

Abstract
A tumor nano-lysate “TNL” vaccine comprised of sonicated 4T1 cells was developed, characterized and implemented for the prevention of triple-negative breast cancer. This study aims to gain a better understanding of the immune response behind the success of the vaccine in vivo, through use of ex vivo and in vivo assays. Here, we analyze the activation of various immune cells isolated from healthy mouse spleens and find that antigen-presenting cells (APCs) such as dendritic cells (DCs) are being activated following 24 h incubation with 1:10 mg TNL/mg splenocytes. These cells were further explored to determine the pathway by which activation is occurring, and it was observed that TNL are phagocytosed by DCs to activate NF-kB and c-Fos pathways, resulting in enhanced cytokine release after 24 h. An in vivo temporal analysis was performed in mice to understand the immune response at 1, 3, 7 and 10 days after one 100 µL dose of TNL consisting of 105 sonicated 4T1 cells via cardiac puncture and splenocyte and peripheral blood mononuclear cell (PBMC) analysis. Changes were observed for up to one week. A multiple dose study was performed comparing mice that were vaccinated with one dose of TNL administered every ten days for 3 doses total, as well as a PBS vehicle control. Survival for TNL-vaccinated mice was enhanced compared to the PBS control, and there was an average delay of 10 days in the onset of metastasis. The differences between the groups at the end of the study demonstrate the potential for TNL as a preventative therapeutic.
Keywords: Cancer vaccines, immunology, antigen-presenting cells
INTRODUCTION
Breast cancer remains an epidemic in the United States, with 1 in 8 women diagnosed with the disease in her lifetime.1 While 5-year survival rates as high as 99% are associated with early stage breast cancers, survival rates for later stage, metastatic breast cancers are reduced to as low as 27%.2 These statistics indicate a need for successful anti-metastatic therapeutics and preventative measures for breast cancer. Current preventative measures include regular screening via breast self-examination (BSE), clinical breast exam (CBE), and mammography after the age of 40.3,4
Currently, there are only five FDA-approved preventative cancer vaccines used in clinical practice, and these protect against sexually transmitted viral infections human papillomavirus (HPV) and hepatitis B (HBV).5 These viruses commonly promote liver, cervical and oral cancers, and when implemented the vaccines have successfully prevented the virus and corresponding disease onset.6–9 In cancer, vaccination is designed to serve as an active immunotherapy strategy to stimulate the patient’s immune system. While strides have been made in research and development of non-viral preventative cancer vaccines, many challenges remain.10
Previously, we developed a tumor nano-lysate (TNL) preventative vaccine for triple-negative breast cancer.11 TNL was fabricated via membrane disruption of 4T1 breast cancer cells using probe sonication, and the vaccine was characterized by size, protein contents, and morphology and analyzed for toxicity in vitro.11 A pilot study was also performed analyzing the effectiveness of the vaccine when used with the 4T1 model in vivo using immunocompetent mice, and for TNL-vaccinated mice, reduced tumor growth was observed, onset of metastasis was delayed, and survival was increased.11 Although TNL-vaccinated mice ultimately developed tumors, the results of the study demonstrated the potential of TNL as a preventative vaccine.11
In this study, we aim to better understand the immunological response to TNL using a variety of in vitro, ex vivo, and in vivo functional assays. We explore the effects of TNL treatment on splenocytes isolated from healthy mice, and further characterize the activation of DCs following a single dose of TNL treatment. We demonstrate that the TNL vaccine more effectively activates DCs compared to potent stimulators, such as CpG and LPS at 4 and 24 h following treatment. This finding is critical considering DCs are vital to connect innate and adaptive immunity and play a key role in T cell priming. We further explore cytokine release from murine DCs following TNL treatment to delineate how this vaccine can enhance the ability of DCs to function as antigen presenting cells (APCs). The preventative effects of the TNL vaccine were tested in vitro and in vivo on 4T1 murine breast cancer cells. Overall, our variety of temporal analyses illustrate the need for booster doses, which is often invaluable with vaccines. These results provide a better understanding of what is occurring in vivo and suggest ways to improve the success of the formulation. The success of this preventative cancer vaccine motivates its future testing and exploration of the immunological response to additional cancer types.
MATERIALS AND METHODS
Cell culture:
Immortalized dendritic cell (DCs) line DC2.4 cells (Sigma-Aldrich Catalog No. SCC142) were grown in culture media consisting of RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 1X L-Glutamine, 1X non-essential amino acids, 1X HEPES buffer solution and 0.0054X β-Mercaptoethanol. Cells were maintained at 37°C with 5% CO2, and experiments performed at ~70–80% confluency.
Reagents:
For preparation of DC2.4 cell culture media, RPMI-1640, FBS, non-essential amino acids and HEPES buffer (Gibco), and β-Mercaptoethanol (Sigma-Aldrich) were used. Hank’s balanced salt solution (HBSS) w/w/o calcium and magnesium was purchased (Gibco). Lipopolysaccharide (LPS) solution was purchased from Thermo Scientific. ODN 1585 - TLR9 ligand (CpG) was purchased from InvivoGen. Anti-mouse PE-CD40 (5C3) was purchased from BD. Anti-mouse PE-MHC (Class I H-2Kk) was purchased from Antibodies-Online.com. Anti-mouse PE-CD70 (FR70), PE-CD80 (16-10A1), PE-I A/I E (M5/114.15.2) (MHC II), and PE mouse anti-Ki-67 (567720) were used for flow cytometry (BD). Anti-mouse PE-CD83 (Michel 17), PE-CD197 (CCR7) (4B12), PE-phospho-NFkB p65 (Ser529) (NFkBp65S529-H3), PE-phospho-c-Fos (AP-1) (Ser32) (cFosS32-BA9), and PE (MA5-36891) were obtained from Thermo Scientific. H-2Ld MuLV gp70 Tetramer-SPSYVYHQF-PE was obtained from MBL International. For immune cell labeling, CD3, CD4 and CD19 were purchased (BD), CD8 was purchased (Thermo Fisher), and CD335, CD68 and CD11c were purchased (BioLegend). Anti-RAC1 (MBS9200589) was purchased through MyBioSource. Proteome Profiler Mouse Cytokine Array Kit, Panel A (ARY006) was used to analyze cytokine release (R&D Systems). VECTASHIELD® Antifade Mounting Medium (H-1000-10) was purchased from Vector Laboratories and used for confocal microscopy. Triton X-100, DAPI (D9542-10MG), Tween® 20, viscous liquid, and Poly-L-lysine solution were purchased from Sigma-Aldrich. ActinRed 555 ReadyProbes Reagent and secondary antibodies Alexa Fluor 488 goat anti-rabbit IgG (H+L) and Alexa Fluor 488 goat anti-mouse IgG (H+L) were purchased from Invitrogen for confocal imaging. 10% Normal Goat Serum (Life Technologies) and 32% paraformaldehyde aqueous solution, electron microscopy grade (Electron Microscopy Sciences) were purchased for imaging. Ficoll-Paque PLUS was purchased from GE Healthcare. Syringe/plungers (BD), cell strainers (Celltreat), and red blood cell (RBC) lysis buffer were purchased for splenocyte isolation. Syringes for mouse injections were purchased from BD.
TNL fabrication:
TNL were fabricated via sonication as previously described.11
Flow cytometry analysis and antibody staining:
For intracellular proteins phospho-NF-κB and - cFos, cells were fixed with 4% paraformaldehyde (PFA) for 10 min. Cells were washed with HBSS and permeabilized with 100% ice cold methanol. Cells were washed again and stained for 15 min in the dark with antibodies fluorescently tagged with PE fluorophore while suspended in 1% BSA. A Guava easyCyte 12HT flow cytometer (Millipore) was used to measure fluorescence intensity, with FlowJo software used for gating and analysis. For extracellular proteins analyzed after 24 h, cells were lifted, washed, and incubated with primary antibodies pre-conjugated with fluorophores in 1% BSA and then washed once more prior to analysis. For RAC1 staining, cells were first stained with the primary antibody and then stained for 15 min with an Alexa 488 secondary antibody and washed prior to analysis via flow cytometry. For these analyses, DC2.4 cells were plated overnight and were treated for 4 h or 24 h with freshly made TNL at a concentration of 1:10 mg TNL/mg DC2.4 (TNLLow) or 2:1 mg TNL/mg DC2.4 (TNLHigh), and equivalent PBS volumes for a vehicle control. When stimulators were used as positive controls, LPS was at a concentration of 10 µg/mL and CpG was at a concentration of 2 µM.
Splenocyte isolation and analysis:
Primary cells were isolated from the spleens of healthy BALB/c mice using a plunger and cell strainer, and red blood cell (RBC) lysis buffer. Splenocytes were counted and incubated with 1:10 mg TNL/mg splenocyte or PBS. T cells were fixed and permeabilized and stained for T cell markers CD3, CD4 and CD8 and intracellular cytokines IL-2, TNF, and IFN. Splenocytes were stained with B cell marker CD19 and activation markers including CD40, CD69, CD86 and majorhistocompatability complexes (MHCs). Additionally, splenocytes were stained for CD68 and CD11c and stained for costimulatory molecules.
Tetramer staining:
Splenocytes were isolated as previously described and plated overnight. Cells were treated for 24 h with PBS, LPS, CpG, or 1:10 mg TNL/mg DC2.4 (TNLLow) or 2:1 mg TNL/mg DC2.4 (TNLHigh). Splenocytes were stained for extracellular marker CD3 and H-2Ld MuLV gp70 Tetramer.
Viability assay:
Splenocytes were isolated as previously described and plated overnight. Cells were treated with PBS or 1:10 TNL/mg splenocytes and 4T1 cells were plated separately. After 24 h, splenocytes were added to 4T1 cells for 24 h at concentrations of 1:10, 1:5, or 2:5 mg splenocytes/mg 4T1 cells. A control of PBS treated-4T1 cells (no-splenocytes) was maintained. Cells were then lifted, centrifuged and resuspended in 150 μL HBSS -/−. 5 μL of annexin v (AV) and 5 μL of propidium iodide (PI) were added to each sample, and unstained and single stained controls were used. Cells were stained for 15 min in darkness and 100 μL HBSS was added to each sample before flow cytometry analysis. Statistical significance was determined using Two-way ANOVA.
Proteome profiler:
DC2.4 cells were treated and plated overnight and treated with TNL at 1:10 mg TNL/mg DC2.4, equal volume of PBS as a vehicle control, or CpG. After 24 h, the supernatant was collected from cell suspensions and centrifuged at 4800 RPM at 4°C for 10 min. Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D Systems) was used for analysis and the manufacturer’s instructions were followed for the procedure. An Odyssey CLx imager was used for 2 min chemiluminescence imaging. Membrane mean pixel density was quantified using Image Studio Lite software. Two replicates were completed for each treatment group.
Confocal microscopy:
DCs were seeded onto glass coverslips previously coated with poly-l-lysine (PLL). After 24 h, DCs were treated with PBS, CpG or TNL. After 4 h or 24 h, cells were fixed with 4% PFA, washed and then permeabilized with 1% Triton. After washing, cells were blocked for 45 min with 5% BSA + 5% goat serum, and then stained with primary antibodies (LAMP2, Syntaxin18) at 1:100 in coating buffer for 1 h. Cells were then stained with a 4 𝜇L Alexa 488 secondary antibody, DAPI nuclear stain and ActinRed 555 for 30 min. Vectashield was added to slides followed by coverslips, and cells were imaged using a Zeiss LSM 900 confocal microscope. Image analysis was performed using FIJI software.
In vivo studies:
The studies were approved by IACUC protocol #M1700009-02. Eight-week-old female BALB/cj (Strain #: 000651) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were monitored by veterinary staff at the Division of Animal Care (DAC) at Vanderbilt University according to institutional guidelines. Mice experienced identical care regimens and housing conditions, and mice were euthanized at humane endpoints, determined by factors such as mobility and tumor size. “One dose” described in each of the in vivo assays is defined by the in vivo pilot study developed in Dombroski, et al., where one dose of TNL consists of 105 sonicated 4T1 cells in 100 μL of PBS.11
In vivo temporal analysis:
Healthy BALB/c mice were injected via tail vein with one dose of TNL (105 sonicated 4T1 cells in 100 μL of PBS) or PBS as a vehicle control. After 1 d, 3 d, and 7 d, n=4 mice from each group (PBS, TNL) were sacrificed and underwent cardiac puncture and spleen resection. Part of each collected blood sample was kept for in-lab analyses and part was given to the Vanderbilt Medical Center Translational Pathology Shared Resource (TPSR) for a complete blood count (CBC). Splenocytes were isolated as described above, counted, and analyzed for extracellular immune cell markers and activation markers. Peripheral blood mononuclear cells (PBMCs) were isolated via differential centrifugation and Ficoll-Paque. Isolated cells were then analyzed for extracellular markers. We continued spleen isolation and PBMC collection and subsequent analysis up to 10 days to gather additional activation and population information on the range of specific immune cell subtypes.
In vivo multi-dose study:
Healthy BALB/c mice were injected with 1 dose of TNL (105 sonicated 4T1 cells in 100 μL of PBS) every 10 d up to 3 doses, or 3 doses of 100 µL PBS at each time point. 10 d after the final dose, mice were inoculated in the anatomical right 4th mammary fat pad with 30,000 4T1-Luciferase cells in 50 µL PBS. Hair was removed from the area surrounding the fat pad prior to inoculation and mice were anesthetized for 5–10 min via continuous inhalation of isoflurane during the administration. Tumors were monitored twice per week via bioluminescent imaging (IVIS Lumina III, PerkinElmer Inc.) and caliper measurements. Subcutaneous injection of 100 μL of D-luciferin (150 mg/kg) was used for imaging. During imaging, mice were anesthetized for 5–10 min via continuous inhalation of isoflurane. Tumor volume was estimated as [length (l) x width (w)2]/2. First signs of observed metastases were considered as bioluminescent signals in a location other than the primary tumor, and these days were recorded. Splenocytes were isolated as previously described and analyzed for immune cell populations and activation markers. n=2 spleens were collected for the PBS group.
Immunohistochemistry (IHC):
After resection, tumors were immediately fixed in 10% neutral buffered formalin (NBF) for 24 h. A board-certified veterinary pathologist examined hematoxylin and eosin (H&E) stained tumor slides and did not find any obvious morphologic differences between treated and control tumors. The Vanderbilt TPSR core routinely processed, paraffin embedded, sectioned at 5 µ.Slides were stained the tumors for CD3m (T cells), CD45r (B cells), or Ki-67 (proliferation) using the Leica Bond-RX staining platform. Briefly, slides were deparaffinized and heat induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution for 5–20 min depending on the primary antibody. Slides were incubated with either anti-CD3 (Cat.# ab16669, Abcam, Cambridge, UK) at 1:250, anti-CD45r (Cat.# 553086, BD Pharmingen, San Diego, CA) at 1:30,000 or anti-Ki-67 (Cat.# ab16667, Abcam, Cambridge, UK) at 1:100 for one hour. The Bond Refine (DS9800, Buffalo Grove, IL, USA) detection system was used for visualization. Slides were then dehydrated and cleared, and coverslips were placed on them. Slides were scanned and uploaded to SlideViewer (3DHistotech) to take snapshots for analysis. FIJI was used to quantify DAB positive staining expression with a Macro developed by A.R.F. Briefly, the color deconvolution function was applied to the snapshots to provide an H&E, DAB, and residual image, so that area fractions (percent positive) can be determined from the DAB image. Data are presented as the percent positive DAB stain ± standard error mean of two sections per tumor per IHC stain.
Statistics:
Data are reported as mean and standard error of the mean. Unless otherwise indicated, statistics were determined using student’s t test. Experiments included at least three independent replicates unless otherwise indicated. *p<0.05, **p<0.01, ***p<0.005, and ****p<0.001 for significance; otherwise, there is no significance. GraphPad Prism software was used to perform statistical analyses and produce figures for this paper.
RESULTS
Antigen-presenting cells are activated by TNL
Splenocytes were isolated and treated with TNL or PBS vehicle control for 24 h. Immune cell populations were analyzed for activation via costimulatory molecule expression (CD40, CD70, CD80, CD83, CD86) or intracellular cytokine staining (IL-2, TNF, IFN). CD3+CD4+ and CD3+CD8+ T cells were observed to not be significantly activated by TNL in vitro (Fig. 1A–B). Similarly, CD68+ macrophages did not have a change in costimulatory molecule expression following incubation with TNL when compared to PBS controls (Fig. 1C). B cells, however, were observed to have significant increases in CD80 and CD83 costimulatory molecule expression and major histocompatibility complexes (MHC) I and II (Fig. 1D). DCs were similarly activated by TNL, as observed by a significant increase in costimulatory molecule expression (Fig. 1E). Given that antigen-presenting cells (APCs) are being activated by TNL, and that DCs are APCs that are often used in successful anti-cancer vaccines, we sought to further explore their response to TNL ex vivo and in vivo.
Figure 1. Cell activation by tumor nano-lysate ex vivo.
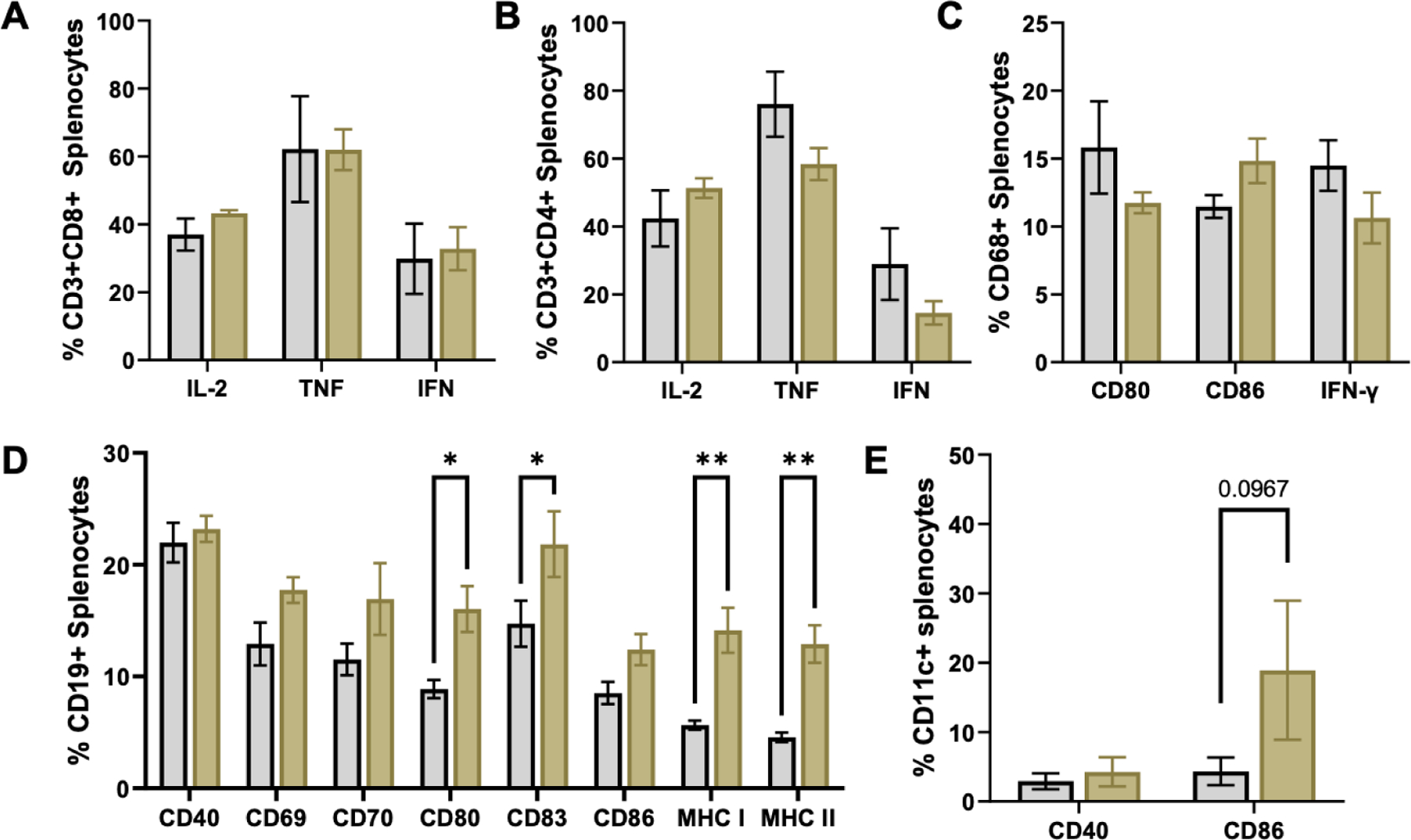
Intracellular cytokine staining of (A) CD3CD8+ and (B) CD3CD4+ splenocytes. (C) Intracellular cytokine and costimulatory molecule staining of CD68+ splenocytes. (D) Costimulatory molecule and MHC staining of CD19+ splenocytes. (E) Costimulatory molecule staining of CD11c+ splenocytes. *p<0.05, **p<0.01.
DCs undergo morphological and metabolic changes in response to TNL
To explore the pathways by which DCs are being activated, DC2.4 immortalized cells were grown in culture and explored for activation of both intracellular and extracellular activation markers. NF-kB phosphorylation and c-Fos phosphorylation are regulated by calcium influx, and in DCs promote cell cycle progression, proliferation and cytokine release.12–15 Ki-67 expression represents an increase in proliferation in a variety of cell types and is often associated with activation of cells.16–18 These activation markers were analyzed for DC2.4 cells treated with LPS, CpG and two concentrations (high and low) of PBS and TNL at 4 h (Fig. 2A) and 24 h (Fig. 2B). Significant increases in phospho-NF-kB and -c-Fos were observed after 4 h for TNL-treated DCs compared to PBS vehicle controls. Changes in c-Fos phosphorylation was observed for up to 24 h. The measure of phospho-NF-kB and -c-Fos for CpG and LPS-treated DCs was similar to the TNL-treated DCs, which provides evidence that the efficacy of the TNL is comparable to that of a potent stimulator.
Figure 2. Analysis of DC activation markers.
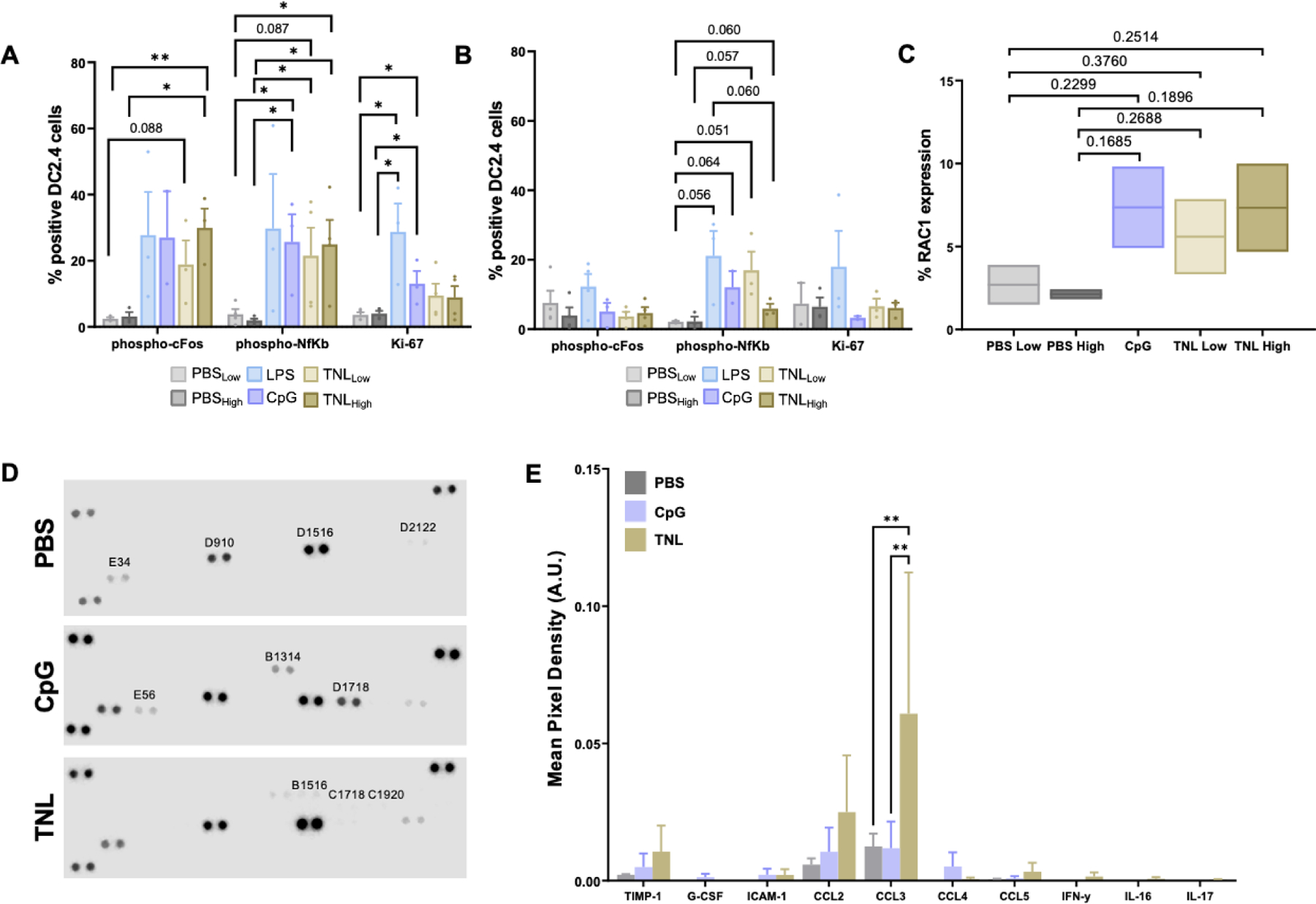
DC2.4 NF-kB phosphorylation, c-Fos phosphorylation and Ki-67 expression after (A) 4 h and (B) 24 h. (C) Percent RAC1 expression in DC2.4 cells after 24 h compared to costimulatory controls. (D) Representative proteome profiler arrays depicting cytokine release. (E) Cytokine and chemokine release in DC2.4 cells after 24 h. *p<0.05, **p<0.01.
Morphological changes in DCs are indicative of the successful uptake and presentation of an antigen, and this process can be observed by changes in RAC1 expression.19 DCs were treated with CpG and two different concentrations (high and low) of PBS and TNL. After 24 h, a trend in enhanced RAC1 expression was observed for CpG- and TNL-treated cells compared to PBS-treated cells (Fig. 2C). This finding indicates that DCs exposed to TNL were experiencing changes indicative of antigen uptake and presentation.
Cytokine and chemokine release is necessary for activated DC trafficking and for T cell priming.20 Using a cytokine array to analyze a panel of cytokines and chemokines, DC2.4 cells were treated with CpG or TNL, or PBS as a vehicle control for TNL. After 24 h, cytokine release was analyzed and a significant increase in CCL3 was observed, as well as trends in other cytokines and chemokines released by TNL-treated cells (Fig. 2D–E). CCL3 has emerged as a potent activator of innate and adaptive immune responses and has been shown to play a key role in the regulation of lymph node homing of DCs. CCL3 also induces antigen-specific T cell responses.21
The activation of the above pathways is essential to DCs successfully activating specific T cells and initiating a preventative immune response. Interestingly, DCs treated with TNL responded similarly to or better than potent stimulators LPS and CpG when treated for the same length of time.
DCs phagocytose TNL and a preventative response is elicited
DCs are activated when they successfully uptake an antigen, and LAMP2 and Syntaxin18 can play a role in this phagocytosis, where LAMP2 is internalized following antigen uptake and Syntaxin18 is essential for ER-mediated phagocytosis.22–24 DC2.4 cells were treated with TNL and PBS as a vehicle control, stained for phagocytosis markers and subsequently imaged using confocal microscopy using a Zeiss LSM 900 (Fig. 3A). While no significant difference was observed in Syntaxin18 expression after 4 h (Fig. 3B), there was a trend in increased expression after 24 h for TNL-treated DCs (Fig. 3C). Similarly, after 24 h, there was a significant increase in LAMP2 expression for TNL-treated DCs (Fig. 3D). These results demonstrated that DCs uptake TNL via phagocytosis.
Figure 3. DC phagocytosis and functional analyses.
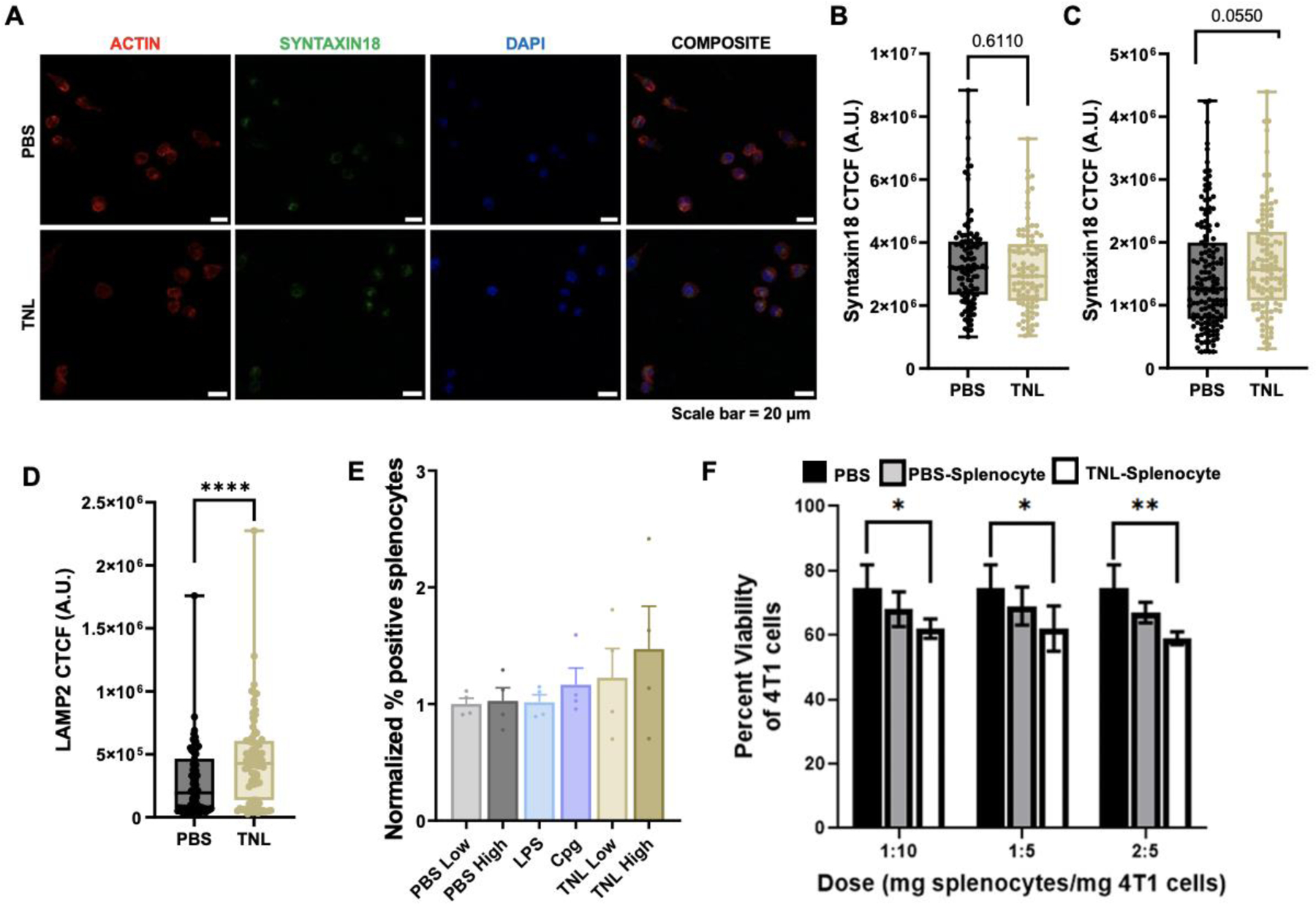
(A) Syntaxin18 24 h representative micrograph. Syntaxin18 expression after (B) 4 h and (C) 24 h. (D) LAMP2 expression after 24 h. (E) CD3+ Tetramer staining. (F) Functional viability assay treating 4T1 breast cancer cells with TNL-treated splenocytes. Scale bar = 50 μm. *p<0.05, **p<0.01, ****p<0.001.
As 4T1 cells express the antigen gp70, a H-2Ld MuLV gp70 tetramer can be used to identify specific DC-activated CD3+ T cells as indicated by CD3+Tetramer+ cells.25–27 When CD3+ splenocytes were treated with LPS, CpG and two different concentrations (high and low) of TNL and PBS as a vehicle control for 24 h, a trend in increased tetramer expression was observed for CpG and both TNL concentrations (Fig. 4E). The results indicated that T cells are specifically activated for the gp70 antigen when treated with TNL without having been exposed to the 4T1 cells.
Figure 4. Temporal analysis of immune response to TNL.
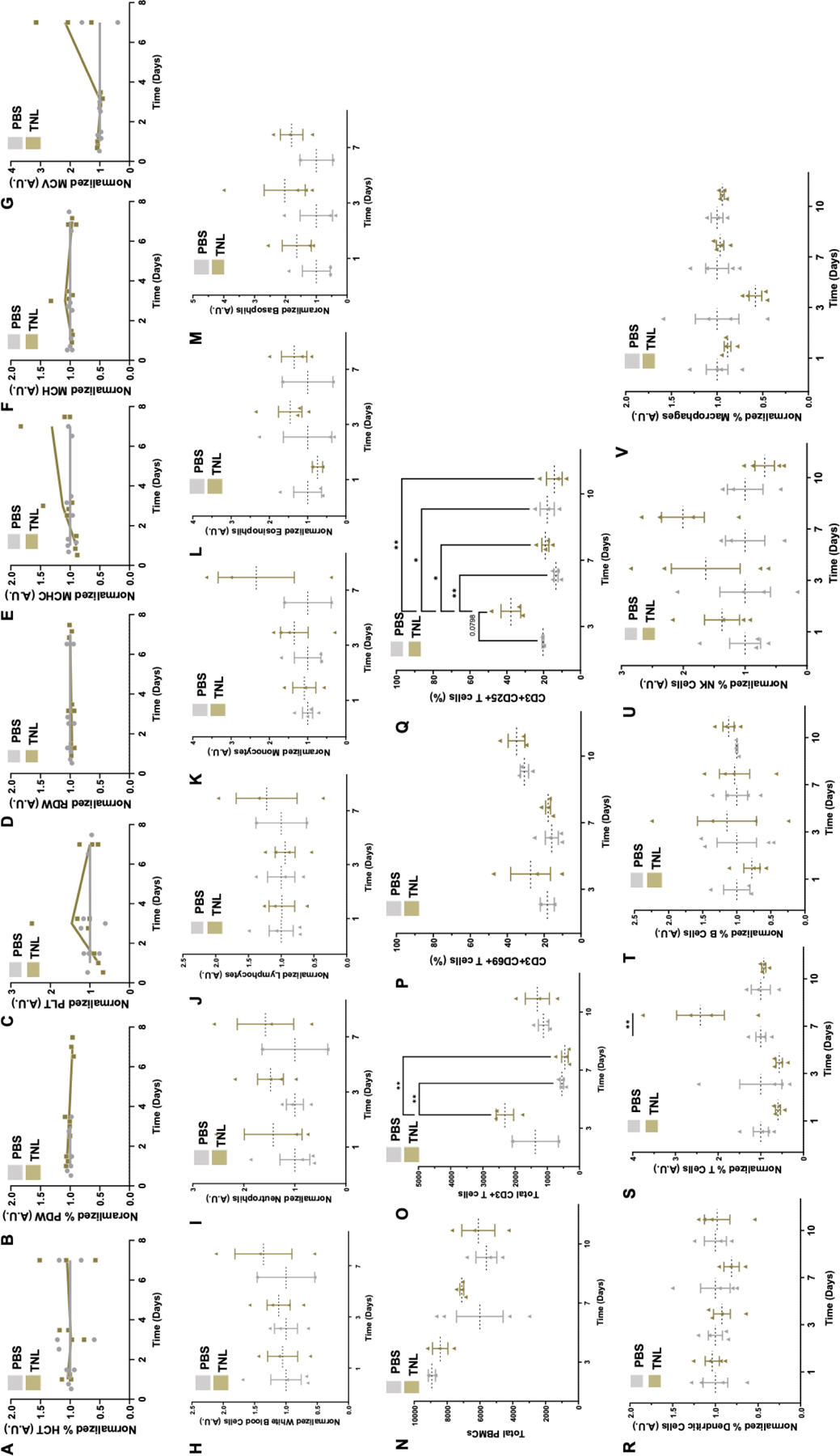
The CBC analyzed general red blood cell (RBC)-related counts including (A) HCT – Hematocrit, (B) %PDW – platelet distribution width, (C) PLT – platelet count, (D) RDW – RBC distribution width size/volume (E) MCHC – mean corpuscular hemoglobin concentration, (F) MCH – mean corpuscular hemoglobin, and (G) MCV – mean corpuscular volume. The CBC also analyzed various white blood cell (WBC) populations including (H) general WBC, (I) neutrophils, (J) lymphocytes, (K) monocytes, (L) eosinophils, and (M) basophils. PBMCs were analyzed for (N) total PBMCs, (O) total CD3+ T cells, and (P) CD69+ and (Q) CD25+ populations. Splenocyte composition was also determined via flow cytometry, marker for (R) DCs, (S) T cells, (T) B cells, (U) NK cells, and (V) macrophages. *p<0.05, **p<0.01.
Splenocytes were treated for 24 h with PBS vehicle control or TNL, and PBS- or TNL-treated splenocytes were used to treat 4T1 cells at three different concentrations (high, mid and low concentrations). After 24 h, 4T1 cells were lifted, and an Annexin V viability assay was performed to analyze 4T1 cell viability. Percent viability was significantly reduced in 4T1s treated with TNL-treated splenocytes compared to PBS-treated 4T1 cells and 4T1s treated with PBS-treated splenocytes (Fig. 3F). This functional study indicates the potential use of TNL as a preventative vaccine.
Initial immune activation is sustained for up to one week
Healthy female BALB/c mice were vaccinated via tail vein injection with one dose of PBS vehicle control or TNL and analyzed over time for a temporal immune response. A complete blood count (CBC) was performed at 1 d, 3 d and 7 d, where blood samples were sent to the Vanderbilt TPSR for general immune cell population counts. No significant differences were observed in red blood cell (RBC)-related counts over time for TNL-treated mice compared to mice that had received the PBS control (Fig. 4A–G). Overall white blood cell (WBC) changes over time between PBS- and TNL-treated mice were largely unchanged (Fig. 4H, L–M). Neutrophils, however, had higher counts for TNL-treated mice overall (Fig. 4I) and lymphocyte and monocyte populations had higher counts for TNL-treated mice at 7 d (Fig. 4J–K).
PBMCs were isolated from whole blood for flow cytometry analysis at 1 d, 3 d, and 7 d. Due to the specificity of immune cell data that were collected, the study continued to 10 d and yielded interesting results. While PBMCs isolated from whole blood had no significant changes in total PBMCs (Fig. 4N), significant differences in CD3+ T cells between PBS- and TNL-treated mice at 3 d were observed, and these counts were significantly reduced after 7 d (Fig. 4O). While there were no significant differences between CD3+CD69+ T cells between groups, there was a significant difference between CD3+CD25+ T cells between population, and these counts were reduced significantly at 7 d and then 10 d (Fig. 4P–Q). These results indicate that T cells were significantly activated by TNL at 3 d, with activation returning to baseline by 10 d.
Splenocyte composition analyses revealed no significant changes in DCs, B cells or macrophages (Fig. 4R, T, V). There was an increase in T cell counts for TNL-treated mice after 7 d and a trend in increased natural killer (NK) cell counts after 3 d and 7 d (Fig. 4S, U).
Multiple dose study demonstrates potential for tumor nano-lysate as a preventative vaccine
Based on the results observed in the activation of various immune cell populations ex vivo after one dose of TNL (Fig 4), we aimed to further understand the adaptive immune response that the TNL vaccine induces through an in vivo study involving breast cancer inoculation in mice.
Healthy mice were treated with one dose of TNL or PBS control once every 10 d, up to 3 doses. 10 d after the final dose, mice were inoculated with 4T1 breast cancer (Fig. 5A). The goal of this in vivo study was to examine the potential of the multiple doses of the TNL vaccine as a preventative measure to delay or prevent the onset of metastasis. Previously, we examined that a single dose of TNL delivered 10 d before tumor inoculation resulted in reduced tumor burden, delayed metastasis, and prolonged survival for TNL-treated mice in the same 4T1 model.11
Figure 5. In vivo multi-dose TNL study.
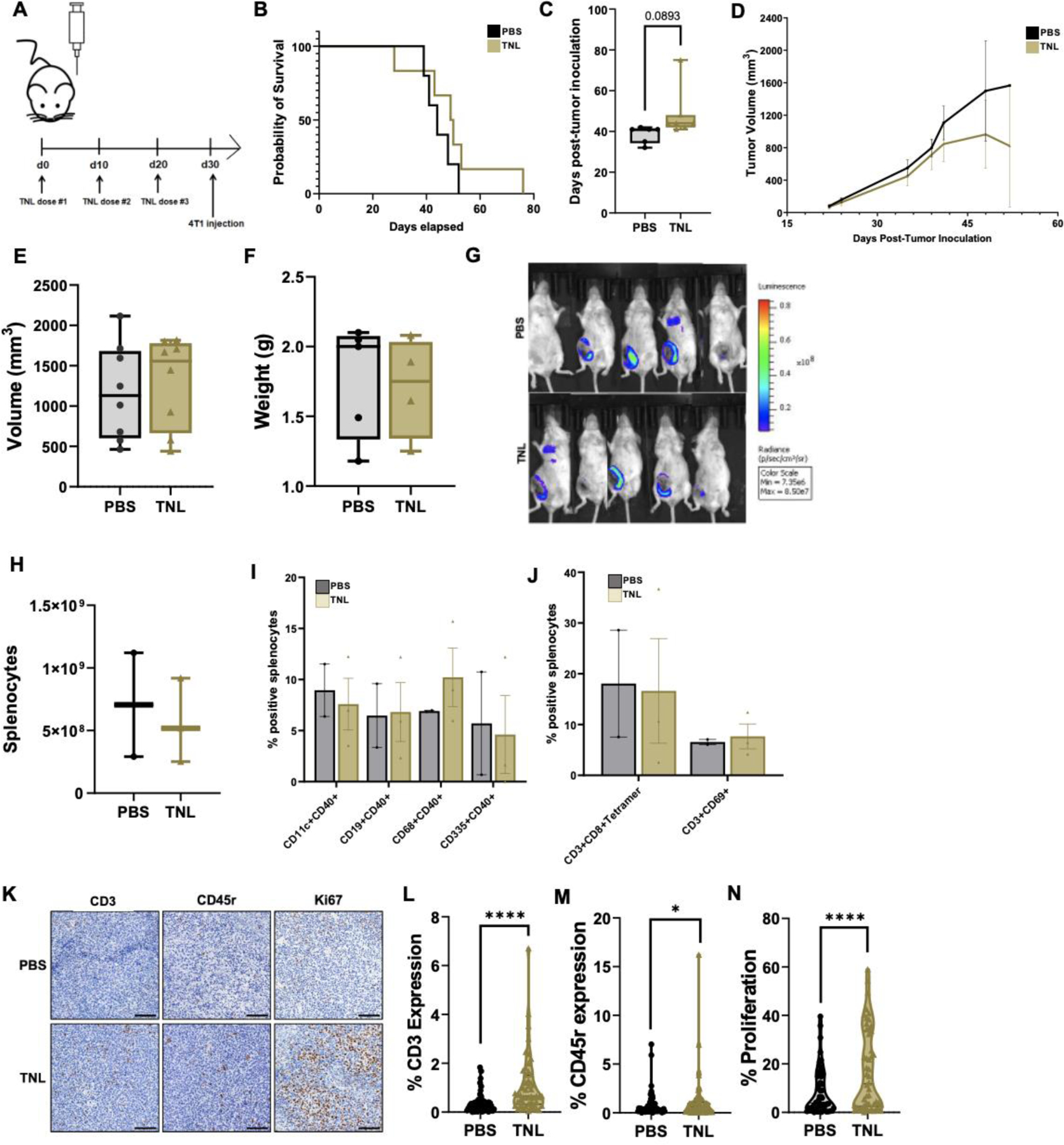
(A) Multi-dose mouse protocol. (B) Survival curves for PBS- and TNL- treated mice. (C) Day of observed metastasis onset following tumor inoculation. (D) Changes in tumor volume over time. (e) Final tumor volume of each mouse. (F) Final tumor weight following resection. (G) Representative bioluminescent imaging (BLI) depicting tumor volume at one sample timepoint. We observed increased luminescence in BLI, indicating increased tumor growth, for PBS-vaccinated mice. (H) Splenocyte population counts. (I) Percent positive splenocyte activation markers and (J) percent positive splenocyte tetramers. (K) Representative immunohistochemistry micrographs. (L) % CD3 expression (M) % CD45r expression and (N) % Proliferation. Scale bar = 100 μm. *p<0.05, ****p<0.001.
Survival for TNL-vaccinated mice was enhanced from an average of 44 to 49.5 d, and onset of metastasis was delayed an average of 10 d (Fig. 5B–C). Tumor volume and growth overtime was slower for TNL-vaccinated mice than PBS-vaccinated mice, although the final tumor volumes and final tumor weights were not statistically significant (Fig. 5D–G). While there was not a significant difference in splenocyte composition post-mortem between the groups, there was a trend in enhanced percent positive CD68+CD40+ splenocytes for the TNL group, indicating activation of macrophages (Fig. 5H–J). IHC was performed to analyze immune cell infiltration at the tumor site and proliferation of tumor cells (Fig. 5K). There was a significant increase in CD3+ T cells and CD45r+ B cells in the tumors of TNL-vaccinated mice compared to controls (Fig. 5L–M). Interestingly, despite the increased immune cell infiltration, the percent of tumor cell proliferation was also significantly enhanced for TNL-vaccinated mice compared to controls (Fig. N).
DISCUSSION
In this study, we expanded upon our previous research and analyzed the immunological response to TNL vaccine both ex vivo and in vivo. Antigen-presenting cells such as DCs were determined to be activated by TNL and were further explored for specific pathways of activation. It was observed that the NF-kB and c-Fos pathways are being significantly activated by TNL, and subsequently cytokine and chemokine release was enhanced. After 24 h, changes in morphology through RAC1 expression occurred, as well as specific markers of TNL antigen phagocytosis. Through functional assays, we saw specific activation of T cells via increased CD3+Tetramer+ cells, and through loss of 4T1 cell viability after treatment with TNL-treated splenocytes.
Temporal analyses using healthy BALB/c mice injected via tail vein by one dose of TNL or a PBS vehicle control revealed sustained activation for one week with increased counts of lymphocytes, monocytes, and neutrophils observed via CBC, and activation of specific CD3+ T cell subsets observed via flow cytometry analysis of PBMCs. The results of the study demonstrated a potential need for multiple “booster” doses after the immune response attenuates after ~10 d. Overall, the multi-dose in vivo study showed reduced tumor burden, delayed onset of metastasis, and increased survival for TNL-vaccinated mice compared to PBS-vaccinated mice. CD3+ T cell and CD45r+ B cell tumor infiltration was also enhanced for TNL-vaccinated mice, indicating a targeted, increased immune response. Increased intertumoral immune cell infiltration has been associated with improved patient prognosis and suggests the potential for this treatment as a successful preventative vaccine.28 Further studies will need to be conducted to better understand the increase in proliferation in the tumor region for TNL-vaccinated mice despite the increase in immune cell infiltration. Although the mice in the TNL treatment group ultimately developed tumors, the overall results from this study demonstrate the potential for TNL on its own as a preventative vaccine.
Understanding the immunogenicity of the TNL vaccine provides insight into the effectiveness of the vaccine in eliciting a preventative response for triple-negative breast cancer in vivo. With the insight gained from this study, we can develop future iterations of the TNL vaccine with enhanced targeting of specific immune cell populations. For instance, the addition of an adjuvant to the TNL formulation as a co-delivery method may aid in augmenting the immune response for further success. Specific biomaterials delivery strategies may also be useful to study and implement in the future for enhanced immune cell targeting. Potential modifications to TNL could include encapsulation, liposomal delivery or PEGylation.
ACKNOWLEDGEMENTS
We acknowledge the Translational Pathology Shared Resource supported by NCI/NIH Cancer Center Support Grant P30CA068485 and the Shared Instrumentation Grant S10 OD023475 for the Leica Bond-RX.
FUNDING STATEMENT
This study was funded by NIH Grant No. CA256054 (M.R.K.) and supported by NSF Graduate Research Fellowship Program Award No. 1937963 (J.A.D).
Footnotes
Publisher's Disclaimer: .This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73(1):17–48. doi: 10.3322/caac.21763 [DOI] [PubMed] [Google Scholar]
- 2.Schairer C, Mink PJ, Carroll L, Devesa SS. Probabilities of Death From Breast Cancer and Other Causes Among Female Breast Cancer Patients. JNCI J Natl Cancer Inst 2004;96(17):1311–1321. doi: 10.1093/jnci/djh253 [DOI] [PubMed] [Google Scholar]
- 3.Monticciolo DL. Current Guidelines and Gaps in Breast Cancer Screening. J Am Coll Radiol 2020;17(10):1269–1275. doi: 10.1016/j.jacr.2020.05.002 [DOI] [PubMed] [Google Scholar]
- 4.Lebron-Zapata L, Jochelson MS. Overview of Breast Cancer Screening and Diagnosis. PET Clin 2018;13(3):301–323. doi: 10.1016/j.cpet.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 5.Tsai HJ. Clinical cancer chemoprevention: From the hepatitis B virus (HBV) vaccine to the human papillomavirus (HPV) vaccine. Taiwan J Obstet Gynecol 2015;54(2):112–115. doi: 10.1016/j.tjog.2013.11.009 [DOI] [PubMed] [Google Scholar]
- 6.Ferber MJ, Montoya DP, Yu C, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003;22(24):3813–3820. doi: 10.1038/sj.onc.1206528 [DOI] [PubMed] [Google Scholar]
- 7.Elrefaey S, Massaro MA, Chiocca S, Chiesa F, Ansarin M. HPV in oropharyngeal cancer: the basics to know in clinical practice. Acta Otorhinolaryngol Ital Organo Uff Della Soc Ital Otorinolaringol E Chir Cerv-facc 2014;34(5):299–309. [PMC free article] [PubMed] [Google Scholar]
- 8.Luo C, Yu S, Zhang J, et al. Hepatitis B or C viral infection and the risk of cervical cancer. Infect Agent Cancer 2022;17(1):54. doi: 10.1186/s13027-022-00466-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer T, Wallace L, Pollock KG, et al. Prevalence of cervical disease at age 20 after immunisation with bivalent HPV vaccine at age 12–13 in Scotland: retrospective population study. BMJ Published online April 3, 2019:l1161. doi: 10.1136/bmj.l1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crews DW, Dombroski JA, King MR. Prophylactic Cancer Vaccines Engineered to Elicit Specific Adaptive Immune Response. Front Oncol 2021;11:626463. doi: 10.3389/fonc.2021.626463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dombroski JA, Jyotsana N, Crews DW, Zhang Z, King MR. Fabrication and Characterization of Tumor Nano-Lysate as a Preventative Vaccine for Breast Cancer. Langmuir 2020;36(23):6531–6539. doi: 10.1021/acs.langmuir.0c00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atsaves V, Leventaki V, Rassidakis GZ, Claret FX. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019;11(7):E1037. doi: 10.3390/cancers11071037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene 2001;20(19):2390–2400. doi: 10.1038/sj.onc.1204383 [DOI] [PubMed] [Google Scholar]
- 14.Ade N, Antonios D, Kerdine-Romer S, Boisleve F, Rousset F, Pallardy M. NF-κB Plays a Major Role in the Maturation of Human Dendritic Cells Induced by NiSO4 but not by DNCB. Toxicol Sci 2007;99(2):488–501. doi: 10.1093/toxsci/kfm178 [DOI] [PubMed] [Google Scholar]
- 15.Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic Cell Survival and Maturation Are Regulated by Different Signaling Pathways. J Exp Med 1998;188(11):2175–2180. doi: 10.1084/jem.188.11.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavanagh LL, Saal RJ, Grimmett KL, Thomas R. Proliferation in Monocyte-Derived Dendritic Cell Cultures Is Caused by Progenitor Cells Capable of Myeloid Differentiation. Blood 1998;92(5):1598–1607. doi: 10.1182/blood.V92.5.1598 [DOI] [PubMed] [Google Scholar]
- 17.Herwig MC, Holz FG, Loeffler KU. Distribution and Presumed Proliferation of Macrophages in Inflammatory Diseases of the Ocular Adnexae. Curr Eye Res 2015;40(6):604–610. doi: 10.3109/02713683.2014.943909 [DOI] [PubMed] [Google Scholar]
- 18.Soares A, Govender L, Hughes J, et al. Novel application of Ki67 to quantify antigen-specific in vitro lymphoproliferation. J Immunol Methods 2010;362(1–2):43–50. doi: 10.1016/j.jim.2010.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benvenuti F, Hugues S, Walmsley M, et al. Requirement of Rac1 and Rac2 Expression by Mature Dendritic Cells for T Cell Priming. Science 2004;305(5687):1150–1153. doi: 10.1126/science.1099159 [DOI] [PubMed] [Google Scholar]
- 20.Blanco P, Palucka A, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev 2008;19(1):41–52. doi: 10.1016/j.cytogfr.2007.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaller TH, Batich KA, Suryadevara CM, Desai R, Sampson JH. Chemokines as adjuvants for immunotherapy: Implications for immune activation with CCL3. Expert Rev Clin Immunol 2017;13(11):1049–1060. doi: 10.1080/1744666X.2017.1384313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leone DA, Rees AJ, Kain R. Dendritic cells and routing cargo into exosomes. Immunol Cell Biol 2018;96(7):683–693. doi: 10.1111/imcb.12170 [DOI] [PubMed] [Google Scholar]
- 23.Hatsuzawa K, Hashimoto H, Hashimoto H, et al. Sec22b Is a Negative Regulator of Phagocytosis in Macrophages. Gruenberg JE, ed. Mol Biol Cell 2009;20(20):4435–4443. doi: 10.1091/mbc.e09-03-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leone DA, Rees AJ, Kain R. Dendritic cells and routing cargo into exosomes. Immunol Cell Biol 2018;96(7):683–693. doi: 10.1111/imcb.12170 [DOI] [PubMed] [Google Scholar]
- 25.Lauder SN, Smart K, Kersemans V, et al. Enhanced antitumor immunity through sequential targeting of PI3Kδ and LAG3. J Immunother Cancer 2020;8(2):e000693. doi: 10.1136/jitc-2020-000693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scrimieri F, Askew D, Corn DJ, et al. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. OncoImmunology 2013;2(11):e26889. doi: 10.4161/onci.26889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci 2015;112(9). doi: 10.1073/pnas.1500712112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melssen MM, Sheybani ND, Leick KM, Slingluff CL. Barriers to immune cell infiltration in tumors. J Immunother Cancer 2023;11(4):e006401. doi: 10.1136/jitc-2022-006401 [DOI] [PMC free article] [PubMed] [Google Scholar]