

Abstract
The CTNNB1 gene, encoding β-catenin, is frequently mutated in hepatocellular carcinoma (HCC, ∼30%) and in hepatoblastoma (HB, >80%), in which DLK1/DIO3 locus induction is correlated with CTNNB1 mutations. Here, we aim to decipher how sustained β-catenin activation regulates DLK1/DIO3 locus expression and the role this locus plays in HB and HCC development in mouse models deleted for Apc (ApcΔhep) or Ctnnb1-exon 3 (β-cateninΔExon3) and in human CTNNB1-mutated hepatic cancer cells. We identified an enhancer site bound by TCF-4/β-catenin complexes in an open conformation upon sustained β-catenin activation (DLK1-Wnt responsive element [WRE]) and increasing DLK1/DIO3 locus transcription in β-catenin-mutated human HB and mouse models. DLK1-WRE editing by CRISPR-Cas9 approach impaired DLK1/DIO3 locus expression and slowed tumor growth in subcutaneous CTNNB1-mutated tumor cell grafts, ApcΔhep HB and β-cateninΔExon3 HCC. Tumor growth inhibition resulted either from increased FADD expression and subsequent caspase-3 cleavage in the first case or from decreased expression of cell cycle actors regulated by FoxM1 in the others. Therefore, the DLK1/DIO3 locus is an essential determinant of FoxM1-dependent cell proliferation during β-catenin-driven liver tumorigenesis. Targeting the DLK1-WRE enhancer to silence the DLK1/DIO3 locus might thus represent an interesting therapeutic strategy to restrict tumor growth in primary liver cancers with CTNNB1 mutations.
Keywords: primary liver cancers, transgenic mice, in vivo CRISPR-Cas9, β-catenin, enhancer site, non-coding RNAs, targeted therapies
Graphical abstract
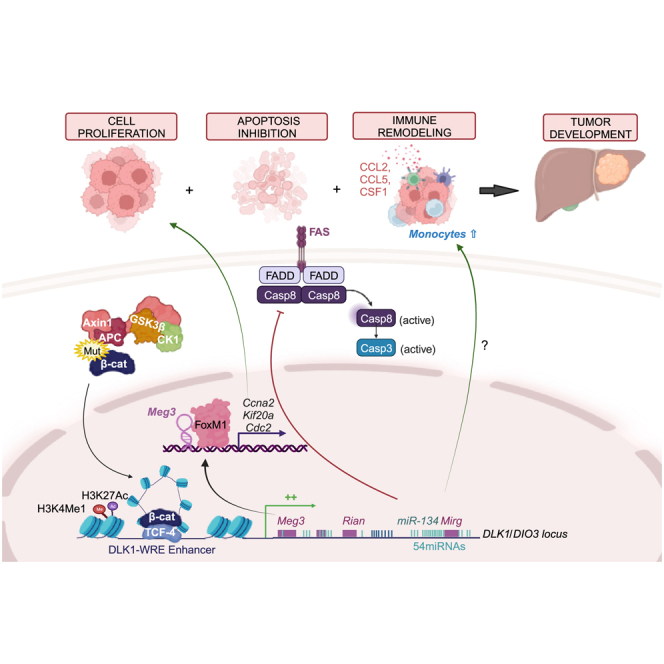
Sanceau and colleagues identified the enhancer site located in the DLK1/DIO3 locus activated by sustained β-catenin signaling and responsible for locus overexpression in CTNNB1-mutated primary liver cancers. Specific targeting of this enhancer limited tumor growth by acting on proliferation, apoptosis, and monocyte recruitment and could represent a promising therapeutic avenue.
Introduction
The two most common primary liver tumors, hepatocellular carcinoma (HCC) in adults and hepatoblastoma (HB) in children, are both characterized by mutations in the Wnt/β-catenin pathway. Somatic point mutations in the CTNNB1 gene, encoding β-catenin, are encountered in approximately 30% of HCC1 and CTNNB1 exon 3 deletions occur in more than 80% of HB.2,3 CTNNB1 mutations prevent β-catenin phosphorylation and its subsequent proteasomal degradation orchestrated by a complex containing APC, AXIN1, GSK3β, and CK1. This leads to β-catenin stabilization and translocation into the nucleus, where, in hepatocytes, it interacts primarily with TCF-4 before recruitment at Wnt responsive elements (WREs).4 This recruitment allows the regulation of a specific gene repertoire acting on metabolic and proliferative pathways.5,6 To finely tune its gene repertoire, β-catenin is able to cooperate with a plethora of histone modifiers and chromatin remodelers,7 numbers of them being mutated in HCC.8
In HB, additional mutations can also affect chromatin modifiers or long non-coding RNAs (lncRNAs) produced from parentally imprinted clusters, such as H19 or MEG3.9,10 The DLK1/DIO3 locus encodes the largest cluster of non-coding RNAs (ncRNAs), including 54 microRNAs (miRNAs), several small nucleolar RNAs (snoRNAs), and lncRNAs (e.g., MEG3, MEG8/RIAN, MEG9/MIRG) expressed from the maternal allele but also encoding paternally expressed RNAs such as DLK1, RTL1, and DIO3 (Figure 1A). The expression of the DLK1/DIO3 locus is mainly regulated by methylation of three differentially methylated regions (DMRs), named DLK1-, IG-, and MEG3-DMRs, with different regulatory functions.11 This imprinted locus is crucial for cell pluripotency12 and liver metabolic adaptation.13 RNAs produced from the DLK1/DIO3 locus are frequently underexpressed in cancers,14 either under- or overexpressed in HCC,15,16 while DLK1/DIO3 locus induction in HB is associated with poor prognosis17,18 and CTNNB1 mutations.18
Figure 1.

The Dlk1/Dio3 locus is induced in mouse HCC- and HB-like tumors driven by β-catenin
(A) Schematic representation of the DLK1/DIO3 locus. (B) In situ hybridization of Meg3 and miR-127 with staining of glutamine synthetase (GS) or active β-catenin in WT and ApcΔhep livers and in ApcΔhep and β-cateninΔExon3 HCC- or HB-like tumors. CV, central vein; PV, portal vein. (C–F) Expression of Rian, Mirg, and miR-127 with error bars representing SEM by RT-qPCR in ApcΔhep tumors (TUM) compared to adjacent non-tumor (NT) tissue (C); in ApcΔhep HCC and HB-like tumors (D); in β-cateninΔExon3 tumors (E); and in DEN tumors without β-catenin activation (F). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.0001; ns, non-significant (Mann-Whitney).
In the present study, we decipher the molecular mechanisms whereby sustained β-catenin activation affects gene expression at the DLK1/DIO3 locus and the role that this activation plays in the development of primary liver cancers. We used two mouse models that develop HCC or HB following oncogenic activation of β-catenin signaling through either inducible and liver-specific loss of function of Apc (ApcΔhep)5,19,20,21,22 or deletion of Ctnnb1-exon 3 (β-cateninΔExon3).23 Using in vivo CRISPR-Cas9 editing, we identified a new regulatory site upstream of Meg3 (DLK1-WRE site) bound by oncogenic β-catenin/TCF-4 complexes and responsible for Dlk1/Dio3 locus induction. We also demonstrated the crucial pro-tumorigenic role of the DLK1/DIO3 locus in the regulation of apoptosis and FoxM1-driven cell cycle progression during liver carcinogenesis mediated by β-catenin in both mouse models and in hepatic cancer cell lines.
Results
The Dlk1/Dio3 locus is induced after sustained β-catenin activation in mouse livers and tumors
Our team has created two mouse models that recapitulate liver cancer development with sustained β-catenin activation, either Cre-Lox-based Apc excision (ApcΔhep) or CRISPR-Cas9 deletion of Ctnnb1-exon 3 (β-cateninΔExon3) (Figures S1A–S1C).23 In both models, two tumor types can emerge from healthy livers: either well-differentiated HCC similar to human G5-G6 HCC or poorly differentiated tumors close to human HB. Human tumors and mouse tumors from both models share dysregulated transcriptional programs.23
RNA sequencing (RNA-seq) and small RNA-seq data showed that coding RNAs and ncRNAs within the Dlk1/Dio3 locus were some of the most significantly overexpressed RNAs in preneoplastic ApcΔhep hepatocytes compared to wild-type (WT) (Tables S1 and S2).5,21 Induction of Meg3 and miR-127 was confirmed by in situ hybridization in ApcΔhep hepatocytes as well as in ApcΔhep and β-cateninΔExon3 HCC and HB-like tumors (Figure 1B). Upregulation of Mirg, Rian, and miR-127 was confirmed by RT-qPCR in ApcΔhep tumors (TUM) relative to adjacent non-tumor tissues (NT) (Figure 1C). It was found stronger in HCC compared to HB (Figure 1D) in agreement with the maintenance of metabolic targets in HCC harboring hepatocyte features.23,24 The locus induction also appeared higher in ApcΔhep HCC compared to β-cateninΔExon3 HCC (compare Figure 1D/1E). It is also noteworthy that strong correlations between RNA expression levels of Rian, Mirg, miR-127, and Glul, a canonical β-catenin target, were found both in ApcΔhep (Figure S2A) and β-cateninΔExon3 tumors (Figure S2B). In diethylnitrosamine (DEN)-induced livers tumors without Glul induction, the expression of Rian, Mirg, and miR-127 was not modified (Figure 1F) and no correlation with Glul was observed (Figure S2C).
Using a Cre-GFP adenovirus, we sorted GFP+ ApcΔhep hepatocytes during the earliest steps of liver tumorigenesis (Figure S1D). RNA-seq data showed that all RNAs within the Dlk1/Dio3 locus were induced between 6 and 15 days after Apc inactivation compared to non-activated GFP− hepatocytes (Figure S3A), similarly to canonical β-catenin targets such as Glul or Axin2 (Figure S3B).
These results indicate that sustained β-catenin activation correlates with coordinated upregulation of ncRNAs within the Dlk1/Dio3 locus in preneoplastic hepatocytes and mouse tumors.
TCF-4/β-catenin complexes bind upstream of Meg3 promoting an enhancer activation
Our next objective was to decipher how β-catenin activation promotes Dlk1/Dio3 locus expression. Our chromatin immunoprecipitation (ChIP) sequencing (ChIP-seq) data targeting the β-catenin cofactor TCF-4 in ApcΔhep hepatocytes showed that TCF-4 bound upstream of Meg3 to a site containing two canonical WRE motifs (named DLK1-WRE) (Figures 2A and S5A). TCF-4 binding was conserved in the human HepG2 cell line with activating CTNNB1 mutations (public dataset GSM782122) (Figures 3A and S5H). TCF-4 did not bind to the DLK1-WRE site in hepatocytes isolated from a mouse model invalidated for β-catenin (β-catΔhep in Figure 2A). Impaired expression of non-coding RNAs within the Dlk1/Dio3 locus was subsequently noticed in β-catΔhep hepatocytes (Tables S1 and S2). ChIP-qPCR targeting the DLK1-WRE site confirmed increased binding of both TCF-4 (Figure 2B) and β-catenin (Figure S4A) in ApcΔhep hepatocytes compared to WT hepatocytes. Assay for transposase-accessible chromatin (ATAC) with sequencing (ATAC-seq) (Figure 2A) and ATAC-qPCR experiments (Figure 2C) indicated an open chromatin configuration at the DLK1-WRE site in ApcΔhep hepatocytes. Identical results were obtained from β-cateninΔexon3 hepatocytes for TCF-4 binding and open chromatin conformation (Figure S4B).
Figure 2.

β-Catenin binding at the DLK1-WRE site opens chromatin and exerts enhancer activity in ApcΔhep hepatocytes
(A) ChIP-seq targeting TCF-4 in WT, ApcΔhep, and β-catΔhep hepatocytes and ATAC-seq data in WT and ApcΔhep hepatocytes. TCF-4 binding site is framed in the blue box (DLK1-WRE) and sites common with 3C in pink. (B, D, F, and G) ChIP-qPCR analysis at the DLK1-WRE site for TCF-4, H3K4me1, and H3K27ac relative to isotype control in ApcΔhep hepatocytes with error bars representing SEM compared to WT (B and D) and compared to ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (F and G). (C and H) ATAC-qPCR analysis at the DLK1-WRE site compared to WT (C) and to ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (H). (E) Relative contact frequencies in arbitrary units (a.u.) between the DLK1-WRE site (blue vertical bar) and 19 genomic sites (small vertical black bars on the map below) measured in 3C experiments performed on WT, ApcΔhep-Rosa26, and ApcΔhep-DLK1/DIO3ΔWRE liver nuclei with error bars representing SEM of six, five, and three biological replicates, respectively. Regions of interest (highlighted in pink) are numbered from 1 to 6. The lower panel illustrates the different chromatin loops distributed into six interaction zones: the darker the pink, the stronger the interaction. Figure made with BioRender. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.0001; ns, non-significant (Mann-Whitney).
Figure 3.

The DLK1/DIO3 locus is induced in human HB in correlation with CTNNB1 mutations and DLK1-WRE opening
(A) Pseudo bulk snATAC-seq aggregated with Cellranger-atac of three human HBs (T) and their adjacent NT tissue (N) at the DLK1-WRE site; the pink panel represents ChIP-seq data targeting TCF-4 in the human HB HepG2 cell line (Gsm782122). (B) RNA-seq expression data for the entire DLK1/DIO3 locus in HB normalized to their adjacent NT tissue (N = 22, cohort 1). A white square in the β-catenin lane indicates HB with intact CTNNB1, a yellow square HB with point mutation in CTNNB1, an orange square HB with CTNNB1 exon 3 deletion. (C) Correlation between RIAN, MIRG, DIO3OS, DIO3, MEG3, miR-411, and miR-136 expressions in cohort 1. (D) RIAN, RTL-1, and MEG3 expression determined by RT-qPCR in primary (N = 83) and recurrent HB (N = 17) versus non-tumor liver (NTL) (N = 100, cohort 2). (E) Expression of DIO3OS, MEG3, and RIAN determined by RT-qPCR in the different subgroups of HB: embryonal (green), fetal hepatocytic 1 (pink), fetal hepatocytic 2 (yellow), and mesenchymal (purple). (F) Expression of RIAN, DIO3OS, and MEG3 determined by RT-qPCR in primary HB with CTNNB1 mutations (CTNNB1mut, N = 76) or with intact CTNNB1 (CTNNB1WT, N = 7). ∗p < 0.05, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001; ns, non-significant (Kruskal-Wallis),with error bars representing SEM.
H3K4me1 and H3K27ac ChIP-qPCR experiments showed that these typical marks of enhancers were both found significantly increased at the DLK1-WRE site in ApcΔhep and β-cateninΔexon3 hepatocytes compared to WT hepatocytes (Figures 2D and S4B). No significant differences were found for the H3K4me3 mark associated with transcriptionally active chromatin at transcription start sites (Figure S4C). Increased TCF-4 binding was also detected at the DLK1-WRE site in ApcΔhep and β-cateninΔexon3 tumors compared to non-tumor tissues (Figure S4D). Remarkably, no modification in DNA methylation profiles and TCF-4 binding was observed at the IG-DMR in ApcΔhep hepatocytes compared to WT hepatocytes (Figures S4E–S4F).22
Interestingly, HNF4α, a transcriptional factor involved in hepatocyte differentiation, bound to the DLK1-WRE site in ApcΔhep hepatocytes and independently of TCF-4 binding since its binding was not affected in ApcΔhep DLK1ΔWRE hepatocytes (Figure S4G). In addition, HNF4α was not co-immunoprecipitated with TCF-4 at the DLK1-WRE site in sequential ChIP experiments (Figure S4H). HNF4α binding favored Meg3 and miR-127 expression since their expression was impaired in HNF4αΔhep hepatocytes (Figure S4I), consistent with previous data.25 HNF4α binding could therefore contribute in the higher expression of the locus observed in HCC (Figure 1D), in which HNF4α expression is maintained in contrast to HB (PRJEB44400 dataset).23
Globally, these results demonstrate that, upon sustained β-catenin activation, a DLK1-WRE site located upstream of Meg3, bound by TCF-4/β-catenin complexes, is in an open configuration and marked by histone modifications typical of active enhancers. We can assume that TCF-4/β-catenin binding at this site promotes the formation of an active enhancer favoring transcription of the entire Dlk1/Dio3 locus.
Besides its transcriptional role, β-catenin is able to bridge distal DNA regions by chromatin looping.7 The tridimensional organization of the DLK1/DIO3 locus is also known to be highly dynamic according to its expression patterns during embryonic development.26,27 We thus investigated whether, following oncogenic TCF-4/β-catenin binding, the activation of the putative DLK1-WRE enhancer could affect the tridimensional organization of the DLK1/DIO3 locus. To this aim, we performed a 3C-qPCR analysis centered on the DLK1-WRE site and covering the region between DLK1 and miR-136. The relative contact frequencies measured all along the locus allow specific interactions to be determined: the higher the frequency, the closer the DNA region is relative to the DLK1-WRE site (vertical blue bar in Figure 2E). In WT hepatocytes (Figure 2E, green dots), six major regions are interacting with the DLK1-WRE site: one located upstream of Dlk1, two regions in the vicinity of the DLK1-WRE site (sites 1 and 2), one site within the Ig-DMR (site 3), one within Meg3 (site 4), and one in the miR-136 region (site 6). In ApcΔhep hepatocytes (Figure 2E, blue dots), binding of TCF-4 to the DLK1-WRE site resulted in drastic DNA loop remodeling. Interactions with sites 1, 2, and 4 were lost, while looping with the Ig-DMR (site 3) and the miR-136 region (site 6) were reinforced. Remarkably, these two loops coincided with two open chromatin regions observed by ATAC-seq in ApcΔhep hepatocytes (sites 3 and 6 highlighted in pink in Figure 2A).
We conclude that, in ApcΔhep hepatocytes, oncogenic β-catenin/TCF-4 complexes bound at the DLK1-WRE site largely modify 3D chromatin organization and favor interactions with coding regions located after the Ig-DMR rather than upstream regions. This important re-organization may thus contribute to upregulate the expression of the non-coding RNAs encoded in the downstream part of the DLK1/DIO3 locus.
The DLK1-WRE regulatory site is conserved in human HB
Consistent with our observations in mouse models and as mentioned above, we identified a TCF-4 binding site upstream of MEG3 in HepG2 cells, a human HB cell line harboring a CTNNB1 deletion (Figures 3A and S5H) (GSM782122). Multiome single-nucleus ATAC-seq (snATAC-seq) experiments conducted on samples from two HB patients harboring CTNNB1 mutations showed that the human DLK1-WRE site was in an open conformation in HB compared to their paired adjacent non-tumor tissues (Figure 3A). The expression of miRNAs, as well as lncRNAs and coding RNAs, from the DLK1/DIO3 locus was increased in both 20 CTNNB1-mutated HB (yellow and orange squares in Figure 3B) and two non-mutated HBs (white square in Figure 3B) compared to their paired adjacent non-tumor tissues (Table S3). Interestingly, a strong correlation was observed between expressions of the different ncRNAs (Figure 3C), supporting the idea of a global induction of this region in human HB. Overexpression of these ncRNAs was confirmed in an independent larger collection of primary (n = 83) and recurrent HB (n = 17) (Figure 3D; Table S4), and regardless of the HB subgroups (Figure 3E). In this collection containing a higher number of non-mutated HBs (n = 7), we found that RIAN and DIO3OS expression was even significantly higher in CTNNB1-mutated HB compared to non-mutated HB, with the same tendency noticed for MEG3 (Figure 3F). Therefore, the upregulation of the DLK1/DIO3 locus appears to be associated with chromatin opening at the DLK1-WRE site in human HB with CTNNB1 mutations.
β-Catenin/TCF-4 binding to DLK1-WRE is required for its optimal enhancer activity
To decipher if the DLK1-WRE site is the key region underlying β-catenin regulation, we designed two small guide RNAs (sgRNAs) to remove the DLK1-WRE site by CRISPR-Cas9 editing (DLK1/DIO3ΔWRE) without potential off-targets (Figure S5A). Then, sgRNAs were integrated into plasmids containing the saCas9 sequence and inverted terminal repeats, allowing in vivo editing using AAV8 particles.28 Once subcloned, these constructs were validated in the murine Hepa1-6 cell line with Ctnnb1 mutation: DNA editing in one stable clone (Figure S5A) was successfully associated with a decrease in Rian, Mirg, Meg3, and Rtl1 expression (Figure S5B), as well as downregulation in DLK1 protein level (Figure S5C).
Once validated, we genetically edited the DLK1-WRE site in vivo with AAV8 constructs in ApcΔhep mice, with the inactive Rosa26 locus as controls (Figure S1A). As expected, DLK1-WRE editing was only detected in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes but not in non-parenchymal cells (Figures S5D and S5E). This resulted in a drastic decrease in all RNAs produced from the Dlk1/Dio3 locus in both RNA-seq and RT-qPCR experiments in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (Tables S5 and S6; Figure S5F), with no impact in WT livers (Figure S5G). Additionally, impairment of TCF-4 binding at the DLK1-WRE site in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (Figure 2F) was associated with a decrease in both H3K4me1 and H3K27ac marks (Figure 2G) and less chromatin opening (Figure 2H). The 3C experiments in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes showed that the 3D-chromatin organization at the DLK1-WRE site got closer to that detected in WT hepatocytes: interactions between the DLK1-WRE site and the site 2 were reinforced, while looping with the site 3 containing the Ig-DMR and the site 6 near miR-136 was lost (pink triangles in Figure 2E).
Altogether, DLK1-WRE site suppression by CRISPR-Cas9 editing in vivo indicates that TCF-4/β-catenin binding at the DLK1-WRE site drives its enhancer activity and the subsequent upregulation of the entire Dlk1/Dio3 locus.
DLK1-WRE site editing in ApcΔhep hepatocytes primarily affects regulators of mitotic entry and progression
Phenotypically, ApcΔhep-DLK1/DIO3ΔWRE livers demonstrated significant decrease in the number of Ki-67+ hepatocytes (Figure 4A) associated with a reduced hepatomegaly20 (Figure 4B) compared to ApcΔhep-Rosa26 controls. Comparisons of RNA-seq data from ApcΔhep-DLK1/DIO3ΔWRE versus ApcΔhep-Rosa26 hepatocytes showed that, among the top 50 most significantly dysregulated genes, several genes implicated in cell proliferation and division were downregulated: Ccna2, Ccnb1, Kif20a, Kif20b, Ckap2, Top2a, Cdc2, Racgap1, Kif4a, Hmmr, Nusap1, and Nuf2 (Table S7, some of which are confirmed by RT-qPCR in Figure 4C). These genes were upregulated in ApcΔhep hepatocytes compared to WT hepatocytes (Table S8). Comparing ApcΔhep-DLK1/DIO3ΔWRE versus ApcΔhep-Rosa26 hepatocytes, Gene Ontology identified genes related to microtubule cytoskeleton, mitotic spindle, cytokinesin, and cyclin B1/CDK1 complex (Figures 4D and 4E) and STRING analysis (https://string-db.org/) unveiled a hub of genes related to mitotic sister chromatid segregation and cyclin-associated events during G2/M transition (Figure 4F).
Figure 4.

DLK1-WRE editing affects ApcΔhep hepatocyte proliferation through inhibition of mitosis and cytokinesis regulators
(A) Percentage of Ki-67+ hepatocytes in WT, ApcΔhep-Rosa26, and ApcΔhep-DLK1/DIO3ΔWRE livers. (B) Percentage of liver to body weight ratio in WT, ApcΔhep-Rosa26, and ApcΔhep-DLK1/DIO3ΔWRE livers. (C) Expression of Top2a, Kif20b, Nuf2, and Nusap1 in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes relative to ApcΔhep-Rosa26 hepatocytes. (D–F) RNA-seq analysis on ApcΔhep−DLK1/DIO3ΔWRE and ApcΔhep-Rosa26 hepatocytes. (D) The histograms summarize ratio obtained with gene set enrichment analysis (GSEA) between the number of genes in the intersection of the query set with a set from MSigDB (k/K), with p value and FDR q-values for each item. (E) Schematic representation of the most significantly deregulated RNAs. (F) Main hub obtained by STRING analysis. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.001; ns, non-significant (Mann-Whitney), with error bars representing SEM.
FoxM1, a typical proliferation-associated transcription factor, is known to regulate the expression of genes involved in G2/M-transition and M-phase progression,29 including Kif20a, Ccna2, Cdc2, or Cenpf. Therefore, we hypothesized that FoxM1 might be the keystone linking the DLK1/DIO3 locus to proliferative actors. While we did not observe any difference in Foxm1 expression (Figure 5A) or nuclear localization (Figure 5B) between all conditions, we noticed that FoxM1 binding was decreased at Ccna2, Kif20a, and Cdc2 promoters in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes compared to ApcΔhep-Rosa26 hepatocytes (Figure 5C). We hypothesized that one lncRNA within the locus, either Meg3, Rian, or Mirg, could act as a guide for FoxM1 at these specific promoter regions. RNA immunoprecipitation (RIP) experiments showed that Meg3 was co-immunoprecipitated with FoxM1 in ApcΔhep-Rosa26 hepatocytes, while this association was undetectable in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (Figure 5D).
Figure 5.

DLK1-WRE editing impairs FoxM1 binding at Ccna2, Kif20a, and Cdc2 promoters
(A) RT-qPCR analysis of Foxm1 expression in ApcΔhep-Rosa26 and ApcΔhep-DLK1/DIO3ΔWRE hepatocytes compared to WT hepatocytes. (B) Number of Foxm1+ nuclei in IHC. (C) Representative images of ChIP-PCR targeting FoxM1 at Ccna2, Kif20a, Cdc2, and Cenpf promoters compared to isotype control in ApcΔhep-Rosa26 and ApcΔhep-DLK1/DIO3ΔWRE hepatocytes and inputs (#1 and 2 for ApcΔhep-Rosa26 hepatocytes, #3 and 4 for ApcΔhep-DLK1/DIO3ΔWREhepatocytes); the lower panel represents the PCR band quantification with ImageJ of all ChIP experiments against FoxM1 relative to isotype control. For the cdc2 promoter in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes, the cropped images are for two different mice analyzed on two gels with the same conditions of exposure. (D) Quantification of Meg3 RNA co-immunoprecipitated with FoxM1 in RIP-qPCR in ApcΔhep-DLK1/DIO3ΔWRE (n = 4) compared to ApcΔhep-Rosa26 hepatocytes (n = 2); data are represented as the relative binding compared to 18S. Figure made with BioRender. ∗p < 0.05, ∗∗p < 0.01; ns, non-significant (Kruskal-Wallis or Mann-Whitney), with error bars representing SEM.
Altogether, these results indicate that the upregulation of the DLK1/DIO3 locus, driven by the β-catenin-dependent DLK1-WRE enhancer, favors FoxM1-mediated cell proliferation in hepatocytes.
DLK1-WRE site editing impairs ApcΔhep HB and HCC growth through cell-autonomous mechanisms and immune remodeling
We studied the effect of DLK1-WRE editing on ApcΔhep and β-cateninΔexon3 tumor development (Figures S1B and S1C). At sacrifice, we obtained 50% HCC- and 50% HB-like tumors (Figures 6A and S6A). Around 50% of tumors were not edited at the DLK1-WRE site as an expected consequence of the probability rule for concomitant invalidation of both Apc and the DLK1-WRE site in the same hepatocyte (ApcΔhep-DLK1/DIO3WT) (Figures 6B and S6B). Retrospective analysis of tumor areas obtained by ultrasonography follow-up21,22 showed that HB-like tumor growth was reduced for ApcΔhep-DLK1/DIO3ΔWRE subgroups (Figure 6C, pink plots) compared to ApcΔhep-Rosa26 (Figure 6C, blue plots) and ApcΔhep-DLK1/DIO3WT (Figure 6C, green plots) HB-like tumors.
Figure 6.

DLK1-WRE site editing slows tumor growth of ApcΔhep HB through decreased expression of mitotic entry regulators
(A) Examples of GS staining of ApcΔhep HB showing a heterogeneous staining with many stromal cells; HB cells losing several metabolic features of mature hepatocytes express low level of GS compared to ApcΔhep HCC cells. (B) Analysis of tumor editing by PCR band quantification with ImageJ in ApcΔhep HB. The upper panel is a representative image obtained from NT tissue and tumors (T). (C) Progression of cumulative tumor areas in ApcΔhep-DLK1/DIO3ΔWRE HB and ApcΔhep-DLK1/DIO3WT HB compared to ApcΔhep-Rosa26 HB with cumulative area at sacrifice indicated in the right panel. (D) Representative images of Ki-67 staining on ApcΔhep HB with high or low proliferation rate (left panel) and quantification of Ki-67+ hepatocytes in percentage for all tumors (right panel). (E) RT-qPCR analysis of Rian and Mirg in ApcΔhep HB compared to their NT tissues. (F) RT-qPCR analysis of Mki67, Ccna2, Nuf2, Top2a, Axin2, Kif20b, Nusap1, Cenpf, and Ckap2 relative to their NT tissues. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001; ns, non-significant (Kruskal-Wallis), with error bars representing SEM.
The slower growth of ApcΔhep-DLK1/DIO3ΔWRE HB-like tumors was associated with less Ki-67+ staining (Figure 6D). Rian, Mirg, Mki67, Ccna2, Nuf2, Top2a, Kif20b, Ckap2, Cenpf, and Nusap1 were underexpressed in ApcΔhep-DLK1/DIO3ΔWRE HB-like tumors compared to ApcΔhep−Rosa26 HB-like tumors and, in most cases, compared to ApcΔhep-DLK1/DIO3WT HB-like tumors (Figures 6E and 6F). Moreover, Axin2 was also decreased in ApcΔhep-DLK1/DIO3ΔWRE HB-like tumors compared to ApcΔhep−Rosa26 HB-like tumors (Figure 6F), which probably reflects decreased β-catenin signaling in these tumors.
In both ApcΔhep and β-cateninΔexon3 HCC (Figures S6C and S7B, respectively), we observed a decrease in tumor growth for DLK1/DIO3ΔWRE HCC (pink/purple plots) compared to Rosa26 HCC (blue plots). DLK1-WRE editing was associated with lower Ki-67 staining (Figure S7C) and decreased Mki67, Top2a, and Kif20b expression levels (Figure S7D) in β-cateninΔexon3-DLK1/DIO3ΔWRE HCC in agreement with the significant reductions in Rian, Mirg, and miR-127 expressions (Figure S7E). In ApcΔhep-DLK1/DIO3ΔWRE HCC, DLK1-WRE editing in the tumors tended to reduce Rian and Mirg expression (Figure S6D) in a non-significant manner, probably because of the higher basal level observed in Dlk1/Dio3 locus expression in ApcΔhep HCC (Figures 1D and 1E). This mild repression of the Dlk1/Dio3 locus was associated with comparable number of Ki67+ cells (Figure S6E) and Mki67, Ccna2, Top2a, and Kif20a expression levels (Figure S6F). However, ApcΔhep-DLK1/DIO3WT tumor size and growth were significantly impaired compared to ApcΔhep-Rosa26 tumors (Figure S6C, green plot). Unfortunately, we could not obtain β-cateninΔexon3-DLK1/DIO3WT HCC to confirm this observation, as a consequence of the concomitant injection and subsequent co-entry of CRISPR-Cas9 constructs into hepatocytes. By comparing ApcΔhep and β-cateninΔexon3 models, we nevertheless observed significant differences in Mki67, Ccna2, and Top2a expression, which were drastically higher in β-cateninΔexon3 non-tumor tissues compared to ApcΔhep non-tumor tissues (Figure S7F). This could suggest that the proliferative index of the adjacent non-tumor tissue could also influence β-catenin-driven HCC growth and efficacy of DLK1-WRE editing.
Next, since oncogenic β-catenin is known to trigger an inflammatory response associated with a significant infiltration of immune cells in ApcΔhep livers,30 we analyzed immune cell proportions by flow cytometry. In both ApcΔhep and β-cateninΔexon3 DLK1/DIO3ΔWRE tumors, monocyte infiltration was reduced as compared to Rosa-26 controls (Figures S8A and S8C), in association with decreased expression in Ccl2, Ccl5, and Csf1 mRNAs, encoding three chemokines essential for monocyte recruitment (Figures S8B and S8D). This suggests that the immune response fostered by oncogenic β-catenin signaling could arise in part downstream of the activation of the DLK1/DIO3 locus and potentially contribute to its tumor-promoting effect.
Altogether, our results show that β-catenin-driven HB growth is mainly dependent on the level of activation of the DLK1/DIO3 locus in the tumor itself, while β-catenin-driven HCC growth seems to be affected by the DLK1/DIO3 level, not only in the tumor but also in the surrounding tissue. They also point to the activation of the DLK1/DIO3 locus as a key intermediate in the oncogenic β-catenin-dependent infiltration of monocytes in the tumors.
Editing of the DLK1-WRE site impairs the pro-tumorigenic capacities of human and mouse hepatic cancer cell lines harboring β-catenin mutations
Finally, we investigated the impact of DLK1-WRE editing in transformed cell lines from mouse and human hepatic cancers harboring β-catenin mutations. The DLK1/DIO3ΔWRE Hepa1-6 clones, characterized in Figures S5A–S5C, were less proliferative than the Rosa26 clones (Figure 7A). This was accompanied by a significant increase in the number of DLK1/DIO3ΔWRE cells in G2/M phase (Figure 7B) together with a decrease in cyclin B1 protein level, a protein required for mitotic initiation (Figure 7C). As observed in ApcΔhep-DLK1/DIO3ΔWRE hepatocytes (Figure 5D), RIP experiments showed that Meg3 co-immunoprecipitation with FoxM1 was decreased in Hepa1-6-DLK1/DIO3ΔWRE clones compared to Hepa1-6-Rosa26 clones (Figure 7D). Consistent with in vitro data, Hepa1-6-DLK1/DIO3ΔWRE clones also exhibited decreased tumorigenic capacity compared to Hepa1-6-Rosa26 clones after subcutaneous allografting into Nu/Nu mice. Tumor progression was significantly slower for Hepa1-6-DLK1/DIO3ΔWRE clones (Figure 7E) with 2.6-fold lower mean tumor volume and 4-fold lower weight at the time of sacrifice (Figure 7F). Impaired DLK1/DIO3ΔWRE tumor progression in Nu/Nu mice was consistent with less Ki-67+ staining (Figure 7G) and more cells harboring cleaved caspase-3 (Figure 7H) compared to Rosa26 tumors. According to Targetscan and Dianalab algorithms, FADD, a pro-apoptotic actor, is a potential target of miR-134 produced within the DLK1/DIO3 locus. We found that Fadd was increased in DLK1/DIO3ΔWRE Hepa1-6 tumors at both the mRNA and protein level (Figures 7I and 7J), and could favor caspase-3 cleavage.
Figure 7.

DLK1-WRE site editing impairs the pro-tumorigenic capacities of murine hepatoma Hepa1-6 cells mutated for Ctnnb1
(A–D) Analysis of DLK1/DIO3ΔWRE Hepa1-6 clones versus Rosa26 control clones. Proliferation rate at 48 h (A); percentage of cells in G2/M phase determined by flow cytometry (B); cyclin B1 and A2 protein level determined by western blot (C); representative quantification of Meg3 RNA co-immunoprecipitated with FoxM1 in RIP-qPCR (n = 2) with data represented as the relative binding compared to 18S (D). (E–K) Analysis of DLK1/DIO3ΔWRE Hepa1-6 tumors versus Rosa26 tumors. Tumor volumes measured every 2 days (E) and tumor weights at sacrifice (F); percentage of Ki67+ tumor cells (G); percentage of tumor cells with cleaved caspase-3 (H); Fadd level determined by RT-qPCR (I); FADD protein level determined by western blot (J); Glul expression determined by RT-qPCR (K). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.0001 (Mann-Whitney),with error bars representing SEM.
The active DLK1-WRE site being conserved in human hepatic cell lines and HB tumors (Figures 3A and S5H), we also generated DLK1/DIO3ΔWRE clones from the Huh6 cell line, a human HB cell line with a CTNNB1 mutation (Figure S5H). Editing of the DLK1-WRE site led to reduced expression of RIAN, MIRG, MEG3, and RTL-1 (Figure S5I). Proliferation of DLK1/DIO3ΔWRE Huh6 clones was slower than that of control clones (Figure 8A). In line with this, a significant increase in the number of DLK1/DIO3ΔWRE cells in G2/M phase was noticed (Figure 8B). DLK1/DIO3ΔWRE clones had a significantly reduced capacity to grow as spheroids (Figure 8C). In vivo, DLK1/DIO3ΔWRE Huh6 clones exhibited decreased tumorigenic capacity compared to control clones after subcutaneous xenografting into Nu/Nu mice (Figure 8D). A 4-fold lower mean tumor volume and weight were observed at sacrifice (Figures 8D and 8E). DLK1/DIO3ΔWRE Huh6 tumors displayed a reduced number of Ki67+ cells (Figure 8F) and decreased MKI67 expression (Figure 8G). Importantly, as previously observed in mouse ApcΔhep hepatocytes (Figure 5C), FOXM1 was found to bind CCNA2, CDC2, and KIF20A promoters in human Huh6 clones (Figure 8H, blue squares) but was not recruited in DLK1/DIO3ΔWRE Huh6 clones (Figure 8H, pink dots). DLK1/DIO3ΔWRE Huh6 tumors also displayed a higher number of cells harboring cleaved caspase-3 (Figure 8I) and increased FADD mRNA and protein levels (Figures 8J and 8K). In both DLK1/DIO3ΔWRE Hepa1-6 (Figure 7K) and Huh6 grafted tumors (Figure 8L), GLUL expression was decreased, in agreement with inhibition of β-catenin signaling in DLK1/DIO3ΔWRE tumors, as previously noticed for mouse HB-like tumors (Figure 6F).
Figure 8.

DLK1-WRE site editing impairs the pro-tumorigenic capacities of human Huh6 HB cells mutated for CTNNB1
(A–C) Analysis of DLK1/DIO3ΔWRE Huh6 clones versus non-edited clones. Proliferation rate at 48 h (A); percentage of cells in G2/M phase determined by flow cytometry (B); number of spheroids (C). (D–L) Analysis of DLK1/DIO3ΔWRE Huh6 tumors versus non-edited tumors. Tumor volumes measured every 2 days (D); tumor weights at sacrifice (E); percentage of Ki67+ tumor cells (F); MKI67 level determined by RT-qPCR (G); representative FOXM1 binding at CCNA2, KIF20A, and CDC2 promoters normalized to isotype control in ChIP-qPCR experiments (n = 2) (H); percentage of tumor cells with cleaved caspase-3 (I); FADD mRNA level determined by RT-qPCR (J); FADD protein level determined by western blot (K); GLUL expression determined by RT-qPCR (L). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.0001 (Mann-Whitney)with error bars representing SEM
In conclusion, the DLK1/DIO3 locus contributes to the pro-tumorigenic capacities of transformed human hepatic cancer cells harboring β-catenin mutations by enhancing their proliferation through the FOXM1 axis and by decreasing FADD-dependent apoptotic programs both in vitro and in vivo.
Discussion
Imprinted loci play major roles in cellular plasticity and cell reprogramming during cancer. More particularly, the DLK1/DIO3 locus, also known as the 14q32.2 cluster, is crucial for cell proliferation and metabolic adaptation in the liver.13 In human and mouse HB, we found that RNAs within the DLK1/DIO3 locus are highly expressed and correlated with oncogenic activation of β-catenin,3,31 as reported by others.18 Here, we have identified a regulatory site, existing in mouse and human, responsible for the DLK1/DIO3 locus induction driven by oncogenic TCF-4/β-catenin complexes and unveiled its functional impact on cell proliferation and apoptosis (Figure S9). We have also shown that dysregulation of the Dlk1/Dio3 locus occurs in mouse HCCs emerging from healthy livers. Importantly, we have demonstrated that part of the oncogenic role of the DLK1/DIO3 locus involves the DLK1-WRE enhancer site in vivo. Altogether, our work provides strong arguments for the therapeutic benefit of a targeted repression of the DLK1-WRE enhancer.
In our study, we have identified the regulatory region activated in the case of sustained β-catenin activation during the very early steps of liver tumorigenesis and determined how this region promotes DLK1/DIO3 locus transcription. This transcriptional regulation requires the binding of oncogenic β-catenin/TCF-4 complexes at the DLK1-WRE site, which then becomes an active enhancer. This site is also engaged in chromatin remodeling and long-range chromatin interactions to bridge the enhancer region with other regulatory sites at the Ig-DMR and in downstream regions. This DNA looping role for β-catenin/TCF-4 complexes echoes two works published by Yochum et al. showing that β-catenin coordinates chromatin looping at an enhancer site upstream of MYC, a canonical β-catenin target in colon cancer.32,33 Furthermore, this regulatory mechanism is also reminiscent of chromatin looping and enhancer-promoter bridging as a way to escape silencing for the imprinted DLK1/DIO3 locus.34 To determine the allele of origin for the regulatory functions that we unveiled at the DLK1-WRE site, we needed to generate hybrids of our transgenic mice. Nevertheless, we did not find any change in DNA methylation status at the Ig-DMR region in ApcΔhep hepatocytes, which is rather in favor of an absence of imprinting loss.
Besides increasing knowledge on the regulation of the DLK1/DIO3 locus, our editing strategy has also deciphered the pro-tumorigenic events subsequent to its activation by β-catenin signaling. Using a CRISPR-Cas9 strategy in mouse, we showed that editing of the DLK1-WRE site inhibited HB and HCC growth in healthy livers. Nevertheless, the antitumor activity of DLK1-WRE editing was the strongest in ApcΔhep HB-like tumors. We also observed a decrease in HCC growth, notably in β-cateninΔexon3 HCC with moderate activation of the Dlk1/Dio3 locus. In ApcΔhep HCC, it appears that tumor growth is highly dependent on Dlk1/Dio3 locus expression in the tumor microenvironment: inhibition of tumor growth of ApcΔhep-DLK1/DIO3WT HCC is similar to that of ApcΔhep-DLK1/DIO3ΔWRE HCC, arguing that an impairment of the DLK1/DIO3 locus in non-tumor tissues could also modulate HCC progression. HCC is the paradigm of inflammation-associated cancer emerging in cirrhotic livers in 80% of cases. The DLK1/DIO3 locus expression has been found to be upregulated in response to several types of stress, particularly in hepatocytes under metabolic disorders35 and lipid overload.36 We have exploited public RNA-seq datasets generated on samples from patients with diseased livers at high risk of HCC (GSE126848 and GSE142530). Interestingly, we found that the expression of RTL1, within the locus, and AXIN2, a canonical target of β-catenin, was upregulated during alcoholic hepatitis and metabolic dysfunction-associated steatohepatitis (MASH). This presumes an activation of β-catenin signaling in diseased livers, as reported by other studies showing that CTNNB1-mutated HCCs are significantly over-represented under MASH context.37,38 These results are also in agreement with data obtained by others in db/db and ob/ob mice,36,39 as well as data generated in our lab with mice fed with choline-deficient diet and methionine-choline-deficient diets, showing an increased expression in Mirg, Rian, and Meg3 (unpublished data).
Besides their roles in hepatocytes, the dysregulation of ncRNAs within the locus has also been reported in immune cells during inflammation, particularly in macrophages.40,41 Here, we exemplified that DLK1-WRE editing in both ApcΔhep and β-cateninΔexon3 tumors impaired monocyte infiltration and expression of Ccl2, Ccl5, and Csf1 encoding chemokines essential for monocyte recruitment. This could open new perspectives regarding the role played by the locus in the control of immune cell populations, with the underlying molecular mechanisms remaining to be deciphered. This also reinforces the interest to impair locus expression during chronic liver diseases preceding HCC.
Our data obtained on DLK1/DIO3ΔWRE hepatocytes and transformed hepatic cell lines revealed that inhibition of cell proliferation and tumor progression was associated with a decrease in the mRNA levels of several actors involved in cytokinesis and G2/M phase, the cell phase during which the levels of β-catenin rise to a peak.42 Interestingly, a hub between cell cycle actors and miRNAs within the DLK1/DIO3 locus has also been inferred by bioinformatics analysis of HB datasets.43 Here, our study supports that the proliferative impact of the DLK1/DIO3 locus in preneoplastic hepatocytes is dependent on the β-catenin-driven DLK1-WRE enhancer and the subsequent redistribution of FoxM1 at the promoters of cell cycle actors. Meg3 appears as a potential guiding partner for FoxM1 at these promoters to regulate their transcription. FOXM1 is involved in the progression of several cancers, including HB,44 and compounds inhibiting FOXM1 have recently demonstrated encouraging antitumor activities.45 This could open new innovative treatment strategies against FOXM1 as an alternative target for the treatment of CTNNB1-mutated cancers.
Altogether, our results unveil how a sustained activation of β-catenin signaling can remodel the epigenetic and chromatin landscape of one of its key oncogenic targets with a subsequent effect on proliferative gene signatures. We put forward the idea that targeting the DLK1-WRE site represents a potent strategy to specifically repress the DLK1/DIO3 locus in liver tumors harboring β-catenin mutations. Regarding the crucial role of β-catenin in tissue homeostasis and repair after injury, many drugs targeting this pathway have failed in treatments because of their toxicities. Targeting downstream events, as we have outlined here with the DLK1/DIO3 locus, appears as a promising option. This could be performed by gene editing therapies with zinc-finger nucleases, which gave promising results in hemophilia B (NCT02695160 ongoing). The development of in silico approaches to predict genome-wide off-targets will open up new horizons for gene editing therapy, which could benefit our proposed strategy against the DLK1/DIO3 enhancer in particular in HB but also probably during chronic liver diseases and HCC.
Material and methods
In vivo CRISPR-Cas9 design and gene editing analysis
sgRNAs against the DLK1-WRE site were designed as previously for β-cateninΔExon3 (Table S9).23 DNA was extracted from edited cells, livers, and tumors and analyzed as previously (Table S9).23 PCR products were run on E-Gel 2% for 10 min (Thermo Fisher, Waltham, MA) and bands quantified with ImageJ.
Murine models
The Apclox/lox model was edited for the DLK1-WRE site with 3.6 × 1011 Vg (viral genomes) of sg2 plus 1.9 × 1011 Vg of sg5 (DLK1/DIO3ΔWRE) 1 month before β-catenin activation as reported elsewhere.23 Intraperitoneal injection of 2 mg of tamoxifen (MP Biomedicals, Irvine, CA) in Apclox/lox TTR-CreERT2 mice resulted in Apc deletion in ≥90% of hepatocytes for preneoplastic studies 6 days post injection (pretumoral ApcΔhep model; Figure S1A).19,20,21 A unique injection of 1 × 109 Vg of an Ad5-Cre-GFP adenovirus led to tumors within 4–6 months,20 which were monitored by 2D-ultrasound (Vevo 2100, Visualsonics, Toronto, Canada), as previously published (Figure S1B).21 The tamoxifen procedure to inactivate β-catenin in mice carrying a biallelic floxed Ctnnb1 gene with loxP sites located between exons 2 and 6 and a TTR-CreTam (βcatΔhep) was reported elsewhere.5,46,47 DEN-induced livers tumors were obtained by intraperitoneal injection of 0.25 mg of DEN (Sigma Aldrich, St. Louis, MO) in 14-day-old male mice. The HNF4αΔhep model was obtained as described earlier.48,49
For the β-cateninΔExon3 model, AAV8 against the DLK1-WRE site and the exon 3 of Ctnnb123 were concomitantly administered and mice were monitored as described above (Figure S1C).
Kinetic hepatocyte sorting was performed after retro-orbital injection of 1.5 × 109 particles of Ad5-Cre-GFP adenovirus, liver perfusion with collagenase as described previously,21 and GFP sorting on an ARIA3 (BD, Franklin Lakes, NJ) (Figure S1D).
Subcutaneous allografts were performed with 2 × 106 cells on both flanks from 5-week-old female Nu/Nu nude mice as reported elsewhere.50 All animal procedures were carried out according to French legal regulations and approved by an ethical committee (agreements 17-082, 14009, and 16420).
Human samples
All samples were recruited in accordance with European and French law and institutional ethical guidelines. For HB cohort 1 (N = 22; Table S3), RNA-seq was performed on total RNA extracted from tumoral and adjacent non-tumor tissues surgically resected from 22 patients using the mirVana kit (Thermo Fisher, Waltham, MA) according to the supplier’s protocol. The RNA integrity number was evaluated by Agilent 2100 Bioanalyzer. Two micrograms were used for generation of each small RNA library with an Illumina TruSeq Small RNA Sample Prep Kit according to standard protocol (Illumina, San Diego, CA). Single-read 50-nt sequencing was performed by MGX-Montpellier GenomiX platform on an Illumina HiSeq 2000 using the sequence-by-synthesis technique. Adapter sequences were trimmed from small RNA reads using the Cutadapt (version 1.4.1) tool (http://code.google.com/p/cutadapt/), retaining reads of the size 16–25 nt. Reads were then mapped to the human hairpin sequences (mirBase version 21) with Bowtie (v1.0). The number of reads mapping in the sense orientation to each hairpin in each patient was used as an input for further analysis. Data were analyzed using DESeq2 package in R studio. The miRNA expression file was loaded in format .txt to obtain a matrix with the value in i-th row corresponding to miRNA names and value in j-th column corresponding to patient samples. We followed the DESeq2 manual,51 performed differential miRNA expression analysis (DE), and produced output files including heatmaps and tables. After the DE analysis, DESeq2 produces a set of values (base mean, log2 fold change, and adjusted p value) for each miRNA between tumoral and non-tumor samples. Once exported, our data were classified in an Excel file and analyzed with the help of miRBase and UCSC Browser (https://genome.ucsc.edu/).
For the HB cohort 2 (N = 100; Table S4), RNA-seq was performed as previously described.3,52,53 Gene expression levels were calculated using the variance stabilizing transformation (VERSUST) and the raw count matrix. Gene-expression-based classification of HB was done as previously described.3 Statistical analysis and data visualization were performed using R software version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria; https://www.R-project.org) and Bioconductor packages.
RNA-seq data from a collection of livers with varying degrees of non-alcoholic fatty liver disease compared with healthy livers from normal-weight individuals (GSE126848) and liver biopsies from 28 patients with alcoholic hepatitis or cirrhosis compared to healthy controls (GSE142530) were retrieved with GEO and analyzed using the R package DESeq2 (v.1.38).51
Cell culture
Hepatocytes were isolated 4 days after tamoxifen injection in Apclox/lox TTR-CreERT2 mice and maintained at 37°C in a humidified atmosphere containing 5% CO2 as reported elsewhere.21 Hepa1-6 cells were obtained from the American Type Culture Collection and Huh6 cells from C. Perret’s lab.1 They were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and 50 U/mL penicillin-streptomycin at 37°C in a humidified atmosphere containing 5% CO2 (Thermo Fisher, Waltham, MA). Stable clones were obtained following co-transfections of sgRNA- and pmax-GFP plasmids, GFP-based sorting (ARIA3), and amplification of selected clones.
Proliferation/cell cycle analysis
Cell proliferation was measured on 8,000 cells with the xCELLigence system (Agilent, Santa Clara, CA) (N = 3), as previously described.21 Cell cycle analysis was performed on 5 × 105 synchronized cells (with 24h FBS deprivation for Huh6 cells and 10 μg/mL colchicine for Hepa1-6 cells), fixed 48 h later in PBS-20%/ethanol-80% at −20°C for 15 min and stained with FxCycle PI/RNAse solution (Thermo Fisher, Waltham, MA) for 30 min at room temperature before analysis (Fortessa, BD).
Sphere formation assay
One thousand Huh6 clones were grown during 14 days onto ultra-low-attachment six-well plates (Corning, Corning, NY) in DMEM/F12 medium supplemented with B27, 20 ng/mL epidermal growth factor (EGF), 20 ng/mL basic fibroblast growth factor (FGF), and 100 μg/mL gentamycin (Thermo Fisher, Waltham, MA) (N = 3).
RNA extraction and RT-qPCR
Levels of miRNAs and mRNAs were determined on total RNA extracted with Trizol reagent (Thermo Fisher, Waltham, MA), as previously reported (Table S9).21
Quantitative chromosome conformation capture (3C-qPCR)
The 3C-qPCR experiment was adapted from Braem et al. and Rebouissou et al.26,54 and conducted on 5 million nuclei isolated from hepatocytes lysed in homogenization buffer with an Ultra-Turrax during 20 min on ice and centrifuged for 1 h at 20,000 × g. The 3C assays were performed with EcoRI digestion, product ligation, and secondary XbaI digestion (N ≥ 3).26 Sample purity and DNA content were determined with internal primers against Gapdh and digestion efficiency with three primer sets (Table S9). For all 3C experiments, qPCR primers used were as previously published with the anchor F16 located at the beginning of the DLK1-WRE site and thus not affected by the CRISPR-Cas9 constructs used.55 Their efficiencies were determined on equimolar amounts of 3C-ligation products generated from BAC RPCI-23 clone 117C15 (Thermo Fisher, Waltham, MA) covering the genome segment between DLK1 and miR-136 genes. The 3C data were normalized to the basal interaction level using the previously published algorithm.26
ATAC-seq and ATAC-qPCR
The 50,000 isolated hepatocytes were transposed for 30 min in a 50 μL reaction mix containing 4.5 μL of transposase (kit #FC-121-103, Illumina, San Diego, CA) and 0.1% digitonin (adapted from Corces et al.56) and the initial protocol was followed after transposition57 for ATAC-seq and ATAC-qPCR experiments (Table S9). For liver samples, omni-ATAC-seq was performed on 50,000 nuclei, isolated as for 3C assays, according to Corces et al.58 in a 50 μL reaction mix with 2.5 μL of transposase, 0.01% digitonin, and 0.1% Tween 20 for 30 min. The following steps were according to the initial protocol.57
Libraries were controlled using a 2100 Bioanalyzer, and an aliquot of each library was sequenced at low depth onto a MiSeq platform to control duplicate level and estimate DNA concentration. Each library was then paired-end sequenced (2 × 100 bp) on a HiSeq instrument to get 40 million read pairs on average. As ATAC-seq libraries are composed in large part of short genomic DNA fragments, and in order to reduce costs, we sequenced our recent libraries on a Nextseq instrument (2 × 38 bp). Our analysis showed that reducing read length to 38 bp does not affect mapping efficiency. Reads were first cleaned using Trimmomatic (removing of adaptors and low-quality bases). Trimmed reads were then aligned to the mouse genome (mm9) using Bowtie2 with the parameter -X2000, and with two mismatches permitted in the seed (default value). The -X2000 option allows fragments <2 kb to align. Duplicated reads were removed with picard-tools. Resulted bam datasets were then converted to BigWig, a coverage track adapted to visualize datasets in UCSC Genome Browser or IGV. Conversion was performed using bamCoverage command from deepTools with the parameters --binSize 10 --normalizeUsing RPKM --extendReads. The parameter --normalizeUsing RPKM is used to normalize each dataset. We selected the normalization method based on reads per kilobase per million mapped reads (RPKM), which calculates the number of reads per bin/number of mapped reads (in millions). The parameter --extendReads allows the extension of reads to fragment size. The default value is estimated from the data (mean of the fragment size of all mate reads).
In human HB, Multiome approach was performed by Integragen SA (Evry, France) on matched non-tumor livers (n = 2) and HBs (n = 3) of two patients, according to the commercial Chromium Single Cell Multiome ATAC + Gene Expression protocol. We used 10X Genomics Cell Ranger ARC 2.0.0 to align snATAC-seq reads to the human genome (GrCh38/hg38) (Roehrig et al. in revision).
ChIP and RIP
ChIP was performed as previously described21 on 25 μg of chromatin with 30 μL of protein A/G(v/v) Dynabeads with antibodies of interest (Table S10). For tumors, samples were homogenized with an Ultra-Turrax in 1% formaldehyde for 1 min. For RIP experiments, beads were reverse crosslinked before RNA isolation by Trizol (Thermo Fisher, Waltham, MA).
RNA-seq/small RNA-seq
RNA-seq and small RNA-seq were performed on 1 μg of total RNA extracted from ApcΔhep-ROSA26 versus DLK1/DIO3ΔWRE hepatocytes (N ≥ 4), respectively, with TruSeq Stranded after ribodepletion and TruSeq Small RNA and sequenced with Nextseq 500 (150b) (Illumina, San Diego, CA).
Fastq files were then aligned using STAR algorithm (version 2.7.6a), on the Ensembl Mus musculus GRCm38 reference release 96. Reads were then counted using RSEM (v1.3.1) and the statistical analyses on the read counts were performed with R (version 3.6.3) and the DESeq2 package (DESeq2_1.26.0) to determine the proportion of differentially expressed genes between two conditions. We used the standard DESeq2 normalization method (DESeq2’s median of ratios with the DESeq function), with a pre-filter of reads and genes (reads uniquely mapped on the genome, or up to 10 different loci with a count adjustment and genes with at least 10 reads in at least three different samples). Following the package recommendations, we used the Wald test with the contrast function and the Benjamini-Hochberg false discovery rate (FDR) control procedure to identify the differentially expressed genes. R scripts and parameters are available on GitHub (https://github.com/BSGenomique/genomic-rnaseq-pipeline/releases/tag/v1.0420). For small RNA-seq data analysis, Fastq files were uploaded on Qiagen geneglobe analysis software for alignment and counting. Then, UMI matrix were used as raw data for our R and DESeq2 pipeline.
Kinetic RNA-seq was performed on HiSeq 4000 in paired end on at least three independent samples of sorted GFP+ ApcΔhep hepatocytes at day 6, 15, and 21 after injection compared to GFP− hepatocytes.
Immunostaining/in situ hybridization
Paraffin-embedded liver sections were treated and labeled as previously21,23 with antibodies and probes of interest (Tables S9 and S10).
Western blot
Experiments were conducted on 20 μg of total proteins as reported elsewhere (Table S10).21
Flow cytometry
Livers and tumors were minced with scissors in DMEM containing collagenase IV (2.5 mg/mL, Sigma Aldrich, St. Louis, MO) and incubated for 30 min at 37°C. Cell suspensions were passed through a 100-μm filter and stained with appropriate antibodies for 30 min on ice as previously (Table S10).59 Data were acquired on a BD LSR Fortessa flow cytometer (BD Franklin Lakes, NJ) and analyzed with FlowJo software. Absolute cell count was calculated as previously with nonfluorescent beads and expressed as a number of cells per milligram of tissue.59
Statistical analysis
We assessed the significance of differences between two groups of samples using Mann-Whitney tests and between three groups of samples using Kruskal-Wallis. p < 0.05 was considered statistically significant. For human samples, difference in gene expression levels, in two or more than two groups, was tested using Wilcoxon or Kruskal-Wallis tests, respectively. Correlation analysis was performed using Pearson r correlation when both variables were normally distributed with the assumptions of linearity and homoscedasticity or Spearman’s rank-order correlation.
Data and code availability
All data were deposited on GEO: RNA-seq and small RNA-seq comparing ApcΔhep ROSA/DLK1/DIO3ΔWRE hepatocytes on GSE206262, ATAC-seq on GSE211930, kinetic RNA-seq on GSE210482), and MeDIP-seq on GSE239777. Others have been previously published (project: PRJNA150641 in European nucleotide archive (ENA).5,21,23
Acknowledgments
We warmly thank for their great help during this project the three platforms Cybio (Karine Bailly and Muriel Andrieu), Genomic (Franck Letourneur and Lucie Audoux), and PIV (Franck Lager, Carmen Marchiol, and Gilles Renault) from Cochin Institute. We warmly thank the animal facilities from the Cordeliers research center and from Saint-Antoine research center (Tatiana Ledent). We also thank Helene Forher-Ting from CHIC platform at Cordeliers research center and INSERM UMR 1089 for AAV production. We warmly thank Katarzina Hooks for small RNA-seq analysis of HB cohort 1, and the two laboratory banks and L2PGH labs from CNRGH for kinetic RNA-seq in mouse. We thank Pr. Zhang for the gift of AAV8-SaCas9 constructs.
This work was supported by and received recognition from the Ligue Nationale Contre le Cancer (Team labellisation), by the Institut National de la Santé et de la Recherche Médicale (INSERM), by the Plan cancer (grant CHROMALIV), the Association Française pour l'Etude du Foie (AAP 2017), the Fondation pour la Recherche Médicale (grant no. DBI20131228566), the Agence Nationale de la Recherche (DLK1-EPILIV 2018–2021), the Institut National Du Cancer (INCa Emergence 2017; grant INCa_TRANSLA_2013_209), and the charity associations Eva pour la Vie, Aidons Marina, and E.S.CA.PE. to MIRCADE team. J.S. was funded by the Université Paris Cité. C.J. was funded by the Ligue Nationale Contre le Cancer. A.R. was supported by the Fondation pour la Recherche Médicale, grant number ECO201906008977.
Author contributions
J.S., investigation, formal analysis, methodology, validation, writing – original draft; L.P., investigation, formal analysis, methodology, validation; C.J., formal analysis, investigation, validation; I.L., investigation; S.C., formal analysis; S.P., investigation; C.D.-M., investigation; C.G., investigation, methodology; A.I., investigation; E.M., investigation; C.D., investigation; S.C., investigation, methodology, formal analysis; A.R., investigation, formal analysis; K.M., formal analysis; B.S.-P., formal analysis; J.-F.D., funding acquisition; S.M-R., resources, formal analysis; T.F., methodology, formal analysis, writing – original draft; C.F.G., resources, formal analysis; J.Z.-R., resources; S.C., conceptualization, funding acquisition, supervision; A.G., investigation, formal analysis, methodology, validation, writing – original draft, conceptualization, funding acquisition, and supervision.
Declaration of interests
Two patents, PCT/EP2023/053419 and EP22305162.4, have been deposited by J.S., L.P., S.C., and A.G.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.01.036.
Supplemental information
References
- 1.de La Coste A., Romagnolo B., Billuart P., Renard C.A., Buendia M.A., Soubrane O., Fabre M., Chelly J., Beldjord C., Kahn A., Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei Y., Fabre M., Branchereau S., Gauthier F., Perilongo G., Buendia M.A. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene. 2000;19:498–504. doi: 10.1038/sj.onc.1203356. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch T.Z., Pilet J., Morcrette G., Roehrig A., Monteiro B.J.E., Molina L., Bayard Q., Trépo E., Meunier L., Caruso S., et al. Integrated Genomic Analysis Identifies Driver Genes and Cisplatin-Resistant Progenitor Phenotype in Pediatric Liver Cancer. Cancer Discov. 2021;11:2524–2543. doi: 10.1158/2159-8290.CD-20-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadigan K.M., Waterman M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gougelet A., Torre C., Veber P., Sartor C., Bachelot L., Denechaud P.D., Godard C., Moldes M., Burnol A.F., Dubuquoy C., et al. T-cell factor 4 and beta-catenin chromatin occupancies pattern zonal liver metabolism in mice. Hepatology. 2014;59:2344–2357. doi: 10.1002/hep.26924. [DOI] [PubMed] [Google Scholar]
- 6.Gougelet A., Colnot S. A Complex Interplay between Wnt/beta-Catenin Signalling and the Cell Cycle in the Adult Liver. Int. J. Hepatol. 2012;2012 doi: 10.1155/2012/816125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosimann C., Hausmann G., Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 2009;10:276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 8.Boyault S., Rickman D.S., de Reyniès A., Balabaud C., Rebouissou S., Jeannot E., Hérault A., Saric J., Belghiti J., Franco D., et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 9.Nagae G., Yamamoto S., Fujita M., Fujita T., Nonaka A., Umeda T., Fukuda S., Tatsuno K., Maejima K., Hayashi A., et al. Genetic and epigenetic basis of hepatoblastoma diversity. Nat. Commun. 2021;12:5423. doi: 10.1038/s41467-021-25430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cairo S., Armengol C., De Reyniès A., Wei Y., Thomas E., Renard C.A., Goga A., Balakrishnan A., Semeraro M., Gresh L., et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer cell. 2008;14:471–484. doi: 10.1016/j.ccr.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 11.da Rocha S.T., Edwards C.A., Ito M., Ogata T., Ferguson-Smith A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 12.Benetatos L., Vartholomatos G., Hatzimichael E. DLK1-DIO3 imprinted cluster in induced pluripotency: landscape in the mist. Cell. Mol. Life Sci. 2014;71:4421–4430. doi: 10.1007/s00018-014-1698-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labialle S., Marty V., Bortolin-Cavaillé M.L., Hoareau-Osman M., Pradère J.P., Valet P., Martin P.G.P., Cavaillé J. The miR-379/miR-410 cluster at the imprinted Dlk1-Dio3 domain controls neonatal metabolic adaptation. EMBO J. 2014;33:2216–2230. doi: 10.15252/embj.201387038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benetatos L., Hatzimichael E., Londin E., Vartholomatos G., Loher P., Rigoutsos I., Briasoulis E. The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell. Mol. Life Sci. 2013;70:795–814. doi: 10.1007/s00018-012-1080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J., Zhang X., Zhang M., Zhu J.D., Zhang Y.L., Lin Y., Wang K.S., Qi X.F., Zhang Q., Liu G.Z., et al. Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis. 2007;28:1094–1103. doi: 10.1093/carcin/bgl215. [DOI] [PubMed] [Google Scholar]
- 16.Luk J.M., Burchard J., Zhang C., Liu A.M., Wong K.F., Shek F.H., Lee N.P., Fan S.T., Poon R.T., Ivanovska I., et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J. Biol. Chem. 2011;286:30706–30713. doi: 10.1074/jbc.M111.229831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honda S., Chatterjee A., Leichter A.L., Miyagi H., Minato M., Fujiyoshi S., Ara M., Kitagawa N., Tanaka M., Tanaka Y., et al. A MicroRNA Cluster in the DLK1-DIO3 Imprinted Region on Chromosome 14q32.2 Is Dysregulated in Metastatic Hepatoblastomas. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.513601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrillo-Reixach J., Torrens L., Simon-Coma M., Royo L., Domingo-Sàbat M., Abril-Fornaguera J., Akers N., Sala M., Ragull S., Arnal M., et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J. Hepatol. 2020;73:328–341. doi: 10.1016/j.jhep.2020.03.025. [DOI] [PubMed] [Google Scholar]
- 19.Benhamouche S., Decaens T., Godard C., Chambrey R., Rickman D.S., Moinard C., Vasseur-Cognet M., Kuo C.J., Kahn A., Perret C., Colnot S. Apc tumor suppressor gene is the "zonation-keeper" of mouse liver. Dev. Cell. 2006;10:759–770. doi: 10.1016/j.devcel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Colnot S., Decaens T., Niwa-Kawakita M., Godard C., Hamard G., Kahn A., Giovannini M., Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA. 2004;101:17216–17221. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gougelet A., Sartor C., Bachelot L., Godard C., Marchiol C., Renault G., Tores F., Nitschke P., Cavard C., Terris B., et al. Antitumour activity of an inhibitor of miR-34a in liver cancer with beta-catenin-mutations. Gut. 2016;65:1024–1034. doi: 10.1136/gutjnl-2014-308969. [DOI] [PubMed] [Google Scholar]
- 22.Gougelet A., Sartor C., Senni N., Calderaro J., Fartoux L., Lequoy M., Wendum D., Talbot J.N., Prignon A., Chalaye J., et al. Hepatocellular Carcinomas With Mutational Activation of Beta-Catenin Require Choline and Can Be Detected by Positron Emission Tomography. Gastroenterology. 2019;157:807–822. doi: 10.1053/j.gastro.2019.05.069. [DOI] [PubMed] [Google Scholar]
- 23.Loesch R., Caruso S., Paradis V., Godard C., Gougelet A., Renault G., Picard S., Tanaka I., Renoux-Martin Y., Perret C., et al. Deleting the beta-catenin degradation domain in mouse hepatocytes drives hepatocellular carcinoma or hepatoblastoma-like tumor growth. J. Hepatol. 2022;77:424–435. doi: 10.1016/j.jhep.2022.02.023. [DOI] [PubMed] [Google Scholar]
- 24.Hu S., Cao C., Poddar M., Delgado E., Singh S., Singh-Varma A., Stolz D.B., Bell A., Monga S.P. Hepatocyte beta-catenin loss is compensated by Insulin-mTORC1 activation to promote liver regeneration. Hepatology. 2023;77:1593–1611. doi: 10.1002/hep.32680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin C., Wang P.Q., Xu W.P., Yang Y., Zhang Q., Ning B.F., Zhang P.P., Zhou W.P., Xie W.F., Chen W.S., Zhang X. Hepatocyte nuclear factor-4alpha reverses malignancy of hepatocellular carcinoma through regulating miR-134 in the DLK1-DIO3 region. Hepatology. 2013;58:1964–1976. doi: 10.1002/hep.26573. [DOI] [PubMed] [Google Scholar]
- 26.Braem C., Recolin B., Rancourt R.C., Angiolini C., Barthès P., Branchu P., Court F., Cathala G., Ferguson-Smith A.C., Forné T. Genomic matrix attachment region and chromosome conformation capture quantitative real time PCR assays identify novel putative regulatory elements at the imprinted Dlk1/Gtl2 locus. J. Biol. Chem. 2008;283:18612–18620. doi: 10.1074/jbc.M801883200. [DOI] [PubMed] [Google Scholar]
- 27.Llères D., Moindrot B., Pathak R., Piras V., Matelot M., Pignard B., Marchand A., Poncelet M., Perrin A., Tellier V., et al. CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol. 2019;20:272. doi: 10.1186/s13059-019-1896-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wierstra I. FOXM1 (Forkhead box M1) in tumorigenesis: overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv. Cancer Res. 2013;119:191–419. doi: 10.1016/B978-0-12-407190-2.00016-2. [DOI] [PubMed] [Google Scholar]
- 30.Anson M., Crain-Denoyelle A.M., Baud V., Chereau F., Gougelet A., Terris B., Yamagoe S., Colnot S., Viguier M., Perret C., Couty J.P. Oncogenic beta-catenin triggers an inflammatory response that determines the aggressiveness of hepatocellular carcinoma in mice. J. Clin. Invest. 2012;122:586–599. doi: 10.1172/JCI43937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Indersie E., Lesjean S., Hooks K.B., Sagliocco F., Ernault T., Cairo S., Merched-Sauvage M., Rullier A., Le Bail B., Taque S., et al. MicroRNA therapy inhibits hepatoblastoma growth in vivo by targeting beta-catenin and Wnt signaling. Hepatol. Commun. 2017;1:168–183. doi: 10.1002/hep4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yochum G.S., Cleland R., Goodman R.H. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol. Cell. Biol. 2008;28:7368–7379. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yochum G.S., Sherrick C.M., Macpartlin M., Goodman R.H. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5' and 3' Wnt responsive enhancers. Proc. Natl. Acad. Sci. USA. 2010;107:145–150. doi: 10.1073/pnas.0912294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kota S.K., Llères D., Bouschet T., Hirasawa R., Marchand A., Begon-Pescia C., Sanli I., Arnaud P., Journot L., Girardot M., Feil R. ICR noncoding RNA expression controls imprinting and DNA replication at the Dlk1-Dio3 domain. Dev. Cell. 2014;31:19–33. doi: 10.1016/j.devcel.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Zhu X., Wu Y.B., Zhou J., Kang D.M. Upregulation of lncRNA MEG3 promotes hepatic insulin resistance via increasing FoxO1 expression. Biochem. Biophys. Res. Commun. 2016;469:319–325. doi: 10.1016/j.bbrc.2015.11.048. [DOI] [PubMed] [Google Scholar]
- 36.de Guia R.M., Rose A.J., Sommerfeld A., Seibert O., Strzoda D., Zota A., Feuchter Y., Krones-Herzig A., Sijmonsma T., Kirilov M., et al. microRNA-379 couples glucocorticoid hormones to dysfunctional lipid homeostasis. EMBO J. 2015;34:344–360. doi: 10.15252/embj.201490464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinyol R., Torrecilla S., Wang H., Montironi C., Piqué-Gili M., Torres-Martin M., Wei-Qiang L., Willoughby C.E., Ramadori P., Andreu-Oller C., et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021;75:865–878. doi: 10.1016/j.jhep.2021.04.049. [DOI] [PubMed] [Google Scholar]
- 38.Rebouissou S., Franconi A., Calderaro J., Letouzé E., Imbeaud S., Pilati C., Nault J.C., Couchy G., Laurent A., Balabaud C., et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology. 2016;64:2047–2061. doi: 10.1002/hep.28638. [DOI] [PubMed] [Google Scholar]
- 39.You L., Wang N., Yin D., Wang L., Jin F., Zhu Y., Yuan Q., De W. Downregulation of Long Noncoding RNA Meg3 Affects Insulin Synthesis and Secretion in Mouse Pancreatic Beta Cells. J. Cell. Physiol. 2016;231:852–862. doi: 10.1002/jcp.25175. [DOI] [PubMed] [Google Scholar]
- 40.Essandoh K., Li Y., Huo J., Fan G.C. MiRNA-Mediated Macrophage Polarization and its Potential Role in the Regulation of Inflammatory Response. Shock. 2016;46:122–131. doi: 10.1097/SHK.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying H., Kang Y., Zhang H., Zhao D., Xia J., Lu Z., Wang H., Xu F., Shi L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015;194:1239–1251. doi: 10.4049/jimmunol.1402088. [DOI] [PubMed] [Google Scholar]
- 42.Hadjihannas M.V., Bernkopf D.B., Brückner M., Behrens J. Cell cycle control of Wnt/beta-catenin signalling by conductin/axin2 through CDC20. EMBO Rep. 2012;13:347–354. doi: 10.1038/embor.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen T., Tian L., Chen J., Zhao X., Zhou J., Guo T., Sheng Q., Zhu L., Liu J., Lv Z. A Comprehensive Genomic Analysis Constructs miRNA-mRNA Interaction Network in Hepatoblastoma. (2021) Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.655703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garnier A., Ilmer M., Kappler R., Berger M. Therapeutic Innovations for Targeting Hepatoblastoma. Anticancer Res. 2016;36:5577–5592. doi: 10.21873/anticanres.11143. [DOI] [PubMed] [Google Scholar]
- 45.Shukla S., Milewski D., Pradhan A., Rama N., Rice K., Le T., Flick M.J., Vaz S., Zhao X., Setchell K.D., et al. The FOXM1 Inhibitor RCM-1 Decreases Carcinogenesis and Nuclear beta-Catenin. Mol. Cancer Ther. 2019;18:1217–1229. doi: 10.1158/1535-7163.MCT-18-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D.H., McMahon A.P., Sommer L., Boussadia O., Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 47.Torre C., Benhamouche S., Mitchell C., Godard C., Veber P., Letourneur F., Cagnard N., Jacques S., Finzi L., Perret C., Colnot S. The transforming growth factor-alpha and cyclin D1 genes are direct targets of beta-catenin signaling in hepatocyte proliferation. J. Hepatol. 2011;55:86–95. doi: 10.1016/j.jhep.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Hayhurst G.P., Lee Y.H., Lambert G., Ward J.M., Gonzalez F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sartor C., Bachelot L., Godard C., Lager F., Renault G., Gonzalez F.J., Perret C., Gougelet A., Colnot S. The concomitant loss of APC and HNF4alpha in adult hepatocytes does not contribute to hepatocarcinogenesis driven by beta-catenin activation. Liver Int. 2019;39:727–739. doi: 10.1111/liv.14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blivet-Van Eggelpoël M.J., Chettouh H., Fartoux L., Aoudjehane L., Barbu V., Rey C., Priam S., Housset C., Rosmorduc O., Desbois-Mouthon C. Epidermal growth factor receptor and HER-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J. Hepatol. 2012;57:108–115. doi: 10.1016/j.jhep.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 51.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bayard Q., Meunier L., Peneau C., Renault V., Shinde J., Nault J.C., Mami I., Couchy G., Amaddeo G., Tubacher E., et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018;9:5235. doi: 10.1038/s41467-018-07552-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirsch T.Z., Negulescu A., Gupta B., Caruso S., Noblet B., Couchy G., Bayard Q., Meunier L., Morcrette G., Scoazec J.Y., et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J. Hepatol. 2020;72:924–936. doi: 10.1016/j.jhep.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 54.Rebouissou C., Sallis S., Forné T. Quantitative Chromosome Conformation Capture (3C-qPCR) Methods Mol. Biol. 2022;2532:3–13. doi: 10.1007/978-1-0716-2497-5_1. [DOI] [PubMed] [Google Scholar]
- 55.Ea V., Sexton T., Gostan T., Herviou L., Baudement M.O., Zhang Y., Berlivet S., Le Lay-Taha M.N., Cathala G., Lesne A., et al. Distinct polymer physics principles govern chromatin dynamics in mouse and Drosophila topological domains. BMC Genomics. 2015;16:607. doi: 10.1186/s12864-015-1786-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015;109:21–29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B., et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tran S., Baba I., Poupel L., Dussaud S., Moreau M., Gélineau A., Marcelin G., Magréau-Davy E., Ouhachi M., Lesnik P., et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity. 2020;53:627–640.e5. doi: 10.1016/j.immuni.2020.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data were deposited on GEO: RNA-seq and small RNA-seq comparing ApcΔhep ROSA/DLK1/DIO3ΔWRE hepatocytes on GSE206262, ATAC-seq on GSE211930, kinetic RNA-seq on GSE210482), and MeDIP-seq on GSE239777. Others have been previously published (project: PRJNA150641 in European nucleotide archive (ENA).5,21,23