

Abstract
Pim‐3, a proto‐oncogene with serine/threonine kinase activity, is aberrantly expressed in malignant lesions, but not in normal tissues, of endoderm‐derived organs, including the pancreas, liver, colon, and stomach. Furthermore, the development of hepatocellular carcinoma is accelerated in mice expressing Pim‐3 transgene selectively in the liver when these mice are treated with a hepatocarcinogen. These observations suggest that a chemical targeting Pim‐3 kinase may be a novel type of anticancer drug. In the present study, we screened low molecular weight chemicals and observed that the phenanthrene derivative T26 potently inhibited Pim‐3 and Pim‐1, but only weakly inhibited Pim‐2. Moreover, T26 markedly inhibited the in vitro growth of human pancreatic cancer cell lines by inducing apoptosis and G2/M arrest. The growth inhibitory effects of T26 were reversed by overexpression of Pim‐3 cDNA in human pancreatic cancer cells, indicating that T26 acts primarily on Pim‐3. Furthermore, T26 inhibited the growth of a human pancreatic cancer cell line in nude mice without causing apparent adverse effects when it was administered after tumor formation was evident. These observations imply that the chemical and its related compounds may be effective for the treatment of cancers in which there is aberrant Pim‐3 expression. (Cancer Sci 2012; 103: 107–115)
Deaths from pancreatic cancer rank fourth among cancer‐related deaths in the US. Most patients are diagnosed at an advanced stage with locally advanced, unresectable, or metastatic disease and are treated with conventional chemotherapy, radiation, or combinations of the two.( 1 ) Existing conventional chemotherapies are more or less non‐selective cytotoxic drugs with systemic adverse effects. Moreover, pancreatic cancer is frequently resistant to conventional chemotherapy and radiotherapy.( 2 ) Thus, an urgent need exists to develop novel treatments for pancreatic cancer based on an understanding of pancreatic carcinogenesis at the molecular level.
Pim‐3 is a member of the proto‐oncogene Pim family that exhibits serine/threonine kinase activity. It was originally identified as a depolarization‐induced gene, namely KID‐1, in the rat pheochromocytoma cell line PC12, but was soon renamed as Pim‐3 owing to its similarity with other Pim family members.( 3 ) Subsequently, Deneen et al. have demonstrated that Pim‐3 gene transcription is enhanced in the Ewing sarcoma breakpoint region/E26 transformation‐specific (EWS/ETS)‐induced malignant transformation of NIH 3T3 cells,( 4 ) suggesting the involvement of Pim‐3 in tumorigenesis. This notion was further substantiated by our observations that the development of hepatocellular carcinoma was accelerated in mice expressing Pim‐3 transgene selectively in the liver after these mice had been treated with a hepatocarcinogen.( 5 ) Moreover, we demonstrated that Pim‐3 expression was enhanced in malignant lesions, but not in normal tissues, of endoderm‐derived organs, including the liver,( 6 ) pancreas,( 7 ) colon,( 8 ) and stomach.( 9 ) Furthermore, we demonstrated that Pim‐3 can inactivate Bad in the human pancreas and colon carcinoma cell lines by phosphorylating Ser112, but not Ser136, ultimately promoting the survival of these cells,( 7 , 8 ) similar to reported observations for Pim‐1( 10 ) and Pim‐2.( 11 ) Because Pim‐3 is selectively enhanced in malignant lesions of endoderm‐derived organs( 6 , 7 , 8 , 9 ) and has potent anti‐apoptotic activity, it may be a novel target for the treatment of cancer of endoderm‐derived organs, particularly the pancreas.
In a previous study in which we conducted a random screening of low molecular weight chemicals, we identified a stemonamide synthetic intermediate, namely T‐2, as a chemical that can inhibit Pim kinases, as well as the in vitro and in vivo growth of a human pancreatic cancer cell line.( 12 ) However, T‐2 inhibited all Pim family members and markedly decreased circulating leukocyte numbers when given to tumor‐bearing animals. Hence, we performed further screening of low molecular weight chemicals in the present study and found that a phenanthrene derivative, namely T26, potently inhibited Pim‐3 and Pim‐1, but only weakly inhibited Pim‐2, and markedly retarded the growth of a human pancreatic cancer cell line in nude mice without causing apparent adverse effects following its administration to mice after tumor formation was evident. These observations imply that the chemical and its related compounds may be effective for the treatment of cancers in which there is aberrant Pim‐3 expression.
Materials and Methods
Cell lines and antibodies. The human pancreatic cancer cell lines PCI35,( 13 ) PCI55,( 13 ) PCI66,( 13 ) PANC‐1,( 14 ) and MiaPaca‐2( 15 ) were cultured in RPMI 1640 medium (Sigma‐Aldrich, St Louis, MO, USA), whereas L3.6pl cells( 16 ) were maintained in minimum essential medium (Invitrogen, Carlsbad, CA, USA). All media were supplemented with 10% FBS (BioWest, Nuaille, France), 50 U/mL penicillin and 50 U/mL streptomycin (Sigma‐Aldrich). The following antibodies were used for western blotting analysis: mouse anti‐human Bad (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit anti‐phosphorylated (p‐) Ser112‐Bad, anti‐caspase‐3, anti‐cleaved caspase‐3, anti‐Cdc25c, anti‐p‐Ser216‐Cdc25c, anti‐cleaved caspase‐9 (Asp330; Cell Signaling Technology, Beverly, MA, USA), anti‐Bcl‐XL (MBL, Nagoya, Japan), anti‐β‐actin antibodies (Sigma‐Aldrich), and mouse anti‐cytochrome c antibodies (BD Biosciences, San Jose, CA, USA).
Preparation of phenanthrene derivatives. Phenanthrene derivatives were synthesized according to procedures reported previously.( 17 , 18 ) The original compound, No. 5, inhibited Pim kinases but exhibited poor water solubility. To improve water solubility, compound No. 5 was modified to generate compounds T19, T19–1, T19–2, and T26 (Fig. 1a). These chemicals were dissolved in 100% dimethyl sulfoxide (DMSO; Sigma‐Aldrich) to a concentration of 100 mmol/L. The chemicals were stable for up to 1 week when dissolved in DMSO.
Figure 1.
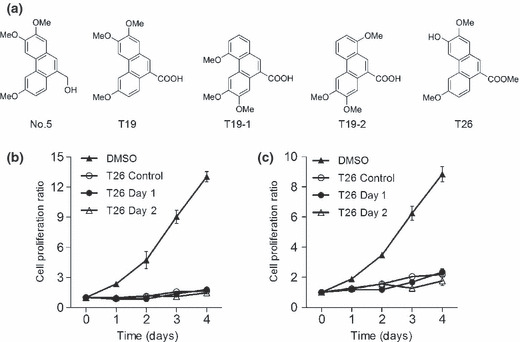
(a) Chemical structure of the phenanthrene derivatives. (b,c) Effects of T26 on in vitro cell proliferation of MiaPaca‐2 (b) and PCI66 cells (c). MiaPaca‐2 and PCI 66 cells (3 × 103 cells/100 μL) were added to each well of 96‐multiwell culture plates and incubated at 37°C for 18 h before T26 at a final concentration of 25 μmol/L was added to the plates. The culture supernatant was replaced with fresh medium 1 or 2 days after the addition of T26. As controls, cells were incubated either in the absence of T26 (0.1% dimethyl sulfoxide; DMSO) or in the presence of T26 (T26 control) for the entire 4 days of culture. Cell proliferation was measured and the ratio of viable cells was determined by comparing the number of viable cells at each time point with the number of viable cells on Day 0.
Assay of Pim kinase activity in a cell‐free system. In vitro kinase assay of Pim‐1, Pim‐2, and Pim‐3 was evaluated as described previously.( 12 ) IC50 values of all three kinases were calculated using logistic regression.
Assay of Akt kinase activity in a cell‐free system. Akt kinase activity was determined based on the capacity of the kinase to phosphorylate Bad at Ser136, as described previously.( 12 ) Either recombinant Akt1 (10 ng; Cell Signaling Technology) or Akt2 (10 ng; Assay Designs, Ann Arbor, MI, USA) were used as the source of the enzyme. The AKT1/2 kinase inhibitor (Sigma‐Aldrich) was used as a control inhibitor.
Assays of platelet‐derived growth factor receptor β chain and protein kinase A kinase in a cell‐free system. Platelet‐derived growth factor receptor (PDGFR) β kinase (Cell Signaling Technology) and protein kinase A (PKA) kinase assays (Assay Designs) were performed according to the manufacturers’ instructions in the presence of either staurosporine (Sigma‐Aldrich) or the chemicals under investigation.
In vitro cell proliferation. Cell suspensions (3 × 103 cells/100 μL) were added to each well of 96‐multiwell culture plates (BD Biosciences) and incubated at 37°C for 18 h. Then, different concentrations of the chemicals (2.5–100 μmol/L) were added to each well and cells were further incubated for specific periods of time (1–4 days) to measure cell proliferation, essentially as described previously.( 12 ) In some experiments, the culture supernatants were replaced with fresh medium at specified times for four days after the addition of the chemical to remove the chemical. The ratio of viable cells at each time point was determined by comparing the number of viable cells at that time to the number of viable cells on Day 0. IC50 values were calculated using logistic regression.
In another series of experiments, MiaPaca‐2 or PCI66 cells in 6‐cm dishes were transfected with pcDNA4 vector (Invitrogen) with or without human full‐length Pim‐3 or Pim‐1 cDNA. Then, 36–40 h after transfection cells were trypsinized and 1 × 103 cells were plated in a 96‐well plate in the presence of 15 μml/L of T26 to determine cell viability one and two days after T26 treatment. In addition, cell aliquots were processed for immunoblotting with anti‐Pim‐3 or anti‐Pim‐1 antibodies to estimate the amount of Pim‐3 or Pim‐1 protein.( 7 )
Transfection of Pim‐3 shRNA. Pim‐3 or control shRNA vector was transfected into PCI66 cells using a jetPrime kit (Funakoshi, Tokyo, Japan) according to the manufacturer’s instructions.
Cell cycle analysis. Cell suspensions (5 × 105/5 mL) were incubated in 6‐cm plates at 37°C for 18 h. Then, cells were treated with different concentrations of T26 (5–25 μmol/L) and were further incubated for 24 h. After incubation, both buoyant and adherent cells were harvested and fixed with 70% ethanol at 20°C. Fixed cells were incubated with 50 μg/mL propidium iodide (PI; Molecular Probes, Eugene, OR, USA) and 5 μg/mL RNase A for 30 min at 37°C. The DNA content was then analyzed on a FACS Canto II system (BD Biosciences) using Cell Quest analysis software (BD Biosciences). The shRNA‐transfected cells were similarly treated and subjected to cell cycle analysis.
Immunofluorescence analysis of mitotic cells. After incubation with 20 μmol/L T26 or 0.1% DMSO for 24 h, cells were cytospun onto a glass slide. Cells were air dried, fixed with 4% paraformaldehyde/PBS for 15 min, and made permeable by treatment with 0.2% Triton X‐100/PBS for 5 min at room temperature. Slides were incubated with 5% donkey serum in 1% BSA/0.2% Triton X‐100/PBS for a further 30 min. Thereafter, slides were incubated with a combination of goat anti‐β‐tubulin and mouse anti‐p‐histone H3Ser10 antibodies overnight at 4°C, followed by incubation with a combination of Alexa Fluor 488‐labeled donkey anti‐goat IgG and Alexa Fluor 594‐labeled donkey anti‐mouse IgG for 1 h at room temperature. Coverslips were mounted using mounting media containing DAPI (Vector Laboratories, Burlingmae, CA, USA). Cells were observed using a confocal microscope (LSM510; Carl Zeiss, Carlsberg, Germany) equipped with a 63× NA1.4 objective lens and Zen 2008 software (Carl Zeiss). At least 40 mitotic cells were observed on each slide and were classified into each phase of mitosis using criteria described previously.( 19 )
Analysis of cell apoptosis. Following treatment with 20 μmol/L T26 for 24–72 h, cells were harvested and were stained using a human annexin V‐FITC apoptosis kit (Bender MedSystem, Vienna, Austria) and PI, as described previously.( 12 ) At least 2 × 104 stained cells were analyzed using the FACS Canto II system (BD Biosciences) for each determination.
Western blotting analysis. For western blotting analysis, 1.5 × 106 of MiaPaca‐2 or PCI66 cells were dispensed into a 100‐mm culture plate and incubated for 18 h before 20 μmol/L T26 was added to the plate. After a further 12–72 h incubation in the presence of T26, both buoyant and adherent cells were suspended 60 μl of Cell Lytic Cell Lysis Reagent (Sigma‐Aldrich) to obtain supernatants for immunoblotting, as described previously.( 12 ) Anti‐β‐actin antibodies were used to confirm that equal amounts of proteins were used for analysis. Signals were detected by using an LAS‐4000 mini CDD camera (GE Healthcare Japan, Hino, Japan). In addition, shRNA‐transfected cells were similarly treated and subjected to western blotting analysis.
Animal experiments. PCI66 cells were suspended in HBSS at a concentration of 2 × 107 cells/mL and 100‐μL cell suspensions were injected subcutaneously into the back of BALB/c nu/nu mice (SLC, Shizuoka, Japan). Fourteen days after injection, either 20 mg/kg T26 or olive oil vehicle was injected intraperitoneally once a day for 5 days, followed by 2 days of cessation. This cycle was repeated three times. Tumor size was measured every 3–4 days using calipers, whereas tumor volume was calculated as follows:
Thirty‐six days after tumor cell injection, tumors were removed and blood samples were collected simultaneously. Serum levels of aspartate aminotransferase (ALT), alanine aminotransferase (AST), creatinine, and blood urea nitrogen (BUN), as well as blood glucose levels, were measured using Fuji DRICHEM 5500V (Fuji Medical System, Tokyo, Japan), whereas blood counts were determined using an automated blood counter (XE‐2100; Sysmex, Kobe, Japan). Tumor tissues were subjected to immunohistochemical analysis using anti‐proliferating cell nuclear antigen (PCNA; BD Biosciences), anti‐cleaved caspase‐3 (Cell Signaling Technology), or anti‐CD31 antibodies (BD Biosciences) to determine the number of PCNA‐ or cleaved caspase‐3‐positive cells and CD31‐postive vascular areas, as described previously.( 5 ) All animal experiments were performed in compliance with the Guidelines for the Care and Use of Laboratory Animals of Kanazawa University.
Statistical analysis. Unless indicated otherwise, data are expressed as the mean ± SD. Differences between mean values were analyzed using one‐way anova, followed by the Tukey‐Kramer test. P < 0.05 was considered significant.
Results
Effects of phenanthrene derivatives on Pim kinase activity in cell‐free system. Random screening identified a phenanthrene derivative, compound No. 5, as a chemical that inhibited all Pim kinases (Table 1). However, low water solubility of No. 5 prompted us to synthesize additional chemicals by modifying the original compound No. 5 (Fig. 1a). Among the new chemicals synthesized, T19–1 and T26 inhibited Pim‐3 and Pim‐1 with a similar potency that was almost fivefold greater than their potency against Pim‐2 activity (Table 1). Because another serine/threonine kinase, namely Akt/protein kinase B (PKB), can phosphorylate a similar set of proteins, including Bad,( 20 , 21 ) we examined the effects of T19–1 and T26 on human Akt/PKB. Approximately 10‐fold higher concentrations of T19–1 and T26 were required to produce a 50% inhibition of Akt1 and Akt2 compared with the specific Akt1 and Akt2 inhibitor AKT1/2 (Table 1). Moreover, an approximately 20‐fold higher concentration of T26 was required to inhibit PKA and PDGFRβ by 50% compared with staurosporine (Table 1).
Table 1.
Inhibitory effects of phenanthrene derivatives on Pim, Akt, protein kinase A, and platelet‐derived growth factor receptor β kinases
| Chemical | IC50 (μmol/L) | ||||||
|---|---|---|---|---|---|---|---|
| Pim‐3 | Pim‐1 | Pim‐2 | Akt‐1 | Akt‐2 | PKA | PDGFRβ | |
| No. 5 | 78.2 | 99.6 | 109.2 | ND | ND | ND | ND |
| T19 | 62.6 | 151.2 | >160 | ND | ND | ND | ND |
| T19‐1 | 1.5 | 3.2 | 164.6 | >320 | >320 | >320 | 213.9 |
| T19‐2 | 74.7 | >320 | >320 | ND | ND | ND | ND |
| T26 | 27.4 | 32.4 | 148.7 | 114.4 | 177.2 | 44.2 | 104.6 |
| Akt1/2 | ND | ND | ND | 10.2 | 29.6 | ND | ND |
| Staurosporine | ND | ND | ND | ND | ND | 2.6 | 0.008 |
Inhibitory effects were investigated up to chemical concentrations of 320 mmol/L and IC50 values were calculated for each of the chemicals.
PKA, protein kinase A; PDGFRβ, platelet‐derived growth factor receptor β; ND, not determined.
Effects of phenanthrene derivatives on in vitro cell proliferation of human pancreatic cancer cell lines. We next examined the effects of phenanthrene derivatives on in vitro cell proliferation of human pancreatic cancer cell lines. T26 efficiently inhibited the in vitro cell proliferation of the human pancreatic cancer cell lines PCI35, PCI55, PCI66, MiaPaca‐2, PANC‐1, and L3.6pl (Table 2), whereas T19–1 and T19–2 failed to do so up to concentrations of 100 μmol/L (data not shown). This could be due to T26 being able to penetrate the cells more efficiently than T19–1 and T19–2. When the cells were incubated with T26 for longer than 24 h, the cells did not regrow again (Fig. 1b,c). Moreover, overexpression of Pim‐3, but not Pim‐1, cDNA reduced the inhibitory effects of T26 on the in vitro cell proliferation of MiaPaca‐2 and PCI66 cells (Fig. 2). Western blotting analysis detected Pim‐3 but not Pim‐1 protein in these pancreatic cancer cell lines (data not shown), consistent with previous observations.( 12 ) Collectively, the data indicate that T26‐induced reductions in cell proliferation may arise, at least in part, from its inhibitory effect on Pim‐3 but not Pim‐1.
Table 2.
Effects of T26 on the in vitro proliferation of human pancreatic cancer cell lines
| Cell line | IC50 (μmol/L) |
|---|---|
| MiaPaca‐2 | 9.1 |
| PCI66 | 17.6 |
| PCI35 | 9.8 |
| PCI55 | 10.3 |
| PANC‐1 | 33.6 |
| L3.6pl | 39 |
IC50 values were determined described in the Materials and Methods.
Figure 2.
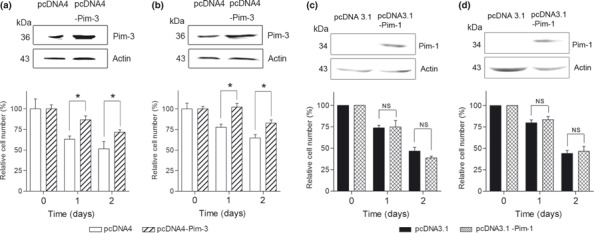
Effects of the forced overexpression of (a,b) Pim‐3 and (c,d) Pim‐1 on cell numbers of MiaPaca‐2 (a,c) and PCI66 (b,d) cells in the presence of T26. Cells were transfected with Pim‐3 or Pim‐1 cDNA or control vectors as described in the Materials and Methods. After transfection, cells were incubated in the presence of 15 μmol/L T26 for the times indicated. Cell numbers were determined and cell lysates were subjected to immunoblotting with anti‐Pim‐3, anti‐Pim‐3, or anti‐β‐actin antibodies, as indicated. Relative cell numbers were determined compared with Day 0 and data are shown as the mean ± SD. Experiments were repeated three times. *P < 0.01.
Effects of T26 on cell cycle progression and apoptotic processes. Because Pim‐3 can promote cell cycle progression by phosphorylating the molecules involved in these processes,( 22 ) we examined the effects of T26 on cell cycle progression. Treatment with T26 over the concentration range 10–25 μmol/L dose‐dependently increased the proportion of the cells present in the G2/M phase and decreased the number in the G0/G1 phase in two human pancreatic cancer cell lines, namely MiaPaca‐2 and PCI66 cells (Fig. 3a–c). Moreover, immunofluorescence analysis of mitotic cells revealed that T26‐treated cells were either in prometaphase or metaphase (Table 3). These observations indicate that T26 induces G2/M arrest, particularly at metaphase. Cdc25C regulates G2/M entry and its inactivation by phosphorylation results in G2/M arrest.( 23 ) T26 treatment increased the amount of phosphorylated Cdc25C and decreased the amount of total Cdc25C (Fig. 4). Similarly, Pim‐3 shRNA increased the amount of phosphorylated Cdc25c and decreased the amount of total Cdc25c (Fig. 5a). Consistent with these observations, Pim‐3 shRNA increased the proportion of cells present in the G2/M phase and decreased the number of cells in the G0/G1 phase (Fig. 5b). Cell cycle analysis revealed that T26 treatment increased the proportion of the cells in sub‐G1 phase (Fig. 3), suggesting that apoptosis was also accelerated by T26 treatment. Indeed, T26 increased the proportion of both early apoptotic (annexin V‐positive and PI‐negative) and late apoptotic (annexin V‐positive and PI‐positive) cells 24 h after the treatment (Fig. 6). Consistent with previous observations,( 7 ) Bad was constitutively phosphorylated at Ser112 in both cell lines (Fig. 4), but not at Ser136 or Ser155 (data not shown). T26 diminished the amount of p‐Ser112‐Bad in these cell lines 24 h after the treatment, but had no effect on the amount of total Bad and Pim‐3 (Fig. 4). Concomitantly, T26 decreased Bcl‐XL, but increased cytochrome c and cleaved caspase‐3 with little effect on total caspase‐3 after 24 h treatment (Fig. 4). Collectively, these observations suggest that T26 can prevent the phosphorylation of Bad, decrease Bcl‐XL, and eventually induce apoptosis and that these effects are dependent on cytochrome c and caspase‐3.
Figure 3.
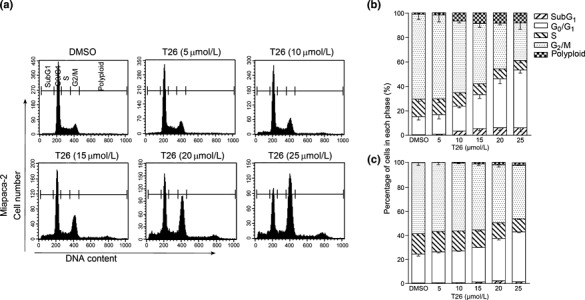
Effects of T26 on cell cycle progression of human pancreatic cancer cell lines. MiaPaca‐2 cells (a,b) or PCI66 cells (c) were incubated with the indicated concentrations of T26 for 24 h, after which cells were subjected to cell cycle analysis. (a) Representative results from three independent experiments are shown. (b,c) Data are expressed as the mean ± 1SD (n = 5). The cells were exposed to 0.1% dimethyl sulfoxide (DMSO).
Table 3.
Effects of T26 (20 μmo/L) on mitotic cells
| Proportion of cells in each phase of mitosis (%) | ||||
|---|---|---|---|---|
| MiaPaca‐2 cells | PCI66 cells | |||
| Control | T26 | Control | T26 | |
| Prophase | 15.6 ± 0.6 | 0 | 14.5 ± 2.0 | 0 |
| Prometaphase | 27.3 ± 0.7 | 57.7 ± 1.9 | 15.9 ± 1.9 | 36.0 ± 2.3 |
| Metaphase | 17.6 ± 1.2 | 42.3 ± 1.3 | 26.7 ± 0.4 | 64.0 ± 1.4 |
| Anaphase | 8.5 ± 0.3 | 0 | 7.3 ± 1.3 | 0 |
| Telophase | 7.3 ± 1.4 | 0 | 8.3 ± 1.8 | 0 |
| Cytokinesis | 23.6 ± 0.7 | 0 | 27.4 ± 2.2 | 0 |
At least 40 mitotic cells were evaluated each time (see Materials and Methods).Data show the mean ± SD calculated from three independent experiment.
Figure 4.
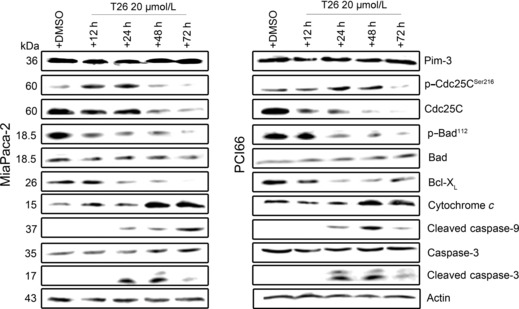
Effects of 20 μmol/L T26 on the molecules involved in cell cycle progression or apoptosis. MiaPaca‐2 and PCI66 cells were incubated in the presence of T26 or 0.1% dimethyl sulfoxide (DMSO) for the times indicated before cell lysates were obtained and subjected to western blotting analysis. Representative results from three independent experiments are shown.
Figure 5.
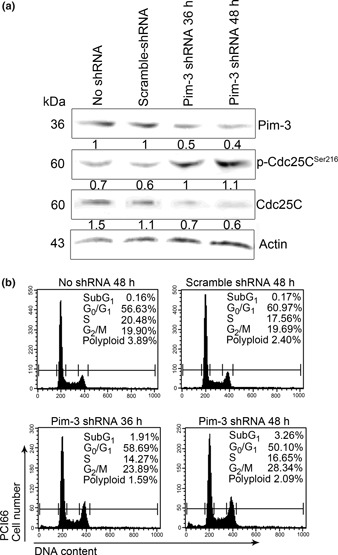
(a) Effects of Pim‐3 shRNA treatment on Cdc25c. PCI66 cells were treated with Pim‐3 shRNA and cell lysates were obtained at the times indicated for western blot analysis. (b) Effects of Pim‐3 shRNA treatment on cell cycle progression. PCI66 cells treated with Pim‐3 shRNA were subjected to cell cycle analysis at the times indicated. Representative results from three independent experiments are shown.
Figure 6.
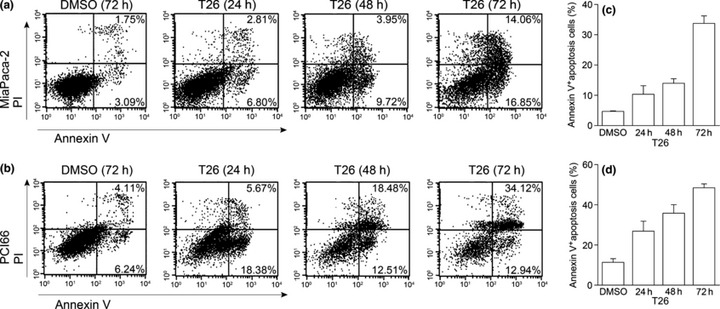
Effects of 20 μmol/L T26 on the apoptotic process of human pancreatic cancer cell lines. MiaPaca‐2 (a,c) or PCI66 cells (b,d) were incubated with T26 or 0.1% dimethyl sulfoxide (DMSO) for the times indicated. The proportion of apoptotic cells was determined as described in the Materials and Methods. Representative results from five independent experiments are shown. (c,d) The proportion of annexin V‐positive cells was determined and is shown as the mean ± 1SD (n = 5).
Effects of T26 on tumor growth in vivo. Finally, we examined the effects of T26 on tumor growth in vivo. In the present study, mice were treated with intraperitoneal injections of T26 starting 14 days after the injection of tumor cells, when the tumors became palpable. Vehicle‐treated animals exhibited progressive tumor growth, whereas T26 treatment markedly retarded tumor growth (Fig. 7a). The tumor disappeared in one of seven mice receiving T26 treatment. Moreover, T26 significantly decreased the number of PCNA‐positive proliferating cells (Fig. 7b,c) and increased the number of cleaved caspase‐3‐positive cells in the tumor (Fig. 7d,e). Furthermore, CD31‐positive vascular areas were significantly reduced by T26 treatment compared with vehicle treatment (Fig. 7f,g). All mice tolerated T26 well, as evidenced by the absence of any loss in appetite (data not shown) or body weight (Fig. S1a). Treatment with T26 had no effect on serum levels of ALT, AST, BUN, and creatinine or on blood glucose levels (Fig. S1b–f) and did not significantly decrease erythrocyte, platelet, and leukocyte numbers (Fig. S1g–i). Thus, T26 treatment retarded tumor growth in vivo by reducing cell proliferation and augmenting the apoptosis of tumor cells without having serious adverse effects on liver and renal function or on the blood count.
Figure 7.
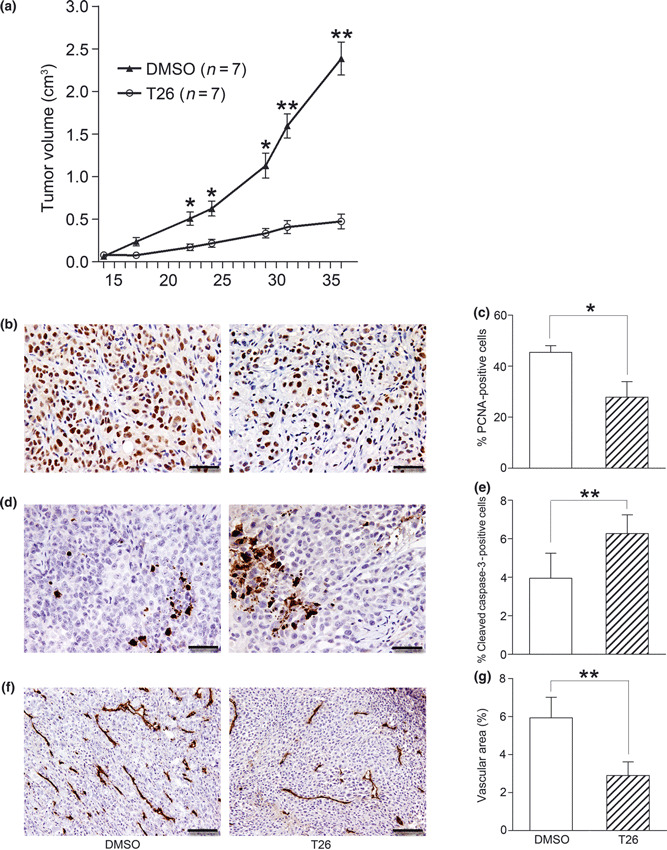
Effects of T26 on in vivo tumor growth. Fourteen days after nude mice had been injected with 2 × 106 PCI66 cells, T26 treatment was initiated as described in the Materials and Methods. (a) Tumor sizes were determined twice a week and are shown here as the mean ± SEM (n = 7). *P < 0.05, **P < 0.01 compared with 0.1% dimethyl sulfoxide (DMSO) control. (b–g) Tumor tissues obtained 36 days after injection of tumor cells were subjected to immunohistochemical analysis using (b,c) anti‐proliferating cell nuclear antigen (PCNA), (d,e) anti‐cleaved caspase‐3, and (f,g) anti‐CD31 antibody. (b,d,f) Representative results from three mice are shown. Bars, 50 μm (b,d); 100 μm (f). (c,e,g) The number of PCNA‐ (c) or cleaved caspase‐3‐positive cells (e) was determined and CD31‐positive areas (g) were measured in five randomly chosen visual fields at a magnification of ×100 using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Data are shown as the mean ± SD (n = 4). *P < 0.05, **P < 0.01.
Discussion
Pim‐3, a proto‐oncogene with serine/threonine kinase activity, is aberrantly expressed in various malignant lesions and inhibition of Pim‐3 expression can cause apoptosis of human pancreatic and colon cancer cells by inactivating the pro‐apoptotic molecule Bad.( 7 , 8 ) Hence, we instigated a search for chemicals that could inhibit Pim‐3 for the treatment of pancreatic cancer and identified the phenanthrene derivative T26 as a Pim‐3 inhibitor, which is totally different from other Pim kinase reported previously. An Akt inhibitor can inhibit Akt kinase activity in a cell‐free system and Akt‐mediated intracellular signaling pathways at micromolar concentrations.( 24 ) Similarly, T26 inhibited both Pim‐3 kinase activity in a cell‐free system and in vitro cell proliferation of human pancreatic cancer cell lines at micromolar concentrations, although most kinase inhibitors inhibit kinase activity in a cell‐free system at nanomolar concentrations. Knockdown of Pim‐3 kinase reversed the changes caused by T26, whereas forced expression of the Pim‐3 gene reversed T26‐induced inhibition of cell proliferation. The Pim‐1 protein was not detected in the human pancreatic cancer cell lines we examined in the present study; thus, T26 inhibits the in vitro proliferation of cancer cells by inhibiting Pim‐3.
We recently demonstrated that the stemonamide intermediate T‐2 can inhibit Pim‐3 activity in a cell‐free system and pancreatic cancer cell growth in vitro and in nude mice.( 12 ) In the present study, T26 inhibited Pim‐3 and Pim‐1 with a similar potency, but only weakly inhibited Pim‐2 activity, in contrast with T‐2, which inhibited these three kinases with a similar potency. Moreover, T26 promoted apoptosis of human pancreatic cancer cells, together with reduced phosphorylation of Bad, enhanced expression of cytochrome c, and enhanced generation of cleaved caspase‐3, as observed for T‐2. T26 inhibited human pancreatic cancer cell growth in nude mice more efficiently than T‐2 and the tumor disappeared in one of seven mice treated with T26. Finally, T26 did not produce any severe adverse effects. Thus, T26 may be a good lead compound in the development of molecular targeting drugs for the treatment of pancreatic cancer with enhanced Pim‐3 kinase activity.( 7 )
In transgenic mice that express human Pim‐3 cDNA selectively in the liver, cell cycle progression in hepatocytes is accelerated,( 5 ) suggesting a potential role for Pim‐3 in cell cycle progression. Indeed, all Pim kinase members can bind to and phosphorylate the CDK inhibitor p27 at its threonine residues and induce the binding of p27 to 14‐3‐3 protein, resulting in its nuclear export and proteasome‐dependent degradation.( 25 ) Moreover, Pim‐1 can promote cell cycle progression by phosphorylating and modulating the functions of the molecules involved in cell cycle progression, particularly those involved in G2/M phase transition, such as Cdc25A, cyclin D1‐associated kinases,( 26 ) Cdc25C‐associated kinase 1 (C‐TAK1),( 27 ) and p21.( 28 ) Given a high sequence identity between Pim‐1 and Pim‐3 in their kinase domains, Pim‐3 can also modulate these molecules. Thus, the inhibitory effects of T26 on cell cycle progression may be ascribed to its effects on Pim‐3. This notion is further supported by our observations that Pim‐1 protein was not detected in the human pancreatic cancer cell lines we used in the present study.
Several groups have claimed that another serine/threonine kinase, namely Akt, may be a good target molecule for the treatment of various cancers because Akt/PKB protein kinases can phosphorylate a similar set of proteins, including Bad, as Pim kinases and eventually promote the growth and survival of tumor cells.( 21 ) Indeed, the Akt inhibitor GSK690693 exhibited potent antitumor activity in preclinical animal experiments.( 21 ) The genetic disruption of each Akt kinase gene results in severe phenotypic changes, such as neonatal mortality, severe growth retardation, and reduced brain size,( 29 , 30 , 31 ) and Akt2 inhibition induces severe hyperglycemia.( 21 ) This may preclude the use of Akt inhibitors for the treatment of pancreatic cancer, which is often complicated by hyperglycemia.( 2 ) In contrast, T26 decreased tumor growth without inducing hyperglycemia. Thus, cancer patients with hyperglycemia may tolerate a Pim‐3 inhibitor better than an Akt inhibitor.
The crystal structure of Pim‐3 has not yet been established, but those of Pim‐1 and Pim‐2, which show high sequence similarity with Pim‐3, have been reported.( 32 , 33 , 34 ) These studies revealed the presence of a unique hinge region that connects the two lobes of the protein kinase domain. As a result, ATP binds to Pim kinases in fundamentally different ways from how it binds to other protein kinases.( 32 , 33 ) Thus, it may be feasible to develop a compound that selectively inhibits Pim kinases and not other serine/threonine kinases.( 35 ) Indeed, several groups have reported small‐molecule inhibitors acting selectively on Pim kinases, including flavonol quercetargetin,( 36 ) imidazole[1,2‐b]pyridazines,( 37 , 38 ) bezylindene‐thiazolidine‐2,4‐dione,( 39 , 40 ) pyrrolo[2,3‐a]carbazole,( 41 ) and stemonamide synthetic intermediates.( 12 ) Herein, we add the phenanthrene derivative T26 to this list.
A deficiency in the Pim‐1 gene specifically reduces the interleukin‐3‐dependent growth of bone marrow‐derived mast cells,( 42 ) whereas loss of the Pim‐2 gene specifically impairs the growth and survival of T lymphocytes in the presence of the mammalian target of rapamycin (mTOR) inhibitor rapamycin.( 43 ) Pim‐3, but not Pim‐1 or Pim‐2, can increase protein synthesis by inhibiting AMP‐dependent protein kinase.( 44 ) Together, these observations suggest that each Pim kinase plays a non‐redundant role in various conditions. This assumption may account for our observations that forced expression of Pim‐3, but not Pim‐1, reversed the growth inhibitory effects of T26. Thus, a specific inhibitor against each Pim family member may be required. Several Pim kinase inhibitors potently inhibit Pim‐1 but only weakly inhibit Pim‐2;( 36 , 37 , 41 ) of these inhibitors, only the effects of pyrrolo[2,3‐a]carbazole on Pim‐3 have been examined and it was shown that this compound inhibits Pim‐3 and Pim‐1 with a comparable potency.( 41 ) Given an extraordinarily similar peptide substrate identity between Pim‐1 and Pim‐3,( 32 ) it may be difficult to obtain an inhibitor that is specific for either Pim‐1 or Pim‐3 kinase. Hence, it remains to be investigated whether a specific inhibitor against each Pim family member will provide any therapeutic advantage over a multi‐Pim kinase inhibitor.
Disclosure
The authors declare no financial conflicts of interest.
Supporting information
Fig. S1. Effects of T26 treatment on various parameters in tumor‐bearing mice.
Supporting info item
Acknowledgments
This work was supported, in part, by the Japan Science and Technology Agency (A‐STEP). The authors thank Dr Takayoshi Okabe (University of Tokyo, Tokyo, Japan) for his invaluable advice.
References
- 1. Van Cutsem E, Aerts R, Haustermans K, Topal B, Van Steenbergen W, Verslype C. Systemic treatment of pancreatic cancer. Eur J Gastroenterol Hepatol 2004; 16: 265–74. [DOI] [PubMed] [Google Scholar]
- 2. Hidalgo M. Pancreactic cancer. N Engl J Med 2010; 362: 1605–17. [DOI] [PubMed] [Google Scholar]
- 3. Konietzko U, Kauselmann G, Scafidi J et al. Pim kinase expression is induced by LTP stimulation and required for the consolidation of enduring LTP. EMBO J 1999; 18: 3359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deneen B, Welford SM, Ho T, Hernandez F, Kurland I, Denny CT. PIM3 proto‐oncogene kinase is a common transcriptional target of divergent EWS/ETS oncoproteins. Mol Cell Biol 2003; 23: 3897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu Y, Wang YY, Nakamoto Y et al. Accelerated hepatocellular carcinoma development in mice expressing the Pim‐3 transgene selectively in the liver. Oncogene 2010; 29: 2228–37. [DOI] [PubMed] [Google Scholar]
- 6. Fujii C, Nakamoto Y, Lu P et al. Aberrant expression of serine/threonine kinase Pim‐3 in hepatocellular carcinoma development and its role in the proliferation of human hepatoma cell lines. Int J Cancer 2005; 114: 209–18. [DOI] [PubMed] [Google Scholar]
- 7. Li YY, Popivanova BK, Nagai Y et al. Pim‐3, a proto‐oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates Bad to block Bad‐mediated apoptosis in human pancreatic cancer cell lines. Cancer Res 2006; 66: 6741–7. [DOI] [PubMed] [Google Scholar]
- 8. Popivanova BK, Li YY, Zheng H et al. Proto‐oncogene, Pim‐3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad‐mediated apoptosis. Cancer Sci 2007; 98: 321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zheng HC, Tsuneyama K, Takahashi H et al. Aberrant Pim‐3 expression is involved in gastric adenoma–adenocarcinoma sequence and cancer progression. J Cancer Res Clin Oncol 2008; 134: 481–8. [DOI] [PubMed] [Google Scholar]
- 10. Aho TL, Sandholm J, Peltola KJ et al. Pim‐1 kinase promotes inactivation of the pro‐apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett 2004; 571: 43–9. [DOI] [PubMed] [Google Scholar]
- 11. Yan B, Zemskova M, Holder S et al. The PIM‐2 kinase phosphorylates BAD on serine 112 and reverses BAD‐induced cell death. J Biol Chem 2003; 278: 45. [DOI] [PubMed] [Google Scholar]
- 12. Li Y‐Y, Wang Y‐Y, Taniguchi T et al. Identification of stemonamidee synthetic intermediates as a novel potent anticancer drug with an apoptosis‐inducing ability. Int J Cancer 2010; 127: 474–84. [DOI] [PubMed] [Google Scholar]
- 13. Yano T, Ishikura H, Kato H et al. Vaccination effect of interleukin‐6‐producing pancreatic cancer cells in nude mice: a model of tumor prevention and treatment in immune‐compromised patients. Jpn J Cancer Res 2001; 92: 83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lieber M, Mazzetta J, Nelson‐Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor‐cell line (panc‐1) from a human carcinoma of the exocrine pancreas. Int J Cancer 1975; 15: 741–7. [DOI] [PubMed] [Google Scholar]
- 15. Yunis AA, Arimura GK, Russin DJ. Human pancreatic carcinoma (MIA PaCa‐2) in continuous culture: sensitivity to asparaginase. Int J Cancer 1977; 19: 128–35. [DOI] [PubMed] [Google Scholar]
- 16. Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999; 1: 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeuchi K, Ishita A, Matsuo J, Ishibashi H. Synthesis of 13a‐methylphenthroindolizidines using radical cascade cylization: synthetic studies toward (+)‐hypoestestatin 1. Tetrahedron 2007; 63: 11. [Google Scholar]
- 18. Cui M, Wang Q. Total synthesis of phenanthro‐quinolizidine alkaloids: (+)‐cryptopleurine, (+)‐boehmeriasin A, (+)‐boehmeriasin B and (+)‐hydroxycryptopleurine. Eur J Org Chem 2009; 2009: 5445–51. [Google Scholar]
- 19. Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensing recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 2001; 152: 669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest 2005; 115: 2618–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rhodes N, Heerding DA, Duckett DR et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 2008; 68: 2366–74. [DOI] [PubMed] [Google Scholar]
- 22. Nawijin M, Alendar A, Berns A. For better or for worse: the role of Pim oncogene in tumorigenesis. Nat Rev Cancer 2011; 11: 23–34. [DOI] [PubMed] [Google Scholar]
- 23. Stark GR, Taylor WR. Control of G2/M transition. Mol Biotechnol 2006; 32: 227–48. [DOI] [PubMed] [Google Scholar]
- 24. Barnett SF, Defeo‐Jones D, Fu S et al. Identification and characterization of pleckstrin‐homology‐domain‐dependent and isoenzyme‐specific Akt inhibitors. Biochem J 2005; 385: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morishita D, Katayama R, Sekimizu K, Tsuruo T, Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down‐regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res 2008; 68: 5076–85. [DOI] [PubMed] [Google Scholar]
- 26. Mochizuki T, Kitanaka C, Noguchi K et al. Physical and functional interactions between Pim‐1 kinase and Cdc25A phosphatase. J Biol Chem 1999; 274: 18. [DOI] [PubMed] [Google Scholar]
- 27. Bachmann M, Hennemann H, Xing PX, Hoffmann I, Möröy T. The oncogenic serine/threonine kinase Pim‐1 phosphorylates and inhibits the activitiy of Cdc25c‐associated kinase 1 (C‐TAK1). J Biol Chem 2004; 279: 48. [DOI] [PubMed] [Google Scholar]
- 28. Wang Z, Bhattacharya N, Mixter PF et al. Phosphorylation of the cell cycle inhibitor p21cip1/WAF1 by Pim‐1 kinase. Biochim Biophys Acta 2002; 1593: 45–55. [DOI] [PubMed] [Google Scholar]
- 29. Chen WS, Xu PZ, Gottlob K et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 2001; 15: 2203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Easton RM, Cho H, Roovers K et al. Role for Akt3/protein kinase Bγ in attainment of normal brain size. Mol Cell Biol 2005; 25: 1869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woulfe D, Jiang H, Morgans A et al. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest 2004; 113: 441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim‐1 kinase. J Biol Chem 2005; 280: 6130–7. [DOI] [PubMed] [Google Scholar]
- 33. Bullock AN, Debreczeni J, Amos AL, Knapp S, Turk BE. Structure and substrate specificity of the Pim‐1 kinase. J Biol Chem 2005; 280: 41. [DOI] [PubMed] [Google Scholar]
- 34. Bullock AN, Russo S, Amos A et al. Crystal structure of the PIM2 kinase in complex with an organoruthenium inhibitor. PLoS ONE 2009; 4: e7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brault L, Gesser C, Bracher F et al. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies. Haematologica 2010; 95: 1004–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holder S, Zemskova M, Zhang C et al. Characterization of a potent and selective small‐molecule inhibitor of the PIM1 kinase. Mol Cancer Ther 2007; 6: 163–72. [DOI] [PubMed] [Google Scholar]
- 37. Pogacic V, Bullock AN, Fedorov O et al. Structural analysis identifies imidazo[1,2‐b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res 2007; 67: 6916–24. [DOI] [PubMed] [Google Scholar]
- 38. Chen LS, Redkar S, Bearss D, Wierda WG, Gandhi V. Pim kinase inhibitor, SGI‐1776, induces apoptosis in chronic lymphocytic leukemia cells. Blood 2009; 114: 4150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xia Z, Knaak C, Ma J et al. Synthesis and evaluation of novel inhibitors of Pim‐1 and Pim‐2 protein kinases. J Med Chem 2009; 52: 74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin YW, Beharry ZM, Hill EG et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T‐cell lymphoblastic leukemia/lymphoma. Blood 2010; 115: 824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akué‐Gédu R, Rossignol E, Azzaro S et al. Synthesis, kinase inhibitory potencies, and in vitro antiproliferative evaluation of new Pim kinase inhibitors. J Med Chem 2009; 52: 6369–81. [DOI] [PubMed] [Google Scholar]
- 42. Domen J, van der Lugt NM, Laird PW et al. Impaired interleukin‐3 response in Pim‐1‐deficient bone marrow‐derived mast cells. Blood 1993; 82: 1445–52. [PubMed] [Google Scholar]
- 43. Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005; 105: 4477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beharry Z, Mahajan S, Zemskova M et al. The Pim protein kinases regulate energy metabolism and cell growth. Proc Natl Acad Sci U S A 2011; 108: 528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effects of T26 treatment on various parameters in tumor‐bearing mice.
Supporting info item