

Abstract
Although the overall survival rate of B cell acute lymphoblastic leukemia (B-ALL) in childhood is more than 80%, it is merely 30% in refractory/relapsed and adult patients with B-ALL. This demonstrates a need for improved therapy targeting this subgroup of B-ALL. Here, we show that the ten-eleven translocation 1 (TET1) protein, a dioxygenase involved in DNA demethylation, is overexpressed and plays a crucial oncogenic role independent of its catalytic activity in B-ALL. Consistent with its oncogenic role in B-ALL, overexpression of TET1 alone in normal precursor B cells is sufficient to transform the cells and cause B-ALL in mice within 3 to 4 months. We found that TET1 protein is stabilized and overexpressed because of its phosphorylation mediated by protein kinase C epsilon (PRKCE) and ATM serine/threonine kinase (ATM), which are also overexpressed in B-ALL. Mechanistically, TET1 recruits STAT5B to the promoters of CD72 and JCHAIN and promotes their transcription, which in turn promotes B-ALL development. Destabilization of TET1 protein by treatment with PKC or ATM inhibitors (staurosporine or AZD0156; both tested in clinical trials), or by pharmacological targeting of STAT5B, greatly decreases B-ALL cell viability and inhibits B-ALL progression in vitro and in vivo. The combination of AZD0156 with staurosporine or vincristine exhibits a synergistic effect on inhibition of refractory/relapsed B-ALL cell survival and leukemia progression in PDX models. Collectively, our study reveals an oncogenic role of the phosphorylated TET1 protein in B-ALL independent of its catalytic activity and highlights the therapeutic potential of targeting TET1 signaling for the treatment of refractory/relapsed B-ALL.
INTRODUCTION
B cell acute lymphoblastic leukemia (B-ALL) accounts for 80% of total ALL cases and often occurs in children and adolescents (1, 2). With contemporary therapies, the 5-year overall survival (OS) rate is more than 80% in patients with childhood ALL but is merely 30 to 40% for adult ALL (2, 3). However, pediatric patients with high-risk ALL often receive intensive chemotherapy (4), which may reduce quality of life because of acute and long-term toxicity and long-term morbidity (5). In addition, patients with refractory B-ALL, such as MLL-rearranged ALL and BCR-ABL1 ALL, and patients with relapsed B-ALL are often resistant to standard chemotherapy and immunotherapy (6–8). Thus, development of improved therapeutics with minimal side effects is urgently needed, especially for the patients with refractory/relapsed B-ALL.
Ten-eleven translocation 1 (TET1) was first identified as a fusion partner of the mixed-lineage leukemia (MLL) gene in patients with acute myeloid leukemia (AML), T cell lymphoma, and precursor B (pre-B) cell ALL (9–11). TET1 was then characterized as a methylcytosine dioxygenase that converts 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), leading to active or passive DNA demethylation (12–16). TET1 plays an important role in embryonic stem cell maintenance and development as a DNA demethylase (14–16). TET1 is down-regulated and functions as a tumor suppressor in various types of solid tumors (17–20). In contrast, we previously showed that TET1 plays an oncogenic role in MLL-rearranged AML (21). More recently, Tet1 deletion was reported to promote B cell lymphoma development in aged mice (over 2 years) (22), although Tet1 deficiency has little effect on the development of B lymphocyte progenitors and pre-B cell populations in young mice (5 to 7 weeks) (23). Thus, it was suggested that TET1 plays a tumor suppressor role in mature B cell malignancies. In clinical oncology, B-ALL and B cell lymphoma are considered to represent two different disease entities with distinct immunophenotypes and expression profiles, along with distinct standard chemotherapies for treatment. B-ALL derives from early (immature) forms of B cells (pro- or pre-B cells) in bone marrow (BM), whereas the blasts of B cell lymphoma are more mature (splenic differentiated B cells) (2, 24, 25). In addition, although various epigenetic modulators—such as writers, erasers, and readers—of DNA, RNA, and histone modifications have been shown to play important roles in many types of cancers (26–29), it is rarely reported that overexpression of a single epigenetic modulator gene could cause malignant transformation. Here, we show that the Tet1 protein, mainly in a short isoform, is highly expressed in patients with B-ALL, and its overexpression alone is sufficient for pre-B cell malignant transformation and the initiation and progression of pre-B cell malignancies in mice. TET1 is required for the development of B-ALL–related oncogene-induced murine B-ALL, such as BCR-ABL1– and NRAS mutant–induced B-ALL, and for the maintenance of human B-ALL in a catalytic activity–independent manner. Protein kinases protein kinase C epsilon (PRKCE) and ATM stabilize TET1 protein through phosphorylation. Inhibitors of ATM and PRKCE—such as AZD0156 and staurosporine, respectively, both tested in clinical trials—exhibited a promising therapeutic efficacy in treating B-ALL in vitro and in vivo. The combination of AZD0156 with staurosporine or vincristine (VCR), a first-line chemotherapy drug, displayed a synergistic effect in treating B-ALL patient–derived xenograft (PDX) models. Pharmacological inhibition of TET1 signaling by targeting its cofactor, signal transducer and activator of transcription 5B (STAT5B), also showed therapeutic potential in treating B-ALL PDX models. Overall, our preclinical animal model studies suggest that pharmacological targeting of TET1 stability or TET1 signaling is a promising strategy for the treatment of refractory/relapsed pre-B cell malignancies.
RESULTS
TET1 promotes B-ALL cell growth/proliferation and normal pre-B cell transformation independent of its catalytic activity
To determine whether TET1 plays a tumor suppressor role in B-ALL similar to that reported in B cell lymphoma (22), we used BCR-ABL1– and NRASG12D-induced B-ALL mouse models (30, 31) coupled with a Tet1 conditional knockout (cKO) (Tet1fl/fl) mouse model (fig. S1A) (23, 32). Tet1 could be effectively knocked out after 4-hydroxytamoxifen (4-OHT) treatment in Tet1fl/fl pre-B cells transduced with Cre-ERT2–internal ribosomal entry site (IRES)–green fluorescent protein (GFP) (Cre) (fig. S1, B and C). We found that induced KO of Tet1 inhibited competitive fitness of BCR-ABL1 and NRASG12D B-ALL cells in in vitro competition growth assays (Fig. 1, A and B, and fig. S1D). In addition, Tet1 KO inhibited the growth/proliferation (fig. S1E) and promoted apoptosis (fig. S1F) of BCR-ABL1 or NRASG12D B-ALL cells. Moreover, induced depletion of Tet1 significantly decreased the number of colonies formed by BCR-ABL1– or NRASG12D-transduced pre-B cells in serial replating (P < 0.001; Fig. 1, C to F). Furthermore, consistent with a previous report (22), mature B cells were accumulated in the BM of Tet1 KO mice [Tet1fl/fl Mx1-Cre mice upon synthetic double-stranded RNA (poly I:C) treatment] compared with that in wild-type mice (Tet1fl/fl mice upon poly I:C treatment) (fig. S1, G and H). In contrast to the inhibited competitive fitness upon inducible KO of Tet1 in BM-derived pre-B cells, Tet1 deficiency in splenic mature B cells induced outgrowth and formed colonies in semisolid gels (fig. S1, I and J). These data imply that Tet1 likely plays distinct roles in the malignant transformations of pre-B and mature B cells.
Fig. 1. TET1 promotes pre-B ALL development independent of its catalytic activity.
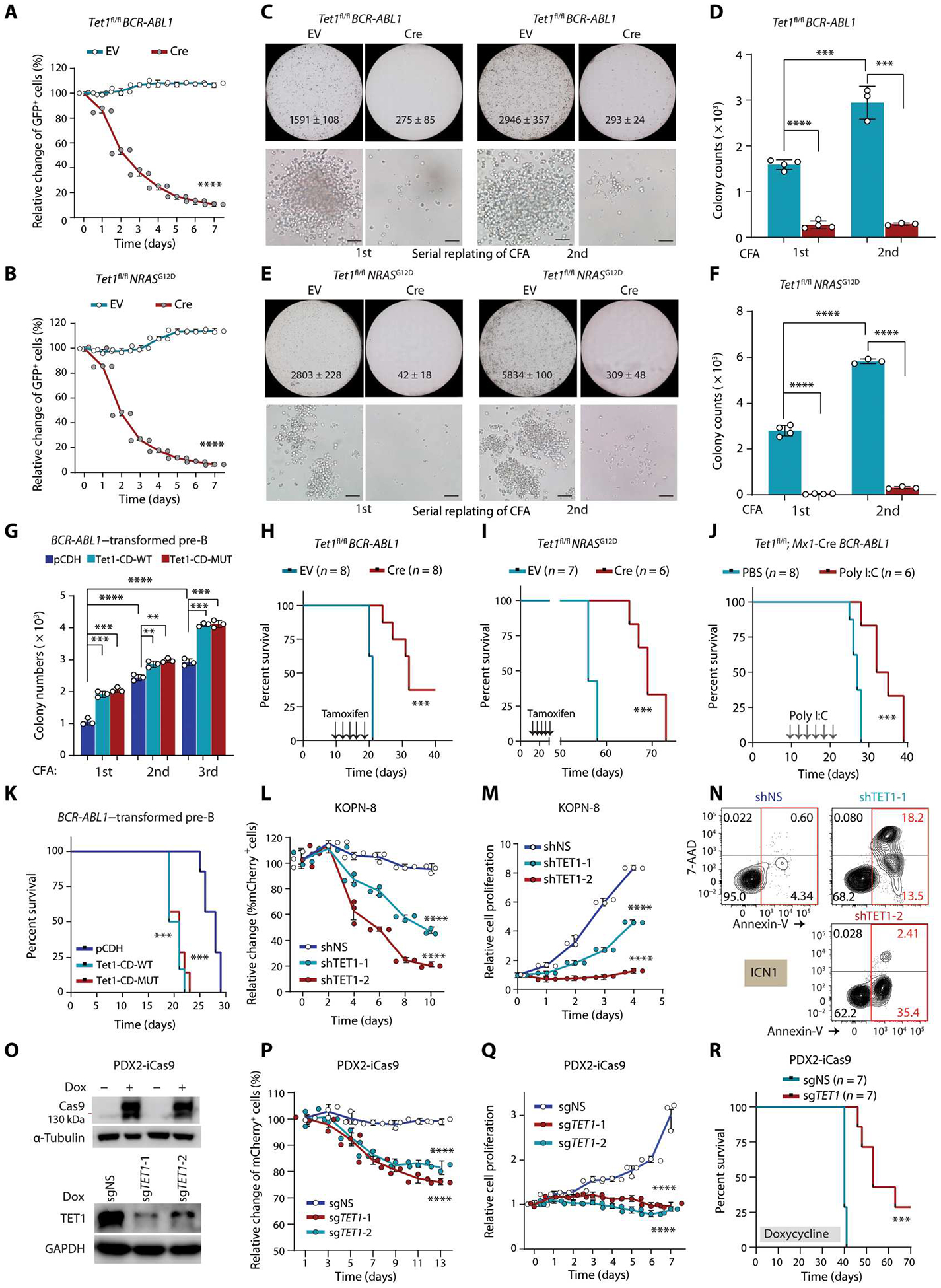
(A and B) Growth competition assays. BCR-ABL1–transduced (A) or NRASG12D-transduced (B) Tet1fl/fl pre-B cells were further transduced with Cre-ERT2-IRES-GFP vectors (Cre) or ERT2-IRES-GFP (EV). Percentages of GFP+ cells were measured by flow cytometry after 4-hydroxytamoxifen (4-OHT) treatment. (C to F) Colony-forming and replating assays (CFAs) and colony number quantification of BCR-ABL1–transduced (C and D) or NRASG12D-transduced (E and F) Tet1fl/fl pre-B cells (further transduced by EV or Cre) upon 4-OHT treatment. Colony count (mean ± SD) of the whole plate and representative images (scale bar, 50 μm) are shown. (G) Colony number quantification of serial CFAs of BCR-ABL1–transduced pre-B cells co-transduced with pCDH (empty vector), wild-type, or catalytically dead mutant murine Tet1-CD (Tet1-CD-WT or Tet1-CD-MUT). (H and I) Kaplan-Meier curves of immunocompromised NSG mice transplanted with BCR-ABL1–transformed (H) or NRASG12D-transformed (I) Tet1fl/fl pre-B cells transduced with either EV or Cre. Tamoxifen was intraperitoneally injected every other day for 5 days starting 10 days after transplantation. (J) Survival curves of immunocompetent C57BL/6 mice transplanted with BCR-ABL1–trans duced Tet1fl/fl Mx1-Cre pre-B cells coupled with poly I:C or PBS injection (intraperitoneally once every other day for five injections) 10 days after transplantation. (K) Survival curves of immunocompetent C57BL/6 mice transplanted with BCR-ABL1–transduced wild-type pre-B cells with or without overexpression of Tet1-CD-WT or Tet1-CD-MUT. (L) mCherry+ cell populations measured by flow cytometry at indicated time points after transduction with lentiviral shRNA vectors expressing mCherry (psi-LVRU6MH) in KOPN-8 cells (carrying MLL-ENL). shNS, non-silencing control. mCherry+ cell populations were measured every 2 days without any selection. (M and N) Cell growth/proliferation (M) and apoptosis (N) assays in B-ALL cells [KOPN-8 or ICN1 (PDX cells carrying BCR-ABL1)] after TET1 KD by shRNAs. (O) Western blotting showing the inducible effects of CRISPR-Cas9 protein in PDX2 (carrying BCR-ABL1)–inducible Cas9-ZsGreen (iCas9) cells after 3 days of induction with doxycycline (1 μg/m1; Dox, top). Validation of induced TET1 KO in PDX2-iCas9 cells after transfection of lentiviral sgRNAs targeting TET1 (bottom). (P) Growth competition assays of human PDX2 B-ALL cells with or without TET1 knockout (KO). ZsGreen+ PDX2-iCas9 cells were transduced with mCherry+ sgRNAs, and percentages of mCherry+ ZsGreen+ double-positive cells in ZsGreen+ cells were analyzed by flow cytometry. (Q) Cell growth/proliferation assays of control and TET1 KO PDX2 cells. (R) Kaplan-Meier curves of PDX2-iCas9 cell xenograft model. Except for survival analyses, other experimental data are representative of at least three independent experiments. Data are shown as means ± SD and assessed by two-tailed Student’s t test (D, F, and G) or two-way ANOVA (A, B, L, M, P, and Q). Log-rank tests were used for survival analyses (H to K and R). **P < 0.01, ***P < 0.001, and ****P < 0.0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
We found that overexpression of either murine wild-type Tet1-catalytic domain (Tet1-CD-WT), which could phenocopy Tet1 when overexpressed, or catalytically dead mutant Tet1-CD (Tet1-CD-MUT), carrying H1652Y and D1654A (12, 33–35), significantly increased the competitive fitness and induced outgrowth of B-ALL cells compared with the pCDH-GFP–overexpressing control cells (P < 0.0001; fig. S2, A and B). Consistently, overexpression of either Tet1-CD-WT or Tet1-CD-MUT increased colony numbers of BCR-ABL1–transduced pre-B cells in semisolid methylcellulose after serial replating (Fig. 1G and fig. S2, C and D). Together, our data suggest that TET1 plays an oncogenic role, instead of the expected tumor-suppressive role, in promoting B-ALL cell growth/proliferation and pre-B cell transformation independent of its catalytic activity.
TET1 promotes B-ALL development and maintenance in vivo independent of its catalytic activity
To investigate the role of TET1 in B-ALL development, we performed BM transplantation (BMT) studies using a Tet1 cKO mouse strain (23, 32) coupled with BCR-ABL1– and NRASG12D-induced murine B-ALL models (30, 31). Our results showed that induced ablation of Tet1 significantly inhibited the engraftment of BCR-ABL1– or NRASG12D-transformed pre-B cells and delayed the onset of B-ALL in vivo, resulting in prolonged OS in the immunocompromised NOD (nonobese diabetic) scid gamma (NSG) recipient mice (P < 0.001; Fig. 1, H and I, and fig. S2E). Similarly, using an immunocompetent C57BL/6 mouse model, we also showed that Tet1 KO significantly inhibited B-ALL development and substantially prolonged OS in the recipient mice; the opposite was true when either Tet1-CD-WT or Tet1-CD-MUT was overexpressed (P < 0.001; Fig. 1, J and K, and fig. S2, F to I).
Constitutive knockdown (KD) of TET1 by short hairpin RNAs (shRNAs) in vitro (fig. S3A) significantly reduced competitive fitness (P < 0.0001; Fig. 1L), inhibited cell proliferation (P < 0.0001; Fig. 1M and fig. S3B), and induced apoptosis (P < 0.001; Fig. 1N and fig. S3C) in multiple human B-ALL cell lines and PDX cells. Similarly, induced TET1 KO (Fig. 1O) significantly attenuated competitive fitness (P < 0.001; Fig. 1P) and proliferation (P < 0.001; Fig. 1Q) of PDX2-iCas9 cells, which are human B-ALL PDX cells (PDX2) transduced with inducible CRISPR-Cas9. Given that TET1 KO in Dependency Map (DepMap) screening data, an online portal to access key cancer dependencies analytical and visualization tool, showed little killing effect on B-ALL cell lines, we compared the single-guide RNAs (sgRNAs) used in DepMap with ours and found that our sgRNAs showed much better TET1 KO efficiency and inhibition of B-ALL cell survival and growth than did the four sgRNAs used in the DepMap (fig. S3, D to F). Moreover, TET1 KO significantly inhibited human B-ALL maintenance and progression in vivo (P < 0.01; Fig. 1R and fig. S3, G and H). There was little change in the mRNA and protein abundance of TET2 upon TET1 KD, indicating that there was no compensational up-regulation of TET2 for TET1 KD in B-ALL cells (fig. S3, I and J). Together, our results demonstrate that TET1 plays a critical tumor-promoting role independent of its catalytic activity in B-ALL initiation, development, and maintenance and is required for B-ALL cell survival and growth/proliferation.
Overexpression of TET1 alone is sufficient to transform normal pre-B cells and initiate B-ALL
It appears to be a paradox that TET1 plays a critical oncogenic role in B-ALL, whereas it is down-regulated at the RNA level in the disease (23). Because protein abundance is the ultimate determinant for the function of a gene, we sought to analyze the protein abundance of TET1 in B-ALL cells. We observed that a short isoform of TET1 protein (TET1-S, ~162 kDa), which has been reported in human solid tumors and mouse adult cells (36, 37), was highly expressed in B-ALL primary patient specimens and cell lines (Fig. 2, A to C). The full-length form of TET1 protein, TET1-FL, which is the main form of TET1 expressed in human embryonic stem cells (hESCs), was expressed at a much lower abundance than TET1-S in human B-ALL samples (Fig. 2, B and C) but at a similar abundance to B-ALL, AML, mantle cell lymphoma (MCL), or healthy controls. Further results from immunoprecipitation (IP) coupled with mass spectrometry (MS) of the TET1-S band from human B-ALL cells verified its identity (Fig. 2D). Whereas TET1-S was highly overexpressed in various subtypes of B-ALL cells, its expression in AML, mature B cell neoplasms, and healthy cells was relatively low (Fig. 2, C, E, and F). These data suggest that TET1-S is specifically highly expressed in human B-ALL.
Fig. 2. The TET1-S is highly expressed in B-ALL, and its overexpression is sufficient to transform pre-B cells.
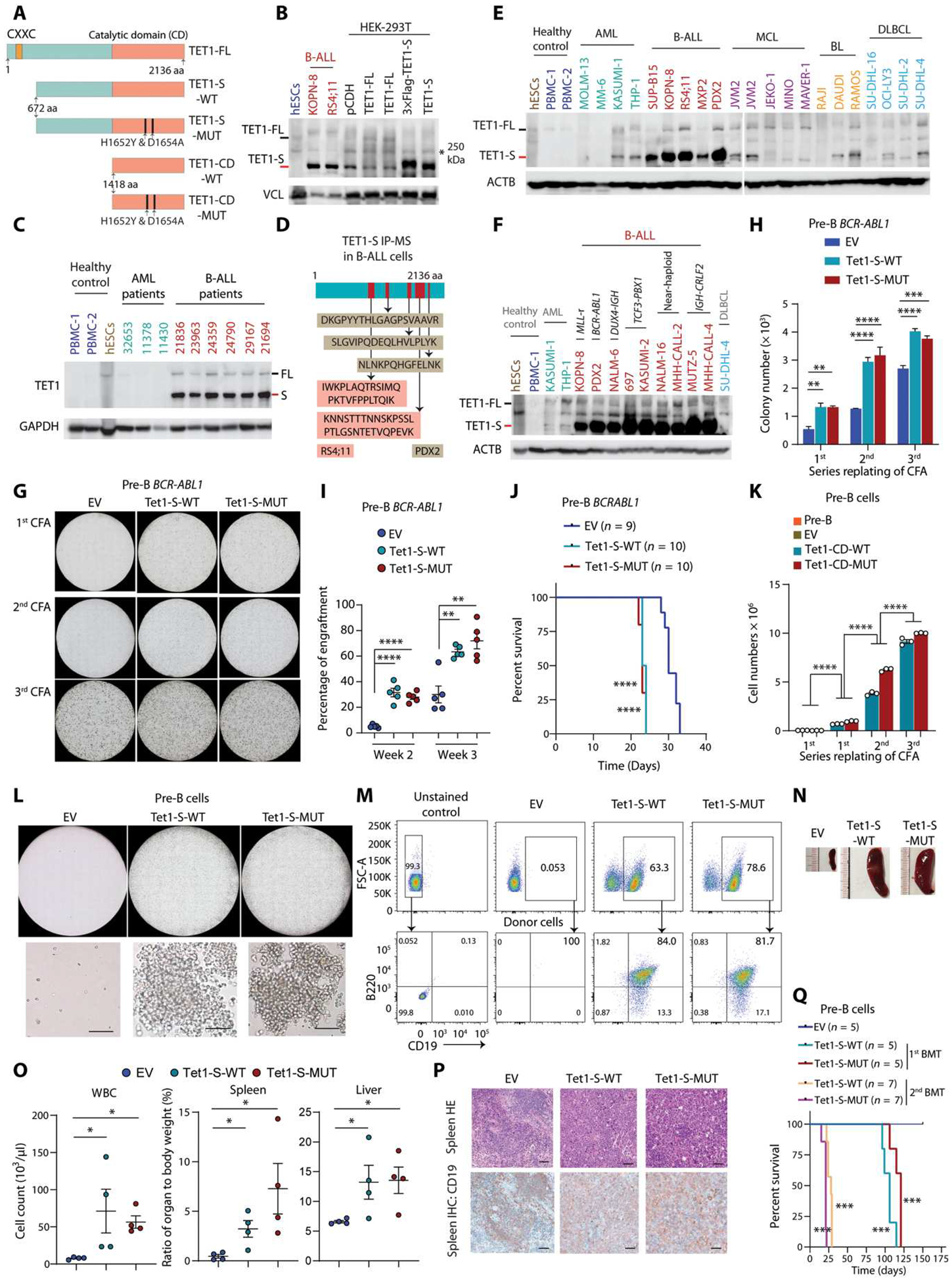
(A) Schematic of full-length (TET1-FL), short isoform (TET1-S), and catalytic domain of TET1 (TET1-CD). (B) Western blots of the TET1-FL (black dash on the left) and TET1-S (red dash on the left) of TET1 in human embryonic stem cells (hESCs), B-ALL cells, and HEK-293T cells with overexpression of empty vector (pCDH), TET1-FL, or TET1-S. Nonspecific bands at 250 kDa are labeled with black stars. Vinculin (VCL) was used as a reference control. (C) Western blotting showing expression of TET1-FL and TET1-S in primary B-ALL patient samples, along with hESCs, peripheral blood mononuclear cells (PBMCs), and AML patient samples as controls. (D) IP with TET1 antibody in B-ALL cell lysate (RS4;11 and PDX2) followed by LC-MS/MS detection and analysis. In-gel protein sample used for LC-MS/MS was collected from the specific band (TET1-S) observed in SDS–polyacrylamide gel electrophoresis gel as shown in (B). (E and F) Western blots showing expression of full-length (black dash) and short isoform (red dash) of TET1 protein in healthy control, different subtypes of B-ALL, and other malignant blood cells [AML, mantle cell lymphoma (MCL), Burkitt lymphoma (BL), and diffuse large B cell lymphoma (DLBCL)]. (G) Representative photomicrographs of serial CFAs. BCR-ABL1–transformed pre-B cells were further transduced with empty vector (EV), Tet-S-WT, or Tet1-S-MUT. (H) Clonal cell number statistics of serial passages of CFAs of BCR-ABL1–transformed pre-B cells further transduced with pCDH, Tet1-S-WT, or Tet1-S-MUT. (I) Engraftment detection by flow cytometry for peripheral blood (PB) samples from C57BL/6 mice transplanted with BCR-ABL1–transformed pre-B cells with or without overexpression of Tet1-S-WT or Tet1-S-MUT at indicated time points. (J) Survival curves of immunocompetent C57BL/6 mice transplanted with BCR-ABL1–transduced wild-type pre-B cells with or without overexpression of Tet1-S-WT or Tet1-S-MUT. (K) Clonal cell number quantification of serial passages of CFAs of pre-B cells transduced with pCDH, Tet1-CD-WT, or Tet1-CD-MUT. (L) Representative photomicrographs of CFAs. Healthy pre-B cells were transduced with EV, Tet-S-WT, or Tet1-S-MUT. Scale bar, 50 μm. (M) Validation of B-ALL by flow cytometry with antibodies of CD45.2 (donor) and murine CD19 and B220. (N) Representative images of spleens from NSG mice transplanted with pre-B cells overexpressing EV, Tet1-S-WT, or Tet1-S-MUT. (O) Cell counts for white blood cells (WBC) and organ weight as a percentage of whole-body weight from leukemic mice at the end time point or from the control group of mice. (P) Representative photomicrographs of splenic tissues stained by hematoxylin and eosin (HE) and immunohistochemistry (IHC) staining for CD19 in mice transplanted by pre-B cells with overexpression of EV, Tet1-S-WT, or Tet1-S-MUT. Scale bar, 50 μm. (Q) Kaplan-Meier curves of NSG mice transplanted with pre-B cells transduced with EV, Tet1-S-WT, or Tet1-S-MUT for two generations of BMT. All experimental data are representative of at least three independent experiments. Data are shown as mean ± SD and assessed by two-tailed Student’s t test (H, I, K, and O). Log-rank test for (J) and (Q). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Our in vitro and in vivo functional studies have shown that TET1 is required for B-ALL oncogenic protein (such as BCR-ABL1 and NRAS-mutant)–mediated normal B cell transformation and leukemogenesis. Given that TET1-S is the major isoform protein in B-ALL cells, we further investigated the function of TET1-S in pre-B ALL development. Our results showed that overexpression of either TET1-S-WT or TET1-S-MUT, similar to that of TET1-CD-WT or TET1-CD-MUT, could significantly promote BCR-ABL1–mediated leukemic cell transformation and B-ALL development (P < 0.01; Fig. 2, G to J). To determine whether ectopic expression of TET1 alone, mimicking endogenous TET1 overexpression, can transform normal pre-B cells, we transduced murine normal pre-B cells with lentiviral vectors carrying wild-type or catalytically mutant Tet1-S or Tet1-CD. As confirmed by DNA dot blot and ultrahigh-performance liquid chromatography coupled with triple quadrupole MS (UHPLC-QQQ-MS) assays, whereas the wild-type Tet1-S or Tet1-CD could increase global 5hmC amounts in pre-B cells, the mutants lost the DNA demethylation activity (fig. S4, A and B). Our functional studies showed that overexpression of Tet1-S or Tet1-CD, either WT or MUT, alone could transform normal pre-B cells into malignant B cells that showed an increased capability of forming colonies after serial replating, whereas vehicle control–transduced normal pre-B cells could not form colonies (Fig. 2, K and L, and fig. S4C). Moreover, NSG mice transplanted with Tet1-S-WT–, Tet1-S-MUT–, Tet1-CD-WT–, or Tet1-CD-MUT–transduced pre-B cells, but not those transplanted with vehicle control–transduced pre-B cells, developed full B-ALL within 3 to 4 months in the first BMT and developed more aggressive B-ALL in the second BMT (Fig. 2, M to Q, and fig. S4, D to J). These results suggest that overexpression of TET1, either TET1-S or TET1-CD, alone is sufficient to transform normal pre-B cells and initiate B-ALL and that this function is independent of its catalytic activity, demonstrating the oncogenic role of TET1 in B-ALL pathogenesis.
TET1 protein is stabilized by ATM and PRKCE-mediated phosphorylation in B-ALL cells
Although TET1 was reported previously to be down-regulated at the RNA level in human B-ALL (23), we showed above that its protein is overexpressed in human B-ALL. There are three possible levels of regulation of TET1 expression, including transcription, translational efficiency, and posttranslational modifications (PTMs) (Fig. 3A). No genomic alterations were detected in TET1 in patients with B-ALL based on published whole-genome sequencing data (fig. S5A). At the transcriptional level, we observed a specific transcriptional start site (TSS) of TET1-S in B-ALL, which is the same as that reported previously (36, 37). In contrast, the usage of the TSS of TET1-FL was observed in hESCs or AML cells but less in B-ALL cells (fig. S5B). Assay for transposase-accessible chromatin using sequencing (ATAC-seq) further showed that the state of chromatin of TET1-S TSS is open, whereas TET1-FL TSS is more condensed in B-ALL cells (fig. S5C). Chromatin IP assay followed by sequencing (ChIP-seq) of transcription factors showed that several B-ALL–associated transcription factors such as recombination activating gene 1 (RAG1), AF4/FMR2 family member 1 (AFF1), and runt-related transcription factor 1 (RUNX1) bound to TET1-S TSS in B-ALL cells; in contrast, in healthy B cells, suppressive transcription factors such as B cell lymphoma 6 (BCL6) potentially bound to TET1 TSS and repressed its transcription (fig. S5C). Our subsequent ChIP–quantitative polymerase chain reaction (qPCR) for RAG1, AFF1, or RUNX1 demonstrated that these transcription factors more preferentially bound to the TSS of TET1-S compared with that of TET1-FL (fig. S5D). Furthermore, mRNA concentrations of TET1-S significantly decreased upon the KD of these individual transcription factors by shRNAs in B-ALL cells (P < 0.05; fig. S5, E and F), whereas TET1-FL concentrations were too low to detect, which is consistent with the deep sequencing data obtained from GSE168625, GSE168752, GSE115656, GSE54641, GSM3563631, GSM1534507, and GSM1534512 (fig. S5B). Consistent with the previous report (23), our reverse transcription qPCR (RT-qPCR) results showed that both TET1-S and especially TET1-FL mRNA expression in B-ALL patient or cell line samples were lower compared with those in other types of blood cancer cells or healthy control cells (Fig. 3, B and C), although B-ALL–specific transcription factors promote TET1-S transcription. Furthermore, TET1 mRNA was associated with a lower stability in B-ALL cells, such as PDX2, than in healthy control peripheral blood mononuclear cells (PBMCs) and AML cells, such as MONOMAC-6 (MM-6) (fig. S5G). TET1-S mRNA expression was much (500- to 100,000-fold) higher than TET1-FL mRNA expression in B-ALL cells (fig. S5H). Overall, our data suggest that TET1-S is the major transcript of TET1 in B-ALL and that its transcription is specifically regulated by RAG1, AFF1, and RUNX1 in B-ALL cells, whereas TET1-FL expression is very low or undetectable.
Fig. 3. TET1 protein is phosphorylated and stabilized by ATM and PRKCE.
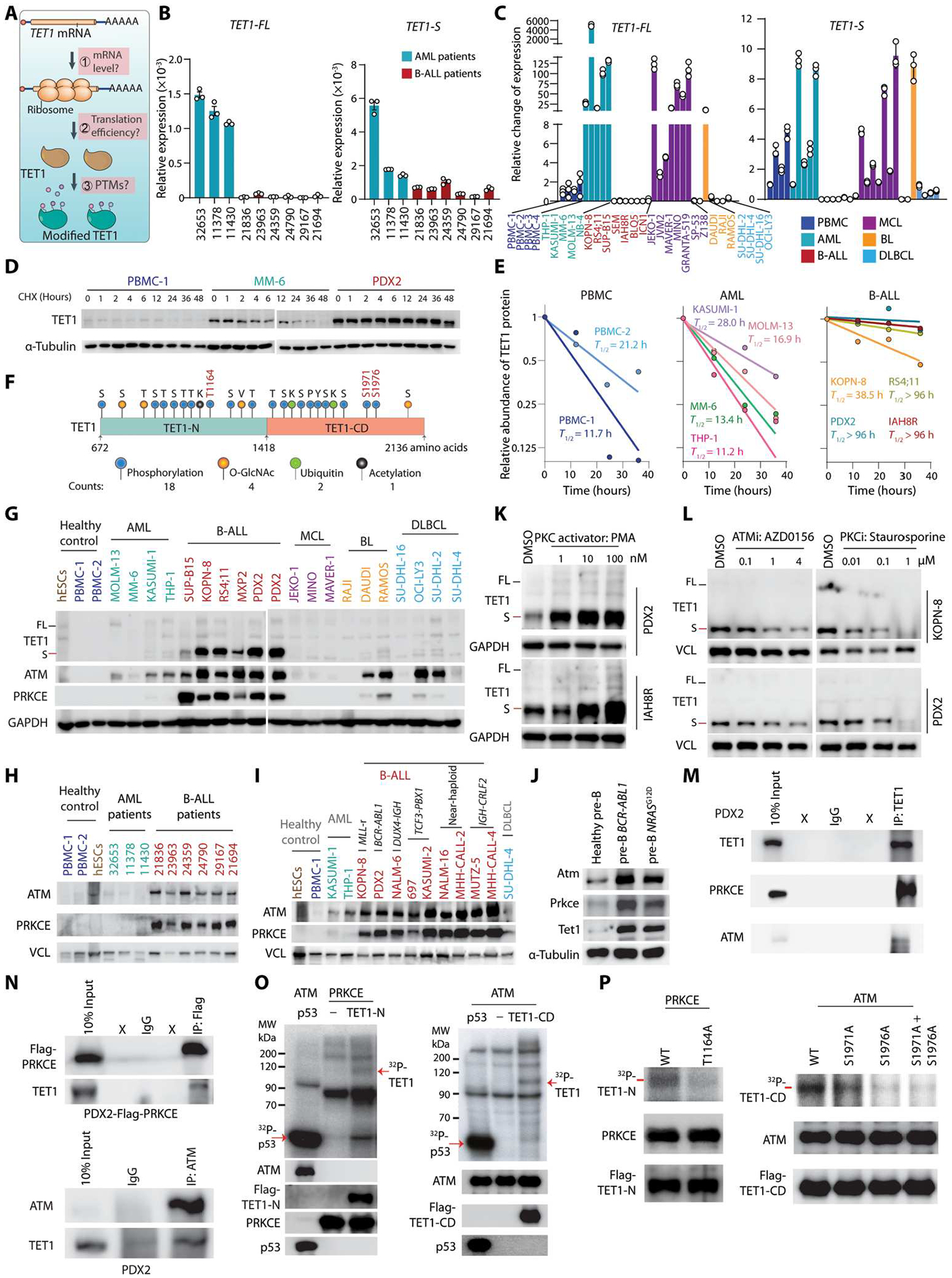
(A) Schematic diagram depicting the potential mechanisms regulating TET1 protein abundance. (B and C) RT-qPCR showing the relative mRNA expression of TET1-FL and TET1-S in B-ALL and AML patient samples (B) or PBMCs, AML, B-ALL, MCL, BL, and DLBCL cell lines (C). RT-qPCR primers were designed to amplify the specific region of TET1 isoforms. Values of 2−ΔCt normalized to ACTB are shown (B). mRNA expression in cell lines was normalized to PBMC-1. (D) TET1 protein stability after CHX treatment at indicated time points in healthy control cells (PBMCs), AML cells (MM-6), and B-ALL cells (PDX2). (E) Quantification of TET1-S protein half-life (T1/2) normalized to reference protein in healthy control cells, AML cells, and B-ALL cells. (F) Predicted and reported PTM sites on TET1 protein (672 to 2136 amino acids). (G to I) Western blots comparing abundance of indicated proteins between healthy controls and B-ALL cell lines or other types of malignant blood cells (G), patient samples (H), and diverse B-ALL subtypes (I). TET1-FL and TET1-S bands were annotated with black and red dashes, respectively (G). (J) Western blots showing expression of Atm, Prkce, and Tet1 in BCR-ABL1– or NRASG12D-transformed pre-B cells compared with healthy pre-B cells. (K and L) TET1 protein abundance [TET1-FL (black dash) and TET1-S (red dash)] after treatment with PKC activator (K) and inhibition of PKC or ATM (L) at gradient doses in B-ALL cells for 24 hours. (M and N) Reciprocal co-immunoprecipitation (co-IP) assays with antibody against ATM and flag-tagged PRKCE. IgG was loaded as negative control; X indicates lanes without loading samples. (O) Kinase assays by 32P-autoradiograph. Active PRKCE or ATM protein was incubated with purified wild-type TET1-N or TET1-CD, respectively. Recombinant p53 was loaded as a positive control. Western blots in parallel as the loading controls. (P) Determination of TET1 phosphorylation at T1164, S1971, and S1976 by 32P-autoradiograph. Western blots in parallel as the loading controls. All experimental data are representative of at least three independent experiments. Data are shown as mean ± SD (B and C). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. For (F), the detailed information of predicted posttranslational modifications of TET1 is listed in table S1.
Given that TET1 mRNA expression (mainly TET1-S) was lower and its protein abundance (mainly TET1-S) was higher in B-ALL samples than in other malignant or normal blood samples, we next performed polysome profiling to check whether TET1 mRNA has a higher translational efficiency. We found that the translational efficiency and distribution of TET1 mRNA in three major translational fractions (nonribosome, 40S–80S, and polysome) are similar between AML and B-ALL cells (fig. S6, A and B). This suggested that its translational efficiency is similar between B-ALL and AML. We found that TET1 protein, mainly TET1-S, was much more stable in B-ALL cells than in PBMCs and AML cells (Fig. 3, D and E, and fig. S6C). Together, our results suggest that TET1 protein stability, rather than mRNA expression or stability or translational efficiency, contributes to the high abundance of TET1 protein in human B-ALL.
To investigate whether TET1 protein stabilization is attributed to PTMs, we listed all the reported and predicted PTMs on human TET1 protein (Fig. 3F). We found that TET1 protein abundance was not affected by selective inhibitors of O-GlcNAcylation, acetylation, or ubiquitin modifications in human B-ALL cells (fig. S6D). Such data leave phosphorylation as the most possible PTM involved in TET1 protein stability regulation, which was also the most abundant modification on TET1 protein (Fig. 3F). Thus, we measured the expression of the top kinases that have the potential to phosphorylate TET1 at multiple sites (table S1). We found that both ATM and the PKC family protein, PRKCE, were overexpressed in human primary B-ALL specimens and B-ALL cell lines relative to healthy controls or other types of malignant blood cells, which was similar to the TET1 expression pattern (Fig. 3, G to I, and fig. S6E). Consistently, overexpression of B-ALL oncogenes, such as BCR-ABL1 and NRSG12D, in murine normal pre-B cells also caused overexpression of Atm, Prkce, and Tet1 (Fig. 3J). In contrast, PKD/PKC mu protein abundance was higher in both B-ALL cells and hESCs, which was different from the TET1 expression pattern (fig. S6E). Moreover, there was no significant change in TET1 protein abundance in B-ALL cells after treatment with CDK1 and CDK5 inhibitor (NG-52) or PKD/PKC mu inhibitor (CID755763) (fig. S6F). Similar to their protein abundances, both ATM and PRKCE mRNA expression was significantly higher in patients with B-ALL than in healthy BM and patients with other blood cancers (P < 0.0001; fig. S6G). This suggested that B-ALL oncoproteins likely directly or indirectly promote their expression at the transcriptional level. Moreover, genomic alterations of ATM and PRKCE are very rare in patients with B-ALL (fig. S6H), indicating that their enzymatic activity should be intact in B-ALL cells. When we treated B-ALL cells with phorbol 12-myristate 13-acetate (PMA), a selective activator of the PKC pathway, we found that TET1 protein abundance increased in a dose-dependent manner (Fig. 3K). Conversely, TET1 protein abundance decreased after treatment with a selective inhibitor of PKC proteins (PKCi, staurosporine) or ATM (ATMi, AZD0156) in B-ALL cells (Fig. 3L). We also found that TET1 protein abundance decreased (fig. S6I, top) without change in mRNA expression (fig. S6I, bottom) at the indicated time point after treatment with ATMi or PKCi in B-ALL PDX cells. Similarly, TET1 protein abundance, but not mRNA expression, decreased upon ATM or PRKCE KD by shRNAs (fig. S6, J and K). Collectively, our data suggest that ATM and PRKCE are critical regulators of TET1 protein stabilization.
To investigate whether ATM and PRKCE directly phosphorylate TET1 and thereby stabilize the protein, we conducted co-IP assays and showed that TET1 protein directly interacts with ATM and PRKCE (Fig. 3, M and N), suggesting that TET1 is a potential phosphorylation substrate of ATM and PRKCE. We then purified PRKCE, TET1 N-terminal (TET1-N), and TET1-CD from human embryonic kidney (HEK)–293T cells (fig. S7A) and demonstrated that TET1-N and TET1-CD were direct substrates of PRKCE and ATM, respectively, by 32P-autoradiograph assays (Fig. 3O). Further kinase assays showed that T1164 and S1971/S1976 on TET1 were directly phosphorylated by PRKCE and ATM, respectively; the mutations in those sites abrogated the corresponding phosphorylation (Fig. 3P). Cotreatment with cycloheximide (CHX) + PMA in B-ALL cells increased the half-lives of TET1 protein compared with treatment with CHX alone (fig. S7B). Conversely, the half-lives of TET1 protein decreased in B-ALL cells upon cotreatment with CHX + ATMi or CHX + PKCi compared with only CHX treatment (fig. S7C). Moreover, the stability of phosphorylation site–mutated TET1 proteins [flag-tagged TET1-N (T1164A) and TET1-CD (S1971A + S1976A)] was decreased compared with the wild-type TET1 proteins in human B-ALL cells (fig. S7, D and E). Furthermore, pre-B cells transformed by TET1-S that carried the mutations T1164A or S1971A + S1976A formed fewer clones than those transduced with TET1-S-WT or TET1-S-MUT (catalytically inactive) in semisolid gels (fig. S7, F and G). Together, our results suggest that B-ALL oncoproteins, such as BCR-ABL1, NRAS mutant, and MLL fusions, directly or indirectly promote the expression of PRKCE and ATM. Increased PRKCE and ATM further enhance the phosphorylation of TET1 protein at T1164, S1971, and S1976 and thereby increase its stability and abundance in B-ALL cells (fig. S7H).
TET1 regulates the expression of target genes involved in the B cell signaling pathway
To explore the molecular mechanism of TET1 in B-ALL independent of its catalytic activity, we profiled the genomic binding sites of TET1 by performing ChIP-seq in PDX2 cells with overexpression of flag-tagged TET1-CD-WT or TET1-CD-MUT (fig. S8, A to C). We saw that both wild-type and mutant TET1 preferentially bound to the genomic regions around the TSS (fig. S8D), which was consistent with previous studies in other tissues (14, 34). The binding sites of TET1-CD-WT and TET1-CD-MUT were highly enriched in gene promoter regions (fig. S8E). This implies that TET1 may transcriptionally regulate the expression of targets by binding to the promoter regions, especially around the TSS of the target genes.
To identify the targets of TET1, we conducted RNA sequencing (RNA-seq) in conditional TET1 KO PDX2-iCas9 cells and found that the target genes, which were bound by both wild-type and mutant TET1 and were also dysregulated upon TET1 KO, were highly enriched in the lymphoblastic leukemia pathway, B cell receptor (BCR) signaling pathway, and cell cycle (fig. S8, F and G). In addition, Gene Ontology (GO) analysis showed that down-regulated TET1-bound genes were highly enriched in mononuclear lymphocyte leukocyte cascade (B cell proliferation, differentiation, activation, lymphocyte homeostasis, etc.) and cell cycle (Fig. 4A and fig. S8H). Gene set enrichment analysis (GSEA) further revealed that up-regulated TET1-bound genes were highly enriched with G2-M checkpoints and apoptosis regulators, whereas down-regulated ones were highly enriched with B cell–related pathways (Fig. 4, B and C, and fig. S8I). Differential gene expression analysis revealed that most dysregulated TET1-bound genes were down-regulated in TET1 KO cells, including cluster of differentiation 72 (CD72) and joining chain of multimeric IgA and IgM (JCHAIN) (Fig. 4, C and D, and fig. S9A), two essential genes related to B cell–specific pathways and B-ALL, as reported previously (38–40).
Fig. 4. TET1 drives B-ALL cell growth/survival by promoting transcription of B cell–specific oncogenic targets, such as CD72 and JCHAIN.
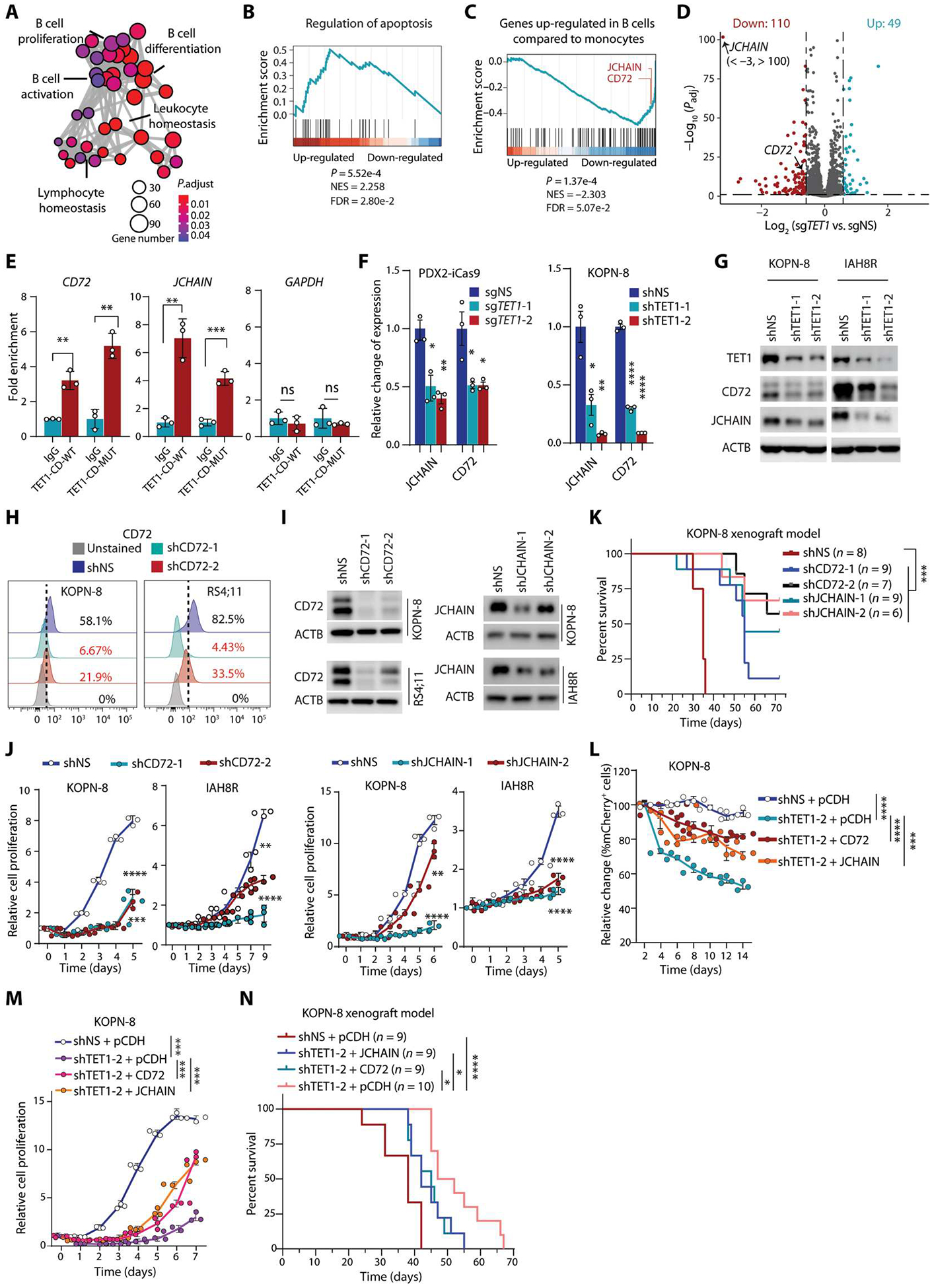
(A) Gene Ontology (GO) analysis of down-regulated genes upon TET1 KO in PDX2 cells, which are enriched in B cell pathways. Size of circle indicates gene number; color shows the adjusted P value. (B and C) Gene Set Enrichment Analysis (GSEA) plots of apoptosis (B) and genes up-regulated in B cells compared with monocytes (GSE29618) (C) using a preranked TET1-CD-WT/MUT–bound gene list weighted by expression fold change in TET1-depleted PDX2 compared with control PDX2 cells. (D) Volcano plot showing expression changes in TET1-CD WT/MUT–bound genes upon TET1 KO in PDX2 cells. Dots in red (down-regulation) and blue (up-regulation) represent significantly differentially expressed genes. (E) ChIP-qPCR showing the binding of both TET1-CD-WT and TET1-CD-MUT on CD72 and JCHAIN genomic loci in PDX2 cells. ChIP-qPCR of GAPDH as a negative control for TET1 ChIP. (F and G) Changes in CD72 and JCHAIN mRNA expression (F) or protein (G) abundance upon TET1 KO or knockdown (KD) in B-ALL cells (PDX2, KOPN-8, and IAH8R). (H) CD72 cell surface expression tested by flow cytometry after CD72 KD in B-ALL cells. (I) Protein abundance changes in CD72 or JCHAIN after shRNA KD. (J) Cell growth/proliferation assays after KD of CD72 or JCHAIN in B-ALL cells. (K) Kaplan-Meier analysis (log-rank test) of NSG mice xenotransplanted with human KOPN-8 B-ALL cells with or without KD of CD72 or JCHAIN. (L) Cell population analysis of mCherry+ versus GFP+ in TET1 KD (shRNA vectors carrying mCherry) KOPN-8 cells after overexpression of CD72-GFP, JCHAIN-GFP, or pCDH-GFP in vitro. (M) Cell growth/proliferation assays of control and TET1 KD KOPN-8 cells with or without overexpression of CD72 or JCHAIN. (N) Kaplan-Meier curves (log-rank test) of KOPN-8–xenotransplanted NSG mice showing the effects of CD72 or JCHAIN overexpression on rescuing TET1 KD–induced inhibition on B-ALL progression. All experimental data are representative of at least three independent experiments. Data are shown as mean ± SD and assessed by two-tailed Student’s t test (E and F) or two-way ANOVA (J, L, and M). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns, not significant.
The specific binding of TET1-CD-WT and TET1-CD-MUT to the promoters of CD72 and JCHAIN was further validated by ChIP-qPCR (Fig. 4E). The mRNA expression and protein abundance of CD72 and JCHAIN were down-regulated upon constitutive KD or cKO of TET1 in B-ALL cells (Fig. 4, F and G, and fig. S9, B and C). Moreover, CD72 and JCHAIN were up-regulated in BM and spleen samples collected from Tet1-CD-WT– and Tet1-CD-MUT–driven B-ALL mice (fig. S9, D and E). In addition, we found that overexpression of TET1 protein with mutant phosphorylation sites in pre-B cells did not increase CD72 or JCHAIN protein abundance, whereas overexpression of wild-type or catalytically inactive mutant TET1 increased the abundance of the two target proteins (fig. S9F). Consistent with the recent reports of CD72 and JCHAIN (38, 39), we also found that their expression in B-ALL cells was higher than that in healthy control and AML cells (fig. S9, G to J). We further showed that growth or proliferation of B-ALL cells was inhibited upon KD of CD72 or JCHAIN by shRNAs in vitro (Fig. 4, H to J). Moreover, our in vivo xenotransplantation studies showed that KD of CD72 or JCHAIN significantly delayed the onset of B-ALL in NSG recipient mice (P < 0.001; Fig. 4K), mimicking the phenotype of TET1 depletion. Furthermore, TET1 KD–induced inhibitory effects on human B-ALL cell proliferation and competitive cell growth could be partially rescued by overexpression of CD72 or JCHAIN (Fig. 4, L and M, and fig. S9, K to N). In vivo mouse model studies also showed that the TET1 KD–induced inhibitory effect on B-ALL development could be partially rescued by overexpression of CD72 or JCHAIN (Fig. 4N). Together, our data indicated that CD72 and JCHAIN are two functionally important direct targets of TET1 in B-ALL.
TET1 recruits STAT5B to regulate expression of downstream targets
Because the short isoform lacking the DNA binding CXXC domain is the main form of TET1 in B-ALL cells, we presumed that one or more transcription factors may function as cofactors that facilitate the binding of TET1 to target loci. To identify such cofactors, we performed co-IP for TET1-CD followed by LC-MS/MS with PDX2 cell lysate. After filtering out nonspecific interactions, we found that STAT5B, a critical transcription factor in B-ALL as reported previously (41–43), potentially interacted with TET1 in B-ALL cells (Fig. 5A and table S2). Subsequent co-IP assays further validated that both wild-type and mutant TET1 proteins directly interacted with STAT5B in B-ALL cells (Fig. 5, B and C). Genome-wide correlation analysis of our ChIP-seq data of STAT5B and TET1-CD revealed that TET1 had a positive correlation of binding signals with STAT5B in PDX2 cells (Fig. 5D). STAT5B binding peaks matched with TET1 binding site peaks, and STAT5B tag density significantly decreased upon TET1 KO in PDX2 cells (P < 0.0001; Fig. 5E). Moreover, STAT5B binding signals on the promoters of most of the genes involved in B cell pathway (see Fig. 4C and fig. S8I, right) were decreased upon TET1 KO (Fig. 5F). ChIP-qPCR results further confirmed that STAT5B binds to the promoter regions of CD72 and JCHAIN and that its binding was decreased upon TET1 KO in PDX2 cells (Fig. 5, G and H). In addition, CD72 and JCHAIN were significantly down-regulated upon KD of STAT5B in B-ALL cells (P < 0.0001; Fig. 5I). Thus, our data suggest that TET1 recruits STAT5B to bind to the promoter regions of CD72 and JCHAIN and activates their transcriptions, which in turn promotes B-ALL development (Fig. 5J).
Fig. 5. TET1 regulates transcription of downstream target genes by recruiting STAT5B in B-ALL cells.
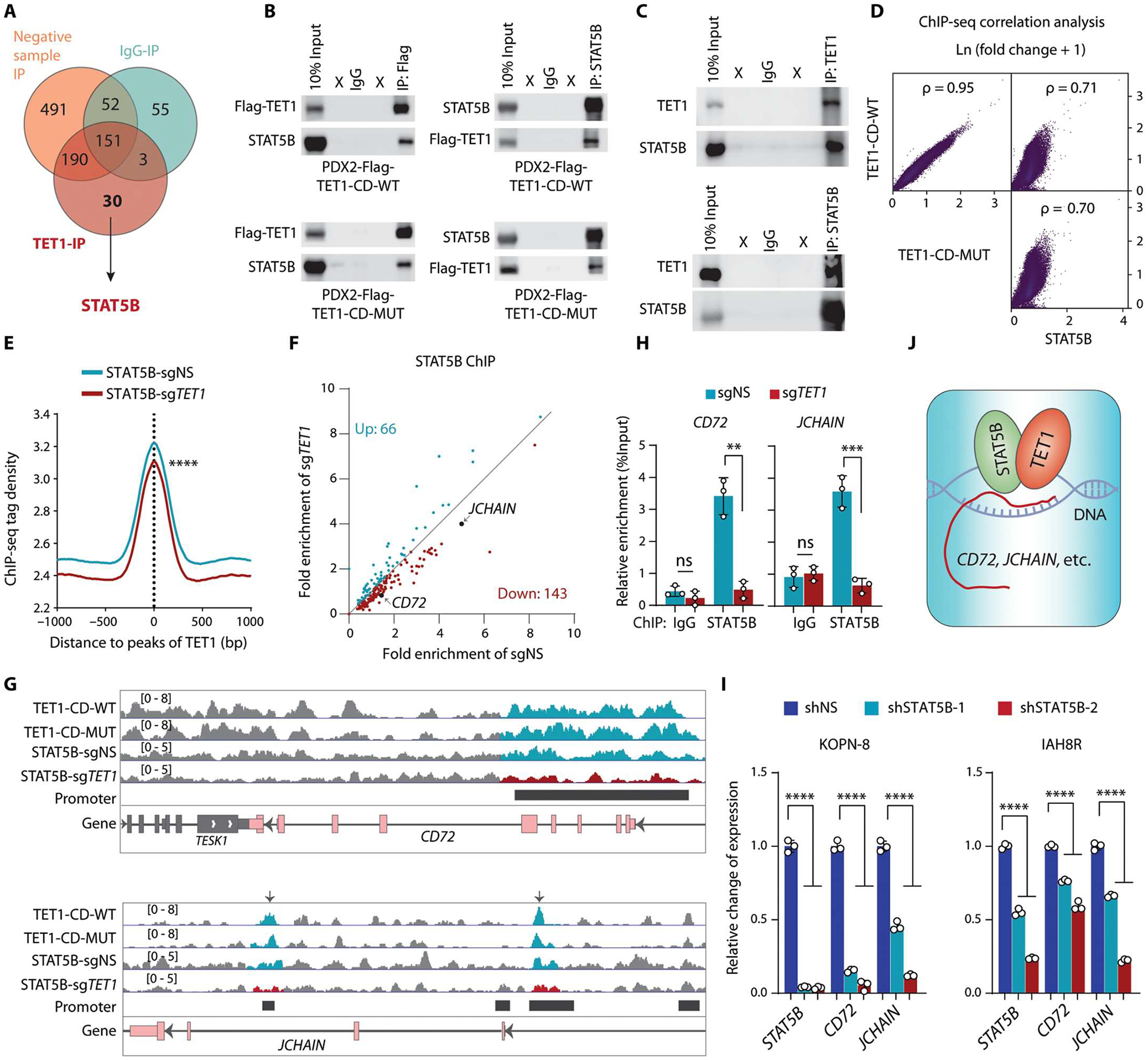
(A) Venn diagram showing candidate proteins that likely interact with TET1, as detected by IP-MS, in PDX2 cells. Negative control IP was conducted with anti-flag antibody in cells without flag-tagged TET1-CD or with control IgG antibody in cells with flag-tagged TET1-CD. (B) Reciprocal co-IP assays examining interaction between TET1-CD (WT and MUT) and STAT5B. (C) Western blotting showing co-IP of TET1 and STAT5B. Reciprocal co-IP assays were conducted using antibodies that recognize the endogenous proteins in PDX2 cells. (D) Genome-wide Spearman’s correlation analysis of the fold change in ChIP-seq signals (IP versus input) between TET1-CD-WT, TET1-CD-MUT, and STAT5B. The P values represent Spearman’s rank correlation coefficients. (E) Distribution and density of the STAT5B ChIP-seq tags around the TET1-bound peaks in PDX2 cells. (F) Scatterplot showing the fold enrichment of STAT5B on the promoter regions of the responsive target genes involved in B cell pathways (see Fig. 4C and extended data fig. 6G) in control and TET1 KO PDX2 cells. The fold enrichment was calculated as IP/input of ChIP-seq counts. (G) Distribution of the fold change in ChIP-seq signals (IP versus input) of TET1 and STAT5B around the promoter regions of CD72 and JCHAIN in PDX2 cells. (H) ChIP-qPCR of STAT5B binding at the promoter region of CD72 and JCHAIN in PDX2 (sgNS and sgTET1) cells. (I) RT-qPCR showing the relative expression of STAT5B, CD72, and JCHAIN upon KD of STAT5B by lentiviral shRNAs in B-ALL cells. (J) Working model of TET1 and STAT5B in the regulation of target gene transcription. All experimental data are representative of at least three independent experiments. Data are shown as mean ± SD and assessed by two-tailed Student’s t test (H and I) or two-sample Kolmogorov-Smirnov test (E). **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns, not significant. For (A), the complete list of proteins identified is shown in table S2.
Targeting STAT5, ATM, or PRKCE inhibits B-ALL
We reported previously that STAT3 and STAT5 bind to the promoter region of TET1 and promote its transcription. Targeting the STAT/TET1 axis using UC-514321 exhibits a potent therapeutic efficacy in treating AML (44). In B-ALL, our results suggest that TET1 interacts with STAT5B and promotes the transcription of their co-targets, such as CD72 and JCHAIN. As expected, STAT5 inhibitors UC-514321 and AC-4-130, a specific SH2 domain inhibitor (45), inhibited B-ALL cell growth with a low median inhibitory concentration (IC50) associated with suppressed expression of CD72 and JCHAIN in vitro (fig. S10, A to C). UC-514321 or AC-4-130 treatment also inhibited human B-ALL progression in mice injected with IAH8R cells (Fig. 6A). Nevertheless, the mRNA and protein abundances of TET1 did not consistently and significantly change in B-ALL cells upon STAT5B KD (fig. S10D), suggesting that STAT5B may not regulate TET1 transcription in B-ALL cells.
Fig. 6. Therapeutic potential of targeting TET1 signaling to treat B-ALL.
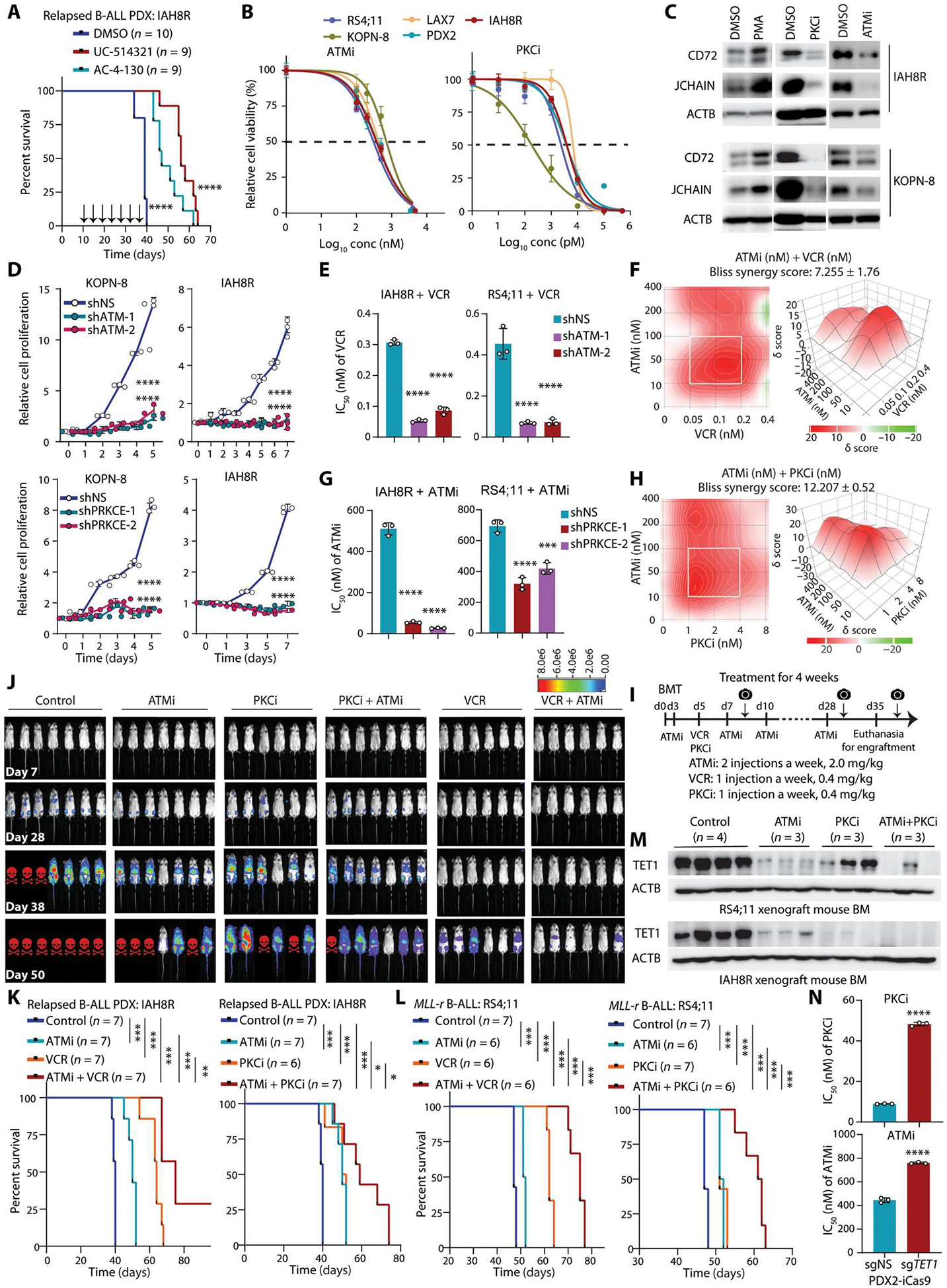
(A) Kaplan-Meier analysis of relapsed B-ALL PDX xenotransplanted NSG mice treated with dimethyl sulfoxide (DMSO), AC-4-130, or UC-514321. Injections were three times a week starting 10 days after transplantation. (B) IC50 of ATM inhibitor (ATMi, AZD0156) or PRKCE inhibitor (PKCi, staurosporine) on inhibition of B-ALL cell survival/growth 72 hours after treatment. (C) Western blots showing the relative changes of abundance of CD72 and JCHAIN after treatment with PMA, ATMi, or PKCi in B-ALL cells. (D) Cell growth/proliferation assays after KD of ATM or PRKCE in B-ALL cells. (E) Changes in IC50 of VCR B-ALL cell line and PDX cells upon ATM KD. (F) Synergistic effects of ATMi + VCR on inhibition of the survival/growth of relapsed B-ALL PDX cells (IAH8R), as determined by the Bliss independence model. Drug combinations with the strongest synergistic effects are outlined with white squares. δ scores represent the percentage of response beyond expectation due to drug interactions. (G) Changes in IC50 of ATMi in B-ALL cell line and PDX cells upon PRKCE KD. (H) Synergistic effects of ATMi + PKCi on inhibition of the survival/growth of relapsed B-ALL PDX cells (IAH8R), as determined by the Bliss independence model. (I) Drug treatment strategy for relapsed PDX and refractory B-ALL models. Camera symbols represent bioluminescence imaging. (J) In vivo bioluminescence imaging of xenotransplanted NSG mice with B-ALL relapse PDX cells (IAH8R) treated with ATMi, PKCi, or VCR alone or in combination. Red skull symbols indicate that those mice were euthanized because of developing B-ALL. (K and L) Kaplan-Meier analysis of relapsed B-ALL PDX (K) or refractory MLL-r B-ALL (L) xenotransplanted NSG mouse models treated intraperitoneally with different drug combinations. The same control and ATMi groups were used in the left and right panels. (M) Western blots showing relative abundance of TET1 in BM samples from the mice carrying IAH8R or RS4;11-induced ALL with or without treatment with the indicated inhibitors. (N) Changes in IC50 values from ATMi or PKCi treatment of PDX2-iCas9 cells upon KO of TET1. Log-rank test was used for the survival curve analyses (A, K, and L). Data are shown as mean ± SD and were assessed by two-tailed Student’s t test (E, G, and N) or two-way ANOVA (D). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Images are representative of three independent experiments (B to H and N).
We next assessed the therapeutic efficacy of inhibitors of ATM (ATMi, AZD0156) and PRKCE (PKCi, staurosporine) in treating B-ALL. As expected, ATMi and PKCi both inhibited survival and growth of human B-ALL cells with a low IC50, associated with an inhibition of expression of CD72 and JCHAIN (Fig. 6, B and C). By contrast, expression of CD72 and JCHAIN increased after treatment with PMA in B-ALL cells (Fig. 6C). Similar to ATMi and PKCi treatment, KD of ATM or PRKCE also significantly inhibited the survival and growth of B-ALL cells (P < 0.0001; Fig. 6D), which phenocopied the effect of TET1 KD. Furthermore, we found that ATM KD or ATMi treatment could sensitize B-ALL cells to VCR, a first-line chemotherapeutic agent for B-ALL treatment (Fig. 6E and fig. S10E); ATMi and VCR showed a synergistic effect on inhibition of the survival and growth of human B-ALL cells (Fig. 6F and fig. S10F). Similarly, PRKCE KD or PKCi treatment decreased the IC50 of ATMi inhibition of B-ALL cell viability and growth. In parallel, ATM KD or ATMi also decreased the IC50 of PKCi (Fig. 6G and fig. S10, G and H). Quantitatively, ATMi and PKCi exhibited a synergistic effect on inhibition of the survival and growth of human B-ALL cells (Fig. 6H and fig. S10I). Moreover, we used relapsed B-ALL PDX that were transfected with lentiviral vectors carrying firefly luciferase and performed BMT followed by drug treatments in transplanted NSG mice. Our results demonstrated that treatment with VCR, ATMi, and PKCi each alone significantly delayed the engraftment, onset, and progression of human relapsed or refractory B-ALL in the recipient mice (P < 0.001; Fig. 6, I to L). Treatment with ATMi + VCR or ATMi + PKCi exhibited a more robust therapeutic efficacy than each alone in treating relapsed or refractory human B-ALL in vivo (Fig. 6, I to L, and fig. S10, J and K). We found that TET1 protein was degraded in the drug-treated mice BMs (Fig. 6M). Induced TET1 KO reduced the sensitivity of B-ALL cells to PKCi and ATMi (Fig. 6N), suggesting that the treatment effects and mechanisms of PKCi and ATMi in B-ALL are through TET1. Overall, our results suggest that targeting TET1 signaling by inhibition of its binding cofactor STAT5 or its stabilization regulators ATM and PKC, alone or in combination with standard chemotherapy such as VCR, is a promising therapeutic strategy for refractory/relapsed B-ALL treatment.
DISCUSSION
It has been reported that Tet1 deficiency only has a minor effect on early B cell development in young mice (23) but causes mature B cell lymphoma in aged mice (22). Mature B cell–specific depletion of both Tet2 and Tet3 in mice disturbs homeostasis, leading to the spontaneous development of germinal center–derived B cell lymphomas (46). Our study suggests that TET1 plays distinct roles in B cell malignancy development at different B cell stages—earlystage B cells versus later-stage B cells. In addition, we showed that ectopic expression of TET1 alone in normal pre-B cells could transform the cells and lead to the development of full B-ALL in vivo within 3 months, highlighting the potent oncogenic function of TET1 in B-ALL pathogenesis. Our study provides an example showing that overexpression of a single epigenetic modulator gene, TET1, could be sufficient to transform normal cells and cause tumorigenesis.
Our data and a previous study (23) showed that TET1 is down-regulated at the RNA level in B-ALL samples relative to healthy controls, which seems to be paradoxical to its oncogenic role in B-ALL. Here, we show that TET1 protein, mainly the short isoform, abundance is high in B-ALL, which is attributed to its increased stability. Because the expression of TET1-FL at both RNA and protein levels is much lower than that of TET1-S in B-ALL, it is TET1-S, not TET1-FL, that is the major TET1 product and plays a critical role in B-ALL. Overall, our study provides a compelling example showing that the protein abundance of a given gene could be markedly different from its RNA expression. Thus, PTM plays an important role in modulating gene expression, suggesting that judging a gene’s pathological role in cancer simply on the basis of its RNA expression could be misleading.
A recent study reported that TET1 promotes T cell–ALL development by maintaining global epigenetic modifications (47). In contrast, our results demonstrate that TET1 promotes B-ALL development independent of its catalytic activity. Our data suggest a working model in which TET1 recruits critical transcription factors to regulate transcription of target genes and play an essential oncogenic role independent of its demethylase activity in cancer.
Given the critical oncogenic role of TET1 protein in B-ALL, it warrants development of highly effective and selective TET1 inhibitors, which are not yet available, for the treatment of B-ALL. Because the oncogenic role of TET1 is independent of its catalytic activity, compounds that target its catalytic activity would not be effective. Thus, development of proteolysis targeting chimera (PROTAC)–based inhibitors (48) to degrade TET1 protein could be an attractive strategy. As an alternative strategy, here, we show that targeting TET1 cofactor STAT5B or its stabilization regulators, PRKCE and ATM, could effectively suppress TET1 signaling and inhibit B-ALL progression. Moreover, suppression of TET1’s phosphorylation to destabilize TET1 protein by both ATM inhibitor (AZD0156) and PRKCE inhibitor (staurosporine) shows a synergistic effect on killing B-ALL cells. In addition, AZD0156 treatment can also synergize with standard chemotherapy agents, such as VCR, in treating B-ALL. Thus, such combinatorial therapy may represent more effective therapeutics for the treatment of refractory and relapsed B-ALL in the clinic, which will lower cumulative drug doses and thus minimize side effects and may substantially prolong survival in the treated patients.
There are some limitations in our study. Considering that TET1 is overexpressed in various subtypes of B-ALL, whether TET1 interacts with other critical transcription factors in non–STAT5B-driven B-ALL subtypes has yet to be further investigated. It is worthwhile to investigate whether the working model of TET1 also plays an important role in other cancer types. Although ATM was considered as a tumor suppressor through the phosphorylation of p53 to maintain genome stability from oncogenic DNA breaks (49), it has been reported to be frequently up-regulated in certain types of cancers, and its selective inhibitor, AZD0156, has been used to treat MLL-r AML and reverse neuroblastoma chemoresistance in mouse models (50–52). However, the underlying molecular mechanisms remain elusive. Our study uncovers a previously unknown mechanism, destabilizing TET1, by which ATM plays a critical oncogenic role in B-ALL, and it would be necessary to determine whether the ATM-TET1 axis also plays an essential role in other ATM-high cancers.
In conclusion, we show here that B-ALL oncogenic proteins promote expression of ATM and PRKCE to stabilize TET1 protein and increase its abundance in pro- or pre-B cells. Then, TET1 recruits the transcription cofactor STAT5B to promote the transcription of B cell–specific oncogenic genes such as CD72 and JCHAIN, contributing to B-ALL initiation and progression. Moreover, targeting TET1 signaling by suppressing its stabilization regulators ATM and PRKCE or its binding cofactor STAT5B exhibits therapeutic efficacy in treating refractory/relapsed B-ALL. Collectively, our findings demonstrate that TET1 plays a critical oncogenic role in early B cell transformation and leukemogenesis independent of its demethylase activity and suggest that targeting TET1 signaling would be an effective therapeutic strategy for refractory/relapsed B-ALL treatment.
MATERIALS AND METHODS
Study design
Our primary objective was to determine whether TET1 plays an essential role in B-ALL initiation and progression. The functions of TET1 were investigated using cKO mouse strains (23, 32) in two different B-ALL mouse models, which were established by BCR-ABL1– and NRASG12D-transformed normal pre-B cells (30, 31). TET1 and its catalytically inactive mutant (12, 33–35) were used to investigate whether TET1 functions in an enzymatic manner in B-ALL. Clinical patient samples and multiple cell lines with different subtypes of B-ALL, lymphoma, and AML were used to assess the relative abundance of TET1 protein. On the basis of the observation of a special high abundance of TET1 protein in B-ALL, in vitro and in vivo functional study assays were further performed using normal pre-B cells with overexpression of TET1. Relapsed B-ALL PDX cells were used in the functional and mechanistic studies. Given the high TET1 protein abundance, but low TET1 mRNA expression in B-ALL, its potential transcriptional, posttranscriptional, translational, and posttranslational regulations were investigated to uncover the mechanism of a highly expressed TET1-S in B-ALL. Then, we performed next-generation sequencing such as RNA-seq, ChIP-seq, and IP-LC-MS/MS to investigate the cofactors and downstream targets of TET1 in B-ALL. We have also tested whether pharmacological destabilization of TET1 using ATM inhibition or PRKCE inhibition, or inhibition of TET1 cofactor STAT5B, holds therapeutic potential in treating B-ALL through preclinical animal model studies, including refractory/relapsed B-ALL PDX models.
For all the in vitro assays, at least three independent biological replicates were conducted. All in vivo transplantation assays were repeated two or three times using different mouse B-ALL models or xenograft models to ensure reproducibility of the observations. All the experiments were randomized and blinded where possible. No statistical method was used to predetermine sample size. The animal number in each group, sample size, and significance analysis value are indicated in the figures or figure legends, and no mice, outliers, or other data points were excluded. The investigators were not blinded to allocation during in vitro experiments and outcome assessment. Mice were randomly allocated to groups by investigators who were not involved in the analysis. Experiments were repeated to ensure reproducibility of the observations.
Murine primary BM and leukemia cells
BM cells were harvested from the mice between 6 and 8 weeks old with any signs of inflammation. BM cells were collected by flushing cavities of femurs and tibiae with regular RPMI 1640 medium with 2% fetal bovine serum (FBS). After filtration through 40-μm cell strainer and depletion of erythrocytes with ammonium chloride solution (STEMCELL Technologies), BM cells were washed with phosphate-buffered saline (PBS) and subsequently cultured in Iscove’s modified Dulbecco’s medium (IMDM; Gibco) with Gluta-MAX supplemented with 20% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), and β-mercaptoethanol (50 μM; Gibco). For interleukin-17 (IL-7)–dependent pre-B cell culture, BM cells were cultured in IMDM supplemented with recombinant mouse IL-7 (10 ng/ml; PeproTech). ALL cells transformed by BCR-ABL1 were cultured in the IL-7–withdrawn IMDM, and ALL cells transduced with NRASG12D-Neo were selected with Geneticin Selective Antibiotic (G418 Sulfate, 1 mg/ml; Thermo Fisher Scientific) and maintained with IL-7–supplemented IMDM. Transduced cells with estrogen-receptor fusion proteins were induced by 4-OHT (1 μg/ml; MilliporeSigma, H7904) or tamoxifen (MilliporeSigma, T5648). After filtration with 40-μm cell strainer, mature splenic B cells were isolated and obtained with CD45R (B220) MicroBeads (Miltenyi Biotec) and then further cultured in IMDM with 20% FBS, penicillin (100 IU/ml), streptomycin (100 μg/ml), and β-mercaptoethanol (50 μM), supplemented with mouse IL-4 (10 ng/ml; PeproTech), human BAFF (100 ng/ml; PeproTech), and mouse CD40L (1 μg/ml; PeproTech).
In vivo BMT
Normal C57BL/6 mice (CD45.2) pre-B cells were cultured and transduced with wild-type or catalytically dead mutant murine Tet1-S (Tet1-S-WT and Tet1-S-MUT) or Tet1-CD (Tet1-CD-WT and Tet1-CD-MUT). After the puromycin selection, 2 million viable cells were immediately intravenously injected into sublethally irradiated 6- to 8-week-old NSG mice. One million viable BM cells from the recipients were collected and used for second BMT. BCRABL1- or NRASG12D-transformed Tet1fl/fl pre-B ALL cells were transduced with MSCV-Cre-ERT2-IRES-GFP (Cre) or MSCV-ERT2-IRES-GFP-EV (empty vector control). GFP+ cells were further sorted by FACSAria II (BD Biosciences); 1 million cells were intravenously injected into sublethally irradiated 6- to 8-week-old NSG mice. After the transplantation, tamoxifen dissolved in corn oil was intraperitoneally injected into the pre-B ALL cell–transplanted recipient mice (75 mg/kg of body weight) every other day five times. For the transplantation in immunocompetent C57BL/6 mice, Tet1fl/fl Mx1-Cre pre-B cells (CD45.2) were transduced with BCR-ABL1. Wild-type pre-B BCR-ABL1 ALL cells were transduced with Tet1-S or Tet1-CD vectors (WT and MUT) or empty vector (pCDH). One million BCR-ABL1 ALL cells, together with 1 million healthy BM cells as helper cells to protect the recipient mice from the side effect of lethal irradiation, were intravenously transplanted into lethally irradiated 6- to 8-week-old C57BL/6 recipient mice (CD45.1). For the cKO model, the dissolvent PBS or poly I:C (R&D Systems Inc.) at a concentration of 10 g/kg of body weight was intraperitoneally injected into the recipient mice (once every other day for five injections) 10 days after transplantation. For xenograft models, KOPN-8 cells were either transfected by lentivirus carrying shRNAs or cotransfected by lentiviral shRNAs and vectors carrying CD72 or JCHAIN. After puromycin selection for 24 hours without change in cell viability, 1 million viable cells were intravenously injected into sublethally irradiated NSG mice. One million PDX2-iCas9-sgRNA+ cells sorted by FACSAria II were tail vein–injected into sublethally irradiated NSG mice. The inducible KO of Tet1 was achieved by feeding a special diet with doxycycline dissolved in water (100 μg/ml). All the mice samples evaluated for engraftment analysis were collected at the same time point after transplantation. Kaplan-Meier survival analyses were performed with GraphPad Prism 9 (GraphPad Software). Log-rank tests were used for statistical analysis. Mice were randomly allocated to groups, and the investigators who conducted the mouse experiments (injection, drug treatment, and monitoring) were blinded to the randomization process.
Bioluminescence imaging
Relapsed PDX B-ALL cells (IAH8R) were transfected with lentiviral vectors carrying firefly luciferase (pLenti-Luciferase-Neo). After the selection of G418 Sulfate (Thermo Fisher Scientific), 1 million viable IAH8R cells were intravenously injected into sublethally irradiated NSG mice. The B-ALL PDX cell progression in vivo was monitored at the times indicated using an in vivo imaging system of bioluminescence and fluorescence (Spectral Instruments Imaging, Tucson, AZ). d-Luciferin (2.5 mg per mouse; GoldBio) in PBS was injected 15 min before imaging. General anesthesia was induced with 5% isoflurane and continued during the procedure with 2% isoflurane introduced through a nose cone.
Liquid chromatography–tandem mass spectrometry
LC-MS/MS was performed in the UCLA Molecular Instrumentation Center following the protocols as below. Protein (1 μg) was injected into UltiMate 3000 nanoLC, which was equipped with a 75 μm × 2 cm trap column packed with C18 3-μm bulk resins (Acclaim PepMap 100, Thermo Fisher Scientific) and a 75 μm × 15 cm analytical column with C18 2-μm resins (Acclaim PepMap RSLC, Thermo Fisher Scientific). The nanoLC gradient was 3 to 35% solvent B (A = H2O with 0.1% formic acid; B = acetonitrile with 0.1% formic acid) over 40 min and from 35 to 85% solvent B in 5 min at a flow rate of 300 nl/min. The nanoLC was coupled with a Q Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The electrospray ionization voltage was set at 1.9 kV, and the capillary temperature was set at 275°C. Full spectra [mass/charge ratio (m/z) 350 to 2000] were acquired in profile mode with resolution 70,000 at m/z 200 with an automated gain control (AGC) target of 3 × 106. The 15 most abundant ions were subjected to fragmentation by higher-energy collisional dissociation (HCD) with a normalized collisional energy of 25. MS/MS spectra were acquired in centroid mode with resolution 17,500 at m/z 200. The AGC target for fragment ions was set at 2 × 104 with maximum injection time of 50 ms. Charge states 1, 7, 8, and unassigned were excluded from MS/MS experiments. Dynamic exclusion was set at 45 s. The raw data of LC-MS/MS were analyzed by searching UniProt TET1_human using Proteome Discoverer version 1.4. The parameters were set up as follows: precursor mass tolerance ±10 parts per million, fragment mass tolerance ±0.02 Th for HCD, up to two miscleavages by semi-trypsin, methionine oxidation as variable modification, and cysteine carbamidomethylation as static modification. False discovery rate was at 1%, and a minimum of one peptide was required for protein identification.
Protein purification
Two 10-cm dishes of HEK-293T cells were transduced by lentiviral vectors carrying 3 × flag-tagged PRKCE or TET1 proteins (TET1-CD, TET1-CD-S1971A, TET1-CD-S1976A, TET1-CD-S1971A/S1976A, TET1-N, and TET1-N-T1164A) and then expanded in six 15-cm tissue-treated cell culture dishes. Cells grown at a confluency of 80 to 90% were lysed with Pierce IP lysis buffer (87787, Thermo Fisher Scientific) supplemented with 1× protease inhibitor cocktail (PI78438, Thermo Fisher Scientific) and 1× phosphatase inhibitor cocktail (PI78426, Thermo Fisher Scientific) for 1 hour on ice in low–protein binding tubes (Eppendorf). Cell lysate was sonicated by Q800R3 (Qsonica) with the settings of 15-s on 30-s off, 10 min on time, 80% amplitude, and then centrifuged with a maximum speed at 4°C for 15 min. Pierce DYKDDDDK Magnetic Agarose (A36797, Thermo Fisher Scientific) was used for IP according to the manufacturer’s instructions. Briefly, 50 μl of well-mixed agarose were placed into a magnetic stand to remove the supernatant. The agarose was washed with IP lysis buffer two times and incubated with the supernatant of cell lysate after spinning down at 4°C overnight. The samples were washed by magnetic stand with washing buffer I [50 mM Tris-HCl (pH 7.4), 250 mM NaCl, and 1 mM EDTA] two times, PBS another two times, and purified water for the last wash. After removing the supernatant, we used 100 μl of Pierce 3 × DYKDDDDK Peptide (1.5 mg/ml; A36805, Thermo Fisher Scientific) to gently elute the purified protein by incubating for 10 min at room temperature in a ThermoMixer device at 1400 rpm. We repeated the elution step and proceeded to Coomassie blue staining to quantity the purified protein using a gradient amount of standard bovine serum albumin (5000007, Bio-Rad).
Autoradiography assay
We followed the procedure reported previously (53, 54) with the following modifications. Purified active human ATM was purchased from MilliporeSigma (14–933), and recombinant human p53 was purchased from R&D Systems (SP-452-020). One microgram of purified TET1 protein (TET1-CD, TET1-CD-S1971A, TET1-CD-S1976A, TET1-CD-S1971A/S1976A, TET1-N, TET1-N-T1164A, and p53) was incubated with kinase protein (ATM or PRKCE) and 10 μCi of [γ-32P]adenosine triphosphate (ATP; NEG002A, PerkinElmer) in 50 μl of kinase buffer [25 mM Tris-HCl (pH 7.5), 5 mM β-glycerophosphate, 2 mM dithiothreitol, 0.1 mM Na3VO4, 10 mM MgCl2, and 0.2 mM ATP]. The reactions were incubated at 30°C for 1 hour and stopped by addition of SDS buffer. Proteins were separated on NuPAGE 4 to 12% gradient gels (Thermo Fisher Scientific) and then dried for 2.5 hours on Whatman 3.0 paper (Cytiva). The signal of phosphorylated proteins was visualized and detected by autoradiography through x-ray film exposure.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 9.3 (GraphPad Software) using unpaired two-tailed Student’s t test or one-way or two-way analysis of variance (ANOVA), as indicated in the figure legends. *P < 0.05 was considered statistically significant. Data are shown as mean ± SD. The number of biological (nontechnical) replicates for each experiment is indicated in the figure legends. Kaplan-Meier analysis was used to calculate the OS with GraphPad Prism 9.3, and log-rank test was used to compare differences between two groups.
Supplementary Material
Acknowledgments:
We thank L. Klemm and M. E. Robinson from Müschen Laboratory for sharing reagent, B-ALL PDX cells, and B cell–related gene sets. We thank Z. Yu and M. Sun from China Medical University for sharing plasmids.
Funding:
This work was supported in part by National Institutes of Health (NIH) grants R01 CA214965 (to J. Chen), R01 CA211614 (to J. Chen), R01 CA178454 (to J. Chen), R01 CA182528 (to J. Chen), R01 CA236399 (to J. Chen), R01 CA271497 (to J. Chen), and T32 CA186895 (to B.T.). J. Chen is supported by City of Hope National Medical Center Cancer Center support grant P30CA33572 and the Simms/Mann Family Foundation. J. Chen is a Leukemia and Lymphoma Society (LLS) Scholar. M.M. is an HHMI Faculty Scholar.
Footnotes
Supplementary Materials
This PDF file includes:
Other Supplementary Material for this manuscript includes the following:
Data files S1 to S6
Competing interests: J. Chen is a scientific advisory board member of Race Oncology. The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All sequencing data (ChIP-seq and RNA-seq) that support the findings of this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE190853 (https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190853). Previous published microarray data of healthy BM and patients with different blood cancers (B-ALL, AML, CML, T-ALL, and MDS) were obtained from the accession code GSE13159 (70). RNA-seq data of human embryonic stem cells, AML cell lines, B-ALL cell lines, and leukemia patient samples were obtained from GSE168625 (71), GSE168752 (72), GSE115656 (73), GSE54641, GSM3563631 (74), GSM1534507, and GSM1534512. The custom Python and R scripts used in this study are available in Zenodo (https://zenodo.org/record/7662436#.Y_Uqj3bMKUk) (75).
REFERENCES AND NOTES
- 1.Siegel R, Naishadham D, Jemal A, Cancer statistics, 2013. CA Cancer J. Clin 63, 11–30 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Terwilliger T, Abdul-Hay M, Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 7, e577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Robison LL, Look AT, Acute lymphoblastic leukaemia. Lancet 371, 1030–1043 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Cooper SL, Brown PA, Treatment of pediatric acute lymphoblastic leukemia. Pediatr. Clin. North Am 62, 61–73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhlen M, Klusmann JH, Hoell JI, Molecular approaches to treating pediatric leukemias. Front. Pediatr 7, 368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel SE, Stock W, Johnson RH, Advani A, Muffly L, Douer D, Reed D, Lewis M, Freyer DR, Shah B, Luger S, Hayes-Lattin B, Jaboin JJ, Coccia PF, DeAngelo DJ, Seibel N, Bleyer A, Pediatric-inspired treatment regimens for adolescents and young adults with philadelphia chromosome-negative acute lymphoblastic leukemia: A review. JAMA Oncol. 4, 725–734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanjuan-Pla A, Bueno C, Prieto C, Acha P, Stam RW, Marschalek R, Menendez P, Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 126, 2676–2685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, Turtle CJ, Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127, 2406–2410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR, TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 17, 637–641 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Ittel A, Jeandidier E, Helias C, Perrusson N, Humbrecht C, Lioure B, Mazurier I, Mayeur-Rousse C, Lavaux A, Thiebault S, Lerintiu F, Gervais C, Mauvieux L, First description of the t(10;11)(q22;q23)/MLL-TET1 translocation in a T-cell lymphoblastic lymphoma, with subsequent lineage switch to acute myelomonocytic myeloid leukemia. Haematologica 98, e166–e168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burmeister T, Meyer C, Schwartz S, Hofmann J, Molkentin M, Kowarz E, Schneider B, Raff T, Reinhardt R, Gokbuget N, Hoelzer D, Thiel E, Marschalek R, The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia: Results from the GMALL study group. Blood 113, 4011–4015 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A, Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8, 200–213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y, Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473, 389–393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K, TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473, 343–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL, Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, Ghesquiere B, Van Dyck L, Boeckx B, Schoonjans L, Hermans E, Amant F, Kristensen VN, Peng Koh K, Mazzone M, Coleman M, Carell T, Carmeliet P, Lambrechts D, Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K, Xu W, Tan L, Hu Y, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang Y, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC Jr., Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG, Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu CH, Peng KL, Kang ML, Chen YR, Yang YC, Tsai CH, Chu CS, Jeng YM, Chen YT, Lin FM, Huang HD, Lu YY, Teng YC, Lin ST, Lin RK, Tang FM, Lee SB, Hsu HM, Yu JC, Hsiao PW, Juan LJ, TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2, 568–579 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Sun M, Song CX, Huang H, Frankenberger CA, Sankarasharma D, Gomes S, Chen P, Chen J, Chada KK, He C, Rosner MR, HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. U.S.A 110, 9920–9925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, Hong GM, Elkahloun AG, Arnovitz S, Wang J, Szulwach K, Lin L, Street C, Wunderlich M, Dawlaty M, Neilly MB, Jaenisch R, Yang FC, Mulloy JC, Jin P, Liu PP, Rowley JD, Xu M, He C, Chen J, TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. U.S.A 110, 11994–11999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H, Lobry C, Mullenders J, King B, Trimarchi T, Aranda-Orgilles B, Liu C, Shen S, Verma AK, Jaenisch R, Aifantis I, TET1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol 16, 653–662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, Cao Z, Shi H, Wang J, Lin L, Chen S, Yuan W, Qin Z, Ni H, Nimer SD, Yang FC, Jaenisch R, Jin P, Xu M, Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep. 13, 1692–1704 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Delabie J, Rimsza LM, Braziel RM, Grogan TM, Campo E, Jaffe ES, Dave BJ, Sanger W, Bast M, Vose JM, Armitage JO, Connors JM, Smeland EB, Kvaloy S, Holte H, Fisher RI, Miller TP, Montserrat E, Wilson WH, Bahl M, Zhao H, Yang L, Powell J, Simon R, Chan WC, Staudt LM; Lymphoma/Leukemia Molecular Profiling Project, Molecular diagnosis of Burkitt’s lymphoma. N. Engl. J. Med 354, 2431–2442 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Celkan T, Guven M, Batar B, Alhaj S, The difference between pre-B cell acute lymphoblastic leukemia and Burkitt lymphoma in relation to DNA damage repair gene polymorphisms in childhood. Leuk. Lymphoma 49, 1638–1640 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Huang H, Weng H, Chen J, m6A modification in coding and non-coding RNAs: Roles and therapeutic implications in cancer. Cancer Cell 37, 270–288 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S, Allis CD, Wang GG, The language of chromatin modification in human cancers. Nat. Rev. Cancer 21, 413–430 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones PA, Issa JP, Baylin S, Targeting the cancer epigenome for therapy. Nature Rev. Genet 17, 630–641 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Jones PA, Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, Qiu YH, von Minden MD, Ernst T, Hochhaus A, Cazzaniga G, Melnick A, Kornblau SM, Graeber TG, Wu H, Jumaa H, Muschen M, PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med 22, 379–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, Zhang QC, Aghajanirefah A, Cosgun KN, Sadras T, Lee J, Mirzapoiazova T, Salgia R, Ernst T, Hochhaus A, Jumaa H, Jiang X, Weinstock DM, Graeber TG, Muschen M, B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173, 470–484.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, Jaenisch R, Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9, 166–175 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin C, Lu Y, Jelinek J, Liang S, Estecio MR, Barton MC, Issa JP, TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 42, 6956–6971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma N, Pan H, Dore LC, Shukla A, Li QV, Pelham-Webb B, Teijeiro V, Gonzalez F, Krivtsov A, Chang CJ, Papapetrou EP, He C, Elemento O, Huangfu D, TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet 50, 83–95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khoueiry R, Sohni A, Thienpont B, Luo X, Velde JV, Bartoccetti M, Boeckx B, Zwijsen A, Rao A, Lambrechts D, Koh KP, Lineage-specific functions of TET1 in the postimplantation mouse embryo. Nat. Genet 49, 1061–1072 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Good CR, Madzo J, Patel B, Maegawa S, Engel N, Jelinek J, Issa JJ, A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 45, 8269–8281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W, Xia W, Wang Q, Towers AJ, Chen J, Gao R, Zhang Y, Yen CA, Lee AY, Li Y, Zhou C, Liu K, Zhang J, Gu TP, Chen X, Chang Z, Leung D, Gao S, Jiang YH, Xie W, Isoform switch of TET1 regulates DNA demethylation and mouse development. Mol. Cell 64, 1062–1073 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Gestrich CK, Oduro KA, Restricted immunoglobulin joining chain (IgJ) protein expression in B lymphoblastic leukemia (B-ALL) based on B-ALL subtype. Blood 136, 7 (2020). [Google Scholar]
- 39.Nix MA, Mandal K, Geng H, Paranjape N, Lin YT, Rivera JM, Marcoulis M, White KL, Whitman JD, Bapat SP, Parker KR, Ramirez J, Deucher A, Phojanokong P, Steri V, Fattahi F, Hann BC, Satpathy AT, Manglik A, Stieglitz E, Wiita AP, Surface proteomics reveals CD72 as a target for in vitro-evolved nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALL. Cancer Discov. 11, 2032–2049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan C, Baumgarth N, Parnes JR, CD72-deficient mice reveal nonredundant roles of CD72 in B cell development and activation. Immunity 11, 495–506 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Chan LN, Murakami MA, Robinson ME, Caeser R, Sadras T, Lee J, Cosgun KN, Kume K, Khairnar V, Xiao G, Ahmed MA, Aghania E, Deb G, Hurtz C, Shojaee S, Hong C, Polonen P, Nix MA, Chen Z, Chen CW, Chen J, Vogt A, Heinaniemi M, Lohi O, Wiita AP, Izraeli S, Geng H, Weinstock DM, Muschen M, Signalling input from divergent pathways subverts B cell transformation. Nature 583, 845–851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, Goloviznina NA, Chen WY, Huan J, LaTocha D, B-ALLabio E, Xiao G, Lee JW, Deucher A, Qi Z, Park E, Huang C, Nahar R, Kweon SM, Shojaee S, Chan LN, Yu J, Kornblau SM, Bijl JJ, Ye BH, Ansel KM, Paietta E, Melnick A, Hunger SP, Kurre P, Tyner JW, Loh ML, Roeder RG, Druker BJ, Burger JA, Milne TA, Chang BH, Muschen M, Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 27, 409–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katerndahl CDS, Heltemes-Harris LM, Willette MJL, Henzler CM, Frietze S, Yang R, Schjerven H, Silverstein KAT, Ramsey LB, Hubbard G, Wells AD, Kuiper RP, Scheijen B, van Leeuwen FN, Muschen M, Kornblau SM, Farrar MA, Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat. Immunol 18, 694–704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang X, Hu C, Ferchen K, Nie J, Cui X, Chen CH, Cheng L, Zuo Z, Seibel W, He C, Tang Y, Skibbe JR, Wunderlich M, Reinhold WC, Dong L, Shen C, Arnovitz S, Ulrich B, Lu J, Weng H, Su R, Huang H, Wang Y, Li C, Qin X, Mulloy JC, Zheng Y, Diao J, Jin J, Li C, Liu PP, He C, Chen Y, Chen J, Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat. Commun 8, 2099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wingelhofer B, Maurer B, Heyes EC, Cumaraswamy AA, Berger-Becvar A, de Araujo ED, Orlova A, Freund P, Ruge F, Park J, Tin G, Ahmar S, Lardeau CH, Sadovnik I, Bajusz D, Keseru GM, Grebien F, Kubicek S, Valent P, Gunning PT, Moriggl R, Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia 32, 1135–1146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shukla V, Samaniego-Castruita D, Dong Z, Gonzalez-Avalos E, Yan Q, Sarma K, Rao A, TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat. Immunol 23, 99–108 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bamezai S, Demir D, Pulikkottil AJ, Ciccarone F, Fischbein E, Sinha A, Borga C, Te Kronnie G, Meyer LH, Mohr F, Gotze M, Caiafa P, Debatin KM, Dohner K, Dohner H, Gonzalez-Menendez I, Quintanilla-Fend L, Herold T, Jeremias I, Feuring-Buske M, Buske C, Rawat VPS, TET1 promotes growth of T-cell acute lymphoblastic leukemia and can be antagonized via PARP inhibition. Leukemia 35, 389–403 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ, Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng Q, Chen J, Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 9, 472–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halazonetis TD, Gorgoulis VG, Bartek J, An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Morgado-Palacin I, Day A, Murga M, Lafarga V, Anton ME, Tubbs A, Chen HT, Ergan A, Anderson R, Bhandoola A, Pike KG, Barlaam B, Cadogan E, Wang X, Pierce AJ, Hubbard C, Armstrong SA, Nussenzweig A, Fernandez-Capetillo O, Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci. Signal 9, ra91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koneru B, Farooqi A, Nguyen TH, Chen WH, Hindle A, Eslinger C, Makena MR, Burrow TA, Wilson J, Smith A, Reddy VP, Cadogan E, Durant ST, Reynolds CP, ALT neuroblastoma chemoresistance due to telomere dysfunction-induced ATM activation is reversible with ATM inhibitor AZD0156. Sci. Transl. Med 13, eabd5750 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen LXT, Mitchell BS, Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc. Natl. Acad. Sci. U.S.A 110, 20681–20686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, Rabidou K, Fang R, Tan L, Xu S, Liu H, Argueta C, Zhang L, Mao F, Yan G, Chen J, Dong Z, Lv R, Xu Y, Wang M, Ye Y, Zhang S, Duquette D, Geng S, Yin C, Lian CG, Murphy GF, Adler GK, Garg R, Lynch L, Yang P, Li Y, Lan F, Fan J, Shi Y, Shi YG, Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C, N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, Sheng Y, Wang Y, Wunderlich M, Zhang B, Dore LC, Su R, Deng X, Ferchen K, Li C, Sun M, Lu Z, Jiang X, Marcucci G, Mulloy JC, Yang J, Qian Z, Wei M, He C, Chen J, METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell 22, 191–205.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, Hu YC, Hüttelmaier S, Skibbe JR, Su R, Deng X, Dong L, Sun M, Li C, Nachtergaele S, Wang Y, Hu C, Ferchen K, Greis KD, Jiang X, Wei M, Qu L, Guan JL, He C, Yang J, Chen J, Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol 20, 285–295 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rappsilber J, Mann M, Ishihama Y, Protocol for micro-purification, enrichment, prefractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Sala SC, Chrast J, Cunningham F, Domenico TD, Donaldson S, Fiddes IT, Girón CG, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P, GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu GC, Wang LG, Han YY, He QY, clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen YW, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li QH, Liu T, Liu XS, Ma LJ, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M, ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK, Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T, deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ianevski A, Giri AK, Aittokallio T, SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 48, W488–W493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, Gilkes A, Chiaretti S, Shurtleff SA, Kipps TJ, Rassenti LZ, Yeoh AE, Papenhausen PR, Liu WM, Williams PM, Foa R, Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol 28, 2529–2537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lv J, Yi Y, Qi Y, Yan C, Jin W, Meng L, Zhang D, Jiang W, Mitochondrial homeostasis regulates definitive endoderm differentiation of human pluripotent stem cells. Cell Death Discov. 8, 69 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sivakumar S, Qi S, Cheng N, Sathe AA, Kanchwala M, Kumar A, Evers BM, Xing C, Yu H, TP53 promotes lineage commitment of human embryonic stem cells through ciliogenesis and sonic hedgehog signaling. Cell Rep. 38, 110395 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Black KL, Naqvi AS, Asnani M, Hayer KE, Yang SY, Gillespie E, Bagashev A, Pillai V, Tasian SK, Gazzara MR, Carroll M, Taylor D, Lynch KW, Barash Y, Thomas-Tikhonenko A, Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 46, 11357–11369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smitheman KN, Severson TM, Rajapurkar SR, McCabe MT, Karpinich N, Foley J, Pappalardi MB, Hughes A, Halsey W, Thomas E, Traini C, Federowicz KE, Laraio J, Mobegi F, Ferron-Brady G, Prinjha RK, Carpenter CL, Kruger RG, Wessels L, Mohammad HP, Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica 104, 1156–1167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou K, Chen Z, Deng X, Chen J, TET1 promotes the initiation and progression of precursor B-cell malignancies independent of catalytic activity and serves as a therapeutic target (2023). 10.5281/zenodo.7662436. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All sequencing data (ChIP-seq and RNA-seq) that support the findings of this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) under accession number GSE190853 (https://ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190853). Previous published microarray data of healthy BM and patients with different blood cancers (B-ALL, AML, CML, T-ALL, and MDS) were obtained from the accession code GSE13159 (70). RNA-seq data of human embryonic stem cells, AML cell lines, B-ALL cell lines, and leukemia patient samples were obtained from GSE168625 (71), GSE168752 (72), GSE115656 (73), GSE54641, GSM3563631 (74), GSM1534507, and GSM1534512. The custom Python and R scripts used in this study are available in Zenodo (https://zenodo.org/record/7662436#.Y_Uqj3bMKUk) (75).