

Abstract
Adoptive cell therapy (ACT) using T cells expressing chimeric antigen receptors (CARs) is an area of intense investigation in the treatment of malignancies and chronic viral infections. One of the limitations of ACT-based CAR therapy is the lack of in vivo persistence and maintenance of optimal cell function. Therefore, alternative strategies that increase the function and maintenance of CAR-expressing T cells are needed. In our studies using the humanized bone marrow/liver/thymus (BLT) mouse model and nonhuman primate (NHP) model of HIV infection, we evaluated two CAR-based gene therapy approaches. In the ACT approach, we used cytokine enhancement and preconditioning to generate greater persistence of anti-HIV CAR+ T cells. We observed limited persistence and expansion of anti-HIV CAR T cells, which led to minimal control of the virus. In our stem cell-based approach, we modified hematopoietic stem/progenitor cells (HSPCs) with anti-HIV CAR to generate anti-HIV CAR T cells in vivo. We observed CAR-expressing T cell expansion, which led to better plasma viral load suppression. HSPC-derived CAR cells in infected NHPs showed superior trafficking and persistence in multiple tissues. Our results suggest that a stem cell-based CAR T cell approach may be superior in generating long-term persistence and functional antiviral responses against HIV infection.
Keywords: CAR T cells, adoptive cell therapy, stem cell gene therapy, HIV, humanized mouse model, nonhuman primate model
Graphical abstract
Kitchen and colleagues use hematopoietic stem cells and T cells to generate anti-HIV CAR T cells and evaluate the efficacy of each approach against HIV infection. Stem-based CAR T cells show superior persistence, trafficking, and viral suppression compared to conventional ex vivo CAR-modified T cells in HIV- and SHIV-infected animals.
Introduction
Recent advancements in chimeric antigen receptor (CAR) T cell immunotherapy have led to the successful treatment of certain B cell malignancies and renewed interest in CAR T cells for a functional cure for chronic diseases such as HIV-1 infection.1,2,3,4,5,6 The first clinical trials in HIV used peripheral T cells expressing a first-generation CD4-based CAR that consisted of the HIV-binding CD4 molecule fused with the CD3 zeta signaling domain (CD4CAR) to target HIV envelope (Env)-expressing cells, resulting in minimal viremia control.4,5 The addition of co-stimulatory domains such as CD28 or 4-1BB to create second-generation CARs was key to improve CAR function and antitumor responses in vivo.7,8 Similarly, there have been new-generation designs of anti-HIV CARs to improve persistence, trafficking, and antiviral efficacy of infused CAR T cells in vivo.9,10,11,12,13,14,15,16,17 Newer strategies featuring second- and third-generation CAR approaches in which T cells express either or both 4-1BB-zeta and CD28-zeta domain structures have been developed for anti-HIV CARs.12,13 In HIV-infected, antiretroviral therapy (ART)-suppressed humanized mice, infused dual CAR T cells rapidly suppressed viral replication and reduced tissue viral burden.13 However, it remains to be determined whether adoptive transfer of anti-HIV CAR T cells can have a significant impact on viral replication long term in the absence of ART. Major challenges for adoptively transferred CAR T cells to be effective therapeutically include augmented proliferation, function, and persistence after infusion.18 Currently, these limitations result in minimal or short-term antiviral or antitumor efficacy, a major barrier that requires further optimization.13,19,20,21,22,23
An alternative to peripheral T cell-derived CAR T cells is the use of CAR-modified hematopoietic stem/progenitor cells (HSPCs) capable of differentiating into CAR-expressing immune cells.24,25,26,27 In animal models of HIV and simian/human immunodeficiency virus (SHIV) infection, we determined that HSPCs modified with CD4CARs differentiated into CAR-expressing T cell, natural killer (NK) cell, B cell, and monocyte lineages.26,27 Importantly, stem cell-derived CD4CAR T cells expanded, trafficked to multiple tissues, suppressed plasma viral loads, and lowered viral burdened in latent reservoir-harboring tissues in humanized bone marrow/thymus/liver (BLT) mice and nonhuman primate (NHPs) animal models.26,27,28 Moreover, we identified a second-generation CD4-based CAR containing 4-1BB (CD4CAR41BB) that outperformed the previous first-generation CD4-based CARs in vivo in multiple ways in an HSPC-based approach. These included greater HSPC engraftment, CAR T cell differentiation, viral suppression, and generation of memory CAR T cells.29 These studies confirmed the safety and feasibility of using HSPCs for generating long-term engraftment and function of CAR-modified cells for HIV immune surveillance and control of viral replication, thus overcoming the lack of long-term persistence and antiviral function of adoptive CAR T cell therapy.
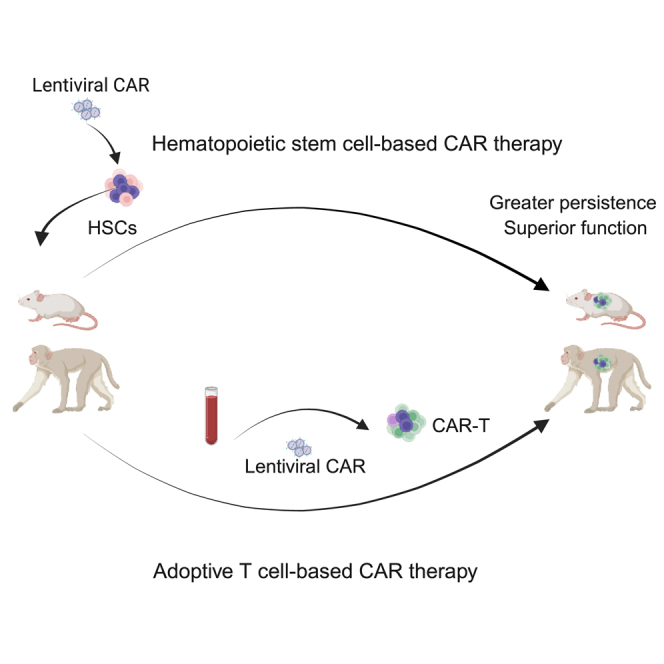
In this study, we compared and evaluated the adoptive-transferred peripheral CAR T cell approach with the HSPC-based approach to target HIV infection. We evaluated different methodologies to enhance the persistence and function of adoptively transferred autologous peripheral T cells modified with first- and second-generation CD4CARs.29,30 We have optimized CAR T cell cell processing and culturing conditions, cytokine administration, and infusion of CAR T cells during ART treatment. We additionally compared proliferation and control of HIV replication in the plasma of infected BLT mice treated with adoptive anti-HIV CAR T cells versus CAR-modified HSPCs. We observed greater proliferation and suppression of plasma viremia in HSPC-derived anti-HIV CAR T cells compared to adoptive anti-HIV CAR T cells in HIV-infected BLT mice. Finally, we observed that SHIV-infected NHPs treated with anti-HIV CAR-modified HSPCs had greater trafficking and persistence of CAR-expressing cells in multiple tissues compared to infected NHPs treated with adoptively transferred anti-HIV CAR T cells. Altogether, these data suggest that stem cell-based CAR T therapy can be a safe and effective approach that yields greater expansion, function, and control of HIV replication in vivo, and furthermore, be a platform that would be highly useful in treating chronic disease conditions that would require better persistence and function of CAR-modified cells.
Results
Culturing in interleukin-7 (IL-7) and IL-15 augments CAR cell persistence in adoptively transferred autologous T cells
Numerous invaluable reports have iteratively optimized ex vivo culturing conditions for improved CAR T cell efficacy and expansion in vivo with autologous peripheral T cells.31,32,33,34 In cancer-specific peripheral CAR T cells and tumor antigen-specific T cells, cytokines such as IL-7 and IL-15 have been shown to be superior to IL-2 in increasing central memory or stem-like memory differentiation, in vivo persistence, and enhanced antitumor efficacy.35,36,37,38,39 We first asked whether these parameters would be similarly beneficial in the context of HIV infection in peripheral T cells using our second-generation CD4CAR41BB (Figure S1A). Healthy human peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3 and anti-CD28 antibodies in the presence of IL-2 or IL-7 and IL-15, and then transduced with CD4CAR41BB lentiviral vectors, which contain two genes that protect transduced cells from infection by the coexpression of C-C chemokine receptor type 5 (CCR5) and HIV long terminal repeat short hairpin RNAs (LTR shRNAs).26,40,41 Cells were then further cultured in the presence of either IL-2 or IL-7/IL-15. After 10 days of culture, CD4CAR41BB T cells cultured in IL-7/IL-15 or IL-2 displayed cytokine responses similar to that of HIV-1 Env-expressing ACH2 cells, confirming that IL-7 and IL-15 do not impede antiviral function (Figure S1B). To determine the benefit of this cell-processing methodology in vivo, we generated autologous CAR T cells using splenocytes from uninfected humanized BLT mice. Splenocytes were activated in the presence of IL-2 or IL-7/IL15, transduced with CD4CAR41BB lentiviruses, followed by culturing with IL-2 or IL-7/IL15 for 7–8 days. We observed similar transduction efficiency and memory populations of CAR-T cells cultured with IL-2 or IL-7 and IL-15 (Figures S1C and S1D). In addition, we observed elevated levels of activation markers CD25 (IL-2R [receptor] α chain) and to a lesser extent CD69 in IL-7/IL-15 cultured CAR T cells (Figures S1E and S1F). We treated HIV-infected BLT mice with these IL-2 or IL-7/IL-15 cultured autologous mock (untransduced) or CD4CAR41BB transduced T cells stained with CellTrace (to track in vivo engraftment and trafficking) at weeks 4, 5, and 7 post-HIV infection at 10 million T cells (∼20% CAR+)/mouse per dose (equivalent to 8 million CAR T cells/kg) (Figures 1A and S1G). As shown in Figure 1B, we observed a minimal impact on plasma viremia after CAR T transfusion as compared to mock T cell controls. After 3 infusions, low-level (∼1%) CAR+ T cells were detected in the peripheral blood among total human CD3+ T cell populations; however, CAR+ levels dropped drastically after 2 weeks postthird infusion (Figure 1C). No differences in CD4/CD8 ratios were observed between mock and CAR T cell-treated mice in blood and spleen (Figures 1D and 1E). We were able to track CellTrace+-infused T cells in the peripheral blood of HIV-infected BLT mice for up to 6 weeks postinfusion at which CellTrace detection levels were low compared to preinfusion levels (Figures S1H and S1G). After 3 infusions, there was a slight increase in the percentages of IL-7/IL-15-cultured CAR T cells in the peripheral blood among CellTrace+-infused T cells compared to IL-2-cultured CAR T cells (Figure S1I). At the end of the study, CAR+ T cells were detectable in the spleen among total human CD3+ T cell populations at low levels (Figure 1F). We were able to detect infused CellTrace+ cells in the spleen and found marginally higher percentages of CAR+ cells from IL-7/IL-15 animals relative to IL-2, especially among CD8+ T cells (Figures 1G, S1J, and S1K). In addition, CAR+ T cells cultured in IL-2 or IL-7/IL-15 were mostly central memory populations at the time of infusion and differentiated in vivo into effector memory (CD45RA−CD62L−) and terminally differentiated effector memory (EMRA) T cells (CD45RA+CD62L−) (Figure 1H). However, at the endpoint of the study, IL-7/IL-15-cultured CAR T cells improved the maintenance of central memory populations and expression of CD27, a marker for memory T cells, relative to IL-2-cultured CAR T cells (Figures 1H and 1I). Furthermore, we observed low levels of programmed cell death protein 1 (PD-1) expression in IL-7/IL-15- and IL-2-cultured CAR T cells (Figure 1J). We also evaluated whether IL-7 and IL-15 culturing led to increased susceptibility to HIV infection in T cells, as reported previously.42,43 Consistent with multistep blockade of viral replications by anti-CCR5 and anti-HIV LTR shRNAs, we observed only minimum infection of CAR T cells that were cultured in either IL-7/IL-15 or IL-2 (Figure S1L). In summary, culturing anti-HIV CAR T cells in IL-7 and IL-15 resulted in modestly improved persistence during chronic HIV infection in vivo when compared to the older methodology with IL-2 alone.
Figure 1.

Culturing anti-HIV CAR T cells in IL-7 and IL-15 increases persistence and central memory phenotype in vivo
(A) Experimental design: humanized BLT mice were infected with HIVNFNSXSL9 for 4 weeks, and then received 10 million autologous T cells transduced with CD4CAR41BB cultured in IL-2 or IL-7 and IL-15 and stained with CellTrace for tracking. A second round of CAR treatment was administered at week 5 of infection and a third round at week 7 of infection. (B) HIV-1 plasma viral loads in infected humanized mice after 3 administrations of CD4CAR41BB T cells cultured in IL-2 or IL-7/IL-15. (C) Representative fluorescence-activated cell sorting (FACS) plot of CD3+CAR+ (GFP+) gating (left) and summary of percentage of human CD3+CAR+ (GFP+) cells in peripheral blood of infected humanized mice after 3 administrations of CD4CAR41BB T cells cultured in IL-2 or IL-7/IL-15 (right). (D) Percentage of CD4/CD8 ratio in peripheral blood of mock- or CAR-treated mice. (E) Percentage of CD4/CD8 ratio in spleens of mock- or CAR-treated mice at endpoint of study. (F) FACS plots representing CD3+ CAR+ (GFP+) gating (left) and summary of percentage of human CD3+ CAR+ (GFP+) cells in spleen at endpoint of study (right). (G) Percentage of CD8+ CAR+ cells in CellTrace+ population in spleen. (H) FACS plots showing memory populations (naive, central memory [CM], effector memory [EM], and terminal differentiated effector memory [EMRA+]) in CAR+ T cells cultured in IL-2 or IL-7/IL-15 before infusion and endpoint of study. (I) FACS plots showing CD27 expression in CAR+ (GFP+) cells cultured in IL-2 or IL-7/IL-15 in spleen. (J) FACS plots showing PD-1 expression in CAR+ (GFP+) cells cultured in IL-2 or IL-7/IL-15 in spleen. Error bars represent SEM. n = 3–4 per group. Significance determined by Mann-Whitney test. (C)–(I) measured by flow cytometry. (H)–(J) FACS plots showing CAR+ T cells from 3 to 4 CAR mice.
In vivo administration of IL-7 and IL-15 reduces cellular exhaustion of autologous peripheral CAR T cells
To compare the impact of IL-7/IL-15 on ex vivo CAR T cell culturing to in vivo efficacy, we next administered cytokines in vivo and measured the persistence and expansion of first-generation and second-generation adoptive CD4CAR T cells. We infected BLT mice with HIV and at weeks 5 and 9 of infection treated mice with 10 million CellTrace-stained mock, CD4CAR, or CD4CAR41BB transduced T cells (∼20%–50% CAR+) cultured in IL-7 and IL-15 with or without IL-7 (200 μg/kg) and IL-15 (200 μg/kg) administration. We administered IL-7 and IL-15 or PBS 2 additional times at weeks 6 and 10 (1 week after CAR T cell administration) (Figure 2A). In vivo administration of CAR T cell and IL-7 and IL-15 did not have a significant impact on the suppression of plasma viral load compared to controls (Figure S2A). CAR+ T cells among human CD45+ populations were detectable in the peripheral blood until 4 weeks following the second mock/CAR T cell treatment (Figures S2B and 2B). Although we did not observe improved expansion or persistence of CAR T cells in mice treated with in vivo IL-7 and IL-15, the levels of CAR T cells were augmented after the second infusion in mice treated with CD4CAR41BB T cells, but not in CD4CAR T cells without a co-stimulatory domain (Figure 2B). At the endpoint of the study, we detected very low levels of CAR T cells in spleen tissue and no significant difference in CAR T cell level comparing different treatment groups (Figure 2C). We were able to detect CellTrace-labeled cells in spleen: within the CellTrace+ population, levels of CD4CAR and CD4CAR41BBB was similar with or without IL-7/IL-15 administration (Figures 2D and 2E). We further investigated the impact of IL-7 and IL-15 administration on human CD8+ CAR(−) T cells. PD-1 levels in CD8+ CAR− T cells were slightly lower in blood and spleen in mice that received IL-7 and IL-15 administrations (Figures 2F and S2C). Moreover, we observed a difference in the expression of the immune activation marker CD38 in CD8+ CAR(−) T cells in mice treated with IL-7 and IL-15; levels were slightly lower in blood and significantly lower in spleen for mock and CD4CAR groups (Figures 2G and S2D). This was also seen in CD4+ CAR(−) T cells where a similar trend of slightly lower PD-1 levels and significantly lower CD38 levels in IL-7- and IL-15-treated groups was observed (Figures S2E and S2F). Similarly, IL-7 and IL-15 treatment led to low levels of PD-1, CD38, and human leukocyte antigen-DR (HLA-DR) expression in CD4CAR+ and CD4CAR41BB+ T cells in blood, spleen, and bone marrow tissues combined from multiple mice relative to CAR+ T cells from PBS-treated groups (Figures 2H and 2I). Altogether, in vivo administration of IL-7 and IL-15 did not have a significant impact on expansion, persistence, and antiviral efficacy for adoptive anti-HIV CAR T cells, but it did reduce the expression of immune activation and exhaustion markers induced by chronic HIV infection.
Figure 2.

Administration of IL-7 and IL-15 decreases chronic immune activation and immune exhaustion markers in adoptively transferred anti-HIV CAR T cells
(A) Experimental design: humanized BLT mice were infected with HIVNFNSXSL9, and at 5 and 9 weeks of infection received i.v. injections of either 10 million mock, CD4CAR, or CD4CAR41BB transduced T cells with or without 200 μg/kg IL-7 and IL-15 intraperitoneally (i.p.). Three more rounds of IL-7 and IL-15 i.p. administrations were given at weeks 6 and 10 of infection. (B) Percentage of CAR+ (GFP+) cells in the human CD45+ population in peripheral blood after CAR T cell and cytokine treatment. (C) Percentage of CAR+ (GFP+) cells in the human CD45+ population in spleen at the endpoint of the study. (D) Representative FACS plot showing CellTrace gating (left) and summary of percentage of CellTrace+ cells in the human CD45+ population in spleen at the endpoint of the study (right). (E) Representative FACS plot showing CAR+ gating in CellTrace+ population (left) and summary of percentage of CAR+ (GFP+) in the CellTrace+ population in spleen at the endpoint of the study. (F) Percentage of human CD8+ PD-1+ T cells (of CAR−population) in peripheral blood (left) and spleen (right). (G) Percentage of human CD8+ CD38+ T cells (of CAR− population) in peripheral blood (left) and spleen (right). (H) FACS plots showing PD-1 expression in CD4CAR and CD4CAR41BB T cells treated with PBS or IL-7 and IL-15 in blood, spleen, and bone marrow. Each FACS plot shows CAR T cells from 3 to 5 mice from each group. (I) FACS plots showing CD38 expression (left) and HLA-DR (right) in CD4CAR and CD4CAR41BB cells treated with PBS or IL-7 and IL-15 in vivo. Each FACS plot shows CAR T cells from blood, spleen, and bone marrow; n = 1–5 mice from each group. ∗p < 0.05; ∗∗p < 0.01; Mann-Whitney test. n = 4–6 per group with 2 independent experiments. Error bars bars represent SEM.
Impacts of first- and second-generation CD4CAR T cells on postanalytic treatment interruption (ATI) viral rebound
We next quantified CD4CAR T cell responses to recrudescent HIV-1 in infected, ART-suppressed humanized mice. We used a triple knockout (TKO) BLT mouse model to facilitate long-term analysis of ART suppression and viral rebound without the complication of graft-versus-host disease that is prominent in conventional NSG-BLT models.44 We injected 10 million total CellTrace-stained mock, CD4CAR, or CD4CAR41BB transduced T cells that were 30% CAR+ into ART-treated mice, and then interrupted ART 2 days later (Figure 3A). We observed no differences in plasma viral rebound in all of the treatment groups (Figure 3B). After an additional 2 rounds of mock or CAR T cell treatments, we observed no effect on plasma viral load in all groups (Figure 3B). We followed CAR+ expansion in the peripheral blood posttreatment and were able to detect CAR T cells among human CD45+ populations above background levels (Figure 3C). We observed the expansion of CD4CAR and CD4CAR41BB T cells only after the second round of CAR treatments; however, CAR levels continued to drop even after the third round of treatments (Figure 3C). Measurement of CAR+ T cells among CellTrace+-infused cells in the peripheral blood revealed that CD4CAR41BB persisted at higher levels than the first-generation CD4CAR T cells (Figure 3D). Among detected CellTrace+-infused cells in the spleen, we observed slightly higher CAR+% among CD4CAR41BB T cells than CD4CAR, showing a similar trend of higher CD4CAR41BB T cell persistence (Figure 3E). In addition, we observed lower viral RNA burden in spleen tissue from CD4CAR41BB-treated mice relative to mock control mice (Figure 3F). These results confirm previously reported findings that the addition of a 4-1BB co-stimulatory domain in anti-HIV CAR design enhances persistence in vivo.12 The findings also confirm a recent report that anti-HIV CAR T cells equipped with a dual CAR design had a minimal impact on plasma viral rebound in a humanized BLT mouse model.13 Thus, mimicking current peripheral blood-based treatment strategies of HIV infection with CAR T cells, we observed limited antiviral effects and CAR T cell persistence in vivo in humanized mouse models.
Figure 3.

CD4CAR 41BB T cells do not reduce or delay plasma viral rebound but lower tissue viral burden
(A) Experimental design: humanized TKO BLT mice were infected with 200 ng HIVNFNSXSL9 for 4 weeks, and then treated with ART for 6 weeks. At week 6 of ART, mice received 10 million mock, CD4CAR, or CD4CAR41BB transduced T cells followed 2 days later by ATI. Two additional rounds of mock or CAR T cell infusion were administered at 14 and 18 weeks postinfection. (B) Average plasma HIV RNA copies per milliliter in mock- or CAR-treated mice measured by RT-PCR. Dotted line indicates limit of detection. Arrows indicate mock or CAR treatment. (C) Representative FACS plots showing CAR+ (EGFR+) among human CD45 gating (left) and summary of percentage of CAR+ (EGFR+) cells in the human CD45+ population in peripheral blood (right). (D) Representative FACS plots showing CD45+ CAR+ (EGFR+) among CellTrace+ population gating and summary of percentage of CD45+ CAR+ cells within the CellTrace+ population in peripheral blood. (E) Percentage of CD45+ CAR+ cells within the CellTrace+ population in spleen at the endpoint of the study. (F) Relative HIV RNA levels in spleen at the endpoint of the study measured by RT-PCR. (C)–(E) measured by flow cytometry. ∗p < 0.05; significance determined 2-way ANOVA (D) and Mann-Whitney test (E and F). Data represent n = 6–7 mice per group. Error bars represent SEM.
Lymphodepletion through cyclophosphamide (CP) preconditioning enhances autologous adoptive T cell engraftment after infusion in SHIV-infected NHP animals
Concurrent with our studies in humanized mice, we also performed studies in NHPs using the pigtailed macaque model of SHIV infection.45 In these studies, we sought to enhance the engraftment and function of autologous T cells using CD4CAR-modified T cells in a model system with intact immune anatomical structures and a more complete immune system than the BLT mouse model. Furthermore, we sought to test clinical methods of preconditioning regimens to enhance engraftment of infused T cells. Preconditioning with lymphodepleting chemotherapy drugs such as CP followed by adoptive T cell transfer has shown to be effective to reduce cytotoxic side effects and improve engraftment and persistence.46,47 We hypothesized that preconditioning NHPs infected with SHIV would enhance the engraftment and persistence of adoptively transferred autologous CD4CAR T cells. Pigtail macaque animals first received stem cell transplantation with HSPCs modified with a Cal-1 lentivirus encoding a fusion inhibitor (mC46) to inhibit infection in HIV-susceptible cells (Table S1).27 This stem cell transplantation was necessary to provide animals with some protection and slow disease progression because no ART was given throughout the study. To evaluate whether CP enhances CAR T cell engraftment and persistence, animals were separated into 2 groups, with group 1 receiving CP and group 2 without CP regimen before CAR T cell treatment (Figure 4A). Following transplantation and recovery, group 1 animals were infected with SHIV virus, and 2 weeks later were preconditioned with a 30-mg/kg CP regimen followed by infusion of CellTrace-stained CD4CAR T cells coexpressing mC46 for protection (C46CD4CAR) (Figure 4A; Table S1). Two animals in group 1 experienced severe adverse events after the CP conditioning (1 before T cell infusion and 1 after) and did not continue with the study. Animals Z14148 and Z14035 continued the study with no apparent side effects after T cell infusion. To better track infused T cells, these animals also received a second round of CellTrace-stained unmodified T cells at 8–9 weeks post-SHIV infection. As a control, group 2 animal z13137 did not receive CP conditioning and received CellTrace-labeled C46CD4CAR T cells at 4 weeks and 8 weeks of SHIV infection (Figure 4A; Table S1). We observed stable lentiviral gene marking (combined Cal-1 and C46CD4CAR gene marking) throughout the study for both groups (Figure 4B). For group 1 animals, after T cell infusion, we observed some expansion of C46CD4CAR gene marking, although it dropped to ≤0.1% 7 weeks after T cell infusion in the peripheral blood (Figure 4C, left). For the group 2 animal, C46CD4CAR gene marking became undetectable in <2 weeks after the first CAR-T cell infusion (Figure 4C, right). At necropsy, we processed lymph nodes and stained with human specific anti-CD4 antibody for the detection of huCD4+ CAR T cells (representative flow shown in Figure 4D). As shown in Figure 4E, we observed low but detectable levels of huCD4+ CAR T cells in multiple lymphoid tissues for group 1 animals. In contrast, the huCD4+ CAR T cell level in the group 2 animal is significantly lower at necropsy across all lymphoid tissues, despite 2 rounds of CAR-T cell infusion.
Figure 4.

CP preconditioning enhances autologous adoptive T cell engraftment after infusion in SHIV-infected NHP animals
Pigtail macaques were first transplanted with autologous HSPCs transduced with Cal-1 vector. Afterward, animals were infected with SHIV-1157ipd3N4 for 6 weeks. Four animals in group 1 were preconditioned with CP, whereas 1 animal in group 2 was not preconditioned before infusion of mock or CAR T cells. To track T cell trafficking, group 1 animals were infused with CellTrace-labeled T cells and the group 2 animal was infused with CellTrace-labeled CAR-T cells 3 days before necropsy. (A) Experimental scheme and time line of NHP adoptive T cell transplant experiment. (B) Lentiviral marking (Cal-1 and CAR) of peripheral blood. (C) CD4zeta CAR gene marking of peripheral blood. (D) Representative FACS plot showing staining for huCD4+ CD4CAR T cells in spleen and jejunum of group 1 animal Z14035. (E) Summary of % huCD4+ cells among CD45+ lymphocytes in lymphoid tissues. (F) Plasma viral load after SHIV infection. Asterisk indicates animals that were sacrificed before completing the study.
To better understand trafficking of infused T cells, group 1 animals were infused with CellTrace-labeled unmodified T cells and the group 2 animal was infused with CellTrace-labeled CAR T cells 3 days before necropsy. As shown in Figure S3A (PBMCs) and S3B (lymphoid tissues and PBMCs), CellTrace+ cells were detected in peripheral blood and lymphoid tissues for group 1 animals that were preconditioned with CP treatment 6–7 weeks before the second infusion. Although the group 2 animal had similar percentage levels of CellTrace+ cells in the peripheral blood as compared to group 1 animals 15 min after infusion (Figure S3B, last column), the CellTrace+ percentage is markedly lower in the blood and lymphoid tissues at necropsy 3 days later (Figures S3A and S3B). CAR T cell engraftment level and proliferation remained low for the nonconditioned group 2 animal. We did not observe a significant difference in plasma viral load between the two groups, although viral load levels gradually decreased over time compared to historical controls (Figure 4F). These results highlight that a conditioning regimen can increase the engraftment and persistence of infused CAR T cells in the short term. However, conditioning during acute unsuppressed SHIV infection may lead to severe adverse effects, as seen with 2 animals in group 1, and therefore should be applied preferentially during ART treatment.
HSPC-derived CAR cells show superior expansion, antiviral efficacy, and persistence versus peripheral T cell-derived CAR cells in HIV-infected humanized BLT mice and SHIV-infected NHPs
We have previously showed that HSPCs can be modified to express a first-generation CD4CAR coexpressing protective antiviral genes and differentiate into multiple CD4CAR-expressing lineages that resulted in persistence, antigen-driven expansion, and reduction in viral burden in a humanized BLT mouse model of HIV infection and a macaque model of SHIV infection.27,30 We also demonstrated that the CD4CAR41BB resulted in superior engraftment, persistence, and antiviral efficacy in vivo in HSPC-modified humanized mice.29 We evaluated whether our first- and second-generation CD4-based CARs would yield similar results in a peripheral T cell-based approach and how this compared to the HSPC-based approach on expansion, impact on plasma viral load, and persistence upon exposure to HIV infection in humanized BLT mice. In these comparative studies, we observed remarkable differences in CAR+ populations in the peripheral blood of HSPC CAR-treated mice after HIV infection versus peripheral CAR T cells (Figure 5). We observed greater proliferation with stem cell-derived CD4CAR and CD4CAR41BB, compared to the decrease in peripheral CD4CAR and CD4CAR41BB T cell populations after infusion in HIV-infected mice (Figure 5A). The differences in expansion were not due to differences in CAR+ engraftment levels, since similar CD4+ and CD8+ CAR+ T cells were observed between both CAR approaches (Figure S4). Both stem cell-derived CD4CAR41BB T cells and peripheral CD4CAR41BB T cell levels remained at higher levels than the first-generation CD4CAR, confirming previous findings that 4-1BB-containing CARs have improved persistence (Figure 5A).12,29 In addition, we observed that HSPC CAR-treated mice had greater reductions in plasma viral load with both CD4CAR and CD4CAR41BB compared to peripheral CD4CAR and CD4CAR41BB peripheral T cell-treated mice (Figure 5B). Similarly, we compared persistence and trafficking in SHIV-infected NHP animals that received CD4CAR-modified stem cells or autologous CAR-modified T cells with or without CP treatment (Figure 5C). We have previously reported the persistence of HSPC-derived CD4CAR cells for >2 years following transplantation in pigtail macaques, whereas we observed peripheral CAR T cells decreased rapidly over weeks following infusion (Figure 4). At necropsy, we observed that NHPs that received CD4CAR-modified HSPCs showed higher levels of detection of CD4CAR cells in multiple tissues compared to animals that received autologous peripheral CD4CAR-modified T cells (Figure 5C). Taken together, these results suggest that HSPC-based CAR therapy generates superior CAR function, antiviral efficacy, and persistence compared to the autologous adoptive CAR T cell treatment approach.
Figure 5.

Anti-HIV CARs derived from HSPCs show superior proliferation and viral suppression compared to adoptive anti-HIV CAR T cells in vivo
Humanized BLT mice were constructed with either unmodified HSPCs or HSPCs modified with CD4CAR or CD4CAR4-BB. After immune reconstitution, mice were infected with HIVNFNSXSL9. Four weeks after infection, mice with unmodified HSPCs were treated with autologous CD4CAR or CD4CAR-41BB T cells cultured in IL-7/IL-15. (A) Average percentage of CAR+ cells among human CD45+ populations in peripheral blood from humanized mice transplanted with HSPCs-anti-HIV CAR or mice treated with 10 million autologous T cells transduced with anti-HIV CAR i.v. (B) Plasma HIV RNA copies from infected mice transplanted with HSPC-anti-HIV CARs at 4 weeks of infection or treated with autologous anti-HIV CAR T cells 4 weeks postinfusion, respectively. (C) Top: average percentage of CD4CAR+ cells in multiple tissues of SHIV-infected pigtail macaque animals transplanted with either unmodified HSPCs or HSPCs modified with CD4CAR. Bottom: average percentage of CD4CAR+ cells in multiple tissues of SHIV-infected pigtail macaque animals treated with autologous CD4CAR+ T cells cultured in IL-2. Error bars represent SEM. n = 4–8 mice per group (representing 2 mouse cohorts). Significance determined by Mann-Whitney test. NS, not significant; ∗∗p < 0.01; ∗∗∗p < 0.001.
Discussion
CAR-based therapeutic approaches are becoming highly promising for combating a number of different disease conditions, with successes in treating liquid tumors. However, success in CAR T therapies in treating solid tumor and persistent malignancies and infections has been challenging. A common challenge in treating these conditions is the lack of functional persistence of heavily processed CAR T cell products in vivo. Ideally, greater functional persistence of CAR-modified T cells would facilitate a more efficacious therapeutic outcome, particularly in chronic disease states like persistent HIV infection, and possibly in other types of chronic viral infections and malignancies. The successful enhancement of HIV antiviral immune responses will require persistence of the responses and wide tissue dissemination of CAR-expressing cells.
In the present study we aimed to improve on the adoptive T cell approach by examining various strategies to enhance proliferation and antiviral functions of CAR-modified peripheral T cells that would lead to viral suppression in vivo. We found that culturing modified CAR T cells with IL-7 and IL-15 modestly improved the persistence of our CD4-based CAR T cells compared to using IL-2 alone. The increase in persistence may have been mediated by the maintenance of naive/central memory populations by IL-7 and IL-15.48 However, the increase in persistence and naive/central memory differentiation had minimal impact on plasma viremia.
To further increase persistence and function, we administered IL-7 and IL-15 after CAR T cell treatments in BLT mice. Initial adoptive transfer of mock or CD4-based CAR T cells and IL-7/IL-15 treatments, consistent with previous reports, did not have a long-term impact on viral suppression.49,50 In addition, it did not have a significant impact on expansion of either first-generation or second-generation CD4-based CAR T cells. IL-7 and IL-15 treatments did, however, decrease the percentage of CD8+ and CAR+ T cells expressing HLA-DR, CD38, and PD-1 from multiple tissues, consistent with prior reports on the impact of IL-7 and IL-15 treatment on immune activation markers and exhaustion markers.50,51,52,53 Collectively, we found that the use of IL-7 and IL-15 reduced T cell exhaustion, but the effects are limited in achieving long-term persistence and reducing viral load.
In an attempt to generate robust proliferation in vivo, we treated ART-suppressed mice with CAR T cells and interrupted ART to increase antigen stimulation. However, we did not observe a prolonged proliferation of CAR T, and viral load rebounded rapidly, even with our second-generation CD4-based CAR T cells. We observed modest decreased viral load in animals receiving CD4CAR41BB T cells but did not observe increased persistence as compared to CD4CAR. Exhaustion of CAR T cells is a major limitation in CAR-based therapies, and we observed the expression of activation and exhaustion marker in our infused anti-HIV CAR T cells as reported by others.13,54 Therefore, designing new adoptive anti-HIV CAR T cell therapies would need to involve novel ways to overcome lack of persistence and HIV-mediated immune dysfunction.
A final strategy to enhance the persistence of adoptive CAR T cells in this study is using lymphodepleting chemotherapy such as CP, which has been used successfully as a preconditioning regimen to engraft and increase the expansion and persistence of anticancer CAR T cells in clinical trials.55,56,57 We found that the CP preconditioning of SHIV-infected animals led to improved levels of infused T cells in multiple tissues as compared to no-preconditioning control animal. However, we also observed severe toxicity effects of CP conditioning in unsuppressed SHIV-infected animals. Two animals (Z14279 and Z14123) experienced severe hemolytic-uremic syndrome (HUS) following conditioning. HUS has been described in people living with HIV, namely in the pre-ART treatment era58,59,60,61,62,63,64; associated thrombocytopenias have also been reported in our SHIV model.65 From these animals we concluded that the combination of HSPC transplantation and CP-mediated immunosuppression during acute SHIV viremia resulted in synergistic nephrotoxicity. These findings are consistent with our previous reports that transplantation-dependent immunosuppression can significantly augment SHIV pathogenesis during acute and early chronic infection.66 These events suggest that preconditioning agents should be considered during ART treatment to avoid adverse events.
Adoptive CAR T cell immunotherapy remains an attractive approach because it is already US Food and Drug Administration approved to treat patients for various types of cancers. Although we have not seen improvement in expansion and persistence with strategies tested in our study, additional strategies need to be examined to enhance adoptive anti-HIV CAR T cell therapy. Coexpression of CXCR5 with an anti-SIV CAR was shown to enhance the trafficking of CAR T cells to B cell follicle locations, but it also demonstrated lack of persistence after 2 to 4 weeks postinfusion.20 Recently, a dual CAR design in which T cells expressed 2 CD4-based CARs independently encoding the CD28 and 4-1BB co-stimulatory domains was used to enhance T cell-based CAR therapy for HIV infection. These dual CAR T cells did proliferate in vivo; however, they did not result in significant plasma viral suppression in the absence of ART.13 In an NHP model of HIV infection, cell-associated HIV-1-Env boost was shown to induce robust anti-HIV CAR T cell proliferation in ART-suppressed animals and delays in viral rebound.21 Our report and those of others together highlight the challenges of achieving adoptive CAR-T cell persistence and continued function for HIV infection.19 Additional combination strategies such as cytokine treatment with antigen boost and checkpoint inhibitor blockade may further improve the functions of infused CAR-T cells.
In summary, similar to observations reported in human peripheral CAR T cell therapy, we saw a rapid decrease in levels of CAR-modified autologous peripheral T cells.67,68 We sought to determine whether an HSPC-based approach can overcome some of the defects in the peripheral CAR T cell approach by comparing the two. We have previously shown that modifying HSPCs with CD4-based anti-HIV CAR can lead to long-term engraftment and successful differentiation of multiple immune subsets, including functional CAR T cells in vivo in humanized mice and in NHPs.26,27,29 Compared to peripheral CAR T cell therapy with the same CD4-based CAR lentivirus, we observed that HSPC-based CAR therapy had clear and evident expansion, persistence, tissue distribution, and suppression of viral replication in both humanized mice and NHPs.27,28,29 The differences in expansion, persistence, tissue distribution, and viral suppression can be due but not limited to several reasons: (1) the potential contribution of other immune cell types expressing the CD4CAR, such as monocytes and NK cells to anti-HIV responses, (2) the continuous replenishment and seeding of a wide distribution of anatomical tissue sites of CD4CAR-expressing cells by self-renewing HSPC-CAR cells from the bone marrow, and (3) the starting naive CAR T cell population capable of generating a proliferative burst after antigen stimulation by HIV infection. Long-term CAR surveillance and detectable presence in multiple HIV-burdened tissues is observed in our stem cell-based CAR therapy studies but not in the T cell-based approach studies. Thus, the stem cell-based approach overcomes the limitations of adoptive CAR T cell where CAR-HSPCs would likely supply a lifetime source of HIV-specific immunity without the need to repeat T cell infusions. However, additional studies are needed to evaluate the safety of the HSPC-CAR approach and the concern for insertional mutagenesis, although stem cell-based gene therapy using lentiviral vectors has maintained a good safety record with no report of insertional mutagenesis.69,70,71,72 In the case of a patient who developed acute myeloid leukemia after 5 years of receiving gene therapy for sickle cell disease using a lentiviral vector, the latest report suggests that mutagenesis was not likely lentiviral vector mediated.73 Another safety concern is the use of CD4-based CAR and its potential to interfere with the immune system. CD4 is the natural receptor for the IL-16 cytokine, and to overcome this concern, we have since developed an optimized D1D2CAR to remove domains 3 and 4 of the CD4 receptor and eliminate IL-16 binding to the CAR.29 There have been successes in recent years in achieving HIV cures with allogeneic hematopoietic stem cell transplants from CCR5Δ32 homozygous donors.74,75 Autologous stem cell-based anti-HIV CAR therapy presents a promising therapy to allow long-term immune surveillance against HIV without the need to replace all HSPCs with CCR5Δ32 donor cells. Additional clinical trials are needed to address the safety, efficacy, and feasibility in HIV+ patients.
Materials and methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (“The Guide”) and was approved by the Institutional Animal Care and Use Committees of the University of California, Los Angeles (protocol 2010-038-31J) and the University of Washington (protocol 3235-01). For humanized mice, all of the surgeries were performed under ketamine/xylazine and isoflurane anesthesia and every effort was made to minimize animal pain and discomfort. Healthy juvenile pigtail macaques were housed at the Washington National Primate Research Center (WaNPRC) under conditions approved by the American Association for the Accreditation of Laboratory Animal Care. NHPs were monitored at least twice daily by animal technicians for basic husbandry parameters (e.g., food intake, activity, stool consistency, overall appearance), as well as daily observation by a veterinary technician and/or veterinarian. Animals were housed in cages approved by “The Guide” and in accordance with Animal Welfare Act regulations. Animals were fed twice daily and were fasted for up to 14 h before sedation. Environmental enrichment included grouping in compound, large activity, or run-through connected cages, perches, toys, food treats, and foraging activities. If a clinical abnormality was noted by WaNPRC personnel, then standard WaNPRC procedures were followed to notify the veterinary staff for evaluation and determination for admission as a clinical case. Animals were sedated by administration of ketamine HCl and/or telazol and supportive agents for balanced anesthesia (e.g., diazepam, midazolam) before all of the procedures. After sedation, animals were monitored according to WaNPRC standard protocols. WaNPRC surgical support staff are trained and experienced in the administration of anesthetics and have monitoring equipment available to assist with electronic monitoring of heart rate, respiration, and blood oxygenation; audible alarms and digital readouts; monitoring of blood pressure, temperature; and so forth. For minor procedures, the presence or absence of deep pain was tested by the toe-pinch reflex, and the absence of response (leg flexion) to this test indicated adequate anesthesia. In cases of general anesthesia, similar monitoring parameters were used and anesthesia was tested by the loss of palpebral reflexes (eye blink). Analgesics (generally buprenorphine with meloxicam or buprenorphine slow release) were provided as prescribed by the clinical veterinary staff for at least 48 h after the procedures and could be extended at the discretion of the clinical veterinarian based on clinical signs.
Humanized BLT mice
BLT mice were constructed as previously reported by Zhen and colleagues.29,30 In brief, CD34+ cells derived from human fetal liver were purified by magnetic cell sorting using CD34 microbeads (Miltenyi). Purified CD34+ cells were left untransduced (BLT only) or transduced with CAR-expressing lentiviruses (BLT HSPC-CAR mice) in retronectin (Takara)-coated plates. Transduction efficiency of CD34+ cells was measured in ex vivo cultures of 0.1 million mock or CAR transduced cells in extension media (IL-3 100 ng/mL, IL-6 100 ng/mL, stem cell factor (SCF) 100 ng/mL in 10% fetal calf serum RPMI 1640) for 7 days. Cells were then analyzed by flow cytometry. On the day of transplant, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SZJ (NOD/SCID/IL-2Rγ−/− or NSG; The Jackson Laboratory) or C57BL/6 Rag2−/−γc−/−CD47−/− (or TKO, The Jackson Laboratory) mice received 2.7 Gy total body irradiation (TBI) and were then transplanted with liver and thymus tissue under the kidney capsule. Next, mice were injected with 0.5 million autologous-untransduced (BLT only) or CAR transduced CD34+ cells (BLT-HSPC CAR). At 8–10 weeks posttransplantation, each mouse was bled retroorbitally, and PBMCs were analyzed by flow cytometry to check human immune reconstitution. Upon reaching >50% human immune cell reconstitution, mice were used for HIV infection studies and the generation of autologous anti-HIV CAR T cells.
Lentivirus production
Lentiviral vectors expressing CD4-based CARs were produced in 293FT cells using calcium phosphate (Clontech). The 293FT cells were transfected with a third-generation packaging system, including an FG12 SIN lentiviral vector expressing first- or second-generation CD4CAR and coexpressing GFP and protective shRNAs, pCMV.R8.2.vpr packaging plasmid, and the porcine cytomegalovirus (pCMV)-vesicular stomatitis virus G or Cocal-Env plasmid. Supernatant was collected 48 h later, 0.45 μm sterile filtered, and concentrated by ultracentrifugation using a Beckman SW32 rotor at 30,000 rpm at 4°C. Supernatant was aspirated, and pellets were resuspended in 1× PBS and stored at −80°C. Lentivirus was titered using 2e5 Jurkat cells transduced with diluted lentivirus and checked for GFP expression 2 days later. We used the following formula to calculate the titer of lentivirus: (2e5 × %GFP)/μL lentivirus × 1,000. We used the dilution that gave close to %50 GFP to calculate titer in international units per milliliter.
Infectious HIV-1 virus production
CCR5-tropic HIVNFNSXSL9 stocks were produced in 293T cells using calcium phosphate and cotransfected with pNFNSXSL9. At 48 h later, supernatant was collected and filtered with 0.45 μm filter and concentrated using the Amicron Ultra-15 Centrifugal Filter Unit (Millipore). The p24 concentration was measured by ELISA.
Plasma viral load
HIV RNA in plasma derived from peripheral blood or heart puncture from mice was isolated using the QIAamp Viral RNA Mini Kit (Qiagen). real time RT-PCR was performed using TaqMan RNA-To-Ct One-Step reagents (Thermo Fisher) with primers HIV-1_F: 5′-CAATGGCAGCAATTTCACCA-3′ and HIV-1_R: 5′-GAATGCCAAATTCCTGCTTGA-3′ and NL4-3 HIV-1 probe: 5′-[6-FAM]CCCACCAACAGGCGGCCTTAACTG[Tamra-Q]-3′.
Cell-associated HIV RNA
HIV RNA was isolated from cells collected from blood and spleen using RNeasy Mini kits (Qiagen). cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). Quantitative real-time PCR was performed using TaqMan Universal PCR master mix (Thermo Fisher) with primers HIV-1_F: 5′-CAATGGCAGCAATTTCACCA-3′; HIV-1_R: 5′-GAATGCCAAATTCCTGCTTGA-3′; HIV-1 probe: 5′-[6-FAM]CCCACCAACAGGCGGCCTTAACTG[Tamra-Q]-3′ and compared to HPRT1 housekeeping gene (primer/probe mix by Thermo Fisher [catalog no. 4331182]).
Lentiviral transduction of T cells in humanized mice
Spleens were collected from uninfected mice and processed for single-cell suspensions. Human splenocytes were stimulated with plate-bound anti-human CD3 (OKT3, Thermo Fisher) and soluble anti-human CD28 (CD28.2, Beckman Coulter) in RPMI 1640 (Thermo Fisher), 1% penicillin-streptomycin, and 10% fetal bovine serum (FBS) supplemented with IL-2 (50 ng/mL, Peprotech) or IL-7 (10 ng/mL, Miltenyi) and IL-15 (10 ng/mL, Miltenyi) for 48–72 h at 37°C, 5% CO2. Afterward, stimulated cells were resuspended at 106/mL in RPMI medium supplemented with IL-2 (50 ng/mL) or IL-7/IL-15 (10 ng/mL) and plated on retronectin-coated wells. Appropriate CAR lentivirus supernatant was added at an MOI of 5–10 and plates were incubated overnight. Mock and transduced cells were replenished with fresh medium supplemented with IL-2 (50 ng/mL) or IL-7/IL-15 (10 ng/mL) every 2–3 days for 7–12 days. Transduction efficiency was measured by GFP or EGFR staining using flow cytometry.
HIV challenge and ART treatment
BLT mice were challenged with 200–500 ng p24 HIVNFNSXSL9 via retroorbital injection. For ART suppression studies, mice were fed ART regimens consisting of tenofovir disproxil-fumarate (TDF, 80 mg/kg), emtricitabine (FTC, 120 mg/kg), and elvitegravir (ELV, 160 mg/kg), beginning 4 weeks after infection. TDF, FTC and ELV were generously supplied by Gilead Sciences. TDF, FTC, and ELV were dissolved in DMSO and mixed with sweetened moist gel meal (DietGel Boost, ClearH2O; Medidrop Sucralose) as described by Mu et al.76
CAR T cell and cytokine treatment
Mock or CAR transduced T cells were stained with CellTrace violet or Far Red cell proliferation kits (Life Technologies) per the manufacturer’s protocol, and then were resuspended in serum-free RPMI. Mice received 10 million mock or CAR (15%–50%) transduced T cells via retroorbital injection. For Figure 2, mice received IL-7 (200 μg/kg, Miltenyi) and IL-15 (200 μg/kg, Miltenyi) via intraperitoneal injection along with mock or CAR T cells that were cultured in IL-7 and IL-15 (10 ng/mL), followed by 3 more rounds on IL-7/IL-15 treatment the following week. For Figure 3, mice received 150 mg/kg CP 24 h before mock or CAR treatment.
NHP HSPC transplantation and T cell adoptive transfer
Six juvenile male pigtail macaques underwent autologous transplantation with Cal-1 lentiviral vector-modified HSPCs, as previously described,77 including TBI-based preparative conditioning, and enrichment of CD34+ HSPCs from bone marrow primed with granulocyte colony-stimulating factor and SCF. The Cal-1 vector78 encoded the mC46 viral fusion inhibitor under the control of the UbC promoter, as well as a nonfunctional (human CCR5-specific) shRNA driven by an H1 promoter.78 The 6 transplanted animals are described in detail in Table S1. Animal Z14160 was euthanized due to acute kidney injury during posttransplantation hematopoietic recovery. Before infection of the remaining 5 animals with SHIV to model the impact of HIV-specific CAR T cells, animals were allowed to recover for >1 year (at least 447 days). This recovery period was included in our study protocol based on our previous findings that TBI-based conditioning may impair the development and retention of virus-specific adaptive immune responses.27,79,80 Over 1 year after HSPC transplantation, animals were infected with 9500 median tissue culture infectious dose of SHIV-1157ipd3N4 via the intravenous (i.v.) route; i.v. infection and plasma viral load assay information has been described previously.27,79,81,82 To provide our virus-specific CD4CAR T cells with the maximal amount of viral antigen, we hypothesized that T cell infusion should occur during peak viremia (i.e., 2–3 weeks postinfection). At 6 and 5 days before T cell infusion, animals received 2 daily doses of 30 mg/kg CP to promote T cell engraftment, with the exception of Z13137, which did not receive a conditioning regimen. Notably, 2 animals (Z14279 and Z14123) experienced severe HUS following conditioning and were euthanized either before (Z14279) or immediately following T cell infusion (Z14123). The remaining 3 animals that proceeded to the CD4CAR T cell study phase (Z14035, Z14148, and Z13137; Table S1) each received 2 T cell infusions. Z14035, Z14148, and Z13137 received CD4CAR-expressing T cells, which were labeled with CellTrace Far Red (Thermo Fisher Scientific). The second CAR T cell infusion for Z14035 and Z14148 was made up of unmodified autologous T cells labeled with CellTrace Far Red, and occurred 47–55 days after the first infusion. The second CAR T cell infusion for Z13137 occurred 22 days after the first infusion, and was made up of dye-labeled, Cal1 CD4CAR-modified cells. Animals were euthanized 2–3 days following infusion of the second T cell dose to quantify the trafficking of dye-labeled cells in vivo. Total and CAR+ cell doses are included in our NHP experimental summary in Table S1. All of the T cell products were manufactured as described previously,21 including isolation of total CD3+ cells and 3-day activation with anti-CD3/CD28 beads (Thermo Fisher), followed by lentiviral vector transduction and expansion for ∼7 days in G-Rex flasks (Wilson Wolf) in the presence of 50 μg/mL IL-2 (Thermo Fisher).
Cytokine assays
T cells from healthy human donors were transduced with CAR-expressing lentiviral vectors, as described above. CAR T cell function was assessed by stimulating 0.5 million mock- or CAR-transduced T cells with 0.5 million ACH2 (Env+) or ACH2 (Env−) cells overnight. The next day, cells were treated with Golgi plug and Golgistop (Thermo Fisher) for 6 h and then stained with anti-human antibodies against tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ) cytokines.
Flow cytometry
Washed cells from blood and tissue were resuspended in 50 μL PBS (4% FBS) and surface stained with anti-human antibodies: CD45 (J33, Beckman Coulter), CD3 (OKT3, Thermo Fisher), CD4 (OKT4, BioLegend), CD4 (RPAT4, Thermo Fisher), CD4 (SFCI12T4D11, Beckman Coulter), CD8 (B9.11, Beckman Coulter), CD8 (SK1, BioLegend), CD45RA (HI100, BD Biosciences), CD45RA (HI100, BioLegend), CD62L (DREG56, Thermo Fisher), CD38 (HIT2, Thermo Fisher), CD25 (BC96, Thermo Fisher), CD25 (M-A251, BioLegend), CD27 (O323, Thermo Fisher), HLA-DR (LN3, Thermo Fisher and BioLegend), PD-1 (J105, Thermo Fisher), PD-1 (EH12.2H7, BioLegend), CD69 (FN50, BioLegend), Tim-3 (F38-2E2, BioLegend), human EGFR (Cetuximab, R&D Systems), and LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Invitrogen). For intracellular protein detection, cells were treated with the BD Cytofix/Cytoperm Cell Permeabilization/Fixation Solution kit and stained with anti-human antibodies: TNF-α (Mab11, Thermo Fisher), IFN-γ (4S.B3, Thermo Fisher), and HIV-1 Core Antigen (KC57, Beckman Coulter). To detect CAR-modified cells in NHPs, we leveraged anti-CD4 antibody clones that specifically recognized extracellular human CD4 (huCD4) on our CAR, but not endogenous NHP CD4. PBMCs and tissue necropsy samples were stained with the following antibodies: anti-huCD4 antibody for the detection and analysis of CD4CAR-modified cells (Beckman Coulter, clone 13B8.2), anti-NHP CD45 (BD Biosciences, clone D058-1283), anti-CD4 (eBiosciences, clone OKT4), anti-CD8 (eBiosciences, clone SK1), anti-CD20 (eBiosciences, clone 2H7), anti-NK2Ga (Beckman Coulter, clone A60797), anti-CD14 (Beckman Coulter, clone IM2707U), anti-CD95 (BD Biosciences, clone DX2), anti-CD28 (BD Biosciences, clone D28.2), and anti-CD3 (BD Biosciences, clone SP34-2). Fluorophore conjugates included BV405, V500, efluor450, fluorescein isothiocyanate, phycoerythrin (PE), PerCP Cy5.5, PE-Cy5, PE-Cy7, ECD (phycoerythrin-Texas Red conjugate), Alexa 700, allophycocyanin (APC), and APC-efluor780. Flow cytometry data were collected on BD LSRII Fortessa using BD FACSDiva Software (BD Biosciences). Flow data were analyzed using FlowJo software.
Statistical analysis
Statistical analysis was performed using Prism software versions 9 and 10. For Figures 1, 2, and 5 in vivo studies, an unpaired Mann-Whitney test was used for nonparametric testing of independent groups. For Figure 3D, a 2-way ANOVA was used to determine the significance of different CAR groups across multiple time points. For Figures 3E and 3F, an unpaired Mann-Whitney test for nonparametric testing was used to determine significance. Statistical significance was determined when p < 0.05.
Data and code availability
All of the data are available in the main text or the supplementary materials.
Acknowledgments
We thank Drs. Romas Geleziunas and Jeff Murry and the people at Gilead for providing the ART drugs used in this study. We thank UCLA Center for AIDS Research (CFAR) Humanized Mouse Core staff research associate Nianxin Zhong for his assistance in the humanized mice work. We thank Drs. Irvin Chen, Jerome Zack, Otto Yang, and An Dong Sung for helpful discussions. This work was funded by the National Institute of Allergy and Infectious Diseases (grants 1R21AI140866 to AZ, R2120200174 to AZ and Jianming Xie (USC), R01AI172727 to AZ); the National Institute on Drug Abuse (grant R01DA-52841 to A.Z.); the American Foundation For AIDS Research (grant 110304-71-RKRL and 110395-72-RPRL to A.Z., and 109577-62-RGRL and 110038-67-RSRL to S.G.K.); the National Cancer Institute (grant 1R01CA239261-01 to S.G.K.); NIH grants P30AI28697 (to the UCLA CFAR Virology Core, Gene and Cell Therapy Core, and Humanized Mouse Core) and U19AI149504 (to S.G.K. and Dr. Irvin Chen [UCLA]); the California Institute for Regenerative Medicine (grant DISC2-10748 to S.G.K.); UPLIFT: UCLA Postdocs’ Longitudinal Investment in Faculty (award K12 GM106996 to M.A.C.); NIH/NIAID UM1 AI126623 and NIH/NHLBI U19 HL156247 (to H.-P.K.); R21AI172582, R01 AI167004, and R01 AI170214 (to C.W.P.); and NIH/ORIP P51 OD010425 and U42 OD011123 (to the Washington National Primate Research Center). This work was also supported by the UCLA AIDS Institute, the James B. Pendleton Charitable Trust, and the McCarthy Family Foundation.
Author contributions
Conceptualization, M.A.C., A.Z., C.W.P., S.G.K., and H.-P.K. Methodology, M.A.C., A.Z., W.M., H.M., V.R., and C.W.P. Investigation, M.A.C., A.Z., W.M., H.M., V.R., and C.W.P. Visualization, M.A.C., A.Z., W.M., and C.W.P. Funding acquisition, S.G.K., A.Z., C.W.P., and H.-P.K. Project administration and supervision, S.G.K. Writing, M.A.C. Review & editing, A.Z., C.W.P., S.G.K., and H.P.K.
Declaration of interests
S.G.K. is a founding member of CDR3. H.-P.K. is or was a consultant to and has or had ownership interests in Rocket Pharmaceuticals, Homology Medicines, VOR Biopharma, and Ensoma.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.02.026.
Supplemental information
References
- 1.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Teachey D.T., Porter D.L., Grupp S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deeks S.G., Wagner B., Anton P.A., Mitsuyasu R.T., Scadden D.T., Huang C., Macken C., Richman D.D., Christopherson C., June C.H., et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 5.Mitsuyasu R.T., Anton P.A., Deeks S.G., Scadden D.T., Connick E., Downs M.T., Bakker A., Roberts M.R., June C.H., Jalali S., et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 6.Mu W., Carrillo M.A., Kitchen S.G. Engineering CAR T Cells to Target the HIV Reservoir. Front. Cell. Infect. Microbiol. 2020;10:410. doi: 10.3389/fcimb.2020.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M., et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hale M., Mesojednik T., Romano Ibarra G.S., Sahni J., Bernard A., Sommer K., Scharenberg A.M., Rawlings D.J., Wagner T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017;25:570–579. doi: 10.1016/j.ymthe.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L., Patel B., Ghanem M.H., Bundoc V., Zheng Z., Morgan R.A., Rosenberg S.A., Dey B., Berger E.A. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J. Virol. 2015;89:6685–6694. doi: 10.1128/JVI.00474-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haran K.P., Hajduczki A., Pampusch M.S., Mwakalundwa G., Vargas-Inchaustegui D.A., Rakasz E.G., Connick E., Berger E.A., Skinner P.J. Simian Immunodeficiency Virus (SIV)-Specific Chimeric Antigen Receptor-T Cells Engineered to Target B Cell Follicles and Suppress SIV Replication. Front. Immunol. 2018;9:492. doi: 10.3389/fimmu.2018.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leibman R.S., Richardson M.W., Ellebrecht C.T., Maldini C.R., Glover J.A., Secreto A.J., Kulikovskaya I., Lacey S.F., Akkina S.R., Yi Y., et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. Plos Pathog. 2017;13 doi: 10.1371/journal.ppat.1006613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maldini C.R., Claiborne D.T., Okawa K., Chen T., Dopkin D.L., Shan X., Power K.A., Trifonova R.T., Krupp K., Phelps M., et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med. 2020;26:1776–1787. doi: 10.1038/s41591-020-1039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan M., Lim L., Holguin L., Han T., Vyas V., Urak R., Miller A., Browning D.L., Echavarria L., Li S., et al. Pre-clinical data supporting immunotherapy for HIV using CMV-HIV-specific CAR T cells with CMV vaccine. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2022;25:344–359. doi: 10.1016/j.omtm.2022.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi J., Ding C., Jiang X., Gao Y. Advances in Developing CAR T-Cell Therapy for HIV Cure. Front. Immunol. 2020;11:361. doi: 10.3389/fimmu.2020.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anthony-Gonda K., Bardhi A., Ray A., Flerin N., Li M., Chen W., Ochsenbauer C., Kappes J.C., Krueger W., Worden A., et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anthony-Gonda K., Ray A., Su H., Wang Y., Xiong Y., Lee D., Block A., Chilunda V., Weiselberg J., Zemelko L., et al. In vivo killing of primary HIV-infected cells by peripheral-injected early memory-enriched anti-HIV duoCAR T cells. JCI insight. 2022;7 doi: 10.1172/jci.insight.161698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietrobon V., Todd L.A., Goswami A., Stefanson O., Yang Z., Marincola F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms221910828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwamoto N., Patel B., Song K., Mason R., Bolivar-Wagers S., Bergamaschi C., Pavlakis G.N., Berger E., Roederer M. Evaluation of chimeric antigen receptor T cell therapy in non-human primates infected with SHIV or SIV. PloS one. 2021;16 doi: 10.1371/journal.pone.0248973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pampusch M.S., Abdelaal H.M., Cartwright E.K., Molden J.S., Davey B.C., Sauve J.D., Sevcik E.N., Rendahl A.K., Rakasz E.G., Connick E., et al. CAR/CXCR5-T cell immunotherapy is safe and potentially efficacious in promoting sustained remission of SIV infection. Plos Pathog. 2022;18 doi: 10.1371/journal.ppat.1009831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rust B.J., Kean L.S., Colonna L., Brandenstein K.E., Poole N.H., Obenza W., Enstrom M.R., Maldini C.R., Ellis G.I., Fennessey C.M., et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood. 2020;136:1722–1734. doi: 10.1182/blood.2020006372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hay K.A., Gauthier J., Hirayama A.V., Voutsinas J.M., Wu Q., Li D., Gooley T.A., Cherian S., Chen X., Pender B.S., et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–1663. doi: 10.1182/blood-2018-11-883710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.López-Cantillo G., Urueña C., Camacho B.A., Ramírez-Segura C. CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.878209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Oliveira S.N., Ryan C., Giannoni F., Hardee C.L., Tremcinska I., Katebian B., Wherley J., Sahaghian A., Tu A., Grogan T., et al. Modification of hematopoietic stem/progenitor cells with CD19-specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum. Gene Ther. 2013;24:824–839. doi: 10.1089/hum.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kao R.L., Truscott L.C., Chiou T.T., Tsai W., Wu A.M., De Oliveira S.N. A Cetuximab-Mediated Suicide System in Chimeric Antigen Receptor-Modified Hematopoietic Stem Cells for Cancer Therapy. Hum. Gene Ther. 2019;30:413–428. doi: 10.1089/hum.2018.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhen A., Kamata M., Rezek V., Rick J., Levin B., Kasparian S., Chen I.S., Yang O.O., Zack J.A., Kitchen S.G. HIV-specific Immunity Derived From Chimeric Antigen Receptor-engineered Stem Cells. Mol. Ther. 2015;23:1358–1367. doi: 10.1038/mt.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhen A., Peterson C.W., Carrillo M.A., Reddy S.S., Youn C.S., Lam B.B., Chang N.Y., Martin H.A., Rick J.W., Kim J., et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. Plos Pathog. 2017;13 doi: 10.1371/journal.ppat.1006753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barber-Axthelm I.M., Barber-Axthelm V., Sze K.Y., Zhen A., Suryawanshi G.W., Chen I.S., Zack J.A., Kitchen S.G., Kiem H.P., Peterson C.W. Stem cell-derived CAR T cells traffic to HIV reservoirs in macaques. JCI insight. 2021;6 doi: 10.1172/jci.insight.141502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhen A., Carrillo M.A., Mu W., Rezek V., Martin H., Hamid P., Chen I.S.Y., Yang O.O., Zack J.A., Kitchen S.G. Robust CAR-T memory formation and function via hematopoietic stem cell delivery. Plos Pathog. 2021;17 doi: 10.1371/journal.ppat.1009404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhen A., Rezek V., Youn C., Rick J., Lam B., Chang N., Zack J., Kamata M., Kitchen S. Stem-cell Based Engineered Immunity Against HIV Infection in the Humanized Mouse Model. J. Vis. Exp. 2016:54048. doi: 10.3791/54048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghassemi S., Nunez-Cruz S., O'Connor R.S., Fraietta J.A., Patel P.R., Scholler J., Barrett D.M., Lundh S.M., Davis M.M., Bedoya F., et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018;6:1100–1109. doi: 10.1158/2326-6066.CIR-17-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petersen C.T., Hassan M., Morris A.B., Jeffery J., Lee K., Jagirdar N., Staton A.D., Raikar S.S., Spencer H.T., Sulchek T., et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv. 2018;2:210–223. doi: 10.1182/bloodadvances.2017011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urak R., Walter M., Lim L., Wong C.W., Budde L.E., Thomas S., Forman S.J., Wang X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer. 2017;5:26. doi: 10.1186/s40425-017-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S.Y., Olsen P., Lee D.H., Kenoyer A.L., Budde L.E., O'Steen S., Green D.J., Heimfeld S., Jensen M.C., Riddell S.R., et al. Preclinical Optimization of a CD20-specific Chimeric Antigen Receptor Vector and Culture Conditions. J. Immunother. 2018;41:19–31. doi: 10.1097/CJI.0000000000000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J., Jin L., Wang F., Zhang Y., Liu B., Zhao T. Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein & cell. 2019;10:764–769. doi: 10.1007/s13238-019-0643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cha E., Graham L., Manjili M.H., Bear H.D. IL-7 + IL-15 are superior to IL-2 for the ex vivo expansion of 4T1 mammary carcinoma-specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res. Treat. 2010;122:359–369. doi: 10.1007/s10549-009-0573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cieri N., Camisa B., Cocchiarella F., Forcato M., Oliveira G., Provasi E., Bondanza A., Bordignon C., Peccatori J., Ciceri F., et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–584. doi: 10.1182/blood-2012-05-431718. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Eerland R., Nuijen B., Heemskerk B., van Rooij N., van den Berg J.H., Beijnen J.H., Uckert W., Kvistborg P., Schumacher T.N., Haanen J.B.A.G., Jorritsma A. Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Hum. Gene Ther. Methods. 2014;25:277–287. doi: 10.1089/hgtb.2014.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le H.K., Graham L., Miller C.H.T., Kmieciak M., Manjili M.H., Bear H.D. Incubation of antigen-sensitized T lymphocytes activated with bryostatin 1 + ionomycin in IL-7 + IL-15 increases yield of cells capable of inducing regression of melanoma metastases compared to culture in IL-2. Cancer Immunol. Immunother. 2009;58:1565–1576. doi: 10.1007/s00262-009-0666-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khamaikawin W., Shimizu S., Kamata M., Cortado R., Jung Y., Lam J., Wen J., Kim P., Xie Y., Kim S., et al. Modeling Anti-HIV-1 HSPC-Based Gene Therapy in Humanized Mice Previously Infected with HIV-1. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2018;9:23–32. doi: 10.1016/j.omtm.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ringpis G.E.E., Shimizu S., Arokium H., Camba-Colón J., Carroll M.V., Cortado R., Xie Y., Kim P.Y., Sahakyan A., Lowe E.L., et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PloS one. 2012;7 doi: 10.1371/journal.pone.0053492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coiras M., Bermejo M., Descours B., Mateos E., García-Pérez J., López-Huertas M.R., Lederman M.M., Benkirane M., Alcamí J. IL-7 Induces SAMHD1 Phosphorylation in CD4+ T Lymphocytes, Improving Early Steps of HIV-1 Life Cycle. Cell Rep. 2016;14:2100–2107. doi: 10.1016/j.celrep.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manganaro L., Hong P., Hernandez M.M., Argyle D., Mulder L.C.F., Potla U., Diaz-Griffero F., Lee B., Fernandez-Sesma A., Simon V. IL-15 regulates susceptibility of CD4(+) T cells to HIV infection. Proc. Natl. Acad. Sci. USA. 2018;115:E9659–E9667. doi: 10.1073/pnas.1806695115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavender K.J., Pang W.W., Messer R.J., Duley A.K., Race B., Phillips K., Scott D., Peterson K.E., Chan C.K., Dittmer U., et al. BLT-humanized C57BL/6 Rag2-/-gammac-/-CD47-/- mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood. 2013;122:4013–4020. doi: 10.1182/blood-2013-06-506949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Younan P.M., Polacino P., Kowalski J.P., Peterson C.W., Maurice N.J., Williams N.P., Ho O., Trobridge G.D., Von Laer D., Prlic M., et al. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood. 2013;122:179–187. doi: 10.1182/blood-2013-01-482224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nissani A., Lev-Ari S., Meirson T., Jacoby E., Asher N., Ben-Betzalel G., Itzhaki O., Shapira-Frommer R., Schachter J., Markel G., Besser M.J. Comparison of non-myeloablative lymphodepleting preconditioning regimens in patients undergoing adoptive T cell therapy. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kochenderfer J.N., Somerville R.P.T., Lu T., Shi V., Bot A., Rossi J., Xue A., Goff S.L., Yang J.C., Sherry R.M., et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017;35:1803–1813. doi: 10.1200/JCO.2016.71.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Surh C.D., Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Sereti I., Dunham R.M., Spritzler J., Aga E., Proschan M.A., Medvik K., Battaglia C.A., Landay A.L., Pahwa S., Fischl M.A., et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood. 2009;113:6304–6314. doi: 10.1182/blood-2008-10-186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mueller Y.M., Do D.H., Altork S.R., Artlett C.M., Gracely E.J., Katsetos C.D., Legido A., Villinger F., Altman J.D., Brown C.R., et al. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J. Immunol. 2008;180:350–360. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hou L., Jie Z., Liang Y., Desai M., Soong L., Sun J. Type 1 interferon-induced IL-7 maintains CD8+ T-cell responses and homeostasis by suppressing PD-1 expression in viral hepatitis. Cell. Mol. Immunol. 2015;12:213–221. doi: 10.1038/cmi.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lévy Y., Sereti I., Tambussi G., Routy J.P., Lelièvre J.D., Delfraissy J.F., Molina J.M., Fischl M., Goujard C., Rodriguez B., et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin. Infect. Dis. 2012;55:291–300. doi: 10.1093/cid/cis383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pellegrini M., Calzascia T., Toe J.G., Preston S.P., Lin A.E., Elford A.R., Shahinian A., Lang P.A., Lang K.S., Morre M., et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144:601–613. doi: 10.1016/j.cell.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Gumber D., Wang L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine. 2022;77 doi: 10.1016/j.ebiom.2022.103941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geyer M.B., Rivière I., Sénéchal B., Wang X., Wang Y., Purdon T.J., Hsu M., Devlin S.M., Palomba M.L., Halton E., et al. Safety and tolerability of conditioning chemotherapy followed by CD19-targeted CAR T cells for relapsed/refractory CLL. JCI insight. 2019;5 doi: 10.1172/jci.insight.122627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kochenderfer J.N., Somerville R.P.T., Lu T., Yang J.C., Sherry R.M., Feldman S.A., McIntyre L., Bot A., Rossi J., Lam N., Rosenberg S.A. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017;25:2245–2253. doi: 10.1016/j.ymthe.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heczey A., Louis C.U., Savoldo B., Dakhova O., Durett A., Grilley B., Liu H., Wu M.F., Mei Z., Gee A., et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017;25:2214–2224. doi: 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Badesha P.S., Saklayen M.G. Hemolytic uremic syndrome as a presenting form of HIV infection. Nephron. 1996;72:472–475. doi: 10.1159/000188916. [DOI] [PubMed] [Google Scholar]
- 59.Gomes A.M., Ventura A., Almeida C., Correia M., Tavares V., Mota M., Seabra J. Hemolytic uremic syndrome as a primary manifestation of acute human immunodeficiency virus infection. Clin. Nephrol. 2009;71:563–566. doi: 10.5414/cnp71563. [DOI] [PubMed] [Google Scholar]
- 60.D'Agati V., Appel G.B. Renal pathology of human immunodeficiency virus infection. Semin. Nephrol. 1998;18:406–421. [PubMed] [Google Scholar]
- 61.Turner M.E., Kher K., Rakusan T., D'Angelo L., Kapur S., Selby D., Ray P.E. A typical hemolytic uremic syndrome in human immunodeficiency virus-1-infected children. Pediatr. Nephrol. 1997;11:161–163. doi: 10.1007/s004670050249. [DOI] [PubMed] [Google Scholar]
- 62.Bachmeyer C., Blanche P., Séréni D., Salmon D., Grateau G., Dreyfus F., Sicard D. Thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome in HIV-infected patients. AIDS (London, England) 1995;9:532–533. [PubMed] [Google Scholar]
- 63.Thompson C.E., Damon L.E., Ries C.A., Linker C.A. Thrombotic microangiopathies in the 1980s: clinical features, response to treatment, and the impact of the human immunodeficiency virus epidemic. Blood. 1992;80:1890–1895. [PubMed] [Google Scholar]
- 64.Mittelman A., Bertram J., Henry D.H., Snyder H.W., Jr., Messerschmidt G.L., Ciavarella D., Ainsworth S., Kiprov D., Arlin Z. Treatment of patients with HIV thrombocytopenia and hemolytic uremic syndrome with protein A (Prosorba column) immunoadsorption. Semin. Hematol. 1989;26:15–18. [PubMed] [Google Scholar]
- 65.Ho O., Larsen K., Polacino P., Li Y., Anderson D., Song R., Ruprecht R.M., Hu S.L. Pathogenic infection of Macaca nemestrina with a CCR5-tropic subtype-C simian-human immunodeficiency virus. Retrovirology. 2009;6:65. doi: 10.1186/1742-4690-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peterson C.W., Haworth K.G., Polacino P., Huang M.L., Sykes C., Obenza W.M., Repetto A.C., Kashuba A., Bumgarner R., DeRosa S.C., et al. Lack of viral control and development of combination antiretroviral therapy escape mutations in macaques after bone marrow transplantation. AIDS (London, England) 2015;29:1597–1606. doi: 10.1097/QAD.0000000000000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cordoba S., Onuoha S., Thomas S., Pignataro D.S., Hough R., Ghorashian S., Vora A., Bonney D., Veys P., Rao K., et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat. Med. 2021;27:1797–1805. doi: 10.1038/s41591-021-01497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li S., Zhang J., Wang M., Fu G., Li Y., Pei L., Xiong Z., Qin D., Zhang R., Tian X., et al. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br. J. Haematol. 2018;181:360–371. doi: 10.1111/bjh.15195. [DOI] [PubMed] [Google Scholar]
- 69.Ferrari G., Thrasher A.J., Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 2021;22:216–234. doi: 10.1038/s41576-020-00298-5. [DOI] [PubMed] [Google Scholar]
- 70.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341 doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341 doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 72.Eichler F., Duncan C., Musolino P.L., Orchard P.J., De Oliveira S., Thrasher A.J., Armant M., Dansereau C., Lund T.C., Miller W.P., et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goyal S., Tisdale J., Schmidt M., Kanter J., Jaroscak J., Whitney D., Bitter H., Gregory P.D., Parsons G., Foos M., et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022;386:138–147. doi: 10.1056/NEJMoa2109167. [DOI] [PubMed] [Google Scholar]
- 74.Gupta R.K., Peppa D., Hill A.L., Gálvez C., Salgado M., Pace M., McCoy L.E., Griffith S.A., Thornhill J., Alrubayyi A., et al. Evidence for HIV-1 cure after CCR5Delta32/Delta32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: a case report. Lancet HIV. 2020;7:e340–e347. doi: 10.1016/S2352-3018(20)30069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Allers K., Hütter G., Hofmann J., Loddenkemper C., Rieger K., Thiel E., Schneider T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- 76.Mu W., Zhen A., Carrillo M.A., Rezek V., Martin H., Lizarraga M., Kitchen S. Oral Combinational Antiretroviral Treatment in HIV-1 Infected Humanized Mice. J. Vis. Exp. 2022;188 doi: 10.3791/63696. [DOI] [PubMed] [Google Scholar]
- 77.Radtke S., Perez A.M., Venkataraman R., Reddy S., Haworth K.G., Humbert O., Kiem H.P., Peterson C.W. Preparation and Gene Modification of Nonhuman Primate Hematopoietic Stem and Progenitor Cells. J. Vis. Exp. 2019;144 doi: 10.3791/58933. [DOI] [PubMed] [Google Scholar]
- 78.Peterson C.W., Haworth K.G., Burke B.P., Polacino P., Norman K.K., Adair J.E., Hu S.L., Bartlett J.S., Symonds G.P., Kiem H.P. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2016;3 doi: 10.1038/mtm.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peterson C.W., Benne C., Polacino P., Kaur J., McAllister C.E., Filali-Mouhim A., Obenza W., Pecor T.A., Huang M.L., Baldessari A., et al. Loss of immune homeostasis dictates SHIV rebound after stem-cell transplantation. JCI insight. 2017;2 doi: 10.1172/jci.insight.91230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reeves D.B., Peterson C.W., Kiem H.P., Schiffer J.T. Autologous Stem Cell Transplantation Disrupts Adaptive Immune Responses during Rebound Simian/Human Immunodeficiency Virus Viremia. J. Virol. 2017;91 doi: 10.1128/JVI.00095-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peterson C.W., Younan P., Polacino P.S., Maurice N.J., Miller H.W., Prlic M., Jerome K.R., Woolfrey A.E., Hu S.L., Kiem H.P. Robust suppression of env-SHIV viremia in Macaca nemestrina by 3-drug ART is independent of timing of initiation during chronic infection. J. Med. Primatol. 2013;42:237–246. doi: 10.1111/jmp.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peterson C.W., Wang J., Deleage C., Reddy S., Kaur J., Polacino P., Reik A., Huang M.L., Jerome K.R., Hu S.L., et al. Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1006956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data are available in the main text or the supplementary materials.