

Abstract
Lipid nanoparticles (LNPs) can be designed to potentiate cancer immunotherapy by promoting their uptake by antigen-presenting cells, stimulating the maturation of these cells and modulating the activity of adjuvants. Here we report an LNP-screening method for the optimization of the type of helper lipid and of lipid-component ratios to enhance the delivery of tumour-antigen-encoding mRNA to dendritic cells and their immune-activation profile towards enhanced antitumour activity. The method involves screening for LNPs that enhance the maturation of bone-marrow-derived dendritic cells and antigen presentation in vitro, followed by assessing immune activation and tumour-growth suppression in a mouse model of melanoma after subcutaneous or intramuscular delivery of the LNPs. We found that the most potent antitumour activity, especially when combined with immune checkpoint inhibitors, resulted from a coordinated attack by T cells and NK cells, triggered by LNPs that elicited strong immune activity in both type-1 and type-2 T helper cells. Our findings highlight the importance of optimizing the LNP composition of mRNA-based cancer vaccines to tailor antigen-specific immune-activation profiles.
Successful deployment of two messenger RNA (mRNA) vaccines, Spikevax (Moderna) and Comirnaty (BioNTech/Pfizer), against SARS-CoV-2 in response to the outbreak of the coronavirus pandemic has clearly validated the safety and efficacy of the lipid nanoparticle (LNP) delivery tool as an mRNA vaccine modality1–3. As a potent vehicle mediating the expression of mRNA encoding an antigen of interest, LNPs have been shown to elicit strong antibody and memory-B-cell responses4. In this case, the antigens are expressed by a combination of immune cells as well as non-immune cells such as myocytes and adipocytes at the injection site. Antigens translated within antigen-presenting cells (APCs) are directly processed and presented on major histocompatibility complex (MHC)-I to CD8+ T cells, which contribute to the activation of cellular immune responses. Antigens translated within non-APCs become internalized by APCs and presented on MHC-II to CD4+ T helper cells. Such a mechanism for the protein translated within non-APCs may also contribute to B-cell differentiation and hence antibody production4–13. The strong potency has been attributed to the adjuvant activity of LNPs, particularly their ability to induce germinal-centre formation and T follicular helper (Tfh) cell responses5–7. In addition, evidence of generated interferon-γ (IFN-γ+) or IL2+ CD8+ T cells as well as CD4+ Th1 cells has also been reported from mRNA LNP vaccines4. These reports support the notion that LNPs optimized for COVID-19 mRNA vaccines may also generate CD4+ Th1 and CD8+ cell-mediated cellular immunity13,14, which may contribute to the Th2 response required for a strong humoral response to generate a high level of neutralizing antibodies15–20. Although Th2 and Th17 responses are essential to generate a potent humoral response and eradicate the extracellular pathogens21–24, an increasing number of reports have demonstrated that combining a robust Th2 response with Th1-mediated cellular immunity can be instrumental in clearing SARS-CoV-2-infected cells and enhancing vaccine efficacy25–30. Th1-biased cytokine secretion activates macrophage- and phagocyte-dependent immunity against viral pathogens, which drives protective, cell-mediated immune responses. Harnessing this Th1-driven cellular component in the vaccine design should contribute to increased effectiveness of infection control, and enhance disease prevention and treatment31,32.
LNPs have been tested previously for the delivery of mRNA vaccines to treat cancer and prevent other infections, including Zika virus33, influenza34, flavivirus35, HIV36 and so on. It has become evident that the induction of a potent antigen-specific immune response requires a specifically tailored immune activation profile. When a delivered antigen is expressed in dendritic cells (DCs) or other APCs, it generates peptide epitopes loaded and displayed in the context of MHC class I molecules, leading to activation of CD8+ T cells9–12,37–42. A strong cytotoxic CD8+ T cell response and a Type-1 T helper cell (Th1) immune response are critical to the design of an effective tumour vaccine, leading to the clearance of intracellular pathogens and cancer cells38. Studies have shown that LNP formulations can be identified to promote transfection and maturation of DCs, macrophages and neutrophils as well as modulate the adjuvant activity for the purpose of potentiating CD8+ T cell response and antitumour efficacy. However, the correlation among the composition, therapeutic efficacy and immune activation profile of mRNA LNPs, particularly the balance between Th1 and Th2 responses and coordination with NK-cell-mediated cell killing, remains elusive.
LNP systems offer distinct advantages in terms of the structural versatility offered by diverse lipid compositions and broad transfection capability across a wide range of cell populations43–53. Most LNP-based nucleic acid delivery tools that are commercially available or investigated in clinical studies consist of four or five lipid components: a helper phospholipid (for example, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)), an ionizable lipid, cholesterol, a PEGylated lipid and a selective organ targeting lipid47,53–55. Recent studies have reported that the choice of lipid components and relative proportions of the lipid ingredients in the formulation greatly influence in vivo transfection efficiency and tissue-specific delivery47,48,52,56,57. Despite the recent advance in mRNA LNP-based vaccines, there is a lack of in-depth analysis on the effect of helper lipid charge and the relative ratios of the LNP components on the transfection of different cell populations at the site of administration, which may play an important role in determining antigen expression levels in APCs, immune activation profile and therapeutic effects. We hypothesized that a distinct immune activation profile may be generated by tailoring LNP composition to modulate transfected cell populations.
Here we screened 1,080 LNP formulations for transfection efficiency in APCs with a goal of inducing a robust cellular immunity, and identified a cohort of formulations with the highest transfection efficiency in bone-marrow-derived dendritic cells (BMDCs) and antigen presentation ability. The selected formulations were further examined for transgene expression levels and immune response induction following subcutaneous (s.c.) or intramuscular (i.m.) injection. We showed the ability to tune the balance of immune stimulation between Th1 and Th2 immune responses by altering the LNP compositions. We also explored the feasibility of using one LNP formulation to elicit both strong Th1 and Th2 immune responses and the effect of such a dual attack in improving antitumour efficacy compared to formulations with Th1-biased immune responses alone. Furthermore, we confirmed the synergistic effect of combining the optimized mRNA LNP vaccine and systemic immune checkpoint blockade therapy in therapeutic melanoma models, and investigated the mechanisms underlying the enhanced immune response and correlations with cellular transfection activity and local immune activation profiles.
Results
LNP screening for mRNA delivery to DCs, antigen presentation and maturation
To generate a 1,080-member LNP library, we used DLin-MC3-DMA as the ionizable lipid, DMG-PEG2000 as the PEGylated lipid, and six helper phospholipids that have previously been used in FDA-approved or experimental LNP formulations48. These were chosen to represent a range of different charges, cationic lipids (1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) and dimethyl dioctadecyl ammonium (DDAB)), zwitterionic lipids (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and DSPC) and anionic lipids (1,2-dimyristoyl-sn-glycero-3-phosphate (14PA) and 1-stearoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (18PG))47,48,51,54,56–58. Using DLin-MC3-DMA, cholesterol, DMG-PEG2000 and one of the six helper lipids, the 1,080 LNP formulations were generated by varying the following parameters: (1) combined molar percentage of DLin-MC3-DMA and helper lipid ranging from 20% to 80%; (2) weight ratio of cholesterol to DMG-PEG2000 ranging from 10 to 500; (3) weight ratio of DLin-MC3-DMA to helper lipid ranging from 1 to 200; and (4) molar ratio of chargeable groups in ionizable lipid to phosphate groups in mRNA (N/P ratio) ranging from 4 to 12. These parameter choices provided us with a sufficiently diverse library of LNP formulations from which we assessed mRNA delivery (Fig. 1a).
Fig. 1 |. In vitro screening of mRNA lipid nanoparticles for the transfection and induction of antigen presentation and maturation in DCs.
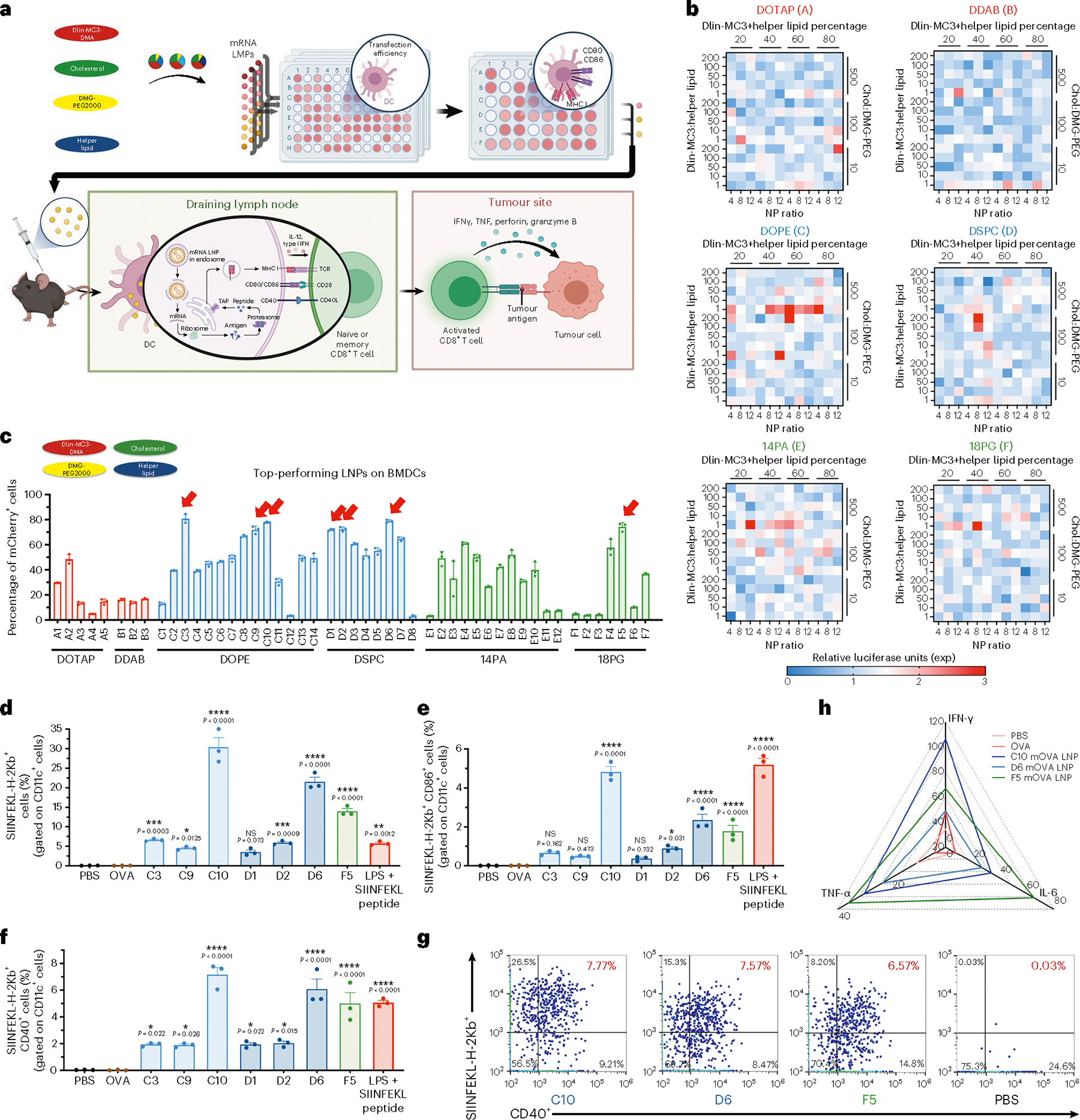
a, Schematic of the screening method and the therapeutic mechanism of mRNA LNP vaccination against a solid tumour. In vitro transfection efficiency was assessed for 1,080 LNP formulations with different helper lipids and component ratios. The top-performing formulations were then tested on BMDCs for transfection and antigen presentation, and in vivo immune responses induced by selected LNPs were assessed. LNPs transfect tissue-resident DCs following s.c. injection, or drain into the neighboring lymph nodes where they transfect APCs including DCs. These APCs translate and process the mRNA into peptides presented on major histocompatibility complex molecules on the cell surface. The lipids also trigger activation pathways that promote co-stimulatory molecule expression and cytokine release. T cells activated by the APCs proliferate and travel to the tumour site to kill cancer cells in an antigen-specific manner. TAP, transporter associated with antigen processing; TCR, T cell receptor; TNF-α, tumour necrosis factor α. b, DC2.4 cells were treated with fLuc mRNA LNPs (1 μg ml−1). The relative luciferase expression after 24 h incubation with fLuc mRNA LNPs is shown in a heat map. c, BMDCs were treated with the 49 top-performing LNPs packaged with mCherry mRNA. The percentage of mCherry+ cells gated on CD11c+ cells after 24 h incubation with mRNA LNPs is shown. LNP formulation details are shown in pie charts with DLin-MC3-DMA in red, cholesterol in green, DMG-PEG2000 in yellow and helper lipids in blue. The top seven formulations, indicated by red arrows, were selected for further study. d–f, Antigen presentation (d), with maturation levels of BMDCs (e,f) were analysed by flow cytometry after 24 h incubation with the seven mOVA-loaded LNPs, PBS, free OVA, or LPS and SIINFEKL peptide. The percentages of SIINFEKL-H-2Kb+ cells (d), additionally positive for CD86 (e) or CD40 (f) gated on CD11c+ cells are shown. g, Representative flow cytometry analysis of SIINFEFL-H-2Kb and CD40 expression on BMDCs treated with the three top-performing LNPs. h, Secretion levels of IFN-γ, TNF-α and IL-6 within the supernatant of BMDCs after 24 h incubation with the three mOVA-loaded LNPs were measured by ELISA and are shown in a radar chart. Data represent the mean ± s.e.m. from a representative experiment (n = 3 (b–g), n = 4 (h) biologically independent samples) of two independent experiments. Data were analysed using one-way ANOVA and Dunnett’s multiple comparisons test against the PBS control group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant.
To select LNP formulations with strong APC-specific transgene expression, we first evaluated the mRNA delivery efficiency of the LNP library in DC2.4 cells (an immortalized murine dendritic cell line) using firefly luciferase (fLuc) mRNA and measured luciferase protein expression (Fig. 1b). With the helper lipid fixed, adjusting the above-mentioned four parameters in the LNP formulations substantially varied the gene expression levels. Next, we validated the transfection efficiency of the top 49 LNP formulations containing mCherry mRNA in BMDCs using flow cytometry analysis (Supplementary Table 1). Results shown in Fig. 1c confirmed the high in vitro transfection efficiency of these LNPs prepared using different helper lipids, DOTAP (Group A), DDAB (Group B), DOPE (Group C), DSPC (Group D), 14PA (Group E) and 18PG (Group F). Seven LNP formulations, including C3, C9, C10, D1, D2, D6 and F5 (indicated by red arrows), achieved high transfection of BMDCs with more than 70% of cells transfected (Supplementary Fig. 1 and Table 2).
A detailed comparative analysis was conducted on the transfection efficiency results of the set of 1,080 LNP formulations to extract the effect of LNP helper lipid and formulation parameters on DC2.4 cell transfection in vitro59. As Supplementary Fig. 2 shows, LNP formulations with relatively high transfection efficiencies were those with (1) high ratios of cholesterol to DMG-PEG2000 approaching 500:1, (2) low ratios of DLin-MC3-DMA to helper lipid approaching 1:1, (3) moderate molar percentages of DLin-MC3-DMA and helper lipid in a range of 40% to 60% and (4) moderate N/P ratios around 8.
For effective mRNA vaccines, both efficient antigen expression and potent immune cell activation are needed to generate a robust immune response. The immunostimulatory effects of the seven selected formulations were therefore tested on BMDCs using mRNA encoding ovalbumin (mOVA), a model antigen protein widely used in vaccine studies. After treatment with mOVA LNP for 72 h, compared with PBS, OVA protein and other formulations (C10, D6 and F5 LNPs) resulted in a markedly elevated expression of the OVA-derived SIINFEKL peptide on MHC-I, indicating antigen presentation (~30% SIINFEKL-H-2Kb+ for C10 LNPs, ~21.5% for D6 LNPs and ~14.0% for F5 LNPs) (Fig. 1d). In addition, higher levels of CD86+SIINFEKL-H-2Kb+ and CD40+SIINFEKL-H-2Kb+ were observed after treatment with these three LNP formulations (Fig. 1e–g). Compared with the positive control group treated with lipopolysaccharides (LPS) and SIINFEKL peptide, F5 LNPs induced a comparable level of CD40+SIINFEKL-H-2Kb+ (~5.0%), whereas C10 and D6 LNPs generated 1.43-fold and 1.22-fold higher levels, respectively. Along with improved maturation of BMDCs, substantially higher secretion levels of inflammatory cytokines IFN-γ, TNF-α and IL-6 were also observed within the supernatants of BMDCs after treatment with the three OVA mRNA-loaded LNPs (C10, D6 and F5) (Fig. 1h and Extended Data Fig. 1). The lack of correlation between OVA peptide presentation on MHC molecules and fLuc/mCherry expression suggests that there may be additional factors influencing antigen presentation beyond protein antigen expression alone, such as cellular metabolic activity, protein expression kinetics, intracellular trafficking and peptide loading onto MHC molecules. Further investigation is needed to better understand the mechanism and its implication for immune responses.
We next evaluated the in vivo delivery efficacy of these candidates in mice following s.c. injection or i.m. injection. To determine whether transfected APCs were present within draining lymph nodes, we delivered Cre-recombinase mRNA (mCre) LNPs to genetically engineered tdTomato (tdTom) reporter (Ai9) mice containing a LoxP-flanked stop cassette that prevents expression of the tdTom protein. This mouse model allows detection of the transfected cells as a result of Cre-recombinase expression, as the expressed recombinase edits out the stop cassette, enabling the expression of fluorescent tdTom. Our results indicated that the three (C10, D6 and F5) LNP formulations yielded substantial levels of tdTom+ lymphocytes and CD11c+ cells in the draining lymph nodes (Fig. 2a, and Supplementary Figs. 3 and 4). Consistent with many reports showing that nanovaccines can efficiently traffic to the draining lymph node after s.c. administration, our results indicated that compared with i.m. injection, a higher percentage of transfected cells was detected in the draining lymph nodes following s.c. injection. On the basis of this finding, the s.c. administration route was selected for further testing.
Fig. 2 |. In vivo assessments of lymph-node-cell transfection and immune activation by the top 3 LNP formulations.
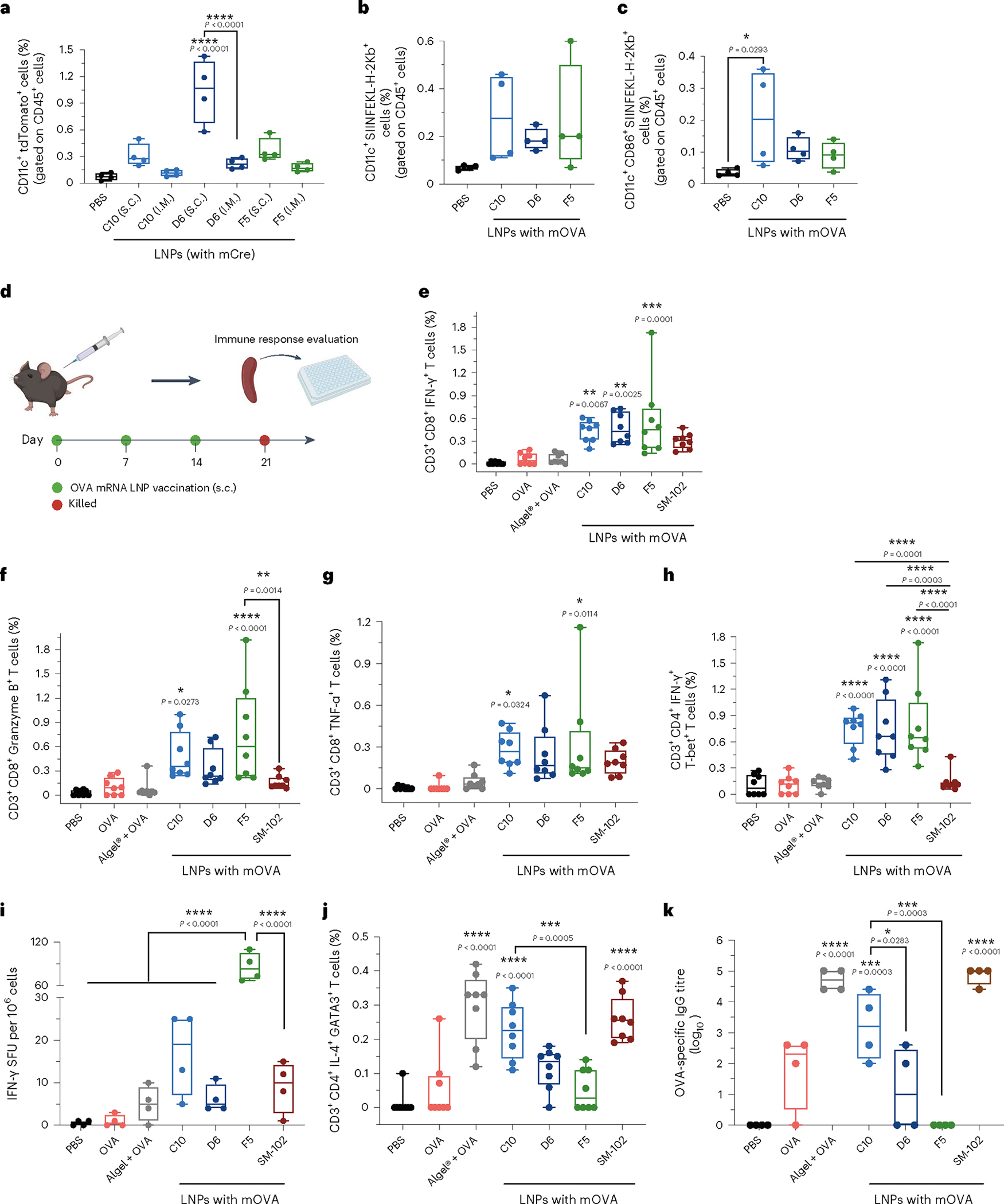
a, Ai9 mice were administered the top 3 LNPs loaded with mCre via i.m. and s.c. injections (10 μg mCre per mouse). Transfection of immune cells in draining lymph nodes was analysed by flow cytometry. Percentages of cells positive for tdTomato as well as CD11c gated on CD45+ cells are shown. Gating strategy is shown in Supplementary Fig. 4. b,c, C57BL/6 mice were administered PBS or C10, D6 or F5 LNPs loaded with mOVA via s.c. injection. DC antigen presentation (b), with maturation levels (c) in the draining lymph nodes were analysed using flow cytometry after 3 d following a single dosage of mRNA LNPs. Cells positive for CD11c and SIINFEKL-H-2Kb (b), as well as CD86 (c) are shown. Gating strategy is shown in Supplementary Fig. 6. d, Timeline for the immune activation experiment. C57BL/6 mice were given three s.c. injections, 1 week apart, of PBS, free OVA protein, or C10, D6 or F5 LNPs loaded with mOVA (10 μg OVA protein or 10 μg mOVA per injection). Mice were killed 1 week after the final injection, and their splenocytes and lymphocytes were isolated for analysis. e–h, C57BL/6 mice were administered with PBS, free OVA protein, OVA protein mixed with aluminum hydroxide gel (Alhydrogel) (1:1), or C10, D6, F5 or SM-102 LNPs loaded with mOVA via s.c. injection (10 μg OVA protein or 10 μg mOVA per injection). Splenocytes were restimulated in vitro with OVA and SIINFEKL peptide (100 μg ml−1 OVA and 2 μg ml−1 SIINFEKL) for 6 h and assessed via flow cytometry and intracellular cytokine staining to determine the percentages of CD3+CD8+IFN-γ+ (e), CD3+CD8+Granzyme B+ (f), CD3+CD8+TNF−α+ (g) and CD3+CD4+IFN−γ+T-bet+ (h) cells. i, Frequency of IFN-γ-producing cells in spot-forming unit (SFU) among restimulated splenocytes, assessed via ELISpot. j, Percentage of restimulated splenocytes positive for CD3, CD4, IL-4 and GATA-3, assessed by flow cytometry and ICS and representing Th2 cells. Gating strategy is shown in Supplementary Figs. 7–12 and 14. k, Titres of OVA-specific IgG antibodies in blood serum on day 21, determined by ELISA. Data represent the mean ± s.e.m. from a representative experiment (n = 4 (a–c,i,k) biologically independent samples of two independent experiments, n = 8 (e–h,j) biologically independent samples of three independent experiments). Data were analysed using one-way ANOVA and Tukey’s multiple comparisons test for a–c, and e–k. For boxplots, the box extends from the 25th to the 75th percentiles and the line in the middle of the box is plotted at the median. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Next, the antigen presentation and maturation levels of APCs in draining lymph nodes were examined after s.c. injection of LNPs (Fig. 2b,c, and Supplementary Figs. 5 and 6). After 3 d following a single dose of mRNA LNPs, we observed an increase in the levels of SIINFEKL-H-2Kb+CD11c+ cells and SIINFEKL-H-2Kb+CD86+CD11c+ cells in the draining lymph nodes of mice that received the three formulations. Specifically, compared with the PBS-treated group, there was a substantial elevation in SIINFEKL-H-2Kb+CD11c+ cells, with 4.0-fold, 2.7-fold, and 3.8-fold higher levels observed for C10, D6 and F5 LNP formulations, respectively. These results indicated effective activation and antigen presentation capacity of the CD11c+ population including dendritic cells in response to the mRNA LNP treatment.
Taken together, the above results demonstrated the identification of the top 49 formulations from an LNP library consisting of 1,080 formulations based on the transfection efficiency in DC2.4 cells. Further evaluations on BMDCs led to the selection of seven leading LNPs, and three of them (C10, D6 and F5) showed potent antigen presentation and immunostimulatory effects both in vivo and in vitro. These three leading formulations were thus selected for further in vivo tests.
Distinct immunological profiles of top-performing LNPs
The vaccination potential of the three lead LNPs was further tested in mice following s.c. injections (three doses at days 0, 7 and 14) (Fig. 2d). For comparison, we included the SM-102 LNP formulation used in the Moderna COVID-19 vaccine (Spikevax) and classic adjuvant aluminum hydroxide gel (Alhydrogel) (mixed with OVA protein at 1:1 ratio) in further experiments. The antigen-specific CD8+ T cell response induced by LNPs was first assessed. Spleens of vaccinated mice were collected on day 21 and homogenized into a cell suspension for ex vivo antigen restimulation. The three LNP-treated groups showed increased frequencies of CD3+CD8+IFN-γ+, CD3+CD8+Granzyme B+ and CD3+CD8+TNF-α+ cell populations (Fig. 2e–g and Supplementary Figs. 7–10). The SM-102 LNP- and Alhydrogel-treated groups did not generate notable antigen-specific cytotoxic T cell response. In comparison to SM-102 LNP, the CD3+CD8+TNF-α+ cell frequencies demonstrated ~1.5-fold increases after treatment with C10, D6 or F5 LNPs. There were ~1.4-fold, 1.6-fold and 1.9-fold increases in CD3+CD8+ IFN-γ + cell frequencies after treatment with C10, D6 and F5 LNPs, respectively, compared with SM-102 LNPs. Similarly, the increases were ~3.0-fold, 2.1-fold and 4.7-fold after treatment with C10, D6 and F5 LNPs, respectively, in terms of CD3+CD8+GranzymeB+ cell frequencies compared with SM-102 LNPs. Along with the potent CD8+ T cell response, an average of fivefold higher numbers of antigen-specific Th1 cells (CD3+CD4+IFN-γ+T-bet+) were observed for all three LNP formulations compared with SM-102 LNPs (Fig. 2h, and Supplementary Figs. 11 and 12). The elevated production of pro-inflammatory cytokines after restimulation with antigen, including IFN-γ, TNF-α and IL-6, also indicated a potent antigen-specific Th1-type response induced by the three LNP formulations (Fig. 2i, Supplementary Fig. 13 and Extended Data Fig. 2).
The frequencies of antigen-specific CD3+CD4+IL-4+GATA-3+ Τh2 cells and CD3+CD4+IL-17+RORγt+ Τh17 cells, and the magnitude of antibody responses generated were further examined for these groups. A substantial number of Th2 cells (18-fold higher than the background, P < 0.0001) was observed in the C10-treated group (Fig. 2j and Supplementary Fig. 14), in contrast to the D6 and F5 groups (2.1-fold and 1.0-fold higher, respectively; P > 0.5). However, limited levels of Th17 immune response were observed across all groups (Supplementary Figs. 15 and 16). In addition, as shown in Fig. 2k and Extended Data Fig. 3, C10 induced a substantially higher OVA-specific IgG titre including both IgG1 and IgG2a subclass titres, indicating a potent humoral response. Conversely, the antibody response generated by D6 was limited, and it was undetectable for F5. For both Alhydrogel and SM-102 LNP groups, high OVA-specific IgG titres were observed at 3 weeks after vaccination. In addition, the biosafety profile of the C10, F5 and D6 formulations was assessed. Throughout the vaccination schedule, no significant differences were observed in terms of body weight or serum cytokine levels of IL-6, TNF-α, TGF-β, IL-12, IFN-γ, IL-10 and IL-4 (Supplementary Figs. 17 and 18).
The above results indicated that after vaccination, all three selected LNPs induced a more potent antigen-specific Th1-type immune response than Alhydrogel and SM-102 LNPs. For the Th2-type response, however, the three leading LNP formulations exhibited distinct profiles. C10 LNPs elicited both potent Th1 and Th2 responses. While D6 and F5 generated a potent Th1 response, they generated moderate or undetectable levels of Th2 responses.
Differential antitumour effects among the top-performing LNPs
Given the strong antigen-specific immune responses induced by the top mRNA LNPs, we examined the efficacy of the three LNP formulations as cancer vaccines in multiple prophylactic and therapeutic tumour models. C57BL/6 mice were immunized on days 0, 7 and 14 with 10 μg of free OVA protein, OVA protein mixed with Alhydrogel, or LNPs containing 10 μg mOVA. On day 21, animals were inoculated s.c. in the right posterior side with 1 × 106 B16-OVA cells (Fig. 3a). All three LNP formulations along with SM-102 LNPs showed stronger tumour growth inhibition with prolonged overall survival times than the free OVA protein and Alhydrogel groups. The median survival time was 40, 32, 30 and 32 d for C10, D6, F5 and SM-102 LNPs, respectively, compared with 15 d for the free OVA protein group and 20 d for Alhydrogel group (Fig. 3b–d). Furthermore, compared with D6 and F5, which generated strong Th1 response only, or SM-102 LNP, which generated strong Th2 response only, C10 LNPs triggering both Th1 and Th2 responses yielded a markedly improved protection effect, with ~40% of the mice remaining tumour-free beyond 60 d. To further validate the observed efficacy of C10 mOVA LNPs and investigate its long-term efficacy, we conducted a tumour rechallenge study with a larger sample size (Fig. 3e). C57BL/6 mice were immunized as described previously, and on day 21, mice were subcutaneously inoculated with 1 × 106 B16-OVA cells on the right posterior side. Survival was monitored for 100 d, and 18 out of 30 mice (60%) remained tumour-free after vaccination with C10 mOVA LNPs (Fig. 3f). These mice were subsequently divided into two groups: one group (n = 9) was rechallenged with 1 × 106 B16-OVA cells on the right posterior side, and the other group (n = 9) was rechallenged with 1 × 106 B16F10 cells on the same side. As illustrated in Fig. 3g,h, 4 out of 9 mice remained tumour-free after being rechallenged with B16-OVA cells, indicating the long-term protective effect provided by C10 mOVA LNPs. Interestingly, a substantial delay in tumour growth was observed in mice challenged with B16F10 cells (Fig. 3i,j), probably due to epitope or antigen spreading, which is a process characterized by the enhancement and diversification of the endogenous T cell response against antigenic epitopes other than the originally targeted epitope60,61. Epitope or antigen spreading usually occurs following initial therapy-mediated tumour destruction, leading to the release of secondary tumour antigens. Additional studies are needed to confirm this hypothesis.
Fig. 3 |. Antitumour efficacy of top mRNA LNP formulations as prophylactic vaccines.
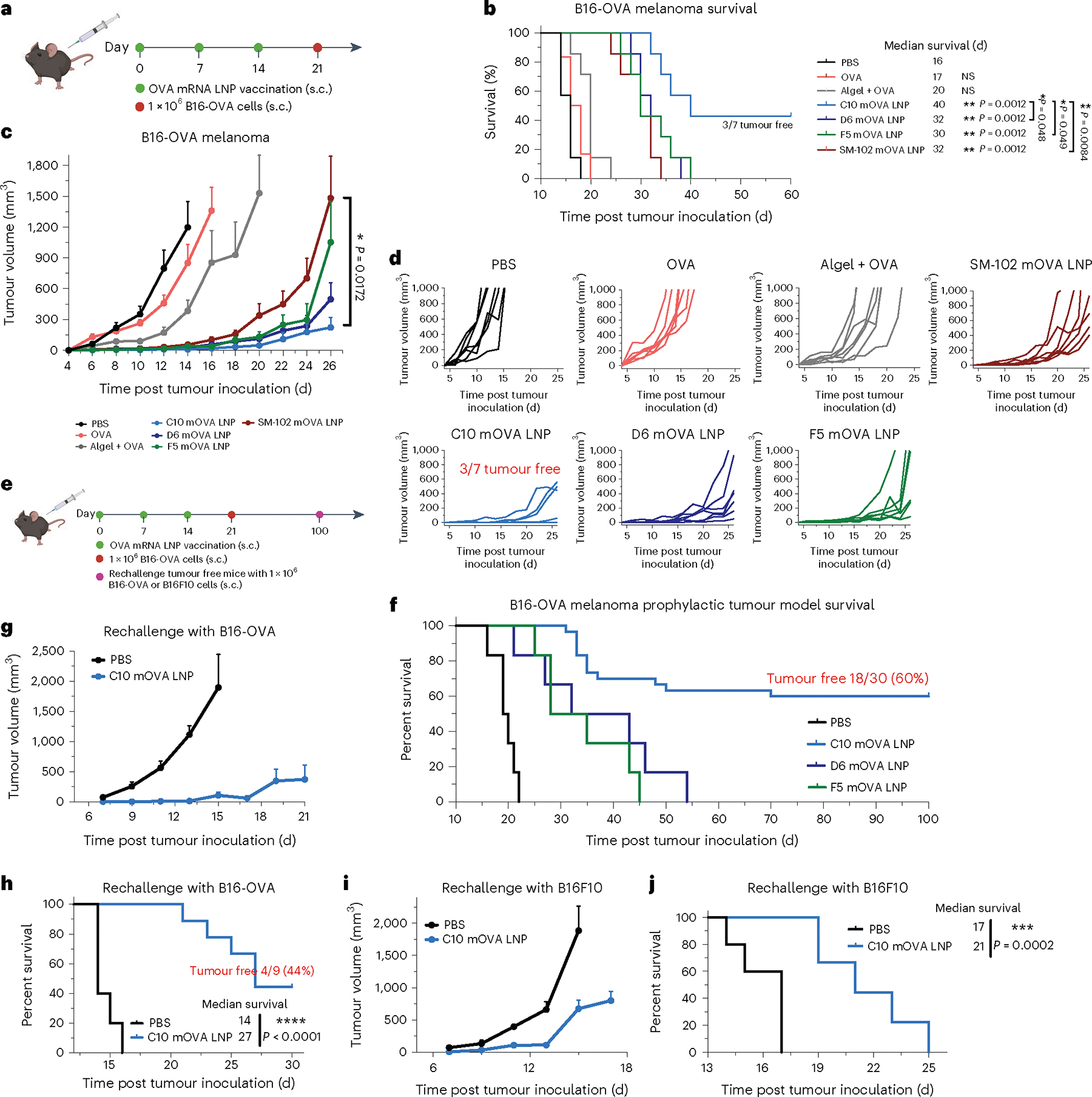
a–d, Schematic and results of a prophylactic vaccination model for OVA-expressing melanoma in C57BL/6 mice. Mice were given three s.c. injections (10 μg mOVA per injection), 1 week apart, of PBS, free OVA protein, OVA protein mixed with Alhydrogel (1:1) or mOVA-loaded C10, D6, F5 or SM-102 LNPs before s.c. inoculation of OVA-expressing melanoma (B16-OVA) cells (a). Survival curves (b), average tumour volume (c) and individual tumour volume (d) over time are shown. e–j, Schematic and results of a prophylactic vaccination and rechallenge model for B16-OVA in C57BL/6 mice. Mice were vaccinated as described above before s.c. inoculation of OVA-expressing melanoma (B16-OVA) cells. After 100 d, the tumourfree mice were rechallenged with either B16-OVA or B16F10 cells, and mice at a similar age were included as a control group injected with PBS (e). Survival curves (f,h,j) and average tumour volume (g,i) are shown. In b–d, data represent mean ± s.e.m. from a representative experiment (n = 7 biologically independent samples) of two independent experiments. In f, data represent mean ± s.e.m.; n = 30 for the C10 mOVA LNP group and for other groups, n = 6 biologically independent samples. In g–j, data represent mean ± s.e.m.; n = 9 biologically independent samples for the C10 mOVA LNP group and n = 5 for the PBS control group. Differences between treatment groups were analysed using one-way ANOVA and Tukey’s multiple comparisons test. Survival curves were compared using log-rank Mantel–Cox test, and the stack of P values were corrected using the Holm-Šídák method for multiple comparisons with α set at 0.05. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We evaluated the therapeutic efficacy of C10 LNPs as a therapeutic vaccine in the B16, MC38 and EG7 tumour models using OVA as the model antigen as well as two other melanoma tumour-associated antigens, tyrosinase-related protein 2 (Trp2) and glycoprotein 100 (Gp100). First, C57BL/6 mice were inoculated s.c. in the right posterior side with 3 × 105 B16-OVA, MC38-OVA or EG7-OVA cells on day 0. On days 4, 11 and 18, the mice were immunized with C10 LNPs containing 10 μg mOVA (Fig. 4a). As demonstrated in Fig. 4b–d, C10 LNPs yielded considerable tumour suppression in the B16-OVA treatment model, with a median survival of 26 d compared with 17 d for the negative control group. When C10 LNPs were given in combination with an immune checkpoint inhibitor (100 μg anti-CTLA-4 monoclonal antibody, given intraperitoneally (i.p.) on days 6, 13, 20 and 27), a synergistic effect was observed, with a prolonged median survival time of 33.5 d. In contrast, no significant tumour suppression was observed for the group treated with only α-CTLA-4 antibody in comparison with the PBS control. The antitumour efficacy was also confirmed in the MC38-OVA and EG7-OVA treatment models (Fig. 4e–h, and Supplementary Figs. 19 and 20). In the MC38-OVA treatment model, C10 LNPs displayed a substantial tumour suppression effect, showing a median survival of 47 d compared with 26 d for the negative control. When C10 LNPs were combined with 100 μg of anti-CTLA-4 mAb using the same protocol described above, the median survival was extended to 53 d. Among all C10 LNP-treated mice with or without anti-CTLA-4 mAb, 42.9% remained tumour-free at 100 d post tumour inoculation. In the EG7-OVA treatment model, C10 LNPs yielded a median survival of 25 d compared with 15 d for the negative control group. Combining C10 LNPs and anti-CTLA-4 mAb also showed synergy, with a prolonged median survival of 37 d; and 1 of 7 mice remained tumour-free at 100 d post tumour inoculation.
Fig. 4 |. Antitumour efficacy of the top mRNA LNP formulations as therapeutic vaccines.
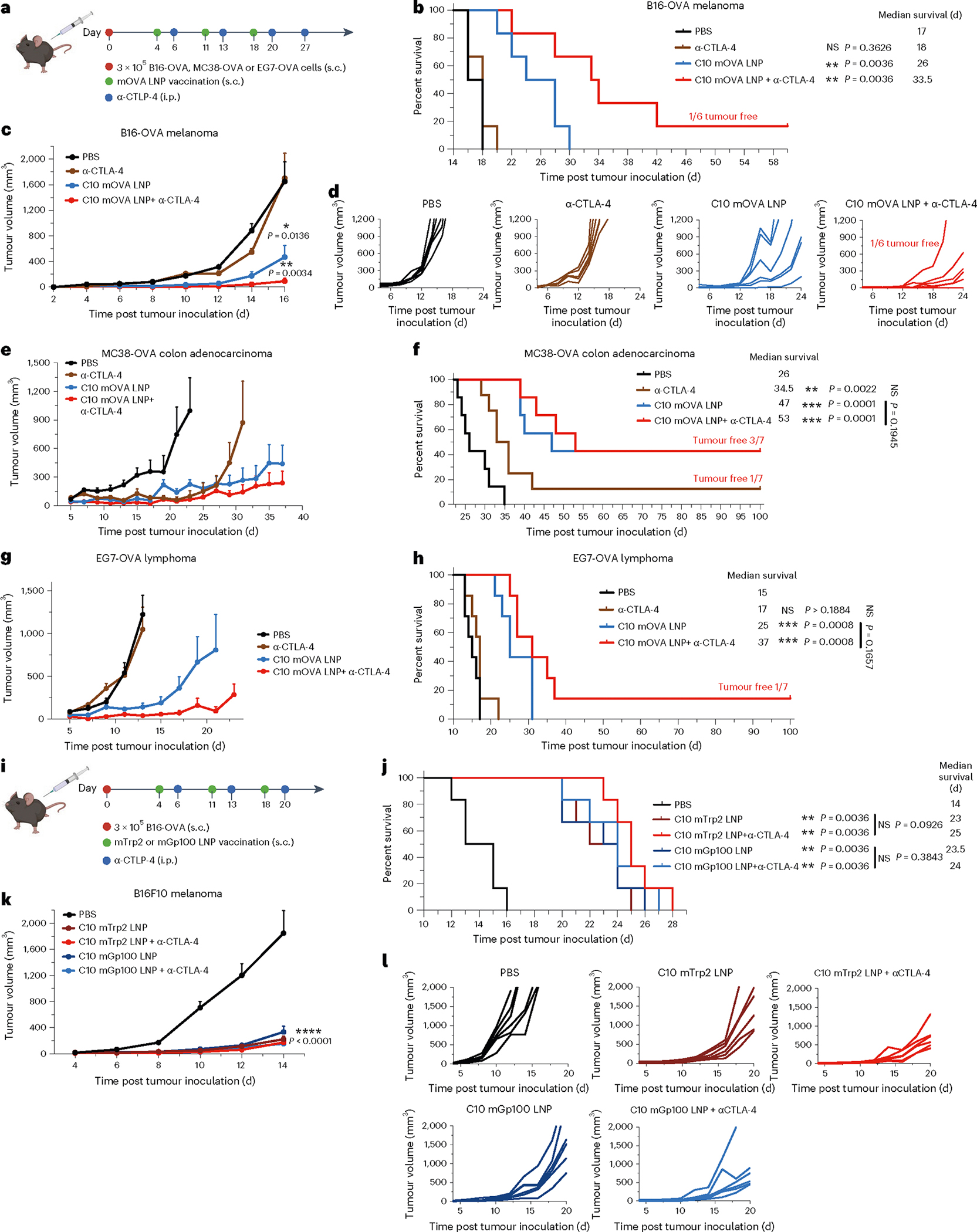
a–h, Schematic and results of a therapeutic vaccination model for B16-OVA, MC38-OVA and EG7-OVA in C57BL/6 mice. Mice were inoculated s.c. with B16-OVA, MC38-OVA or EG7-OVA cells and then given three s.c. injections, 1 week apart, of mOVA-loaded C10 (10 μg mOVA per injection) or PBS. Two groups received a repeated anti-CTLA-4 mAb (100 μg per i.p. injection) treatment alone or in combination with the LNPs (a). Survival curves (b,f,h), average tumour volumes (c,e,g) and individual tumour volumes (d) are shown. i–l, Schematic and results of a therapeutic vaccine against melanoma-associated antigens for melanoma in C57BL/6 mice. Mice were inoculated s.c. with B16F10 cells and then given three s.c. injections, 1 week apart, of PBS or C10 LNP loaded with mRNA encoding Trp2 (mTrp) or Gp100 (mGp100) (10 μg mRNA per injection). Two groups received the anti-CTLA-4 mAb (100 μg per i.p. injection) treatment in combination with LNP treatment (i). Survival curves (j), average tumour volumes (k) and individual tumour volumes (l) are shown. Data represent mean ± s.e.m. with n = 7 (e–h) and n = 6 (a–d, i–l) biologically independent samples. Differences between treatment groups were analysed using one-way ANOVA and Tukey’s multiple comparison tests. Survival curves were compared using log-rank Mantel–Cox test and P values were corrected using the Holm-Šídák method for multiple comparisons with α set at 0.05. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; αCTLA-4, anti-CTLA-4 mAb.
The C10 LNPs were next tested in the same mouse model using clinically relevant tumour antigens Trp2 and Gp100 instead of the model antigen OVA (Fig. 4i). C57BL/6 mice were inoculated subcutaneously in the right posterior side with 3 × 105 B16F10 cells on day 0. On days 4, 11 and 18, the mice were vaccinated with C10 LNPs containing 10 μg of mRNA encoding either Trp2 or Gp100. The strong antitumour effect was also observed by using these two antigens, showing substantially prolonged median survival times of 23 and 23.5 d for C10-mTrp2 LNPs and C10-mGp100 LNPs, respectively (Fig. 4j–l). However, no significant improvement was observed when combining these C10 formulations with α-CTLA-4 antibody treatment. The observed synergistic effect between the C10 LNP and anti-CTLA-4 mAb with the mOVA model antigen, but not with Trp2 and mGp100 tumour antigens, raises interesting questions. Differences in antigen immunogenicity, antigen processing, T cell receptor affinity and the tumour microenvironment may contribute to the difference. Further investigation is warranted to understand the underlying mechanisms and optimize antigen selection for effective combination therapies.
Long-term protection correlated with dual attack by T cells and NK cells
To further understand the mechanisms of mRNA LNP vaccine efficacy, cell depletion experiments were conducted for C10 and F5 LNPs, which induced distinct immunological profiles on the B16-OVA melanoma model (Fig. 5a). As shown in Fig. 5b–g, depletion of T (CD3+) cells, NK (NK1.1+) cells or B (CD20+) cells markedly reduced the survival advantage conferred by C10 mOVA LNPs. In contrast, for F5-mOVA LNPs, the antitumour effect was abolished only when T cells were depleted. Removal of NK cells or CD20+ B cells did not significantly alter the tumour suppression effect induced by F5-mOVA LNPs.
Fig. 5 |. A coordinated attack by T cells and NK cells was responsible for long-term protection.
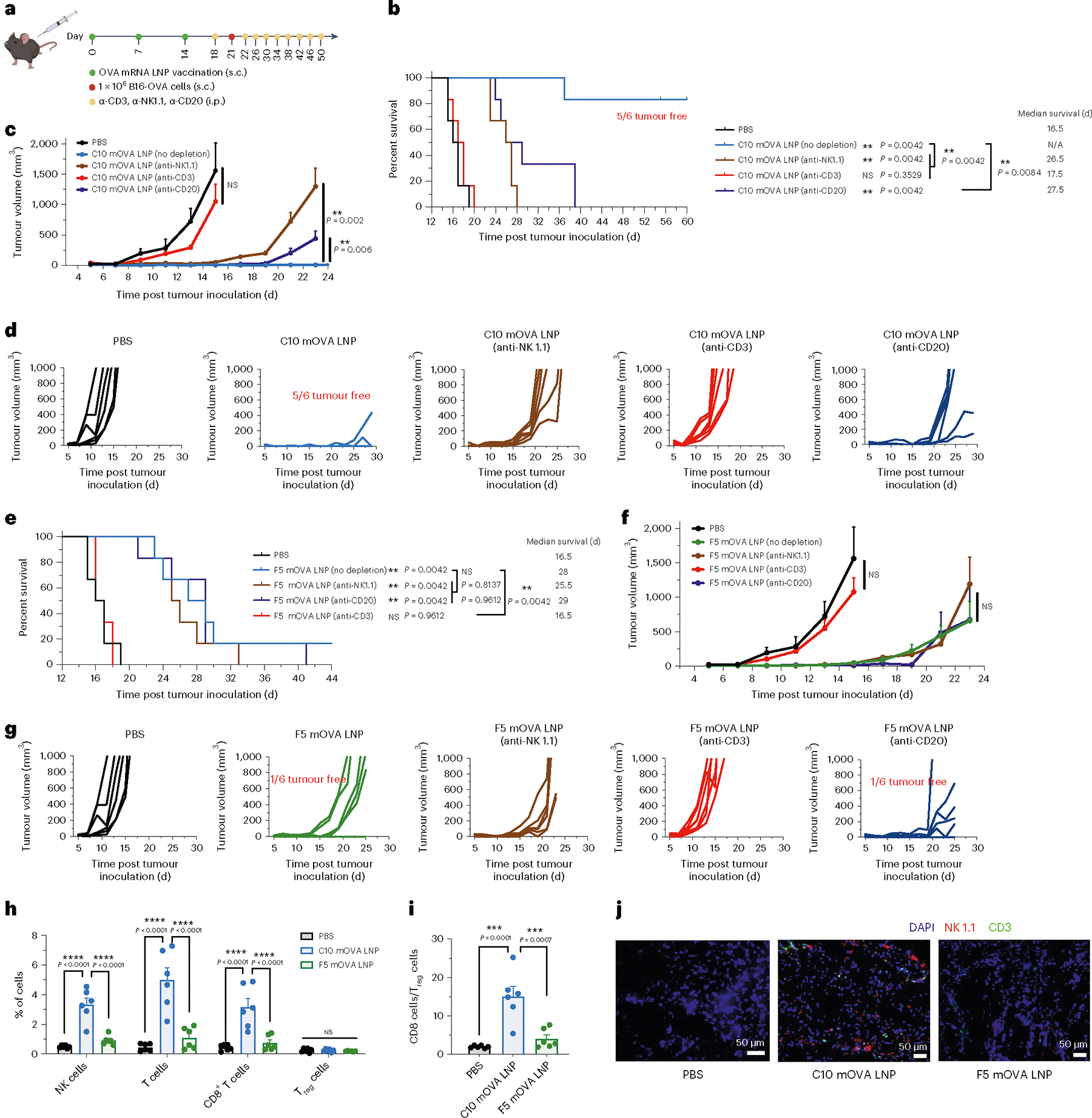
a–g, Schematic and results of cell depletion experiments in the prophylactic vaccination model for OVA-expressing melanoma in C57BL/6 mice. Mice were given three s.c. injections, 1 week apart, of PBS or mOVA-loaded C10 or F5 LNPs (10 μg mOVA per injection) before s.c. inoculation of B16-OVA cells, and antibody for cell depletion were injected every 4 d (i.p., 200 μg per mice) (a). Survival curves (b,e), average tumour volumes (c,f) and individual tumour volumes (d,g) over time are shown (n = 6 biologically independent mice per group). h, Tumour-infiltrating immune cells including NK cells, T cells, CD8+ T cells and Treg cells were determined by flow cytometry on day 22 post tumour inoculation (n = 6 per group). i, Ratio of CD8+ T cell percentage to CD4+FoxP3+CD25+ Treg cell percentage on day 22 post tumour inoculation (n = 6 per group). j, Immunofluorescent analysis of CD3 T cell and NK-cell infiltration of tumour section on day 22 post tumour inoculation. Blue, DAPI; green, CD3; red, NK 1.1. Data represent the mean ± s.e.m. Differences between treatment groups were analysed using one-way ANOVA and Tukey’s multiple comparisons test. Survival curves were compared using log-rank Mantel–Cox test and P values were corrected using the Holm-Šídák method for multiple comparisons with α set at 0.05. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Multiple effector lymphocyte populations were statistically enriched in tumours from mice immunized with C10 LNPs at 22 d post tumour inoculation, with a 6.9-fold enrichment in NK cells, an 11.2-fold enrichment in T cells and a 7.4-fold enrichment in CD8+ T cells compared with the PBS control group (Fig. 5h). In addition, C10 mOVA LNPs established a markedly higher CD8-to-regulatory T cell (Treg) ratio in the tumour: 7.8- and 3.7-fold higher than in the PBS group and F5-mOVA LNPs, respectively (Fig. 5i). Furthermore, as shown in Fig. 5j, higher numbers of NK cells and T cells were observed in tumours from the mice treated with C10 mOVA LNPs, whereas no such enrichment effect was detected in mice treated with F5-mOVA LNPs.
These results collectively showed that C10 LNPs are capable of eliciting both Th1 and Th2 immune responses, serving a dual role in promoting strong cytotoxic T lymphocyte responses and Th1 responses against cancer, and facilitating humoral and Th2 responses, which contribute to the antitumour effect probably through activation of NK cells via the antibody-dependent cellular cytotoxicity pathway62,63. Coordination of these cell populations and responses orchestrated by C10 LNPs provides a highly effective approach to potentiate cancer vaccines.
Potential pathways for inducing Th1-only or Th1-plus-Th2 immune responses
As shown in Fig. 6a, when the antigen is translated in the cytosol of DCs and the processed peptides are loaded onto MHC-I molecules, potent cytotoxic T cell response along with Th1 immune response can be initiated. On the other hand, a Th2 response requires the antigen to be expressed and released by non-APCs, and then internalized and processed by DCs, macrophages or B cells to be presented in the context of MHC-II. Although in some circumstances, antigens from the extracellular environment can be presented on MHC class I molecules via cross-presentation pathways, the compositions of mRNA LNPs have limited effect on this process. To explore the mechanism of biased Th1 vs Th2 responses generated by GFP mRNA-containing LNPs with different compositions, we examined the local transfection process following s.c. injection. First, we assessed the overall transfection efficiency of the three LNPs by quantifying the expression of luciferase protein using a luciferase-encoding mRNA. As shown in Supplementary Fig. 21a, C10, D6 and F5 LNPs all showed high but transient transgene expression levels at the injection site, although F5 LNPs showed slightly lower expression at 6 h. Beyond this timepoint, differences in luciferase expression level and duration were non-significant. Luciferase expression levels among all major organs measured at 48 h after injection were not statistically different. More than 90% of the total luciferase expression was localized at the injection site, confirming that transgene expression was predominantly confined to the site of administration (Supplementary Fig. 21b). Furthermore, at 24 h post injection, we quantified GFP expression levels in various cell types at the injection sites using flow cytometry (Fig. 6b–c and Supplementary Fig. 22). After a single injection of D6 or F5 LNPs, ~45% of the transfected cells in the local tissue were immune cells, which was more than twofold higher than for C10 LNPs. The ratio of non-immune cells to immune cells among the GFP-expressing cells was 4.5 in the C10 group, which was 2.9-fold and 3.6-fold higher than for the D6 and F5 groups, respectively. These results indicated considerable differences among LNPs with different compositions in terms of cell types transfected. In addition, we measured the immunostimulatory cytokines at the injection site following LNP administration. C10 LNP injection induced similar (P > 0.05) levels of Th1 cytokines IFN-γ and TNF-α at the injection site at 4 h and 24 h post injection, compared with D6 or F5 LNP injection (Extended Data Fig. 4). More importantly, the average levels of a Th2 cytokine IL-4 in C10 injected sites were ~1.6-fold and fourfold higher at 4 h and 24 h post injection, respectively, than in the group treated with PBS. These results are consistent with the cellular evidence that C10 is capable of inducing strong Th1 and Th2 responses. This unique property of C10 correlates well with its ability to effectively transfect both dendritic cells and non-APCs at the injection site, which is critical to initiating both types of antigen presentation processes and eliciting subsequent Th1/Th2 response profiles. These contrasting results among the 3 LNP formulations, even though they all generated a strong Th1 response, highlight the importance of LNP formulation design and cell targeting in modulating the immune response.
Fig. 6 |. Local transfection, cellular uptake and endosomal escape of mRNA LNPs.
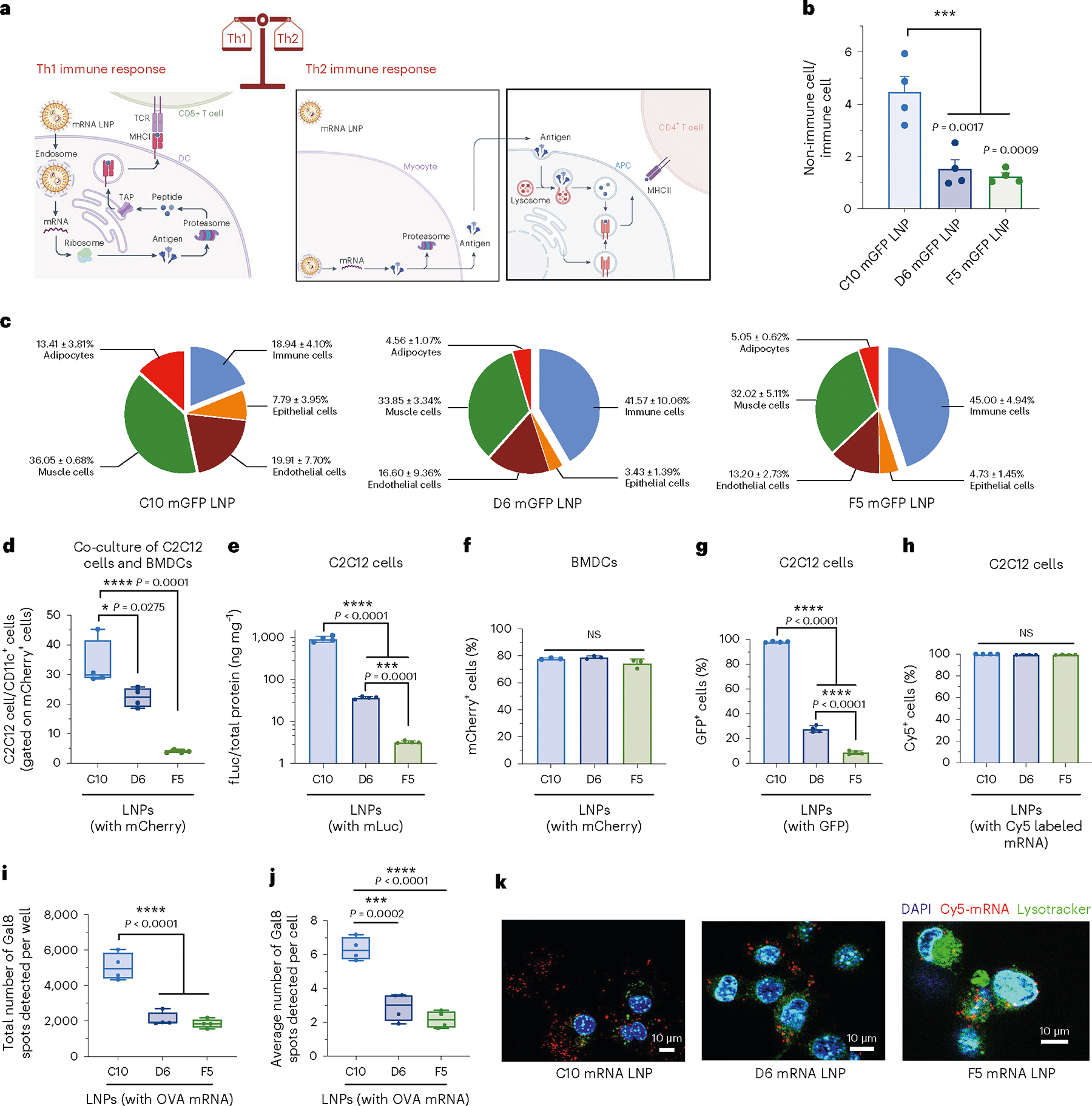
a, Schematic of different immune responses induced by mRNA LNPs. Transfected APCs translate, process and present antigen epitopes on MHC-I molecules to CD8+ T cells, while transfected non-APCs such as myocytes translate and release antigen for APCs to internalize and present antigen epitopes on MHC-II molecules to helper T cells. Some exogenous antigens are uptaken and presented on MHC-I molecules by the cross-presentation pathway. b,c, Makeup of transfected cells at the injection sites at 24 h post injection with GFP mRNA (mGFP)-loaded C10, D6 and F5 formulations. Flow cytometry was used to determine the ratios of non-immune and immune cells (b) and the relative abundance of each cell type (c). Immune cells, CD45+; epithelial cells, CD326+; endothelial cells, CD31+; muscle cells, desmin+; adipocytes, CD45−CD31−CD36+. d–h, In vitro evaluation of transfection or uptake efficiency by formulations C10, D6 and F5 in C2C12 cells and BMDCs. Flow cytometry was used to determine the ratios of mCherry mRNA-transfected C2C12 cells to transfected BMDCs in in vitro co-culture (d), transfection efficiency of LNPs containing fLuc mRNA in pure C2C12 cell culture (e), transfection efficiency of LNPs containing mCherry mRNA in pure BMDCs (f), transfection efficiency of LNPs containing mGFP in pure C2C12 cells (g) and uptake efficiency of LNPs containing Cy5-labelled mRNA in C2C12 cells (h). i,j, Quantitative Cellomics HCA of endosomal escape by different LNPs using C2C12-Gal8-GFP cells. Total number of Gal8 spots per well (i) or average number of Gal8 spots per cell as an indication of endosomal escape level (j). k, LysoTracker was used to identify the co-localization of fluorescent labelled lysosomes (Lysotracker) and LNPs containing Cy5-labelled mRNA in C2C12 cells in vitro. Data represent the mean ± s.e.m. from a representative experiment (n = 4 biologically independent samples for b–j) of two independent experiments. For boxplots, the box extends from the 25th to the 75th percentiles and the line in the middle of the box is plotted at the median. *P < 0.05, ***P < 0.001, ****P < 0.0001.
To further investigate the differences between the transfection efficiencies in non-immune cells versus immune cells, we co-cultured C2C12 cells (a mouse myoblast cell line) and BMDCs (1:1) and quantified in vitro delivery to both cell types using flow cytometry to detect mCherry expression. Results revealed that the ratio of C2C12 cells to BMDCs among transfected cells reached as high as 33.41 for C10 LNPs, 22.21 for D6 and only 3.94 for F5 (Fig. 6d). When only C2C12 cells were transfected with LNPs containing fLuc mRNA, the average fLuc expression level in C2C12 cells transfected with C10 was 25- and 289-fold higher than those transfected with D6 and F5 LNPs, respectively (Fig. 6e). However, when a pure BMDC culture was transfected with LNPs containing mCherry mRNA, similar transfection efficiencies were observed for C10, D6 and F5 (Fig. 6f). We further examined the transfection efficiency together with cellular uptake using LNPs containing 50% mGFP labelled with Cy5 and 50% unlabelled mGFP. While nearly 100% Cy5+ cells (that is, cellular uptake) were observed for all three groups, only ~27.7% and 8.9% of C2C12 cells were transfected with D6 and F5, respectively, in contrast to 98.1% of cells transfected with C10, probably caused by different endosomal escape capabilities among the three LNP formulations in C2C12 myoblasts (Fig. 6g–h). To assess the endosomal escape activity of the 3 LNP formulations, we generated a genetically modified C2C12 cell line expressing galectin-8 (Gal8) fused with GFP. As demonstrated previously64,65, Gal8 protein, distributed throughout the cytosol, binds to cell membrane glycans exposed upon endosomal vesicle damage, leading to the aggregation and formation of GFP spots. The average number of GFP-Gal8 spots per cell measured using Cellomics high-content analysis (HCA) in fixed cells after a 4-h treatment with C10 LNPs (Fig. 6i,j) was 2.2-fold and 2.9-fold higher than with D6 and F5 LNPs, respectively, indicating an enhanced capability for endosomal escape and cytosolic delivery. To verify these findings, we used LysoTracker Green DND-26 to visualize lysosomes in C2C12 cells at 4 h after LNP transfection. We observed a lack of fluorescence co-localization between C10 LNPs (red) and lysosomes (green), confirming more efficient lysosomal escape and cytosolic delivery by C10 than by D6 and F5 (Fig. 6k).
Discussion
The use of LNPs as non-viral gene carriers has advanced rapidly over the past few years, as evidenced by the approval of multiple LNP-based COVID-19 vaccines and one small interfering RNA therapy11,66. Safety following repeated LNP dosing provides strong momentum for extending the utility of LNPs to therapeutic vaccines and other gene delivery applications1,4,30. Previous reports on LNP-mediated gene delivery revealed that both the choice of lipids and their molar ratios can drastically influence the encapsulation efficiency of nucleic acid payload, transfection efficiency and cell/tissue-targeting profiles45,47,52. In this study, we screened LNP compositions to interrogate the role of the carriers themselves in polarizing therapeutic immune responses. We identified 3 top-performing LNPs from a 1,080-candidate library on the basis of transfection efficiency in BMDCs. These 3 LNPs induced comparably potent antigen-specific Th1 responses following three doses by s.c. injection but substantially different Th2 responses. All 3 formulations showed substantial levels of efficacy in tumour suppression and markedly prolonged survival in a prophylactic model of OVA-expressing melanoma in C57BL/6 mice. The best candidate, C10 LNPs, however, showed the strongest potency in slowing tumour growth and extending survival when tested in therapeutic melanoma models using mRNA encoding OVA, Trp2 or Gp100 antigens.
Previous reports on cancer-vaccine delivery systems demonstrated that a potent Th1 immune response is essential to antitumour efficacy67. Using LNP-delivered mRNA vaccine optimized in this study, we revealed that incorporating a strong Th2 response with a strong Th1 response further enhanced the tumour-suppression efficiency by comparing the two representative LNP formulations that induced Th1-only or Th1-plus-Th2 immune responses. The results confirmed that coordinated immune responses by various cell populations, including T cells, NK cells and B cells, generated by C10-mRNA LNPs provided more effective and comprehensive protection against tumour challenge, and that multiple effector cell populations involved in both Th1 and Th2 responses collectively contributed to long-term protection. T cell-mediated immunity greatly inhibits the growth of tumours, but many tumours effectively evade the immune system and progress under the immune pressure via multiple mechanisms, including loss of MHC Class I expression and development of an immunosuppressive tumour microenvironment. Our data suggested that with effective induction of Th2 immune responses, long-term antitumour protection could be achieved by making use of the innate immune cells, such as NK cells, providing antitumour cytotoxicity activated by antibodies linked to target cells. Our data showed that a coordinated action of NK cells and B cells played a critical role in terms of the long-term protection against tumour.
Previous reports on LNP-enabled gene delivery systems argue that LNP composition influences tissue-targeting and transfection46,48,49,54. We revealed that by tuning the composition of LNP formulations, we were able to alter the transgene level delivered by LNP-mediated mRNA vaccines in different cell types in vivo. Further, the biases in cell type-specific gene expression were shown to alter the immune activation profile of these formulations. LNPs with strong transfection efficiencies in APCs can generate potent cytotoxic T cell and Th1 responses. By varying the composition, LNPs may also show strong transfection efficiency in non-APC cell types such as myoblasts, thereby aiding Th2 responses. These findings further highlight the need for a rational screening approach when evaluating LNP formulations for antigen-specific therapeutic vaccines and other genetic medicine applications. This study showed that it is feasible to modulate Th1- vs Th2-biased responses by varying the composition of LNPs to alter their preferential transfection properties in APCs and non-APC cell types. This synthetic tuning approach creates opportunities for devising new mRNA LNP-based immunotherapy strategies that can be intricately tailored to specific disease targets, tissues and cell types.
The three selected LNP candidates generated different levels of Th2 responses that correlate with different transfection abilities in non-APC cells such as myoblasts. Among them, C10 LNPs with a zwitterionic helper lipid DOPE showed potent Th1-plus-Th2 responses correlated with their higher transfection activities in both DCs and myoblasts, whereas F5 LNPs with an anionic helper lipid showed strong transfection activity only in DCs with consequently Th1-skewed responses. These results showed that altering the composition of LNP formulations allowed for preferential transfection activity across various cell types, as illustrated here by myoblast cells vs DCs. Different endosomal escape capabilities of different LNP formulations were probably the determining factor for the difference in transfection efficiency. This cell type-preferential transfection strategy might be a potential tool for modulating the balance of antigen-specific immune stimulation between Th1 and Th2 responses.
In this study, we completed the LNP sample preparation using small-volume pipette mixing for the LNP library generation and screening experiments. Larger-scale LNP production for selected formulations used for in vivo studies was achieved using our previously established flash nanocomplexation (FNC) method68–70. We compared the quality metrics of the 3 top LNP formulations (C10, D6 and F5) prepared by these two methods by assessing their Z-average size, size distribution via polydispersity index, average zeta potential and encapsulation efficiency. Data shown in Supplementary Figs. 23–25 confirmed that the qualities of the LNPs prepared by these two methods were comparable; only a minor difference was observed in encapsulation efficiency for D6 formulations.
Overall, we have reported a composition-screening approach that allowed us to identify the best-performing mRNA LNPs for APC-specific transgene expression that showed a strong Th1 immune response against tumour antigens in a melanoma mouse model. Among the top LNP candidates, C10 showed both potent Th1 and Th2 responses that further enhanced therapeutic efficacy compared with a Th1-skewed response against melanoma antigens. The data indicate that coordinated T-cell, NK-cell and B-cell responses were responsible for enhanced antitumour efficacy. In addition, tuning the composition of LNP formulations altered the transgene level delivered by LNP-mediated mRNA vaccines in different cell types in vivo. This study thus shows a potential strategy to tailor antigen-specific immune-activation profiles generated by tuning LNP composition. It may provide a versatile vaccine-development approach that can be applied to a variety of diseases and leveraged to expand the utility of mRNA LNP-based immunotherapies.
Methods
Materials
DLin-MC3-DMA was purchased from MedKoo Biosciences. DSPC, DOPE, DOTAP, DDAB, 18PG, 14PA and DMG-PEG-2000 were obtained from Avanti Polar Lipids. Cholesterol was from Sigma-Aldrich. B16F10 cells (CRL-6475) were purchased from American Type Culture Collection (ATCC). DC2.4 cells and B16-OVA (expressing model antigen, OVA, with a transmembrane domain) were kindly provided by the lab of Prof. Jonathan Schneck. Reporter lysis buffer and luciferin assay solution were purchased from Promega. All mRNA was purchased from TriLink BioTechnologies and capped using the TriLink CleanCap proprietary co-transcriptional capping method. This method results in the naturally occurring Cap 1 structure with high capping efficiency. The mRNAs are polyadenylated, modified with 5-methoxyuridine and optimized for mammalian systems. They were purified by standard purification consisting of two silica-column steps. The purity was confirmed via agarose gel analysis. d-luciferin was purchased from Gold Biotechnology and Alhydrogel was purchased from InvivoGen.
Cell culture and high-throughput screening for transfection studies
For monolayer culture studies, DC2.4 cells were seeded into 96-well plates at a cell density of 10,000 cells per well 1 d before transfection. LNPs were pipetted into RPMI medium at a final concentration of 1 μg ml−1 of mRNA. For example, 8 μl of an LNP suspension at 25 μg ml−1 of mRNA was pipetted into the 200 μl culture medium in each well. The transgene expression was analysed following 24 h incubation. When characterizing luciferase as the reporter, cells were lysed by reporter lysis buffer (Promega) using two freeze–thaw cycles, with the lysate characterized by a luminometer upon addition of luciferin assay solution (Promega) against a standard curve generated using luciferase samples (Promega).
LNP synthesis and characterization
LNPs were synthesized by directly adding an organic phase containing the lipids to an aqueous phase containing the mRNAs in a 96-well plate or 1.5 ml microcentrifuge tubes for high-throughput screening. To prepare the organic phase, a mixture of DLin-MC3 DMA, cholesterol (Sigma-Aldrich), DMG-PEG2000 (Avanti) and a helper lipid selected from a group consisting of DOTAP, DDAB, DOPE, DSPC, 14PA and 18PG (Avanti) was dissolved in ethanol. For SM-102 LNP preparation, a mixture of SM-102, DSPC, cholesterol and PEG-DMG at a molar ratio of 50:10:38.5:1.5 was prepared. To prepare the aqueous phase, corresponding mRNA (fLuc mRNA, GFP mRNA, mCherry mRNA, Cre mRNA, OVA mRNA, Trp2 mRNA or Gp100 mRNA) was prepared in 25 mM magnesium acetate buffer (pH 4.0, Fisher). All mRNA samples were stored at −80 °C and thawed on ice before use. For in vitro screening, LNPs were incubated with cells without dialysis. For larger-scale LNP production, the aqueous and ethanol phases prepared were mixed at a 3:1 ratio in a flash nanocomplexation (FNC) device using syringe pumps48, purified by dialysis against deionized water using a 100-kDa molecular weight cut-off (MWCO) cassette (Thermo Fisher) at 4 °C for 24 h and stored at 4 °C before injection. The size, polydispersity index and zeta potentials of LNPs were measured using dynamic light scattering (ZetaPALS, Brookhaven Instruments). Diameters are reported as the intensity mean average.
Animals and primary cells
All animal procedures were performed under an animal protocol approved by the Johns Hopkins Institutional Animal Care and Use Committee (protocol no. MO21E193). Male and female C57BL/6 mice (6–8 weeks old) were purchased from the Jackson Laboratory. Male Ai9 mice (6–8 weeks old) were bred in Johns Hopkins Animal Facilities and randomly grouped. The mice were supplied with free access to pelleted feed and water. The pelleted feed generally contained 5% fibre, 20% protein and 5–10% fat. The mice usually ate 4–5 g of pelleted feed (120 g kg−1 body weight) and drank 3–5 ml of water (150 ml kg−1 body weight) per day. The temperature of the mouse rooms was maintained at 18–26 °C (64–79 °F) at 30–70% relative humidity, with a minimum of 10 room air changes per hour. Standard shoebox cages with corncob as bedding were used to house the mice.
The LNPs were given through s.c. (right flank) or i.m. (right quadriceps) injection at a predetermined dose per mouse. The LNP suspensions were concentrated to 200 μg ml−1 for s.c. injection or 400 μg ml−1 for i.m. injection of mRNA by an Amicon Ultra-2 centrifugal filter unit with an MWCO of 100 kDa. For experiments in Ai9 mice, the Cre mRNA LNP formulations were prepared as described above and administered via s.c. or i.m. injections at an mRNA dose of 10 μg per mouse. After 7 d, mice were killed and the draining lymph nodes were collected for flow cytometry analysis.
Antibodies, cell isolation and staining for flow cytometry
Antibodies used in this study are: PE-Cyanine 7 anti-mouse CD40 (BioLegend, 124622); PerCP-Cyanine 5.5 anti-mouse CD80 (BioLegend,104722); FITC, APC, Brilliant Violet 750 anti-mouse CD11c (BioLegend, 117306, 117310, 117357); Brilliant Violet 421 anti-mouse CD86 (BioLegend, 105032); PE anti-mouse SIINFEKL-H-2KB (ThermoFisher, 12574382); FITC, Brilliant Violet 605, Brilliant Violet 421 anti-mouse CD45 (BioLegend, 103108, 103140, 103134); APC, Brilliant Violet 421 anti-mouse CD3 (BioLegend, 100236, 100228); FITC, APC, Brilliant Violet 750 anti-mouse CD8 (BioLegend, 100706, 100712; BD Biosciences, 747502); PerCP-Cyanine 5.5, PE-Cy7 anti-mouse CD4 (BioLegend, 100540, 100422); PE-Cy7 anti-mouse Granzyme B (BioLegend, 372214); Brilliant Violet 650, APC-Cy7 anti-mouse IFN-γ (BioLegend, 505832, 505850); Brilliant Violet 421, PerCP-eFluor 710 anti-mouse IL-4 (Thermo Fisher, 46-7041-82); APC anti-mouse IL-17A (BioLegend, 506916); PE-Cyanine 7 anti-mouse TNF-α (BioLegend, 506324); Brilliant Violet 711 anti-mouse T-bet (Biolegend, 644820); PE anti-mouse ROR γt (BD Biosciences, 562607); Alexa Fluor 488 anti-mouse GATA-3 (Biolegend, 653808); and Alexa Fluor 488 anti-mouse TNF-α (BioLegend, 506338). All antibodies were diluted at a ratio of 1:100 before use. LIVE/DEAD fixable aqua dead cell stain kit was used to determine the viability of cells. eBioscience Foxp3/Transcription Factor Staining buffer set (Thermo Fisher, 00-5523-00) was used for intracellular staining.
For isolation, restimulation and staining of spleen cells, the spleen was removed and minced using a sterile blade and homogenized in 250 μl of digestion medium (45 U μl−1 collagenase I, 25 U μl−1 DNase I and 30 U μl−1 hyaluronidase). The suspension was transferred into a 15 ml tube containing 5–10 ml of digestion medium and then filtered through a 70 μm filter and washed once with PBS. Cells were pelleted at 300 × g for 5 min at 4 °C, resuspended in 5 ml of red blood cell lysis buffer (BioLegend) and then incubated on ice for 5 min. Cells were then pelleted at 300 × g for 5 min at 4 °C and washed two times with PBS. The collected cells were then seeded into 12-well plates using RPMI-1640 media. Splenocytes were restimulated in vitro with OVA (InvivoGen, vac-pova) and SIINFEKL peptide (InvivoGen, vac-sin) (10 μg ml−1 OVA and 2 μg ml−1 SIINFEKL) for 12 h. After restimulation, cells were collected and centrifuged at 300 × g for 5 min. Cell pellet was washed with staining buffer three times and stained with antibodies against surface markers (total volume 100 μl) for 30 min in the dark at 4 °C. The stained cells were washed twice with 1 ml PBS, and then fixed and permeabilized using the eBioscience Foxp3/Transcription Factor Staining buffer set (Thermo Fisher, 00–5523-00). Then, cells were stained with anti-IFN-γ or other antibodies against intracellular cytokines. Flow data were acquired on a Sony SH800S or Attune NXT flow cytometer and analysed using FlowJo software.
For isolation and staining of lymph node cells, isolated lymph nodes were mechanically digested through 70 μm nylon cell strainers to prepare single-cell suspensions. The cell suspension was washed once with PBS via centrifugation (300 × g) for 5 min. Then, the cells were resuspended in 100 μl of staining buffer and stained with antibodies (total volume 100 μl) for 20 min in the dark at 4 °C. The stained cells were washed twice with 1 ml PBS and resuspended in 300 μl of staining buffer for flow cytometry analysis. Flow data were acquired on a Sony SH800S and Attune NXT flow cytometer and analysed using FlowJo software.
Enzyme-linked immunosorbent spot (ELISpot) assay
Multiscreen filter plates (Millipore-Sigma, S2EM004M99) were coated with antibodies specific for IFN-γ (BD Biosciences, 551881) and blocked following manufacturer protocols. Then, 1 × 105 isolated splenocytes were plated per well and stimulated with SIINFEKL peptide (2 μg ml−1 SIINFEKL) for 24 h. All tests were performed in duplicate or triplicate and included assay positive controls as well as cells from a reference donor with known reactivity. Spots were visualized with mouse IFN-γ detection antibody (BD Biosciences, 551881), followed by incubation with streptavidin-HRP (BD Biosciences, 557630) and AEC substrate (BD Biosciences, 551951). Plates were then sent to the SKCCC Immune Monitoring Core for analysis.
Enzyme-linked immunosorbent assay (ELISA)
For antibody detection, groups of C57BL/6 mice were immunized with different vaccines on days 0, 7 and 14. On day 21, 100 μl of blood sample was drawn from the tail vein and levels of antigen-specific IgG in the serum were measured by ELISA. For ELISA, flat-bottomed 96-well plates (Nunc) were precoated with OVA protein at a concentration of 2 μg protein per well in 100 mM carbonate buffer (pH 9.6) at 4 °C overnight, which were then blocked with 10% fetal bovine serum (FBS) in PBS-Tween (PBS-T). Serum obtained from immunized animals were diluted 100 times in PBS-T (PBS-0.05% Tween) (pH 7.4) and then in fourfold serial dilution. The undiluted and diluted serum were added to the wells and incubated at 37 °C for 2 h. Horseradish peroxidase-conjugated goat anti-mouse IgG (Southern Biotech Associates, 1013–05) was used at a dilution of 1:5,000 in PBS-T-10% FBS for labelling. After adding the horseradish peroxidase substrates, optical densities were determined at a wavelength of 450 nm in an ELISA plate reader (Bio-Rad). A sample was considered positive if its absorbance was twice as much as or higher than the absorbance of the negative control.
For cytokine detection, cell supernatants of BMDCs and splenocytes were obtained, and levels of IFN-γ, TNF-α and IL-6 were measured by ELISA. Supernatants were diluted at 1:5. ELISAs were performed using uncoated ELISA kits (Invitrogen) following manufacturer protocols. Optical densities were determined at a wavelength of 450 nm in an ELISA plate reader (Bio-Rad).
BMDC isolation, activation and antigen presentation assay
A mouse was killed and transferred to a clean bench. The mouse was disinfected with 70% ethanol. The skin and muscle on the legs were carefully removed to separate the femur and tibia. The proximal and distal ends of each bone were cut with a pair of scissors. The bones were flushed with full medium (RPMI-1640, supplemented with 10% FBS and 1% penicillin/streptomycin). Two to three ml of medium was flushed from each side for each bone. The cell-containing medium was filtered through a 70 μm cell strainer and the filtrate was collected. The cell suspension was centrifuged at 200 × g for 10 min at room temperature and the supernatant was discarded. The cells were resuspended in 10 ml full medium and the cell concentration was determined. The cell suspension was diluted to a concentration of 3 × 106 cells per ml. The cells were plated in ultra-low-attachment-surface Petri dishes at 10 ml per dish (100 mm × 15 mm). Two ml of 40 ng ml−1 granulocyte-macrophage colony-stimulating factor (GM-CSF) was added in full medium to each well to a final GM-CSF concentration of 20 ng ml−1. The cells were cultured at 37 °C and 5% carbon dioxide. Half of the GM-CSF-containing medium was replaced every 2 d. On day 6, non-adherent and loosely adherent immature dendritic cells were collected. The cell suspension was centrifuged at 200 g for 10 min at room temperature and then the supernatant was discarded. The cells were plated at 5 × 105 cells per well in a 24-well plate.
BMDCs were incubated with 1 μg ml−1 OVA mRNA in various LNPs formulations or with PBS, free OVA (InvivoGen, vac-pova), LPS (Sigma-Aldrich, L6529) or SIINFEKL peptide (InvivoGen, vac-sin) in complete medium for 24 h at 37 °C with 5% CO2; LPS + SIINFEKL peptide was used as a dendritic cell activation positive control. After co-incubation, BMDCs were collected, washed with FACS buffer (1% BSA, 10% FBS in PBS) and then stained on ice with fluorophore-labelled antibodies against CD45, CD11c, CD40, CD80, CD86 and SIINFEKL/H-2Kb monoclonal antibody.
Immunization and tumour therapy experiments
Mice aged 6–8 weeks were injected subcutaneously with B16-OVA cells (1 × 106 in prophylactic and depletion studies and 3 × 105 in therapeutic studies) or 3 × 105 B16F10 melanoma cells into the right flank. In therapeutic studies, vaccinations began when tumour sizes were less than 50 mm3 (on day 4 after tumour inoculation). Animals were immunized by subcutaneous injection of different LNP formulations containing 10 μg OVA mRNA, mTrp2 or mGp100 as described in the main text. A total of three doses were given. For combinatorial immunotherapy, some groups were intraperitoneally injected with 100 μg checkpoint inhibitor (anti-CTLA-4 mAb) at days 6, 13, 20 and 27 dose for OVA-expressing melanoma after inoculation. Tumour growth was measured three times a week using a digital caliper and calculated as 0.5 × length × width × width. Mice were euthanized when the tumour volumes reached 2,000 mm3.
Quantitative endosomal escape assessments by Cellomics
The C2C12 cell line expressing GFP-coupled galectin-8 (GFP-Gal8) was generated through transfection using plasmids encoding Super PiggyBac Transposase from System Biosciences and Piggybac-transposon-GFP-Gal8 from Addgene (plasmid 127191) according to our previously reported protocol64,65. The transfection process involved the use of the poly(ethylenimine) carrier (Polyplus). The transfected cells were subsequently sorted three times using an SH800 cell sorter (Sony) and cultured in DMEM supplemented with 10% FBS at a density of 100,000 cells per well. The engineered cells were treated with LNPs as described above for 4 h, washed three times with PBS and fixed using a 4% paraformaldehyde solution. The cells were then stained with Hoechst 33342 and washed three times with PBS. For analysis, the plates were imaged using CellInsight CX7 high-content analysis (HCA) technology from Thermo Fisher at ×20 magnification, resulting in a resolution of 1,104 × 1,104 pixels per field, corresponding to an area of 501.2 × 501.2 μm2. A total of 30 fields were analysed within each well and the results were averaged across all cells and fields within a well. The manufacturer-supplied programme was used for analysis, employing laser/filter sets of Channel 1: 386/440 nm and Channel 2: 485/521 nm, with fixed exposure times. During the analysis, cell nuclei and GFP-Gal8 spots were identified using appropriate smoothing and thresholding settings, which were visually confirmed to ensure accurate recognition in the sample images.
Characterization of the encapsulation efficiency of mRNA LNP formulations
The Quant-it RiboGreen assay (ThermoFisher, R11490) was employed to assess the encapsulation efficiency of the LNP formulations. To begin, LNPs were first treated with 0.5% w/v Triton X-100 (Sigma-Aldrich, T8787) to disrupt the LNP structure and release the mRNA. Both the treated LNPs and the untreated LNPs were then diluted to a concentration below 1 μg ml−1 mRNA. Subsequently, they were mixed with an equal volume of 0.5% v/v RiboGreen assay solution, which was previously diluted 200-fold. A standard curve was constructed using a series of free mRNA solutions ranging from 0.1 to 1.0 μg ml−1 mRNA, with or without 0.5% w/v Triton X-100, ranging from 0.1 to 1.0 μg ml−1 mRNA. The concentrations of free unencapsulated mRNA in LNP samples and the total released mRNA in the formulation were determined by measuring the fluorescence signal (excitation: 480 nm, emission: 520 nm) of the samples against their corresponding standard curve and used to calculate the encapsulation efficiency (EE) for each LNP formulation according to the following equation. This bulk fluorescent reading provided quantification of the mRNA concentrations in the LNP formulations.
Depletion studies
Depletions of immune cells were done using antibodies against NK1.1 (clone PK136, BioXCell), CD3 (clone 145–2C11, BioXCell) and CD20 (clone MB20–11, BioXCell) at 200 μg i.p. every 4 d. All depletion antibodies dosing was initiated at 3 d before tumour inoculation and continued every 4 d.
Statistical analysis
A two-tailed Student’s t-test or a one-way analysis of variance (ANOVA) was performed when comparing two groups or more than two groups, respectively. Survival curves were compared using log-rank Mantel–Cox test and P values were corrected using the Holm-Šídák method for multiple comparisons with α set at 0.05. Statistical analysis was performed using Microsoft Excel and Prism 8.0 (Graph-Pad). A difference was considered significant if P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Extended Data
Extended Data Fig. 1 |. Secretion levels of cytokines within the supernatant of BMDCs after 24 h of incubation.
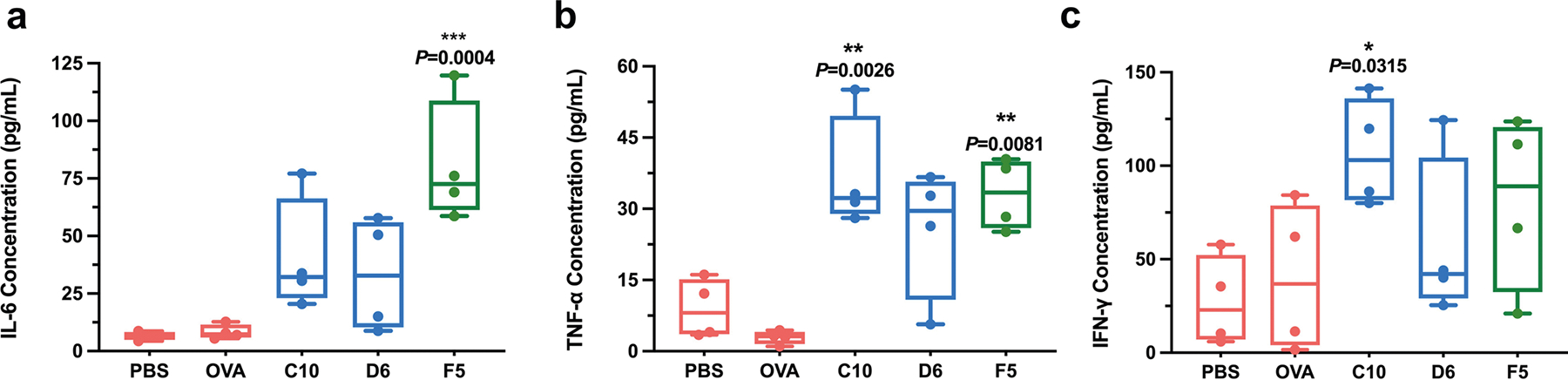
Secretion levels of IL-6 (a), TNF-α (b) and IFN-γ (c), within the supernatant of BMDCs after 24 h incubation with the three mOVA-loaded LNPs were measured by ELISA. Data are represented as the mean ± s.e.m. Data were analyzed using one-way ANOVA and Dunnett’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001; NS, not significant; BMDC, bone marrow derived dendritic cell; ELISA, enzyme-linked immunoassay.
Extended Data Fig. 2 |. IL-6 and TNF-α secretion levels from antigenstimulated splenocytes.
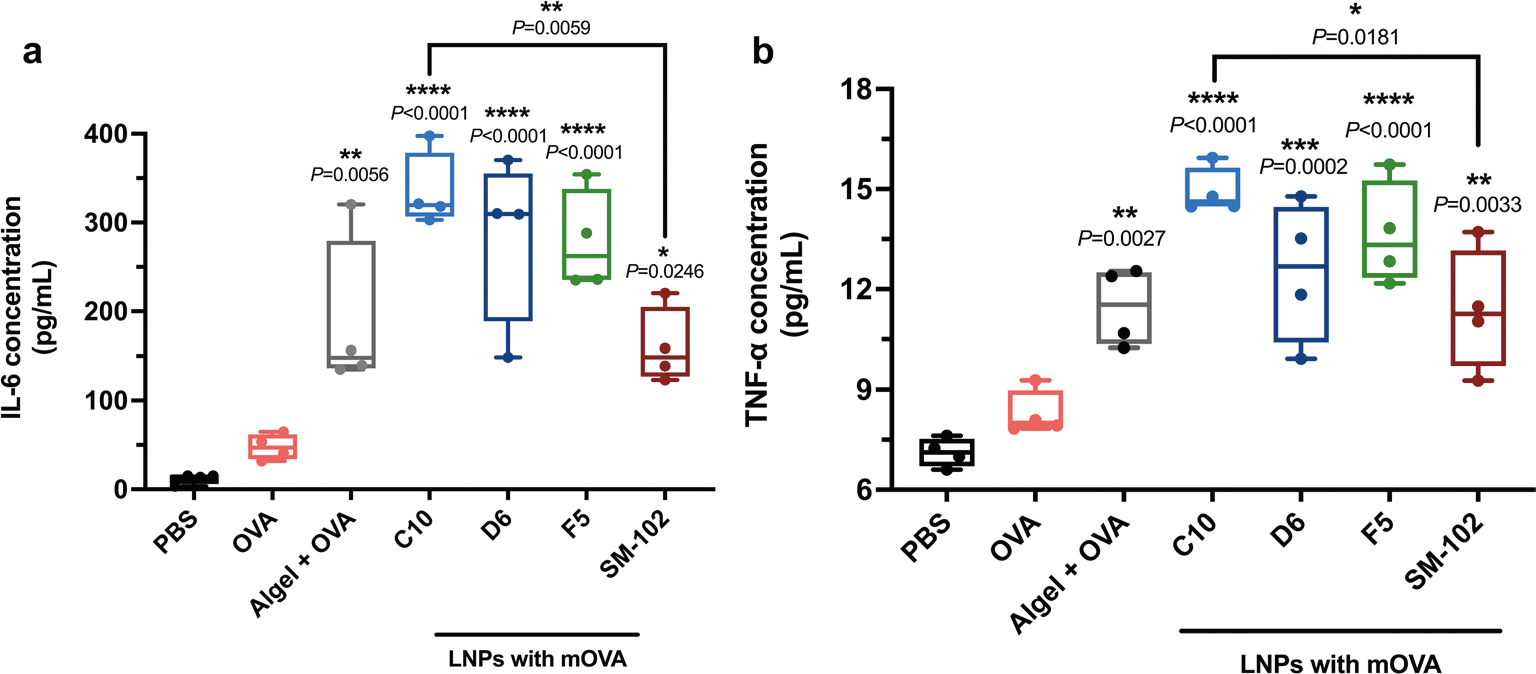
Splenocytes were isolated from vaccinated mice and restimulated in vitro with OVA and SIINFEKL peptide (100 μg ml−1 OVA and 2 μg ml−1 SIINFEKL) for 72 h. Secretion levels of IL-6 (a) and TNF-α (b) within the supernatant of were measured by ELISA. ‘Algel+OVA’ stands for Alhydrogel®+OVA group. Data represent the mean ± s.e.m. from a representative experiment (n = 4 biologically independent samples) of two independent experiments. Data were analyzed using one-way ANOVA and Dunnett’s multiple comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, not significant; BMDC, bone marrow derived dendritic cell; ELISA, enzyme-linked immunoassay.
Extended Data Fig. 3 |. Titres of OVA-specific IgG subclass antibody in blood-serum samples collected on day 21 following immunization.
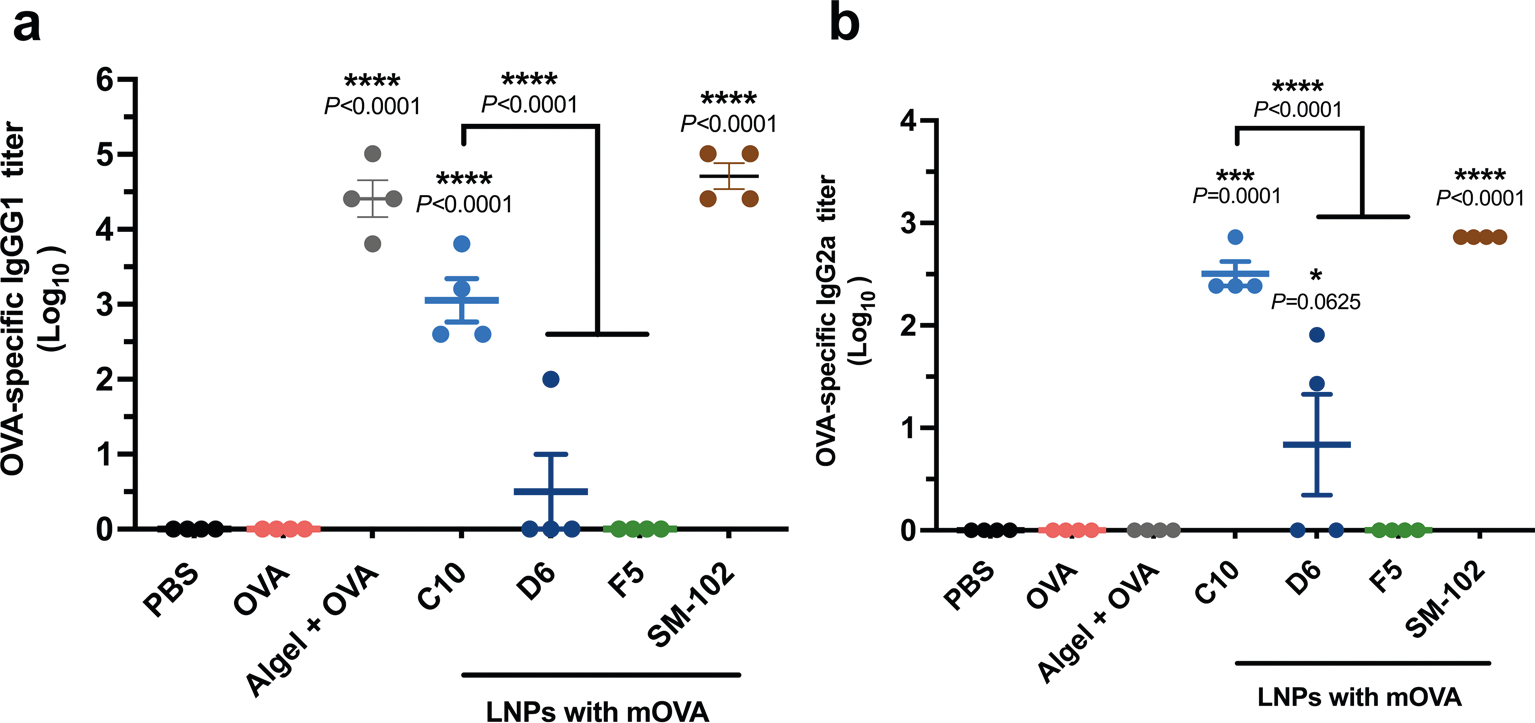
IgG1 (a) and IgG2a (b) antibodies in blood serum on day 21 were determined by ELISA. ‘Algel+OVA’ stands for Alhydrogel®+OVA group. Data represent the mean ± s.e.m. from a representative experiment (n = 4 biologically independent samples) of two independent experiments. Data were analyzed using one-way ANOVA and Dunnett’s multiple comparisons test. ****P < 0.0001; ELISA, enzyme-linked immunoassay.
Extended Data Fig. 4 |. Cytokine levels in the local injection site measured at 4 h or 24 h post-administration of the top-performing LNPs.
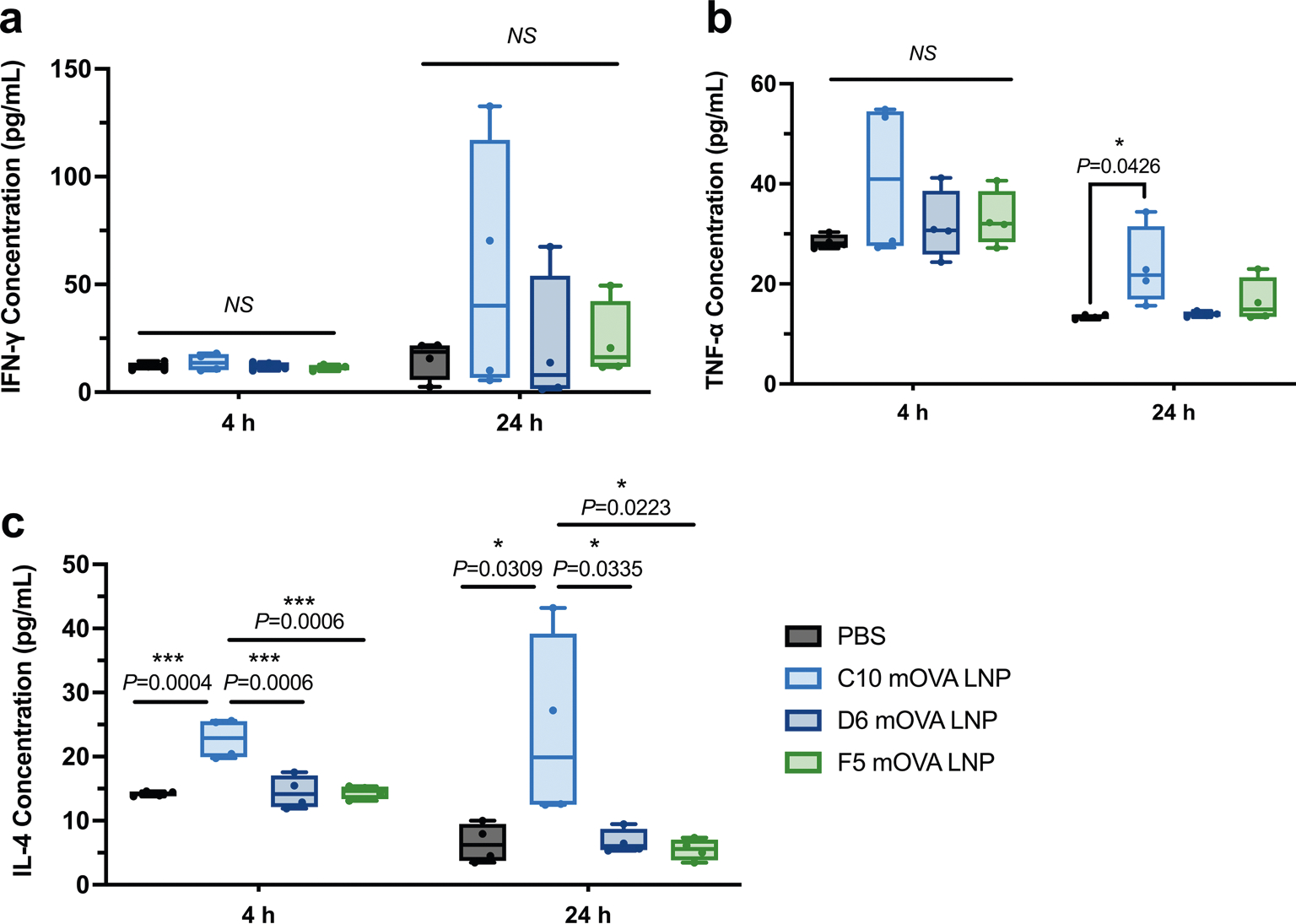
ELISA was employed to quantify the cytokine levels, including IFN-γ (a), TNF-α (b), and IL-4 (c), at the local injection site using OVA-encoding mRNA after the administration of three formulations (C10, D6, and F5) at 4 h and 24 h. The local injection sites were collected, homogenized, and subjected to tissue lysis to extract the proteins. The resulting lysate was centrifuged to separate the insoluble cellular debris. The protein concentration in each sample was determined using the BCA assay and normalized accordingly. Data represent the mean ± s.e.m. with n = 4 biologically independent samples. Data were analyzed using one-way ANOVA and Tukey’s multiple comparisons test. *P < 0.05; ***P < 0.001; NS, not significant.
Supplementary Material
Acknowledgements
S.C.M. and H.-Q.M. disclose support for the research described in this study from the National Institutes of Health (U01AI155313). J.P.S., J.J.G. and H.-Q.M. also disclose support for the publication of this study from the National Institutes of Health (P41EB028239).
Footnotes
Competing interests
H.-Q.M., Y.Z., J.M, R.S, L.C., I.V. and S.K.R. are co-inventors of a pending patent application (PCT/US2023/016938, filed in March 2023) covering the LNP formulation described in this paper. The patent was filed through Johns Hopkins Technology Ventures and is managed by it. The other authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41551-023-01131-0.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551023-01131-0.
Peer review information Nature Biomedical Engineering thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are available for research purposes from the corresponding authors on reasonable request. Source data for the figures are provided with this paper.
References
- 1.Huang Q, Zeng J & Yan J COVID-19 mRNA vaccines. J. Genet. Genomics 48, 107–114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang S, Zhu Y, Zhang L & Zhang Z Recent advances in delivery systems for genetic and other novel vaccines. Adv. Mater. 34, e2107946 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Wouters OJ et al. Challenges in ensuring global access to COVID-19 vaccines: production, affordability, allocation, and deployment. Lancet 397, 1023–1034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahin U et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 595, 572–577 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Wang Z et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 592, 616–622 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lederer K et al. SARS-CoV-2 mRNA vaccines foster potent antigen-specific germinal center responses associated with neutralizing antibody generation. Immunity 53, 1281–1295.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alameh MG et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner JS et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature 596, 109–113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou X, Zaks T, Langer R & Dong Y Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahin U, Karikó K & Türeci Ö mRNA-based therapeutics – developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pardi N, Hogan MJ, Porter FW & Weissman D mRNA vaccines – a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowalski PS, Rudra A, Miao L & Anderson DG Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trougakos IP et al. Adverse effects of COVID-19 mRNA vaccines: the spike hypothesis. Trends Mol. Med. 28, 542–554 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laczkó D et al. A single immunization with nucleoside-modified mRNA vaccines elicits strong cellular and humoral immune responses against SARS-CoV-2 in mice. Immunity 53, 724–732.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Painter MM et al. Rapid induction of antigen-specific CD4+ T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 54, 2133–2142.e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lozano-Rodríguez R et al. Cellular and humoral functional responses after BNT162b2 mRNA vaccination differ longitudinally between naive and subjects recovered from COVID-19. Cell Rep. 38, 110235 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reichmuth AM, Oberli MA, Jaklenec A, Langer R & Blankschtein D mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 7, 319–334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassett KJ et al. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release 335, 237–246 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Pilkington EH et al. From influenza to COVID-19: lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater. 131, 16–40 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao L, Zhang Y & Huang L mRNA vaccine for cancer immunotherapy. Mol. Cancer 20, 41 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaradoss A et al. Immune profile and responses of a novel dengue DNA vaccine encoding an EDIII-NS1 consensus design based on Indo-African sequences. Mol. Ther. 30, 2058–2077 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bretscher PA On the mechanism determining the Th1/Th2 phenotype of an immune response, and its pertinence to strategies for the prevention, and treatment, of certain infectious diseases. Scand. J. Immunol. 79, 361–376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bretscher P On analyzing how the Th1/Th2 phenotype of an immune response is determined: classical observations must not be ignored. Front. Immunol. 10, 1234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Prete G The concept of type-1 and type-2 helper T cells and their cytokines in humans. Int. Rev. Immunol. 16, 427–455 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Duarte LF et al. Immune profile and clinical outcome of breakthrough cases after vaccination with an inactivated SARS-CoV-2 vaccine. Front. Immunol. 12, 742914 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyriakidis NC, López-Cortés A, González EV, Grimaldos AB & Prado EO SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. npj Vaccines 6, 28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, Phase 1/2 clinical trial. Lancet Infect. Dis. 21, 181–192 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulligan MJ et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586, 589–593 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Polack FP et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alameh MG, Weissman D & Pardi N Messenger RNA-based vaccines against infectious diseases. Curr. Top. Microbiol. Immunol. 440, 111–145 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Ewer KJ et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a Phase 1/2 clinical trial. Nat. Med. 27, 270–278 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Jeyanathan M et al. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 20, 615–632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richner JM et al. Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindgren G et al. Induction of robust B cell responses after influenza mRNA vaccination is accompanied by circulating hemagglutinin-specific ICOS+ PD-1+ CXCR3+ T follicular helper cells. Front. Immunol. 8, 1539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanBlargan LA et al. An mRNA vaccine protects mice against multiple tick-transmitted flavivirus infections. Cell Rep. 25, 3382–3392.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardi N et al. Characterization of HIV-1 nucleoside-modified mRNA vaccines in rabbits and rhesus macaques. Mol. Ther. Nucleic Acids 15, 36–47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahin U et al. COVID-19 vaccine BNT162b1 elicits human antibody and T(H)1 T cell responses. Nature 586, 594–599 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Miao L et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 37, 1174–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Oberhardt V et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 597, 268–273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oberli MA et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 17, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karmacharya P, Patil BR & Kim JO Recent advancements in lipid-mRNA nanoparticles as a treatment option for cancer immunotherapy. J. Pharm. Investig. 52, 415–426 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guevara ML, Persano F & Persano S Advances in lipid nanoparticles for mRNA-based cancer immunotherapy. Front. Chem. 8, 589959 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cullis PR & Hope MJ Lipid nanoparticle systems for enabling gene therapies. Mol. Ther. 25, 1467–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Sun C, Wang C, Jankovic KE & Dong Y Lipids and lipid derivatives for RNA delivery. Chem. Rev. 121, 12181–12277 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lokugamage MP et al. Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs. Nat. Biomed. Eng. 5, 1059–1068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Miao L, Satterlee A & Huang L Delivery of oligonucleotides with lipid nanoparticles. Adv. Drug Del. Rev. 87, 68–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dilliard SA, Cheng Q & Siegwart DJ On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl Acad. Sci. USA 118, e2109256118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel S, Ryals RC, Weller KK, Pennesi ME & Sahay G Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 303, 91–100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel SK et al. Hydroxycholesterol substitution in ionizable lipid nanoparticles for mRNA delivery to T cells. J. Control. Release 347, 521–532 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Swingle KL et al. Amniotic fluid stabilized lipid nanoparticles for in utero intra-amniotic mRNA delivery. J. Control. Release 341, 616–633 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Y et al. Multi-step screening of DNA/lipid nanoparticles and co-delivery with siRNA to enhance and prolong gene expression. Nat. Commun. 13, 4282 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 15, 8099–8107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng X & Lee RJ The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Del. Rev. 99, 129–137 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Kulkarni JA et al. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine 13, 1377–1387 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Dobrowolski C et al. Nanoparticle single-cell multiomic readouts reveal that cell heterogeneity influences lipid nanoparticle-mediated messenger RNA delivery. Nat. Nanotechnol. 17, 871–879 (2022). [DOI] [PubMed] [Google Scholar]
- 57.Xue L et al. Rational design of bisphosphonate lipid-like materials for mRNA delivery to the bone microenvironment. J. Am. Chem. Soc. 144, 9926–9937 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Zhang H et al. Rational design of anti-inflammatory lipid nanoparticles for mRNA delivery. J. Biomed. Mater. Res. A 110, 1101–1108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bannigan P et al. Machine learning models to accelerate the design of polymeric long-acting injectables. Nat. Commun. 14, 35 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gulley JL et al. Role of antigen spread and distinctive characteristics of immunotherapy in cancer treatment. J. Natl Cancer Inst. 109, djw261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Z et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med. 27, 515–525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo Nigro C et al. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann. Transl. Med. 7, 105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang W, Erbe AK, Hank JA, Morris ZS & Sondel PM NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 6, 368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu Y et al. Size-controlled and shelf-stable DNA particles for production of lentiviral vectors. Nano Lett. 21, 5697–5705 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thurston T et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akinc A et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Badrinath S et al. A vaccine targeting resistant tumours by dual T cell plus NK cell attack. Nature 606, 992–998 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He Z et al. Size-controlled lipid nanoparticle production using turbulent mixing to enhance oral DNA delivery. Acta Biomater. 81, 195–207 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Hu H et al. Flash technology-based self-assembly in nanoformulation: from fabrication to biomedical applications. Mater. Today 42, 99–116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu Y et al. Kinetic control in assembly of plasmid DNA/polycation complex nanoparticles. ACS Nano. 13, 10161–10178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are available for research purposes from the corresponding authors on reasonable request. Source data for the figures are provided with this paper.