

Abstract
Heat shock protein 90 (Hsp90), a conserved molecular chaperone for a specific set of proteins critical for signal transduction including several oncogenic proteins, has been recognized as a promising target for anticancer therapy. Hsp90 inhibition also sensitizes cancer cells to DNA damage. However, the underlying mechanisms are not fully understood. Here, we provide evidence that Hsp90 is a general regulator of phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) family proteins, central regulators of stress responses including DNA damage. Inhibition of Hsp90 causes a reduction of all PIKK and suppresses PIKK‐mediated signaling. In addition, Hsp90 forms complexes with RUVBL1/2 complex and Tel2 complex, both of which have been shown to interact with all PIKK and control their abundance and functions. These results suggest that Hsp90 can form multiple complexes with the RUVBL1/2 complex and Tel2 complex and function in the regulation of PIKK, providing additional rationale for the effectiveness of Hsp90 inhibition for anticancer therapy, including sensitization to DNA damage. (Cancer Sci 2012; 103: 50–57)
Heat shock protein 90 (Hsp90) is an evolutionarily conserved molecular chaperone that plays a critical role in cellular homeostasis through stabilization and activation of several “client proteins” involved in a variety of cellular processes, including signal transduction, transcriptional regulation and cellular stress responses.( 1 , 2 ) Hsp90 works cooperatively with the Hsp40‐Hsp70 chaperone system in an ordered pathway, where Hsp90 binds to a client protein at the late stage of protein folding and facilitates its stability, structural maturation and assembly of a complex.( 3 ) Hsp90 is an ATPase, and ATP binding/hydrolysis‐driven conformational changes of Hsp90 are required for its chaperone activity.( 4 , 5 , 6 ) In this Hsp90 chaperone cycle, Hsp90 forms dynamic complexes with co‐chaperones/factors that regulate the Hsp90 ATPase cycle and the client interactions with Hsp90.( 2 , 7 ) The dynamic and transient nature of the Hsp90 chaperone complexes and the unstable character of Hsp90 clients have hindered the establishment of a comprehensive picture of the Hsp90 chaperone system.
Geldanamycin and its derivatives, such as 17‐allylamino‐17‐desmethoxygeldanamycin (17‐AAG), compete with ATP binding and inhibit Hsp90 chaperone activity, leading to degradation of client proteins.( 8 , 9 ) Importantly, these Hsp90 inhibitors selectively kill cancer cells compared to normal cells. This selectivity means that Hsp90 is a crucial facilitator of oncogene addiction and cancer cell survival and a molecular target for cancer therapy.( 10 ) The mechanisms of the cancer selectivity of Hsp90 inhibitor are not fully understood but are partly explained by the observation that the bulk of Hsp90 exists in active Hsp90 complexes in cancer cells, whereas most Hsp90 in normal cells exists in a latent, uncomplexed state.( 11 ) Therefore, analyses of the biochemical nature of the specific Hsp90‐client complexes in cancer cells are critical for understanding of the cancer specificity of Hsp90 inhibitors.
In addition to the selective killing of cancer cells, Hsp90 inhibitors sensitize cancer cells to radiation and DNA damaging agents. Hence, the combination of the Hsp90 inhibition and radiation/DNA damaging therapeutic drugs is a promising strategy for anticancer therapy.( 12 , 13 ) Although multiple mechanisms participate in the radio‐sensitization caused by Hsp90 inhibition, impaired DNA repair pathways seem to be responsible for at least part of that. For example, an Hsp90 inhibitor 17‐AAG blocks homologous recombination repair induced by DNA double‐strand breaks (DSB) in prostate or lung cancer cells but not normal fibroblasts.( 14 ) 17‐AAG also impairs the Fanconi anemia (FA) DNA repair pathway, whose activation requires ataxia telangiectasia mutated (ATM)‐and Rad3‐related (ATR)‐mediated phosphorylation of FANCA, one of the FA proteins.( 15 , 16 ) Another Hsp90 inhibitor, 17‐N‐dimethylaminoethylamino‐17‐demethoxygeldanamycin (17DMAG), compromises the DSB repair through an impairment of ionizing radiation (IR)‐responsive activation of DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs) and ATM.( 17 )
The phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) family includes ATM, DNA‐PKcs and ATR, as well as suppressor with morphological effect on genitalia 1 (SMG‐1), mammalian target of rapamycin (mTOR) and transformation/transcription domain‐associated protein (TRRAP) in mammals. SMG‐1 is an essential factor of nonsense‐mediated mRNA decay (NMD), one of the mRNA quality control systems,( 18 , 19 ) and TRRAP regulates transcription as a shared component of histone acetyl transferase complexes.( 20 ) SMG‐1 and TRRAP are also involved in DNA damage signaling and repair.( 21 , 22 , 23 ) mTOR senses nutrient status and coordinates cellular translational activity and cell growth/proliferation.( 24 )
Recent studies have revealed the existence of common regulators of all PIKK members, RuvB‐like 1 (RUVBL1), RUVBL2 and telomere maintenance 2 (Tel2). RUVBL1 and RUVBL2 are conserved ATPases belonging to the ATPases with associated diverse cellular activities (AAA+) family.( 25 ) They form a complex (RUVBL1/2) and participate in diverse cellular processes, including transcription, RNA modification, telomere maintenance and DNA repair.( 26 ) RUVBL1/2 interacts with all PIKK members and controls the PIKK abundance at least at the mRNA level.( 27 ) RUVBL1/2 also regulates functional complex formation of SMG‐1 during NMD.( 27 ) Tel2 interacts with all PIKK and regulates the stability of PIKK proteins.( 28 ) Tel2 also functions in the recruitment of Tel1 (ATM ortholog in S. cerevisiae) to DNA damage sites and the Rad3/ATR‐mediated DNA damage response.( 29 , 30 , 31 ) We recently found that Hsp90 inhibition causes the downregulation of PIKK members,( 27 ) as observed for knockdown/knockout of either RUVBL1/2 or Tel2. This raises an intriguing possibility that at least a part of the Hsp90‐induced sensitization to DNA damage can be attributed to the reduction and/or inactivation of these PIKK. However, it has been reported, controversially, that Hsp90 inhibition does not affect the abundance of PIKK.( 17 , 32 )
In this study, we first evaluated the effect of the Hsp90 inhibition on PIKK abundance and PIKK‐mediated signaling. We confirm that Hsp90 inhibition decreases the abundance of all PIKK proteins (ATM, ATR, DNA‐PKcs, mTOR, SMG‐1 and TRRAP). Importantly, Hsp90 inhibition severely compromises PIKK‐mediated signaling pathways. In addition, Hsp90 physically interacted with RUVBL1/2, Tel2, and their associating proteins. Both RUVBL1/2 and Tel2 interacted with two evolutionarily conserved Hsp90 co‐factors, NOP17 and RPAP3. These results strongly support the notion that Hsp90 can form diverse protein complexes with RUVBL1/2 and/or Tel2, and that it acts as a general PIKK regulator.
Materials and Methods
Plasmids, antibodies, siRNA and inhibitor. pcDNA5/FRT/TO/NTAP‐GST and pcDNA5/FRT/TO/NTAP‐SMG‐10 have been described previously.( 27 ) pcDNA5/FRT/TO/NTAP‐NOP17 and pcDNA5/FRT/TO/NTAP‐RPAP3 were constructed by the cloning of each cDNA fragment to the pcDNA5/FRT/TO/NTAP vector using standard methods.
Anti‐Tel2 and anti‐Tti2 antisera were generated against recombinant human Tel2 (aa 624–691) or human Tti2 (aa 8–108) fused to glutathione S‐transferase (GST). The anti‐SMG‐1, Upf1, SMG‐10 and Phosho‐Upf1 (clone 3B8 or 8E6) antibodies have been described previously.( 19 , 33 , 34 , 35 ) Antibodies to Hsp90α/β (#4874; Cell Signaling Technology, Beverley, MA, USA), RUVBL1 (#BMR00431; Biomatrix, Chiba, Japan and #sc‐15259; Santa Cruz Biotechnology, Santa Cruz, CA, USA), RUVBL2 (#612482; BD Transduction Laboratories, Franklin Lakes, NJ, USA), RNA polymerase II subunit 5 (RPB5) (#S3157_EP; Euromedex, Souffelweyersheim, France), DNA‐PKcs (#A300‐519A; Bethyl Laboratories, Montgomery, TX, USA), ATM (#2873; Cell Signaling Technology), ATR (#ab1; Calbiochem, Darmstadt, Germany), mTOR (#2972; Cell Signaling Technology), TRRAP (#A301‐132A; Bethyl), Akt (#9272; Cell Signaling Technology), Chk1 (#sc‐8408; Santa Cruz), P‐Chk1 (Ser345) (#2348; Cell Signaling Technology), Chk2 (#2662; Cell Signaling Technology), P‐Chk2 (Thr68) (#2661; Cell Signaling Technology), p70 S6K (#sc‐230; Santa Cruz), P‐p70 S6K (Thr389) (#9205; Cell Signaling Technology), P‐S/TQ ATM/ATR substrate (#2851; Cell Signaling Technology), JNK (#15701A; BD Pharmingen, San Diego, CA, USA), unconventional prefoldine RPB5 interactor (URI) (#A301‐164‐1; Bethyl), nucleolar protein 17 (NOP17) (#H00055011‐M05; Abnova, Taipei, Taiwan), RNA polymerase II associated protein 3 (RPAP3) (#H00079657‐B01P; Abnova), Hsp70 (#SPA‐810; Stressgen, Victoria, BC, Canada), Myosin IIa (#M8064; Sigma, St. Louis, MO, USA), GAPDH (#ab8245; Abcam, Cambridge, MA, USA), nuclear cap binding protein subunit 1, 80kDa (CBP‐80) (#10349‐1‐AP; Protein Tech Group, Chicago, IL, USA), β‐actin (#A1978; Sigma) and α‐tubulin (#T6199; Sigma) were obtained commercially.
The following siRNA target sequences were used: RUVBL1, siGENOME duplex D‐008977‐02 (Dharmacon, Lafayette, CO, USA); RUVBL2, siGENOME duplex D‐012299‐03 (Dharmacon); SMG‐10, siGENOME duplex J‐014188‐05 (Dharmacon); Tti2, Hs_Tti2_2 HP Validated siRNA SI00401660 (Qiagen, Valencia, CA, USA); Tel2, Hs_KIAA0683_4 HP Validated siRNA SI00454909 (Qiagen); URI, Hs_C19orf2_4 HP Validated siRNA SI00322462 (Qiagen); RPB5, Hs_POLR2E_3 HP Validated siRNA SI00689073 (Qiagen); NOP17, Hs_FLJ20643_4 HP Validated siRNA SI00120148 (Qiagen); RPAP3, Hs_FLJ21908_3 HP Validated siRNA SI00399875 (Qiagen); DNA‐PKcs, #1: Hs_PRKDC_ 8_HP Validated siRNA SI02663633 (Qiagen), #2: Hs_ PRKDC_2_HP Validated siRNA SI00093079 (Qiagen); ATM, #1: Hs_ATM_4_HP Validated siRNA SI00000847 (Qiagen), #2: Hs_ATM_14_HP Validated siRNA SI03068506 (Qiagen); ATR, #1: Hs_ATR_12_HP Validated siRNA SI02664347 (Qiagen), #2: Hs_ATR_2_HP Validated siRNA SI00023107 (Qiagen); mTOR, #1: Hs_FRAP1_7 _HP Validated siRNA SI03023587 (Qiagen), #2: Hs_FRAP1_8 _HP Validated siRNA SI03064985 (Qiagen); SMG‐1, #1: Mm_2610207I05Rik_4 HP Validated siRNA SI02765546 (Qiagen), #2: GTGTATGTGCGCCAAAGTA; TRRAP, #1: Hs_TRRAP_2_HP Validated siRNA SI00052591 (Qiagen), #2: Hs_TRRAP_3_HP Validated siRNA SI00052598 (Qiagen); and NS siRNA, All Star Negative Control siRNA (Qiagen).
17‐allylamino‐17‐desmethoxygeldanamycin (17‐AAG) (Sigma) was used.
Cell culture and transfection. HeLa TetOff cells (TaKaRa Clontech, Shiga, Japan) and Flp‐In T‐Rex HEK293 cells (Invitrogen, Carlsbad, CA, USA) were grown in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Tet‐inducible SBP streptavidin binding peptide (SBP)‐tagged FlpIn T‐Rex HEK 293 stable cells have been described previously.( 27 ) siRNA transfections were performed in 12‐well or 6‐well plates using siLentFect (BioRad, Hercules, CA, USA) according to the manufacturer’s protocol, and cells were harvested 60 h later. Plasmid transfections were performed in 15‐cm dishes using Lipofectamine LTX (Invitrogen), according to the manufacturer’s protocol, and cells were harvested 36 h later.
Affinity purification and mass spectrometry. Affinity purification and mass spectrometry analysis were performed as described previously.( 27 )
Immunoprecipitation and western blot analysis. HeLa TetOff cells were lysed in T‐buffer (20 mM Hepes‐NaOH at pH 7.5, 150 mM NaCl, 0.05% Tween 20, 2.5 mM MgCl2, 0.5 mM DTT, 100 nM okadaic acid [Calbiochem], protease inhibitor cocktail [Roche Applied Science, Indianapolis, IN, USA], phosphatase inhibitor cocktail [Roche] and 100 μg/mL RNaseA [Qiagen]). The soluble fractions were incubated with antibodies for 1 h at 4°C with gentle rotation. Subsequently, the soluble fractions were incubated with Dynabeads protein G (Invitrogen) for an additional 1 h at 4°C with gentle rotation. After washing with RNase(−) lysis buffer, the immunocomplexes were boiled in SDS sample buffer, and analyzed by western blotting. All proteins in western blot experiments were detected with Lumi‐Light plus western blotting substrate (Roche) or Immobilon Western (Millipore, Billerica, MA, USA) and quantified with a Lumino‐Imager, LAS‐3000, and Science Lab 2001 Image Gauge software (Fuji Photo Film, Tokyo, Japan).
Results
Heat shock protein 90 inhibition causes reduction of all phosphatidylinositol 3‐kinase‐related protein kinase and phosphatidylinositol 3‐kinase‐related protein kinase‐mediated stress signaling. To re‐examine the role of Hsp90 on the abundance of PIKK proteins, we treated HeLa TetOff cells with 17‐AAG, an Hsp90 inhibitor, and incubated for 12 or 24 h. We showed that Hsp90 inhibition caused apparent downregulation of ATM, ATR and DNA‐PKcs (Fig. 1A). The different effect of Hsp90 inhibition on PIKK abundance from previous reports might reflect the difference in sensitivity to Hsp90 of each cell line.( 17 , 32 ) Moreover, the Hsp90 inhibition attenuates IR‐induced Chk2 phosphorylation at Thr68, which is mediated by ATM and induces cell cycle checkpoint to response DNA damage (Fig. 1B). In addition, IR‐induced phosphorylations of putative ATM/ATR substrates were decreased under the Hsp90 inhibition (Fig. 1E, lower panel). Consistent with these results, the inhibition of Hsp90 mediates impairment of IR‐induced cell cycle checkpoints and significant delay of DNA repair.( 17 ) Furthermore, Hsp90 inhibition reduced the protein amount of other PIKK, mTOR, SMG‐1 and TRRAP (Fig. 1A). In addition, suppression of ATR‐mediated Chk1 phosphorylation at Ser345 accompanied by a significant reduction of Chk1 was observed, which is consistent with a previous study (Fig. 1C).( 36 ) These results suggest that at least a part of the sensitization to DNA damage caused by Hsp90 inhibition results from the reduction of PIKK and impaired PIKK‐mediated DNA damage responses. We further investigated the effects of Hsp90 inhibition on mTOR‐mediated or SMG‐1‐mediated signaling. mTOR controls cell size/proliferation and translation activity in response to the nutrient status through the phosphorylation of p70S6 kinase.( 24 ) The Hsp90 inhibition reduced the mTOR‐mediated phosphorylation of p70S6 kinase at Thr389, with moderate reduction of p70S6 kinase (Fig. 1D). Because mTOR is activated by Akt, one of the Hsp90 clients shown in Figure 1A, we cannot exclude the possibility that the reduced phosphorylation of p70S6 kinase is a consequence of the downregulation of Akt. The SMG‐1‐mediated Upf1 phosphorylation at Ser1078/1096 is essential for nonsense‐mediated mRNA decay, a quality control system that selectively degrades aberrant mRNA with premature termination codons.( 18 ) The Hsp90 inhibition obviously decreased the SMG‐1‐mediated Upf1 phosphorylation (Fig. 1E, upper panel). No apparent reduction of α‐tubulin was observed with the Hsp90 inhibition (Fig. 1F). Taken together, these results indicate that Hsp90 is required for the maintenance of all PIKK proteins and PIKK signaling.
Figure 1.
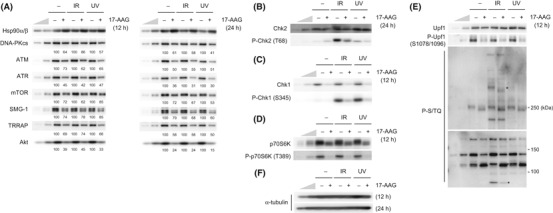
Inhibition of heat shock protein 90 (Hsp90) activity decreases the abundance of all phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) proteins and the downstream signaling. (A–F) HeLa TetOff cells were treated with vehicle or 2 μM 17‐allylamino‐17‐desmethoxygeldanamycin (17‐AAG) for 12 or 24 h, then the cells were untreated, treated with 10 Gy IR, or 100 J/m2 of UV, and incubated for 1 h. Total cell lysates were analyzed by western blotting with the indicated antibodies. To estimate the protein abundance, 33 and 11% of the 17‐AAG, IR and UV‐untreated samples were loaded. The anti‐P‐S/TQ antibody recognizes phosphorylated serine or threonine in the SQ motif, potential phosphorylation sites by ATM/ATR/SMG‐1/DNA‐PKcs (E, lower panel). Asterisks indicate some examples of phosphoproteins affected by the 17‐AAG treatment (E, lower panel). ATM, ataxia telangiectasia mutated; ATR, ATM‐and Rad3‐related; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; mTOR, mammalian target of rapamycin; SMG‐1, suppressor with morphological effect on genitalia 1; TRRAP, transformation/transcription domain‐associated protein.
Heat shock protein 90 physically interacts with the common regulators of phosphatidylinositol 3‐kinase‐related protein kinase, RUVBL1/2 and Tel2. Previous studies have revealed that PIKK members can be regulated by common factors, the RUVBL1 and RUVBL2 complex (RUVBL1/2) and Tel2.( 27 , 28 ) The physical interactions between RUVBL1 and Hsp90 and between RUVBL1 and Tel2 led us to hypothesize that Hsp90 is involved in the PIKK regulation together with RUVBL1/2 and/or Tel2.( 27 ) To evaluate this possibility, we examined physical interactions among Hsp90, RUVBL1/2 and Tel2.
We confirmed co‐purification of Hsp90 with RUVBL1 from a HEK 293‐cell extract stably expressing SBP‐tagged RUVBL1 (Fig. 2A). (27) In addition, endogenous Tel2 co‐immunoprecipitated Hsp90 from HeLa TetOff cell extract (Fig. 2B).
Figure 2.
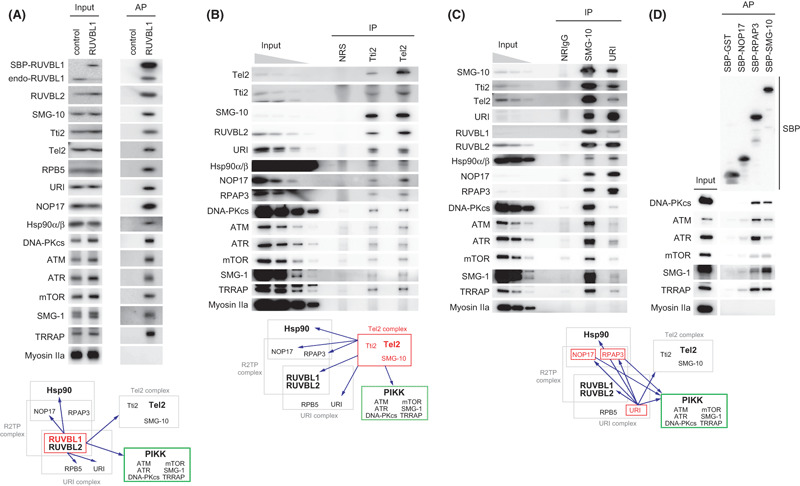
Heat shock protein 90 (Hsp90) interacts with two other phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) regulators, RUVBL1/2 and Tel2 and their associated proteins. (A) RUVBL1 interacted with Hsp90, a Hsp90 co‐factor (NOP17), URI complex and Tel2 complex. Tet‐inducible streptavidin‐binding peptide (SBP)‐tagged RUVBL1 stable HEK 293 cells or control cells, which express tag peptides only, were treated with 1 ng/mL doxycycline for 3 days. Cytoplasmic cell extracts were affinity purified with streptavidin sepharose, and biotin‐eluted fractions were analyzed by western blotting with the indicated antibodies. (B,C) Protein interactions of Tel2, SMG‐10, Tti2 and URI. HeLa TetOff cells were immunoprecipitated with anti‐Tti2, anti‐Tel2 antiserum or normal rabbit serum (NRS) (B), or anti‐SMG‐10, URI, or normal rabbit IgG (NRIgG) (C). The immunoprecipitates were analyzed by western blotting with the indicated antibodies. Input: 1, 0.33, 0.11 and 0.037% (B) or 0.5, 0.17 and 0.06% (C) of the amount immunoprecipitated. (D) RPAP3 interacts with all PIKK. HeLa TetOff cells were transfected with pcDNA5/FRT/TO/NTAP‐GST, pcDNA5/FRT/TO/NTAP‐NOP17, pcDNA5/FRT/TO/NTAP‐RPAP3 or pcDNA5/FRT/TO/NTAP‐SMG‐10. The cell extracts were subjected to affinity purification with streptavidin sepharose 36 h later and analyzed by western blotting with indicated antibodies. ATM, ataxia telangiectasia mutated; ATR, ATM‐and Rad3‐related; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; mTOR, mammalian target of rapamycin; NOP17, nucleolar protein 17; RPB5, RNA polymerase II subunit 5; SMG‐1, suppressor with morphological effect on genitalia 1; TRRAP, transformation/transcription domain‐associated protein.
Tel2 form an evolutionally conserved complex with SMG‐10 (also known as Tti1) and Tti2.( 37 , 38 , 39 ) In our experiments, tight associations among Tel2, SMG‐10 and Tti2 were also observed (Fig. 2B,C). As with Tel2, both SMG‐10 and Tti2 co‐immunoprecipitated RUVBL1/2 and Hsp90, indicating that the Tel2 complex associates with RUVBL1/2 and Hsp90 (Fig. 2B,C).
RUVBL1/2 has been identified in a complex named R2TP, containing Tah1 (RPAP3 in mammals) and Pih1 (NOP17 in mammals),( 40 ) and URI‐prefoldin complex together with RPB5 (see the schemes in Fig. 2).( 41 ) Because the Tel2 complex components interact with RUVBL1, as confirmed in Figure 2A, we probed the antibodies against these RUVBL1 interacting proteins,( 27 , 42 ) NOP17, RPAP3, RPB5 and URI, for the purified RUVBL1 complex and Tel2, SMG‐10 or Tti2 immunoprecipitates. As expected, all of them co‐purified and co‐immunoprecipitated with SBP‐RUVBL1 and endogenous Tel2, SMG‐10 and Tti2, respectively (Fig. 2A–C). To confirm these protein interactions, endogenous URI immunoprecipitate from HeLa TetOff cell extract was analyzed. As shown in Figure 2C, URI co‐immunoprecipitated with Hsp90, RUVBL1/2, Tel2, and their interacting proteins. These results indicate that Hsp90, RUVBL1/2, Tel2, SMG‐10, Tti2, NOP17, RPAP3 and URI can physically interact and form complexes.
Because both RUVBL1/2 and Tel2 are common PIKK binding proteins, we investigated whether RUVBL1/2‐interacting and Tel2‐interacting proteins also associate with PIKK. We probed PIKK antibodies to purified SBP‐RUVBL1 complexes (Fig. 2A), immunoprecipitates of endogenous Tel2, Tti2 (Fig. 2B), SMG‐10 and URI (Fig. 2C), and purified SBP‐NOP17, PARP3 and SMG‐10 complexes (Fig. 2D). The results indicate that all protein tested in this study can interact with all PIKK, although the significances differ among precipitated proteins (Fig. 2). Taken together, Hsp90, RUVBL1/2, Tel2, SMG‐10, Tti2, NOP17, RPAP3 and URI can physically interact with PIKK, probably as a part of complex.
RUVBL1/2, Tel2 and their associated proteins are involved in phosphatidylinositol 3‐kinase‐related protein kinase signaling. The physical association of the RUVBL1/2‐associated and Tel2‐associated proteins with PIKK suggests the possibility that they are involved in the regulation of PIKK abundance and/or PIKK signaling, like RUVBL1/2 and Tel2. To test this, we analyzed knockdown effects of each molecule. As shown in Figure 3A, the siRNA efficiently knocked down each molecule. We found close relationships among RUVBL1/2, Tel2 and their associated proteins (Fig. 3A). For instance, SMG‐10 and Tti2 interdependently regulated the other protein abundance. NOP17 also depended on RPAP3 for its abundance. Knockdown of RUVBL1, RUVBL2 or Tel2 also decreased the abundance of SMG‐10, Tti2 and NOP17. NOP17 was also reduced by the knockdown of Tel2. Tel2 was decreased by the knockdown of RUVBL1, RUVBL2, SMG‐10, Tti2, RPB5 or NOP17. The abundance of RUVBL1 and RUVBL2 was decreased to <50% by the knockdown of RPB5 or NOP17. These observations indicate an interdependent relationship among RUVBL1/2, Tel2 and their associated proteins. It might reflect the change of their protein stability, based on the previous reports.( 38 , 43 )
Figure 3.
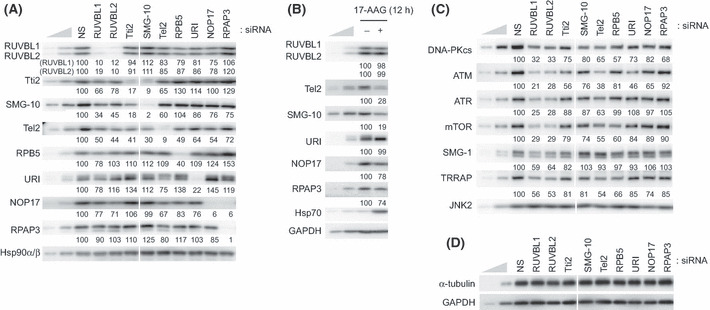
Knockdown of RUVBL1/2 and Tel2 interacting proteins partially affects the phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) abundance. (A,C,D) HeLa TetOff cells were transfected with the indicated siRNA. Sixty hours after transfection, total cell lysates were analyzed by western blotting with the indicated antibodies. Same knockdown samples were used in panels A, C, D in Figure 3, and panels A and B in Figure 4. Mean values of the relative protein levels compared to the nonsilencing (NS) control from two independent experiments are shown under the blots. (B) HeLa TetOff cells were treated with vehicle or 2 μM 17‐AAG for 12 h and total cell lysates were analyzed by western blotting with the indicated antibodies. To estimate the protein abundance, 33 and 11% of the NS sample (A,C,D) or untreated sample (B) were loaded. ATM, ataxia telangiectasia mutated; ATR, ATM‐and Rad3‐related; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; mTOR, mammalian target of rapamycin; SMG‐1, suppressor with morphological effect on genitalia 1; TRRAP, transformation/transcription domain‐associated protein.
In addition, Hsp90 inhibition clearly decreased the abundance of Tel2 and SMG‐10 (Fig. 3B), suggesting a possibility that the Tel2 complex components are Hsp90 clients. Note that any knockdown of RUVBL1/2‐associated and Tel2‐associated proteins did not decrease the abundance of Hsp90 (Fig. 3A).
Consistent with previous reports, knockdown of RUVBL1, RUVBL2 or Tel2 decreased the abundance of PIKK (Fig. 3C).( 27 , 28 ) However, a slight reduction of DNA‐PKcs and ATM was observed by the knockdown of Tti2, SMG‐10, URI or NOP17 in this transient knockdown experiment (Fig. 3C). Long‐term knockdown of SMG‐10 or Tti2 probably results in the downregulation of all PIKK, as reported in recent report.( 38 ) RPB5 knockdown also modestly affects the abundance of DNA‐PKcs, mTOR and TRRAP; however, it might influence transcription activity, given that RPB5 is an RNA polymerase subunit (Fig. 3C).( 44 )
In contrast, knockdown of RUVBL1/2‐associated and Tel2‐associated proteins partially affected PIKK signaling. For example, the relative level of the mTOR‐mediated phosphorylation of p70S6 kinase at Thr389 without additional stimulation was decreased by the knockdown of URI, NOP17 or RPAP3, in addition to the knockdown of RUVBL1 or RUVBL2 (Fig. 4A). The relative level of SMG‐1‐mediated Upf1 phosphorylation at Ser1078/1096 was reduced only by the RUVBL1, RUVBL2 or RPB5 knockdown but not affected by others (Fig. 4B). However, the phosphorylation of a 140‐kDa protein detected by a phospho‐S/TQ antibody, which recognizes phosphorylated ATM/ATR substrates, was reduced by the knockdown of Tti2 and SMG‐10, in addition to the knockdown of RUVBL1, RUVBL2 or Tel2 (Fig. 4B). Furthermore, strong reduction of the phosphorylation of a 110‐kDa protein detected by the phospho‐S/TQ antibody was observed by the knockdown of RPB5, but not others (Fig. 4B). In analogy to the situation with Hsp90 inhibition, Chk1 is downregulated by the knockdown of RUVBL1/2, Tel2 and their associated proteins (1, 4). The relative level of ATR‐mediated phosphorylation of Chk1 at Thr345 caused by UV irradiation was slightly decreased by the knockdown of RUVBL2, Tti2 and NOP17 (Fig. 4C). In contrast, knockdown of RUVBL1/2‐associated and Tel2‐associated proteins had different effects on the relative level of ATM‐mediated phosphorylation of Chk2 at Thr68 in response to IR. Whereas knockdown of RUVBL1 or RPB5 decreased the Chk2 phosphorylation, knockdown of Tti2, SMG‐10, URI, NOP17 or RPAP3 enhanced it (Fig. 4D). These results suggest that RUVBL1/2‐associated or Tel2‐associated proteins are involved in the PIKK signaling and that their degree of contribution differs according to each PIKK and its substrate. However, we cannot exclude the possibility that the different effect is resulting from knockdown efficiency of each molecule.
Figure 4.
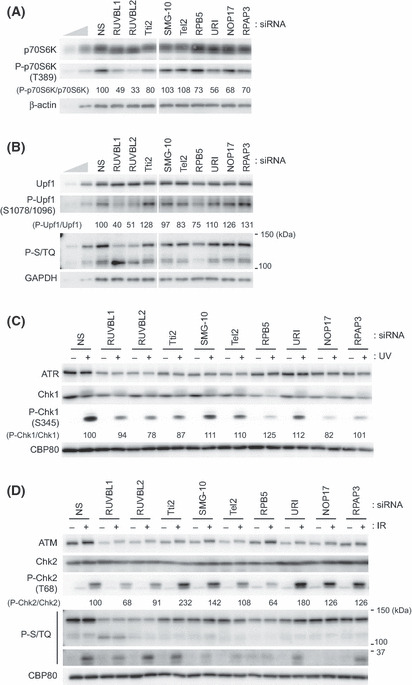
Knockdown of RUVBL1/2 and Tel2 interacting proteins affects the phosphatidylinositol 3‐kinase‐related protein kinase (PIKK) signaling. (A,B) HeLa TetOff cells were transfected with the indicated siRNA. Sixty hours after transfection, total cell lysates were analyzed by western blotting with the indicated antibodies. Same knockdown samples were used in panels A,C,D in Figure 3, and panels A and B in Figure 4. To estimate protein abundances, 33 and 11% of the nonsilencing (NS) control were loaded. (C,D) HeLa TetOff cells were transfected with the indicated siRNA. Sixty hours later, cells were untreated, treated with 100 J/m2 of UV (C), or 10 Gy IR (D), and incubated for 1 h. Total cell lysates were analyzed by western blotting with the indicated antibodies. The anti‐P‐S/TQ antibody recognizes phosphorylated serine or threonine in the SQ motif, potential phosphorylation sites by ATM/ATR/SMG‐1/DNA‐PKcs (B,D). Relative phosphorylation levels of p70S6K (A), Upf1 (B), Chk‐1 (C), Chk‐2 (D) were indicated under each blot. ATM, ataxia telangiectasia mutated; ATR, ATM‐and Rad3‐related; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; SMG‐1, suppressor with morphological effect on genitalia 1.
RUVBL2 and Hsp90 are substrates of S/TQ‐directed protein kinases,( 45 ) ATM, ATR, DNA‐PKcs and SMG‐1. This suggests possible mutual regulatory mechanisms among PIKK. We analyzed knockdown effects of each PIKK member on other PIKK protein abundance. As shown in Figure 5, knockdown of DNA‐PKcs or mTOR caused downregulation of ATM.
Figure 5.
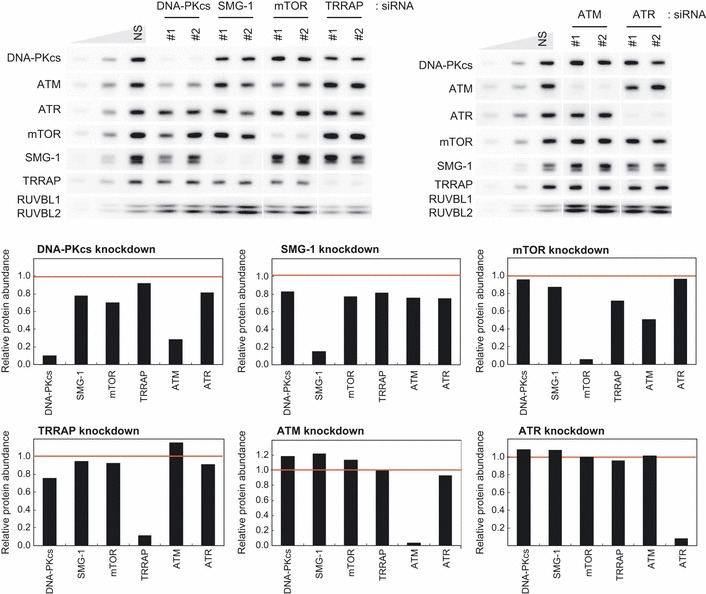
Knockdown of DNA‐PKcs or mTOR reduces the ATM abundance. HeLa TetOff cells were transfected with two independent siRNA targeted to each phosphatidylinositol 3‐kinase‐related protein kinase (PIKK). Sixty hours after the transfections, total cell lysates were analyzed by western blotting with the indicated antibodies. To estimate the PIKK abundance, 33 and 11% of the nonsilencing (NS) control sample were loaded. The relative protein levels of each PIKK compared with the NS control levels were graphed. The values are an average of the data from two siRNA from two independent experiments. ATM, ataxia telangiectasia mutated; ATR, ATM‐and Rad3‐related; DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; mTOR, mammalian target of rapamycin; SMG‐1, suppressor with morphological effect on genitalia 1; TRRAP, transformation/transcription domain‐associated protein.
Discussion
In this study, we showed that Hsp90 regulates all PIKK members and PIKK‐mediated signaling (Fig. 1). This finding highlights the importance of Hsp90 as a key regulator of signal transduction and a target of cancer therapy. ATM, ATR and DNA‐PKcs are critical regulators of DNA damage response and repair,( 46 , 47 , 48 ) and other PIKK members, SMG‐1 and TRRAP, are also involved in these processes.( 21 , 22 , 23 ) DNA repair pathway has been studied as a target for cancer therapy in combination with DNA‐damaging drugs.( 49 ) Moreover, NMD inhibition, including SMG‐1 inactivation, has attracted attention as a novel anti‐cancer strategy by induction of tumor immunity.( 50 , 51 ) mTOR controls cell proliferation and angiogenesis, and mTOR inhibitors have been developed as antitumor agents.( 52 ) Hsp90 is required for the maintenance of all PIKK and PIKK‐mediated signaling, as we have shown here. Therefore, Hsp90 inhibition is expected to yield additive effects of all PIKK disruption‐mediated defect of cellular function mentioned above. Thus, our results support the effectiveness of Hsp90 inhibition for cancer therapy and provide new insight into the mechanisms of the anticancer effects of Hsp90 inhibition, including the sensitization to DNA damage. Because co‐chaperone expression can affect the sensitivity of cancer cells to Hsp90 inhibitors, putative Hsp90 co‐factor complex (see below) might be an additional molecular target to enhance the efficacy of Hsp90 inhibitors in cancer therapy. ( 53, 54
In addition to the regulation of the PIKK family by Hsp90, we demonstrated the relationship between Hsp90 and PIKK common regulators RUVBL1/2 and Tel2. Hsp90 physically interacts with RUVBL1/2 and Tel2 (Fig. 2A,B). Inhibition of any molecule causes PIKK reduction and suppression of PIKK signaling (1, 3, 4).( 27 , 28 ) These observations give rise to the idea that RUVBL1/2 and Tel2 function in PIKK regulation together with Hsp90. One possibility is that RUVBL1/2 and Tel2 act as Hsp90 co‐factors. In support of this idea, both RUVBL1/2 and Tel2 interact with two Hsp90 co‐factors, Pih1 (NOP17 in mammals) and Tah1 (RPAP3 in mammals) (Fig. 2A,B),( 40 ) which bind to Hsp90 and affect its ATPase activity.( 55 , 56 ) Because all of RUVBL1, Tel2, SMG‐10, Tti2 and URI interact with Hsp90, RPAP3 and NOP17, and share interactors (Fig. 2), a large protein complex is expected to act with Hsp90. Indeed, it was recently reported that RUVBL1/2, RPAP3 and URI interact with Tel2 through NOP17 in a phospho‐dependent manner of Tel2, and this interaction is required for the maintenance of SMG‐1 and mTOR.( 57 )
In contrast, we observed different knockdown effects of each molecule on the PIKK signalling, as shown in Figure 4. This result might reflect the different roles of each molecule, such as a positive or negative regulator in the co‐factor complex, or a different degree of contribution to each PIKK. It is also possible that multiple co‐factor complexes function with Hsp90. In support of this possibility, the interaction of Tel2 complex with R2TP/URI complex is dispensable for Tel2 to regulate the stability of ATM, ATR and DNA‐PKcs.( 57 ) In this view, further investigations are required to clarify the role of each molecule in the Hsp90 function and the specificity on each PIKK.
Intriguingly, Hsp90, RUVBL1/2, RPAP3 and NOP17 participate in other cellular processes, including small nucleolar ribonucleoprotein complex (snoRNP) maturation( 43 , 58 ) and telomerase complex assembly.( 59 , 60 ) Furthermore, RPB5 is a subunit of RNA polymerases( 44 ) and all of RUVBL1/2, RPAP3, NOP17 and URI not only physically associate with RNA polymerase II transcription machinery( 42 ) but also are involved in their assembly, together with Hsp90.( 61 ) Therefore, R2TP/URI complex might broadly function with Hsp90 and such functions are not limited to PIKK. However, the involvement of the Tel2 in these processes has not been reported. Considering that Tel2 directly interacts with ATM and mTOR( 28 ) and that Tel2 is depend on the Tel2 complex formation for its abundance (Fig. 3A),( 39 ) Tel2‐containing complex is likely to be a mediator complex specialized for PIKK to link them to Hsp90.
As another possibility, RUVBL1/2 or Tel2 complex itself is an Hsp90 client and the reduction of PIKK induced by the Hsp90 inhibition is an indirect effect of the inactivation of RUVBL1/2 and/or Tel2 complex. In fact, Hsp90 inhibition causes apparent reduction of Tel2 and SMG‐10 (Fig. 3B), and the interaction between Tel2 and PIKK is depend on Hsp90.( 38 ) However, the physical associations between Hsp90 and at least two PIKK, DNA‐PKcs( 62 ) and SMG‐1,( 27 ) support that PIKK are direct Hsp90 clients.
As for the control of the PIKK abundance, some differences have been observed between RUVBL1/2 and Tel2. Whereas RUVBL1/2 affects the mRNA abundance of PIKK proteins except for SMG‐1,( 27 ) Tel2 affects the stability of PIKK proteins but does not affect their mRNA abundance.( 28 ) This might be one reason for the strong effect of RUVBL1/2 knockdown on PIKK abundance. Moreover, mutual regulatory mechanisms exist at least among DNA‐PKcs, ATM, mTOR and SMG‐1 (Fig. 5).( 63 ) Therefore, PIKK abundance is probably regulated by multiple mechanisms, including Hsp90‐mediated pathway.
In addition to the maintenance of PIKK abundance, RUVBL1/2, Tel2 and SMG‐10 regulate the functional complex formation of ATR, mTOR and SMG‐1.( 27 , 64 , 65 ) Hsp90 inhibition also impairs the interaction between ATM and MRN complex without affecting ATM abundance.( 17 ) Given these observations, Hsp90 potentially participate in the PIKK signaling in a positive manner through facilitating assembly of PIKK protein complexes not only during the PIKK protein synthesis. Further investigation is required to reveal the precise roles of Hsp90 and its putative co‐factor complex containing RUVBL1/2 and Tel2 in PIKK regulation.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
We thank Ms. Rie Kurata, Ms. Reiko Muramatsu, Ms. Yumi Bamba, Dr. Yukiko Okada‐Katsuhata and Mr. Kei Kutsuzawa for technical support. This work was supported, in part, by grants from the Japan Society for the Promotion of Science (to S.O. and N.I.), from the Ministry of Education, Culture, Sports, Science and Technology of Japan (a Grant‐in‐Aid for Scientific Research [A] [to S.O.], Young Scientists [A] [to A.Y.], Scientific Research on Innovative Areas “RNA regulation” [to A.Y.] and Scientific Research on Innovative Areas “Functional machinery for noncoding RNAs” [to A.Y.]), from the Japan Science and Technology Corporation (to A.Y. and S.O.), Takeda Science Foundation (to S.O.), Mitsubishi foundation (to S.O.), Uehara memorial foundation (to S.O.), and the Yokohama Foundation for Advancement of Medical Science (to A.Y.). N.I. is a Research Fellow of the Japan Society for the Promotion of Sciences.
References
- 1. McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 2007; 131: 121–35. [DOI] [PubMed] [Google Scholar]
- 2. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11: 515–28. [DOI] [PubMed] [Google Scholar]
- 3. Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone‐mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 2004; 5: 781–91. [DOI] [PubMed] [Google Scholar]
- 4. Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem 2008; 283: 18473–7. [DOI] [PubMed] [Google Scholar]
- 5. Panaretou B, Prodromou C, Roe SM et al. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J 1998; 17: 4829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J Cell Biol 1998; 143: 901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hahn JS. The Hsp90 chaperone machinery: from structure to drug development. BMB Rep 2009; 42: 623–30. [DOI] [PubMed] [Google Scholar]
- 8. Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90‐geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 1997; 89: 239–50. [DOI] [PubMed] [Google Scholar]
- 9. Schulte TW, Neckers LM. The benzoquinone ansamycin 17‐allylamino‐17‐demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol 1998; 42: 273–9. [DOI] [PubMed] [Google Scholar]
- 10. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010; 10: 537–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kamal A, Thao L, Sensintaffar J et al. A high‐affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425: 407–10. [DOI] [PubMed] [Google Scholar]
- 12. Kabakov AE, Kudryavtsev VA, Gabai VL. Hsp90 inhibitors as promising agents for radiotherapy. J Mol Med 2010; 88: 241–7. [DOI] [PubMed] [Google Scholar]
- 13. Camphausen K, Tofilon PJ. Inhibition of Hsp90: a multitarget approach to radiosensitization. Clin Cancer Res 2007; 13: 4326–30. [DOI] [PubMed] [Google Scholar]
- 14. Noguchi M, Yu D, Hirayama R et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17‐allylamino‐17‐demethoxygeldanamycin. Biochem Biophys Res Commun 2006; 351: 658–63. [DOI] [PubMed] [Google Scholar]
- 15. Oda T, Hayano T, Miyaso H, Takahashi N, Yamashita T. Hsp90 regulates the Fanconi anemia DNA damage response pathway. Blood 2007; 109: 5016–26. [DOI] [PubMed] [Google Scholar]
- 16. Collins NB, Wilson JB, Bush T et al. ATR‐dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 2009; 113: 2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 2006; 66: 9211–20. [DOI] [PubMed] [Google Scholar]
- 18. Yamashita A, Kashima I, Ohno S. The role of SMG‐1 in nonsense‐mediated mRNA decay. Biochim Biophys Acta 2005; 1754: 305–15. [DOI] [PubMed] [Google Scholar]
- 19. Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S. Human SMG‐1, a novel phosphatidylinositol 3‐kinase‐related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense‐mediated mRNA decay. Genes Dev 2001; 15: 2215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murr R, Vaissiere T, Sawan C, Shukla V, Herceg Z. Orchestration of chromatin‐based processes: mind the TRRAP. Oncogene 2007; 26: 5358–72. [DOI] [PubMed] [Google Scholar]
- 21. Brumbaugh KM, Otterness DM, Geisen C et al. The mRNA surveillance protein hSMG‐1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell 2004; 14: 585–98. [DOI] [PubMed] [Google Scholar]
- 22. Murr R, Loizou JI, Yang YG et al. Histone acetylation by Trrap‐Tip60 modulates loading of repair proteins and repair of DNA double‐strand breaks. Nat Cell Biol 2006; 8: 91–9. [DOI] [PubMed] [Google Scholar]
- 23. Robert F, Hardy S, Nagy Z et al. The transcriptional histone acetyltransferase cofactor TRRAP associates with the MRN repair complex and plays a role in DNA double‐strand break repair. Mol Cell Biol 2006; 26: 402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma XM, Blenis J. Molecular mechanisms of mTOR‐mediated translational control. Nat Rev Mol Cell Biol 2009; 10: 307–18. [DOI] [PubMed] [Google Scholar]
- 25. Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure – diverse function. Genes Cells 2001; 6: 575–97. [DOI] [PubMed] [Google Scholar]
- 26. Jha S, Dutta A. RVB1/RVB2: running rings around molecular biology. Mol Cell 2009; 34: 521–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Izumi N, Yamashita A, Iwamatsu A et al. AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense‐mediated mRNA decay. Sci Signal 2010; 3: ra27. [DOI] [PubMed] [Google Scholar]
- 28. Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K‐related protein kinases. Cell 2007; 131: 1248–59. [DOI] [PubMed] [Google Scholar]
- 29. Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ. HCLK2 is essential for the mammalian S‐phase checkpoint and impacts on Chk1 stability. Nat Cell Biol 2007; 9: 391–401. [DOI] [PubMed] [Google Scholar]
- 30. Anderson CM, Korkin D, Smith DL et al. Tel2 mediates activation and localization of ATM/Tel1 kinase to a double‐strand break. Genes Dev 2008; 22: 854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shikata M, Ishikawa F, Kanoh J. Tel2 is required for activation of the Mrc1‐mediated replication checkpoint. J Biol Chem 2007; 282: 5346–55. [DOI] [PubMed] [Google Scholar]
- 32. Ohji G, Hidayat S, Nakashima A et al. Suppression of the mTOR‐raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J Biochem 2006; 139: 129–35. [DOI] [PubMed] [Google Scholar]
- 33. Kashima I, Yamashita A, Izumi N et al. Binding of a novel SMG‐1‐Upf1‐eRF1‐eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense‐mediated mRNA decay. Genes Dev 2006; 20: 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohnishi T, Yamashita A, Kashima I et al. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG‐5 and hSMG‐7. Mol Cell 2003; 12: 1187–200. [DOI] [PubMed] [Google Scholar]
- 35. Yamashita A, Izumi N, Kashima I et al. SMG‐8 and SMG‐9, two novel subunits of the SMG‐1 complex, regulate remodeling of the mRNA surveillance complex during nonsense‐mediated mRNA decay. Genes Dev 2009; 23: 1091–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem 2003; 278: 52572–7. [DOI] [PubMed] [Google Scholar]
- 37. Hayashi T, Hatanaka M, Nagao K et al. Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 2007; 12: 1357–70. [DOI] [PubMed] [Google Scholar]
- 38. Takai H, Xie Y, de Lange T, Pavletich NP. Tel2 structure and function in the Hsp90‐dependent maturation of mTOR and ATR complexes. Genes Dev 2010; 24: 2019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hurov KE, Cotta‐Ramusino C, Elledge SJ. A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev 2010; 24: 1939–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao R, Davey M, Hsu YC et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 2005; 120: 715–27. [DOI] [PubMed] [Google Scholar]
- 41. Gstaiger M, Luke B, Hess D et al. Control of nutrient‐sensitive transcription programs by the unconventional prefoldin URI. Science 2003; 302: 1208–12. [DOI] [PubMed] [Google Scholar]
- 42. Jeronimo C, Forget D, Bouchard A et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol Cell 2007; 27: 262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boulon S, Marmier‐Gourrier N, Pradet‐Balade B et al. The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J Cell Biol 2008; 180: 579–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woychik NA, Liao SM, Kolodziej PA, Young RA. Subunits shared by eukaryotic nuclear RNA polymerases. Genes Dev 1990; 4: 313–23. [DOI] [PubMed] [Google Scholar]
- 45. Matsuoka S, Ballif BA, Smogorzewska A et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316: 1160–6. [DOI] [PubMed] [Google Scholar]
- 46. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9: 616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lavin MF. Ataxia‐telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008; 9: 759–69. [DOI] [PubMed] [Google Scholar]
- 48. Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA‐PK. Oncogene 2005; 24: 949–61. [DOI] [PubMed] [Google Scholar]
- 49. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8: 193–204. [DOI] [PubMed] [Google Scholar]
- 50. Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense‐mediated mRNA decay. Nature 2010; 465: 227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. El‐Bchiri J, Guilloux A, Dartigues P et al. Nonsense‐mediated mRNA decay impacts MSI‐driven carcinogenesis and anti‐tumor immunity in colorectal cancers. PLoS ONE 2008; 3: e2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene 2006; 25: 6436–46. [DOI] [PubMed] [Google Scholar]
- 53. Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17‐allylamino‐17‐demethoxygeldanamycin. Cancer Res 2008; 68: 1188–97. [DOI] [PubMed] [Google Scholar]
- 54. Gray PJ Jr, Stevenson MA, Calderwood SK. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res 2007; 67: 11942–50. [DOI] [PubMed] [Google Scholar]
- 55. Millson SH, Vaughan CK, Zhai C et al. Chaperone ligand‐discrimination by the TPR‐domain protein Tah1. Biochem J 2008; 413: 261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Eckert K, Saliou JM, Monlezun L et al. The Pih1‐Tah1 cochaperone complex inhibits Hsp90 molecular chaperone ATPase activity. J Biol Chem 2010; 285: 31304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horejsi Z, Takai H, Adelman CA et al. CK2 phospho‐dependent binding of R2TP complex to TEL2 is essential for mTOR and SMG1 stability. Mol Cell 2010; 39: 839–50. [DOI] [PubMed] [Google Scholar]
- 58. Zhao R, Kakihara Y, Gribun A et al. Molecular chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates snoRNA accumulation. J Cell Biol 2008; 180: 563–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Holt SE, Aisner DL, Baur J et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev 1999; 13: 817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE. Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 2008; 132: 945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boulon S, Pradet‐Balade B, Verheggen C et al. HSP90 and its R2TP/Prefoldin‐like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol Cell 2010; 39: 912–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Falsone SF, Gesslbauer B, Tirk F, Piccinini AM, Kungl AJ. A proteomic snapshot of the human heat shock protein 90 interactome. FEBS Lett 2005; 579: 6350–4. [DOI] [PubMed] [Google Scholar]
- 63. Peng Y, Woods RG, Beamish H et al. Deficiency in the catalytic subunit of DNA‐dependent protein kinase causes down‐regulation of ATM. Cancer Res 2005; 65: 1670–7. [DOI] [PubMed] [Google Scholar]
- 64. Kaizuka T, Hara T, Oshiro N et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem 2010; 285: 20109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rendtlew Danielsen JM, Larsen DH, Schou KB et al. HCLK2 is required for activity of the DNA damage response kinase ATR. J Biol Chem 2009; 284: 4140–7. [DOI] [PubMed] [Google Scholar]