JA; Raboisson P; Chaltin P; Marchand A; Neyts J; Deval J; Vandyck K, The Substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro Are Selected by a Protease Inhibitor In Vitro and Confer Resistance To Nirmatrelvir. mBio 2023, e0281522. DOI: 10.1128/mbio.02815-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y; Lewandowski EM; Tan H; Zhang X; Morgan RT; Zhang X; Jacobs LMC; Butler SG; Gongora MV; Choy J; Deng X; Chen Y; Wang J, Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. bioRxiv 2022. DOI: 10.1101/2022.06.28.497978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moghadasi SA; Heilmann E; Khalil A; Nnabuife C; Kearns F; Ye C; Moraes SN; Costacurta F; Esler M; Aihara H; von Laer D; Martinez-Sobrido L; Palzkill T; Amaro RE; Harris R, Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. bioRxiv 2022. DOI: 10.1101/2022.08.07.503099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Oliveira V; Ibrahim M; Sun X; Hilgenfeld R; Shen J, H172Y mutation perturbs the S1 pocket and nirmatrelvir binding of SARS-CoV-2 main protease through a nonnative hydrogen bond. Res Sq 2022. DOI: 10.21203/rs.3.rs-1915291/v1. [DOI] [Google Scholar]
- 24.Zhou Y; Gammeltoft KA; Ryberg LA; Pham LV; Tjornelund HD; Binderup A; Duarte Hernandez CR; Fernandez-Antunez C; Offersgaard A; Fahnoe U; Peters GHJ; Ramirez S; Bukh J; Gottwein JM, Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci Adv 2022, 8 (51), eadd7197. DOI: 10.1126/sciadv.add7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heilmann E; Costacurta F; Moghadasi SA; Ye C; Pavan M; Bassani D; Volland A; Ascher C; Weiss AKH; Bante D; Harris RS; Moro S; Rupp B; Martinez-Sobrido L; von Laer D, SARS-CoV-2 3CL(pro) mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Sci Transl Med 2023, 15 (678), eabq7360. DOI: 10.1126/scitranslmed.abq7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali A; Bandaranayake RM; Cai Y; King NM; Kolli M; Mittal S; Murzycki JF; Nalam MNL; Nalivaika EA; Ozen A; Prabu-Jeyabalan MM; Thayer K; Schiffer CA, Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2 (11), 2509–2535. DOI: 10.3390/v2112509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shafer RW; Schapiro JM, HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev 2008, 10 (2), 67–84. [PMC free article] [PubMed] [Google Scholar]
- 28.Kozisek M; Bray J; Rezacova P; Saskova K; Brynda J; Pokorna J; Mammano F; Rulisek L; Konvalinka J, Molecular analysis of the HIV-1 resistance development: enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J Mol Biol 2007, 374 (4), 1005–16. DOI: 10.1016/j.jmb.2007.09.083. [DOI] [PubMed] [Google Scholar]
- 29.Ragland DA; Whitfield TW; Lee SK; Swanstrom R; Zeldovich KB; Kurt-Yilmaz N; Schiffer CA, Elucidating the Interdependence of Drug Resistance from Combinations of Mutations. J Chem Theory Comput 2017, 13 (11), 5671–5682. DOI: 10.1021/acs.jctc.7b00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider-Nachum G; Flynn J; Mavor D; Schiffer CA; Bolon DNA, Analyses of HIV proteases variants at the threshold of viability reveals relationships between processing efficiency and fitness. Virus Evol 2021, 7 (2), veab103. DOI: 10.1093/ve/veab103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azam M; Latek RR; Daley GQ, Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 2003, 112 (6), 831–43. DOI: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 32.Kurt Yilmaz N; Swanstrom R; Schiffer CA, Improving Viral Protease Inhibitors to Counter Drug Resistance. Trends Microbiol 2016, 24 (7), 547–557. DOI: 10.1016/j.tim.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma L; Boucher JI; Paulsen J; Matuszewski S; Eide CA; Ou J; Eickelberg G; Press RD; Zhu LJ; Druker BJ; Branford S; Wolfe SA; Jensen JD; Schiffer CA; Green MR; Bolon DN, CRISPR-Cas9-mediated saturated mutagenesis screen predicts clinical drug resistance with improved accuracy. Proc Natl Acad Sci U S A 2017, 114 (44), 11751–11756. DOI: 10.1073/pnas.1708268114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagenaar TR; Ma L; Roscoe B; Park SM; Bolon DN; Green MR, Resistance to vemurafenib resulting from a novel mutation in the BRAFV600E kinase domain. Pigment Cell Melanoma Res 2014, 27 (1), 124–33. DOI: 10.1111/pcmr.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flynn JM; Samant N; Schneider-Nachum G; Bakan DT; Yilmaz NK; Schiffer CA; Moquin SA; Dovala D; Bolon DNA, Comprehensive fitness landscape of SARS-CoV-2 M(pro) reveals insights into viral resistance mechanisms. Elife 2022, 11. DOI: 10.7554/eLife.77433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou J; Lewandowski E; Hu Y; Lipinski A; Morgan R; Jacobs L; Zhang X; Bikowitz M; Langlais P; Tan H; Wang J; Chen Y; Choy J, A yeast-based system to study SARS-CoV-2 Mpro structure and to identify nirmatrelvir resistant mutations. Res Sq 2022. DOI: 10.21203/rs.3.rs-1942964/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinen T; Hamada K; Taguchi A; Asami Y; Shiomi K; Hayashi Y; Usui T, Multidrug Sensitive Yeast Strains, Useful Tools for Chemical Genetics. In The Yeast Role in Medical Applications, Abdulkhair WMH, Ed. IntechOpen: 2016. [Google Scholar]
- 38.Suzuki Y; St Onge RP; Mani R; King OD; Heilbut A; Labunskyy VM; Chen W; Pham L; Zhang LV; Tong AH; Nislow C; Giaever G; Gladyshev VN; Vidal M; Schow P; Lehar J; Roth FP, Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat Methods 2011, 8 (2), 159–64. DOI: 10.1038/nmeth.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers B; Decottignies A; Kolaczkowski M; Carvajal E; Balzi E; Goffeau A, The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J Mol Microbiol Biotechnol 2001, 3 (2), 207–14. [PubMed] [Google Scholar]
- 40.Toussi SS; Neutel JM; Navarro J; Preston RA; Shi H; Kavetska O; LaBadie RR; Binks M; Chan PLS; Demers N; Corrigan B; Damle B, Pharmacokinetics of Oral Nirmatrelvir/Ritonavir, a Protease Inhibitor for Treatment of COVID-19, in Subjects With Renal Impairment. Clin Pharmacol Ther 2022, 112 (4), 892–900. DOI: 10.1002/cpt.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kosuge M; Furusawa-Nishii E; Ito K; Saito Y; Ogasawara K, Point mutation bias in SARS-CoV-2 variants results in increased ability to stimulate inflammatory responses. Sci Rep 2020, 10 (1), 17766. DOI: 10.1038/s41598-020-74843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Administration, U. S. F. a. D., Emergency Use Authorization for Paxlovid [Fact Sheet for Healthcare Providers]. 2021.
- 43.Shaqra AM; Zvornicanin SN; Huang QYJ; Lockbaum GJ; Knapp M; Tandeske L; Bakan DT; Flynn J; Bolon DNA; Moquin S; Dovala D; Kurt Yilmaz N; Schiffer CA, Defining the substrate envelope of SARS-CoV-2 main protease to predict and avoid drug resistance. Nat Commun 2022, 13 (1), 3556. DOI: 10.1038/s41467-022-31210-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prabu-Jeyabalan M; Nalivaika E; Schiffer CA, Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002, 10 (3), 369–81. DOI: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- 45.Romano KP; Laine JM; Deveau LM; Cao H; Massi F; Schiffer CA, Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J Virol 2011, 85 (13), 6106–16. DOI: 10.1128/JVI.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki Y; Oishi K; Nakano H; Nagayama T, A strong correlation between the increase in number of proline residues and the rise in thermostability of five Bacillus oligo-1,6-glucosidase. Applied Microbiology and Biotechnology 1987, 26, 546–551. [Google Scholar]
- 47.Watanabe K; Masuda T; Ohashi H; Mihara H; Suzuki Y, Multiple proline substitutions cumulatively thermostabilize Bacillus cereus ATCC7064 oligo-1,6-glucosidase. Irrefragable proof supporting the proline rule. Eur J Biochem 1994, 226 (2), 277–83. DOI: 10.1111/j.1432-1033.1994.tb20051.x. [DOI] [PubMed] [Google Scholar]
- 48.Tan J; Verschueren KH; Anand K; Shen J; Yang M; Xu Y; Rao Z; Bigalke J; Heisen B; Mesters JR; Chen K; Shen X; Jiang H; Hilgenfeld R, pH-dependent conformational flexibility of the SARS-CoV main proteinase (M(pro)) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J Mol Biol 2005, 354 (1), 25–40. DOI: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi J; Sivaraman J; Song J, Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J Virol 2008, 82 (9), 4620–9. DOI: 10.1128/JVI.02680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng SC; Chang GG; Chou CY, Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys J 2010, 98 (7), 1327–36. DOI: 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrila J; Bacha U; Freire E, Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 2006, 45 (50), 14908–16. DOI: 10.1021/bi0616302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L; Lin D; Sun X; Curth U; Drosten C; Sauerhering L; Becker S; Rox K; Hilgenfeld R, Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368 (6489), 409–412. DOI: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu F; Boross PI; Wang YF; Tozser J; Louis JM; Harrison RW; Weber IT, Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J Mol Biol 2005, 354 (4), 789–800. DOI: 10.1016/j.jmb.2005.09.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schock HB; Garsky VM; Kuo LC, Mutational anatomy of an HIV-1 protease variant conferring cross-resistance to protease inhibitors in clinical trials. Compensatory modulations of binding and activity. J Biol Chem 1996, 271 (50), 31957–63. DOI: 10.1074/jbc.271.50.31957. [DOI] [PubMed] [Google Scholar]
- 55.Sternke M; Tripp KW; Barrick D, Consensus sequence design as a general strategy to create hyperstable, biologically active proteins. Proc Natl Acad Sci U S A 2019, 116 (23), 11275–11284. DOI: 10.1073/pnas.1816707116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pollock DD; Goldstein RA, Strong evidence for protein epistasis, weak evidence against it. Proc Natl Acad Sci U S A 2014, 111 (15), E1450. DOI: 10.1073/pnas.1401112111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shafer RW; Vuitton DA, Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed Pharmacother 1999, 53 (2), 73–86. DOI: 10.1016/s0753-3322(99)80063-8. [DOI] [PubMed] [Google Scholar]
- 58.Abed Y; Pizzorno A; Bouhy X; Boivin G, Role of permissive neuraminidase mutations in influenza A/Brisbane/59/2007-like (H1N1) viruses. PLoS Pathog 2011, 7 (12), e1002431. DOI: 10.1371/journal.ppat.1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hurt AC; Hardie K; Wilson NJ; Deng YM; Osbourn M; Gehrig N; Kelso A, Community transmission of oseltamivir-resistant A(H1N1)pdm09 influenza. N Engl J Med 2011, 365 (26), 2541–2. DOI: 10.1056/NEJMc1111078. [DOI] [PubMed] [Google Scholar]
- 60.Ottoz DS; Rudolf F; Stelling J, Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res 2014, 42 (17), e130. DOI: 10.1093/nar/gku616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gietz RD; Schiestl RH, Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2007, 2 (1), 38–41. DOI: 10.1038/nprot.2007.15. [DOI] [PubMed] [Google Scholar]
- 62.Nalam MN; Ali A; Altman MD; Reddy GS; Chellappan S; Kairys V; Ozen A; Cao H; Gilson MK; Tidor B; Rana TM; Schiffer CA, Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol 2010, 84 (10), 5368–78. DOI: 10.1128/JVI.02531-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoffman RL; Kania RS; Brothers MA; Davies JF; Ferre RA; Gajiwala KS; He M; Hogan RJ; Kozminski K; Li LY; Lockner JW; Lou J; Marra MT; Mitchell LJ Jr.; Murray BW; Nieman JA; Noell S; Planken SP; Rowe T; Ryan K; Smith GJ 3rd; Solowiej JE; Steppan CM; Taggart B, Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J Med Chem 2020, 63 (21), 12725–12747. DOI: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunter JD, “Matplotlib: A 2D Graphics Environment”. Computing in Science and Engineering 2007, 9 (3), 90–95. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next generation sequencing data has been deposited to the NCBI short read archive (PRJNA933538).