


Abstract
Coronaviruses are a diverse subfamily of viruses containing pathogens of humans and animals. This subfamily of viruses replicates their RNA genomes using a core polymerase complex composed of viral non-structural proteins: nsp7, nsp8 and nsp12. Most of our understanding of coronavirus molecular biology comes from betacoronaviruses like SARS-CoV and SARS-CoV-2, the latter of which is the causative agent of COVID-19. In contrast, members of the alphacoronavirus genus are relatively understudied despite their importance in human and animal health. Here we have used cryo-electron microscopy to determine structures of the alphacoronavirus porcine epidemic diarrhea virus (PEDV) core polymerase complex bound to RNA. One structure shows an unexpected nsp8 stoichiometry despite remaining bound to RNA. Biochemical analysis shows that the N-terminal extension of one nsp8 is not required for in vitro RNA synthesis for alpha- and betacoronaviruses. Our work demonstrates the importance of studying diverse coronaviruses in revealing aspects of coronavirus replication and identifying areas of conservation to be targeted by antiviral drugs.
Graphical Abstract
Graphical Abstract.

Introduction
The coronavirus (CoV) subfamily is composed of four genera: the alpha-, beta-, gamma- and deltaCoVs. Across all these genera there are diverse human and animal pathogens that cause a wide range of disease severities (1). Since 2002, three betaCoVs have emerged from animal reservoirs and caused human epidemics or pandemics: SARS-CoV, MERS-CoV and SARS-CoV-2 (2–5). The most recent, SARS-CoV-2, emerged in 2019 and is the causative agent of the COVID-19 pandemic (5,6). There are four endemic common-cold causing human CoVs: of these there are two alphaCoVs, HuCoV-229E and HuCoV-NL63, and two betaCoVs, HuCoV-HKU1 and HuCoV-OC43 (1). In 2021, a recombined feline/canine alphaCoV, named CCoV-HuPn-2018, was identified in samples from human patients with pneumonia in Malaysia and the United States (7–9). CCoV-HuPn-2018 is the only recorded human emergent alphaCoV in recent years and exemplifies the persistent threat of alphaCoV spillover. In the alphaCoV genus there are several porcine pathogens including transmissible gastroenteritis virus (TGEV), swine enteric alphacoronavirus (SeACoV), and porcine epidemic diarrhea virus (PEDV) (10,11). Since 2010, PEDV has had detrimental impacts on the global swine industry, with outbreaks having 70–100% unweaned piglet fatality rates at swine farms despite vaccination efforts (12–14).
CoVs are positive-sense, single-stranded RNA viruses with large, ∼30 kb genomes (1,15). To replicate their genomes, CoVs encode an RNA-dependent RNA polymerase (RdRP), termed non-structural protein 12 (nsp12) (16–18). Nsp12 also contains the nidovirus RdRP-associated nucleotidyltransferase (NiRAN) domain at its N terminus, which has been shown to be involved in mRNA capping (19,20). In vitro studies of betaCoVs have shown that viral replication factors nsp7 and nsp8 interact with nsp12 to form an active and processive polymerase complex (21). The SARS-CoV-2 complex is the fastest known RdRP with nucleotide addition rates up to 170 nt/s in vitro (22). The first structure of a CoV polymerase complex from the betaCoV, SARS-CoV revealed a subunit stoichiometry of one nsp12, two nsp8, and one nsp7 where nsp7 and one nsp8 form a heterodimer (19). The nsp8 that directly interacts with nsp12 is denoted as nsp8F (fingers), and the nsp8 that interacts with nsp7 and nsp12 is denoted as nsp8T (thumb). In this paper we refer to this complex of nsp12, nsp7 and nsp8 as the CoV core polymerase complex.
Recent SARS-CoV-2 core polymerase structures have provided key insights into betaCoV replication. Nearly all structures of SARS-CoV-2 complexes shown to be enzymatically active or bind RNA in vitro had the same 1:2:1 nsp7:nsp8:nsp12 stoichiometry as SARS-CoV (23,24). An exception is a cryo-EM structure of the SARS-CoV-2 polymerase complex stalled by remdesivir that remained bound to RNA but lacked any density for nsp8T (25). Several SARS-CoV-2 polymerase structures determined with longer dsRNA substrates have shown each nsp8’s N-terminal extension bound to upstream dsRNA as it exits the polymerase active site, which has been hypothesized to promote processivity through a ‘sliding-pole’ mechanism (23,24). Addition of the viral RNA helicase, nsp13, to core polymerase complexes showed that each nsp8 subunit independently scaffolds a nsp13 (two nsp13s bind a single core complex) (20,26,27). The nsp13 associated with nsp8T was shown to be capable of binding to the 5′ end of template RNA prior to the RNA entering the polymerase active site (27). Nsp13’s association with the template RNA produced a conundrum, where nsp12 and nsp13 are oriented such that the polymerase and helicase translocate in opposite directions. Further biochemical and structural work demonstrated that this nsp13 can stimulate template backtracking of the polymerase, during which the 3′ end of nascent RNA extrudes through the nsp12 NTP channel (28). While this nsp13 has been implicated in backtracking, the triggers and regulation of backtracking are unknown.
To date, there is limited knowledge of CoV polymerase biochemistry and structural biology outside the sarbecovirus subgenus (SARS-CoV and SARS-CoV-2). To our knowledge, in vitro demonstration of robust nsp12 polymerase activity has yet to be demonstrated for an alpha-, gamma- or deltaCoV. Similarly, outside of betaCoVs there is limited structural and biochemical information for nsp7 and nsp8. A crystal structure of the alphaCoV feline coronavirus (FCoV) nsp7–nsp8 complex revealed that alpha- and betaCoV nsp7s and the nsp8 head domains have high structural homology (29). Previous native mass spectrometry experiments of alpha- and betaCoV nsp7–nsp8 complexes determined that each genera's cofactors form homo- and hetero-oligomers, although the oligomers sampled by each genus varied in cofactor ratio and/or prevalence of particular oligomers (30). Across CoV genera there is sequence conservation (>40%) of the nsps involved in RNA synthesis (i.e. nsp7, nsp8 and nsp12) (31). Sequence and available structural homology suggest shared mechanisms among these viruses to replicate their RNA genomes. A recent publication showed that SARS-CoV-2 replication complexes assembled with either a truncated nsp8F or nsp8T, lacking their respective RNA binding domains, were capable of processive RNA synthesis similar to the wildtype complex (32). These results challenge the aforementioned ‘sliding-pole’ hypothesis, further displaying the need for more biochemical and structural characterization of CoV polymerases and their replication factors.
Though our knowledge of SARS-CoV and SARS-CoV-2 replication mechanics has improved over the last two decades, the narrow structural biology focus on two closely related betaCoVs limits our understanding of CoV replication across the virus subfamily. To address this gap, we studied the polymerase core complex of the alphaCoV PEDV using biochemistry and structural biology. Our structure of the PEDV core polymerase complex bound to an RNA primer-template pair without nsp8T shows the lability of nsp8T to participate in this complex and additional mutagenesis and biochemistry demonstrate that the nsp8T N-terminal helical extension is not necessary for either alpha- or betaCoV RNA synthesis activity in vitro. The identification of conserved mechanisms and structural motifs between alpha- and betaCoVs will allow for the development of broadly acting CoV therapeutic strategies.
Materials and methods
Expression construct design
DNA encoding PEDV and SARS-CoV-2 nsps were codon optimized and synthesized (Genscript). PEDV protein sequences correspond to GenBank AKJ21892.1. SARS-CoV-2 protein sequences correspond to GenBank UHD90671.1. PEDV nsp7 and nsp8 genes were cloned into pET46 and pET45b expression vectors, respectively. PEDV nsp7 was cloned with a C-terminal TEV protease site and hexahistidine tag. PEDV nsp8 has an N-terminal hexahistidine tag and TEV protease site. SARS-CoV-2 nsp7 and nsp8 were both cloned into pET46 with N-terminal hexahistidine tags, and enterokinase and TEV protease sites. Both PEDV and SARS-CoV-2 nsp12 genes were cloned into pFastBac with C-terminal TEV protease sites and double Strep II tags.
Expression plasmids for nsp12 point mutants were produced using mutagenesis on the pFastBac plasmids. The nsp8-nsp7 (nsp8L7) fusion constructs were produced by overlap PCR creating a GSGSGS peptide linker between nsp8 and nsp7 and inserted into pET46 expression vectors with N-terminal hexahistidine tags, and enterokinase and TEV protease sites. Truncation constructs were produced using Kinase-Ligase-DpnI (KLD) cloning. All open reading frames on the DNA plasmids were verified by Sanger sequencing.
Recombinant protein expression
Recombinant nsp7 and nsp8: Replication factor proteins were expressed in Rosetta 2pLysS E. coli cells (Novagen). Cultures were grown at 37°C and induced at an OD600 of 0.6–0.8 with isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 500 μM. After growing for 16 h at 16°C, cells were harvested by centrifugation and resuspended in replication factor wash buffer (10 mM Tris–Cl pH 8.0, 300 mM sodium chloride, 30 mM imidazole and 2 mM dithiothreitol (DTT)). Cells were lysed in a microfluidizer (Microfluidics) and lysates cleared via centrifugation. Replication factors were purified using Ni-NTA agarose beads (Qiagen), eluting with 300 mM imidazole. Eluted protein was digested with 1% (w/w) TEV protease overnight while dialyzing (10 mM Tris–Cl pH 8.0, 300 mM sodium chloride and 2 mM DTT) at 4°C. Digested protein was flowed back over Ni-NTA agarose beads to remove undigested protein, and further purified using a Superdex 200 Increase 10/300 GL column (Cytiva) in 25 mM Tris–Cl pH 8.0, 300 mM sodium chloride and 2 mM DTT. Fractions containing the protein of interest were concentrated using ultrafiltration. Concentrated protein was aliquoted, flash frozen in liquid nitrogen, and stored at −80°C. Replication factor protein yields for 1 L of cells was 10–40 mg.
Recombinant nsp12s: pFastBac plasmids carrying the nsp12 gene and DH10Bac Escherichia coli (Life Technologies) were used to produce recombinant bacmids for each gene. Bacmids were transfected into Sf9 cells (Expression Systems) with Cellfectin II (Life Technologies) to produce recombinant baculoviruses, which were twice amplified using Sf9 cells. Amplified baculoviruses were used to infect Sf21 cells (Expression Systems) for protein expression. After two days of incubation at 27°C, cells were collected via centrifugation and pellets resuspended in wash buffer (25 mM HEPES pH 7.4, 300 mM sodium chloride, 1 mM magnesium chloride, and 5 mM DTT) with an added 143 μl of BioLock (IBA) per 1 L of culture. Cells were lysed using a microfluidizer (Microfluidics) and lysates cleared via centrifugation. Protein was affinity purified using Streptactin superflow agarose (IBA) and eluted with wash buffer that contained 2.5 mM desthiobiotin. Protein was further purified via size exclusion chromatography with a Superdex 200 Increase 10/300 GL column (Cytiva) in 25 mM HEPES pH 7.4, 300 mM sodium chloride, 100 μM magnesium chloride, and 2 mM tris(2-carboxyethyl)phosphine (TCEP). Fractions containing nsp12 were pooled and concentrated using ultrafiltration. Concentrated protein was aliquoted, flash frozen in liquid nitrogen and stored at −80°C. Average protein yield for 1 L of culture was 3–5 mg.
Preparation of RNA substrates
RNA oligos were purchased from Integrated DNA Technologies. Primer RNAs were modified with a 5′ fluorescein to monitor the RNA by gel electrophoresis.
Pair 1:
Primer RNA: CAUUCUCCUAAGAAGCUAUUAAAAUCACA
Template RNA: AAAAAGGGUUGUGAUUUUAAUAGCUUCUUAGGAGAAUG
Pair 2:
Primer RNA: CAUUCUCCUAAGAAGCUAUUAAAUCACAGAUU
Template RNA: CAGUGUCAUGGAAAAACAGAAAAAUCUGUGAUUUUAAUAGCUUCUUAGGAGAAUG
RNA template was always held in slight excess of primer (primer:template ratio of 1:1.2). RNA oligos were annealed in 2.5 mM HEPES pH 7.4, 2.5 mM potassium chloride and 0.5 mM magnesium chloride and heated at 95°C for 5 min, then allowed to cool slowly back to 25°C for 75 min before either being used immediately or stored at −20°C.
Native mass spectrometry
Component concentrations were, unless stated otherwise, 10.4 μM nsp12, 20.8 μM nsp7, 31.2 μM nsp8 (or nsp8-nsp7 fusion), and 12.5 μM RNA duplex (Pair 1). Proteins were combined in native-MS buffer (10 mM Tris–Cl pH 8.0, 100 mM ammonium acetate, 2 mM magnesium chloride, and 1 mM DTT) and incubated at 25°C for 15 min. RNA duplex was then added, and reactions were incubated at 25°C for an additional 15 min. SARS-CoV-2 T853R and PEDV V848R complexes (homologous mutations) were prepared at half the normal protein and RNA concentrations due to low yields of the mutant nsp12 proteins.
All samples were buffer exchanged into 100 mM ammonium acetate (NH4OAc) with 2 mM MgCl2. First, 350 μl 100 mM NH4OAc with 2 mM MgCl2 was added to a 100 kDa molecular weight cut-off Amicon Ultra-0.5 Centrifugal Filter Unit (Millipore Sigma), followed by 15 μl of ∼10 μM polymerase sample. The samples were centrifuged at 14 000 × g for 10 min to load the sample, followed by two rinses with 400 μl of the NH4OAc/MgCl2 solution, also centrifuged at 14 000 × g for 10 min. Finally, the filter was inverted into a new catch tube and centrifuged at 1000 × g for 2 min to recover the filtered and concentrated sample. Samples were diluted using the same NH4OAc/MgCl2 solution to roughly 1–5 μM for introduction to the mass spectrometer via electrospray ionization.
Standard wall borosilicate tubing (1.20 mm o.d., 0.69 mm i.d., Sutter Instrument) was pulled using a P-1000 Micropipette Puller (Sutter Instrument) to a tapered tip of ∼3–5 μm diameter. Each sample was loaded into a pulled glass capillary and 1.1–1.3 kV was applied to the sample using a platinum wire inserted into the back of the capillary. Charged droplets entered a Q Exactive UHMR Hybrid Quadrupole Orbitrap Mass Spectrometer (ThermoFisher Scientific) via a heated inlet capillary. The heated capillary was set to 200–250°C to minimize solvent adduction to analyte ions. Additional removal of adducts was accomplished using in-source trapping with a range of injection voltages typically between 150 and 300 V. The voltage was tuned for each sample to maximize adduct removal with minimal dissociation of the ionized complex. For these experiments the vacuum was set to a value of 10 resulting in a pressure readout of 2e-4 mbar and the S-lens radiofrequency level was set to a value of 200. All reported spectra are an average of 50 scans collected with the Orbitrap mass analyzer at a resolution setting of 6250. Charge states were assigned manually by minimizing the standard deviation of masses calculated by peaks within a single charge state distribution. The average mass and standard deviation are reported for major distributions in each spectrum.
In vitro primer extension assay
Assay conditions were 10 mM Tris–Cl pH 8.0, 10 mM sodium chloride, 2 mM magnesium chloride, and 1 mM DTT with a typical reaction volume of 20 μl. Protein final concentrations were 500 nM nsp12, 1.5 μM nsp7 and 1.5 μM nsp8 (nsp8-nsp7 fusion and truncation proteins were also at 1.5 μM). Duplex RNA (Pair 1) final concentration was 250 nM. Prior to use, proteins were diluted in assay buffer. Diluted proteins were then combined and incubated at 25°C for 15 min, duplex RNA was added and reactions incubated at 25°C for an additional 15 min. Reactions were initiated by the addition of NTPs to a final concentration of 40 μM and reactions ran for 1 min (at 25°C for SARS-CoV-2 polymerases, or 30°C for PEDV polymerases) before being halted by addition of two volumes of sample loading buffer (95% (v/v) formamide, 2 mM ethylenediaminetetraacetic acid (EDTA) and 0.75 mM bromophenol blue). Samples were heated at 95°C then analyzed using denaturing urea-PAGE (8 M urea, 15% polyacrylamide) run in 1× TBE (89 mM Tris–Cl pH 8.3, 89 mM boric acid, 2 mM EDTA). Gels were imaged using a Typhoon FLA 9000 (GE Healthcare) to identify fluorescein signals. Extension was quantified using ImageJ (33).
Sample preparation and grid freezing for CryoEM
For the complex lacking nsp8T:
PEDV polymerase complexes were prepared at a total protein concentration of 1 mg/ml with a ratio of 2:2:1:1.2 nsp7:nsp8:nsp12:RNA duplex (Pair 1). The complex was assembled in 25 mM HEPES pH 7.5, 50 mM sodium chloride, 2 mM magnesium chloride and 2 mM DTT. Proteins were diluted in buffer then immediately combined and incubated at 25°C for 15 min before RNA was added and incubated at 25°C for another 15 min. Samples were stored on ice prior to grid freezing.
Samples were prepared for structural analysis using UltraAuFoil R1.2/1.3 300 mesh grids (Quantifoil) and a Vitrobot Mark IV (ThermoFisher Scientific). Grids were freshly glow discharged using a GloQube Plus (Quorom) for 20 seconds with a current of 20 mA in an air atmosphere, creating a negative charge on the grid surface. Immediately before applying sample to grids, 0.5 μl N-dodecyl-β-d-maltoside (DDM) was added to samples at a final concentration of 60 μM. 3.5 μl of sample was spotted onto grids before double-sided blotting and plunge freezing in liquid ethane. Samples were blotted for 4 s at a blot force of −15 with chamber conditions of 100% humidity and 4°C.
For the complete complex with nsp8T:
PEDV polymerase complex were prepared at a total protein concentration of 1 mg/ml with a ratio of 2.2:2.2:1:1.2 nsp7:nsp8:nsp12:RNA duplex (Pair 2). The complex was assembled using the same buffer and protocol as above.
Samples were frozen on Quantifoil R1.2/1.3 200 mesh grids using a Vitrobot Mark IV (ThermoFisher Scientific). Grids were freshly glow discharged as described above. Right before blotting, 0.5 μl of 3-([3-cholamidopropyl] dimethylammonio)-2-hydroxy-1-propanesulfonate (CHAPSO) was added to samples at a final concentration of 6 mM. 3 μl of sample was spotted onto grids before double-sided blotting for 8 s at a blot force of −6 with chamber conditions of 100% humidity and 4°C. Following blotting, samples were plunge frozen in liquid ethane.
Cryo-EM data collection, processing and model building
For the complex lacking nsp8T:
SerialEM was used for data collection on a Titan Krios 300 keV transmission electron microscope (ThermoFisher Scientific) (34). Movies were collected on a K3 direct electron detector (Gatan) in CDS mode with a GIF quantum energy filter slit width of 20 eV and a stage tilt of 25°. Data was collected at a magnification of 105 000×, a pixel size of 0.834 Å, and a defocus range of −0.75 to −1.75 μm at a step size of 0.5 μm.
Data processing was performed using cryoSPARC v3.3.1 (35). After patch motion correction and patch CTF estimation, particle picking was performed using blob picker with a diameter of 125–175 Å and a minimum separation between particles of 12.5 Å. Particles were extracted with a box size of 256 pixels and subjected to 2D classification with a window radius of 0.33 – 0.66 for 100 classes with 40 online-EM iterations and a batch size of 1000. Force max over poses/shifts was turned off to improve visualization of the complex during 2D classification. Three initial models were created ab-initio and further used for heterogenous refinement. Particles from two maps that looked like polymerase complexes were pooled and used for non-uniform refinement under default parameters. The 3D reconstruction was used for 3D variability analysis (36) and further 3D classification before a final non-uniform refinement and 3D reconstruction (Supplementary Table S1, Supplementary Figures S1 and S2). Any 3D jobs run in cryoSPARC were done under default parameters unless noted otherwise.
To build the PEDV polymerase complex coordinate model, a SARS-CoV-2 polymerase complex structure (PDB ID: 7CYQ) was docked into the cryo-EM map in Coot and regions of the protein (including whole chains of nsp8, nsp9, and nsp13) and RNA that lacked map density were removed (20,37). Mutation of protein chains to PEDV sequences, further model building, and validation was performed in Coot. The PEDV model was refined using real space refinement in Phenix (38). Refinement in Phenix and model building/validation in Coot was an iterative process to produce a coordinate model. Final model adjustments were made with ISOLDE (39).
For the complete complex with nsp8T
EPU was used for data collection on a Talos Arctica 200 keV transmission electron microscope (ThermoFisher Scientific). Movies were collected on a K3 direct electron detector (Gatan) in CDS mode with a GIF quantum energy filter slit width of 20 eV and no stage tilt. Data was collected at a magnification of 79 000×, a pixel size of 1.064 Å, and a defocus range −0.5 to −2.0 μm at a step size of 0.5 μm (Supplementary Table S1).
Data was processed using cryoSPARC v4.1.0 (35). Motion correction, CTF estimation, particle picking, particles extraction, 2D classification, ab-initio model generation, 3D classification, and non-uniform refinement were done using matching parameters as above to produce the final 3D reconstruction (Supplementary Table S1, Supplementary Figures S1 and S2). No 3D variability analysis was used in producing this 3D reconstruction.
To build the coordinate model, our initial PEDV model was docked into the cryo-EM map in ChimeraX, then we superimposed a SARS-CoV-2 polymerase complex structure (7KRP) with our PEDV model using the Matchmaker function in ChimeraX (28,40). Regions except for nsp8T and the nsp8F N-terminal extension were deleted from the SARS-CoV-2 model. The reduced model was saved in relation to our PEDV model in space. In Coot our PEDV model and the SARS-CoV-2 nsp8 coordinates were merged, nsp8 protein chains were mutated to PEDV sequences, and initial refinement was performed (37). Refinement in Phenix and model building/validation in Coot was an iterative process to produce a coordinate model, the final adjustments for which were made with ISOLDE (38,39).
Figure making
Images of protein models and maps used for figures were made in ChimeraX (40). Bar graphs were produced in Microsoft Excel. Figures were assembled in Adobe Illustrator.
Results
Assembly of an active PEDV polymerase complex
To study the PEDV polymerase complex we recombinantly expressed and purified nsp7, nsp8, and nsp12 (Supplementary Figure S3). Assembly of these proteins with a short RNA duplex revealed a complex weight of 185 120 Da (+/- 70 Da) by native mass spectrometry (Table 1, Supplementary Figure S4). This mass is equivalent to one nsp12, one nsp7, two nsp8s and one RNA duplex. Using an in vitro primer extension assay we saw polymerase activity in the presence of all three nsps (Figure 1A and Supplementary Figure S5). The necessity of nsp7, nsp8 and nsp12 for robust primer extension, and the observed nsp binding stoichiometry are shared between PEDV, SARS-CoV and SARS-CoV-2 (17,19,21,23).
Table 1.
Native mass spectrometry of coronavirus polymerase complexes
| Sample (expected mass in Da) | Major peak(s) (Da) | Major complex(es): | Minor peak(s) (Da) | Minor complex(es): |
|---|---|---|---|---|
| PEDV nsp12 + 8 + 7 + RNA (184 293) | 185 120 ± 70 | Intact polymerase complex | 109 450 ± 40 | Free nsp12 |
| PEDV nsp12 + 8 + RNA (152 529) | 109 600 ± 30 | Free nsp12 | None | None |
| PEDV nsp12-A382R + 8 + 7 + RNA (184 293) | 109 533 ± 60 185 120 ± 30 | Free nsp12, Intact polymerase complex | 218 880 ± 50 | Dimeric nsp12 |
| PEDV nsp12-V848R + 8 + 7 + RNA (184 293) | 185 200 ± 200 | Intact polymerase complex | 370 700 ± 600 109 630 ± 200 | Dimeric polymerase complex, free nsp12 |
| PEDV nsp12 + 8 + 8L7 + RNA (183 913) | 184 670 ± 60 | Intact polymerase complex | 109 420 ± 30 | Free nsp12 |
| SARS-CoV-2 nsp12 + 8 + 7 + RNA (185 245) | 186 240 ± 70 | Intact polymerase complex | 110 680 ± 20 | Free nsp12 |
| SARS-CoV-2 nsp12 + 8 + RNA (154 011) | 110 870 ± 90 | Free nsp12 | 221 320 ± 40 | Dimeric nsp12 |
| SARS-CoV-2 nsp12-L387R + 8 + 7 + RNA (185 245) | 186 400 ± 100 | Intact polymerase complex | 110 900 ± 100 22 400 ± 100 | Free nsp12, excess nsp8 and/or RNA |
| SARS-CoV-2 nsp12-T853R + 8 + 7 + RNA (185 245) | 186 190 ± 50 | Intact polymerase complex | 22 300 ± 50 | Excess nsp8 and/or RNA |
| SARS-CoV-2 nsp12 + 8 + 8L7 + RNA (185 603) | 186 270 ± 40 | Intact polymerase complex | 110 600 ± 40 | Free nsp12 |
Major and minor species from native mass spectrometry experiments are listed for each complex tested with their respective (±) standard deviations. The complexes best explained by the determined masses are listed next to each mass. Intact polymerase complexes are composed of one nsp7, two nsp8s, one nsp12, and one RNA duplex. Individual protein or RNA molecular weight are as follows for PEDV: 21 962 Da – RNA, 10 076 Da – nsp7, 21 688 Da – nsp8, 108 879 Da – nsp12, 31 384 Da – nsp8L7. Molecular weights for SARS-CoV-2 are as follows: 21 962 Da – RNA, 9296 Da – nsp7, 21 938 Da – nsp8, 110 111 Da – nsp12, 31 592 Da – nsp8L7. (See also Supplementary Table S2 and Supplementary Figure S4). The average and standard deviation for each complex were calculated from assigned spectral peaks in observed charge state distributions, where each spectrum is an average of 50 individual spectra.
Figure 1.

Assembly of an active PEDV polymerase complex. A 29 nt RNA primer with a 5′ fluorophore is annealed to a 38 nt template and extended in the presence of CoV polymerase complexes. Combinations of nsp7, nsp8, and nsp12 were tested for PEDV (A) and SARS-CoV-2 (B). (C, E) 3.3 and 3.4 Å cryo-EM reconstruction of the PEDV core polymerase complex with (E) and without (C) nsp8T, respectively. (D, F) Coordinate models of the PEDV core polymerase complexes docked into their corresponding electron density maps colored by chain.
Structure of PEDV core polymerase complex
Our initial attempts at cryo-EM structure determination using a short RNA and DDM yielded a model of the PEDV core polymerase complex (nsp7, nsp8, nsp12) bound to a short RNA primer-template pair in a post-translocated state (Figure 1C and D). Our final map has a resolution of 3.3 Å (Supplementary Table S1, Supplementary Figure S1). This cryo-EM map contains density for most of nsp12, including the NiRAN and polymerase domains. We resolved most of nsp7 (residues 2–62), the C-terminal portion of nsp8F (residues 79–192), and a full turn of dsRNA exiting the polymerase active site. We lack well-resolved density for the nsp8F N-terminal helical extension. 3D variability analysis showed that the dsRNA leaving the active site has flexibility, likely leading to the lack of density in the final reconstruction (Supplementary Video S1) (36). Unexpectedly, this reconstruction lacked any density for nsp8T, differing with most RNA-bound CoV polymerase structures as well as the PEDV polymerase native mass spectrometry data described above. To obtain the structure of the intact complex observed by native mass spectrometry we screened different detergents and grid preparation strategies. We found that the combined addition of the detergent CHAPSO and a longer RNA substrate promoted complex stability and allowed the determination of the complete PEDV core polymerase complex structure. Our more complete map has a resolution of 3.4 Å and lacks orientation bias. Importantly, in addition to previously resolved protein and RNA regions, the improved map has density for the N-terminal extension of nsp8F and most of nsp8T which was previously absent entirely.
Comparison of PEDV and SARS-CoV-2 polymerase core complex models
PEDV and SARS-CoV-2 nsp7, nsp8 and nsp12 protein sequences have significant sequence identity (nsp12 58.6%, nsp8 43.1% and nsp7 41.8%) (Supplementary Figure S6). The overall architecture of the PEDV polymerase core complex is very similar to published SARS-CoV-2 models with a nsp12 RMSD value of 0.964 Å (Figure 2A, Supplementary Figure S7) (28). Comparison of our PEDV models’ RdRP and NiRAN active sites with SARS-CoV-2 structures revealed highly conserved active site structures (Supplementary Figure S7). This structural conservation suggests that antivirals targeting SARS-CoV-2, such as Remdesivir or the recently studied dual-purpose AT-527, could be effective against PEDV and other alphaCoVs (41–43).
Figure 2.

Structural differences between PEDV and SARS-CoV-2 nsp12s. (A) Overlay of SARS-CoV-2 (PDB: 7KRP) and PEDV (8URB) nsp12. The black box highlights the loop region depicted in (B). (B) PEDV nsp12 loop (residues 249–268) buries extra surface area on nsp8F compared to SARS-CoV-2 (PDB: 7KRP). (C) PEDV nsp12 841–849 lacks helical definition in the absence of nsp8T, while it has a short helix when the nsp8T N-terminal extension is present. For each panel except (C) PEDV and SARS-CoV-2 models are superimposed with PEDV being colored in darker shades. In (C), nsp12 from our complex without nsp8T is colored in grey. Superimposition of models was achieved using the Matchmaker function in ChimeraX (40).
A major difference in the PEDV polymerase models is an altered loop conformation (PEDV nsp12 residues 249–268) that binds the nsp8F head domain in PEDV but not in SARS-CoV-2 complexes (Figure 2B). This conformational difference increases nsp8F’s buried surface area by 115 Å2 in the PEDV polymerase core complex. This PEDV nsp12 loop sequence is well conserved within alphaCoV but not across CoV genera (Supplementary Figure S6). Based on this conservation, we predict that the observed PEDV nsp12 loop conformation to interact with nsp8F is shared among and specific to alphaCoVs.
A small difference between the two PEDV models is that in the model without nsp8T the PEDV nsp12 residues 841–849 lack helical definition that is present in the complete core complex reconstruction (Figure 2C). Helical definition in this region is also observed in SARS-CoV-2 models with resolved nsp8T N-terminal helical extensions (PDB IDs: 6YYT, 7KRN, 7KRO, 6XEZ), but not in SARS-CoV (PDB ID: 6NUR) and SARS-CoV-2 nsp12 structures (PDB IDs: 7CYQ, 7CXM) without well resolved nsp8T extensions region suggesting that association of the nsp8T N-terminal helical region promotes helical secondary structure in this nsp12 region.
A PEDV nsp12 structure model bound to RNA without nsp8T
Missing nsp8T density in our initial reconstructed map was unexpected. As previously mentioned, there is a cryoEM structure of a betaCoV polymerase complex without nsp8T that could bind RNA, so while our missing nsp8T was surprising, it is not unprecedented (25). Prior structural work for betaCoV polymerases has proposed that both nps8T and nsp8F participate in CoV RNA synthesis, acting as ‘sliding-poles’ that guide dsRNA out of the polymerase active site (23). Our native mass spectrometry results also contrast this PEDV polymerase structural model's stoichiometry (Table 1). These observations led us to hypothesize that nsp8T’s interactions with the polymerase complex are weaker than other replication factors (nsp8F and nsp7), and perhaps the vitrification process during grid preparation caused nsp8T disassociation. Despite the lability of nsp8T during grid freezing, the PEDV nsp12 polymerase remained bound to nsp7, nsp8F, and primer/template RNA in contravention to proposed models for polymerase complex assembly and activity (23). We therefore hypothesized that nsp8T may not be required for CoV polymerase RNA binding and/or RNA synthesis.
Prior studies have shown that MERS-CoV and SARS-CoV-2 nsp12 and nsp8 complexes can extend RNA in vitro (44,45). As the association of nsp8T with the core complex is largely through binding to nsp7, even modest polymerase activity in the absence of nsp7 suggests a strong role for nsp8F in stimulating polymerase activity, and the dispensability of nsp8T for in vitro RNA synthesis. Under our reaction conditions, PEDV and SARS-CoV-2 polymerases require all three proteins (nsp12, nsp7, nsp8) for in vitro polymerase activity (Figure 1A and B). To further dissect nsp8F and nsp8T’s contributions to promoting polymerase activity, we designed homologous PEDV and SARS-CoV-2 nsp12 single amino acid mutations at each nsp8’s protein-protein interfaces. These mutations were designed to be highly disruptive to hydrophobic protein-protein interfaces through the substitution of nsp12 surface residues with arginine to block interactions with regions of the nsp8s (PEDV nsp12 A382R, V384R, V842R, V848R and V894R and SARS-CoV-2 nsp12 A375R, L387R, L389R, I847T and T853R) (Figure 3A, B, and Supplementary Figure S8).
Figure 3.

Effects of replication factor disrupting nsp12 mutations on polymerase activity. Sites of nsp12 mutations designed to disrupt replication factor interactions for (A) PEDV and (B) SARS-CoV-2 are shown as orange spheres. The effects of PEDV (C) and SARS-CoV-2 (D) nsp12 mutations were evaluated using in vitro RNA primer extension assays. Technical triplicates of reactions were run and percent activity of wildtype nsp12 is presented. Error bars indicate standard deviation of the triplicates. All reactions, except PEDV nsp12-V842R, were significantly (P< 0.05) different compared to the wildtype control. Significance was determined using an unpaired t-test for each mutant complex.
Native mass spectrometry (Table 1, Supplementary Figure S4) of select mutant complexes showed full complex stoichiometry, 1:2:1:1 nsp7:nsp8:nsp12:RNA (∼186 kDa mass), for all mutants tested. The only exception being PEDV A382R where there were two major species: nsp12 alone and intact core polymerase complexes, indicating that nsp12 was either completely free or completely bound by nsp7 and nsp8 replication factors. These data suggest that the disruptive mutations are only effective in blocking the nsp12-nsp8 subdomain interactions and are not sufficient to fully prevent nsp8 subunit association with nsp12. This is congruent with the extended nature of nsp8 and the presence of both an N-terminal helical extension and a C-terminal head domain that bind distinct sites in coronavirus core polymerase complexes for both nsp8F and nsp8T. Hence nsp12 mutations disrupting the binding of one region of nsp8 may not preclude association of that nsp8 with the core complex. The effects of these mutations on polymerase activity were examined using an in vitro primer extension assay (Figure 3C, D and Supplementary Figure S9).
Nsp12 mutants designed to disrupt the association of PEDV nsp8F N-terminal helical extension, (nsp12 C370R) or nsp8F C-terminal head domain (nsp12 A382R or V384R) both resulted in complexes that either completely or mostly lost RNA extension activity (Figure 3C and Supplementary Figure S9). In contrast, mutant complexes designed to disrupt association of nsp8T N-terminal helical extension (nsp12 V842R, V848R or V849R) retained RNA synthesis activity (Figure 3C). These results indicate the importance of nsp8F for PEDV RNA synthesis and demonstrate that association of the nsp8T N-terminal helical extension with nsp12 is not required for stimulation of RNA synthesis in vitro.
To determine if these findings are specific to the PEDV polymerase core complex or a shared feature among CoVs, we tested homologous mutations for SARS-CoV-2 nsp12. Like PEDV, SARS-CoV-2 nsp12 mutants designed to disrupt association of the nsp8F N-terminal extension (nsp12 A375R) or nsp8F C-terminal head domain (nsp12 L387R or L389R) resulted in inactive polymerase complexes (Figure 3D and Supplementary Figure S9). However, nsp12 mutations designed to disrupt association of the nsp8T N-terminal extension (I847R or T853R) produced polymerase complexes with activity resembling that of the wild-type protein (Figure 3D). Similar results for mutagenesis across both PEDV and SARS-CoV-2 core polymerase complexes suggest shared roles for nsp8s across the coronavirus subfamily. These data suggest that both the nsp8F N-terminal extension and the C-terminal head domain play essential roles in stimulating polymerase activity beyond simply facilitating protein-protein interactions while the nsp12 interaction with nsp8T N-terminal extension appears non-essential for stimulating RNA synthesis.
Nsp8T N-terminal RNA binding domain is not required to stimulate RNA synthesis activity
Recent work showed that a nsp8–nsp7 fusion protein (nsp8L7) can efficiently function as a nsp12 replication factor and allows the contributions of each nsp8 of a core complex's polymerase activity to be delineated (32). We produced SARS-CoV-2 and PEDV nsp8L7 fusion proteins with six-residue linker regions (Figure 4A, Supplementary Figure S3). Both SARS-CoV-2 and PEDV nsp8L7 fusion proteins could stimulate their respective polymerase core complex activities in the presence of free nsp8 (Figure 4B,C and Supplementary Figure S5). While SARS-CoV-2 nsp8L7 stimulated polymerase activity similar to wildtype nsp7 and nsp8, PEDV nsp8L7 only resulted in 20% stimulation compared to wildtype subunits. Since PEDV nsp12 + nsp8 reactions have no RNA extension (Figure 4B and Supplementary Figure S5) these data show that the PEDV nsp8L7 fusion protein can function as a cofactor, albeit with limited activity compared to wildtype proteins. We predict that the PEDV nsp8L7 reduction in polymerase stimulation is due to a reduced dissociation of the nsp8L7 fusion protein from oligomeric states into forms available for binding nsp12 leading to defects in polymerase assembly (32). Therefore, only a fraction of the available nsp12 may form full complexes as compared to SARS-CoV-2 nsp8L7 complexes leading to the reduced overall observed activity. Differences in the oligomeric states of alpha- and betaCoV nsp7-nsp8 cofactor complexes have been previously identified, supporting our prediction that PEDV nsp8L7 behaves differently than SAR-CoV-2 nsp8L7 (30). The prediction of incomplete PEDV core complex formation with nsp8L7 is supported by native mass spectrometry where a significant amount of PEDV nsp12 alone is observed (Table 1, Supplementary Figure S4). In support of nsp8F being required for RNA synthesis in vitro and in validation of the fusion protein strategy to delineate nsp8 contributions, PEDV and SARS-CoV-2 nsp8L7 fusion proteins are not sufficient to stimulate CoV RNA synthesis in the absence of isolated nsp8 (Figure 4B and C).
Figure 4.
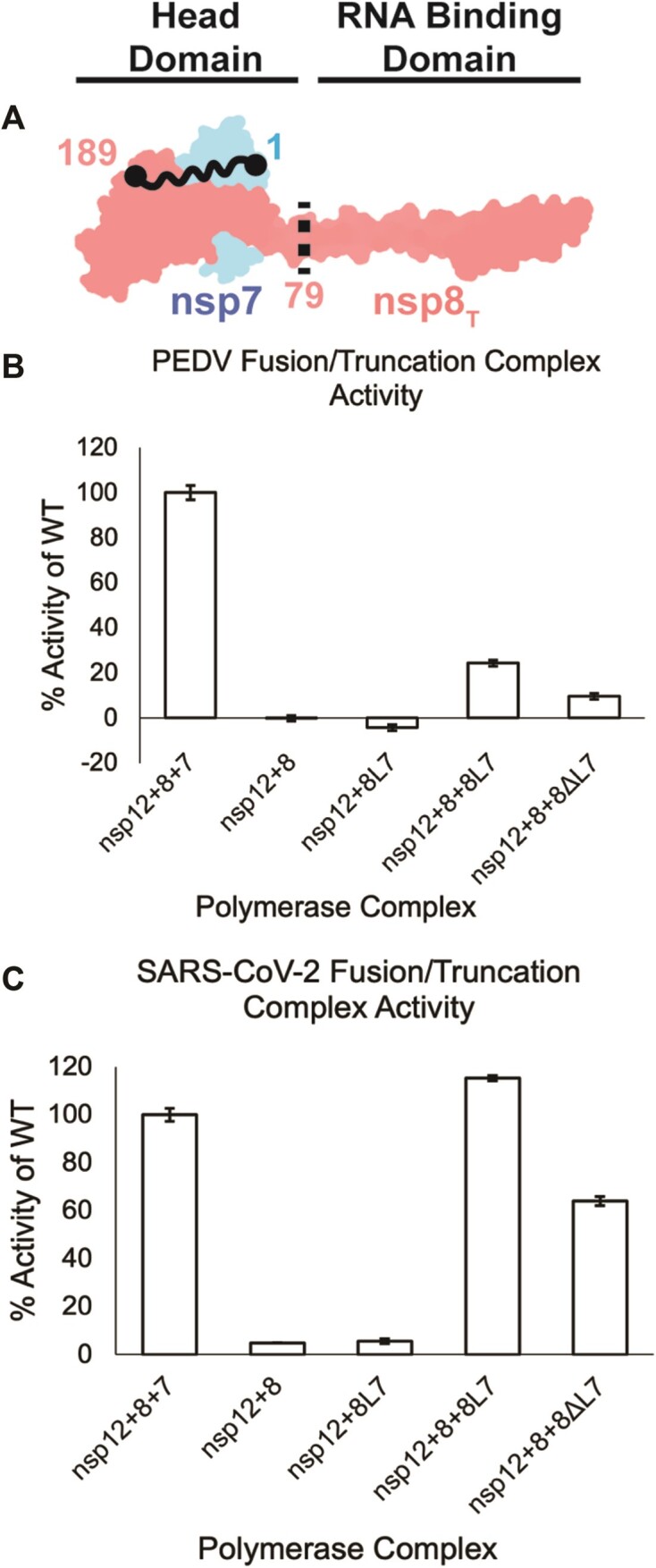
nsp8T’s N-terminal domain is not required for RNA synthesis. (A) Model of nsp7 (blue) and nsp8T (red) heterodimer (PDB ID: 7KRP). The black dots and squiggly line depict the nsp8 and nsp7 termini that are fused in nsp8L7. The dashed black line is the site of truncation of nsp8ΔL7, removing the nsp8T N-terminal 79 amino acid RNA binding domain. (B) PEDV and (C) SARS-CoV-2 polymerase complex activity using the nsp8L7 and nsp8ΔL7 replication factors. Reactions were run in triplicate and percent activity was compared to a wildtype nsp7 + nsp8 + nsp12 complex. Error bars indicate standard deviation of the triplicates. Each reaction was determined to be significantly different than all other complexes tested (P< 0.05) using an unpaired t-test.
Using the PEDV and SARS-CoV-2 nsp8L7 fusion constructs, we produced 79 residue N-terminal truncations of nsp8T (nsp8ΔL7) which lack the previously described RNA binding region of the protein (Figure 4A, Supplementary Figure S3) (23). Both PEDV and SARS-CoV-2 nsp8ΔL7 stimulated polymerase primer extension activity in vitro (Figure 4 and Supplementary Figure S5). Compared to polymerase stimulation using the nsp8L7 fusion constructs, PEDV and SARS-CoV-2 nsp8ΔL7 each had an approximately 50% decrease in activity from their respective nsp8L7 reactions. These results indicate that the nsp8T N-terminal region is not required for stimulation of RNA synthesis in vitro. Our results confirm similar prior results for SARS-CoV-2, while expanding the conclusions to PEDV (32).The differences in PEDV and SARS-CoV-2 polymerase activities could be attributed to the replication factor proteins’ unique interactions across the CoV genera (30). While the magnitude of the ability of PEDV and SARS-CoV-2 nsp8ΔL7 truncations to stimulate polymerase activity vary, that both truncation proteins can stimulate some polymerase activity while reactions lacking nsp7 do not, indicates that nsp8ΔL7 is a functional replication factor and that the N-terminal extension of nsp8T is not required for stimulation of alpha- or betaCoV RNA synthesis in vitro.
Discussion
We have established that the PEDV core polymerase complex assembles into an active polymerase complex using a similar replication factor stoichiometry as was previously seen in betaCoVs. The structure of the PEDV core polymerase complex reveals an overall similar architecture to betaCoV complexes with one large conformational difference in PEDV nsp12 residues 249–269 to interact with nsp8F that we predict to be conserved among alphaCoVs. Additionally, we used biochemistry and mutagenesis to show that for both PEDV and SARS-CoV-2, nsp8F is required for in vitro RNA synthesis while the N-terminal extension of nsp8T is not.
Since the observed structural differences in the PEDV and SARS-CoV-2 core polymerase complexes lie outside of the common antiviral drug design targets (the polymerase and NiRAN active sites), we predict that antivirals targeting these shared sites would be effective against both alpha- and betaCoV. For example, structures have shown that SARS-CoV-2 nsp12 residue S861 is likely important for the effectiveness of the nucleotide analogue Remdesivir (46). The proposed mechanism is that S861 sterically classes with the 1′-cyano group of the antiviral, impairing RNA elongation. For PEDV, the residue S856 is conserved in both sequence and space to S861 of SARS-CoV-2 (Supplementary Figure S7), indicating that Remdesivir could be used to treat alphaCoV infections (47).
After the initial observation of a labile nsp8T interaction within the RNA-bound PEDV polymerase complex, we sought to explore the contribution of the nsp8 N-terminal extensions in simulating RNA synthesis activity. In vitro analyses with mutant PEDV and SARS-CoV-2 nsp12s and truncated nsp8T (nsp8ΔL7) established that nsp8T’s N-terminal extension is not required to stimulate in vitro RNA synthesis. Similar in vitro work with truncated nsp8s has already established that the RNA binding domains of each nsp8 are not required for processive RNA synthesis for SARS-CoV-2, but rather play important roles in complex assembly and RNA binding (32). In some published studies, SARS-CoV-2 and MERS-CoV complexes of just nsp12 and nsp8 are sufficient to produce some polymerase activity in vitro (44,45). The absence of nsp7 from these reactions indicates that nsp8T is not absolutely required for in vitro RNA synthesis, further supporting our conclusions. Recent structural work has proposed that the N-terminal extensions of nsp8F and nsp8T act as sliding poles to promote RNA synthesis and processivity (23). The sliding pole hypothesis provided a mechanistic explanation for prior observations that the mutation of a positively charged residue (K58A) in SARS-CoV nsp8’s N-terminal extension was lethal to virus replication in vivo and reduced RNA synthesis in vitro (21). Our work was able to build upon these observations, by delineating the contributions of the two nsp8s and further confirm nsp8F’s importance in RNA synthesis. We propose a revision to this model where the nsp8T N-terminal extension is not essential to stimulate RNA synthesis but may instead have other important roles in RNA synthesis.
We hypothesize that nsp8T may act as a scaffolding protein to regulate the engagement of other viral factors like the nsp13 RNA helicase to promote RNA backtracking (Figure 5). Previous work has shown that the viral helicase, nsp13, binds template RNA entering the active site and can cause polymerase backtracking on primer-template RNA (27,28). In a backtracked polymerase state, the 3′ end of the nascent RNA extrudes out of the polymerase NTP entry channel. While a function for backtracking within viral replication remains unclear, one hypothesis is that it mediates proofreading of mis-incorporated 3′ nucleotides during CoV RNA synthesis. The nsp13 responsible for backtracking RNA binds the polymerase core complex via the nsp8T N-terminal extension. As a potential model for viral RNA proofreading, we propose that aberrant nucleotide incorporation stalls elongation, allows engagement of the nsp8T N-terminal extension with nsp12 and the RNA allowing the backtracking nsp13 RNA helicase to bind template RNA and induce a backtracked state. 3′ nucleotides of the backtracked RNA could then be excised by the viral nsp14 exonuclease removing the mis-incorporated nucleotides.
Figure 5.

Model for coronavirus backtracking and proofreading. Stalling of polymerase complexes by misincorporation is hypothesized to promote the engagement of nsp8T’s N-terminal helical extension with RNA allowing the recruitment of additional viral factors for backtracking or proofreading. Whether or not nsp8T is always associated with the complex is unknown.
Our current work is focused on the multiple roles of nsp8 in alpha and beta-CoV RNA synthesis. To date, there are no reports of active gamma- or deltaCoV polymerase complexes in vitro, or published structures of polymerases from these two genera. Sequence conservation of the nsps involved in genome replication (i.e. nsp7, nsp8 and nsp12) across all CoV genera indicates that these proteins have conserved roles in replication (48). To our knowledge nsp7 does not directly interact with RNA during replication, but it serves multiple important roles to support CoV RNA synthesis. Beyond nsp7’s role as a replication factor for nsp12 and scaffolding protein for nsp8T, nsp7 also de-oligomerizes nsp8 allowing monomeric nsp8 to interact with nsp12 as nsp8F (21,30,49). Furthermore, the interactions between nsp7 and nsp8 oligomers are known to vary across the CoV species (30). These differences are possible explanations for the different activities of nsp8L7 and nsp8ΔL7 containing complexes seen in our work.
This work expands the diversity of CoV polymerase structural biology to the alphaCoVs. Our work shows a high level of structural conservation among CoV replication machinery and has allowed for the generation of hypotheses for CoV RNA synthesis that span more than a single virus genus. Such work is essential not only for a greater understanding of CoV biology but also in preparing for the threat of emerging CoV.
Supplementary Material
Acknowledgements
We would like to thank the Cryo-EM Research Center in the Biochemistry Department at University of Wisconsin-Madison for technical and staff support. We would like to thank Dr Olve Peersen for his suggestions and insight for use of the nsp8–nsp7 fusion construct.
Author contributions: T.K.A. and R.N.K. designed the project. T.K.A., P.J.H. and K.J.C. designed and completed experiments. K.W.L. and J.J.C. designed and completed native mass spectrometry sample preparation, experimentation, data analysis, and figure making. T.K.A. and R.N.K. wrote the paper. K.W.L. and J.J.C. provided comments and edits on the paper. All authors approved of the final manuscript.
Contributor Information
Thomas K Anderson, Biochemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA; Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA; Center for Quantitative Cell Imaging, University of Wisconsin-Madison, Madison, WI 53706, USA.
Peter J Hoferle, Biochemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA; Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA; Center for Quantitative Cell Imaging, University of Wisconsin-Madison, Madison, WI 53706, USA.
Kennan J Chojnacki, Biochemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA; Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA; Center for Quantitative Cell Imaging, University of Wisconsin-Madison, Madison, WI 53706, USA.
Kenneth W Lee, Biomolecular Chemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA.
Joshua J Coon, Biomolecular Chemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA; Department of Chemistry, University of Wisconsin-Madison, Madison, WI 53715, USA; Morgridge Institute for Research, Madison, WI 53715, USA.
Robert N Kirchdoerfer, Biochemistry Department, University of Wisconsin-Madison, Madison, WI 53706, USA; Institute for Molecular Virology, University of Wisconsin-Madison, Madison, WI 53706, USA; Center for Quantitative Cell Imaging, University of Wisconsin-Madison, Madison, WI 53706, USA.
Data availability
The PEDV core polymerase complex electron density maps have been deposited in the Electron Microscopy Data Bank for the complex lacking nsp8T (EMDB: 29779) and the complete complex (EMDB: 42488). Coordinate models have been deposited in the Protein Data Bank for the complex lacking nsp8T (PDB: 8G6R) and the complete complex (PDB: 8URB).
Limitations of study
We acknowledge that our study uses short RNA substrates, short extension reactions, and has been done exclusively in vitro and so does not model processive CoV RNA synthesis. This limits our ability to make conclusions on nsp8T’s importance for processive RNA synthesis, and replication in vivo. We believe studies using longer RNA substrates, and in vivo reverse genetics will allow our hypotheses and conclusions developed here to be evaluated in the context of processive replication and virus replication in vivo but are beyond the scope of the current work.
Our work only studies the core polymerase complex (nsp7, nsp8 and nsp12) but our conclusions include hypotheses about interactions with other viral proteins (i.e. nsp13 and nsp14). While our current work is unable to directly assess these hypotheses, this work provides a foundation for further study.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
NIH/NIAID, USDA [AI123498, AI158463, USDA WIS03099 to R.N.K.]; NIH [R35GM118110 to J.J.C.]. Funding for open access charge: Federal grants.
Conflict of interest statement. None declared.
References
- 1. Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015; 1282:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003; 331:991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012; 367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 4. de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C., Enjuanes L., Fouchier R.A., Galiano M., Gorbalenya A.E., Memish Z.A. et al. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013; 87:7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020; 579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Silva C.S., Mullis L.B., Pereira O., Saif L.J., Vlasova A., Zhang X., Owens R.J., Paulson D., Taylor D., Haynes L.M. et al. Human Respiratory coronaviruses detected In patients with influenza-like illness in Arkansas, USA. Virol. Mycol. 2014; 2014:004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiu L., Binder R.A., Alarja N.A., Kochek K., Coleman K.K., Than S.T., Bailey E.S., Bui V.N., Toh T.H., Erdman D.D. et al. A RT-PCR assay for the detection of coronaviruses from four genera. J. Clin. Virol. 2020; 128:104391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vlasova A.N., Diaz A., Damtie D., Xiu L., Toh T.H., Lee J.S., Saif L.J., Gray G.C. Novel canine coronavirus isolated from a hospitalized patient with pneumonia in East Malaysia. Clin. Infect. Dis. 2022; 74:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yan Q., Liu X., Sun Y., Zeng W., Li Y., Zhao F., Wu K., Fan S., Zhao M., Chen J. et al. Swine Enteric Coronavirus: diverse pathogen-host interactions. Int. J. Mol. Sci. 2022; 23:3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Q., Wang H.Y. Porcine enteric coronaviruses: an updated overview of the pathogenesis, prevalence, and diagnosis. Vet. Res. Commun. 2021; 45:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y., Chen Y., Zhou J., Wang X., Ma L., Li J., Yang L., Yuan H., Pang D., Ouyang H. Porcine Epidemic Diarrhea Virus: an updated overview of virus epidemiology, virulence variation patterns and virus-host interactions. Viruses. 2022; 14:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stevenson G.W., Hoang H., Schwartz K.J., Burrough E.R., Sun D., Madson D., Cooper V.L., Pillatzki A., Gauger P., Schmitt B.J. et al. Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Invest. 2013; 25:649–654. [DOI] [PubMed] [Google Scholar]
- 14. Sun R.Q., Cai R.J., Chen Y.Q., Liang P.S., Chen D.K., Song C.X. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg. Infect. Dis. 2012; 18:161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brian D.A., Baric R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005; 287:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S.G., Ding J. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 2003; 31:7117–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. te Velthuis A.J., Arnold J.J., Cameron C.E., van den Worm S.H., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent. Nucleic Acids Res. 2010; 38:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Snijder E.J., Decroly E., Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv. Virus Res. 2016; 96:59–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirchdoerfer R.N., Ward A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019; 10:2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan L., Ge J., Zheng L., Zhang Y., Gao Y., Wang T., Huang Y., Yang Y., Gao S., Li M. et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate State in cap synthesis. Cell. 2020; 184:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Subissi L., Posthuma C.C., Collet A., Zevenhoven-Dobbe J.C., Gorbalenya A.E., Decroly E., Snijder E.J., Canard B., Imbert I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E3900–E3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seifert M., Bera S.C., van Nies P., Kirchdoerfer R.N., Shannon A., Le T.T., Meng X., Xia H., Wood J.M., Harris L.D. et al. Inhibition of SARS-CoV-2 polymerase by nucleotide analogs from a single-molecule perspective. eLife. 2021; 10:e70968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020; 584:154–156. [DOI] [PubMed] [Google Scholar]
- 24. Yin W., Mao C., Luan X., Shen D.D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020; 368:1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bravo J.P.K., Dangerfield T.L., Taylor D.W., Johnson K.A. Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol. Cell. 2021; 81:1548–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan L., Zhang Y., Ge J., Zheng L., Gao Y., Wang T., Jia Z., Wang H., Huang Y., Li M. et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020; 11:5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J., Malone B., Llewellyn E., Grasso M., Shelton P.M.M., Olinares P.D.B., Maruthi K., Eng E.T., Vatandaslar H., Chait B.T. et al. Structural basis for helicase-polymerase coupling in the SARS-CoV-2 replication-transcription complex. Cell. 2020; 182:1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malone B., Chen J., Wang Q., Llewellyn E., Choi Y.J., Olinares P.D.B., Cao X., Hernandez C., Eng E.T., Chait B.T. et al. Structural basis for backtracking by the SARS-CoV-2 replication-transcription complex. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2102516118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiao Y., Ma Q., Restle T., Shang W., Svergun D.I., Ponnusamy R., Sczakiel G., Hilgenfeld R. Nonstructural proteins 7 and 8 of feline coronavirus form a 2:1 heterotrimer that exhibits primer-independent RNA polymerase activity. J. Virol. 2012; 86:4444–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krichel B., Bylapudi G., Schmidt C., Blanchet C., Schubert R., Brings L., Koehler M., Zenobi R., Svergun D., Lorenzen K. et al. Hallmarks of alpha- and betacoronavirus non-structural protein 7+8 complexes. Sci. Adv. 2021; 7:eabf1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma M., Yang Y., Wu L., Zhou L., Shi Y., Han J., Xu Z., Zhu W. Conserved protein targets for developing pan-coronavirus drugs based on sequence and 3D structure similarity analyses. Comput. Biol. Med. 2022; 145:105455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campagnola G., Govindarajan V., Pelletier A., Canard B., Peersen O.B. The SARS-CoV nsp12 polymerase active site is tuned for large-genome replication. J. Virol. 2022; 96:e0067122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schneider C.A., Rasband W.S., Eliceiri K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005; 152:36–51. [DOI] [PubMed] [Google Scholar]
- 35. Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017; 14:290–296. [DOI] [PubMed] [Google Scholar]
- 36. Punjani A., Fleet D.J. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 2021; 213:107702. [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol. 2018; 74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018; 74:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.A.-O., Ferrin T.A.-O. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shannon A., Fattorini V., Sama B., Selisko B., Feracci M., Falcou C., Gauffre P., El Kazzi P., Delpal A., Decroly E. et al. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 2022; 13:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., Smith E.C., Case J.B., Feng J.Y., Jordan R. et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018; 9:e00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gordon C.J., Tchesnokov E.P., Woolner E., Perry J.K., Feng J.Y., Porter D.P., Gotte M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020; 295:6785–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gordon C.J., Tchesnokov E.P., Feng J.Y., Porter D.P., Gotte M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020; 295:4773–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pourfarjam Y., Ma Z., Kim I.K. ATP enhances the error-prone ribonucleotide incorporation by the SARS-CoV-2 RNA polymerase. Biochem. Biophys. Res. Commun. 2022; 625:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J., Farnung L., Siewert A., Hobartner C., Cramer P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021; 12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krentz D., Zenger K., Alberer M., Felten S., Bergmann M., Dorsch R., Matiasek K., Kolberg L., Hofmann-Lehmann R., Meli M.L. et al. Curing cats with feline infectious peritonitis with an oral multi-component drug containing GS-441524. Viruses. 2021; 13:2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grellet E., L’Hote I., Goulet A., Imbert I. Replication of the coronavirus genome: a paradox among positive-strand RNA viruses. J. Biol. Chem. 2022; 298:101923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Biswal M., Diggs S., Xu D., Khudaverdyan N., Lu J., Fang J., Blaha G., Hai R., Song J. Two conserved oligomer interfaces of NSP7 and NSP8 underpin the dynamic assembly of SARS-CoV-2 RdRP. Nucleic Acids Res. 2021; 49:5956–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PEDV core polymerase complex electron density maps have been deposited in the Electron Microscopy Data Bank for the complex lacking nsp8T (EMDB: 29779) and the complete complex (EMDB: 42488). Coordinate models have been deposited in the Protein Data Bank for the complex lacking nsp8T (PDB: 8G6R) and the complete complex (PDB: 8URB).