

Abstract
Metabolic reprogramming is central to malignant transformation and cancer cell growth. How tumours use nutrients and the relative rates of reprogrammed pathways are areas of intense investigation. Tumour metabolism is determined by a complex and incompletely defined combination of factors intrinsic and extrinsic to cancer cells. This complexity increases the value of assessing cancer metabolism in disease-relevant microenvironments, including in patients with cancer. Stable-isotope tracing is an informative, versatile method for probing tumour metabolism in vivo. It has been used extensively in preclinical models of cancer and, with increasing frequency, in patients with cancer. In this Review, we describe approaches for using in vivo isotope tracing to define fuel preferences and pathway engagement in tumours, along with some of the principles that have emerged from this work. Stable-isotope infusions reported so far have revealed that in humans, tumours use a diverse set of nutrients to supply central metabolic pathways, including the tricarboxylic acid cycle and amino acid synthesis. Emerging data suggest that some activities detected by stable-isotope tracing correlate with poor clinical outcomes and may drive cancer progression. We also discuss current challenges in isotope tracing, including comparisons of in vivo and in vitro models, and opportunities for future discovery in tumour metabolism.
Introduction
Metabolic reprogramming is a hallmark of cancer and source of therapeutic targets1,2. Since Otto Warburg’s experiments describing aerobic glycolysis in tumours, it has been a goal of cancer research to identify pathways that support cancer progression. As techniques to analyse metabolism have improved in scope and sensitivity, it has become possible to characterize tumour metabolism in increasing detail.
This Review focuses on stable-isotope tracing to analyse tumour metabolism in vivo (Fig. 1a). The most abundant elemental constituents of biomolecules are hydrogen, carbon, nitrogen and oxygen. Each one has stable-isotopic forms with extra neutrons compared with the most abundant form; for example, 2H, 13C, 15N and 18O. Owing to their extra neutrons, molecules containing these stable isotopes can be readily distinguished from molecules lacking them. Tracing experiments use molecules chemically synthesized so that one or more positions contain a much higher fraction of the stable isotope than would occur naturally. For example, glucose can be synthesized so that all six carbon positions are essentially 100% 13C. Molecules synthesized in this manner are commonly said to be ‘labelled’ with the stable isotope. With the exception of some biochemical reactions being substantially slowed by replacement of 1H with 2H (ref. 3), heavy-labelled metabolites are processed similar to their unlabelled counterparts. Thus, tracking the fate of stable isotopes provides information about endogenous metabolism.
Fig. 1 |. Basic concepts of isotope labelling.
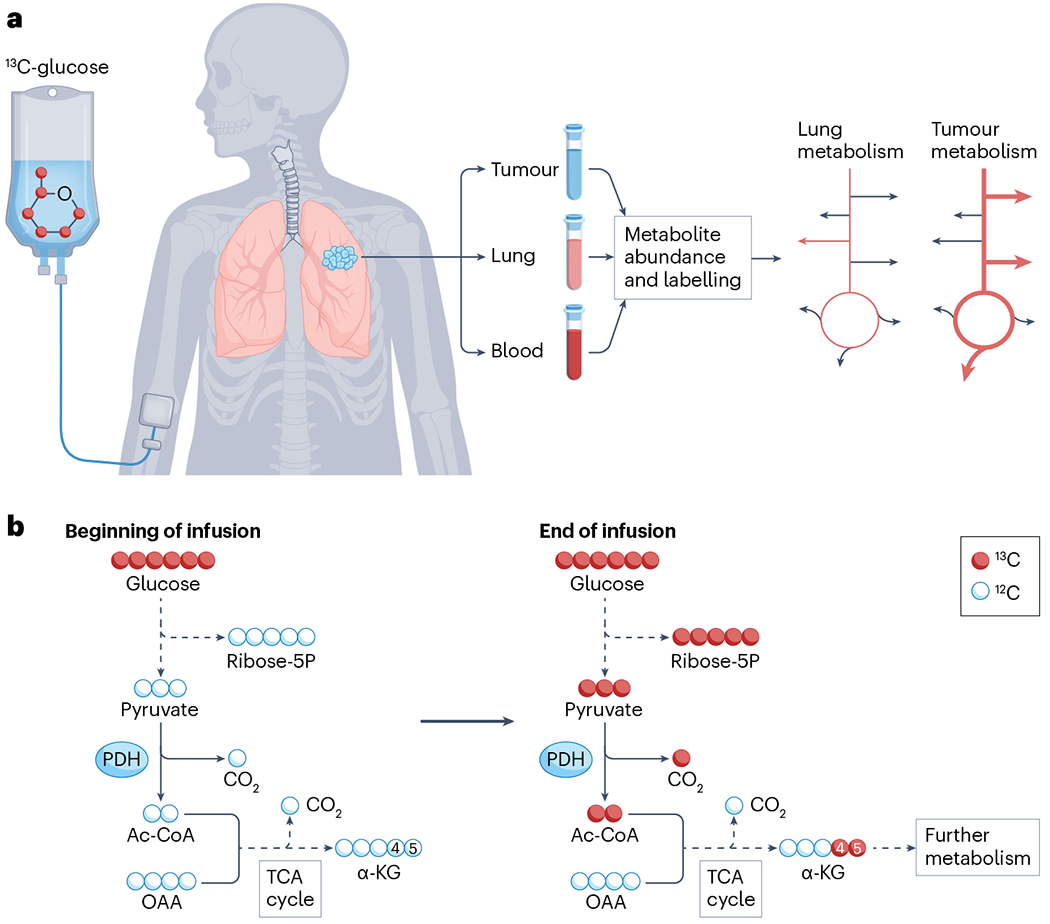
a, Workflow of stable-isotope tracing experiments in patients with cancer, using lung cancer as an example. The labelled tracer, in this case uniformly labelled [U-13C]glucose, is provided by feeding or, as shown here, by intravenous infusion. The tracer is then metabolized along with the endogenous unlabelled nutrient, spreading heavy atoms into downstream metabolites in a stereotyped way depending on the reactions that take place. Blood is sampled during the infusion, and the tumour and adjacent lung tissue are sampled immediately at resection. The duration of the infusion is variable, but most applications in human cancer have used infusions of at least 2 h. Metabolites are extracted and analysed for abundance and 13C labelling by mass spectrometry or NMR. Although mass spectrometry detects differences in atomic mass resulting from the presence of isotope labels, NMR detects the nuclear spin of certain isotopes. Labelling reports information about relative pathway utilization between the tumour mass and non-malignant lung tissue. The pathway illustration at the right indicates enhanced 13C labelling (heavier red arrows) in glycolysis, the tricarboxylic acid (TCA) cycle and other pathways supplied by glucose. b, Isotope labelling detail. At the beginning of the infusion, all metabolites other than the glucose tracer are unlabelled. Over time, metabolites in glycolysis, the pentose phosphate pathway and the TCA cycle become labelled in positions that depend on the labelling state of the precursor, in this case, [U-13C]glucose, and the pathways used in the tissue. For example, if pyruvate dehydrogenase (PDH) is active, pyruvate is converted to acetyl-CoA (Ac-CoA) and subsequently α-ketoglutarate (α-KG) is labelled in positions 4 and 5 on the first turn of the TCA cycle, and the label is distributed to other positions through further metabolism. If pyruvate carboxylase is also active, the oxaloacetate (OAA) pool becomes labelled and results in 13C at positions 1, 2 and 3 of α-KG on the first turn of the TCA cycle (not shown). Further metabolism in the TCA cycle leads to more complex labelling patterns downstream of both PDH and pyruvate carboxylase.
In stable-isotope tracing, labelled nutrients are introduced into a biological system and metabolism within the system transfers the label to downstream metabolites. The position and extent of metabolite labelling provide information about metabolic activity (Fig. 1b). Stable-isotope tracing reports pathway activities and is thus conceptually different from metabolomics, which reports metabolite abundance and does not require the addition of tracers (reviewed elsewhere4–6). Both techniques have become prominent in cancer research over the past 15 years.
Building on work in the 1920s by George de Hevesy with radioactive isotopes7, Schoenheimer and Rittenberg8 pioneered the systematic use of stable isotopes to study metabolism (Box 1). Stable isotopes do not undergo radioactive decay and can therefore be safely used in relatively large quantities. Stable-isotope tracing coupled to modern detection methods provides more precise information about the fate of the tracer than radioactive tracing. These benefits have resulted in stable isotopes largely displacing radioisotopes for tracer studies in mice and humans.
Box 1. Timeline of stable-isotope tracing to probe cancer metabolism.
Nearly 100 years after their discovery, stable isotopes remain highly informative tools to study metabolism in vivo. This timeline highlights historical benchmarks in the development of isotope tracing in vivo and technological advancements that aided the analysis of isotope tracing data, leading to the use of 13C to analyse cancer metabolism in patients, starting in 2009.
1913: Frederick Soddy coins the term ‘isotopes’, in which atoms can be identical chemically, but contain unique atomic weights
1919: Francis Aston discovers naturally occurring stable isotopes of neon161
1923: George Hevesy uses radioactive tracers to monitor lead metabolism in plants162 and later investigates metabolism in animals163,164
1929: 13C is discovered by Arthur King and Raymond Birge165
1932: Harold Urey discovers deuterium (2H)166
1935: Deuterated water (2H2O) is used to measure fatty acid synthesis in mice by Schoenheimer and Rittenberg8
1940s: Radioisotope studies begin to outpace stable isotopes, largely due to superior signal/noise ratios in detection
1943: Hevesy wins Nobel Prize for the use of isotopes as tracers
1947: Improved resolution on mass spectrometers enables better detection of stable isotopes167
1953: Development of quadrupole mass spectrometry further enhances the ability to measure stable isotopes168
1950s: Multiple investigators combine gas chromatography with mass spectrometry
1960s: A stable-isotope renaissance begins, with multiple groups using stable isotopes to study metabolism. Numerous studies in the 1960–1980s assess central metabolic pathways in humans
1987: Mathematical models are developed to capitalize on positional 13C analysis by NMR to assess tricarboxylic acid cycle metabolism in the heart13
1997: 2H positional labelling in glucose differentiates between gluconeogenesis and glycogenolysis169
2009: First use of [13C]glucose in patients with non-small-cell lung cancer19
2013: First use of hyperpolarized [1-13C]pyruvate in patients with cancer29
Within the basic workflow of a stable-isotope tracing experiment (Fig. 1a), isotope labelling is analysed by mass spectrometry or NMR. The primary advantage of NMR is that it provides positional information for isotope labels; this type of information is less reliably obtained with mass spectrometry, although increasingly attainable through tandem mass spectrometry9,10. NMR is non-destructive and can be performed in vivo through magnetic resonance spectroscopy (MRS), which in some cases can be integrated with MRI. Mass spectrometry has superior sensitivity and is readily combined with metabolomics. Improvements in both technologies have helped drive the expansion of stable-isotope applications11.
Classical work with stable isotopes allowed investigators to assess metabolism in isolated organs and to develop mathematical approaches to translate isotopic labelling into metabolic fluxes12–15. Stable-isotope administration to humans has been used since 1934, when Hevesy and Hofer used deuterium oxide to estimate the size of the whole-body water pool and rate of water elimination7. Landmark human studies with stable isotopes since then have studied diverse processes, including maintenance of blood glucose during fasting16; the role of endurance training on lactate metabolism17 and the effects of fatty liver disease on the hepatic tricarboxylic acid (TCA) cycle18.
Applying stable-isotope tracers to study cancer in humans is a relatively recent development (Box 1), but its use in cancer research has already influenced our understanding of metabolic reprogramming (Table 1). In vivo tracer studies provide information about which pathways and which nutrients are used in the native tumour microenvironment, which involves a metabolic milieu and complex set of cell–cell interactions that cannot be recapitulated in cell culture. Therefore, the insights that arise from in vivo tracer studies cannot be achieved from metabolomics or from analysis of cultured cells. This Review discusses methods to perform and interpret isotope labelling experiments in cancer, and discoveries emerging from these approaches since the first use of 13C tracers in humans with cancer was reported in 2009 (ref. 19). We also discuss the limitations of isotope tracing, open questions and priorities for future research.
Table 1 |.
Isotope tracing studies in patients with cancer
| Study | Cancer | Tracer(s) | Key conclusions |
|---|---|---|---|
| Fan et al., 2009 (ref. 19) | NSCLC | [U-13C]glucose | Labelling of glycolytic and TCA cycle intermediates in tumours exceeds labelling in lung Labelling patterns in TCA cycle intermediates are consistent with PC |
| Maher et al., 2012 (ref. 39) | Glioma, brain metastases | [U-13C]glucose | Glycolysis, TCA cycle turnover, anaplerosis and glutamine synthesis are active in these tumours in vivo Relatively low labelling of acetyl-CoA feeding the TCA cycle, suggesting that tumours oxidize other fuels in addition to glucose |
| Mashimo et al., 2014 (ref. 43) | Glioma, brain metastases | [1,2-13C]acetate | Blood acetate provides an acetyl-CoA source for the TCA cycle in human glioblastoma and NSCLC metastases to the brain |
| Sellers et al., 2016 (ref. 27) | NSCLC | [U-13C]glucose | Tumours express high levels of PC, and labelling provides strong evidence of enhanced PC contribution to the TCA cycle in tumours relative to adjacent lung |
| Hensley et al., 2016 (ref. 36) | NSCLC | [U-13C]glucose | Treatment-naive NSCLCs display extensive metabolic heterogeneity Regional metabolic heterogeneity exists within the same tumour Some metabolic characteristics correlate with regional perfusion assessed by MRI |
| Faubert et al., 2017 (ref. 33) | NSCLC | [U-13C]glucose, [U-13C]lactate | Lactate from the blood enters lung tumours and contributes to TCA cycle metabolism |
| Courtney et al., 2018 (ref. 37) | ccRCC | [U-13C]glucose, [U-13C]acetate | The contribution of glucose to the TCA cycle is low in ccRCC relative to adjacent kidney and tumours from the lung and brain |
| Gonsalves et al., 2020 (ref. 42) | Multiple myeloma | [5-13C]glutamine, [U-13C]glutamine | The contribution of glutamine to the TCA cycle in CD138+ bone marrow cells is higher in multiple myeloma than in the premalignant lesion MGUS |
| Johnston et al., 2021 (ref. 35) | Extracranial solid tumours in children | [U-13C]glucose | Intra-operative [U-13C]glucose infusions are safe and informative in children as young as 4 months Glucose provides carbon to the TCA cycle in neuroblastoma, sarcoma and other paediatric malignancies |
| Ghergurovich et al., 2021 (ref. 38) | Triple-negative breast cancer | [1,2-13C]glucose | Isotope labelling indicates metabolism of glucose within tumours to amino acids and pentose phosphate pathway intermediates Within tumours, ribose phosphate is largely produced by the oxidative rather than non-oxidative pentose phosphate pathway |
ccRCC, clear cell renal cell carcinoma; CD138, cluster of differentiation 138 (also known as syndecan-1); MGUS, monoclonal gammopathy of undetermined significance; NSCLC, non-small-cell lung cancer; PC, pyruvate carboxylase; TCA, tricarboxylic acid.
Methodological considerations
In some ways, cancer is an ideal disease to study with stable isotopes. Altered metabolism is a hallmark of cancer, and therapies targeting these pathways are in clinical use or development20–24. Standard of care often involves tissue procurement by biopsy or surgical tumour resection, making it possible to sample metabolites from the tumour and adjacent, non-malignant tissue. Molecular heterogeneity among tumours in patients begs the question of whether metabolic features discoverable by stable-isotope tracing can help define actionably distinct cancer subsets.
Labelled nutrient administration
To date, most studies involving patients with cancer that used stable isotopes have relied on 13C-labelled nutrients administered intravenously (Table 1). A few studies have given oral 2H2O to monitor DNA synthesis in cancer cells25,26. For tracers administered intravenously, an important choice in study design is delivery kinetics: bolus versus continuous infusion. Some early 13C-tracing studies in patients with cancer provided [U-13C]glucose, a ‘uniformly’ labelled form of glucose that contains 13C at all six carbon positions, as a bolus before or during tumour resection19. In non-small-cell lung cancer (NSCLC), these studies revealed excess labelling from [U-13C]glucose into glycolytic and TCA cycle intermediates in tumours relative to adjacent lung. TCA cycle labelling resulted in part from induction of pyruvate carboxylation within the tumour19,27 (Fig. 2a).
Fig. 2 |. Metabolic rewiring in different kinds of human cancer.
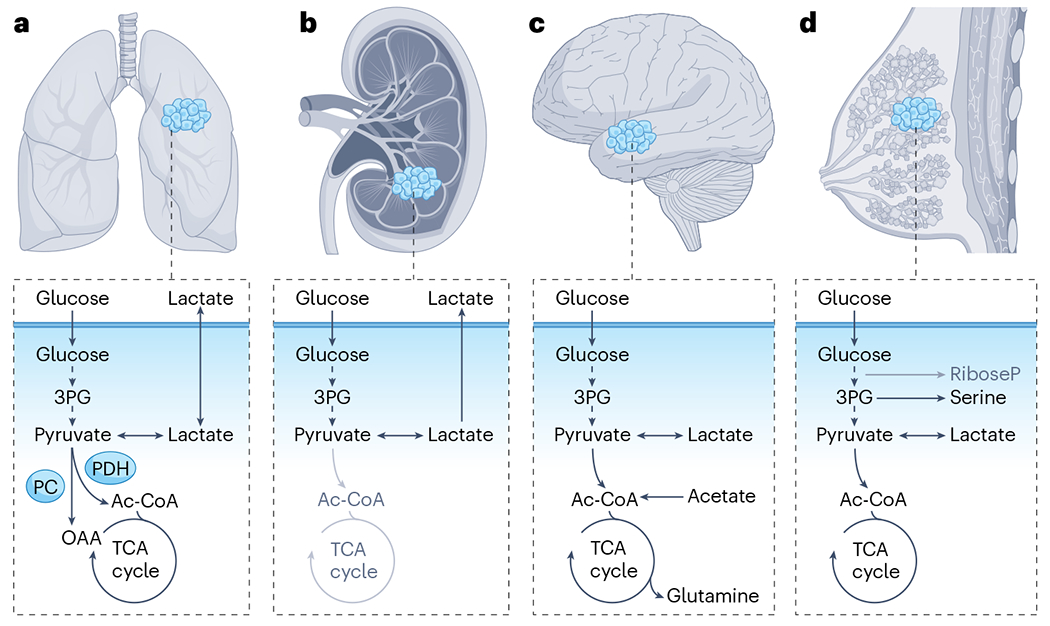
Stable-isotope tracing with 13C-labelled nutrients has been performed in patients with several types of cancer. The pathways illustrated in dark text reflect labelling features observed through stable-isotope tracing in tumours. Those in grey text appear to be suppressed in tumours relative to adjacent, non-malignant tissue. a, In non-small-cell lung cancer, both [13C]glucose and [13C]lactate are oxidized in the tricarboxylic acid (TCA) cycle. Labelled pyruvate that is derived from either tracer enters the TCA cycle through pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC). b, Clear cell renal cell carcinomas display a Warburg-like metabolic phenotype, with prominent contributions of circulating glucose to glycolytic intermediates but suppressed contribution to the TCA cycle. c, In the brain, both glioblastoma and brain metastases oxidize [13C]glucose within the TCA cycle, can use [13C]acetate as a TCA cycle fuel and synthesize glutamine from either substrate. d, In triple-negative breast cancer, [13C]glucose-derived carbons fuel multiple pathways, including the TCA cycle, serine biosynthesis and lactate, indicating local production of these intermediates within the tumour. Ac-CoA, acetyl-CoA; OAA, oxaloacetate; 3PG, 3-phosphoglycerate.
The rapid timescale of boluses allows application of 13C tracers in which the nuclear spins of 13C atoms have been transiently displaced far from equilibrium into a hyperpolarized state, and this hyperpolarization markedly increases the intensity of MRI signals28. Experiments capitalizing on 13C hyperpolarization are most feasible for carbons with no adjacent hydrogens, which reduce persistence of the hyperpolarized state. The carboxylic acid carbon of pyruvate is advantageous because neither it nor the adjacent carbonyl carbon is attached to hydrogens; consequently, [1-13C]pyruvate, in which the carbon at position 1 is labelled with 13C, is the predominant hyperpolarized tracer in practice29–31. The passage of the hyperpolarized carbon into lactate, alanine, bicarbonate or other metabolites can be monitored noninvasively by MRI over an approximately 60-s timescale. These experiments involve an extremely high dose of tracer and could be considered to measure the capacity of the tissue to transfer carbon from the labelled nutrient into downstream metabolites.
For stable-isotope tracers without hyperpolarization, continuous infusions have the advantage of easier and more quantitative interpretation of the labelling data. Specifically, continuous infusions result in steady-state labelling of both the circulating tracer and downstream metabolites. This enables quantitative assessment of tumour metabolite sources. For example, from steady-state data, it is straightforward to calculate ‘How much of a metabolite of interest comes from the nutrient tracer?’. Quantitatively, this fractional contribution to the metabolite of the circulating tracer is calculated by dividing the tumour metabolite labelling by the circulating tracer labelling. Owing to the ready metabolic interpretation of steady-state labelling data, continuous infusions have become the most common way of administering stable-isotope tracers.
To accelerate steady-state circulating tracer labelling, continuous infusions are often preceded by a bolus, which aims to quickly raise circulating enrichment close to the eventual steady-state level, thereby shortening the required infusion duration. Ideally, this is done with minimal perturbation to endogenous metabolism, by limiting the bolus size and infusion rate. Steady-state labelling in the blood of the infused metabolite in the range of 10–30% offers a good blend of enough label to detect and limited metabolic perturbation32.
Tumour sampling
Samples for measuring tumour metabolite labelling can be acquired by needle biopsy or surgical resection. The acquisition procedure imposes complications that should be considered when interpreting the data. Patients may be stressed, for example, during awake biopsy, or anaesthetized and mechanically ventilated. Some resections involve a period of tissue ischaemia as blood vessels supplying the tumour are ligated. Additionally, there may be unavoidable delays between tissue resection and freezing. Such delays impact some metabolic measurements more than others. For example, the ATP/AMP ratio changes rapidly and is sensitive to hypoxia, whereas TCA cycle labelling changes more slowly. In limited comparisons, [13C]glucose-derived labelling data from resected human lung tumours are similar to biopsies obtained without ischaemia33, and also similar to results from analogous experiments in non-anaesthetized mice, in which tumours can be freeze-clamped with minimal ischaemia34. Moreover, if the samples are kept cold, 13C labelling in TCA cycle intermediates persists for several minutes after resection, providing flexibility in the operating room as the samples are processed35. The stability of metabolite labelling, although not guaranteed in every context, helps make clinical tracing studies feasible and informative.
Catabolism in tumours of patients with cancer
A goal of isotope tracing in patients with cancer has been to define which nutrients supply metabolic pathways in tumours, and how these differ from adjacent tissue. Most studies have focused on glucose, although lactate, acetate and glutamine have also been used (Table 1). Oxidation of pyruvate derived from circulating [U-13C]glucose has been observed in all tumours analysed, including gliomas, brain metastases, breast tumours, NSCLC and clear cell renal cell carcinomas (ccRCCs) in adults (Fig. 2a–d) and paediatric extracranial solid malignancies, including neuroblastoma and sarcoma19,35–39.
The most extensive comparisons of labelling between tumour and adjacent tissue to date have been in patients with NSCLC who have heterogeneous but generally higher labelling of TCA cycle intermediates in tumours relative to adjacent lung. The extent of labelling correlates inversely with regional perfusion, where low perfusion yields high labelling36. These findings reflect higher glucose contribution to the TCA cycle in NSCLC relative to adjacent lung. In ccRCC, the contribution of glucose to the TCA cycle is low relative to adjacent kidney37 (Fig. 2b). This may reflect the metabolic effects of von Hippel–Lindau (VHL) tumour suppressor inactivation in these tumours, as VHL loss imposes a pseudohypoxic state that promotes glycolysis and suppresses pyruvate oxidation40. These relative enrichment studies are helpful in showing how carbon sources differ between tumours and the organs from which they arise. But keep in mind that carbon sources differ among organs, and this should be considered when interpreting the biological meaning of a relative increase or decrease in labelling within a tumour.
Glutamine metabolism is regulated by oncogenes, and glutamine is avidly consumed by most cancer cell lines41. One study infused [U-13C] glutamine into patients to assess metabolism in bone-marrow-derived plasma cells42. CD138+ multiple myeloma cells had higher labelling in TCA cycle intermediates than non-malignant CD138− cells from the same marrow. By contrast, pre-malignant plasma cells from patients with monoclonal gammopathy of undetermined significance had equivalent labelling of TCA cycle intermediates as CD138− cells. These findings suggest that glutamine becomes a more prominent carbon source for the TCA cycle as plasma cells progress from monoclonal gammopathy of undetermined significance to multiple myeloma.
Acetyl-CoA (Ac-CoA) is an important metabolic node that feeds many pathways. Most mitochondrial Ac-CoA flows into the TCA cycle, whereas cytosolic Ac-CoA supports lipid synthesis and nuclear Ac-CoA supports histone acetylation. In patients with gliomas or brain metastases, [U-13C]glucose infusions demonstrated that Ac-CoA enrichment is proportional to, but lower than, glucose enrichment39, implying that these tumours make Ac-CoA from other substrates in addition to glucose, for example, from fat or acetate. Infusions with [U-13C] acetate in human gliomas revealed robust labelling of the TCA cycle, reflecting tumour uptake of extracellular acetate and its assimilation into Ac-CoA43 (Fig. 2c).
Pyruvate/lactate metabolism
The textbook pathway of glucose catabolism involves glycolysis producing cellular pyruvate, which enters the mitochondria and is converted into acetyl-CoA and TCA cycle intermediates. Alternatively, cells may convert glucose to lactate, which they can excrete into the bloodstream. It is becoming clear that lactate excretion is far from a unique property of cancer cells or hypoxic tissues. Instead, it is a major metabolic activity in the healthy body, as reflected by rapid labelling of circulating lactate from infused [13C]glucose44. This lactate secretion is balanced by lactate uptake and oxidation to pyruvate, as reflected by extensive use of lactate as a TCA cycle fuel in most tissues, with many cells presumably both excreting and consuming lactate to no net effect. Lactate uptake and secretion are mediated by monocarboxylate transporters (MCTs), which also transport pyruvate, coupled to the reversible interconversion between lactate and pyruvate by lactate dehydrogenase (LDH). Consistent with the frequent overexpression of MCT and LDH isoforms in human cancer45,46, several clinical studies using hyperpolarized [1-13C]pyruvate revealed the increased appearance of hyperpolarized lactate in breast, prostate and renal tumours relative to adjacent, non-malignant tissue29–31,47.
After infusing patients with lung cancer with [U-13C]glucose, some tumours were observed to have higher 13C enrichment in lactate relative to glycolytic metabolites such as 3-phosphoglycerate and phosphoenolpyruvate36. This is inconsistent with the tumours making all of their lactate from the infused glucose. Because lactate labelling in the blood exceeded 3-phosphoglycerate labelling in tumours, an alternative possibility is that the infused, labelled glucose was being converted elsewhere in the body into circulating lactate, which was then taken up by the tumour. Infusing patients with [U-13C]lactate proved this hypothesis, directly demonstrating lactate uptake and oxidation in tumours33. This does not necessarily indicate net consumption of lactate by the tumours, merely that lactate consumption occurs, even though lactate secretion occurs in parallel33.
In a cohort of NSCLC patients, high labelling in lactate relative to glycolytic intermediates, indicating the high contribution of circulating lactate, correlated with cancer recurrence and distant metastasis, indicating clinical aggressiveness of tumours with this labelling property33. The contribution of circulating lactate to lactate within cancer cells depends both on tumour perfusion by systemic blood and on expression of MCTs. In this human NSCLC study, MCT expression correlated positively with high relative lactate labelling, and in mice bearing NSCLC cell line xenografts, MCT1 is required for tumours to show higher lactate than glycolytic intermediate labelling from infused [13C]glucose33. One interpretation is that the capacity to rapidly excrete or take up lactate, mediated by MCT1, provides metabolic flexibility that promotes cancer progression. Consistent with this, among patient-derived melanoma xenografts, those with more prominent lactate uptake metastasized more frequently, and even though it had minimal effect on subcutaneous tumour growth, MCT1 blockade reduced metastasis48. Thus, there is some evidence in the context of both lung cancer and melanoma that lactate transport between tumours and the circulation promotes cancer progression. Whether this process can be safely targeted, or used to stratify patients to different therapies, is an important open question.
In triple-negative breast cancer, a different pattern of lactate labelling from [13C]glucose was observed, with lactate labelling in the tumour exceeding lactate labelling in the bloodstream38 (Fig. 2d). This implies that the tumour lactate was made locally from intratumoral glycolysis and that tumour production of lactate outpaced exchange with the circulation38,49. The opposite findings in some NSCLCs suggest that, at least from the perspective of lactate, lung tumour perfusion is adequate to rapidly exchange metabolites between the systemic circulation and the tumours33.
Anabolism in tumours of patients with cancer
Although Warburg metabolism is often considered the primary metabolic hallmark of cancer, some of the greatest therapeutic successes against cancer have come instead from targeting tumour anabolism. Many chemotherapeutic agents interfere with nucleotide synthesis, and thereby alter DNA and RNA synthesis, including antifolates such as pemetrexed and methotrexate, and nucleoside analogues such as 5-fluorouracil and gemcitabine50–53. Despite the importance of such drugs, we still do not have a quantitative understanding of how fast tumours synthesize cellular building blocks such as amino acids, proteins, nucleotides, and lipids or which pathways they use to make them. Isotope tracing provides a direct way to address such questions, but current tracing experiments in patients with cancer only begin to cover the scope of anabolic reactions important for tumours.
Amino acids
Protein production from amino acids is required for cell division, as protein accounts for two-thirds of the dry weight of a cell54. Of the 20 common amino acids, 9 are essential, whereas 11 others can be synthesized endogenously. If cancer cells synthesized their own non-essential amino acids, targeting these synthesis routes could be a therapeutic strategy. Alternatively, if cancer cells (but not healthy tissues) are auxotrophic for a particular non-essential amino acid, restricting the blood level of the amino acid could be well tolerated and therapeutic.
Tracing studies show that, among non-essential amino acids, some are mainly synthesized within tumours, whereas others are acquired from the blood and ultimately from the diet. Many tumours synthesize alanine from [13C]glucose and [13C]lactate, with the contribution of de novo synthesis varying widely. Across published studies, 15–70% of alanine was made from these tracers19,27,33,36,37,39. Serine and glycine displayed some labelling from [13C]glucose in many tumour types, although most of the serine pool was unlabelled35,38,39. The reliance of tumours on circulating serine and glycine is consistent with the therapeutic benefits of serine-free and glycine-free diets in mice55 and argues for examining such diets in patients. Unlike serine, most glutamate was produced de novo in tumours, with many breast, brain and lung tumours also producing most glutamine internally19,38,39,56. This is consistent with preclinical evidence that glutamine synthetase promotes tumour growth57, although contrasting with the concept of exogenous glutamine as a primary tumour fuel. The capacity for tumours to acquire non-essential amino acids from the circulation, and ultimately the diet, is a barrier to targeting amino acid biosynthetic pathways to treat cancer, but perhaps this could be overcome by combining pathway inhibitors with diets lacking selected non-essential amino acids.
Proteins
As protein synthesis is a major metabolic task required for cell division54, inhibiting protein synthesis could in theory kill tumour cells, and there is clinical evidence supporting this concept58. However, few studies have carefully assessed whether tumours synthesize protein faster than healthy tissues. Protein synthesis rate can be measured by infusing a labelled essential amino acid, then isolating protein from a biopsy and measuring what fraction of protein contains labelled amino acids59. Two such studies showed that brain tumours in patients produce protein faster than healthy brain60,61. Another two studies came to differing conclusions about whether colorectal cancers in patients make protein faster than non-malignant colon62,63. We recently showed that mouse pancreatic tumours synthesized protein slowly relative to healthy pancreas, but faster than other healthy tissues such as lung and quadriceps64. Increased protein synthesis could drive dependency on amino acid supply, consistent with the antitumour activity of glutamine synthetase deficiency or serine-free and glycine-free diets mentioned earlier. There is also preclinical support for the antitumour benefit of lowering the levels of other amino acids, for example, by cysteinase therapy or diets lacking asparagine, proline or valine65–69. It would be valuable to assess protein synthesis in more cancer types to identify tumours with the highest rates of protein synthesis. An interesting candidate is multiple myeloma, which synthesizes and secretes large amounts of immunoglobulin protein and might respond therapeutically to protein synthesis inhibitors or amino acid deprivation70.
Nucleotides
Drugs targeting de novo nucleotide synthesis have been key components of therapy in lung, pancreatic, colorectal and other cancers50–53, yet we know surprisingly little about the metabolic pathways used to produce nucleotides in human tumours. Nucleotides can be synthesized de novo or salvaged from nucleosides and nucleobases from the blood or microenvironment. Given these alternate possibilities, knowing which tumours use which pathways might enable better treatment. De novo nucleotide synthesis can be measured by infusing 13C, 15N or 2H building blocks such as glucose, glutamine and glycine and measuring nucleotide labelling71,72, whereas salvage can be measured using isotope-labelled nucleobases such as hypoxanthine or nucleosides such as thymidine or cytidine73–76. Several decades ago, tritium-labelled thymidine was used to measure tumour proliferation in patients with cancer, suggesting that tumours salvage nucleosides73–75. By contrast, genes encoding folate cycle enzymes required for de novo nucleotide synthesis are among the most frequently over-expressed genes in human tumours77,78. Antifolate drugs are widely used, suggesting that de novo nucleotide synthesis may be dominant in many tumours. Ideally, infusion studies could identify tumours relying on de novo synthesis versus on salvage and this could guide therapy. So far, there are no approved strategies to inhibit nucleotide salvage, although at least one is in clinical trials and common therapeutics such as gemcitabine and 5-fluorouracil hijack salvage pathways79. In addition, infusing labelled nucleosides and nucleobases in patients who fail to respond to nucleotide synthesis inhibitors could reveal whether upregulation of nucleoside salvage is a route to resistance.
Lipids, fatty acids and cholesterol
Similar to nucleotides, lipids, including triglycerides and membrane phospholipids, can be synthesized de novo or assembled from circulating fatty acids, but little is known about which route predominates in human cancer. Other than liver and adipose, most healthy tissues take up fat from the circulation80, suggesting that de novo fat synthesis would distinguish most tumours from their tissue of origin and could potentially be a targetable pathway. De novo fat synthesis can be assessed by infusing 13C fat precursors, including glucose or acetate81,82, or by infusing deuterated water, which labels new fat molecules in part via NADPH regardless of the carbon source83. Fatty acid synthesis has been monitored using stable-isotope tracing80,84,85 or using a similar strategy with radioactive tracers82 in mouse models of cancer, including brain80, liver82,84, breast86 and lung87 tumours. However, such studies have not been carried out in patients with cancer. Several human tumour types upregulate enzymes involved in de novo fat synthesis, providing indirect evidence that this pathway may be active. For example, in ccRCC, which accumulates abundant lipids, expression of acetyl-CoA carboxylase and fatty acid synthase (FASN) portend poor outcomes88. FASN and ATP-citrate lyase are over-expressed in breast cancer, including metastases81,85. It would be valuable to carry out isotope-tracing studies to test whether fatty acid synthesis occurs in human tumours, although it is important in such studies to concomitantly measure blood enrichment of fatty acids, to eliminate the possibility that labelled fatty acids in tumours were synthesized in another tissue and imported from the blood80,86. FASN inhibition has shown antitumour activity in some preclinical models and early-stage clinical trials89,90, suggesting that high rates of fatty acid synthesis could serve as a biomarker for tumours vulnerable to this strategy.
Cholesterol, a key structural element of cell membranes, can also be synthesized or scavenged. Synthesis of cholesterol can be tracked using [13C]-labelled or [2H]-labelled acetate tracers or deuterated water91, whereas salvage can be measured using [13C]-labelled or [2H]-labelled cholesterol92. All tissues have the enzymes to synthesize cholesterol de novo, although the liver and intestines exhibit the highest constitutive synthesis rate93. Cholesterol synthesis and salvage have not been measured in human tumours, but data in mouse models suggest that blocking cholesterol synthesis and/or salvage might suppress tumour growth, at least in some tumour types. In mice, both hepatomas and leukaemias were found to produce cholesterol94, and blocking cholesterol synthesis slowed cancer growth in a leukaemia model95. Perhaps most intriguingly, statins, cholesterol synthesis inhibitors that are widely used to prevent cardiovascular disease, are reported to decrease prostate cancer risk96,97. Additionally, cholesterol salvage was required for the growth of lymphoma98, glioblastoma99 and ccRCC100 xenografts in mice. Thus, it would be valuable to use tracers to measure cholesterol synthesis and salvage in human tumours to assess which cancers merit cholesterol synthesis inhibition and which might respond to cholesterol uptake blockers.
From cells and mice to patients with cancer
Rapid glucose catabolism has stood the test of time as a hallmark of malignancy. It is observed in cultured cells, animal models and human tumour tissue cultivated ex vivo, as well as in patients with cancer, as determined by 18-fluorodeoxyglucose (18FDG)-PET imaging, which is extensively used in clinical oncology to diagnose and track progression of many solid tumour types, including lung, colorectal, ovarian and prostate cancers101. Other aspects of cancer cell metabolism in vitro are less conserved in vivo. This section reviews the current knowledge about metabolic activities that have been assessed in cells, mice and/or humans, discussing where reductionist models have translated well and where they have fallen short. These comparisons are currently limited by the paucity of tracers that have been applied to patients with cancer and the extensive metabolic heterogeneity among tumour types and patients.
Conserved metabolic features
The contribution of glucose to the TCA cycle is robust among cell lines and tumours in mice and humans. Experiments using [13C]glucose in cultured cancer cells demonstrated that glucose makes a substantial contribution to TCA cycle metabolites, such as citrate, succinate and malate, typically between 10% and 30% and often higher for citrate34,102,103. Healthy mouse tissues and tumours similarly showed 10–30% labelling of succinate and malate from glucose, with little difference between mouse tumours, cell lines and healthy tissues44,104 (Fig. 3a and Supplementary Table 1). Note that succinate and malate are typically in fast exchange and thus show similar labelling enrichments in cells, mice and human tissues. Likewise, in a range of human tumours, 9–29% of TCA cycle carbon was derived from glucose33,35–37 (Fig. 3a). Note that although the contribution of glucose is substantial, these data indicate that more carbon comes from other fuels.
Fig. 3 |. Shared and divergent metabolic properties in cultured cells, mice and patients.
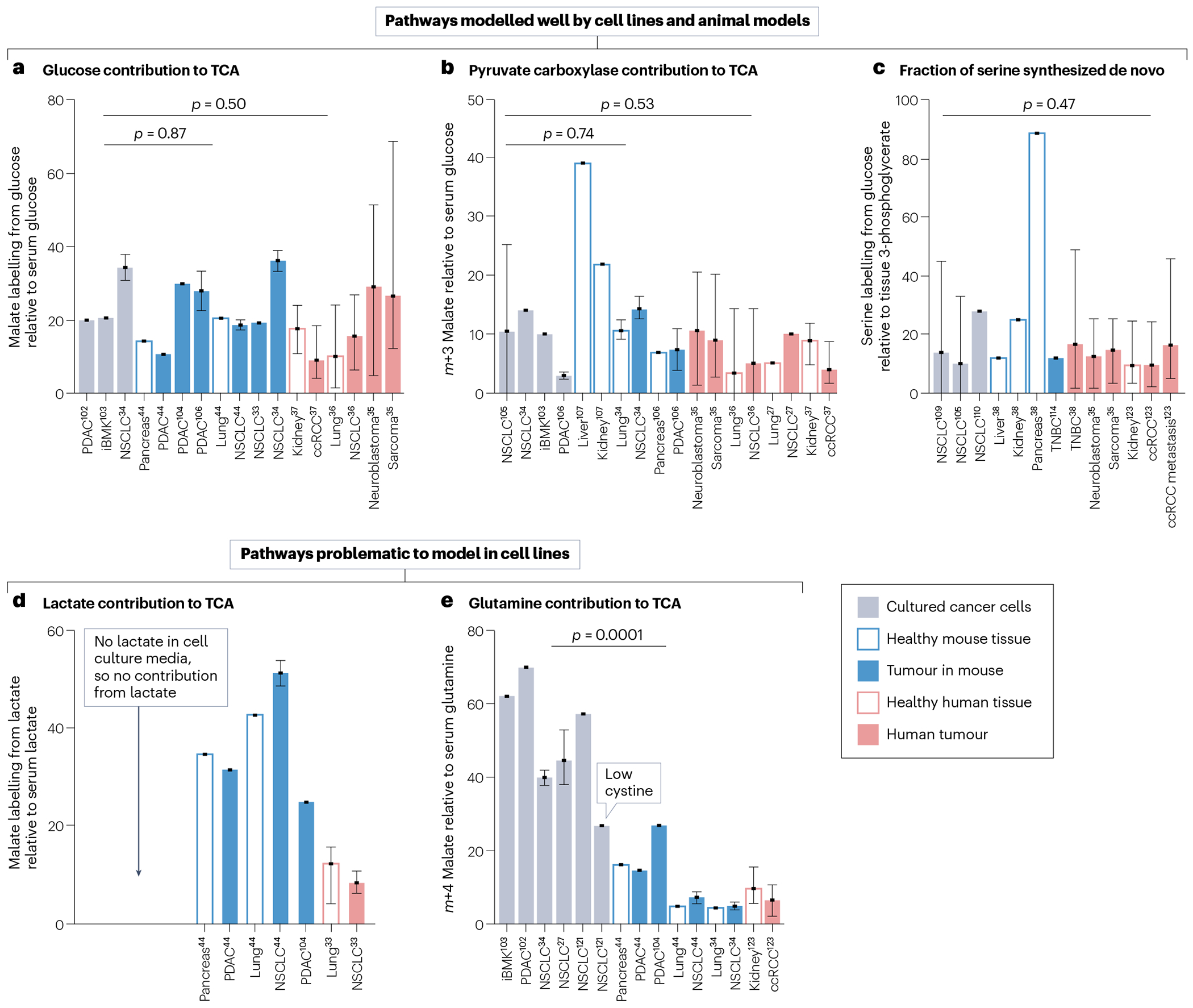
Comparison of data from published studies is shown. In all graphs, bars represent means of different cell lines, tumour models or individual patients, and error bars represent highest and lowest values in the study. P-values are from two-tailed t-tests comparing cell lines with mouse tumours or cell lines with patient samples. Neither of these comparisons includes healthy tissues. The sources of data in this figure are summarized in Supplementary Table 1. a, Contribution of glucose to the tricarboxylic acid (TCA) cycle, calculated as the fraction of carbons in malate labelled from [U-13C]glucose. For mouse and patient tissues and tumours, this is normalized to the fraction of carbons labelled in serum glucose. p = 0.87, cell lines versus mouse tumours and p = 0.50, cell lines versus human patient tumours. b, Pyruvate carboxylase contribution to the TCA cycle, calculated as the fraction of m + 3 malate from [U-13C]glucose. For mouse and patient tissues and tumours, this is normalized to the fraction of carbons labelled in serum glucose. p = 0.74, mouse tumours compared with cell lines; p = 0.53, human tumours compared with cell lines. c, Fraction of serine synthesized from [13C]glucose. For mouse and patient tissues and tumours, this is normalized to the fraction of carbons labelled in tissue 3-phosphoglycerate. p = 0.47 comparing cell lines with human patient tumours. d, Contribution of lactate to the TCA cycle, calculated as the fraction of carbons in malate labelled from [U-13C]lactate, normalized to the fraction of carbons labelled in serum lactate. e, Contribution of glutamine to the TCA cycle, calculated as the fraction of carbons in malate labelled from [U-13C]glutamine. For mouse and patient tissues and tumours, this is normalized to the fraction of carbons labelled in serum glutamine. p = 0.001, comparing cell lines with mouse tumours, excluding the cell line grown with low cystine. ccRCC, clear cell renal cell carcinoma; iBMK, immortalized baby mouse kidney epithelial cell line; NSCLC, non-small-cell lung cancer; PDAC, pancreatic adenocarcinoma; TNBC, triple-negative breast cancer.
The relative anaplerotic contribution of pyruvate carboxylase in patients is also similar across cell lines, mice and patients with cancer (Fig. 3b). Pyruvate carboxylase can be tracked using labelled glucose or lactate tracers that generate [1-13C]pyruvate. This 13C-labelled carbon is lost in the pyruvate dehydrogenase reaction as CO2, but enters the TCA cycle via pyruvate carboxylase. However, such tracing experiments have not been performed in patients with cancer, as [U-13C]glucose provides information about a wider variety of reactions. Using data from [U-13C]glucose studies, pyruvate carboxylase contribution to TCA intermediates can instead be estimated using m + 3 labelling in TCA cycle intermediates. The drawback is that a significant fraction of tissue pyruvate is m + 2 or m + 1 labelled in this scheme, so the approach underestimates the contribution of pyruvate carboxylase. A panel of more than 80 human NSCLC cell lines displayed variable pyruvate carboxylase contribution to the TCA cycle, with a mean of 11% (ref. 105). Mouse lung and pancreas tumours displayed 7–14% contribution34,106 (Fig. 3b). Human NSCLCs, sarcomas, neuroblastomas and ccRCCs displayed 3.4–10% contribution (Fig. 3b). Note that in gluconeogenic tissues such as liver and kidney, the pyruvate carboxylase contribution is much higher than in tumours107,108, whereas in more purely catabolic tissues such as muscle it is lower107,108. The consistent ‘intermediate’ pyruvate carboxylase contribution in tumours could reflect tumours engaging in substantial anabolic metabolism but using multiple anaplerotic routes in addition to pyruvate carboxylase; amino acids are a logical alternative source of anaplerotic nutrients that should be further studied.
Another feature of metabolism that appears conserved between cell lines and human tumours is de novo synthesis of serine from glucose. Enzymes from this pathway are amplified in many tumours38. [13C]glucose tracing in panels of cancer cell lines has reported wide variations in de novo serine/glycine synthesis, with 0–45% of these pools produced from glucose105,109,110, and similar variability is observed in human tumours (Fig. 3c). In tumours of patients with paediatric neuroblastomas and sarcomas, 2–25% of serine was synthesized from [13C]glucose35. All the values uniformly normalize tumour serine labelling to tumour labelling of 3-phosphoglycerate, to account for labelling dilution of glycolytic intermediates from glycogen111 (Fig. 3c and Supplementary Table 1). In patients with triple-negative breast cancer, there was a similarly wide variability in the fraction of serine derived from glucose, with 2–49% of serine synthesized de novo38. Although serine de novo synthesis has not been extensively reported in mouse tumours112–114, this pathway varies widely among healthy mouse tissues, from 12% of total serine in liver to 89% in pancreas38. These observations suggest that different cell types – including healthy organs in mice and human cancer cells in culture and in vivo – have intrinsic differences in their reliance on de novo serine/glycine synthesis and that these differences result in highly variable labelling from [13C]glucose.
Discrepancies between cell lines and tumours
Conventional culture conditions were developed to maximize cell proliferation, not to mimic physiology. Nutrient-rich medium and monolayer growth on stiff matrices exert large effects on metabolism115–117. Thus, some metabolic features displayed in culture are not observed in vivo and vice versa.
Standard tissue culture media do not contain lactate. Therefore, with the exception of a few dedicated experiments adding lactate118,119, in vitro studies capture aerobic glycolysis with lactate secretion, but not lactate usage, even though circulating lactate is a substantial TCA cycle substrate in both tumour mouse models and patients with cancer33,44,48 (Fig. 3d). Although TCA cycle labelling from infused [13C]glucose is indisputable, distinguishing whether glucose or lactate is the direct tumour TCA cycle substrate in vivo is complicated by the fact that infusion of [13C]glucose leads to extensive labelling of blood lactate. Therefore, TCA cycle labelling from infused [13C]glucose can occur via lactate even in tissues that do not consume glucose. Conversely, infused [13C] lactate can extensively label the TCA cycle via rapid exchange between circulating lactate and tumour pyruvate, even if the net direction of lactate transport in the tumour is outward.
Tissue culture and mouse models disagree on the magnitude of role of glutamine as a fuel for malignant cells. As cell culture was being optimized in the 1950s, Eagle120 found that HeLa cells and fibroblasts grew best in high concentrations of glutamine; therefore, media formulations such as DMEM and RPMI use 2–4 mM glutamine, higher than the 0.6 mM present in blood121,122. In cultured cancer cells, glutamine contributes 40–80% of TCA cycle carbon, much higher than the 4–20% observed in tumours in mice34,44,102,121 (Fig. 3e). This difference is caused in part by high levels of cystine in culture media. Cystine indirectly drives glutamine consumption because it exchanges with glutamate via the SLC7A11 transporter, and glutamine supplies the cellular glutamate pool in cultured cells121. Decreasing media cystine reduces the glutamine contribution to the TCA cycle in culture, yielding results more similar to tumours in mice (see bar labelled ‘low cystine’ in Fig. 3e). There is very little information about glutamine metabolism in human tumours, but a recent preprint suggested that glutamine contribution in the TCA cycle of kidney cancer aligns more closely to mouse models than to cell lines, with 7% contribution to malate in human kidney cancer, 5–27% in different mouse tumour types and 40–70% in cell lines123.
The robust catabolism of glutamine in so many cancer cell lines inspired the clinical development of inhibitors of glutaminase, a mitochondrial enzyme that converts glutamine to glutamate, for cancer therapy. So far, however, clinical trials involving glutaminase inhibitors have not been successful124. The large discrepancy between the contribution of glutamine to TCA cycle metabolism in cultured cells versus tumours in vivo may contribute to the lack of efficacy of these drugs. In mice, even tumours that do consume glutamine seem to possess enough metabolic flexibility to evade glutaminase inhibition125. Interestingly, compounds which broadly inhibit several glutamine-consuming enzymes in addition to glutaminase have shown remarkable efficacy in several mouse models of cancer22,126,127. In tumours that require glutamine catabolism, blocking multiple glutamine-using enzymes simultaneously might achieve therapeutic benefit.
These studies emphasize the limitations of non-physiological culture conditions to predict the metabolism of tumours in vivo. Nevertheless, cultured cell models are undeniably important to assess liabilities and study mechanisms of metabolic regulation. So how can cell culture be modified to better reflect tumour metabolism in patients? Several groups have devised ‘physiological’ media formulations that match nutrients available in blood and better reflect some aspects of cancer metabolism115,128. These media also contain physiological levels of vitamins and minerals, which influence metabolism129–131. Such formulations are helpful, but the concentrations of constituents drift as cells consume nutrients and excrete waste. In vivo biology can be modelled further by using flow-based Nutrostat systems in which the levels of nutrients and waste products are held at consistent levels over time132,133. Further development of such systems holds potential for making cell culture systems more metabolically accurate.
Limitations of stable-isotope tracing
Several caveats should be considered when interpreting data from stable-isotope tracing studies in patients with cancer. An important issue is that tracer infusions so far have measured nutrient contributions to metabolites, but not absolute metabolic fluxes (Fig. 4a). This distinction is similar to the difference between which fuel a car uses versus how fast the fuel is consumed. In cancer studies involving human participants, the tumour is typically sampled only once. When this single biopsy timepoint is at labelling steady state, it can reliably reveal fractional contributors (for example, glucose to TCA metabolites) and pathway relative rates (for example, anaplerosis relative to citrate synthase). But without additional information, such as absolute rates of nutrient import and secretion or pre-steady-state accumulation rates of isotope labelling, one cannot reliably measure absolute enzyme velocities.
Fig. 4 |. Limitations of stable-isotope tracing studies in patients with cancer.
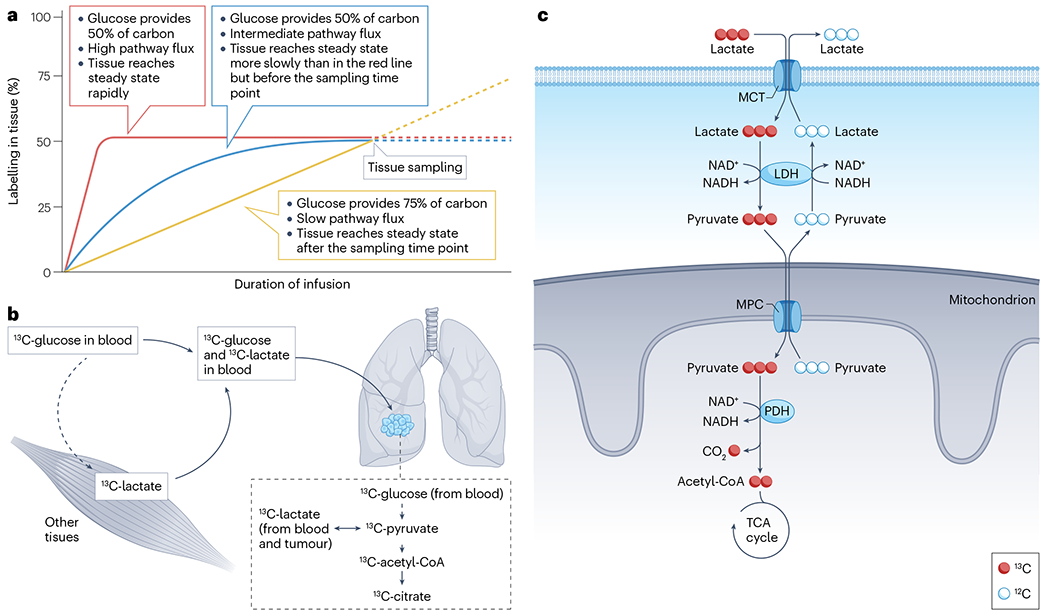
a, It is not possible to determine absolute metabolic flux (that is, absolute rates of pathways) from steady-state isotope labelling alone. In an isotope tracing experiment using [13C]glucose as the tracer, metabolite labelling depends on several factors, including the speed of turnover and the relative contribution of glucose to the pool. As shown in the graph, similar labelling at the time of sampling can occur even when the metabolic properties of the tissue differ. b, Secondary tracers, that is, labelled metabolites in the circulation arising from metabolism of the initial tracer, must be considered when analysing metabolite labelling in the tumour. In this example, [13C]glucose is introduced into the blood and is the initial source of all 13C labelling that ensues. However, as [13C]glucose circulates, it is taken up by the tissues such as muscles and converted to other metabolites, particularly [13C]lactate, which may then be released into the blood. Over time, circulating lactate becomes appreciably labelled, allowing 13C to enter tumour cells as either [13C]glucose or [13C]lactate. Therefore, 13C labelling within tumour cells may arise either from glycolysis using [13C]glucose as a substrate or from uptake of blood-borne [13C]lactate. c, Exchange fluxes contribute to 13C labelling patterns. In the illustration, circulating [13C]lactate is taken up by tumour cells and contributes to labelling of pyruvate, acetyl-CoA and tricarboxylic acid (TCA) cycle intermediates (red pathway). However, several steps of this pathway are rapid and bidirectional. These include lactate transport by monocarboxylate transporters (MCTs) on the plasma membrane, interconversion of lactate and pyruvate by lactate dehydrogenase (LDH) and pyruvate transport into the mitochondrial matrix by the mitochondrial pyruvate carrier (MPC). Metabolite labelling therefore does not mean that tumour cells have a net consumption of lactate, because lactate efflux and other exchange reactions may equal or exceed lactate uptake and metabolism. Note that PDH is irreversible, so labelling of acetyl-CoA and TCA cycle intermediates implies bona fide flux through the PDH reaction.
Another challenge is that prolonged infusions lead to ‘secondary tracer’ labelling in the bloodstream134 (Fig. 4b). As discussed earlier, [13C]glucose infusion leads to extensive labelling in blood lactate owing to glycolysis and lactate release by many tissues, and similar labelling can occur in other glucose-derived metabolites, including serine, glutamine, pyruvate, alanine and others44,107,135. Therefore, labelled metabolites extracted from tumour tissue could have been produced from the infused tracer within the tumour, or could have arisen from uptake of secondary tracers from the bloodstream. Recognizing the contribution of secondary tracers can lead to important insights, as described earlier for lactate metabolism in human lung cancers33,36. But secondary tracers complicate the interpretation of labelling data. The source of label can be clarified to some extent by comparing labelling of a metabolite of interest between the blood and tumour. When labelling in the tumour exceeds labelling in the blood, the simplest explanation is that at least part of the metabolite pool was produced in the tumour.
Related to these concerns is that label transfer from one metabolite to another does not necessarily require a net flux in the direction of the labelling recipient metabolite. Rapid exchanges involving metabolite transport and reversible enzymatic reactions can transfer an isotopic label along a pathway even when net flow is in the other direction. Lactate metabolism is a relevant example (Fig. 4c). As illustrated, 13C on lactate in the bloodstream is transferred to TCA cycle metabolites within the tumour, but whether this involves net lactate consumption and oxidation by the tumour depends on the velocities of all reactions between extracellular lactate and intracellular TCA cycle intermediates. Most of these reactions, particularly lactate transport and LDH, are rapid and highly reversible, and their net directions depend on factors such as compartmentalized metabolite concentrations and redox ratios that are difficult to quantify in human tumours. Other highly reversible processes of interest in human cancer include exchanges between serine and glycine and between amino acids and ketoacids catalysed by aminotransferases, so caution will be necessary when interpreting labelling transfer along these pathways.
The immense clinical and biological complexity of cancer must also be considered when interpreting labelling data. Demographic factors, including the age and sex of the patient, systemic factors such as obesity and diabetes, and clinical oncology factors such as grade, stage and genetics of the tumour and its previous exposure to therapy may influence the metabolic state and therefore impact the results of an isotope infusion experiment44,136–140. Circadian rhythms also influence systemic metabolism141, so it is conceivable that the time of day may impact the results of isotope infusion experiments. Caution is therefore advised when comparing results among subjects or studies in which these factors differ enough to contribute to labelling differences.
Future priorities and challenges
Many questions in human cancer metabolism are poised to be addressed with stable-isotope tracers in the coming years (Fig. 5). Currently, most isotope tracer studies have used [13C]glucose and inform about pathways supplied by glucose19,36 (Table 1). Infusing other nutrients such as [13C]glutamine or fatty acids would reveal their contributions to bioenergetics107. [13C]acetate could be used to probe fatty acid synthesis and [13C]serine to measure de novo nucleotide synthesis. Nutrients labelled with 15N could address aspects of cancer metabolism not accessible with 13C tracers, including amidotransferase reactions, the urea cycle and others142–144.
Fig. 5 |. Future directions of research in cancer metabolism in vivo.
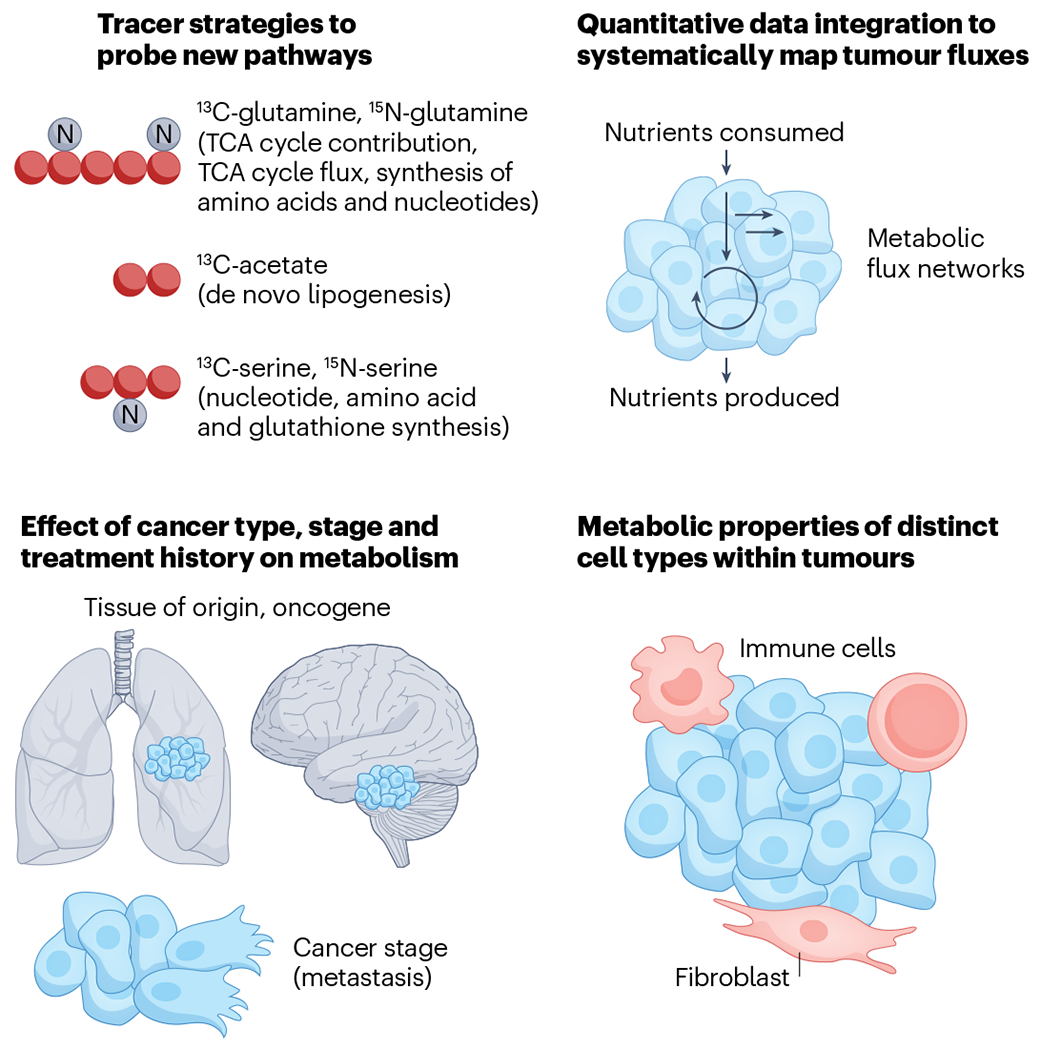
Upper left, using additional stable-isotope tracers such as [13C]glutamine, [15N]glutamine, [13C]acetate, [13C]serine and [15N]serine to probe cancer metabolism would reveal new information about tumour nutrient preferences. Performing glucose or glutamine infusions and sampling early timepoints could reveal tricarboxylic acid (TCA) cycle flux. Upper right, applying quantitative frameworks such as metabolic flux analysis to integrate multiple data types could help determine metabolic fluxes in tumours. Metabolic flux analysis is a quantitative framework that integrates metabolic data, such as nutrient production and consumption measured by sampling tumour draining veins and nutrient contributions measured by tracer infusion, using a mass balance constraint to infer other unmeasured fluxes. Lower left, future tracing studies should assess metabolism in more tumour types and at different clinical stages, with an emphasis on determining how stage and treatment status alter metabolic fluxes. For example, it is unknown whether pre-cancerous lesions, primary tumours and metastases differ in their metabolism. Additionally, if chemotherapy alters metabolic fluxes in cancer cells, this could uncover synthetic lethalities that might improve therapeutic responses over the current standard of care. Lower right, measuring the metabolism of specific cell types within tumours, including immune cells and fibroblasts, is an important future direction. This could potentially be achieved by fast sorting of different cell types from tumours or by spatial mass spectrometry analysis of tumour slices.
Quantifying metabolic flux rates in tumours is an important goal of the cancer metabolism field because these rates may contain clinically valuable information not captured by steady-state infusions. A recent study addressed this issue in mice by performing brief infusions of respiratory fuels such as [13C]lactate and [13C]glutamine, followed by tissue sampling at multiple time points. This allowed central fluxes to be quantified, revealing that glycolytic flux is elevated and TCA cycle flux is suppressed in tumours compared with highly metabolically active healthy tissues64. These flux measurements are consistent with the classical description of the Warburg effect, but the findings imply that low TCA cycle flux occurs because tumours inactivate energy-demanding processes of differentiated tissues. Thus, although the tumours display the Warburg effect, the mechanism is quite different from Warburg’s concept of impaired capacity for respiration in cancer cells145. The fact that metastatic tumours in mice activate TCA cycle flux relative to primary tumours also argues against permanent disabling of the TCA cycle64. It will be important to test whether this is also true in humans. Repeated sampling may be possible in patients with some forms of cancer, although it is unlikely that such sampling could be performed with the level of exquisite time resolution achievable in mice. Kinetic imaging data, such as the time-dependent appearance of PET tracers or metabolites labelled with hyperpolarized 13C, may provide helpful information in the analysis of metabolic rates in human cancer.
As data measuring metabolism of different nutrients in different pathways accumulate, integrating such data sets using quantitative frameworks such as metabolic flux analysis will help map overall tumour metabolism54,146. Metabolic flux analysis incorporates both metabolite labelling from different nutrients and flux balance to calculate relative fluxes of different pathways. Fluxes can be further informed by additional data types, such as tumour influxes and effluxes measured on the basis of arteriovenous sampling, which is feasible for tumours with major draining veins147,148. Such arteriovenous sampling could also address the important question of whether tumours are net lactate consumers or producers. The few previous arteriovenous studies of cancer suggest that some tumours do produce net lactate149,150. In general, generating a map of the metabolism of a tumour using quantitative approaches could calculate pathway fluxes that are not easily addressed by experiments with single tracer infusions.
A major question is what accounts for the metabolic heterogeneity observed among tumour types, patients or even different regions of the same tumour36,38,56. Metabolic fluxes can be governed by nutrient availability or enzyme activity. Within these two broad categories, nutrient availability can be altered by perfusion, blood nutrient levels, nutrient transporter expression and transporter post-translational modification, whereas enzyme activity encompasses expression, post-translational modification and allosteric regulation of enzymes. Measuring metabolic activity using isotope tracing in parallel with blood metabolite levels, and with tumour enzyme and transporter expression levels using RNA sequencing and/or proteomics, will help identify the factors driving tumour metabolism within each patient and tumour. Applying spatial measurement techniques, such as immuno-histochemistry or spatial RNA sequencing151 to measure enzyme levels, and spatial mass spectrometry to measure metabolite levels152, may clarify the drivers of metabolic heterogeneity within tumours. Such approaches may also provide information about metabolite transfer among cells within the tumour microenvironment. This is of interest because preclinical models have provided examples of metabolites produced within one subset of cells being taken up to support metabolism in other cells119,153. A complete understanding of metabolic heterogeneity will require that we identify such processes. A related challenge is to understand the effect of cancer stage on metabolism, because identifying unique metabolic dependencies in metastatic tumours or in tumours with acquired drug resistance could have substantial therapeutic value123.
Tumours are composed of multiple cell populations, including fibroblasts, immune cells and endothelial cells, in addition to tumour cells, and the individual contributions of these cell types are challenging to measure. In some tumours, such as pancreatic adenocarcinoma, stromal cells may outnumber cancer cells and may dominate the metabolic picture154. These non-malignant cells contribute to the pace of tumour growth, so understanding their biology is critical155,156. Different cell types within tumours may display distinct metabolic properties106,157. In mice, malignant cells have higher pyruvate carboxylase contribution than fibroblast or immune cells in the microenvironment of pancreatic adenocarcinomas106. In a syngeneic colon adenocarcinoma model, 18FDG-PET positivity stems from tumour-associated macrophages as well as from malignant cells, with approximately 30% of signal contributed by macrophages, whereas 18F-glutamine uptake is largely confined to malignant cells157. These findings advance our understanding of nutrient consumption within complex tumours. Similar studies could be carried out in human tumour biopsy samples, either by digestion and sorting of cell types106,157, or with imaging mass spectrometry64,158.
Conclusions and perspectives
Can human isotope tracing studies attain relevance to clinical oncology? We envision a few ways that these studies can integrate with clinical research and practice. Isotope tracing provides a different view of cancer biology than genetic, transcriptomic or metabolomic profiling. Thus, as we aggregate isotope labelling data from more patients with distinct types of cancer, correlating these features with clinical outcomes may provide predictive information that otherwise could not be obtained. Isotope tracing studies are cumbersome, but once a pathway with predictive value is identified, it may be possible to develop scalable ways to assess that pathway in many patients at multiple centres. A priority should be assessing whether the correlation between avid lactate use and poor outcomes in NSCLC that can be measured by stable-isotope infusion, biopsy and mass spectrometry translates into a correlation between high pyruvate utilization and poor outcomes measured by hyperpolarized MRI without the need for biopsy, as in patients with prostate and renal cancer30,31. In the longer term, novel PET or hyperpolarized tracers could be designed to image clinically informative fluxes initially uncovered by stable isotopes159.
We can also use isotope tracing to better understand imaging techniques already used clinically. A tumour with elevated FDG uptake on PET is assumed to be ‘glycolytic’. But FDG-PET studies are performed after fasting, so it is unclear whether FDG avidity reflects particularly impressive glucose usage or just a failure to respond to fasting. Furthermore, in patients with NSCLC assessed by FDG-PET and [U-13C]glucose infusions, strength of the PET signal does not correlate with labelling of glycolytic intermediates160. Stable-isotope tracing should help us better understand the metabolic implications of imaging with FDG, hyperpolarized [13C]pyruvate and other tracers.
Finally, there should be a role for isotope tracing in clinical trials assessing metabolic therapies. Several inhibitors against metabolic enzymes have been developed over the past decade, but so far, robust therapeutic responses have been hard to come by, for example, for inhibitors of indoleamine 2,3-dioxygenase-1 (IDO1), fatty acid synthesis90 and glutaminase124. It is unclear whether therapeutic failures result from poor target engagement, metabolic compensation by the tumour or simply that the tumour does not use the pathway blocked by the drug. Stable-isotope infusions would be valuable here, as they could help identify patients most or least likely to benefit from an experimental drug by assessing utilization of the pathway and possibly confirming effective target blockade. Isotope studies could also help map resistance mechanisms. For example, when anti-nucleotide chemotherapies fail, metabolic tracing might reveal which alternate metabolic pathways are used to maintain tumour growth, and these could perhaps be targeted in the future. Thus, isotope tracing is poised to contribute to the fight against cancer.
Supplementary Material
Acknowledgements
R.J.D. is supported by the Howard Hughes Medical Institute and by US National Cancer Institutes grants R35CA22044901, P50CA196516-06A1 and 2P50CA070907-21A1.
B.F. is supported by US National Cancer Institutes grant R00CA237724.
Glossary
- 18-Fluorodeoxyglucose-PET
(18FDG-PET). 18Fluorodeoxyglucose (FDG) is an analogue of glucose conjugated with a radioisotope of fluorine (18F). FDG is imported into tissues and tumours using the same transporters that import glucose, for example, GLUT1, and the tracer is retained after phosphorylation by hexokinase. The localized accumulation of 18F can be imaged by positron emission tomography. FDG uptake is increased in many tumours, so this technique can be used to diagnose and stage multiple cancers.
- Acetyl-CoA carboxylase
The first enzyme of fatty acid synthesis, which catalyses the carboxylation of acetyl-CoA to produce malonyl-CoA for fatty acid synthesis.
- Anaplerotic contribution
Metabolic pathways that contribute to recover or replenish catalytic intermediates, particularly in the TCA cycle. Examples include the use of glutamine or pyruvate to provide 4-carbon and 5-carbon intermediates to the TCA cycle.
- Antifolates
Drugs that inhibit folate-using enzymes, including three of the enzymes required for nucleotide biosynthesis. Several chemotherapeutic agents, such as pemetrexed and methotrexate, are antifolates.
- Fatty acid synthase
(FASN). A multifunctional enzyme that catalyses several reactions required to produce fatty acids from precursors, including malonyl-CoA and acetyl-CoA.
- Folate cycle
A metabolic pathway producing one-carbon metabolites for use in numerous biochemical reactions, including nucleotide synthesis.
- Fractional contribution
Refers to the fraction of a metabolite pool arising from a particular precursor. In stable-isotope tracing experiments, transfer of the isotope from the labelled precursor such as [13C]glucose to a metabolite, for example, pyruvate, provides information about the fractional contribution of that tracer to that metabolite.
- Hyperpolarization
A molecular state in which the nuclear spin is polarized well beyond thermal equilibrium, increasing the ability to detect the nucleus by NMR. Hyperpolarization of 13C nuclei can be achieved by transferring to 13C the high polarization state of free electrons contained in a radical.
- Imaging mass spectrometry
An analytical technique that enables visualization of the spatial distribution of metabolites within a tissue sample.
- Lactate dehydrogenase
(LDH). A tetrameric, reversible NAD(H)-dependent enzyme that interconverts pyruvate and lactate.
- Metabolic flux analysis
An algorithm to calculate metabolic fluxes in cultured cells, which takes as inputs the consumption and production rates of media nutrients, 13C labelling of cellular metabolites from 13C isotope tracers such as glucose and glutamine and the known reaction structure of metabolic pathways.
- Metabolic fluxes
The rates at which metabolites flow through biochemical reactions.
- Monocarboxylate transporters
A family of membrane proteins that transport monocarboxylates such as lactate, pyruvate and ketone bodies across the plasma membrane.
- Monoclonal gammopathy of undetermined significance
A condition characterized by the accumulation of abnormal monoclonal immunoglobulin in the blood. Monoclonal gammopathy of undetermined significance can be a precursor to multiple myeloma or other disorders.
- Nuclear spins
The intrinsic angular momentum of an atomic nucleus.
- Nucleoside analogues
Synthetic compounds that structurally mimic endogenous nucleosides and are used as chemotherapeutic agents. These analogues can be incorporated into the DNA of rapidly dividing cells, disrupting DNA replication.
- Nutrostat
Cell culture system designed to maintain consistent levels of extracellular nutrients and other metabolites by providing a consistent source of fresh medium to the culture.
- Pyruvate carboxylation
A mitochondrial anaplerotic activity in which the enzyme pyruvate carboxylase converts pyruvate to oxaloacetate.
- Von Hippel–Lindau (VHL) tumour suppressor
A tumour suppressor whose physiological role is to target the α subunits of hypoxia-inducible factors (HIF1α and HIF2α) for degradation when oxygen is present. In VHL-deficient tumours such as ccRCC, HIF1α and HIF2α are stabilized regardless of oxygen availability. The resulting ‘pseudohypoxic’ state causes chronic expression of genes involved in the hypoxic response.
- Warburg metabolism
Named after Otto Warburg and also known as aerobic glycolysis, this classic metabolic phenotype of tumours and proliferating cells involves the brisk conversion of glucose to lactate in the presence of sufficient oxygen to oxidize glucose to CO2.
Footnotes
Competing interests
R.J.D. is a founder and adviser for Atavistik Bioscience and an adviser for Agios Pharmaceuticals, Vida Ventures and Droia Ventures. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41568-023-00632-z.
Data availability
The data supporting the findings displayed in Fig. 3 are available in Supplementary Table 1.
References
- 1.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Pavlova NN & Thompson CB The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies PSW Stable isotopes: their use and safety in human nutrition studies. Eur. J. Clin. Nutr 74, 362–365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alseekh S et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu W, Bennett BD & Rabinowitz JD Analytical strategies for LC-MS-based targeted metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci 871, 236–242 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson CH, Ivanisevic J & Siuzdak G Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol 17, 451–459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myers WG Georg Charles de Hevesy: the father of nuclear medicine. J. Nucl. Med 20, 590–594 (1979). [PubMed] [Google Scholar]
- 8.Schoenheimer R & Rittenberg D Deuterium as an indicator in the study of intermediary metabolism. Science 82, 156–157 (1935). [DOI] [PubMed] [Google Scholar]; This paper reports the first use of a stable isotope — deuterium — to trace metabolism in vivo, focusing on fatty acid and sterol metabolism in mice.
- 9.Alves TC et al. Integrated, step-wise, mass-isotopomeric flux analysis of the TCA cycle. Cell Metab. 22, 936–947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi J, Grossbach MT & Antoniewicz MR Measuring complete isotopomer distribution of aspartate using gas chromatography/tandem mass spectrometry. Anal. Chem 84, 4628–4632 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson DJ Historical and contemporary stable isotope tracer approaches to studying mammalian protein metabolism. Mass. Spectrom. Rev 37, 57–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a detailed historical review of stable isotopes, emphasizing the influence of technology in the development and use of tracer studies, with a focus on protein metabolism.
- 12.Malloy CR, Sherry AD & Jeffrey FM Evaluation of carbon flux and substrate selection through alternate pathways involving the citric acid cycle of the heart by 13C NMR spectroscopy. J. Biol. Chem 263, 6964–6971 (1988). [PubMed] [Google Scholar]
- 13.Malloy CR, Sherry AD & Jeffrey FM Carbon flux through citric acid cycle pathways in perfused heart by 13C NMR spectroscopy. FEBS Lett. 212, 58–62 (1987). [DOI] [PubMed] [Google Scholar]; With the perfused heart as a model, this paper used isotope labelling with [13C]acetate to determine relative fluxes of anaplerosis and TCA cycle turnover.
- 14.Chance EM, Seeholzer SH, Kobayashi K & Williamson JR Mathematical analysis of isotope labeling in the citric acid cycle with applications to 13C NMR studies in perfused rat hearts. J. Biol. Chem 258, 13785–13794 (1983). [PubMed] [Google Scholar]
- 15.Russell RR 3rd et al. Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J. Clin. Invest 100, 2892–2899 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothman DL, Magnusson I, Katz LD, Shulman RG & Shulman GI Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 254, 573–576 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Bergman BC et al. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol 87, 1684–1696 (1999). [DOI] [PubMed] [Google Scholar]
- 18.Sunny NE, Parks EJ, Browning JD & Burgess SC Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14, 804–810 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan TW et al. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 8, 41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study, the first to infuse [13C]glucose into patients with cancer and assess labelling in metabolites extracted from the tumour, concluded that lactate, alanine and intermediates related to the TCA cycle were more enriched in lung tumours than in adjacent non-malignant lung.
- 20.Vander Heiden MG & DeBerardinis RJ Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 10.1126/science.aaw5473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leone RD et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein EM et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood 133, 676–687 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sallan SE et al. Influence of intensive asparaginase in the treatment of childhood non-T-cell acute lymphoblastic leukemia. Cancer Res. 43, 5601–5607 (1983). [PubMed] [Google Scholar]
- 25.Hayes GM et al. Regional cell proliferation in microdissected human prostate specimens after heavy water labeling in vivo: correlation with prostate epithelial cells isolated from seminal fluid. Clin. Cancer Res 18, 3250–3260 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Calissano C. et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood 114, 4832–4842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellers K. et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest 125, 687–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurhanewicz J. et al. Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia 13, 81–97 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson SJ et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med 5, 198ra108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper imaged the metabolism of a hyperpolarized 13C-labelled nutrient in humans for the first time, focusing on hyperpolarized [1-13C]pyruvate in prostate cancer.
- 30.Granlund KL et al. Hyperpolarized MRI of human prostate cancer reveals increased lactate with tumor grade driven by monocarboxylate transporter 1. Cell Metab. 31, 105–114.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ursprung S. et al. Hyperpolarized 13C-pyruvate metabolism as a surrogate for tumor grade and poor outcome in renal cell carcinoma — a proof of principle study. Cancers (Basel) 10.3390/cancers14020335 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartman CR, TeSlaa T & Rabinowitz JD Quantitative flux analysis in mammals. Nat. Metab 3, 896–908 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faubert B. et al. Lactate metabolism in human lung tumors. Cell 171, 358–371.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidson SM et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston K. et al. Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med 2, 395–410 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hensley CT et al. Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Courtney KD et al. Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo. Cell Metab. 28, 793–800.e2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghergurovich JM et al. Local production of lactate, ribose phosphate, and amino acids within human triple-negative breast cancer. Med 2, 736–754 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maher EA et al. Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR Biomed. 25, 1234–1244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reported the first use of intra-operative steady-state infusions of [13C] glucose in patients with cancer and concluded that glucose contributes a minority of acetyl-CoA for the TCA cycle in high-grade gliomas and brain metastases in vivo.
- 40.Chappell JC, Payne LB & Rathmell WK Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Invest 129, 442–451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altman BJ, Stine ZE & Dang CV From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 749 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Gonsalves WI et al. In vivo assessment of glutamine anaplerosis into the TCA cycle in human pre-malignant and malignant clonal plasma cells. Cancer Metab. 8, 29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mashimo T. et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui S. et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payen VL, Mina E, Van Hee VF, Porporato PE & Sonveaux P Monocarboxylate transporters in cancer. Mol. Metab 33, 48–66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altenberg B & Greulich KO Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 84, 1014–1020 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Woitek R. et al. Hyperpolarized carbon-13 MRI for early response assessment of neoadjuvant chemotherapy in breast cancer patients. Cancer Res. 81, 6004–6017 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tasdogan A. et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Canaveras JC, Chen L & Rabinowitz JD The tumor metabolic microenvironment: lessons from lactate. Cancer Res. 79, 3155–3162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farber S & Diamond LK Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med 238, 787–793 (1948). [DOI] [PubMed] [Google Scholar]
- 51.Seley-Radtke KL & Yates MK The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir. Res 154, 66–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ducker GS & Rabinowitz JD One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chattopadhyay S, Moran RG & Goldman ID Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol. Cancer Ther 6, 404–417 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Antoniewicz MR A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med 50, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baksh SC et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat. Cell Biol 22, 779–790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tardito S. et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol 17, 1556–1568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bott AJ et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 29, 1287–1298.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pemmaraju N. et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N. Engl. J. Med 380, 1628–1637 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Wagenmakers AJ Tracers to investigate protein and amino acid metabolism in human subjects. Proc. Nutr. Soc 58, 987–1000 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Willemsen AT et al. In vivo protein synthesis rate determination in primary or recurrent brain tumors using L-[1-11C]-tyrosine and PET. J. Nucl. Med 36, 411–419 (1995). [PubMed] [Google Scholar]
- 61.Bustany P. et al. Brain tumor protein synthesis and histological grades: a study by positron emission tomography (PET) with C11-l-methionine. J. Neurooncol 3, 397–404 (1986). [DOI] [PubMed] [Google Scholar]
- 62.Garlick PJ, Wernerman J, McNurlan MA & Heys SD Organ-specific measurements of protein turnover in man. Proc. Nutr. Soc 50, 217–225 (1991). [DOI] [PubMed] [Google Scholar]
- 63.Hartl WH, Demmelmair H, Jauch KW, Koletzko B & Schildberg FW Effect of glucagon on protein synthesis in human rectal cancer in situ. Ann. Surg 227, 390–397 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartman CR et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 614, 349–357 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports a method for quantifying metabolic fluxes in tumours in mice and concludes that TCA cycle flux is low in primary tumours owing to low demand for ATP, but is higher in metastatic tumours.
- 65.Badgley MA et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Knott SRV et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maddocks OD et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thandapani P. et al. Valine tRNA levels and availability regulate complex I assembly in leukaemia. Nature 601, 428–433 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sahu N. et al. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab. 24, 753–761 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Kumar SK et al. Multiple myeloma. Nat. Rev. Dis. Prim 3, 17046 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Ducker GS et al. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 24, 640–641 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Ali ES et al. The mTORC1-SLC4A7 axis stimulates bicarbonate import to enhance de novo nucleotide synthesis. Mol. Cell 82, 3284–3298.e7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sulkes A, Livingston RB & Murphy WK Tritiated thymidine labeling index and response in human breast cancer. J. Natl Cancer Inst 62, 513–515 (1979). [DOI] [PubMed] [Google Scholar]
- 74.Johnson HA, Rubini JR, Cronkite EP & Bond VP Labeling of human tumor cells in vivo by tritiated thymidine. Lab. Invest 9, 460–465 (1960). [PubMed] [Google Scholar]
- 75.Clarkson B. et al. Studies of cellular proliferation in human leukemia. IV. Behavior of normal hemotopoietic cells in 3 adults with acute leukemia given continuous infusions of 3H-thymidine for 8 or 10 days. Cancer 26, 1–19 (1970). [DOI] [PubMed] [Google Scholar]
- 76.Pareek V, Tian H, Winograd N & Benkovic SJ Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nilsson R. et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 10, 119–130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehrmohamadi M, Liu X, Shestov AA & Locasale JW Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 9, 1507–1519 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT05055609 (2022). [DOI] [PubMed]
- 80.Zhang Z. et al. Serine catabolism generates liver NADPH and supports hepatic lipogenesis. Nat. Metab 3, 1608–1620 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferraro GB et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2, 414–428 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Comerford SA et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Z, Chen L, Liu L, Su X & Rabinowitz JD Chemical basis for deuterium labeling of fat and NADPH. J. Am. Chem. Soc 139, 14368–14371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mendez-Lucas A. et al. Identifying strategies to target the metabolic flexibility of tumours. Nat. Metab 2, 335–350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Szutowicz A, Kwiatkowski J & Angielski S Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 39, 681–687 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ookhtens M, Kannan R, Lyon I & Baker N Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am. J. Physiol 247, R146–R153 (1984). [DOI] [PubMed] [Google Scholar]
- 87.Svensson RU et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med 22, 1108–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Falchook G. et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. eClinicalMedicine 34, 100797 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rohrig F & Schulze A The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16, 732–749 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Bloch K & Rittenberg D On the utilization of acetic acid for cholesterol formation. J. Biol. Chem 145, 625–636 (1942). [Google Scholar]
- 92.Ostlund RE Stable Isotopes in Human Nutrition: Laboratory Methods and Research Applications 157–173 (CABI Publishing, 2003). [Google Scholar]
- 93.Osono Y, Woollett LA, Herz J & Dietschy JM Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J. Clin. Invest 95, 1124–1132 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Siperstein MD Regulation of Cholesterol Biosynthesis in Normal and Malignant Tissues Vol. 2, 65–100 (Elsevier, 1970). [Google Scholar]
- 95.Rashkovan M. et al. Intracellular cholesterol pools regulate oncogenic signaling and epigenetic circuitries in early T-cell precursor acute lymphoblastic leukemia. Cancer Discov. 12, 856–871 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boudreau DM, Yu O & Johnson J Statin use and cancer risk: a comprehensive review. Expert Opin. Drug Saf 9, 603–621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Platz EA et al. Statin drugs and risk of advanced prostate cancer. J. Natl Cancer Inst 98, 1819–1825 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Garcia-Bermudez J. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guo D. et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 1, 442–456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Riscal R. et al. Cholesterol auxotrophy as a targetable vulnerability in clear cell renal cell carcinoma. Cancer Discov. 11, 3106–3125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhu A, Lee D & Shim H Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin. Oncol 38, 55–69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yuan M. et al. Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat. Protoc 14, 313–330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fan J. et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol 9, 712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang L. et al. Ketogenic diet and chemotherapy combine to disrupt pancreatic cancer metabolism and growth. Med 3, 119–136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen PH et al. Metabolic diversity in human non-small cell lung cancer cells. Mol. Cell 76, 838–851.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lau AN et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. eLife 10.7554/eLife.56782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hui S. et al. Quantitative fluxomics of circulating metabolites. Cell Metab. 32, 676–688 e674 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Landau BR et al. 14C-labeled propionate metabolism in vivo and estimates of hepatic gluconeogenesis relative to Krebs cycle flux. Am. J. Physiol 265, E636–E647 (1993). [DOI] [PubMed] [Google Scholar]
- 109.DeNicola GM et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet 47, 1475–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kottakis F. et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.TeSlaa T. et al. The source of glycolytic intermediates in mammalian tissues. Cell Metab. 33, 367–378.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ngo B. et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 10, 1352–1373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Piskounova E. et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pacold ME et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol 12, 452–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cantor JR et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park JS et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 578, 621–626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang L. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen YJ et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol 12, 937–943 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sonveaux P. et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest 118, 3930–3942 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eagle H. The specific amino acid requirements of a mammalian cell (strain L) in tissue culture. J. Biol. Chem 214, 839–852 (1955). [PubMed] [Google Scholar]
- 121.Muir A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 10.7554/eLife.27713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lagziel S, Gottlieb E & Shlomi T Mind your media. Nat. Metab 2, 1369–1372 (2020). [DOI] [PubMed] [Google Scholar]
- 123.Bezwada D. et al. Mitochondrial metabolism in primary and metastatic human kidney cancers. Preprint at bioRxiv 10.1101/2023.02.06.527285 (2023). [DOI] [Google Scholar]
- 124.Tannir NM et al. Efficacy and safety of telaglenastat plus cabozantinib vs placebo plus cabozantinib in patients with advanced renal cell carcinoma: the CANTATA randomized clinical trial. JAMA Oncol. 10.1001/jamaoncol.2022.3511 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oh MH et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest 130, 3865–3884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kaushik AK et al. In vivo characterization of glutamine metabolism identifies therapeutic targets in clear cell renal cell carcinoma. Sci. Adv 8, eabp8293, 10.1126/sciadv.abp8293 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vande Voorde J. et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 5, eaau7314(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guarecuco R. et al. Dietary thiamine influences l-asparaginase sensitivity in a subset of leukemia cells. Sci. Adv 10.1126/sciadv.abc7120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee WD et al. Tumor reliance on cytosolic versus mitochondrial one-carbon flux depends on folate availability. Cell Metab. 33, 190–198.e6 (2021). [DOI] [PubMed] [Google Scholar]
- 131.Leney-Greene MA, Boddapati AK, Su HC, Cantor JR & Lenardo MJ Human plasma-like medium improves T lymphocyte activation. iScience 23, 100759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Birsoy K. et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DeBerardinis RJ et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl Acad. Sci. USA 104, 19345–19350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Previs SF & Kelley DE Tracer-based assessments of hepatic anaplerotic and TCA cycle flux: practicality, stoichiometry, and hidden assumptions. Am. J. Physiol. Endocrinol. Metab 309, E727–E735 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Marin-Valencia I. et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 15, 827–837 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Petersen KF et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ying H. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Friedlander AL et al. Training-induced alterations of carbohydrate metabolism in women: women respond differently from men. J. Appl. Physiol 85, 1175–1186 (1998). [DOI] [PubMed] [Google Scholar]
- 140.Viale A. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Woelders T. et al. Machine learning estimation of human body time using metabolomic profiling. Proc. Natl Acad. Sci. USA 120, e2212685120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spinelli JB et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim J. et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat. Metab 2, 1401–1412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Poillet-Perez L. et al. Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Warburg O. On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 146.Orth JD, Thiele I & Palsson BO What is flux balance analysis. Nat. Biotechnol 28, 245–248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jang C. et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. 30, 594–606.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wolfe RR & Chinkes DL Isotope Tracers in Metabolic Research: Principles and Practice of Kinetic Analysis Vol. 2 (Wiley, 2004). [Google Scholar]; This is an excellent resource on the principles and applications of stable-isotope tracing.
- 149.Holm E. et al. Substrate balances across colonic carcinomas in humans. Cancer Res. 55, 1373–1378 (1995). [PubMed] [Google Scholar]
- 150.Xiong N. et al. Using arterial-venous analysis to characterize cancer metabolic consumption in patients. Nat. Commun 11, 3169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stahl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 152.Cornett DS, Reyzer ML, Chaurand P & Caprioli RM MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods 4, 828–833 (2007). [DOI] [PubMed] [Google Scholar]
- 153.Sousa CM et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kleeff J. et al. Pancreatic cancer. Nat. Rev. Dis. Prim 2, 16022 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Zhu Y. et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338.e326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ozdemir BC et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reinfeld BI et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper used radioisotopes to study nutrient uptake among different cell types in the tumour microenvironment and reported that myeloid cells exceed cancer cells in their ability to take up glucose, with the reverse occurring for glutamine uptake.
- 158.Wang L. et al. Spatially resolved isotope tracing reveals tissue metabolic activity. Nat. Methods 19, 223–230 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.DeBerardinis RJ & Keshari KR Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell 185, 2678–2689 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kernstine KH et al. Does tumor FDG-PET avidity represent enhanced glycolytic metabolism in non-small cell lung cancer. Ann. Thorac. Surg 109, 1019–1025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aston FW The constitution of atmospheric neon. Philos. Mag 39, 449–455 (1920). [Google Scholar]
- 162.Hevesy G. The absorption and translocation of lead by plants: a contribution to the application of the method of radioactive indicators in the investigation of the change of substance in plants. Biochem. J 17, 439–445 (1923). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hahn LA, Hevesy GC & Lundsgaard EC The circulation of phosphorus in the body revealed by application of radioactive phosphorus as indicator. Biochem. J 31, 1705–1709 (1937). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hevesy G. Adventures in Radioisotope Research: The Collected Papers of George Hevesy in Two Volumes Vol. 1 (Pergamon Press, 1962). [Google Scholar]
- 165.King AS An isotope of carbon, mass 13. Nature 124, 127 (1929). [Google Scholar]
- 166.Urey HC, Brickwedde FG & Murphy GM A hydrogen isotope of mass 2. Phys. Rev. J 39, 164–165 (1932). [Google Scholar]
- 167.Nier AO The development of a high resolution mass spectrometer: a reminiscence. J. Am. Soc. Mass Spectrom 2, 447–452 (1991). [DOI] [PubMed] [Google Scholar]
- 168.Steinwedel PWAH Notizen: Ein neues Massenspektrometer ohne Magnetfeld. Z. Naturforsch. Teil. A 8, 448–450 (1953). [Google Scholar]
- 169.Chandramouli V. et al. Quantifying gluconeogenesis during fasting. Am. J. Physiol 273, E1209–E1215 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings displayed in Fig. 3 are available in Supplementary Table 1.