

Summary:
The gene amplification of Sox2 at 3q26.33 is a common event in squamous cell carcinomas (SCCs) of lung, esophagus, and several other cancers. Here, we show that the expression of LSD1/KDM1 histone demethylase is significantly elevated in Sox2-expressing lung SCCs. LSD1-specific inhibitors selectively impair the growth of Sox2-expressing lung SCC cells but not that of Sox2-negative cells. Sox2 expression is associated with the sensitivity towards LSD1 inhibition in lung, breast, ovarian, and other carcinoma cells. Inactivation of LSD1 reduces Sox2 expression, promotes G1 cell cycle arrest, and induces genes for differentiation by selectively modulating the methylation states of histone H3 at lysines 4 (H3K4) and 9 (H3K9). Reduction of Sox2 further suppresses Sox2-dependent lineage-survival oncogenic potential, elevates tri-methylation of histone H3 at lysine 27 (H3K27), and enhances growth arrest and cellular differentiation. Our studies suggest that LSD1 serves as a selective epigenetic target for the therapy of Sox2-expressing cancers.
Graphical Abstract
Introduction:
Lung cancer is the most frequent cause of cancer death worldwide (Jemal et al., 2009; Jones et al., 2006). Squamous cell carcinoma of the lung is a major form of frequent and aggressive lung cancer (Jones et al., 2006). Recent studies show that the gene amplification of Sox2 that encodes a high mobility group domain-containing transcription factor is the most frequent and common event in squamous cell carcinomas of the lung, esophagus, and oral cavity at 3q26.33 (Bass et al., 2009; Hussenet et al., 2010; Maier et al., 2011). It is also amplified in a fraction of small-cell lung carcinomas (Rudin et al., 2012) and glioblastoma multiforme (GBM)(Alonso et al., 2011). Sox2 is a master regulator of pluripotent embryonic stem cells (ESCs) and adult neural stem cells (Episkopou, 2005; Okita and Yamanaka, 2006). It reprograms somatic cells into the induced pluripotent stem cells (iPSCs) with Oct4, Klf4, and c-Myc or with Oct4, Lin 28, and Nanog (Takahashi et al., 2007; Yu et al., 2007). Sox2 also plays an essential role in the morphogenesis and homeostasis of the esophageal, tracheo-bronchial and bronchiolar epithelia (Que et al., 2007). Sox2 acts as a lineage-survival oncogene for the expression of pluripotent stem cell signatures and the lineage-specific genes in lung squamous cell carcinomas (Bass et al., 2009). Ectopic expression of Sox2 causes the oncogenic transformation of normal tracheobronchial epithelial cells (Bass et al., 2009). Sox2 is frequently expressed in other types of poorly differentiated and aggressive human cancers (Ben-Porath et al., 2008). In lung adenocarcinomas, Sox2 expression is associated with poor prognosis (Sholl et al., 2010). In stem cell-like ovarian cancer cells, Sox2 is also co-expressed with Oct4 and Lin28 (Peng et al., 2008; Zhong et al., 2010). In breast carcinomas, Sox2 expression is required for mammosphere formation as part of stem cell-like properties (Leis et al., 2012). In colon cancer, Sox2 expression correlates with lymph-node and distant metastases (Neumann et al., 2011). In these cancers, Sox2 appears to confer certain progenitor/stem cell properties to carcinoma cells to allow them to differentiate.
Histone methylation is a major covalent modification of histones that provides the structural and functional characteristics of chromatin to epigenetically define gene expression patterns in a cell (Klose and Zhang, 2007; Shi, 2007). LSD1 (also known as KDM1, AOF2, or BHC110) is a highly conserved flavin adenine dinucleotide (FAD)-dependent lysine-specific demethylase that was initially found to specifically remove mono- and dimethyl groups from methylated histone H3 at lysine 4 (H3K4) to suppress gene expression. In prostate cancer cells, it also demethylates the repressive mono- and di-methylated lysine 9 (H3K9) in an androgen receptor-dependent manner (Metzger et al., 2005; Shi et al., 2004). Recent studies indicate that LSD1 is an essential epigenetic regulator of pluripotency in ESCs (Adamo et al., 2011; Whyte et al., 2012). We have previously shown that the levels of LSD1 are significantly elevated in cells derived from pluripotent germ cell tumors such as teratocarcinoma, embryonic carcinoma, and seminoma cells (Wang et al., 2011). Our newly developed novel LSD1 inhibitors selectively inhibited the growth of ESCs and pluripotent germ tumor cells (Wang et al., 2011). LSD1 is also essential for maintaining the oncogenic potential of MLL-AF9 leukemia stem cells and acute myeloid leukemia (Harris et al., 2012; Schenk et al., 2012). However, it remains unclear the critical downstream targets of LSD1 in ESCs and cancer cells. Here we report that Sox2 serves as the critical target of LSD1 in a wide variety of human carcinoma cells.
Results:
LSD1 expression is elevated in human squamous cell lung carcinomas that over-express Sox2
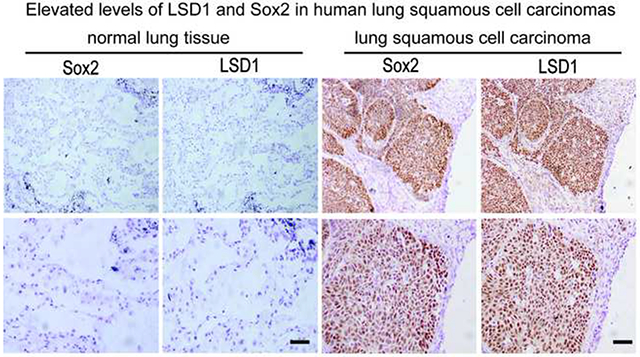
Our previous studies indicated that germ tumor cells such as teratocarcinoma and embryonic carcinoma cells expressed elevated levels of LSD1. They were also highly sensitive to our novel LSD1-specific inhibitors that non-covalently interact with LSD1 (Wang et al., 2011). Since these cells usually express pluripotent stem cell proteins Oct4, Sox2, and Lin28, which are also expressed in other human cancers (Leis et al., 2012; Peng et al., 2008; Zhong et al., 2010) we examined LSD1 in various human cancer tissues that also co-express some of pluripotent stem cell markers. In 13 independent cases of human lung squamous cell carcinoma (SCC) patient samples, we observed highly elevated levels of Sox2 and LSD1 in 5 poorly differentiated cases, moderate Sox2 and LSD1 increases in 7 moderately differentiated cancer cases, and low LSD1 expression in a single Sox2-negative and moderately differentiated cancer (Figure 1A, supplementary Figure S1A and Table S1). Statistical analysis revealed a significant correlation between Sox2 and LSD1 expression (Pearson's correlations: R2=0.4372 and P =0.014). In contrast, Sox2 expression was low or non-detectable in all 17 cases of lung adenocarcinoma carcinomas. In lung adenocarcinoma samples, only 2 cases of poorly differentiated cancers had moderately increased levels of LSD1, while LSD1 was low in remaining 15 moderately differentiated cancers. As a control, the surrounding normal lung tissues expressed undetectable levels of LSD1 and Sox2 proteins (Figure 1A).
Figure 1. LSD1 and Sox2 expression in lung squamous cell carcinomas and lung carcinoma cells that express Sox2 are selectively sensitive to LSD1 inactivation.

A. LSD1 and Sox2 expression in lung squamous cell carcinomas. One example of serial tissue sections from clinical lung SCC patients (N=13) immunostained with anti-Sox2 or LSD1 antibodies. LSD1 and Sox2 were strongly stained in pathological tissues (right) while low or non-detectable in normal lung tissue (left) surrounding the pathological areas. Scale bar, 100 microns. Lower panels represent magnified images (4x). See also Figure S1A and S1B.
B. Sox2 and LSD1 expression in human lung SCC NCI-H520, lung adenocarcinoma A549, lung carcinoma NCI-H1437 and H1299 cells. CUL1 and histone H3: loading control.
C. LSD1 inactivation specifically inhibited the growth of H520 and A549 cells but not that of H1437 or H1299 cells. Active growing lung cancer cells were treated with 50 μM LSD1 inhibitor CBB1007 for 30 hours or were transfected with 50 nM control luciferase (Luc) or LSD1 siRNAs for 60 hours. Cell growth was examined under microscopy.
D. Percentages of cell viability treated with LSD1 inhibitors or siRNAs compared to that of control cells using the MTT assay. Data are presented as mean ± SD. The statistical differences between inhibitor-treated and control groups were analyzed by one-way ANOVA. Scale bar: 100 microns. ** indicates p < 0.01.
Our finding that the levels of LSD1 are significantly elevated in Sox2-expressing SCCs is consistent with reported studies. For example, using the public reported tumor microarray data from Oncomine (www.oncome.org), our search found that in 21 SCC cases reported by Bhattacharjee et al. (Bhattacharjee et al., 2001), Sox2 was over-expressed in 18 cases while LSD1 was co-over-expressed in 16 cases. In another 6 small cell lung carcinoma cases with elevated LSD1, Sox2 was also over-expressed in 4 of them. In 17 cases of esophagus carcinomas (Hu et al., 2010) with high LSD1 levels, Sox2 was over-expressed in 14 cases. The over-expression of Sox2 and LSD1 was also found in other cancers. For example, in 19 cases of large cell lung carcinomas (Hou et al., 2010), 16 cases over-expressed LSD1 while 15 of them over-expressed Sox2. In cervical squamous cell carcinomas (Scotto et al., 2008), Sox2 is over-expressed in 50% of 84 cases and LSD1 was co-elevated in 48% of all cases. In 122 cases of ductal breast carcinomas (Sorlie et al., 2003), Sox2 was over-expressed in about 52% of them and LSD1 was co-over-expressed with Sox2 in about 49% of all cases. In a single undifferentiated breast carcinoma case, both Sox2 and LSD1 were over-expressed. Our studies suggest that there is a significant correlation between Sox2 and LSD1 expression in a wide array of human cancers.
Lung SCC cells that contain amplified Sox2 gene or other lung carcinoma cells that express Sox2 are particularly sensitive to LSD1 inactivation
Our observation that the LSD1 level is elevated in Sox2-expressing lung SCCs prompted us to investigate the functional relationship between Sox2 and LSD1 in lung carcinomas. For this purpose, we used human SCC NCI-H520 cells that contain the Sox2 gene amplification at 3q26.33 and human lung adenocarcinoma NCI-H1437 cells that do not express Sox2 (Bass et al., 2009)(Figure 1B). H520 and H1437 cells were treated with LSD1 inhibitors for 24-30 hours and cell growth was analyzed. We found that LSD1 inhibitor CBB1007 selectively and specifically inhibited the growth of H520 cells but had no detectable effects on H1437 cells (Figure 1C and 1D).
We wondered whether the elevated expression of Sox2 is responsible for such sensitivity towards LSD1 inhibitors in lung cancer cells. To identify additional Sox2 or other pluripotent stem cell-specific gene expression signatures in carcinoma cells, we searched the microarray mRNA data of the Cancer Genome Anatomy Project database (http://cgap.nci.nih.gov/Microarray/GeneList) in 60 cancer cell lines collected by National Cancer Institute. Using such an approach, we found about 1/3 of these cancer cells express Sox2 or other pluripotent stem cell proteins. Treatment of A549, a Sox2-expressing human lung adenocarcinoma cell derived from adenocarcinomic alveolar basal epithelial cells, and H1299, a Sox2-negative human non-small cell lung carcinoma cell (Figure 1B), with LSD1 inhibitors revealed that A549 cells are highly sensitive to LSD1 inhibitors while H1299 cells are not (Figure 1C and 1D). We also ablated the expression of LSD1 using specific siRNAs in H520, H1437, A549, and H1299 cells and these studies showed that ablation of LSD1 phenocopied the selective growth inhibitory effects of LSD1 inhibitors on H520 and A549 cells but not on that of H1437 and H1299 cells (Figure 1C and 1D). Our results indicate that lung carcinoma cells that express Sox2 are particularly sensitive to LSD1 inactivation but not the cells that are Sox2-negative. In addition, both Sox2-expressing H520 and A549 cells appeared to express higher levels of LSD1 (Figure 1B).
Breast and ovarian carcinoma cells can be distinguished by their sensitivity towards LSD1 inhibition
Although LSD1 inhibitors specifically target lung carcinoma cells that over-express Sox2, it remains unclear whether lung carcinoma cells are uniquely sensitive to LSD1 inhibition. Search of the Cancer Genome Anatomy Project database and published reports revealed that several breast and ovarian carcinoma cells may also express key pluripotent stem cell proteins Oct4, Sox2, Lin28, Nanog, and Sall4 either alone or in combination (Leis et al., 2012; Peng et al., 2008; Zhong et al., 2010). We therefore examined a panel of ovarian, breast, and other carcinoma cells for their response towards LSD1 inhibition (Supplementary Table S2). While the growth of some cells was not inhibited by LSD1 inhibitors, such as human ovarian carcinoma cells OVCAR8, Hs38.T, and ES-2 and breast carcinoma cells MDA-MB-231, BT549, and SK-BR-3, several of them are highly sensitive to LSD1 inhibitors, such as ovarian carcinoma cells OVCAR3, A2780, SKOV-3, and IGROV-1, ovarian teratocarcinoma PA-1 cells, and breast carcinoma cells MDA-MB-468, T47D, and MCF-7 (Figure 2A, 2B, and S1C, S1D, S2A-C). The selective effects of LSD1 inhibitors were confirmed by ablation of LSD1 using LSD1 siRNAs. Analysis of cell cycle effects by Flow-cytometry (FACS) revealed that loss of LSD1 induced significant G1 cell cycle arrest in cancer cells that are sensitive to LSD1 inhibition, such as A549, T47D, and IGROV1, which was associated with decreased expression of cell cycle regulatory proteins c-Myc and various cyclins, while such an arrest was not observed in cancer cells such as H1437 that are not sensitive to LSD1 inhibitors (Figure 2E, 3G and S3A).
Figure 2. LSD1 inactivation-induced growth inhibition is associated only with Sox2 expression in lung, breast, ovarian, and other types of carcinoma cells.

A. The effects of LSD1 inhibitor CBB1007 and siRNAs on the growth of ovarian carcinoma OVCAR-8, OVCAR-3, A2780 and breast carcinoma MDA-MB-231, MDA-MB-468, and T47D cells. See also Figure S1C and D, S2A-C, S2E and F, and S7B-D.
B and C. Quantitative analyses of the sensitivity of a panel of breast, ovarian, and other human carcinoma cells to LSD1 inhibitors (B) and LSD1 siRNA-mediated ablation (C). The statistical differences between inhibitor-treated and control groups were analyzed by one-way ANOVA. *: p < 0.05 and **: p < 0.01.
D. Analysis of the expression of Oct4, Sox2, Nanog, Lin28, Sall4, and other proteins in lung, breast, ovarian, teratocarcinoma, embryonic carcinoma, and other carcinoma cells as indicated. Cells that are sensitive to LSD1 inactivation are indicated by *. Among pluripotent stem cell proteins, only the expression of Sox2 correlates with the growth-inhibitory effects of LSD1 inactivation. See also Table S2 for summary.
E. Ablation of LSD1 by siRNAs induces G1 cell cycle arrest in Sox2-expressing A549, T47D, and IGROV1 cells but not in Sox2-negative H1437 carcinoma cells. The distributions of cell cycle population: A549 cells, Luc siRNA: G0/G1: 54.64%, S: 28.42%, G2/M: 16.94% and A549, LSD1 siRNA: G0/G1: 74.81%, S: 24.55%, G2/M: 0.65%. IGROV1 cells, Luc siRNA: G0/G1: 55.48%, S: 27.48%, G2/M: 17.04% and IGROV1, LSD1 siRNA: G0/G1: 78.07%, S: 13.37%, G2/M: 8.56%.
T47D cells, Luc: G0/G1: 65.87%, S: 17.91%, G2/M: 16.22% and T47D, LSD1 siRNA: G0/G1: 76.34%, S: 11.49%, G2/M: 12.17%.
H1437 cells, Luc siRNA: G0/G1: 39.53%, S: 23.30%, G2/M: 37.17% and H1437 cells, LSD1 siRNA: G0/G1: 37.28%, S: 24.97%, G2/M: 37.75%. See also Figure S3A.
Figure 3. LSD1 regulates Sox2 expression through modulating bivalent H3K9 and H3K4 methylations.

A. Inactivation of LSD1 by LSD1 inhibitors or siRNAs caused the downregulation of Sox2. Indicated carcinoma cells were treated with 50 μM LSD1 inhibitors CBB1007 and CBB1003 for 30 hours or 50 nM of Luc or LSD1 siRNAs for 60 hours. The protein levels of LSD1, Sox2, Lin28, CUL1, and histone H3 were analyzed by Western blotting. See also Figure S4B.
B. The mRNA levels of Sox2 in A were analyzed by real time quantitative RT-PCR. Data are presented as mean ± SD. **: p < 0.01.
C and D. Indicated cells were transfected with 50 nM Luc or LSD1 siRNAs or treated with 50 μM CBB1007 as in A or indicated time course for T47D cells (C) or other cells (D). The changes in methylations of H3K4, H3K9, and H3K27 were examined by Western blotting with specific antibodies. See also Figure S2C and D, and S7F.
E. Chromatin immunoprecipitation (ChIP) assays for the presence of LSD1, H3K4me1/2 and H3K9me2 on the Sox2 gene in A549 cells. Chromatin-associated proteins were cross-linked to chromatin, sonicated (average 500-1,000 base pairs DNA), and immunoprecipitated with control rabbit IgG, anti-LSD1 or H3K9me2 antibodies as indicated. The DNA fragments were purified and used for real time quantitative PCR with various primers along the Sox2 promoter. Data are presented as mean ± SD. The statistical differences of increased H3K4 and H3K9 methylations along the Sox2 gene between inhibitor-treated and control groups were analyzed by one-way ANOVA. *: p < 0.05 and **: p < 0.01. See also Figure S6C.
F. Re-ChIP assays for the co-presence of H3K9Me2 and H3K4Me2 on the Sox2 gene in A549 cells. Top panels: Chromatin fragments were first immunoprecipitated with anti-H3K9me2 antibodies or control IgG. The fragments were eluted with H3K9me2 peptide and re-immunoprecipitated with anti-H3K4me2 antibodies. Bottom panels: the chromatin fragments were first immunoprecipitated with anti-H3K4me2 and then anti-H3K9me2 antibodies. Real-time quantitative PCR ratios reflected the relative enrichment to the Input of the indicated histone methylations on Sox2 after sequential immunoprecipitations. Data are presented as mean ± SD of at least two independent assays.
G. Effects of LSD1 inactivation as in D on the protein levels of c-Myc and cyclins.
Sox2 is the only pluripotent stem cell protein whose expression correlates with the sensitivity towards LSD1 inactivation in carcinoma cells
Our previous studies indicate that the sensitivity to LSD1 inactivation usually associates with cells derived from germ cell tumors that express pluripotent stem cell proteins (Wang et al., 2011). As some breast and ovarian cancer cells in our collection were reported to express pluripotent stem cell proteins such as Oct4, Sox2, Lin28, Nanog, and/or Sall4 (Leis et al., 2012; Peng et al., 2008; Zhong et al., 2010), a direct correlation between the expression of these proteins and the sensitivity towards LSD1 inactivation has not been established. To determine the mechanism by which various carcinoma cells are sensitive to LSD1 inactivation, we analyzed the expression of known pluripotent stem cell proteins in our collected cell lines and correlated their expression with the sensitivity towards LSD1 inactivation, using teratocarcinoma/embryonic carcinoma F9 and NTERA-2 cells and cervical carcinoma HeLa cells as controls (Figure 2D and Table S2)(Wang et al., 2011).
Several notable findings were revealed using this approach. First, we found that Oct4, Lin28, Sall4 and Nanog were each expressed in breast and ovarian carcinoma IGROV1, A2780, and T47D cells, and all teratocarcinoma/embryonic carcinoma F9, PA-1, and NTERA-2 cells (Figure 2D). We did not find a single cell line that expresses these pluripotent stem cell proteins independent of Sox2, suggesting the importance of Sox2. Second, Sox2 was the only pluripotent stem cell protein that is expressed alone and independent of other pluripotent stem cell proteins in SKOV-3, OVCAR-3, MCF-7, MDA-MB-361, MDA-MB-468, MDA-MB-453, NCI-H520 and A549 cells, again indicating that Sox2 is unique for these carcinoma cells. Third, most importantly, our analyses revealed that all Sox2-expressing carcinoma cells were sensitive to LSD1 inactivation while all Sox2-negative cancer cells were insensitive (Figure 2D and Table S2). Consistently, we also found that K562, a human myelogenous leukemia cell, and G401, a human rhabdoid tumor derived cell, also expressed Sox2 and both were sensitive to LSD1 inhibition while MDA-MB-435S, a melanoma cell that does not express Sox2, is insensitive (Figure 2D, S2E and F, and Table S2). While K652 cells only express Sox2, G401 cells also express Lin28 and Sall4. Our studies thus indicate that there is a strong correlation between Sox2 expression and sensitivity towards LSD1 inactivation.
LSD1 inactivation of LSD1 suppresses Sox2 expression and increases the mono- and di-methylations of H3K9 and tri-methylation of H3K27 only in Sox2-expressing carcinoma cells
Analysis of the effects of LSD1 inactivation revealed that LSD1 inactivation consistently reduced the expression of Sox2 in H520, A2780, T47D (Figure 3A and 3B), and other Sox2-expressing cancer cells (Figure S4B). In contrast, LSD1 inactivation did not change the levels of Lin28, another pluripotent stem cell protein, which is often co-expressed with Sox2 in a fraction of carcinoma cells (Figure 2D, 3A, and S4B). Our results suggest that LSD1 is required for the expression of Sox2 in these carcinoma cells.
As LSD1 is a histone demethylase that removes the mono- and di-methyl groups from methylated H3K4 (H3K4me1/me2)(Shi et al., 2004), we found that LSD1 inactivation caused a dose-dependent increases of H3K4me1/me2 in both Sox2-postive and negative cells (Figure 3C, 3D, S2C and D), indicating that the inhibitors specifically blocked the LSD1 demethylase activity in all cancer cells.
Because LSD1 interacts with the androgen receptor to act as a H3K9 specific demethylase to remove the mono- and di-methyl groups from methylated H3K9 (H3K9me1/me2) in certain prostate cancer cells (Metzger et al., 2005), we also examined whether LSD1 inactivation affected H3K9me1/me2 as well as the tri-methylation of histone H3 at lysine 27 (H3K27me3) which is not a target of LSD1. Strikingly, we found that LSD1 inactivation induced global increases of both H3K9me1/me2 and H3K27me3 only in Sox2-expressing carcinoma cells but not in Sox2-negative cancer cells such as H1299 or H1437 (Figure 3C and D). Kinetic analysis of induction revealed that the increases of both H3K4me1/2 and H3K9me1/2 occurred early (<1 hour) and simultaneously while H3K27me3 was induced at much later time (between 4-8 hours, Figure 3C) after LSD1 inhibition, suggesting that the increases of H3K4me1/2- and H3K9me1/2 are likely a direct consequence of LSD1 inhibition while H3K27me3 elevation may occur as a secondary event.
LSD1 directly binds to the transcriptional regulatory region of Sox2 to regulate the bivalent H3K4 and H3K9 methylation
As LSD1 inactivation reduced Sox2 expression, we tried to determine whether Sox2 is a direct target of LSD1 by using the chromatin immunoprecipitation (ChIP) assay (Whyte et al., 2012) to determine if LSD1 binds to the Sox2 gene. Our ChIP analysis revealed that LSD1 is enriched on the transcriptional regulatory region (−3.0 to −4.0 kb) of the Sox2 gene (Figure 3E), in a region that was reported to act as a distal enhancer for Sox2 expression in breast cancer cells (Leis et al., 2012). This enrichment of LSD1 appears to be specific for Sox2 as no enrichment was observed on Lin28, Klf4, or the pericentromeric heterochromatin region (Dovey et al., 2010)(Figure 7G and S6D), which are not regulated by LSD1.
Figure 7. Inactivation of LSD1 induces the expression of differentiation genes by increased methylation of H3K4 but not methylated H3K9 and suppression of cyclin A gene by increased H3K4me1/2 and H3K9me2.

A-C. A549, NCI-H520, H1437, A2780, and T47D cells were transfected with 50 nM luciferase or LSD1 siRNAs for 48 hours or treated with 50 μM LSD1 inhibitors CBB1003 and 1007 for 30 hours. The effects LSD1 inactivation on the induction of FOXA2 (A) or on various differentiation genes FOXA2, HNF4A, BMP2, EOMES, and Sox17 (B and C) were analyzed by Western blotting (A) or real time quantitative RT PCR (B and C). See also Figure S5A.
D. Indicated lung, breast, ovarian, and other carcinoma cells were treated with 50 μM CBB1003 or CBB1007 for 30 hours and induced expression of FOXA2 mRNAs was monitored, quantified, and compared between control and inhibitor-treated cells using real time quantitative RT-PCR. *: p < 0.05 and **: p < 0.01. See also Figure S5B.
E-G. ChIP assays for the presence of LSD1, H3K9me2, H3K4me1 and H3K4me2 on the regulatory regions of differentiation gene FOXA2 with or without LSD1 inhibitors for 30 hours (E, see also Figure S6A-C) or on the cyclin A gene (F, see also Figure S3B and S6C) and the pericentromeric heterochromatin region SAT2 and BMP2 gene (G, see also Figure S6D) after LSD1 inactivation in A549 cells. Chromatin fragments were immunoprecipitated with control rabbit IgG, anti-LSD1, H3K9me2, H3K4me1 and H3K4me2 specific antibodies as indicated. The DNA fragments were purified and used for real time quantitative PCR with various primers along the FOXA2 and cyclin A genes or the SAT2 and BMP2 regions. Data are presented as mean ± SD.
To determine whether LSD1 binding is associated with the demethylase activity on Sox2, the presence and changes of the characteristic H3K4me1/me2 and H3K9me1/me2 on Sox2 after LSD1 inactivation were examined. Repeated ChIP analyses in Sox2-expressng cells such as A2780 and A549 cells consistently revealed that H3K9me2 (we did not have good ChIP-grade anti-H3K9me1 antibodies) and H3K4me1/me2 were present on the Sox2 regulatory region and inhibition of LSD1 caused significantly increased levels of both H3K9me2 and H3K4m1/me2 on Sox2 (Figure 3E). Reciprocal re-ChIP of H3K9me2 or H3K4me2-enriched chromatin fragments revealed that H3K9me2 and H3K4me2 co-existed on the same Sox2 regulatory fragment (Figure 3F). Although H3K27me3 was also induced on Sox2, the major site is located further down the gene within the coding region (+2.0, Figure S6C). Thus, our results indicate that the Sox2 regulatory region is directly regulated by the bivalent H3K4 and H3K9 methylations by LSD1 demethylase. Sox2 downregulation after LSD1 inactivation is likely to be directly caused by increased repressive H3K9 methylations on the Sox2 gene, even though H3K4me1/2 also increased on Sox2 (see below).
Our ChIP analysis also indicated that LSD1 also binds to the cyclins A, B, and D1 genes, and inactivation of LSD1 caused the increased levels of H3K4me1/me2, H3K9me2, and H3K27me3 on the cyclin promoters (Figure 7F and S3B and S6C). Thus, increased levels of H3K9me2 on the cyclin genes may also repress the expression of cyclins which may contribute to the G1 cell cycle arrest after LSD1 inactivation (Figure 2E, 3G, and S3A and B).
Loss of Sox2 phenocopies the growth-inhibitory effects of LSD1 inactivation on carcinoma cells
Although Sox2 acts as an amplified lineage-survival oncogene in lung SCCs (Bass et al., 2009), the role of Sox2 in other carcinoma cells remains largely uncharacterized. As LSD1 inactivation reduced Sox2 expression, we further investigated the role of Sox2 in regulating cancer cell growth. Ablation of Sox2 expression using specific siRNAs consistently showed that it caused the G1 cell cycle arrest and growth inhibition in Sox2-expressing carcinoma cells that are sensitive to LSD1 inactivation, but not that of Sox2-negative cancer cells (Figure 4A, 4B, 4E, and S4A and C). Loss of Sox2 also downregulated c-Myc and cyclins A, B, and D1, and induced the expression of genes for differentiation including FOXA2, HNF4A, BMP2, EOMES, and Sox17 (Figure 4C and D). However, loss of Sox2 only increased the levels of trimethylated H3K27, but not that of H3K4 and H3K9 methylations, suggesting that induction of H3K27 trimethylation after LSD1 inactivation might be an indirect consequence of Sox2 downregulation (Figure 3C, 3D, and 4C). ChIP analysis revealed that Sox2 inactivation induced elevated H3K27me3 on Sox2 and cyclin promoters after Sox2 ablation (Figure 4F), suggesting increased H3K27me3 on these genes suppressed their expression. Thus, our data indicates that Sox2 serves as a primary and key direct target of LSD1 inactivation for growth inhibition and differentiation because downregulation of Sox2 further amplifies and enhances the effects of LSD1 inactivation through the increased levels of H3K27me3. Our observation is consistent with previous reports of critical thresholds and phenotypes associated with haploid insufficiency and hypomorphic mutations of Sox2 in animals and human diseases (Episkopou, 2005). Mutations of human Sox2 that compromise one allele of the Sox2 genes were shown to cause the anophthalmia-esophageal-genital (AEG) syndrome and exhibited neurological phenotypes including seizures (Fantes et al., 2003; Williamson et al., 2006), while the hypomorphic deletion of the enhancer of the mouse Sox2 genes, which reduced Sox2 mRNA and protein levels by 20-30% of wild-type levels, exhibited lower birth frequency and neurological phenotypes in the mouse (Ferri et al., 2004).
Figure 4. Inactivation of Sox2 causes G1 cell cycle arrest, inhibits cell growth of Sox2-expressing carcinoma cells and induces the genes for differentiation.

A. A549, NCI-H1437, T47D, and IGROV1 cells were transfected with 50 nM luciferase or Sox2 siRNAs for 60 hours and cell growth of control and Sox2-ablated cells was examined using microscopy. See also Figure S4C.
B. The cell cycles after Sox2 inactivation were analyzed by FACS. Cell cycle distribution of cells: A549 cells, Luc siRNA: G0/G1: 54.68%, S: 30.52%, G2/M: 14.80%, and A549, LSD1 siRNA: G0/G1: 76.40%, S: 23.41%, G2/M: 0.19%.
H1437 cells, Luc siRNA: G0/G1: 39.69%, S: 23.11%, G2/M: 37.20% and H1437 cells, LSD1 siRNA: G0/G1: 39.39%, S: 25.10%, G2/M: 35.51%.
IGROV1 cells, Luc siRNA: G0/G1: 57.29%, S: 27.32%, G2/M: 15.39%; IGROV1, LSD1 siRNA: G0/G1: 74.62%, S: 12.87%, G2/M: 12.52%. T47D cells, Luc: G0/G1: 66.02%, S: 17.27%, G2/M: 16.71%, T47D, LSD1 siRNA: G0/G1: 74.65%, S: 14.48%, G2/M: 10.86%. See also Figure S4A.
C. The effects of Sox2 inactivation in B on c-Myc, cyclins, methylated H3K4, H3K9, and H3K27 proteins were analyzed by Western blotting.
D. Induction of differentiation genes FOXA2, HNF4A, BMP2, EOMES, and Sox17 by Sox2 deficiency in A549, H520, A2780, and T47D cells using Western blotting and real time quantitative RT-PCR.
E. Indicated lung, breast, ovarian, and other carcinoma cells were transfected with 50 nM luciferase or Sox2 siRNAs for 48 hours and cell growth was monitored by the MTT assay. *: p < 0.05 and **: p < 0.01.
F. ChIP arrays on A549 cells with or without Sox2 siRNA-mediated ablation as in B for increases of H3K27me3 on Sox2 and cyclin A, B and D1 genes.
Sox2 is involved in mediating the growth inhibitory effects of LSD1 inactivation
To further determine whether reduced expression of Sox2 is responsible for the growth inhibition caused by LSD1 inactivation, we tried to ectopically express Sox2 in Sox2-expressing carcinoma cells. In both Sox2-expressing ovarian A2780 and lung A549 carcinoma cells, stable and ectopic expression of Sox2 led to a significant resistance of these cells towards LSD1 inhibition, as compared with that of the control cells (Figure 5A). Co-inactivation of Sox2 and LSD1 in Sox2-expressing cancer cells by siRNAs or LSD1 inhibitors also did not reveal any additive or synergetic effects on growth inhibition induced by the loss of LSD1 or Sox2 alone (Figure 5D and E), suggesting that LSD1 and Sox2 act in the same pathway to control cell growth.
Figure 5. Sox2 is a target of LSD1 inactivation in Sox2-expressing carcinoma cells.

A. Ectopic expression of Sox2 conferred resistance to LSD1 inhibitors. Human Sox2 cDNA was tagged with Flag-epitope at amino terminus and stably expressed in A2780 or A549 cells using the retroviral pMSCV vector. The control and Flag-Sox2 expressing cells were treated with various concentrations of CBB1007 for 30 hours and cell viability were assayed and compared.
B. Sox2-expressing A549 carcinoma cells were separated by serial dilution into single cells. The single-cell clones were expanded and two representative clones were shown. The mRNA levels of Sox2 and LSD1 in the small and large clones were analyzed by real time quantitative RT-PCR.
C. The responses of single small and large clones of A549 cells in B to various concentrations of CBB1007 were examined.
D and E. Sox2-expressing A549, T47D, or A2780 cells were treated with 50 μM CBB1007 for 30 hours (D) or transfected with 50 nM luciferase, LSD1, Sox2, and LSD1+Sox2 siRNAs for 48 hours (E) and the effects on their growth were monitored and quantified.
LSD1 was shown to interact with several proteins, such as CoREST, in various cells (Shi et al., 2005). To confirm that the effects of LSD1 inactivation are mediated through its interaction with cellular proteins, we also ablated the expression of CoREST. We found that loss of CoREST caused the same selective growth inhibitory effects and induction of H3K4, H3K9, and H3K27 methylations on Sox2-expressing cancer cells but not that of Sox2-negative cancer cells as LSD1 inactivation (Figure S7). It is likely that LSD1 acts through the CoREST complex to selectively regulate the growth of Sox2-expressing cancer cells, although loss of CoREST slightly reduced the level of LSD1 protein, possibly because of their in vivo association.
As Sox2-expressing carcinoma cells may be heterogeneous in Sox2 expression and different cells may vary in their responses to LSD1 inactivation, we examined the clonal response of Sox2-expressing carcinoma cells. To isolate single cells, we used serial dilution to separate A549 cells. The isolated single cell was clonally expanded and characterized. We found that the isolated single cell clones exhibited different growth rates represented by their differences in colony size (Figure 5B). Examination of Sox2 expression in various clones revealed that smaller clones usually contain much higher levels of Sox2 expression than the larger clones (Figure 5B). Notably, the smaller clones also express more LSD1 and are more sensitive to LSD1 inhibitors than that of larger clones that express less Sox2 and LSD1 (Figure 5B and C). These studies again underscore the correlation between Sox2 and LSD1 expression and indicate that cancer cells that express higher levels of Sox2 are more sensitive to LSD1 inhibition.
Loss of LSD1 suppresses Sox2-dependent lineage-specific gene expression and reduced Sox2 binding and repression on genes for differentiation
In lung SCCs, Sox2 acts as a lineage-survival oncogene that is required for the expression of lineage-specific genes such as TP63 and KRT6A (Bass et al., 2009). Loss of either LSD1 or Sox2 led to the downregulation of TP63 and KRT6A expression in Sox2-expressing H520 and A549 but not in Sox2-negative H1299 lung carcinoma cells (Figure 6A and B). We further used ChIP assays to determine whether LSD1 inactivation affects the ability of Sox2 to bind the promoters of TP63 and KRT6A. In H520 cells, we found that Sox2 directly binds to the promoters of TP63 and KRT6A genes (Figure 6C) and inhibition of LSD1 significantly reduced the binding of Sox2 to these lineage-specific genes (Figure 6D).
Figure 6. LSD1 inactivation impairs Sox2 regulation on the expression of lineage-specific genes and genes for differentiation.

A and B. Lineage-specific genes TP63 (A) and KRT6A (B) were downregulated after inactivation of LSD1 or Sox2. Lung carcinoma NCI-H520, A549 and H1299 cells were either treated with 50 μM CBB1003 for 30 hours or were transfected with Luc, LSD1 or Sox2 specific siRNAs for 48 hours as indicated. The mRNA levels of TP63 and KRT6A were measured by real-time quantitative RT-PCR. The downregulation of TP63 and KRT6A occurred in Sox2-ablated H520 and A549 cells but not in H1299 cells. The statistical differences between experimental and control groups were analyzed by one-way ANOVA. *: p < 0.05 and **: p < 0.01.
C. ChIP analysis of Sox2 binding to the promoters of TP63, KRT6A, FOXA2, and Sox17 genes using control IgG and specific anti-Sox2 antibodies in NCI-H520 cells. *: p < 0.05 and **: p < 0.01.
D. LSD1 inhibitor CBB1007 reduced Sox2 binding to TP63, KRT6A, FOXA2, and Sox17 genes in H520 cells.
Our studies also revealed that ablation of Sox2 induced the expression of differentiation genes such as FOXA2 and Sox17 (Figure 4D). ChIP assays indicated that Sox2 directly binds to these promoters and inactivation of LSD1 significantly decreased Sox2 binding to these promoters (Figure 6C and 6D), suggesting Sox2 normally acts as a repressor of these differentiation genes. Thus, our data confirm that LSD1 inactivation directly acts on a Sox2-dependent transcriptional program to reduce the lineage-survival oncogene function of Sox2 and to impair Sox2-mediated repression of differentiation genes.
Loss of LSD1 induces the expression of genes for differentiation through selectively increasing the levels of methylated H3K4 but not methylated H3K9 or H3K27 on the promoters
LSD1 inactivation in germ tumor cells or ESCs induced the expression of genes for differentiation (Wang et al., 2011). To determine whether the function of LSD1 is preserved in Sox2-expressing carcinoma cells, we analyzed the effects of LSD1 inactivation on the expression of differentiation genes. We found that LSD1 inactivation led to the induction of differentiation genes such as FOXA2, HNF4A, BMP2, EOMES, and Sox17, only in Sox2-expressing cancer cells but not in Sox2-negative cancer cells (Figures 7A-D and S5). Although loss of LSD1 caused the G1 cell cycle arrest (Figure 2E and S 3A), the induction of differentiation genes did not appear to be the consequence of LSD1 inactivation-induced G1 cell cycle arrest, as arresting Sox2-expressing A549 cells in the G1/S border alone did not promote the induction of differentiation genes, nor the suppression of Sox2 expression (Figure S3C).
Because LSD1 inactivation promotes the primary global methylations of H3K4me1/2, H3K9me1/2 and H3K27me3 in Sox2-expressing cancer cells (Figure 3C and D) and the increased levels of these methylations on the promoters of Sox2 and cyclin genes (Figure 3E, 7F, S3B and S6C), we examined these methylations on the regulatory regions of differentiation genes. In sharp contrast to the increased H3K4, H3K9, and H3K27 methylations on the Sox2 or cyclin genes after LSD1 inactivation, LSD1 inhibition consistently increased only the mono- and di-methylations of H3K4 on the regulatory regions of FOXA2, BMP2, and Sox17 genes but not H3K9 and H3K27 methylations in Sox2-expressing carcinoma cells (Figure 7E, and S6A and B). Thus, our studies reveal a novel mechanism by which LSD1 inactivation suppresses the expression of Sox2 and cyclins by increased H3K9 and H3K27 methylations on their regulatory regions/promoters, whereas it causes the induction of differentiation genes by selectively elevating the levels of H3K4me1/me2 on differentiation genes but not methylated H3K9 and H3K27. The effects of LSD1 inhibitors on differentiation may be enhanced by Sox2 downregulation, which de-represses Sox2-mediated suppression of differentiation genes and causes further cellular differentiation (Figure 6).
Discussion:
Although we previously found that LSD1 inactivation selectively inhibited the growth of pluripotent ESCs and germ tumor cells (Wang et al., 2011), the mechanism by which these cells are selectively sensitive to LSD1 inhibitors remains unclear. In this study, we found that tissue-specific carcinoma cells can be divided into two groups based on their sensitivity towards LSD1 inactivation. While some sensitive carcinoma cells express Oct4, Lin28, and/or Sall4, we found all carcinoma cells that are sensitive to LSD1 inactivation consistently express Sox2, often alone and independent of other known pluripotent stem cell proteins (Figure 1, 2, S1-3 and Table S2). This correlation strongly suggests that the growth inhibitory effects of LSD1 inhibitors are dependent on Sox2, but not other pluripotent stem cell proteins. Our finding that Sox2 is the sole pluripotent stem cell protein expressed in a wide array of lung, breast, ovarian and other cancer cells suggests that Sox2 is a master regulator of growth, differentiation, and survival of these cancers.
LSD1 primarily acts as a histone demethylase that removes the methyl groups from H3K4me1/me2 to suppress gene expression (Shi et al., 2004). Only in some prostate cancer cells, LSD1 can bind to androgen receptor in a ligand-dependent way to remove the methyl groups from H3K9me1/me2 to activate gene expression (Metzger et al., 2005). Strikingly, we found that LSD1 inactivation induced a global increase of both H3K4me1/me2 and H3K9me1/me2 in Sox2-expressing cancer cells (Figure 3), which form bivalent chromatin domains on the regulatory regions of Sox2 and cell cycle regulatory genes such as cyclins to suppress their expression (Wang et al., 2009). Although trimethylated H3K27 was also induced, it appears that this increase is caused by Sox2 downregulation after LSD1 inactivation, as loss of Sox2 only induced the increase of trimethylated H3K27 but not methylated H3K4 and H3K9 (Figure 4C). However, this increase in H3K27me3 appears to be critical for response to LSD1 inactivation, since inactivation of Sox2 produced the same growth inhibitory effects as that of LSD1 inhibition in cancer cells (Figure 4 and S4). In contrast, our studies revealed that LSD1 inactivation only caused the increased levels of H3K4me1/me2 but not H3K9me1/2 and H3K27me3 on the regulatory regions of differentiation genes to induce their expression (Figure 7 and S6). Thus, our work suggests the following mechanism that LSD1 inactivation causes the selective increased methylations of H3K4 and H3K9 on the Sox2 and cell cycle genes but only induces increased H3K4 methylation on the promoters of differentiation genes, resulted in the downregulation of Sox2 and cyclins and induction of genes for differentiation. The downregulation of Sox2 consequently reduces lineage-specific oncogenic potential and further promote cellular differentiation. A key event of Sox2 downregulation by LSD1 inactivation is the induction of H3K27 tri-methylation, which further reduces the expression of Sox2 and cell cycle genes to amplify the growth inhibitory and differentiation-promoting effects of LSD1 inactivation. Our studies collectively indicate that LSD1 is a key regulator of Sox2 expression, cell cycle progression, and cellular differentiation in ESCs and Sox2-expressing cancer cells and suggest that LSD1 serves as a specific and selective target for the therapy of Sox2-expressing cancers.
Experimental Procedures:
Cell lines and Cell Culture
Lung, breast, ovarian, and other carcinoma cells were either obtained from American Type Culture Collection (ATCC) or from the DTP and DCTD Tumor Repository of National Cancer Institute as listed in supplementary Table S2. They were cultured according to instructions and treated with various concentrations of LSD1 inhibitors for 24-30 hours for cell viability or expression assays (Wang et al., 2011). Additional procedures are described in Supplementary Information.
Western Blot, Immunohistochemistry, and Antibodies
Log-phase growing cancer cells were directly lysed in a buffer containing 0.5% SDS. Proteins in the lysates were equalized and analyzed by Western blotting using specific antibodies as described previously (Wang et al., 2011). For immunohistochemical staining, rabbit monoclonal anti-LSD1 and Sox2 antibodies were obtained from Cell Signaling and immunostaining was conducted as previously described (Wang et al., 2011). Additional experimental methods are described in Supplementary Information.
Small RNA Interference
Cells were transfected with 50 nM siRNAs using DharmaFECT reagent 1 (Dharmacon) for 48-60 hours as described previously (Wang et al., 2011). The siRNA sequences are: human LSD1: GGAAGAAGAUAGUGAAAAC and human Sox2: CGCUCAUGAAGAAGGAUAA.
Chromatin Immunoprecipitation (ChIP) Assays
The ChIP assays were carried out according to published protocols (Whyte et al., 2012). After cross-linking chromatin proteins with formaldehyde, chromatin DNA was sonicated to average 500-1,000 base pairs and used for immunoprecipitation by specific antibodies. DNA was isolated for quantitative real time PCR after reversing the cross-linking on immunoprecipitated chromatin fragments. The real-time PCR primers are shown in supplementary Table S3. The ChIP grade anti-H3K4me2 (Ab32356), H3K4me1 (Ab8895), H3K9me2 (Ab1220), and LSD1 (Ab17721) antibodies were from Abcam. Re-chromatin immunoprecipitation (Re-ChIP) for co-occupancy of methylated H3K4 and H3K9 was conducted as previously described (Geisberg and Struhl, 2004).
Statistical Analyses
Statistical analysis was performed using the GraphPad Prism v4.0 software as described previously (Wang et al., 2011). Data are presented as mean ± SD. One-way ANOVA was used for comparisons.
Supplementary Material
Highlights:
LSD1 levels are elevated in Sox2-expressing lung squamous cell carcinomas.
Sox2 confers the sensitivity towards LSD1 inhibition in cancer cells.
LSD1 controls Sox2 and differentiation through selective H3K4 and H3K9 methylation.
Sox2 amplifies the effects of LSD1 inactivation by H3K27 methylation.
Acknowledgement:
This work was supported by grants from Fong Shu Fook Tong Foundation and Joyce M. Kuok Foundation, Shenzhen SZSITIC grant (JC201104210125A), National Natural Science Foundation of China (NSFC30971616 and 21133002), National Institutes of Health (R01CA989550), and cancer research funds from Carper and Elegbede to University of Nevada, Las Vegas.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, and Izpisua Belmonte JC (2011). LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol 13, 652–659. [DOI] [PubMed] [Google Scholar]
- Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, Lopez de Munain A, Sampron N, et al. (2011). Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS One 6, e26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. (2009). SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, and Weinberg RA. (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. (2001). Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98, 13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey OM, Foster CT, and Cowley SM (2010). Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A 107, 8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Episkopou V. (2005). SOX2 functions in adult neural stem cells. Trends Neurosci 28, 219–221. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, et al. (2003). Mutations in SOX2 cause anophthalmia. Nat Genet 33, 461–463. [DOI] [PubMed] [Google Scholar]
- Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S , Pandolfi PP, Sala M, DeBiasi S, et al. (2004). Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131, 3805–3819. [DOI] [PubMed] [Google Scholar]
- Geisberg JV, and Struhl K (2004). Quantitative sequential chromatin immunoprecipitation, a method for analyzing co-occupancy of proteins at genomic regions in vivo. Nucleic Acids Res 32, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. (2012). The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487. [DOI] [PubMed] [Google Scholar]
- Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, et al. (2010). Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One 5, e10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N, Clifford RJ, Yang HH, Wang C, Goldstein AM, Ding T, Taylor PR, and Lee MP (2010). Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma. BMC Genomics 11, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussenet T, Dali S, Exinger J, Monga B, Jost B, Dembele D, Martinet N, Thibault C , Huelsken J, Brambilla E, et al. (2010). SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One 5, e8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, and Thun MJ (2009). Cancer statistics, 2009. CA Cancer J Clin 59, 225–249. [DOI] [PubMed] [Google Scholar]
- Jones DR, Daniel TM, Denlinger CE, Rundall BK, Smolkin ME, and Wick MR (2006). Stage IB nonsmall cell lung cancers: are they all the same? Ann Thorac Surg 81, 1958–1962; discussion 1962. [DOI] [PubMed] [Google Scholar]
- Klose RJ, and Zhang Y (2007). Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 8, 307–318. [DOI] [PubMed] [Google Scholar]
- Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, and Martin AG (2012). Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 31, 1354–1365. [DOI] [PubMed] [Google Scholar]
- Maier S, Wilbertz T, Braun M, Scheble V, Reischl M, Mikut R, Menon R, Nikolov P, Petersen K, Beschorner C, et al. (2011). SOX2 amplification is a common event in squamous cell carcinomas of different organ sites. Hum Pathol 42, 1078–1088. [DOI] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, and Schule R (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439. [DOI] [PubMed] [Google Scholar]
- Neumann J, Bahr F, Horst D, Kriegl L, Engel J, Luque RM, Gerhard M, Kirchner T, and Jung A (2011). SOX2 expression correlates with lymph-node metastases and distant spread in right-sided colon cancer. BMC Cancer 11, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, and Yamanaka S (2006). Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr Stem Cell Res Ther 1, 103–111. [DOI] [PubMed] [Google Scholar]
- Peng S, Maihle NJ, and Huang Y (2008). Pluripotency factors Lin28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 29, 2153–2159. [DOI] [PubMed] [Google Scholar]
- Que J, Okubo T, Goldenring JR, Nam KT, Kurotani R, Morrisey EE, Taranova O, Pevny LH, and Hogan BL (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134, 2521–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med 18, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, et al. (2008). Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer 47, 755–765. [DOI] [PubMed] [Google Scholar]
- Shi Y. (2007). Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet 8, 829–833. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, and Casero RA (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, and Shi Y (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19, 857–864. [DOI] [PubMed] [Google Scholar]
- Sholl LM, Barletta JA, Yeap BY, Chirieac LR, and Hornick JL (2010). Sox2 protein expression is an independent poor prognostic indicator in stage I lung adenocarcinoma. Am J Surg Pathol 34, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. (2003). Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 100, 8418–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, et al. (2011). Novel Histone Demethylase LSD1 Inhibitors Selectively Target Cancer Cells with Pluripotent Stem Cell Properties. Cancer Research 71, 7238–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, and Zhao K (2009). Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, and Young RA (2012). Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, et al. (2006). Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet 15, 1413–1422. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Zhong X, Li N, Liang S, Huang Q, Coukos G, and Zhang L (2010). Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J Biol Chem 285, 41961–41971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.