


Abstract
Arginine and glutamate rich 1 (ARGLU1) is a poorly understood cellular protein with functions in RNA splicing and transcription. Computational prediction suggests that ARGLU1 contains intrinsically disordered regions and lacks any known structural or functional domains. We used adenovirus Early protein 1A (E1A) to probe for critical regulators of important cellular pathways and identified ARGLU1 as a significant player in transcription and the DNA damage response pathway. Transcriptional effects induced by ARGLU1 occur via enhancement of promoter-proximal RNA polymerase II pausing, likely by inhibiting the interaction between JMJD6 and BRD4. When overexpressed, ARGLU1 increases the growth rate of cancer cells, while its knockdown leads to growth arrest. Significantly, overexpression of ARGLU1 increased cancer cell resistance to genotoxic drugs and promoted DNA damage repair. These results identify new roles for ARGLU1 in cancer cell survival and the DNA damage repair pathway, with potential clinical implications for chemotherapy resistance.
Graphical Abstract
Graphical Abstract.

Introduction
ARGLU1 stands for arginine and glutamate rich 1, a small intrinsically disordered cellular protein (IDP) of 273 amino acids, with AlphaFold prediction suggesting an α-helix at residues 93–255 that has not been experimentally confirmed (Figure 1). ARGLU1 was initially described as a direct binding partner of the Mediator subunit 1, where it was shown to be required for oestrogen-receptor mediated gene transcription and breast cancer cell growth (1). Additionally, ARGLU1 expression was shown to be elevated by another tumour virus protein, the Epstein-Barr virus EBNALP protein, in EBV-associated lymphoma cells (2). More recently, ARGLU1 was further implicated in transcriptional regulation and splicing when it was shown to play a critical role in stress hormone signaling and development (3,4). Interestingly, a recent report demonstrated that ARGLU1 deletion leads to global splicing alterations and neuronal deficiencies (4). Critically, this recent study showed that ARGLU1 deletion leads to prolonged mitosis, p53 activation, and apoptosis. Importantly, others have shown a role for ARGLU1 in regulating cell growth and cancer, firstly when it was shown that ARGLU1 contributes to hypertrophic scar formation by promoting fibroblast growth (5); and secondly when it was shown to inhibit tumour growth in a patient-derived xenograft model of gastric cancer (6). Lastly, a recent study has shown that ARGLU1 is involved in head and neck squamous cell carcinoma as a potential negative regulator of the Notch signaling pathway (7). The Wellcome Sanger Cancer Dependency Map labels ARGLU1 as a pan-cancer core fitness protein with high Target Priority Score for many cancers (Supplemental Figure S1), indicating strong therapeutic potential. Together, these findings paint a picture of ARGLU1 as a multi-functional protein that plays a role in multiple cellular processes, a common feature of IDPs in cells (8).
Figure 1.

ARGLU1 is a disordered protein with similarities to BRD4 ET domain. (A) PrDOS prediction of disorder propensity for human ARGLU1. (B) Sequence alignment of ARGLU1 residues 101–273 and BRD4 residues 600–682, the ET domain. (C) AlphaFold prediction of ARGLU1 structure and comparison to predicted BRD4 ET domain structure and PDB-deposited BRD4 ET domain structure, colour indicating confidence strength of predicted structures as indicated in the inset colour legend. The α-helix in ARGLU1 is predicted to be from residues 93–255.
ARGLU1 was previously shown to bind to JMJD6, an arginine demethylase and lysine hydroxylase, during studies that identified anti-pause enhancers that drive promoter-proximal pause-release of RNA polymerase II during transition of the polymerase from initiating to elongating (9). This study demonstrated that pause-release is enhanced by recruitment of JMJD6 via BRD4, a bromodomain-containing protein, and anti-pause enhancers. Unfortunately, the role of ARGLU1 in this process was not explored. In addition to regulation by anti-pause enhancers, general pausing of RNA polymerase II is an important regulatory mechanism that adds another layer of transcriptional regulation (10). Significantly, DNA damage will induce RNA polymerase II stalling and inhibit transcription until the damage is mended via transcription-coupled repair mechanisms (11). At this stage, the role of ARGLU1 in these processes was not explored nor previously reported.
Human adenovirus (HAdV) has been a useful tool for the identification of key players and cellular mechanisms regulating a multitude of cellular pathways involved in cell growth, differentiation, immune response, transcription and many others (12,13). We have effectively used HAdV E1A as a fine molecular probe to identify key players of cellular regulatory pathways, many of which play important roles in cancer, including FUBP1 (14), Nek9 (15), DREF (16), Ku70 (17) and RuvBL1 (18). As the first protein that is expressed during HAdV infection (19), E1A is tasked with reprogramming the host cell to enable viral replication (12). Beyond the well-studied species C viruses, there are many other adenovirus species that are poorly understood with some of them being considerably more oncogenic (20). We were particularly interested in E1A-binding partners of type B7 HAdV, which is well known to be much more pathogenic and belongs to the more oncogenic species B adenoviruses (21). Mass spectrometry was initially used to identify ARGLU1 as a binding partner of E1A from HAdV-B7.
The present study investigated the role of ARGLU1 in RNA polymerase II promoter-proximal pausing and the DNA damage response pathway, while also looking at the influence of ARGLU1 on HAdV replication. We demonstrate that ARGLU1 binds to E1A from species B and C HAdVs, and that knockdown of ARGLU1 increased virus growth and viral gene expression. More importantly, we have discovered that ARGLU1 affects the interaction between BRD4 and JMJD6 via binding of ARGLU1 to JMJD6 and displacement of BRD4. We further advance our understanding of ARGLU1 function by demonstrating that ARGLU1 promotes cancer cell survival when cells are exposed to two genotoxic drugs and promotes DNA damage repair, possibly via enhancement of promoter-proximal pausing of RNA polymerase II. Together, our study implicates ARGLU1 in cancer cell chemoresistance and shows that it does so via promotion of DNA damage repair, with critical implications for treatment.
Materials and methods
Antibodies
Rat monoclonal anti-hemagglutinin (anti-HA) clone 3F10 was obtained from MilliporeSigma (cat. 11867423001); antibodies for RNA polymerase II (CTD, CTD-S2p, CTD-S5p), BRD4, and HEXIM1 were obtained from Abcam (cats. ab26721, ab238146, ab5408, ab128874, ab240647 respectively); anti-mCherry and anti-GFP antibodies came from Takara Bio (cats. 632543 and 632592, respectively); anti-JMJD6 antibody used for immunoprecipitation and chromatin immunoprecipitation (ChIP) of endogenous JMJD6 was obtained from Proteintech (cat. 16476-1-AP-150UL) and for western blot (clone D3O3N) from Cell Signaling Technologies (cat. 60602S). Secondary antibodies were purchased from Jackson ImmunoResearch. Mouse monoclonal M73 antibody was grown in house and was previously described (22). Antibody for human ARGLU1 was newly generated by Pacific Immunology using the following peptide sequence: Cys-KEEQKIILGKGKSRPKLSFSLKTQD. The antibody was affinity purified using peptide columns before use.
Cloning of ARGLU1 cDNA
Total RNA from HT1080 (HT) cells was used as template in one-step RT-PCR reaction using OneTaq One-Step RT-PCR Kit from NEB (cat. E5215S). ARGLU1 was cloned in-frame with an N-terminal HA-tag into pCAN-HA plasmid using EcoRI and XhoI. Plasmid was sequenced and verified to express. Primer sequences used are listed in the primers table below.
Plasmids
Cloning of pCAN-HA-ARGLU1 is described above. Plasmid for the expression of FLAG-tagged JMJD6, MSCV-CMV-CMV-Flag-HA-JMJD6, was obtained from Addgene (cat. 31358) and was previously described (23), it expresses a FLAG and HA-tagged JMJD6.
Transfections
Media was changed on cells 20 min prior to transfection. Transfections were prepared by mixing 1 ml of serum-free DMEM, 10 μg of total plasmid DNA, and 20 μl of linear 1 mg/ml solution of polyethyleneimine 25kDa reagent from Polysciences (cat. 23966-2). The complexes were then added to the cells and incubated for 24–48 h. Note that reagent quantities assume application to a 10 cm plate of cells (fill volume 10 ml). For other plate/well sizes, reagent quantities were altered proportionally to the fill volume.
Cell and virus culture
HT1080 (ATCC# CCL-121) cells were grown in Dulbecco's modified Eagle's medium (MilliporeSigma) supplemented with 5% fetal bovine serum, streptomycin and penicillin (Corning). Unless otherwise specified, all virus infections were carried out at a multiplicity of infection (MOI) of 10 in serum-free medium for 1 hour after which fresh complete media was added without removal of the infection media. ARGLU1-overexpressing HT1080-ARGLU1 (HT-A) cells were generated by transfecting linearized pCAN-HA-ARGLU1 into HT-1080 cells, allowing the cells to recover for 24 h and then selecting with 800 μg/ml of Geneticin (Invitrogen) until no further cell death was observed, approximately 3 weeks. The stable pool was tested for HA-ARGLU1 expression by western blot and immunofluorescence, and used as a stable pool for all future experiments. HT1080 cells were chosen for this as they were able to overexpress ARGLU1. We have also tested overexpression in A549 cells, but stable pools generated in these cells did not express HA-tagged ARGLU1.
Viruses
Viruses used in the study were Ad.CMV, Ad.ARGLU1, and Ad.ISceI and these were generated in-house by homologous recombination as previously described (24). HAdV dl309 was previously described (25), dl520 and pm975 were also previously described (26,27). HAdV-B7 Gomen strain (ATCC# VR-7) was obtained from ATCC.
Co-immunoprecipitation
Ten-centimeter plates were transfected with the indicated plasmid, then harvested 24 h later, and lysed in 1 ml of NP-40 lysis buffer (0.5% NP-40, 50 mM Tris [pH 7.8], 150 mM NaCl) supplemented with a protease inhibitor cocktail (MilliporeSigma). Cell nuclei were pelleted, protein concentration was determined via the Bradford assay, and 1 mg of total cell lysate was used for immunoprecipitation and a fraction was saved for the input western blot. Immunoprecipitation tubes were then nutated with the indicated antibody and 12.5 μL (packed bed volume) of Protein A Sepharose beads (MilliporeSigma) for 1 h at 4°C. The bead, antibody, and protein complexes were pelleted using a microcentrifuge and washed with 1 ml of NP-40 lysis buffer three times, then boiled in 2× sample buffer and 100 mM dithiothreitol (DTT). Immunoprecipitation and input samples were then resolved on a gradient 4–12% denaturing Invitrogen Bis–Tris Bolt Plus mini protein gel and visualized using western blot.
Western blot
All protein samples were boiled in sample buffer with 100 mM DTT at 100°C for 10 min. Samples were resolved on Invitrogen Bis-Tris Bolt Plus 4–12% protein gel in MOPS buffer or MES buffer (both from Invitrogen) depending on the size of the target protein. Gels were transferred to PVDF membrane via Genscript eBlot L1 blot transfer apparatus using the default protocol. Membranes were blocked for 1 h in 5% skim milk powder in TBST. Primary antibodies were applied in 3% BSA in TBST, shaking, overnight at 4°C. Horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) were applied in blocking buffer, diluted 1:100 000. Proteins were visualized using either X-ray film or Azure C600 digital imager using Luminata Forte ECL reagent (MilliporeSigma). Band quantification was performed using AzureSpot Pro software version 1.3-531.
siRNA knockdowns
Knockdowns were performed using the siLentFect Lipid Reagent for RNAi (BioRad) according to manufacturer specifications. Specific assays were performed at times indicated in the figure legends. siRNA for ARGLU1, JMJD6, and ERCC6 were obtained from Life Technologies (siRNA IDs s30129, s23289, s4806).
Virus growth assay
HT1080 cells were transfected with siRNA targeting ARGLU1 or negative control siRNA for 24 h. Cells were then infected with dl309 at MOI 10 in serum-free media. Virus was adsorbed for 1 h at 37°C under 5% CO2, and then complete media was added to the cells. At the indicated time points, the infected cells were harvested and then freeze-thawed three times. Non-encapsulated DNA, including free viral genomic DNA, was degraded by treating the lysates with DNAse I for 1 h at 37°C, followed by DNase I inactivation using the TurboDNAfree kit from Invitrogen. Subsequently, all proteins were degraded using 1 hour proteinase K treatment at 42°C to liberate encapsulated viral genomic DNA. Viral genomic DNA was purified using BioBasic DNA purification kit. Viral particles were quantified by quantitative real-time PCR, with a serial dilution of pXC1 plasmid (having the E1 region cloned in it) serving as the standard curve as previously described (24) using the BioRad CFX96 real-time thermocycler (BioRad) as described below in the real-time gene expression analysis section.
Statistical analysis
Statistical analysis for all experiments was performed with GraphPad Prism v5 software using a two-tailed Student's t-test or ANOVA with Tukey's test where appropriate. P-values of ≤0.05 were considered statistically significant. In all cases, mean is shown with error bars representing standard deviation of all biological and technical replicates. The number of biological replicates is indicated with an ‘n’ in the figure legends.
Real-time gene expression analysis
HT or HT-A cells grown in 6-well plates (∼1 million cells per well) were lysed directly on the plate after media removal using TRIzol Reagent (MilliporeSigma) according to manufacturer's instructions. Total RNA was then treated with TURBO DNA-free kit (Invitrogen) to remove contaminating DNA according to manufacturer's instructions. RNA was quantified using A260 and quality determined using A260/A280 ratio, then 1 μg of total RNA was used for cDNA generation with SuperScript VILO reverse transcriptase master mix (Invitrogen) according to manufacturer's guidelines, using random hexanucleotides for priming. Next, 2% of total cDNA was used for real-time expression analysis using the BioRad CFX96 real-time thermocycler with BioRad SsoAdvanced Universal SYBR Green Supermix in a total reaction volume of 10 μl with the standard cycling program as specified by the manufacturer for this reagent using BioRad CFX Manager software version 3.1 for data acquisition. For each sample, at least three biologicals and two technical replicates were analysed unless otherwise specified in a figure legend. Analysis of expression data was carried out using the percentage of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) method with raw Cq values for GAPDH serving as a quality control and samples showing Cq values greater than 25 were re-processed. GAPDH was chosen as we did not observe its raw Cq values fluctuate significantly in our experimental systems. Specificity of amplicons was verified using melt curve analysis. To ensure no contaminating DNA was present, RNA samples were also subject to qPCR analysis without prior reverse transcriptase reaction. Post-acquisition data analysis was performed using Excel for Office 365 version 16.82 for Mac.
PCR primers
The table below lists the PCR primers used in quantitative PCR analysis and ARGLU cDNA cloning, where possible primers span intron/exon boundaries to prevent amplification from genomic DNA. Primers for expression analysis and ChIP of viral genes were previously described (15,28,29).
| Primers used in the study | ||
|---|---|---|
| Gene | Forward | Reverse |
| GAPDH | GAGTCAACGGATTTGGTCGT | TTGATTTTGGAGGGATCTCG |
| ARGLU1 | TGGTAGCAAAAAGGGTGGAG | TGTGCTTCTGCAATTTTTCG |
| MCM4 | TTGAAGCCATTGATGTGGAA | GGCACTCATCCCCGTAGTAA |
| PCNA | GAAGCACCAAACCAGGAGAA | TCACTCCGTCTTTTGCACAG |
| CCNE2 | GGAACCACAGATGAGGTCCAT | TCACTGCAAGCACCATCAGT |
| ALDOA | TGTGCATCAGTAGACAG | AGTCAGCTCGTCTCTG |
| RHOB | AGAGTGTGTGGCTGTGTGCT | TGAAGGCGACATCTGATG |
| IFIT1 | AAAAGCCCACATTTGAGGTG | GAAATTCCTGAAACCGACCA |
| ALDOA for Pol II ChIP | TCCACGGACTCTCCGTTATT | CCTCCCTGGCTCCTTCTCTA |
| RHOB for Pol II ChIP | TGGTTGGAGCTGTTGTCTTG | GTTCTCTCGCTGCGCTTC |
| MCM4 for ChIP | CCGAGCGAGGCCTACTTCT | GGACAGTGCCGCTTCTTTCA |
| PCNA for ChIP | CTGGCTGCTGCGCGA | CACCACCCGCTTTGTGACT |
| ALDOAp for ChIP | TCTCTGCTTCGAGATCAAGCTC | GTGGGGAAAGGCTCTTCAGG |
| RHOBp for ChIP | GGGAGTTTGCCAGGAAGAGG | TTCCGAGACAGGCTTCACTC |
| ALDOAape for ChIP | TCCATTGACCTAATTGCCTCTT | TTGACATCCAGGAGGGAGAC |
| RHOBape for ChIP | AACACATTTCCCTGGCATTC | CAGGAGGAAACAGGACAAGC |
| ARGLU1 cloning primers | ACTGATGAATTCATGGGCCGGTCTCGGAGCCG | ACTGTCCTCGAGTTAATCCTGGGTTTTTAATG |
Doubling time determination
HT or HT-A cells were plated at 50 000 cells/well in 6-well dishes. Twenty-four hours later, cells were imaged every 6–24 h using the ImageXpress Micro 4 high content imager and automatically counted using the MetaXpress software suite version 6.7.0.211. Doubling time was determined as indicated using the formula in the Figure 3C, while growth rate of the cells was determined using the formula: growth rate = ln(final cell count/initial cell count)/time.
Figure 3.
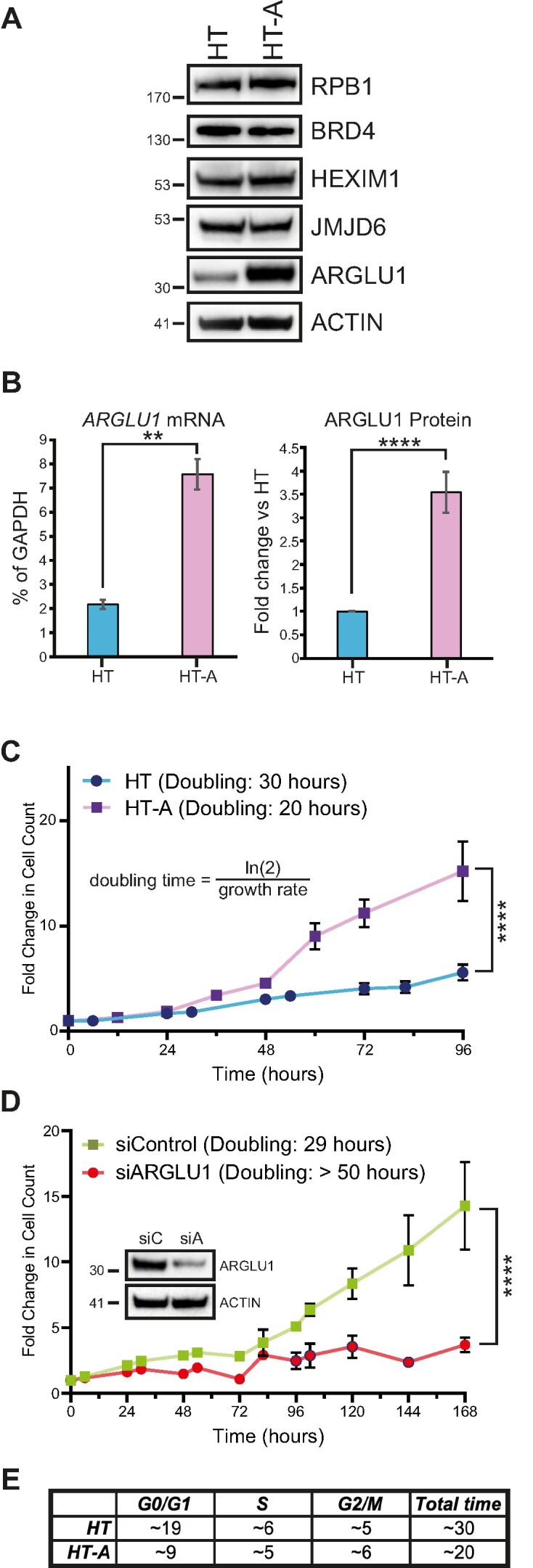
ARGLU1 promotes cell growth by accelerating the cell cycle. (A) HT and HT-A cells were lysed in NP-40 lysis buffer and 20 μg of total protein was loaded per well into an SDS polyacrylamide gel, resolved, and blotted for the endogenous levels of the indicated proteins. ACTIN was used as a loading control. (B) mRNA and protein quantification of ARGLU1 levels in HT and HT-A cells. mRNA was extracted from HT or HT-A cells using the TRIzol reagent, converted to cDNA using VILO master mix and quantified using qPCR with GAPDH serving as an internal control; ** P ≤ 0.01; n = 3. Protein was quantified from three independent western blots for ARGLU1 using AzureSpot software and ACTIN as a loading control; **** P ≤ 0.0001; n = 3. (C) HT and HT-A cells were plated at 50 000 cells/well in 6-well plates and allowed to recover for 24 h, which was then set as time 0. Cells were then imaged every 6–12 h for 96 h and counted using the ImageXpress Micro 4 imager and MetaXpress software with the cell counting module; **** P ≤ 0.0001; n = 3. (D) HT cells were transfected with siRNA targeting ARGLU1 (siARGLU1) or a negative control siRNA (siControl). Cells were then allowed to recover for 24 h, which was set as time 0. Cells were then imaged as in (B) over 168 h; **** P ≤ 0.0001; n = 3. Inset shows expression of endogenous ARGLU1 following knockdown at the 72 h time point, siC - siRNA negative control, siA -siRNA targeting ARGLU1. (E) HT or HT-A cells were subjected to double-thymidine block as described in materials and methods. To quantify the DNA content of the cells, cells were fixed and stained with propidium iodide every 2 h following release from the thymidine block. DNA content was then quantified on a flow cytometer as described in materials and methods. The duration of each phase of the cell cycle was estimated in hours using FlowJo software.
Cell cycle analysis
Double-thymidine block was performed essentially as previously described (30). Briefly, HT and HT-A cells were plated at 1.5 million cells in 10 cm plates in 5% FBS DMEM and incubated at 37°C overnight. Thymidine was added to a final concentration of 2 mM and cells were incubated for 18 h. Cells were washed with PBS, media replaced, and incubated for 9 h. Thymidine was added again to a final concentration of 2 mM and cells incubated for 18 h. Cells were released from thymidine block by PBS rinse and media replacement. Timepoints were harvested every 2 h for 36 h with T0 harvested directly after thymidine block release. Harvested samples were then stained using propidium iodide as described below.
Flow cytometry
Flow cytometry was performed essentially as previously described (31). Briefly, cells were washed with PBS and then fixed in ice cold 70% ethanol for 10 min on ice. Fixed cells were washed again in PBS and resuspended in 1 ml of propidium iodide staining solution (4 mM sodium citrate, 0.1% Triton X-100, 75 μM propidium iodide, 10 μg/ml RNase A) and incubated at 37ºC for 10 min. Cells were assayed for DNA content using a Cytoflex LX flow cytometer (Beckman Coulter) and analyzed using FlowJo software version 10.9.0.
Proteomic analysis
HT or HT-A cells were harvested and lysed in PBS using sonication and 300 μg of total protein were precipitated via trichloroacetic acid prior to analysis. Protein was sent to the Southern Alberta Mass Spectrometry Centre for 2D-LC-MS/MS shotgun proteomics analysis where raw reads were interpreted into Scaffold data. Results were analyzed broadly by Heatmapper software (32) and in detail by Cytoscape's (33) STRING function and clusterMaker app (34). The Markov Clustering Algorithm (MCL) was then used to analyze functional interactions by topologically partitioning the STRING graph using a Markov based flow simulation using the Cytoscape clusterMaker app. All proteomics data has been deposited into PRIDE database under accession # PXD046329 and DOI: 10.6019/PXD046329, while MS data for affinity purification of ARGLU1 and HAdV-B7 E1A associated proteins has been deposited under accession # PXD050223 and DOI: 10.6019/PXD050223.
Focus forming assay
Two hundred thousand HT or HT-A cells were plated in 6-well plates and allowed to reach 90% confluency at which point 5 μM or 50 nM bleomycin or actinomycin D was added, respectively, and kept on for the next 24 h. Media was then changed, and cells were allowed to recover and form foci for 7 days for bleomycin-treated cells and 21 days for actinomycin D-treated cells. For hygromycin B control, cells were treated with 350 μg/ml of hygromycin B for 24 h, drug was removed and replaced with fresh media, and foci were allowed to form for 21 days. Media was then removed from the cells, the cells were rinsed with PBS twice and fixed using 4% formaldehyde in PBS for 10 min. Plates were subsequently rinsed with PBS two more times for 10 min, then stained with crystal violet for 10 min. After staining, cells were repeatedly rinsed with PBS until the removed PBS ran clear.
Comet assay
Comet assay was performed using the Comet Assay Kit from Abcam (cat. ab238544) according to manufacturer's instructions. Comets were resolved in alkaline electrophoresis buffer prior to staining and imaged using ImageXpress Micro 4 high content imager (Molecular Devices) using MetaXpress software version 6.7.0.211. When (+)-JQ1 (Selleckchem, cat. S7110) was used at 500 nM, it was applied to cells 16 h prior to inducing DNA damage and assaying comet length. Comets were measured and quantified using ImageJ software with OpenComet plug-in (35) or TriTek CometScore 2.0.
Transcription reporter assay
U2OS-2-6-3 cells were plated in 6-well plates at 300 000 cells/well. The next day, cells were infected with Ad.CMV or Ad.ARGLU1 at an MOI of 200, induced to express the mCherry-LacI-FokI nuclease with 4-hydroxytamoxifen and Shield-1 with 1 μM each, and induced to express the reporter CFP gene with 2 μg/ml of doxycycline. Twenty-four hours later, cells were lysed in NP-40 lysis buffer and CFP fluorescence was measured using FlexStation 3 plate reader (Molecular Devices) and normalized to total protein level as determined by the Bradford assay.
Immunofluorescence
HT or HT-A cells were plated at low density (∼40 000 cells per chamber) on chamber slides (Nalgene Nunc) and subsequently infected as described above. Twenty-four hours after infection, cells were fixed in 4% formaldehyde, permeabilized in 0.1% Triton, blocked in blocking buffer (1% normal goat serum, 1% BSA, 0.2% Tween-20 in PBS), and stained with specific primary antibodies. After staining and three 10-min washes in PBS-T, slides were mounted using Prolong Gold with DAPI (Invitrogen) and imaged using Zeiss LSM700 confocal laser scanning microscope. Images were analyzed using Zeiss ZEN software package version 8. For U2OS-2-6-3 and U2OS-2-6-5 cells, cells were induced to produce DNA damage 24 h prior to fixation and staining as above and as previously described (36). For U2OS-EJ5-DSB and U2OS-DR-GFP, 24 h after plating, cells were infected with Ad.CMV, Ad.ARGLU1, and/or Ad.I-SceI at a multiplicity of infection of 200 for 24 h prior to assaying for GFP expression using ImageXpress Micro 4 high content imager (Molecular Devices).
Chromatin immunoprecipitation
ChIP was carried out essentially as previously described (29). HT or HT-A cells were infected with HAdV5 strain dl309 at a MOI of 10 and harvested 24 h after infection for ChIP analysis of infected cells. Uninfected cells were plated on 10 cm plates and ChIPs were performed 24 h later, on approximately 10 million cells. Cells were double-fixed first with 2 mM disuccinimidyl glutarate and then with 1% formaldehyde, and harvested for ChIP analysis as previously described (37). For quantitative ChIP analysis, digital PCR was performed on 3% of total DNA eluted from the ChIP using BioRad QX200 ddPCR EvaGreen Supermix using BioRad QX200 droplet digital PCR instrument with AutoDG, which provides absolute quantification without the need for a standard curve and then converted to % of input based on absolute quantification of the input. A minimum of three biological replicates were analysed for each condition, unless specified otherwise in a figure legend, with no technical replicates as these are not needed for digital PCR. Data acquisition was performed using QX Manager Standard Edition version 1.2.345 and analysed using QuantaSoft Analysis Pro version 1.0.596 with automatic threshold setting. Post-acquisition analysis was performed using Microsoft Excel for Office 365 version 16.82 for Mac.
Results
ARGLU1 is an intrinsically disordered protein with a predicted structured C-terminus
Structural data for ARGLU1 is not available. However, computational prediction by AlphaFold (38) and similar tools provide limited insight into the structural domains within the protein. PrDOS (39) predicts that most of ARGLU1 is likely to be unstructured, with a stretch of amino acids within the C-terminus having a predicted structure (Figure 1A). The structured region falls within the AlphaFold-predicted α-helix, which is projected to be between residues 93 and 255. A previous study of anti-pause enhancers has identified ARGLU1 as a binding partner of JMJD6 (9) and we have also observed this via mass spectrometry of ARGLU1-associated protein complexes (see below). Our identification of JMJD6 associated with ARGLU1 was performed in HT1080 (henceforth referred to as HT) cells transfected with HA-tagged ARGLU1 and immunoprecipitated using anti-HA antibody 12CA5. In this purification, three peptides matched JMJD6 with >99% confidence: CGEDNDGYSVK, DDSPLYIFDSSYGEHPK, FFTDDLFQYAGEK. JMJD6 is recruited to the anti-pause enhancer complex via interactions with the extraterminal (ET) domain of BRD4 (9,40) and while investigating the sequence of ARGLU1 we observed a curious similarity between the C-terminus of ARGLU1 and the BRD4 ET domain. To determine whether there was similarity, we performed sequence alignment between BRD4 ET domain (residues 600–682) and ARGLU1 C-terminus (residues 101–273) (Figure 1B). This alignment showed that the residues required for the interaction of BRD4 ET domain with JMJD6 are also conserved in ARGLU1. This observation suggests that ARGLU1 may interact with JMJD6 via the same region of JMJD6 that binds BRD4, serving as a competitor of BRD4 binding to JMJD6, and potentially a regulator of RNA polymerase II pausing. Structural comparison of the predicted ARGLU1 structure and BRD4 ET domain reveals presence of α-helices, showing some potential structural conservation (Figure 1C).
ARGLU1 binds to HAdV E1A and inhibits virus growth and gene expression
Mass spectrometry analysis of complexes associated with HAdV-B7 E1A found ARGLU1 as a potential binding partner. Using transfected HA-tagged HAdV7 E1A immunoprecipitated with anti-HA 12CA5 monoclonal antibody from HT cells, identified a single peptide that matched to human ARGLU1 protein (peptide sequence: QLLEELER) with greater than 99% confidence that the identified protein is ARGLU1. To confirm this interaction, we performed a co-immunoprecipitation of HAdV-C5, and -B7 E1As with ARGLU1 (Figure 2A) as well as with HAdV-C5 E1A isoforms of 289 residues (R)—expressed from mutant pm975—and 243R—expressed from mutant dl520—(Figure 2B), which are the two isoforms of E1A most abundant early in infection and differ only by the presence of conserved region 3 (CR3) in the larger isoform (16,41). ARGLU1 bound to E1As from both species of HAdV with greater affinity for HAdV-B7 E1A. ARGLU1 interacted with the two major E1A isoforms (289R and 243R) from HAdV-C5, with perhaps slightly weaker association with E1A243R (Figure 2B). Since ARGLU1 was found to interact with Mediator subunit MED1 (1), it is possible that CR3 enhances E1A association with ARGLU1 as it also binds the Mediator complex via MED23 (42). Mapping of the ARGLU1 binding site on HAdV-C5 E1A during infection and with transfected HA-tagged ARGLU1 showed that there is a loss of binding with E1A mutants dl1102 and dl1103, which map to residues 26–49 (Figure 2C). Taking the sequence that maps to these residues in HAdV-C5 E1A (Figure 2D) and performing a protein BLAST with it identified several proteins involved in transcription, chromatin remodeling and histone methylation, with two proteins showing particularly high degree of conservation; Lysine Methyltransferase 2D (KMT2D) and Arginine-Glutamic Acid Dipeptide Repeats (RERE). Sequence alignment of this area of E1A with these two proteins showed a high degree of protein identity and conservation. Interestingly, a similar sequence was found in JMJD6 (Figure 2D). These observations suggested to us that ARGLU1 may play a role in transcription regulation that E1A may be disrupting, in line with previously reported functions of ARGLU1 (1,3,6).
Figure 2.

ARGLU1 binds to E1A N-terminus and is a negative regulator of HAdV growth and gene expression. (A) HT cells were infected with HAdV-C5 dl309 or HAdV-B7 Gomen strain, immunoprecipitated 24 h after infection for E1A using M73 monoclonal antibody for HAdV-C5 and custom rabbit polyclonal antibody for HAdV-B7 E1A. Associated endogenous ARGLU1 was detected with a custom rabbit polyclonal anti-ARGLU1 antibody. (B) HT cells were transfected with HA-ARGLU1 and infected with the indicated dl309 HAdV-C5 or mutants pm975 expressing predominantly E1A289R or dl520 expressing predominantly E1A243R. Immunoprecipitations were performed 24 h after infection/transfection for E1A using M73 anti-E1A hybridoma and the associated HA-ARGLU1 was detected with anti-HA rat monoclonal antibody 3F10. (C) HT cells were infected with the indicated HAdV-C5 dl309-based deletion mutants and transfected with a plasmid expressing HA-ARGLU1, 24 h later cells were lysed and immunoprecipitated for E1A using the M73 monoclonal antibody. Associated HA-ARGLU1 was detected using rat anti-HA monoclonal antibody 3F10. (D) Multiple sequence alignment of the region in E1A that interacts with ARGLU1 and proteins with similar sequences. E1A-C5 refers to E1A from HAdV-C5 while E1A-B7 refers to HAdV-B7 E1A. (E) HT cells were transfected with siRNA targeting ARGLU1 mRNA (siARGLU1) or a negative control siRNA (siControl). Twenty-four hours later cells were infected with HAdV-C5, virus was allowed to replicate for 24 and 48 h prior to quantification. Viral particle counts were determined by quantifying encapsulated viral genomes, * P ≤ 0.05; n = 3. Bottom panel shows efficiency of ARGLU1 knockdown by siRNA at the 3 time points examined, time 0 is the time of virus infection and occurs 24 h after siRNA transfection. (F) Cells treated in the same way as in (B) above were harvested at the indicated time points for total RNA extraction using the TRIzol reagent. Time 0 represents uninfected cells. Total RNA was converted to cDNA using VILO reverse transcriptase master mix and used in qPCR to assay viral gene expression normalized to GAPDH mRNA levels; * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001; n = 3. (G) Same as (D) except indicated cellular mRNAs were quantified; * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001; n = 3.
We have previously characterized the effects of deletion of E1A in the two mutants that have impaired binding to ARGLU1 (dl1102 and dl1103) in primary lung fibroblasts WI-38 (43). Interestingly, we observed that mutant dl1102 initially grew better than the in vitro phenotypically wild-type (wt) virus strain dl309, with this mutant growing particularly well 48 and 72 h after infection and expressing its genes to higher levels than wt HAdV-C5, while mutant dl1103 grew much worse than wt despite expressing its genes at a similar level to the wt virus (43). To determine whether knockdown of ARGLU1 affects viral replicative cycle in a similar way to deletion of its interaction region in E1A, we performed siRNA-mediated ARGLU1 knockdown studies. ARGLU1 was knocked-down via siRNA transfection followed by infection and determination of viral titer 24 and 48 h after infection with HAdV-C5 dl309 (Figure 2E). Depletion of ARGLU1 had an overall enhancing effect on virus growth, similar to what we previously observed with the mutant dl1102 (43), with viral titers increasing approximately 2-fold with ARGLU1 knockdown versus siControl-treated cells. The enhanced virus growth correlated with enhanced viral gene expression when ARGLU1 was knocked down (Figure 2F), similarly to what we previously have observed in E1A mutants that no longer bind ARGLU1 (43). Under conditions of ARGLU1 knockdown, levels of viral mRNAs were significantly higher for most genes with the exception of E2, which showed little effect. We also investigated the effect on select cell cycle genes, commonly upregulated during HAdV infection (44) (Figure 2G). PCNA, CCNE2 and MCM4 are expressed in cycling HT cells, but should still be upregulated during infection. Similarly to what we observed for viral genes, knockdown of ARGLU1 resulted in enhanced expression of the cell cycle-associated genes during infection that we investigated. Interestingly, levels of PCNA and MCM4 transcripts were reduced in ARGLU1 knockdown cells at the time of infection before any viral proteins were made (Figure 2G), suggesting that ARGLU1 may play some role in activating their expression. As was the case with viral genes, these cellular genes were upregulated in WI-38 cells when they were infected with ARGLU1-binding deficient mutant dl1102 versus wt, but were slightly downregulated with mutant dl1103 (43). Collectively, these results indicate that ARGLU1 is a negative regulator of viral growth and a repressor of viral gene expression.
ARGLU1 enhances cellular growth rate and accelerates the cell cycle
We generated a stable HT1080-derived cell line that constitutively expresses HA-tagged ARGLU1, henceforth referred to as HT-A (Figure 3A). We initially assessed the levels of ARGLU1 in these cells, as well as other cellular proteins pertinent to the present study (Figure 3A and B). Levels of ARGLU1 protein and mRNA were approximately 4-fold higher in HT-A cells as compared to the parental HT cells. Levels of other proteins tested; RNA polymerase II subunit 2A (RPB1), BRD4, HEXIM1 and JMJD6; were not significantly altered by ARGLU1 overexpression (Figure 3A). During routine culturing, we observed that HT-A cells grew at a quicker pace than the parental HT cells. We therefore proceeded to determine the doubling time and the duration of cell cycle phases in HT-A and HT cells (Figure 3C–E). HT cells doubled every 30 h, whereas HT-A cells doubled every 20 h. Conversely, when endogenous ARGLU1 was knocked down via siRNA transfection (Figure 3C), we observed a greatly extended doubling time of over 50 h. In fact, we observed very little growth, suggestive of growth arrest when ARGLU1 was knocked down (Figure 3D). To determine the duration of the cell cycle phases we employed double thymidine block to arrest HT and HT-A cells, cells were released, and DNA content was determined using propidium iodide staining and flow cytometry (Figure 3E and Supplementary Figure S2A). We observed a substantial reduction in the duration of G1 and a slight reduction in the length of S-phase in HT-A cells, G2/M phase was also slightly extended in HT-A cells versus HT. Gross morphology of ARGLU1-overexpressing HT-A cells was not significantly altered as compared to HT cells (Supplementary Figure S2B). Overall, these findings indicate that ARGLU1 promotes cell growth and accelerates the G1- and S-phases of the cell cycle.
Proteomic analysis of ARGLU1-overexpressing HT cells reveals a role in DNA damage response
Faster cell cycling observed in ARGLU1-overexpressing HT-A cells suggested that the protein may play a role in cell cycle regulation. To investigate, on a global scale, the effects of ARGLU1 overexpression we performed shotgun proteomics analysis comparing protein levels in HT cells, versus HT-A cells overexpressing ARGLU1. Overall, 1599 proteins were upregulated when ARGLU1 was overexpressed, 795 were downregulated, and 141 were unchanged (Supplementary spreadsheet). Functional analysis using gene ontology (GO) grouping identified a significant number of cellular pathways that were functionally enriched when ARGLU1 was overexpressed (Table 1 and Supplementary Figure S3, Supplementary Tables S1–S3, and Supplementary spreadsheet). Of those pathways, several GO biological processes involved in cell cycle and cell division were enriched, in agreement with our cell cycle and growth analysis results (Figure 3). Interestingly, several GO biological processes involved in nucleic acid metabolism, DNA damage response, and DNA repair were also significantly enriched in ARGLU1-overexpressing HT-A cells. Other processes of interest enriched by ARGLU1 overexpression included RNA metabolic processes, telomere biology, transcription by RNA polymerase II, p53-regulated processes, general stress response, and some metabolic processes. Overall, proteomic analysis suggests that ARGLU1 overexpression promotes cell cycle and stress response, including response to DNA damage.
Table 1.
Gene ontology analysis of biological processes most affected by ARGLU1 overexpression in HT-A cells as compared to parental HT cells
| GO biological processes | Fold enrichment | P-value (–log10) |
|---|---|---|
| Macromolecule biosynthetic process (GO:0 009 059) | 1.86 | 24.18 |
| Cellular response to stress (GO:0 033 554) | 1.8 | 22.48 |
| Mitotic cell cycle (GO:0 000 278) | 2.38 | 21.58 |
| Mitotic cell cycle process (GO:1 903 047) | 2.28 | 16.06 |
| Response to stress (GO:0 006 950) | 1.4 | 15.18 |
| Positive regulation of protein metabolic process (GO:0 051 247) | 1.66 | 14.27 |
| DNA damage response (GO:0 006 974) | 1.94 | 14.13 |
| Regulation of cellular localization (GO:0 060 341) | 1.81 | 13.74 |
| Cell division (GO:0 051 301) | 2.14 | 13.45 |
| Chromosome organization (GO:0 051 276) | 2.21 | 12.89 |
| Regulation of programmed cell death (GO:0 043 067) | 1.59 | 12.54 |
| Regulation of apoptotic process (GO:0 042 981) | 1.6 | 12.51 |
| Negative regulation of programmed cell death (GO:0 043 069) | 1.74 | 11.23 |
| DNA repair (GO:0 006 281) | 2.03 | 11.12 |
| Negative regulation of apoptotic process (GO:0 043 066) | 1.74 | 10.93 |
| Regulation of apoptotic signaling pathway (GO:2 001 233) | 2.11 | 9.28 |
| Positive regulation of chromosome organization (GO:2 001 252) | 3.05 | 7.80 |
| Negative regulation of apoptotic signaling pathway (GO:2 001 234) | 2.3 | 7.46 |
| Programmed cell death (GO:0 012 501) | 1.51 | 7.13 |
| Apoptotic process (GO:0006915) | 1.5 | 6.51 |
ARGLU1 promotes cancer cell resistance to DNA damaging drugs and enhances DNA damage repair
Proteomic analysis suggested that ARGLU1 may affect DNA damage repair or response. To investigate this possibility, we initially determined whether ARGLU1 overexpression promotes resistance of HT-A cells to DNA damaging drugs bleomycin and actinomycin D. Bleomycin induces double-stranded DNA breaks (DSBs) via binding to GC-rich regions and inducing single-strand breaks at the 3′–4′ bonds in deoxyribose. Breaks on both strands eventually lead to DSBs (45). Actinomycin D inhibits DNA-dependent RNA polymerases, inducing DNA single-stranded breaks via stalled replication forks during S-phase and during stalled transcription (46). We initially determined the lethal concentrations of each drug to HT cells, with 50 μM of bleomycin and 50 nM of actinomycin D killing most HT cells. Next, we treated the cells for 24 h with each drug and allowed them to recover for 7 days following bleomycin treatment and 21 days after actinomycin D treatment to form foci (Figure 4A and B). When ARGLU1 was overexpressed, we observed a significant increase in drug-resistant foci for cells treated with either bleomycin or actinomycin D. To rule out the possibility that ARGLU1 overexpression led to a general resistance to cytotoxic drugs, we determined whether it affects hygromycin B resistance, which is an atypical aminoglycoside antibiotic that interferes with protein translation (47). For this purpose, HT and HT-A cells were treated with 350 μg/ml of hygromycin B, which is a lethal dose for HT cells, for 24 h and then allowed to recover and form colonies for 21 days (Supplementary Figure S4A). We observed no significant difference in the number of colonies formed by HT or HT-A cells following hygromycin B treatment (Supplementary Figure S4B). These colony forming assays suggest that ARGLU1 promotes resistance of cancer cells to genotoxic drugs.
Figure 4.

ARGLU1 promotes HT cells resistance to genotoxic drugs and enhances DNA damage repair. (A) HT or HT-A cells were plated at 200 000 cells/well in 6-well plates and treated with 50 μM bleomycin or 50 nM actinomycin D for 24 h. The cells were then washed and allowed to grow for 7 days for bleomycin-treated and 21 days for actinomycin D-treated conditions before colonies were fixed and visualised with crystal violet stain. (B) Quantification of A; *** P ≤ 0.001, **** P ≤ 0.0001; n = 6 wells. (C) HT or HT-A cells were plated at 50 000 cells/well and grown in 6-well plates for 24 h, cells were then harvested and assayed for DNA damage using the comet assay as described in materials and methods; ** P ≤ 0.01, n = 48 comets; a.u. – arbitrary units. (D) HT or HT-A cells were plated at 50 000 cells/well and 24 h later were treated with 5 μM bleomycin for 15 min. Cells were then allowed to recover in bleomycin free medium for the indicated time in minutes, harvested and subjected to comet assay as described in the materials and methods; * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001; n = 25 comets. (E) Representative images of comets from (D) at t = 30 min of recovery after bleomycin removal. (F) HT or HT-A cells treated as in (D) were also pre-treated with 500 nM of the inhibitor JQ1 for 16 h prior to 15-min treatment with 5 μM bleomycin and comet assay analysis at the indicated time points with t = 0 indicative of no recovery and t = 30 indicative of 30 min recovery after drug removal. **** P ≤ 0.0001; n = 48 comets; a.u. – arbitrary units. Right panel indicates the ratio of HT treated with bleomycin over HT pre-treated with JQ1 and treated with bleomycin from the left panel 30 min after bleomycin removal, comparing to HT-A results analyzed in the same manner. (G) HT cells were plated in 6-well plates at 50 000 cells/well and 24 h later transfected with siRNA targeting ARGLU1 (siA) or control siRNA (siC). Forty-eight hours later, the cells were harvested and analyzed by comet assay; ** P ≤ 0.01, n = 30 comets; a.u. – arbitrary units. Bottom panel shows a western blot of ARGLU1 knockdown in these cells 48 h after siRNA transfection. (H) U2OS-EJ5-GFP (NHEJ) or U2OS-DR-GFP (HR) were plated in 6-well plates at 100 000 cells/well and 24 h later infected with Ad.I-SceI and Ad.ARGLU1 to overexpress ARGLU1 or Ad.CMV as a negative control at an MOI of 200. Cells were imaged 24 h after infection using ImageXpress Micro 4 high content imager to quantify the number of GFP-positive cells, Hoechst 33342 dye was used as a nuclear counterstain to visualize all cells; *** P ≤ 0.001, **** P ≤ 0.0001; n = 3 wells and 36 fields per well were imaged. Bottom panel shows sample expression of HA-ARGLU1 and HA-I-SceI from the adenovirus vectors. (I) U2OS-2-6-5 cells were induced to express the mCherry-LacI-FokI nuclease with 18 h application of Shield-1 and 4-hydroxytamoxifen and either infected at an MOI of 200 with Ad.ARGLU1 (right panel) or not (left panel), and stained for mCherry to visualize the site of DNA damage, HA for Ad.ARGLU1-infected cells to visualize HA-ARGLU1, or endogenous ARGLU1 with rabbit polyclonal antibody to ARGLU1 as indicated. DAPI was used as a nuclear stain. Representative images of the two observed ARGLU1 phenotypes are shown, with chevron indicating the site of DNA damage. Observed phenotype frequency is also indicated based on imaging of 30 random cells. (J) U2OS-2-6-3 cells were induced to express the mCherry-LacI-FokI nuclease as in panel I and, at the same time, induced to express the CFP reporter with addition of 2 μg/ml of doxycycline prior to harvest and imaging as described in the materials and methods. Twenty-four hours prior to DNA damage induction, the cells were infected with either Ad.ARGLU1 to overexpress ARGLU1 or Ad.CMV as a control at an MOI of 200. Twenty-four hours after DNA damage induction and reporter induction, the cells were harvested and fluorescence was quantified using Molecular Devices FlexStation 3 plate reader and normalized to total protein level. Fluorescence is represented as percentage of CFP-induced and Ad.CMV-infected U2OS-2-6-3 cells; **** P ≤ 0.0001; n = 3. Bottom panel shows expression of HA-ARGLU1 in these cells. (K) Representative images of U2OS-2-6-3 cells following DNA damage induction and infected with Ad.ARGLU1 to overexpress HA-ARGLU1, stained with GFP to show MS2-YFP localization to a transcriptional focus and HA to show ARGLU1 overexpression. Chevron indicates a transcriptional focus and asterisk marks ARGLU1 overexpressing cell. (L) Quantification of cells treated as in (K); ** P ≤ 0.01, n = 20 fields of view. (M) HT or HT-A cells were transfected with a plasmid expressing HA-Ubiquitin. Twenty-four hours later, the cells were treated with 5 μM of bleomycin for 15 min and then lysed in NP-40 lysis buffer supplemented with N-ethylmaleimide and immunoprecipitated for RNA polymerase II using anti-RNA polymerase II C-terminus domain antibody. Immunoprecipitates and inputs were resolved on SDS polyacrylamide gel and probed for HA (ubiquitin), RNA polymerase II (RPB1), ARGLU1, and ACTIN as a loading control. Short – refers to a short exposure; long – refers to a long exposure.
To investigate whether the enhanced resistance to genotoxic drugs caused by ARGLU1 overexpression was due to enhanced DNA damage repair we firstly investigated the endogenous levels of DNA damage observed in HT and HT-A cells via the comet assay (Figure 4C). HT-A cells had less endogenous DNA damage than HT cells, suggesting ARGLU1 may be promoting DNA repair. To further investigate this possibility, we carried out comet assays on HT and HT-A cells transiently treated with bleomycin and allowed to recover for 30 and 60 min (Figure 4D and E). HT-A cells were able to repair most of the bleomycin-induced DNA damage within 30 min after the drug was removed, whereas HT cells required twice as long to reach a similar level of DNA repair.
Since we observed that ARGLU1 had some similarity to BRD4 within the BRD4 ET domain, we conjectured that some of the enhanced DNA damage repair may occur through release of otherwise unavailable BRD4 by outcompeting it for binding to other factors. Higher levels of free BRD4 could promote DNA repair as has been previously reported (48). Therefore, we performed the comet assay under conditions where BRD4 activity was inhibited via the inhibitor JQ1 in HT cells and ARGLU1-overexpressing HT-A cells (Figure 4F). Inhibition of BRD4 via JQ1 led to a significant reduction in DNA damage repair following bleomycin-induced DNA breaks in HT-A cells, while having no effect on untreated HT-A cells, and minimal effect on the parental HT cells as illustrated by the ratio of bleomycin treated over bleomycin and JQ1-treated cells (Figure 4F). These results suggest that, at least partly, the enhanced DNA damage repair observed in HT-A cells is occurring through BRD4.
Since overexpression of ARGLU1 led to enhanced DNA damage repair, we wanted to determine whether reduction of ARGLU1 protein levels via siRNA-mediated knockdown impaired DNA damage mending. For this purpose, HT cells were transfected with siRNA targeting ARGLU1 mRNA and assayed for DNA damage repair after bleomycin treatment 30 min after removal of the drug (Figure 4G). Knockdown of ARGLU1 protein levels led to a ∼50% inhibition of DNA damage repair following bleomycin treatment as compared to cells treated with a control siRNA that does not affect any human protein levels.
To determine whether the enhanced DNA damage repair observed in HT-A cells occurred via homologous recombination (HR) or non-homologous end joining (NHEJ) we utilized a U2OS-based reporter cell line system previously developed by Dr Jeremy Stark (49,50). Two U2OS-based cell lines were used, U2OS-EJ5-GFP that reports the efficiency of NHEJ and U2OS-DR-GFP, which reports the efficiency of HR. In each case, a DSB is generated by expression of I-SceI nuclease, and upon subsequent repair, GFP is expressed and can be quantified as readout of DSB repair. Each cell line was induced to generate a DSB via expression of I-SceI endonuclease by transducing the cells with an adenovirus vector expressing the nuclease (Ad.I-SceI). To induce ARGLU1 overexpression, cells were also transduced with an adenovirus vector expressing ARGLU1 (Ad.ARGLU1), while a negative control empty virus vector (Ad.CMV) was used as a control in place of Ad.ARGLU1. We observed that ARGLU1-overexpression enhanced the efficiency of both NHEJ and HR (Figure 4H). Moreover, we investigated whether ARGLU1 localized to a DSB induced by the expression of FokI nuclease in the reporter cell line U2OS-2-6-5, which expresses an mCherry-FokI-LacI nuclease fusion protein that is localized to DNA via an array of LacO sites, obtained from Dr Roger Greenberg (36). Induction of the nuclease, via Shield-1 and 4-hydroxytamoxifen administration, induces a single DSB that can be visualized with mCherry. We investigated the localization of endogenous ARGLU1 and HA-tagged ARGLU1 in these cells following induction of a DSB (Figure 4I). We observed two populations of cells, cells that showed co-localization of ARGLU1 and the DSB, and those that did not, in roughly equal proportion. We did not observe differences in staining between endogenous ARGLU1 detected with anti-ARGLU1 antibody and HA-tagged ARGLU1, detected with anti-HA antibody, and no signal was detected with anti-HA antibody when ARGLU1 was not transfected (Supplementary Figure S5).
We also wanted to determine whether ARGLU1 affects transcription around the site of a DSB, since we observed it generally reduced gene transcription (Figure 2F and G). For this purpose, we used the U2OS-2-6-3 cell line also provided by Dr Roger Greenberg that has a tetracycline-inducible CFP reporter near the DSB (51). U2OS-2-6-3 cells were transduced with either Ad.CMV or Ad.ARGLU1, induced to produce a DSB (with the same treatment as U2OS-2-6-5 cells) and express the CFP reporter with the addition of 2 μg/ml of doxycycline for 24 h prior to measurement of fluorescence (Figure 4J). We observed that ARGLU1 reduced overall transcription of the reporter by approximately 20% in the absence of a DSB and this was decreased by over 40% when a DSB was introduced, indicating that ARGLU1 promotes the inhibition of gene expression proximal to a site of DNA damage. We visually confirmed this by utilizing the same U2OS-2–6-3 cells that also constitutively express MS2-YFP protein, which binds to and recognizes nascent CFP transcripts that carry an array of MS2 repeats. We observed that there was a significant reduction in the number of MS2-YFP foci in cells overexpressing ARGLU1 (Figure 4K and L).
Previous studies have shown that RNA polymerase II undergoes transcription-coupled and DNA damage-dependent ubiquitination, with ubiquitination being indicative of stalled RNA polymerase II (11,52,53). Therefore, we wanted to determine whether in the presence of high levels of ARGLU1 protein RNA polymerase II is more ubiquitinated. To do this, HT or HT-A cells were transfected with HA-tagged ubiquitin and 24 h later treated with bleomycin for 15 min and then immunoprecipitated for RNA polymerase II, resolved and blotted for HA and RNA polymerase II (Figure 4M). RNA polymerase II was significantly more ubiquitinated in HT-A versus HT cells with bleomycin treatment having no detectable global effect on RNA polymerase II levels or ubiquitination.
We also wanted to determine whether the inhibition of BRD4 or knockdown of the DNA damage repair protein, ERCC6, previously shown to play a critical role in transcriptionally-coupled DNA damage repair (54–56), would affect the growth rate of HT or HT-A cells. To do this, HT or HT-A cells were treated with JQ1 for 24 h prior to imaging, and then imaged for 96 h (Supplementary Figure S6A), we observed a significant reduction in growth of JQ1 treated cells versus control treated, with HT-A cells inhibited in growth to a rate that was below that of untreated HT cells (Supplementary Figure S6A). Knockdown of ERCC6 had little effect on growth of HT or HT-A cells, only reducing growth later in the time course (Supplementary Figure S6B) and with no significant difference. These results suggest that although BRD4 contributes to the accelerated growth rate of HT-A cells, inhibition of transcriptionally-coupled DNA damage repair by knockdown of ERCC6 had only a minimal effect.
ARGLU1 enhances promoter-proximal RNA polymerase II pausing and interferes with BRD4 binding to JMJD6
We observed that upon ARGLU1 knockdown, some viral and cellular genes were upregulated (Figure 2F and G) and RNA polymerase II underwent greater degree of ubiquitination when ARGLU1 levels were high (Figure 4M), suggesting that ARGLU1 may be a negative regulator of transcription under certain circumstances. Furthermore, the similarity between the ARGLU1 C-terminus and the BRD4 ET domain, with JMJD6-binding residues within the ET domain conserved in ARGLU1, suggested that ARGLU1 may play a role in promoter-proximal RNA polymerase II pausing. To investigate this possibility, we examined the degree of paused and elongating RNA polymerase II at cellular promoters regulated by anti-pause enhancers, which promote RNA polymerase II pause-release (9). We chose two genes, previously reported to be regulated by an anti-pause enhancer, RHOB and ALDOA (9). Chromatin immunoprecipitations were performed in HT or HT-A cells using anti-phospho-serine 5 or anti-phospho-serine 2 RNA polymerase II C-terminal domain (CTD) antibodies, with phosphorylation of serine 2 indicative of elongating polymerase while phosphorylation of serine 5 indicative of paused polymerase. We looked at the region within 500 bp of the transcription start site (TSS). In HT-A cells, we observed significant reduction in elongating RNA polymerase II versus parental HT cells on promoter proximal regions of RHOB and ALDOA, while the degree of paused polymerase was enhanced within the same regions (Figure 5A). Although the enrichment values from the ChIP were low, they were considerably higher than background and much higher than for a non-transcribed gene (Supplementary Figure S7). Expression of these genes was also significantly down-regulated when ARGLU1 was overexpressed (Figure 5A). To determine whether this occurs with viral and cellular genes not reported to be regulated by an anti-pause enhancer, we investigated the state of RNA polymerase II on the viral E4 promoter and cellular PCNA and MCM4 promoters, all of which were upregulated when ARGLU1 was knocked down (Figure 2F and G). We observed no significant difference in the degree of elongating RNA polymerase II at any of these three genes, but we did observe a significant enhancement in the amount of paused polymerase. Interestingly, only the expression of E4 was significantly affected by overexpression of ARGLU1, whereas PCNA and MCM4 were not (Figure 5B). We also investigated what effect knockdown has on the expression of RHOB and ALDOA, and both genes were significantly upregulated by knockdown of ARGLU1 (Figure 5C).
Figure 5.

ARGLU1 enhanced promoter proximal RNA polymerase II pausing. (A) Top panel; ChIPs of elongating (phosphorylated at the CTD at S2) or paused (phosphorylated at the CTD at S5) RNA polymerase II at the RHOB and ALDOA promoter-proximal region (+1 to +500 bp) in HT or HT-A cells; * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001; n = 4. IgG negative control consisted of rabbit anti-rat antibody. Bottom panel; expression levels of RHOB and ALDOA in HT or HT-A cells; ** P ≤ 0.01; n = 3. (B) Top panel, ChIPs of elongating (phosphorylated at the CTD at S2) or paused (phosphorylated at the CTD at S5) RNA polymerase II at the E4, PCNA and MCM4 promoter region in HT or HT-A cells. For the viral E4 gene the cells were infected for 24 h, for PCNA and MCM4 genes the cells were uninfected; ** P ≤ 0.01, n = 4. IgG negative control consisted of rabbit anti-rat antibody. Bottom panel; expression of E4orf3 in HAdV-C5 dl309-infected cells HT or HT-A cells, PCNA and MCM4 in uninfected HT and HT-A cells, cells were infected for assaying of viral E4orf3 expression, and uninfected for PCNA and MCM4; **** P ≤ 0.0001; n = 3. (C) HT cells were transfected with siRNA targeting ARGLU1 (siA) or a control siRNA (siC), mRNA levels of ALDOA, RHOB and ARGLU1 were assayed 48 h after siRNA transfection by extracting total RNA using TRIzol reagent, converting it to cDNA using VILO master mix reverse transcriptase and performing qPCR; * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001; n = 3. (D) HT cells were transfected with the indicated plasmids to express FLAG-tagged JMJD6, HA-ARGLU1, and HAdV-C5 E1A. Cells were then lysed and immunoprecipitated for FLAG using M2 FLAG resin to precipitate JMJD6 and associated proteins. Western blots were then performed for BRD4, HEXIM1, HA-ARGLU1, E1A and FLAG-JMJD6 on resolved immunoprecipitations and inputs, HA-ARGLU1 and FLAG-JMJD6 were detected using anti-HA rat monoclonal antibody 3F10, FLAG-JMJD6 is double-tagged with HA and FLAG epitopes, the remaining proteins were detected using their specific antibodies described in the materials and methods. (E) HT cells were transfected with a plasmid to express FLAG-tagged JMJD6 and increasing concentrations (0, 0.5, 1, 2 and 5 μg) of a plasmid expressing HA-ARGLU1. Twenty-four hours later, cells were lysed and immunoprecipitated for FLAG using M2 FLAG resin to precipitate JMJD6 and associated proteins. Western blots were then performed for ARGLU1, BRD4, HEXIM1, HA-ARGLU1, and FLAG-JMJD6 on resolved immunoprecipitations and inputs (as in D). ACTIN was used as a loading control. (F) HT or HT-A cells were subjected to ChIP analysis with the indicated antibodies to assess occupancy of ARGLU1, BRD4, JMJD6 and HEXIM1 at the promoters (P; –1000 to +1) and anti-pause enhancers (ape) of RHOB and ALDOA genes. Non-specific rabbit anti-rat immunoglobulin (IgG) was used as a negative control; * P ≤ 0.05, ** P ≤ 0.01, **** P ≤ 0.0001; n = 3. (G) HT-A cells were transfected with an siRNA targeting JMJD6 (siJ) or a negative control siRNA (siC) for 48 h, at which time total RNA was extracted using TRIzol method, converted to cDNA using VILO master mix, and analyzed for the levels of RHOB and ALDOA mRNA by qPCR. GAPDH was used as a normalization control and the data is represented as percentage of GAPDH. Bottom blot shows knockdown of JMJD6 48 h after siRNA transfection. (H) HT cells were left untreated or transfected with a control siRNA (siC) or ARGLU1 siRNA (siA) for 48 h. Cells were then lysed and immunoprecipitated for JMJD6. Immunoprecipitates were resolved on SDS polyacrylamide gel and probed for BRD4. Density of BRD4 bands was quantified using AzureSpot software and normalized to total BRD4 and ACTIN loading controls and is represented as fold increase over untreated HT cells. Inputs are shown for BRD4, JMJD6, ARGLU1 and ACTIN.
The inhibition of BRD4 via JQ1, the presence of BRD4 ET-like sequences within the C-terminus of ARGLU1, similar to those responsible for the interaction of BRD4 with JMJD6, and detection of JMJD6 in immunoprecipitates of ARGLU1, collectively suggested that some activities of ARGLU1 may be mediated via modulation of the interaction between JMJD6 and BRD4. To determine what effect ARGLU1 had on the association between BRD4 and JMJD6, we performed a co-immunoprecipitation of JMJD6 from HT cells transfected with FLAG-JMJD6 alone or in combination with ARGLU1, and ARGLU1 and E1A (Figure 5D). Overexpression of ARGLU1 nearly depleted all BRD4 associated with JMJD6 regardless of E1A presence. E1A had a slight positive effect on the association of BRD4 and JMJD6, but it was not able to prevent ARGLU1-driven displacement of BRD4 from JMJD6. HEXIM1, which is a negative regulator of RNA polymerase II pause-release, was also found in the complex with JMJD6 and was only slightly reduced in association with JMJD6 when ARGLU1 was overexpressed. To determine whether the displacement of JMJD6 from BRD4 was ARGLU1-level dependent we performed a titration of ARGLU1 plasmid by transfection into HT cells together with constant levels of FLAG-JMJD6 plasmid (Figure 5E) and immunoprecipitation using anti-FLAG antibody. Increasing levels of ARGLU1 led to the dissociation of BRD4 and JMJD6 complex at the two highest levels of ARGLU1 expression.
ARGLU1-induced dissociation of BRD4 from JMJD6 suggests that the observed enhanced promoter-proximal RNA polymerase II pausing may be driven by loss of JMJD6 from the promoter. To investigate this possibility, we performed ChIP for ARGLU1, BRD4, JMJD6 and HEXIM1 at the promoter and anti-pause enhancer of the genes RHOB and ALDOA in HT and HT-A cells (Figure 5F). We did not observe significant differences in BRD4 promoter occupancy, however, at the anti-pause enhancer the levels of BRD4 were significantly enhanced when ARGLU1 was overexpressed. Interestingly, levels of JMJD6 were significantly reduced at the promoter and the anti-pause enhancer, while levels of HEXIM1 were significantly increased at both. ARGLU1 was also detected at the promoter and anti-pause enhancers, with its occupancy significantly increased in HT-A cells as compared to HT.
Depletion of JMJD6 was previously shown to reduce transcription of reporter constructs containing the anti-pause enhancers found in RHOB and ALDOA (9). If ARGLU1 interferes with anti-pause enhancer function by sequestering away JMJD6, as our data suggests, then depletion of JMJD6 should have no effect on the transcription of RHOB or ALDOA in cells overexpressing ARGLU1. To test for this eventuality, we performed qPCR analysis of RHOB and ALDOA expression 48 h after JMJD6 knockdown via siRNA (Figure 5G). No significant difference in expression of these genes was found in HT-A cells after JMJD6 was knocked down.
Lastly, we wanted to determine what effect depletion of endogenous ARGLU1 in HT cells would have on BRD4 association with JMJD6. To investigate this, HT cells were transfected with siRNA targeting ARGLU1 and 48 h later immunoprecipitated for endogenous JMJD6 and associated BRD4 was detected by western blot (Figure 5H). Knockdown of ARGLU1 detectably enhanced the association of BRD4 with JMJD6, while negative control siRNA had a minimal effect on this interaction.
Collectively, these results suggest that ARGLU1 promotes promoter-proximal RNA polymerase II pausing, possibly via displacement of JMJD6 from BRD4 by competitive binding to JMJD6, which likely blocks the BRD4 binding site on JMJD6.
Discussion
The present study advances our understanding of the role of ARGLU1 in the cell and its impact on the adenovirus replicative cycle. Although we show that ARGLU1 inhibits human adenovirus replication by negatively affecting viral gene expression, the role of ARGLU1 in the cell appears much more significant as we provide evidence linking ARGLU1 to cell cycle regulation, the DNA damage response pathway, and cancer cell chemoresistance. Overexpression of ARGLU1 promoted cell growth, while knockdown inhibited cell growth. Likewise, we observed that ARGLU1 overexpression enhances cancer cell resistance to the genotoxic drugs bleomycin and actinomycin D. Our results show that this enhanced resistance is a consequence of enhanced DNA damage repair driven by ARGLU1, plausibly by freeing BRD4 from JMJD6 to promote DNA mending. ARGLU1 was found to localize to DSBs and it inhibited transcription of a reporter proximal to the break. ARGLU1-mediated transcriptional inhibition was likely mediated by enhanced RNA polymerase II pausing driven by ARGLU1 displacement of JMJD6 from BRD4, inhibiting RNA polymerase II pause-release. Proteomic analysis of ARGLU1-overexpressing cells identified a number of significantly enriched pathways, including those involved in DNA damage response, DNA damage repair, and cell cycle regulation. We summarize our findings in Figure 6.
Figure 6.

Under normal conditions, release signals will recruit JMJD6 to BRD4, allowing RNA polymerase II to release from paused state. However, when ARGLU1 is present, it will compete with BRD4 for binding to JMJD6, preventing recruitment of JMJD6 and JMJD6-associated removal of pause-enhancing histone marks, retaining HEXIM1 at the promoter and keeping RNA polymerase II in paused state.
ARGLU1 has not previously been directly linked with the DNA damage response pathway. Previous studies have implicated it mainly in the regulation of splicing and transcriptional control (1,3,4). The present study further extends our understanding of ARGLU1 function in transcription and links this poorly studied protein to the DNA damage response pathway (Figure 4D) and cancer cell resistance to genotoxic drugs (Figure 4A). A previous study correlated low ARGLU1 expression with poor outcomes in gastric cancer (6). Interestingly, this earlier study also showed that ARGLU1 enhanced expression of several mismatch repair genes by binding to and potentiating the recruitment of transcription factors Sp1 and YY1 to the promoters of these mismatch repair genes (6), potentially indirectly linking ARGLU1 with mismatch repair. The Cancer Genome Atlas shows that ARGLU1 is altered in a number of cancers, especially rectal adenocarcinoma, testicular cancer, uterine carcinosarcoma, ovarian, and colon, amongst many others. GENT2 reveals that ARGLU1 mRNA levels are upregulated in several cancers over neighbouring normal tissues, including prostate, pharynx, pancreas, soft tissue sarcoma, colon, HPV-positive head & neck squamous cell carcinoma, and kidney (57,58). Downregulation of ARGLU1 mRNA was less frequent, observed only in a few types of cancer including adipose tissue cancer and head & neck cancer. Our results clearly indicate that ARGLU1 promotes cancer cell survival and accelerates cell growth (Figures 3C and 4A), which would lead to poorer patient outcomes and potentially drug resistance. Overexpression of ARGLU1 accelerated the cell cycle by 30%, particularly shortening the G1 and S-phases (Figure 3E). While at the same time, it enhanced resistance of HT1080 cells to genotoxic drugs bleomycin and actinomycin D (Figure 4A). It is plausible that the two processes are linked, and accelerated cell cycle of cells overexpressing ARGLU1 may be due to enhanced DNA damage repair as we observed significantly less endogenous DNA damage in the faster growing HT-A cells overexpressing ARGLU1 as compared to the parental HT1080 cells (Figure 4C).
Interestingly, in addition to many cell cycle regulators, overexpression of ARGLU1 enhanced expression of a number of proteins involved in DNA damage response (Table 1 and Supplementary Excel spreadsheet). It is possible that beyond enhancing the expression of these proteins, ARGLU1 promotes DNA repair by increasing promoter-proximal RNA polymerase II pausing, which would then allow cells to repair their damaged DNA more efficiently before resuming transcription. This is supported by our observations of enhanced RNA polymerase II pausing proximal to promoters when ARGLU1 is overexpressed (Figure 5A). This appears to be caused by blocking of JMJD6 binding to BRD4, which would relieve RNA polymerase II pausing (9). Instead, by binding to JMJD6, the BRD4 binding site on JMJD6 is occupied by ARGLU1, preventing this association. Indeed, we observed enhanced occupancy of the negative regulator of elongation, HEXIM1, and reduced occupancy of the positive regulator of elongation, JMJD6, on genes regulated by anti-pause enhancers (Figure 5F). In support of transcriptional role for ARGLU1, previous reports indicate that it associates with promoter-bound factors (3,6). BRD4 levels were not significantly changed at the promoter, but they were enhanced at the anti-pause enhancer, perhaps as a response to increased levels of paused polymerase (Figure 5F). Moreover, when we blocked BRD4 bromodomains with the inhibitor JQ1, we observed reduced DNA repair efficiency in ARGLU1-overexpressing HT-A cells (Figure 4F), suggesting that, at least in part, the enhanced DNA repair observed when ARGLU1 is overexpressed may be through BRD4. Potentially, this could occur via BRD4 being more available for DNA repair duties rather than transcriptional activities, as may happen when BRD4 is associated with JMJD6. Indeed, it was recently demonstrated that BRD4 plays an important role in transcriptionally-coupled DNA damage repair (59). Additionally, previous (3) mass spectrometry of ARGLU1-associated proteins identified numerous proteins involved in RNA polymerase II regulation and transcription (HEXIM1, POLR1E, SUPT4H1, PIAS2 and others) and proteins involved in DNA damage repair (RAD54L2, XRCC6, PIAS4, HMGB1 and others), supporting our findings and further solidifying a role for ARGLU1 in the DNA damage response pathway.
Previous studies have indicated that ARGLU1 may also play a role as a transcriptional activator (1,3), whereas we observed that on certain genes it functions as a repressor. Interestingly, our proteomics data shows that when ARGLU1 is overexpressed, more proteins are upregulated than downregulated (1599 up and 795 down), which correlates well with what was previously reported using transcriptomics under conditions of ARGLU1 knockdown (3). It is likely that transcriptional repression or activation by ARGLU1 will depend on the cellular and promoter context, and this is not uncommon for transcriptional regulators, such as Snail1 that, depending on the context, can be an activator or a repressor (60). Depending on this context, different factors may interact with ARGLU1 driving its activity as an activator or as a repressor. Interestingly, we observed that on RHOB and ALDOA promoters, ARGLU1 functions to repress transcription when it was overexpressed (Figure 5A), but it had no effect on transcription of PCNA or MCM4 (Figure 5B). Interestingly, expression of PCNA and MCM4 was reduced when ARGLU1 was knocked down outside of adenovirus infection (Figure 2G), suggesting an activating role for ARGLU1 on these two genes and indicating that repression is not a universal function of ARGLU1. ALDOA and RHOB both contain anti-pause enhancers (9), whereas we are not aware of those for PCNA and MCM4, suggesting that perhaps for genes regulated by anti-pause enhancers, ARGLU1 may function more as a repressor than an activator. In the context of DNA damage, which previous studies have not investigated, ARGLU1 may function more universally as a transcriptional repressor to enable more efficient DNA repair, as we have observed. Together, these findings suggest that ARGLU1 is a dual-function transcriptional regulator, that functions either as a repressor or an activator in a context-dependent manner. Future studies may shed light on the exact molecular mechanism governing this behaviour.
In the context of adenovirus infection the role of ARGLU1 is paradoxical. On one hand, ARGLU1 appears to promote the cell cycle while on the other it seems to inhibit virus growth and viral gene expression (Figures 3C, 2E and F). It would appear that targeting of ARGLU1 by E1A is intended to prevent RNA polymerase II pausing and enhance overall viral gene expression. Secondary effects associated with DNA repair and cell cycle may also play a role. While ARGLU1 knockdown seems to arrest uninfected cells (Figure 3D), the virus replicated better under these conditions (Figure 2E), suggesting that adenovirus bypasses any cell cycle arrest associated with ARGLU1 knockdown. Interestingly, one viral E1A mutant that does not bind to ARGLU1 (dl1102, deleting amino acids 26–35 in HAdV-C5 E1A289R) was previously observed to grow to greater titers and express some of its genes at higher levels than wt HAdV-C5 (43). It is unclear why loss of ARGLU1 binding by E1A would lead to higher viral gene expression, but one possibility is that E1A inadvertently brings ARGLU1 to viral promoters, driving transcriptional inhibition potentially by inhibiting RNA polymerase II elongation. Beyond this, co-immunoprecipitation experiments suggest that E1A slightly enhances the association between BRD4 and JMJD6 (Figure 5D), which would promote transcription (9), but it was unable to overcome ARGLU1-mediated block to the interaction between JMJD6 and BRD4. Nevertheless, our study suggests that ARGLU1 functions to repress transactivation from viral promoters as most viral genes express at significantly higher levels when ARGLU1 is knocked down and we also detected more paused RNA polymerase II on the viral E4 promoter when ARGLU1 was overexpressed.
In conclusion, our study further advanced our understanding of the role of ARGLU1 in the cell and during adenovirus infection. Specifically, we show that ARGLU1 is a target for adenovirus E1A from species B and C viruses and it plays a role as a negative regulator of viral gene expression and viral replication. Significantly, our study implicates ARGLU1 in regulation of RNA polymerase II promoter-proximal pausing and the repair of DNA damage in our experimental systems. Importantly, we show that ARGLU1 enhances resistance of a cancer cell line to genotoxic drugs which may have clinical implications. Lastly, we show that ARGLU1 interacts with JMJD6, preventing the association of JMJD6 with BRD4, potentially contributing to RNA polymerase II pausing and DNA repair. Despite significant advancements in our understanding of ARGLU1 presented here, much remains unknown about this poorly understood, yet important, protein. Critically, the role of ARGLU1 in DNA damage repair and cancer cell drug resistance may have important implications for patients.
Supplementary Material
Acknowledgements
LF was supported by the University of Manitoba Graduate Fellowship, RH was supported by a graduate scholarship from Research Manitoba. We are grateful to Dr Roger Greenberg for U2OS-2-6-3 and U2OS-2-6-5 cells, and Dr Jeremy Stark for U2OS-EJ5-GFP and U2OS-DR-GFP cells. The authors thank Dr Joe Mymryk for critical feedback on the manuscript and Dr Christine Zhang for technical assistance with flow cytometry. We also thank Fazia Ait Zenati for assistance with figure design. We are thankful to the anonymous reviewers for their high quality, constructive criticisms and comments that significantly improved our manuscript, and we are grateful to the editor for finding high quality reviewers. PP thanks Stanisława Pełka for invaluable support and assistance and Ryszard Pełka for inspiration and curiosity, and for their critical feedback on the manuscript.
Author contributions: SB: methodology, data analysis, visualization; NA: methodology, data analysis, visualization; LF: methodology, data analysis; DG: methodology, data analysis; JR: methodology, data analysis; RH: methodology, data analysis, visualization; PP: conceptualization, funding, methodology, data analysis, visualization, writing, revisions.
Contributor Information
Scott Bachus, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Nikolas Akkerman, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Lauren Fulham, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Drayson Graves, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Rafe Helwer, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Jordan Rempel, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Peter Pelka, Department of Microbiology, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada; Department of Medical Microbiology and Infectious Diseases, University of Manitoba, 45 Chancellor's Circle, Buller Building Room 427, Winnipeg, MB R3T 2N2, Canada.
Data availability
Proteomics data has been deposited into the PRIDE database under accession numbers: PXD046329 and PXD050223; and DOIs: 10.6019/PXD046329 and 10.6019/PXD050223, respectively.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Natural Sciences and Engineering Research Council of Canada [RGPIN/05366-19 to PP]; Canadian Institutes of Health Research [PJT-173376 to PP]. Funding for open access charge: Canadian Institutes of Health Research [PJT-173376].
Conflict of interest statement. None declared.
References
- 1. Zhang D., Jiang P., Xu Q., Zhang X Arginine and glutamate-rich 1 (ARGLU1) interacts with mediator subunit 1 (MED1) and is required for estrogen receptor-mediated gene transcription and breast cancer cell growth. J. Biol. Chem. 2011; 286:17746–17754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Portal D., Zhao B., Calderwood M.A., Sommermann T., Johannsen E., Kieff E. EBV nuclear antigen EBNALP dismisses transcription repressors NCoR and RBPJ from enhancers and EBNA2 increases NCoR-deficient RBPJ DNA binding. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:7808–7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Magomedova L., Tiefenbach J., Zilberman E., Le Billan F., Voisin V., Saikali M., Boivin V., Robitaille M., Gueroussov S., Irimia M. et al. ARGLU1 is a transcriptional coactivator and splicing regulator important for stress hormone signaling and development. Nucleic Acids Res. 2019; 47:2856–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao F., Huang S., Liu J., Tan C., Xu M., Wang D., Huang M., Zhu Y., Huang X., He S. Deletion of ARGLU1 causes global defects in alternative splicing in vivo and mouse cortical malformations primarily via apoptosis. Cell Death. Dis. 2023; 14:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li L., Han W., Chen Y., Chen Y. MiR-3613-3p inhibits hypertrophic scar formation by down-regulating arginine and glutamate-rich 1. Mol. Cell. Biochem. 2021; 476:1025–1036. [DOI] [PubMed] [Google Scholar]
- 6. Li F., Li J., Yu J., Pan T., Yu B., Sang Q., Dai W., Hou J., Yan C., Zang M. et al. Identification of ARGLU1 as a potential therapeutic target for gastric cancer based on genome-wide functional screening data. EBioMedicine. 2021; 69:103436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi J., Yu X., Li G., Zhao X., Chen J., Fang Y., Yang Y., Wang T., Xu T., Bian L. et al. DTL promotes head and neck squamous cell carcinoma progression by mediating the degradation of ARGLU1 to regulate the Notch signaling pathway. Int. J. Biol. Macromol. 2024; 259:129184. [DOI] [PubMed] [Google Scholar]
- 8. Wright P.E., Dyson H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015; 16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu W., Ma Q., Wong K., Li W., Ohgi K., Zhang J., Aggarwal A., Rosenfeld M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013; 155:1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Core L., Adelman K. Promoter-proximal pausing of RNA polymerase II: A nexus of gene regulation. Genes Dev. 2019; 33:960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van den Heuvel D., van der Weegen Y., Boer D.E.C., Ogi T., Luijsterburg M.S. Transcription-coupled DNA repair: from mechanism to human disorder. Trends Cell Biol. 2021; 31:359–371. [DOI] [PubMed] [Google Scholar]
- 12. Pelka P., Ablack J.N., Fonseca G.J., Yousef A.F., Mymryk J.S. Intrinsic structural disorder in adenovirus E1A: A viral molecular hub linking multiple diverse processes. J. Virol. 2008; 82:7252–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. King C.R., Zhang A., Tessier T.M., Gameiro S.F., Mymryk J.S. Hacking the cell: network intrusion and exploitation by adenovirus E1A. mBio. 2018; 9:e00390-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frost J.R., Mendez M., Soriano A.M., Crisostomo L., Olanubi O., Radko S., Pelka P. Adenovirus 5 E1A-mediated suppression of p53 via FUBP1. J. Virol. 2018; 92:e00439-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jung R., Radko S., Pelka P. The dual nature of Nek9 in adenovirus replication. J. Virol. 2015; 90:1931–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radko S., Jung R., Olanubi O., Pelka P. Effects of adenovirus type 5 E1A isoforms on viral replication in arrested human cells. PLoS One. 2015; 10:e0140124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frost J.R., Olanubi O., Cheng S.K., Soriano A., Crisostomo L., Lopez A., Pelka P. The interaction of adenovirus E1A with the mammalian protein Ku70/XRCC6. Virology. 2016; 500:11–21. [DOI] [PubMed] [Google Scholar]
- 18. Olanubi O., Frost J.R., Radko S., Pelka P. Suppression of type I interferon signaling by E1A via RuvBL1/pontin. J. Virol. 2017; 91:e02484-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crisostomo L., Soriano A.M., Mendez M., Graves D., Pelka P. Temporal dynamics of adenovirus 5 gene expression in normal human cells. PLoS One. 2019; 14:e0211192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berk A.J. Fields Virology. 2013; 6th edn.Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health. [Google Scholar]
- 21. MacNeil K.M., Dodge M.J., Evans A.M., Tessier T.M., Weinberg J.B., Mymryk J.S. Adenoviruses in medicine: innocuous pathogen, predator, or partner. Trends Mol. Med. 2023; 29:4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harlow E., Franza B.R. Jr, Schley C Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 1985; 55:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rahman S., Sowa M.E., Ottinger M., Smith J.A., Shi Y., Harper J.W., Howley P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011; 31:2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bachus S., Akkerman N., Fulham L., Graves D., Stephanson C., Memon H., Miller M.S., Pelka P. Adenovirus 5 vectors expressing SARS-CoV-2 proteins. mSphere. 2022; 7:e0099821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones N., Shenk T. Isolation of deletion and substitution mutants of adenovirus type 5. Cell. 1978; 13:181–188. [DOI] [PubMed] [Google Scholar]
- 26. Haley K.P., Overhauser J., Babiss L.E., Ginsberg H.S., Jones N.C. Transformation properties of type 5 adenovirus mutants that differentially express the E1A gene products. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:5734–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montell C., Fisher E.F., Caruthers M.H., Berk A.J. Resolving the functions of overlapping viral genes by site-specific mutagenesis at a mRNA splice site. Nature. 1982; 295:380–384. [DOI] [PubMed] [Google Scholar]
- 28. Radko S., Koleva M., James K.M., Jung R., Mymryk J.S., Pelka P. Adenovirus E1A targets the DREF nuclear factor to regulate virus gene expression, DNA replication, and growth. J. Virol. 2014; 88:13469–13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pelka P., Ablack J.N., Torchia J., Turnell A.S., Grand R.J., Mymryk J.S. Transcriptional control by adenovirus E1A conserved region 3 via p300/CBP. Nucleic Acids Res. 2009; 37:1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen G., Deng X. Cell synchronization by double thymidine block. Bio. Protoc. 2018; 8:e2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pelka P., Scime A., Mandalfino C., Joch M., Abdulla P., Whyte P. Adenovirus E1A proteins direct subcellular redistribution of Nek9, a NimA-related kinase. J. Cell. Physiol. 2007; 212:13–25. [DOI] [PubMed] [Google Scholar]
- 32. Babicki S., Arndt D., Marcu A., Liang Y., Grant J.R., Maciejewski A., Wishart D.S. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016; 44:W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morris J.H., Apeltsin L., Newman A.M., Baumbach J., Wittkop T., Su G., Bader G.D., Ferrin T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinf. 2011; 12:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gyori B.M., Venkatachalam G., Thiagarajan P.S., Hsu D., Clement M.V. OpenComet: an automated tool for comet assay image analysis. Redox. Biol. 2014; 2:457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shanbhag N.M., Greenberg R.A. The dynamics of DNA damage repair and transcription. Methods Mol. Biol. 2013; 1042:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian B., Yang J., Brasier A.R. Two-step cross-linking for analysis of protein-chromatin interactions. Methods Mol. Biol. 2012; 809:105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Zidek A., Potapenko A. et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ishida T., Kinoshita K. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007; 35:W460–W464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konuma T., Yu D., Zhao C., Ju Y., Sharma R., Ren C., Zhang Q., Zhou M.M., Zeng L. Structural mechanism of the oxygenase JMJD6 recognition by the extraterminal (ET) domain of BRD4. Sci. Rep. 2017; 7:16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Graves D., Akkerman N., Bachus S., Pelka P Differential splicing of human adenovirus 5 E1A RNA expressed in cis versus in trans. J. Virol. 2021; 95:e02081-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boyer T.G., Martin M.E., Lees E., Ricciardi R.P., Berk A.J. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature. 1999; 399:276–279. [DOI] [PubMed] [Google Scholar]
- 43. Costa R., Akkerman N., Graves D., Crisostomo L., Bachus S., Pelka P. Characterization of adenovirus 5 E1A exon 1 deletion mutants in the viral replicative cycle. Viruses. 2020; 12:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pelka P., Miller M.S., Cecchini M., Yousef A.F., Bowdish D.M., Dick F., Whyte P., Mymryk J.S. Adenovirus E1A directly targets the E2F/DP-1 complex. J. Virol. 2011; 85:8841–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dorr R.T. Bleomycin pharmacology: mechanism of action and resistance, and clinical pharmacokinetics. Semin. Oncol. 1992; 19:3–8. [PubMed] [Google Scholar]
- 46. Lu D.F., Wang Y.S., Li C., Wei G.J., Chen R., Dong D.M., Yao M. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int. J. Clin. Exp. Med. 2015; 8:1904–1911. [PMC free article] [PubMed] [Google Scholar]
- 47. Borovinskaya M.A., Shoji S., Fredrick K., Cate J.H. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA. 2008; 14:1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barrows J.K., Lin B., Quaas C.E., Fullbright G., Wallace E.N., Long D.T. BRD4 promotes resection and homology-directed repair of DNA double-strand breaks. Nat. Commun. 2022; 13:3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gunn A., Stark J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012; 920:379–391. [DOI] [PubMed] [Google Scholar]
- 50. Gunn A., Bennardo N., Cheng A., Stark J.M. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J. Biol. Chem. 2011; 286:42470–42482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Janicki S.M., Tsukamoto T., Salghetti S.E., Tansey W.P., Sachidanandam R., Prasanth K.V., Ried T., Shav-Tal Y., Bertrand E., Singer R.H. et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004; 116:683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee K.B., Wang D., Lippard S.J., Sharp P.A. Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakazawa Y., Hara Y., Oka Y., Komine O., van den Heuvel D., Guo C., Daigaku Y., Isono M., He Y., Shimada M. et al. Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell. 2020; 180:1228–1244. [DOI] [PubMed] [Google Scholar]
- 54. Fousteri M., Mullenders L.H. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008; 18:73–84. [DOI] [PubMed] [Google Scholar]
- 55. van Hoffen A., Natarajan A.T., Mayne L.V., van Zeeland A.A., Mullenders L.H., Venema J. Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res. 1993; 21:5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ui A., Chiba N., Yasui A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020; 111:1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park S.J., Yoon B.H., Kim S.K., Kim S.Y. GENT2: an updated gene expression database for normal and tumor tissues. BMC Med Genomics. 2019; 12:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salnikov M., Gameiro S.F., Zeng P.Y.F., Barrett J.W., Nichols A.C., Mymryk J.S. The HPV induced cancer resource (THInCR): a suite of tools for investigating HPV-dependent human carcinogenesis. mSphere. 2022; 7:e0031722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lam F.C., Kong Y.W., Huang Q., Vu Han T.L., Maffa A.D., Kasper E.M., Yaffe M.B. BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat. Commun. 2020; 11:4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcia de Herreros A. Dual role of Snail1 as transcriptional repressor and activator. Biochim. Biophys. Acta Rev. Cancer. 2024; 1879:189037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data has been deposited into the PRIDE database under accession numbers: PXD046329 and PXD050223; and DOIs: 10.6019/PXD046329 and 10.6019/PXD050223, respectively.