

Abstract
PURPOSE
High-grade neuroendocrine carcinoma (HGNEC) of the lung is an aggressive cancer with a complex biology. We aimed to explore the prognostic value of genetic aberrations and poly(ADP-ribose) polymerase-1 (PARP1) expression in HGNEC and to establish a novel prognostic model.
MATERIALS AND METHODS
We retrospectively enrolled 191 patients with histologically confirmed HGNEC of the lung. Tumor tissues were analyzed using PARP1 immunohistochemistry (IHC; N = 191) and comprehensive cancer panel sequencing (n = 102). Clinical and genetic data were used to develop an integrated Cox hazards model.
RESULTS
Strong PARP1 IHC expression (intensity 3) was observed in 153 of 191 (80.1%) patients, and the mean PARP1 H-score was 285 (range, 5-300). To develop an integrated Cox hazard model, our data set included information from 357 gene mutations and 19 clinical profiles. When the targeted mutation profiles were combined with clinical profiles, 12 genes (ATRX, CCND2, EXT2, FGFR2, FOXO1, IL21R, MAF, TGM7, TNFAIP3, TP53, TSHR, and DDR2) were identified as prognostic factors for survival. The integrated Cox hazard model, which combines mutation profiles with a baseline model, outperformed the baseline model (incremental area under the curve 0.84 v 0.78; P = 8.79e-12). The integrated model stratified patients into high- and low-risk groups with significantly different disease-free and overall survival (integrated model: hazard ratio, 7.14 [95% CI, 4.07 to 12.54]; P < .01; baseline model: 4.38 [2.56 to 7.51]; P < .01).
CONCLUSION
We introduced a new prognostic model for HGNEC that combines genetic and clinical data. The integrated Cox hazard model outperformed the baseline model in predicting the survival of patients with HGNEC.
INTRODUCTION
Pulmonary high-grade neuroendocrine carcinomas (HGNECs) are a distinct type of lung cancer that includes small cell lung cancer (SCLC) and large cell neuroendocrine carcinoma (LCNEC). SCLC and LCNEC share common clinicopathologic characteristics, such as aggressive behavior, high proliferation rate, neuroendocrine immunohistochemistry (IHC), strong smoking association, male predominance, and poor prognosis. Although HGNEC is linked to Notch pathway activation and TP53 and RB1 inactivation caused by smoking-induced DNA damage,1,2 the lack of molecular targets and proper prognostic models is a major hurdle for the treatment of lethal HGNEC. Current SCLC treatments are stage-dependent, yet outcomes vary within stages while prognostic models for LCNEC remain scarce owing to rarity and heterogeneity.3
CONTEXT
Key Objective
What is the prognostic relevance of genetic alterations in high-grade neuroendocrine carcinoma (HGNEC) of the lungs?
Knowledge Generated
We integrated clinicopathologic characteristics, including poly (ADP-ribose) polymerase-1 immunohistochemistry expression, with mutation profiles to develop a prognostic model and identified 12 genes as prognostic factors for survival. The integrated Cox hazard model, which combines mutation profiles with a baseline model, demonstrated superior predictive performance compared with the baseline model.
Relevance
The new prognostic model effectively stratified patients into high- and low-risk groups, which may offer valuable insights for managing HGNEC patients.
SCLC exhibits recurrent genetic aberrations in DNA damage response (DDR) pathways, including poly(ADP-ribose) polymerase (PARP) and ataxia telangiectasia mutated.4-7 PARP facilitates single-strand DNA break repair via base excision. Inhibiting PARP leads to single-strand DNA breaks accumulation, potentially causing double-strand DNA breaks when DNA replication forks stall.8 These DDR pathway aberrations are theoretically plausible therapeutic targets for HGNEC. PARP inhibitors, effective in tumors with deficient homologous repair pathways such as ovarian, breast, and prostate cancers,8-10 have shown promise in early-phase clinical trials in SCLC.11-15 However, established predictive biomarkers such as BRCA1/2 mutation and homologous recombination deficiency scoring metrics do not consistently predict PARP inhibitor sensitivity in SCLC.16 Schlafen family member 11 overexpression is a potential biomarker for predicting response to veliparib treatment, although its correlation is imperfect, and mechanisms underlying nonresponsiveness remain unclear.13
In this study, we used patient tumor tissues to investigate genomic aberrations and aimed to develop a prognostic model with both clinical and genetic variables to predict survival of HGNEC. Second, clinical significance of PARP1 expression in HGNEC was evaluated using IHC.
MATERIALS AND METHODS
Study Design and Participants
This retrospective study analyzed archival formalin-fixed paraffin-embedded (FFPE) specimens from 191 patients with pulmonary HGNEC treated at the National Cancer Center of Korea between March 2001 and April 2014. Owing to limited prior data, an exploratory design was adopted. Specimens and data collection followed the research protocols approved by the National Cancer Center Ethics Committee (NCC2014-0156) with an exemption from informed consent after deidentifying information. Patient demographics, TNM staging, smoking status, and survival outcomes (overall survival [OS] and disease-free survival [DFS]) were extracted from medical records. Histological types followed the 2004 WHO classification, and neuroendocrine differentiation was confirmed using IHC markers (chromogranin, synaptophysin, and NCAM [CD56]). Tumor node metastasis staging was based on the seventh edition of the Lung Cancer Staging System.17 The recurrence or metastases were confirmed using chest computed tomography, brain magnetic resonance imaging, bone scan, and/or positron emission tomography.
IHC Analysis of PARP1 Expression
IHC staining for PARP1 was performed on 191 tumor samples using Ventana Medical Systems. Sections (5 μm) from the FFPE blocks were assessed with total PARP1 antibody (Thermo Fisher, Freemont, CA) on the Benchmark XT platform with Cell Conditioning 1 for 64 minutes, pre-peroxidase inhibition, and primary antibody incubation for 16 minutes at 37°C. PARP1 protein expression was detected using an OptiView DAB IHC Detection Kit (Ventana Medical Systems, Tuscan, AZ), with tissues counterstained with Mayer's hematoxylin and blue reagent for 4 minutes.
Observers (J.H.L. and G.-K.L.), blinded to the clinicopathological data, independently assessed tumor cores and PARP1 protein expression using a previously reported method. Positive PARP1 expression in individual cells was defined as a brown nuclear reaction, whereas cytoplasmic PARP1 expression was deemed nonspecific. PARP1 expression was evaluated using the multiplicative quick score method, which accounts both staining intensity (scored from 0 to 3: 0, no staining; 1, weak; 2, intermediate; and 3, strong) and the proportion of positive cells (scored on a scale of 0%-100%). The quick score was calculated by multiplying the proportion score by the intensity score, yielding in value ranging from 0 to 300.
Mutation Analysis
Genomic DNA underwent multiplex polymerase chain reaction (PCR) targeting 409 cancer-related genes using the Ion AmpliSeq Comprehensive Cancer Panel (Thermo Fisher, MA). This panel consists of approximately 16,000 primer pairs covering 409 known cancer-associated genes. Each sample used 10 ng of genomic DNA to prepare barcoded libraries using IonXpress barcoded adapters. Libraries, combined to a concentration of 3 ng/mL, underwent emulsion PCR with the Ion Torrent OneTouchTM 2 System and were sequenced on an Ion Proton sequencer using Ion P1 chips. Sequencing reads were aligned to 409 genes on the basis of Human Genome version 19 using Sequence Pilot v4.2.0. Read depth and amplicon coverage uniformity were also assessed. Bioinformatic analysis was performed as previously described.18
Integrated Cox Hazard Model
A data set comprising 102 samples and 376 variables (19 clinical and 357 genetic) was used to construct prognostic models. Cox hazard models were fitted using the coxph function in survival R-package. To prevent overfitting, the lasso method with leave-one-out cross-validation was used on the Cox models, aiding in selection of the most pertinent variables with nonzero estimates.19 For the evaluation of feature importance, the selection-adjusted statistics were calculated20 for the lasso model and compared with the unadjusted statistics for a multivariate Cox model fitted with the selected variables. The rank correlation of the two statistics was 1.0, and hence, the latter is reported as the proportion of the Wald statistics to the sum of all statistics, approximating the adequacy index.21 The final lasso model predicted a risk score xTβ, calculated as a linear combination of the model's coefficient vector β and the observed profile x for a test sample that was not used during training. The lasso model performance was evaluated using the time-dependent area under the receiver operating characteristic curve from 10 to 90 months.22 The models were compared using areas under the time-dependent AUC(t) curves (incremental AUC [iAUC]) calculated with the survcomp R-package and the Wilcoxon rank sum test. A 95% CI for AUC(t) was calculated using bootstrapping with 500 random samplings with replacement for each time point while maintaining the same sample size.23 The integrated model's accuracy was compared with the baseline Cox model, fitted solely to the clinical profiles using the same procedures. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated for both models. Significant correlations among selected features were depicted as solid lines of varying width (P < .05). Pearson's correlation and r2 were reported for continuous variables. Fisher's exact test or the chi-square test was used for categorical variables. The Kruskal-Wallis test was used for mixed pairs. Correlations between risk scores and other variables were also evaluated.
Statistical Analysis
Fisher's exact test was used to evaluate the relationship between PARP1 expression subgroups (low or high) and clinical variables. The Mann-Whitney U test analyzed differences in PARP1 expression scores across clinical variables. All reported P values were two-sided (P < .05). Survival curves were created using the Kaplan-Meier method. Multivariate analysis employed the Cox regression model. Statistical analyses used SPSS 25 software (IBM, Chicago, IL). To determine the optimal PARP1 expression score cutoff for survival prediction, we applied a maximally selected rank statistics test with the Maxstat R-package.
For the meta-analysis of gene expression, a standard mean difference was calculated as (tumor-normal) normalized by a pooled standard deviation using Hedges' G as the summary effect size because it statistically corrects for variance.24 For the significance test, Wald statistics and CIs were calculated on the basis of a standard normal distribution.
Enriched Pathway Analysis
All protein interactions with our markers were analyzed using STRING, a database of known and predicted functional associations from other interaction databases.25 Using the default STRING score while excluding gene fusion and mere co-occurrence, the analysis focused on known associations, with maximum of five functional partners considered per gene. Network nodes were grouped according to enriched biological processes or pathways, using a rigorous false discovery rate (FDR) cutoff of 0.001.
RESULTS
Patients' Characteristics
Table 1 summarizes the characteristics of the 191 patients included in this study. Among them, 120 (62.8%) had SCLC and 71 (37.2%) had LCNEC. The median age at diagnosis was 66 years (range, 36-89 years), predominantly male (90.7%) and ever-smokers (81.4%). Never-smokers were more common among patients with SCLC (24.2%) than patients with LCNEC (9.9%). Patients with SCLC had worse baseline performance status and were diagnosed at more advanced stages (limited stage 29, 24.2%; extensive stage 91, 75.8%; stage I-III 39, 32.5%; stage IV 81, 67.5%) compared with patients with LCNEC (stage I-III, 53, 74.6%; stage IV 18, 25.4%; P < .01). Frontline treatment varied by histological type and stage. Genomic data were available for 102 of 191 patients (Data Supplement, Fig S1), with similar characteristics to the original cohort.
TABLE 1.
Characteristics of Patients
| Characteristic | Total (N = 191) | Patients With Genetic Information (n = 102) |
|---|---|---|
| Histology, No. (%) | ||
| SCLC | 120 (62.8) | 63 (61.8) |
| LCNEC | 71 (37.2) | 39 (38.2) |
| Age at diagnosis, median (range) | 66 (36-89) | 67 (36-85) |
| Sex, No. (%) | ||
| Male | 173 (90.6) | 94 (92.2) |
| Female | 18 (9.4) | 8 (7.8) |
| ECOG performance, No. (%) | ||
| 0 | 22 (11.5) | 13 (12.7) |
| 1 | 123 (64.4) | 59 (57.8) |
| 2 | 30 (15.7) | 19 (18.6) |
| 3 | 16 (8.4) | 11 (10.8) |
| Smoking status, No. (%) | ||
| Never-smoker | 36 (18.8) | 21 (20.6) |
| Smoker | 155 (81.2) | 81 (79.4) |
| Smoking pack-years, median (range) | 44 (1-214) | 45 (1-214) |
| TNM stage, No. (%) | ||
| 1 | 25 (13.1) | 20 (19.6) |
| 2 | 12 (6.3) | 7 (6.9) |
| 3 | 55 (28.8) | 28 (27.5) |
| 4 | 99 (51.8) | 47 (46.1) |
| First treatment, No. (%) | ||
| Surgery | 44 (25.7) | 29 (30.5) |
| Chemoradiation | 34 (15.8) | 9 (9.5) |
| Chemotherapy | 95 (60.8) | 57 (60.0) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; LCNEC, large cell neuroendocrine carcinoma; SCLC, small cell lung cancer.
PARP1 Expression and Its Clinical Correlations
PARP1 protein expression was assessed in 191 tissue samples, and Figure 1 shows representative images. Most tumors showed strong nuclear staining in tumor cells (intensity 3 = 153/191, 80.1%), whereas a small percentage showed low PARP1 expression (intensity 1 = 2/191, 1.0%; intensity 2 = 36/191, 18.8%). The median extent of PARP1 expression was 95% (range, 5%-100%), and the median H-score was 285 (range, 5-300). The mean PARP1 H-scores were higher in SCLC than in LCNEC, but the difference was not statistically significant (263.5 v 249.7; P = .14; Fig 1D, left). In the combined cohort of SCLC and LCNEC tumors, no association was found between the mean PARP1 H-score and baseline characteristics such as age, sex, smoking history, and disease stage. However, in patients with extensive-stage SCLC initially treated with chemotherapy, those with stable or progressive disease had a higher mean PARP1 H-score (n = 12, 276.3) than those with complete or partial response (n = 62, 254.7; P = .02; Fig 1D, right). Survival outcomes were analyzed separately by histology, as Cox regression analysis identified histology and stage as statistically significant prognostic factors. Within various subgroups stratified by histology and stage, segregating patients into high or low PARP1 expression subgroups using the maximal χ2 method yielded no prognostic significance.
FIG 1.
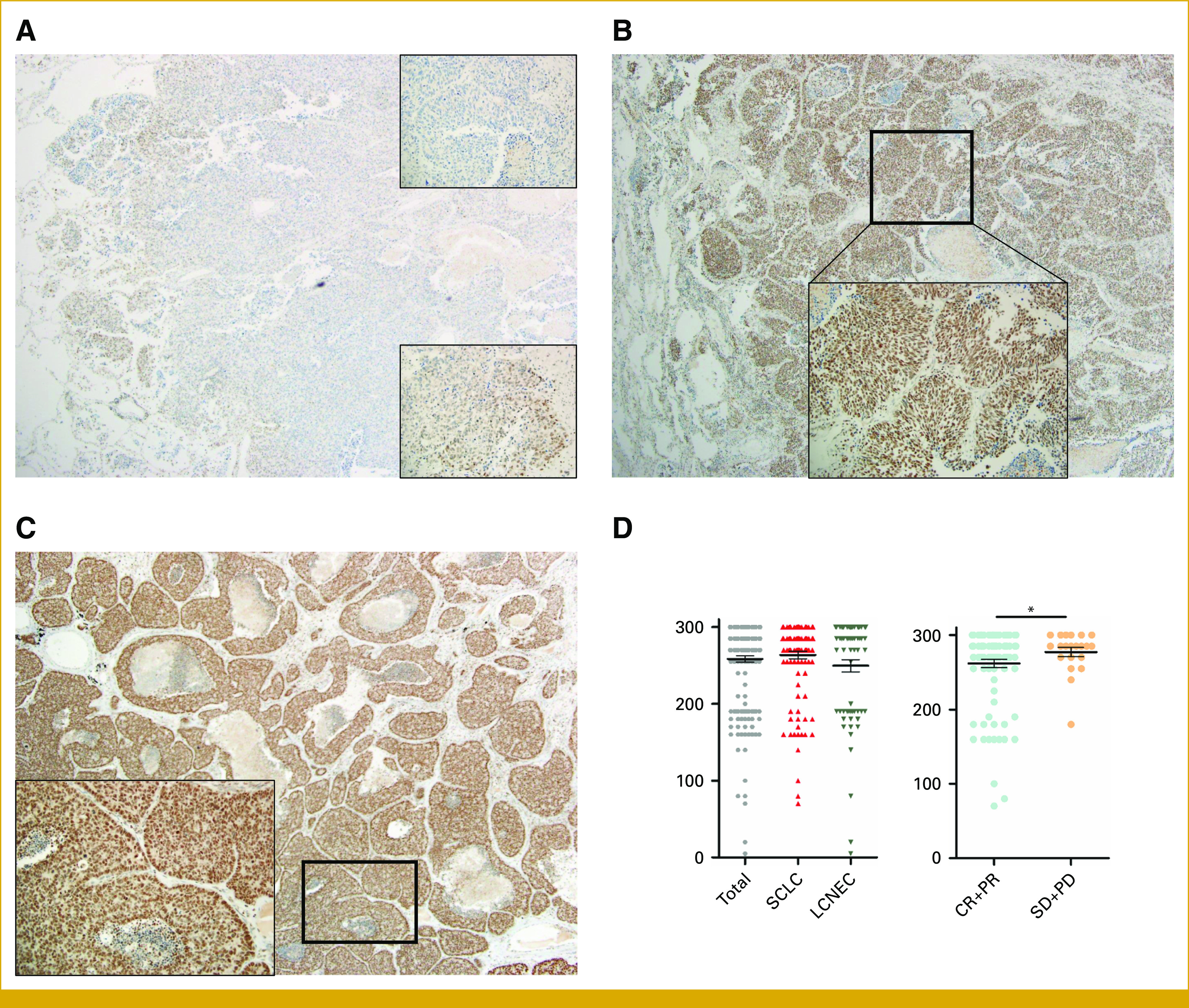
Expression of PARP1 immunohistochemistry. (A) Intensity 1, extent 5%, ×40 (upper right, ×200). No PARP1 expression in tumor compared with internal control (positively stained lymphocytes; lower right, ×200) focal area of PARP1 expressing tumors with intensity 1. (B) Intensity 2, extent 95%, ×40 (right, ×200) strong PARP1 expression in most tumor cells, but the intensity is lower than internal control (positively stained lymphocytes). (C) Intensity 3, extent 100%, ×40 (right, ×200) strong PARP1 expression in all tumor cells, the intensity is same or slightly lower than internal control (positively stained lymphocytes). (D) Scatter dot plots showing PARP1 score by histology (left) and response to chemotherapy (right). * P < .05. CR, complete response; LCNEC, large cell neuroendocrine carcinoma; PARP1, poly (ADP-ribose) polymerase-1; PD, progressive disease; PR, partial response; SCLC, small cell lung cancer; SD, stable disease.
Genetic Aberrations Identified From Targeted Sequencing
Of 191 HGNEC specimens, 107 were suitable for genetic analysis (Data Supplement, Fig S1). Of the 43 LCNEC specimens, we identified a relatively high prevalence of nonsynonymous mutations in SYNE1 (33/43, 77%), CIC (32/43, 74%), CREBBP (27/43, 63%), and SEPT9 (25/43, 58%). By contrast, we identified a relatively high prevalence of nonsynonymous mutations in PPP2R1A (35/64, 55%), ARID1A (33/64, 52%), TRRAP (31/64, 48%), and NOTCH2 (11/64, 17%) in SCLC. Mutations exhibiting statistically significant differences in frequency on the basis of histology are presented in the Data Supplement (Table S1). Several dominant mutations were identified (Data Supplement, Table S2), including the P854L mutation in the FLT3 tyrosine kinase domain, which has not been previously reported in the COSMIC database.
Prognostic Model Through Integration of Genomic and Clinical Data
PARP1 IHC expression alone showed limited correlation with clinical factors, and we investigated whether the addition of the mutational status of genomic data to multimodal data would help with outcome prediction. We constructed an integrated Cox hazard model using a data set of 357 gene mutational profiles and 19 clinical profiles from 102 individuals. The 19 clinical variables encompassed age, sex, smoking history, smoking status, performance status, comorbidity scores, histology, stage, treatment type, and response to chemotherapy. Four of these variables (histology, stage, age, and PARP1 H-score) were identified as clinical prognostic factors and incorporated into a baseline model. Combining the targeted mutation profiles with the clinical profiles revealed 11 genes (ATRX, CCND2, EXT2, FGFR2, FOXO1, IL21R, MAF, TGM7, TNFAIP3, TP53, and TSHR) as prognostic factors for OS and three genes (DDR2, MAF, and TGM7) for DFS (Fig 2A) through the feature selection (See “Integrated Cox hazard model”). To assess the predictive performance, we used the time-dependent AUC(t), as prognostic models guide decisions across multiple disease time points.20 The integrated model consistently exhibited superior predictive performance over the baseline model at each time point (Table 2), as reflected in the area under the AUC(t) (iAUC) summary.24 Moreover, the integrated model outperformed the baseline model in true-positive fraction at a fixed false-positive fraction of 20% (or sensitivity for a fixed 1–specificity; Data Supplement, Fig S2). For OS in the baseline model (Fig 3A, blue), performance declined over time, reaching a maximum AUC(t) of 0.90 at 32 months and 0.73 at the final time point. By contrast, the integrated model (orange) maintained consistent performance with a lesser decline (max. AUC(t) of 0.90 and 0.85). The iAUC's of the competing models were significantly different (0.84 v 0.78; Wilcoxon test; P = 8.79e-12). A similar trend was observed for DFS (iAUC of 0.85 v 0.81; P = 8.03e-10; Fig 3B). The Data Supplement (Table S3) provides additional metrics, such as PPV and NPV, for comprehensive comparison.
FIG 2.
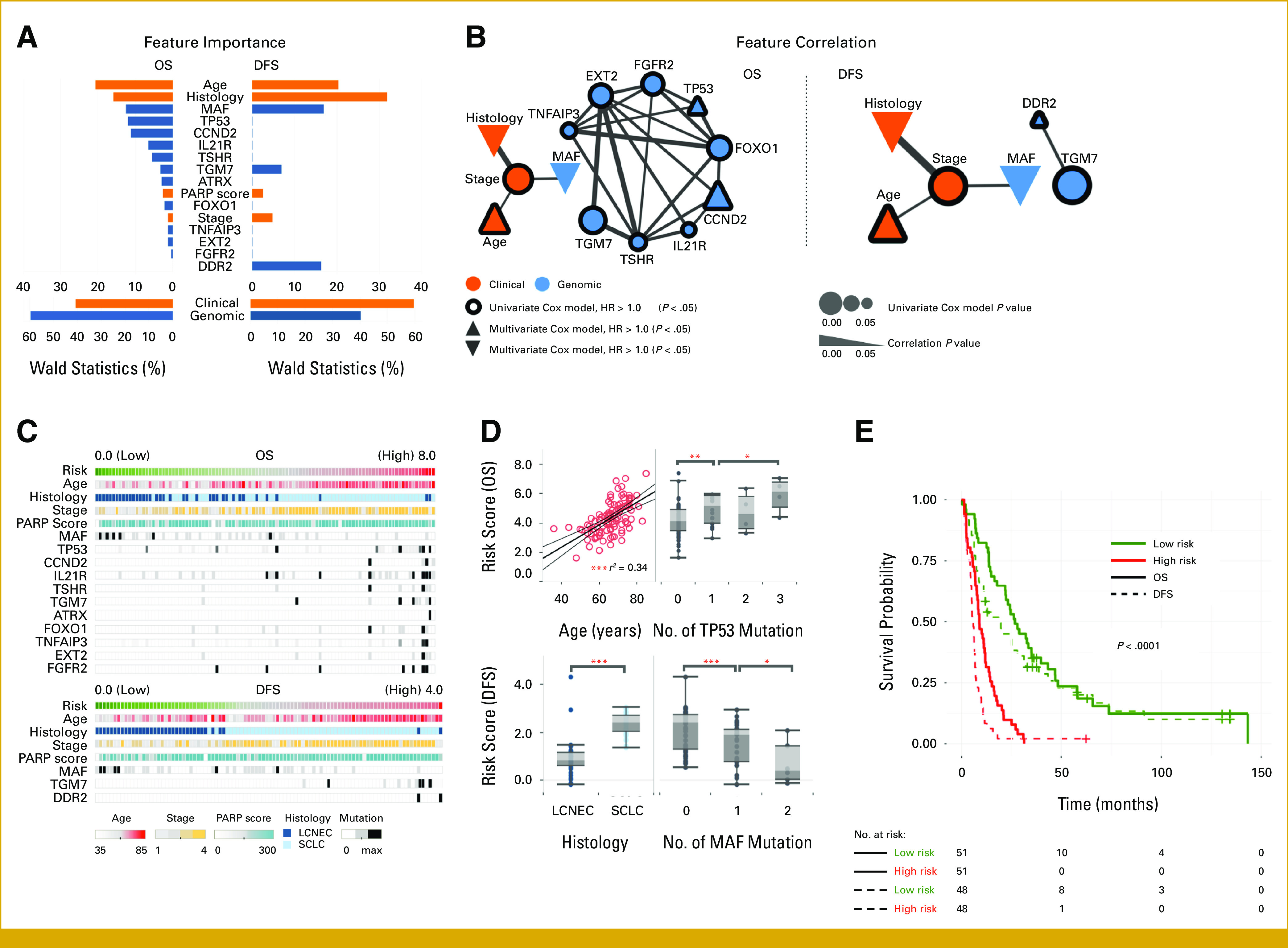
Importance of clinical and genomic features selected in profiles of patients with lung cancer (n = 102) and their correlation. (A) Top, feature importance in multivariate Cox models (percentage of the Wald statistics); bottom, total importance for each feature group. (B) A node represents a feature, and its size is proportional to significance of univariate Cox model (P < .05). A line represents significant correlation of a pair of features (P < .05), and its width is proportional to significance. Features are highlighted with border and shape according to univariate and multivariate Cox HR, respectively. (C) Risk score and clinical and genomic phenotypes of patients with lung cancer. (D) Relations of risk score with important prognostic features. Risk is associated with age (linear correlation r = 0.58). Higher mutation burden on TP53 is associated with higher risk for OS. LCNEC and MAF mutations are associated with lower risk for DFS. (E) Kaplan-Meier curves of survival groups for high- and low risk patients, categorized by the median risk score. *P < .05, **P < .01, ***P < .001. DFS, disease-free survival; HR, hazard ratio; LCNEC, large cell neuroendocrine carcinoma; OS, overall survival; SCLC, small cell lung cancer.
TABLE 2.
Time-Dependent Performance of Baseline and Integrated Risk Models With Respect to AUC at Each Year
| Survival | AUC(t) (95% CI) | |||||
|---|---|---|---|---|---|---|
| Model | t = 1 year | t = 2 year | t = 3 year | t = 4 year | t = 5 year | |
| OS | Baseline | 0.75 (0.62 to 0.86) | 0.85 (0.72 to 0.93) | 0.87 (0.7 to 0.94) | 0.83 (0.64 to 0.92) | 0.78 (0.56 to 0.92) |
| Integrative | 0.82 (0.71 to 0.90) | 0.88 (0.77 to 0.95) | 0.88 (0.76 to 0.96) | 0.85 (0.69 to 0.95) | 0.86 (0.69 to 0.95) | |
| DFS | Baseline | 0.89 (0.74 to 0.95) | 0.87 (0.78 to 0.94) | 0.88 (0.76 to 0.94) | 0.82 (0.71 to 0.92) | 0.82 (0.66 to 0.93) |
| Integrative | 0.90 (0.78 to 0.96) | 0.89 (0.81 to 0.96) | 0.89 (0.78 to 0.96) | 0.85 (0.73 to 0.94) | 0.82 (0.70 to 0.93) | |
Abbreviations: DFS, disease-free survival; OS, overall survival.
FIG 3.
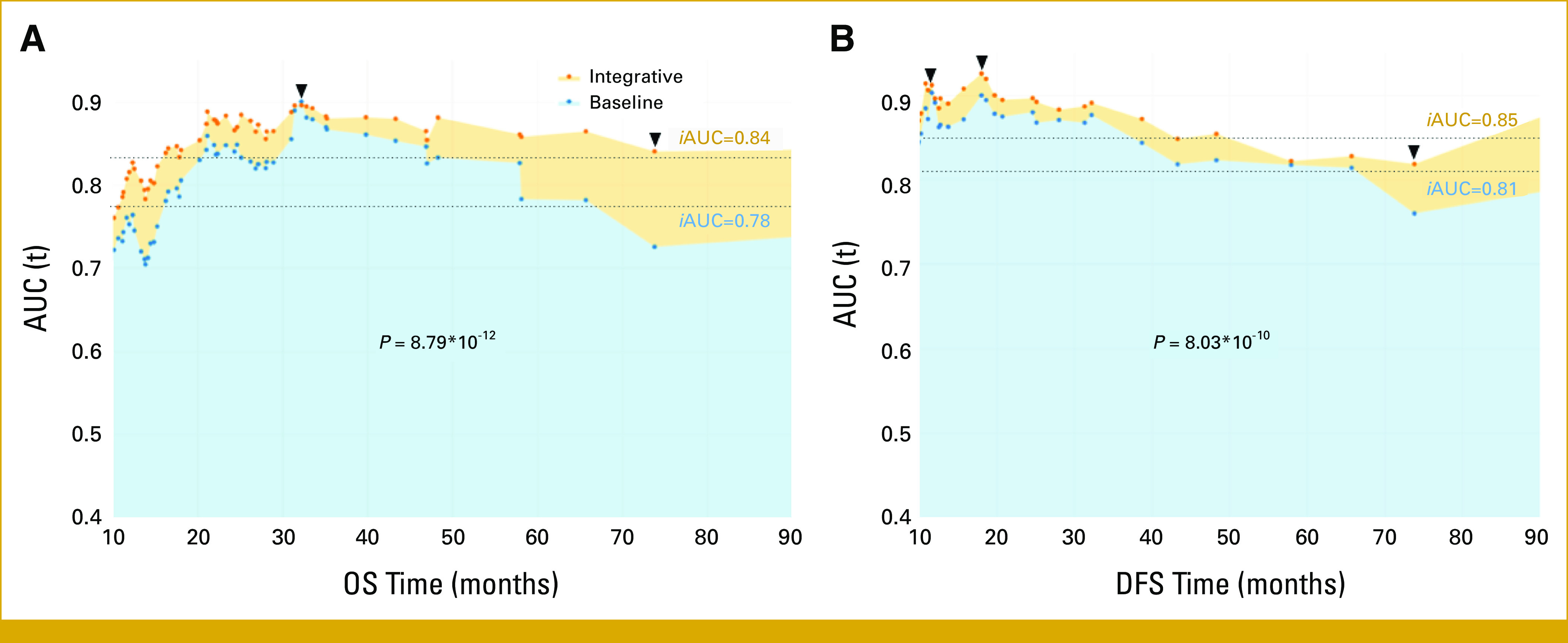
Time-dependent AUC(t) was measured from 10 months to 90 months at every event time, reflecting the performance of predicting (A) OS and (B) DFS at different time points for the integrative risk model (with genomic and clinical variables) and the baseline model (solely with clinical variables). iAUC values summarize the AUC(t) with P value of Wilcoxon rank sum test for the difference between AUC(t). DFS, disease-free survival; iAUC, incremental AUC; OS, overall survival.
Figure 2E demonstrates the robust ability of the integrated model to generate scores strongly correlated with patient outcomes. Patients were stratified into high- or low-risk groups on the basis of the median score, and survival analysis for OS and DFS was conducted. The integrated model outperformed the baseline model for both OS (P = 1e-10) and DFS (P = 8e-09) by assigning high scores to patients with early events, compared with the baseline model for OS (P = 6e-10) and DFS (P = 3e-08). Adjusting the cut threshold with the Maxstat R-package optimized for survival outcome association further improved performance (P = 9e-12 v 3e-11 and P = 3e-13 v 1e-08 for OS and DFS, respectively). In addition, the results of the univariate Cox hazard model with dichotomized scores favored the integrated approach (P = 2e-10 v 7e-10 and P = 8e-12 v P = 8e-08 for OS and DFS, respectively). The Data Supplement (Table S4) provides a comprehensive comparison of these results. Consistent with previous research, stage and age were identified as prognostic factors,26,27 and our cohort exhibited higher risk among elderly patients (linear correlation, r = 0.58), many TP53 mutations, or SCLC histological type (Fig 2D). Conversely, patients with many MAF mutations tend to have lower disease stages and better survival rates. This prompts an exploration of the interactions among prognostic variables. A systematic examination of variable pairs revealed significant correlations, forming a network (Fig 2B). The network showcased two clusters, genomic and clinical, suggesting their independence yet complementarity. Within the clinical cluster, the stage was significantly correlated with age (P = .017), histology (P = 9.6e-08), and MAF mutations (P = .007). Notably, the OS and DFS correlation network exhibited a quadruplet structure, underscoring their crucial roles in predicting survival outcomes in HGNEC. The correlation was most discernible in the spectrum of risk scores, arranged in an ascending order (Fig 2C).
Meta-Analysis of Gene Expression
We computed the standardized mean difference of gene expression across two data sets from previous studies on lung neuroendocrine carcinoma (NEC)27,28 to assess tumor expression distinctions from normal. Among the 13 genes, nine were significantly altered in tumors (Fig 4A; P < .005). Genes were ranked by importance (Fig 2A). The most crucial genes were upregulated (TP53, IL21R, TSHR, TGM7, and PARP1), whereas less critical genes were downregulated (ATRX, FOXO1, TNFAIP3, and DDR2). Notably, PARP1 showed substantial upregulation in NEC (P = 4.6e-21) and various tumor types across 20 studies,27-44 including TCGA lung adenocarcinoma and squamous cell carcinoma (P = 2.5e-58; Data Supplement, Fig S3). The high PARP1 expression in the meta-analysis aligned with our results (IHC intensity of 3 in 80.1% of samples). Dysregulated genes in lung NEC are highlighted (*) in Figure 4B.
FIG 4.
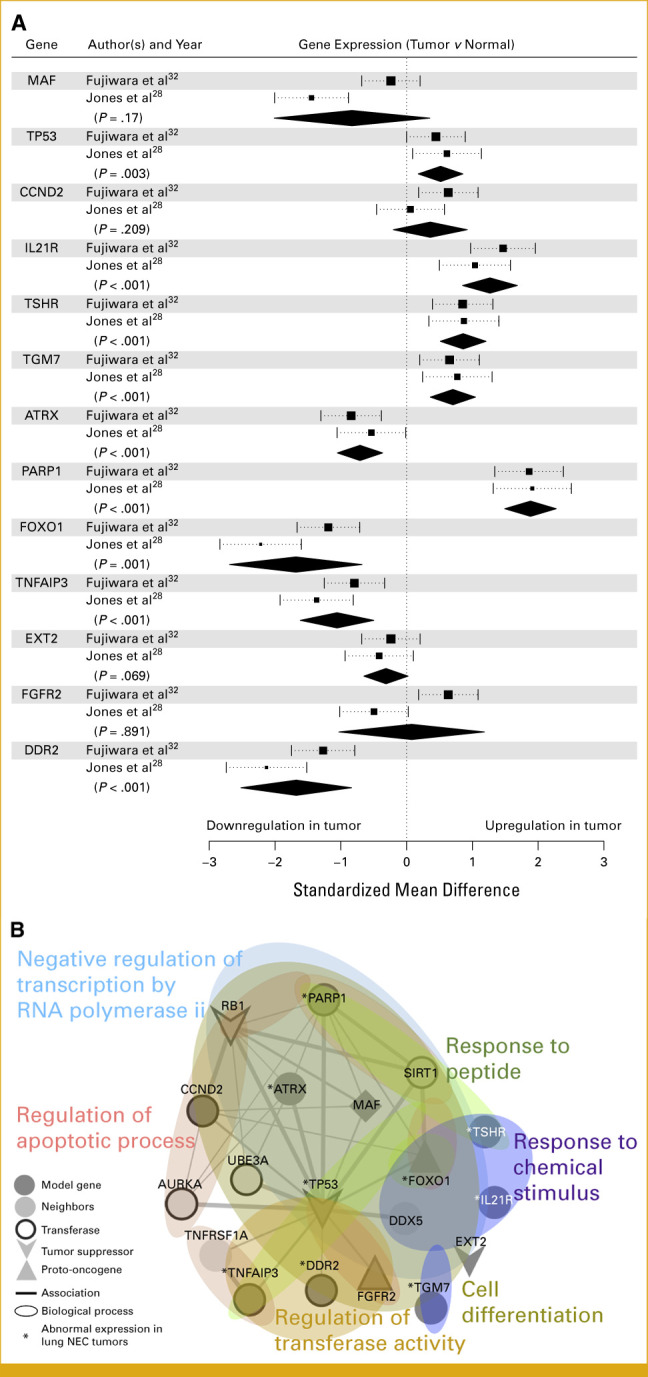
(A) Forest plot shows the difference in gene expression (square) with 95% CI for lung HGNEC samples in two different studies. A summary diamond is added to show a pooled difference for each gene selected in this study, and its P value is highlighted (95% CI is depicted by length of the diamond). (B) Association (line) among prognostic genes and their neighbors (node). Line width is in proportion to confidence score (>0.4). Genes are colored according to enriched biological processes, and their tumorigenic roles are represented by node shape (MAF is both tumor suppressor and oncogene). Genes for transferase are depicted with thick border. (A) The genes of abnormal expression in tumors are highlighted with (*). HGNEC, high-grade neuroendocrine carcinoma.
Enrichment Analysis of Marker Genes
In this study, we aimed to identify biological processes influencing HGNEC risk. Genes from our models and their functional partners from the STRING database25 were pooled. Figure 4B shows the gene ontology analysis results for the 17 pooled genes, revealing functional links, especially with TP53, within biological processes affecting HGNEC risk. Several gene clusters engaged in cellular regulation, responses, and/or differentiation (FDR <0.001). Notably, mutated genes (MAF, FOXO1, DDX5, TP53, and ATRX) were significantly involved in negative regulation of transcription by RNA polymerase II (FDR = 0.00012, GO:0000122). Mutations in this pathway, including the tumor suppressors TP53, RB1, and MAF, might be associated with high PARP1 expression via dysregulated expression of ATRX, FOXO1, and TP53 in HGNEC. Some mutations in tumor suppressor EXT2 involved in cell differentiation (FDR = 5.22e-05, GO:0045595), and others related to regulation of apoptotic process (FDR = 0.00025, GO:0042981) may disrupt apoptosis through high PARP1 and low TNFAIP3 expression. Additionally, mutations in genes involved in response to chemical stimulus (FDR = 0.00018, GO:0070887) were linked to altered expression (TSHR and IL21R overexpression, FOXO1 downregulation), implying a potential abnormal response to toxic chemicals in 81 (78.6%) ever-smoker patients in our cohort. The risk was also associated with mutations in transferase/kinase regulators (eg, TNFAIP3 and DDR2), crucial in signaling pathways (FDR = 0.003, GO:0051338). Mutations causing constitutive kinase activation or disrupting normal FGFR2 maturation, internalization, and degradation can induce aberrant signaling (UniProt: Q91147). Many transferase partners (AURKA, UBE3A, TNFRSF1A, SIRT1, and DDX5) are connected to this pathway through the hub protein TP53. Notably, the proto-oncogene MAF (BZIP transcription factor) is expressed in monocytes and macrophages, acting as both a positive and negative regulator of cytokine-encoding gene expression. It is also a critical controller of immunosuppressive M1/M2 macrophage polarization and functions in cancer. Deletion of MAF resulted in reduced tumor burden with enhanced antitumor T-cell immunity in lung cancer, providing a partial explanation for the correlation of hypermutations with low-stage (P = .007) and low-risk scores (Figs 2B-2D). MAF had the largest effect size in the risk model (Fig 4A). These results suggest that MAF's mutational burden could serve as a marker for effective antitumor T-cell immunity in HGNEC. Further studies are necessary to determine the connections from TSHR, IL21R, EXT2, or TGM7 to the enriched pathways in lung HGNEC.
DISCUSSION
HGNEC, an aggressive lung cancer subtype with extraordinarily complex biology but lacking an established prognostic model,45,46 may benefit from integrating with SCLC and LCNEC because of the potential biological similarities. Our data-driven modeling, involving clinicians and biological researchers, offers a novel method for identifying the best prognostic predictors. Despite insights into the interconnected nature of prognostic factors in HGNEC survival, practical constraints and costs limit widespread clinical application. Further validation across diverse cohorts is required to ascertain the biological and clinical implications of our findings.
This study leveraged patient-level real-world genomic and clinicogenomic data sets to evaluate rare mutations. Through various regression analyses and the LASSO-Cox model, we identified a set of 12 genes and PARP1 expression levels as components of an integrated prognostic model. Mutations in CCND2, FOXO1, IL2R, INFAIP3, MAF, and TGM7 were identified as prognostic factors for HGNEC survival. Although these mutations are not standard cancer hallmark genes,47 meta-analysis and GO enrichment analysis revealed their associations with immune cell signaling, cytokines, genomic instability, and metabolic derangement in cancer cells. Particularly, TGM7 and MAF mutations affected both DFS and OS. TGM7 gene encodes a transglutaminase involved in protein stabilization48-50 and potentially influencing carcinogenesis and the tumor microenvironment, including epithelial-mesenchymal transition and macrophage infiltration into the tumor.51,52 MAF, known as a transcriptional modulator, has been linked to multiple myeloma and lymphoma, tumor-stroma interactions, and lymphocyte regulation, suggesting its significance in tumorigenesis, metastasis, and manipulation of the tumor microenvironment.53,54 Liu et al55 evaluated c-MAF as a controller of immunosuppressive macrophages in lung cancer and suggested that c-MAF is a potential target for effective tumor immunotherapy. Further exploration of TGM7 and MAF roles in HGNEC is warranted to better understand their implications.
Previous studies have linked high PARP1 expression to poor prognosis in non–small cell lung cancer (NSCLC).56,57 In lung NEC, meta-analysis indicated high PARP1 expression in HGNEC,27,28 yet the underlying mechanisms for universally high PARP1 expression compared with NSCLC4 remain unknown. Given emerging evidence of DDR pathways defects in HGNEC, high PARP1 expression may offer avenues for novel therapies. Although PARP1 expression alone did not correlated with patient survival in our study, it correlated in our integrated prognostic model. GO analysis suggested that mutations in various signaling pathways may contribute to increase PARP1 expression in HGNEC, warranting further investigation.
In conclusion, our study emphasizes the importance of integrating genetic aberrations, high PARP1 expression, and clinical variables into prognostic models for HGNEC. However, the small sample size and retrospective design may limit the generalizability of our findings, highlighting the need for validation and refinement in larger cohorts. Further research is necessary to identify additional prognostic biomarkers for HGNEC.
DISCLAIMER
The study sponsor had no role in the study design; collection, analysis, and interpretation of data; writing of the report; or decision to submit the paper for publication.
SUPPORT
Supported by the National Cancer Center Research Grant (NCC-2210550 to J.-Y.H.).
H.S.K. and J.K.K. equally contributed.
DATA SHARING STATEMENT
The data underlying this article will be shared on reasonable request by the corresponding author.
AUTHOR CONTRIBUTIONS
Conception and design: Hye Sook Kim, Ji-Youn Han
Financial support: Ji-Youn Han
Administrative support: Ji-Youn Han
Provision of study materials or patients: Geon-Kuk Lee, Ji-Youn Han, Young Joo Lee
Collection and assembly of data: All authors
Data analysis and interpretation: Hye Sook Kim, Jong Kwang Kim, Jeong Hyeon Lee, Geon-Kuk Lee, Ji-Youn Han
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Young Joo Lee
Consulting or Advisory Role: Roche, MSD, Yuhan
Ji-Youn Han
Stock and Other Ownership Interests: Yuhan
Honoraria: AstraZeneca, Takeda, Novartis, Merck, Janssen, Pfizer, Yuhan
Consulting or Advisory Role: Bristol Myers Squibb, Novartis, Pfizer, Merck, Abion, J Ints Bio, Takeda, Janssen, Lantern Pharma
Research Funding: Roche, Pfizer, Ono Pharmaceutical, Takeda
No other potential conflicts of interest were reported.
REFERENCES
- 1.Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM) : A genomics-based classification of human lung tumors. Sci Transl Med 5:209ra153, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyoshi T, Umemura S, Matsumura Y, et al. : Genomic profiling of large-cell neuroendocrine carcinoma of the lung. Clin Cancer Res 23:757-765, 2017 [DOI] [PubMed] [Google Scholar]
- 3.Yang Q, Xu Z, Chen X, et al. : Clinicopathological characteristics and prognostic factors of pulmonary large cell neuroendocrine carcinoma: A large population-based analysis. Thorac Cancer 10:751-760, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byers LA, Wang J, Nilsson MB, et al. : Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov 2:798-811, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George J, Lim JS, Jang SJ, et al. : Comprehensive genomic profiles of small cell lung cancer. Nature 524:47-53, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross JS, Wang K, Elkadi OR, et al. : Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol 67:772-776, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wakuda K, Kenmotsu H, Serizawa M, et al. : Molecular profiling of small cell lung cancer in a Japanese cohort. Lung Cancer 84:139-144, 2014 [DOI] [PubMed] [Google Scholar]
- 8.Audeh MW, Carmichael J, Penson RT, et al. : Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 376:245-251, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Gelmon KA, Tischkowitz M, Mackay H, et al. : Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12:852-861, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Mateo J, Carreira S, Sandhu S, et al. : DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373:1697-1708, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lallo A, Frese KK, Morrow CJ, et al. : The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 24:5153-5164, 2018 [DOI] [PubMed] [Google Scholar]
- 12.Owonikoko TK, Dahlberg SE, Khan SA, et al. : A phase 1 safety study of veliparib combined with cisplatin and etoposide in extensive stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E2511). Lung Cancer 89:66-70, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietanza MC, Waqar SN, Krug LM, et al. : Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol 36:2386-2394, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas A, Mian I, Tlemsani C, et al. : Clinical and genomic characteristics of small cell lung cancer in never smokers: Results from a retrospective multicenter cohort study. Chest 158:1723-1733, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wainberg ZA, Rafii S, Ramanathan RK, et al. : Safety and antitumor activity of the PARP inhibitor BMN673 in a phase I trial recruiting metastatic small-cell lung cancer (SCLC) and germline BRCA-mutation carrier cancer patients. J Clin Oncol 32, 2014. (suppl 15; abstr 7522) [Google Scholar]
- 16.Lok BH, Gardner EE, Schneeberger VE, et al. : PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res 23:523-535, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Travis W, Brambilla E, Müller-Hermelink HK, et al. : Pathology and Genetics: Tumours of the Lung, Pleura, Thymus and Heart. Lyon, France, IARC, 2004 [Google Scholar]
- 18.Han JY, Kim SH, Lee YS, et al. : Comparison of targeted next-generation sequencing with conventional sequencing for predicting the responsiveness to epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) therapy in never-smokers with lung adenocarcinoma. Lung Cancer 85:161-167, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Tibshirani R: The lasso method for variable selection in the Cox model. Stat Med 16:385-395, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Taylor J, Tibshirani R: Post-selection inference for ℓ1-penalized likelihood models. Can J Stat 46:41-61, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrell FE: Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis. Springer, pp 207, 2001 [Google Scholar]
- 22.Heagerty PJ, Lumley T, Pepe MS: Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 56:337-344, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Kohavi R: A Study of Cross-Validation and Bootstrap for Accuracy Estimation and Model Selection. Montreal, Canada, IJCAI, 1995, pp 1137-1145 [Google Scholar]
- 24.Hedges LV, Olkin I: Statistical Methods for Meta-Analysis. Academic Press, pp 78, 1985 [Google Scholar]
- 25.https://string-db.org
- 26.Wang S, Yang L, Ci B, et al. : Development and validation of a nomogram prognostic model for SCLC patients. J Thorac Oncol 13:1338-1348, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong J, Zheng Q, An T, et al. : Nomogram to predict cause-specific mortality in extensive-stage small cell lung cancer: A competing risk analysis. Thorac Cancer 10:1788-1797, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones MH, Virtanen C, Honjoh D, et al. : Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet 363:775-781, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Beer DG, Kardia SL, Huang CC, et al. : Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 8:816-824, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Bhattacharjee A, Richards WG, Staunton J, et al. : Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98:13790-13795, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network : Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519-525, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiwara T, Hiramatsu M, Isagawa T, et al. : ASCL1-coexpression profiling but not single gene expression profiling defines lung adenocarcinomas of neuroendocrine nature with poor prognosis. Lung Cancer 75:119-125, 2012 [DOI] [PubMed] [Google Scholar]
- 33.Hou J, Aerts J, den Hamer B, et al. : Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One 5:e10312, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabbout M, Garcia MM, Fujimoto J, et al. : ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res 19:3383-3395, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landi MT, Dracheva T, Rotunno M, et al. : Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 3:e1651, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu TP, Tsai MH, Lee JM, et al. : Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol Biomarkers Prev 19:2590-2597, 2010 [DOI] [PubMed] [Google Scholar]
- 37.The Cancer Genome Atlas Research Network : Comprehensive molecular profiling of lung adenocarcinoma. Nature 511:543-550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okayama H, Kohno T, Ishii Y, et al. : Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res 72:100-111, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Rousseaux S, Debernardi A, Jacquiau B, et al. : Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med 5:186ra66, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Palencia A, Gomez-Morales M, Gomez-Capilla JA, et al. : Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int J Cancer 129:355-364, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Selamat SA, Chung BS, Girard L, et al. : Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res 22:1197-1211, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su LJ, Chang CW, Wu YC, et al. : Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8:140, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xi L, Feber A, Gupta V, et al. : Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Res 36:6535-6547, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu K, Ganesan K, Tan LK, et al. : A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet 4:e1000129, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan H, Shi X, Xiao D, et al. : Nomogram prediction for the survival of the patients with small cell lung cancer. J Thorac Dis 9:507-518, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baty F, Facompre M, Kaiser S, et al. : Gene profiling of clinical routine biopsies and prediction of survival in non-small cell lung cancer. Am J Respir Crit Care Med 181:181-188, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Grenard P, Bates MK, Aeschlimann D: Evolution of transglutaminase genes: Identification of a transglutaminase gene cluster on human chromosome 15q15. Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J Biol Chem 276:33066-33078, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Rafnar T, Sulem P, Besenbacher S, et al. : Genome-wide significant association between a sequence variant at 15q15.2 and lung cancer risk. Cancer Res 71:1356-1361, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin TA, Gomez K, Watkins G, et al. : Expression of breast cancer specific gene-1 (BCSG-1/gamma-synuclein) is associated with tumour grade but not with clinical outcome of patients with breast cancer. Oncol Rep 16:207-212, 2006 [PubMed] [Google Scholar]
- 50.Earp MA, Raghavan R, Li Q, et al. : Characterization of fusion genes in common and rare epithelial ovarian cancer histologic subtypes. Oncotarget 8:46891-46899, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho SY, Oh Y, Jeong EM, et al. : Amplification of transglutaminase 2 enhances tumor-promoting inflammation in gastric cancers. Exp Mol Med 52:854-864, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao L, Shao M, Schilder J, et al. : Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 31:2521-2534, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Hurt EM, Wiestner A, Rosenwald A, et al. : Overexpression of c-MAF is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 5:191-199, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Annunziata CM, Hernandez L, Davis RE, et al. : A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 117:2396-2404, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu M, Tong Z, Ding C, et al. : Transcription factor c-MAF is a checkpoint that programs macrophages in lung cancer. J Clin Invest 130:2081-2096, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michels J, Vitale I, Galluzzi L, et al. : Cisplatin resistance associated with PARP hyperactivation. Cancer Res 73:2271-2280, 2013 [DOI] [PubMed] [Google Scholar]
- 57.Michels J, Adam J, Goubar A, et al. : Negative prognostic value of high levels of intracellular poly(ADP-ribose) in non-small cell lung cancer. Ann Oncol 26:2470-2477, 2015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying this article will be shared on reasonable request by the corresponding author.