

Abstract
Agents that cause DNA damage have been widely used as anticancer drugs because DNA lesions can initiate DNA checkpoints that induce cell death. The results presented here indicate that QS‐ZYX‐1‐61, a derivative of VP‐16, was significantly more potent than VP‐16 in suppressing the viability of A549 cells. Treatment of cells with QS‐ZYX‐1‐61 led to a DNA damage response and a dramatic increase of apoptosis. Our results also suggest that QS‐ZYX‐1‐61 may be a topoisomerase (topo) II targeting agent, as evidenced by relaxation assay and induction of reversible cleavable complexes. Moreover, blocking of p53, topo IIα, and topo IIβ greatly protected against caspase‐3 activation, poly‐ADP‐ribose polymerase cleavage, and cell growth inhibition, indicating that QS‐ZYX‐1‐61 acts through these proteins. These results support our conclusion that QS‐ZYX‐1‐61 has potential as an anticancer agent because it causes accumulation of DNA cleavable complexes, with downstream consequences that include double‐strand breaks and DNA damage response signaling for apoptosis. Taken together, our results indicate that QS‐ZYX‐1‐61 is a novel DNA damaging agent and displays an outstanding activity that could be worthy of further investigation. (Cancer Sci 2012; 103: 80–87)
Many clinically approved anticancer agents target DNA.( 1 , 2 , 3 , 4 ) These DNA‐targeting agents can be classified as alkylating agents, antimetabolites, topoisomerase inhibitors, or radiomimetics.( 1 , 5 ) Topoisomerases are essential for the modification of DNA topology and have been established molecular targets for several decades.( 1 , 6 , 7 ) Topoisomerases relax the helical supercoiling of DNA that is generated during replication, transcription, and chromatin remodeling.( 8 , 9 )
Topoisomerase II (topo II) catalyzes an ATP‐dependent reaction in which one DNA double helix is passed through another.( 6 ) There are two topo II isozymes, topo IIα (170 kDa) and topo IIβ (180 kDa). The expression of topo IIα is high in the S phase and peaks in the late S/G2 phase of the cell cycle, whereas topo IIβ is expressed at a constant level throughout the cell cycle.( 10 )“Topo II poisons”, which are highly cytotoxic, stimulate and stabilize the formation of topo II–DNA cleavable complexes, leading to accumulation of these cleavable complexes.( 11 , 12 ) In contrast, “topo II catalytic inhibitors” such as ICRF‐187, disrupt enzyme activity without stabilizing the cleavable complex.( 13 , 14 ) Etoposide (VP‐16) is a topo II poison that has been used in the chemotherapy of non‐small‐cell lung cancer (NSCLC) and other cancers.( 15 , 16 , 17 , 18 , 19 ) In previous studies it has been shown that etoposide induces double‐strand breaks (DSBs) and triggers the DNA damage response (DDR).( 20 , 21 ) Agents that induce DNA damage in cells can trigger a complex network known as the “checkpoint pathways”, thereby promoting cell cycle delay or arrest and allowing more time for DNA repair.( 22 , 23 ) This intricate signaling network can be turned on by activation of ataxia telangiectasia mutated (ATM) protein kinase, which phosphorylates numerous downstream substrates.( 24 )
Lung cancer is the leading cause of cancer death worldwide. The standard dual agent chemotherapy improves survival rate.( 25 , 26 ) Many clinical trials have tested the combination of cisplatin and etoposide with other drugs in treatment of NSCLC( 27 ), and found the overall survival rate has not been improved significantly. QS‐ZYX‐1‐61, a derivative of etoposide (VP‐16), was synthesized in Dr. Kuo‐Hsiung Lee’s Natural Products Research Laboratories (University of North Carolina, Chapel Hill, NC, USA). In previous studies, a series of 4′‐O‐demethylepipodophyllotoxins have been developed( 28 , 29 ) and it was found that various C4 substitutions had important roles in the activity profiles of VP‐16 analogues.( 28 ) In the present study, we propose that QS‐ZYX‐1‐61, a novel derivative of etoposide with a better performance, triggers a DDR followed by apoptosis in A549 cells. Our data suggest that QS‐ZYX‐1‐61 is a novel DNA damage agent that could be applied as targeted therapeutic drug for NSCLC.
Materials and Methods
Cell line and reagents. A549 (p53 wild‐type) lung adenocarcinoma cells, NCI‐H226 (p53 mutant) lung squamous carcinoma cell line, and H1299 (p53 null) human NSCLC cell line were obtained from ATCC (Manassas, VA, USA). Cells were maintained in 10% FBS‐supplemented RPMI‐1640 medium (Gibco, Grand Island, NY, USA) and 1% penicillin–streptomycin (Gibco) at 37°C in a humidified incubator containing 5% CO2. QS‐ZYX‐1‐61, 4β‐[(4″‐benzylpiperdin‐4‐yl) amino]‐4′‐O‐demethyl‐epipodophyllotoxin (Fig. 1A), was obtained from Professor Kuo‐Hsiung Lee (Natural Products Research Laboratories). Figure 1(B) shows the chemical structure of VP‐16, which was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies against various proteins were obtained from poly‐ADP‐ribose polymerase (PARP), Bcl‐2, Bcl‐xL, Bax, Chk2, p21, puma, and topo IIβ, and anti‐mouse and anti‐rabbit IgGs were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). P‐ATM (Ser1981), p‐Chk2 (Thr68), p‐p53 (Ser15), γH2AX, caspase 8, and caspase 9 were obtained from Cell Signaling Technology (Danvers, MA, USA). Topoisomerase IIα, topo I, and p53 were obtained from BD Biosciences (San Jose, CA, USA). Pan‐actin was from Millipore (Billerica, MA, USA). Caspase 3 and survivin were obtained from Imgenex (San Diego, CA, USA) and Novus (Littleton, CO, USA), respectively.
Figure 1.
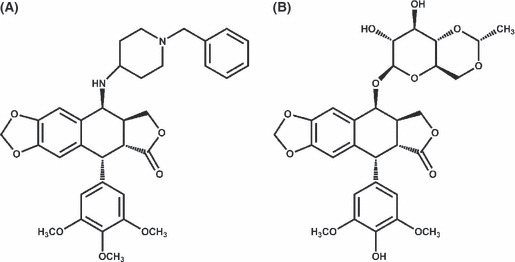
(A) Chemical structure of QS‐ZYX‐1‐61 (4β‐[(4″‐benzylpiperdin‐4‐yl) amino]‐4′‐O‐demethyl‐epipodophyllotoxin). (B) Chemical structure of etoposide (VP‐16).
Cell viability assay. Cell viability was determined using the MTT assay and carried out as described previously.( 30 )
Flow cytometry analysis. Cells were seeded in six‐well plates (2.5 × 105/well) and treated with DMSO or QS‐ZYX‐1‐61 at various concentrations for indicated times. Cells were washed with PBS, fixed in ice‐cold 70% ethanol at −20°C overnight, and stained with propidium iodide (80 μg/mL) containing Triton X‐100 (0.1%, v/v) and RNase A (100 μg/mL) in PBS. DNA content was analyzed with the FACScan and CellQuest software (Becton Dickinson, Mountain View, CA, USA).
Immunoblotting. A549 cells were seeded in dishes and allowed to attach overnight. The cells were treated with QS‐ZYX‐1‐61 at indicated concentrations. After the indicated exposure time, cells were lysed and the immunoblotting was carried out as described previously.( 30 ) Quantification of the density of bands was carried out using Gel‐Pro Analyzer (version 3.1; Media Cybernetics, Bethesda, MD, USA).
Comet assay. Cells were treated for 30 min in 12 wells (1.5 × 105 cells/well) and the assay was carried out according to the published protocol.( 30 )
Topoisomerase II relaxation assay. The reaction was done according to the protocol from TopoGEN (Port Orange, FL, USA). After incubation at 37°C for 30 min, the reaction was terminated by adding 5 mL stopping buffer. The reaction products were analyzed by electrophoresis on 1% agarose gel using a Tris‐borate/EDTA buffer at 1 V/cm, stained by ethidium bromide, and photographed using a short‐wavelength UV lamp (ChemiImager 5500; Alpha Innotech, Santa Clara, CA, USA).
Band depletion assay. The cells were lysed with SDS sample buffer (4% SDS/2%‐mercaptoethanol, 20% glycerol, 125 mM Tris HCl, pH 6.8) and were separated by 7.5% SDS/PAGE. The following procedures were the same as for the immunoblotting protocol.
Apoptosis assay. Drug‐induced apoptotic cell death was assessed using the Cell Death Detection ELISA kit (Roche Diagnostics, Besel, Switzerland). Cells were treated with QS‐ZYX‐1‐61 for 24 h. Both floating and adherent cells were collected and the assay was done according to the manufacturer’s instructions.
Stable expression of shRNA. Stable expressing topo IIα and IIβ shRNA A549 cells were kindly provided by Dr. T.K. Li (Department of Microbiology, National Taiwan University, Taipei, Taiwan). Cells were cultured in RPMI‐1640 medium containing 10% FBS and 0.1 μM puromycin (Sigma‐Aldrich).
Small interfering RNA transfection. The siRNA for p53, topo IIα, IIβ, and negative control were purchased from Invitrogen (Carlsbad, CA, USA). Transfection was done using Lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions. Following transfection, cells were allowed to recover for 24 h then treated for another 24 h.
Statistics and data analysis. Each experiment was carried out at least three times, and representative data are shown. Data in bar graphs are given as the means ± SEM. Means were checked for statistical difference using the Student's t‐test and P‐values < 0.05 were considered significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Effect of QS‐ZYX‐1‐61 on cell proliferation and cell cycle distribution. The cell growth inhibition activities of QS‐ZYX‐1‐61 were assessed in three human NSCLC cell lines, A549, H1299, and NCI‐H226. Etoposide (VP‐16) was included as a positive control DNA‐damaging agent. Cells were grown in the absence or presence of different concentrations of QS‐ZYX‐1‐61 and VP‐16 for 48 h, and cytotoxicity was measured by the MTT assay. Figure 2(A,B) shows that QS‐ZYX‐1‐61 and VP‐16 induced cell death in a concentration‐dependent manner. Notably, cells were much more sensitive to QS‐ZYX‐1‐61 than VP‐16 (IC50 of QS‐ZYX‐1‐61: A549, 0.71 ± 0.07 μM; H1299, 1.66 ± 0.11 μM; NCI‐H226, 2.38 ± 0.95 μM; IC50 of VP‐16: A549, 81.56 ± 1.84 μM; H1299, 56.95 ± 3.27 μM; NCI‐H226, 49.98 ± 1.92 μM). Next, we investigated the mechanism of cell growth repression by QS‐ZYX‐1‐61 by measuring cell cycle distribution with flow cytometry and propidium iodide staining in A549 cells. Figure 2(C,D) shows that QS‐ZYX‐1‐61 induced significant accumulation of sub‐G1 phase, suggesting that QS‐ZYX‐1‐61 causes apoptosis in A549 cells.
Figure 2.
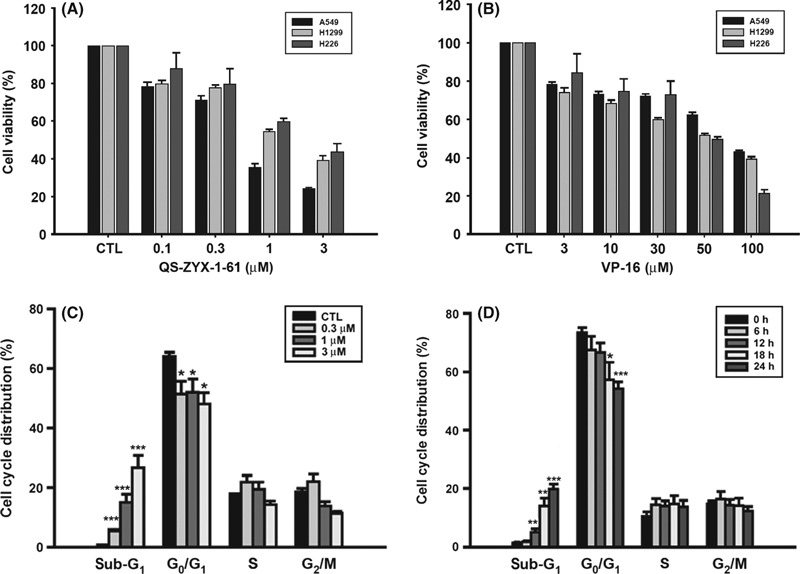
Effect of QS‐ZYX‐1‐61 on viability and cell cycle distribution in non‐small‐cell lung cancer cells. (A, B) Concentration‐dependent effect of QS‐ZYX‐1‐61 and VP‐16 on suppression of A549, H1299, and NCI‐H226 cells. Cells were exposed to QS‐ZYX‐1‐61 (A) or VP‐16 (B) at the indicated concentrations in 10% FBS‐supplemented RPMI‐1640 for 48 h, and the cell viability was assessed by MTT assay. CTL, control. Concentration‐dependent (C) and time‐dependent (D) effects of QS‐ZYX‐1‐61 on flow cytometric analysis of apoptotic cell death in A549 cells. Cells were treated with DMSO or QS‐ZYX‐1‐61 at the indicated concentrations in 10% FBS‐containing RPMI‐1640 for 24 h (C) or treated with a single dose of QS‐ZYX‐1‐61 (1 μM) for indicated times (D). The cells were analyzed by flow cytometry after staining with propidium iodide. Asterisks indicate significant difference between control and QS‐ZYX‐1‐61‐treated cells. *P < 0.05; **P < 0.01; ***P < 0.001.
QS‐ZYX‐1‐61 induces apoptosis in A549 cells. Next, we evaluated the effect of QS‐ZYX‐1‐61 on the induction of apoptosis by use of an enzyme immunoassay for histone‐associated DNA fragments. Figure 3(A) shows that QS‐ZYX‐1‐61 increased the amount of histone‐associated DNA fragments in a concentration‐dependent manner. Apoptosis may occur through caspase‐dependent or caspase‐independent mechanisms. Thus, we evaluated the activation of caspase‐3, caspase‐8, caspase‐9, and PARP cleavage after treatment of cells with QS‐ZYX‐1‐61 and VP‐16. Figure 3(B,C) shows that QS‐ZYX‐1‐61 induced caspase‐3, caspase‐8, caspase‐9, and cleavage of PARP in a concentration‐ and time‐dependent manner. Notably, the cells were much more sensitive to QS‐ZYX‐1‐61 than VP‐16 (Fig. 3B).
Figure 3.
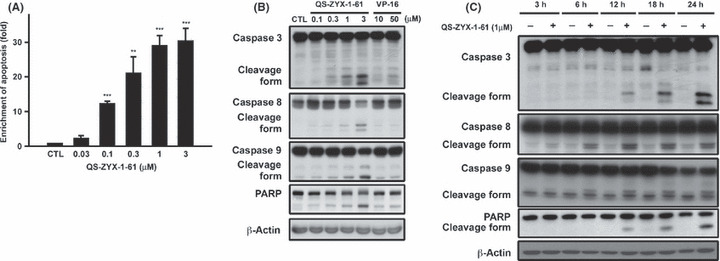
QS‐ZYX‐1‐61 induced cell death in A549 non‐small‐cell lung cancer cells in a dose‐dependent manner. (A) Effect of QS‐ZYX‐1‐61 on drug‐induced DNA fragmentation in A549 cells. Cells were treated with DMSO or QS‐ZYX‐1‐61 at various concentrations for 24 h. Formation of cytoplasmic DNA was quantitatively measured by a cell death detection ELISA kit. Concentration‐dependent (B) and time‐dependent (C) effects of QS‐ZYX‐1‐61 on cleavage of caspases and poly‐ADP‐ribose polymerase (PARP) in A549 cells by immunoblotting analysis. Protein lysates from 24 h‐treated A549 cells were immunoblotted for indicated proteins. Asterisks indicate significant difference between control (CTL) and QS‐ZYX‐1‐61‐treated cells. **P < 0.01; ***P < 0.001.
Role of P53 in QS‐ZYX‐1‐61‐induced apoptosis. Next, we evaluated the effect of this drug on p53 by treatment of A549 cells with individual agents at various concentrations for 24 h, followed by immunoblotting with various antibodies against p53 and its target proteins. Figure 4(A) shows that QS‐ZYX‐1‐61 increased the level of p53 protein, although the p53 mRNA level remained unchanged (data not shown). Moreover, QS‐ZYX‐1‐61 treatment increased the levels of phospho‐p53 (Ser15) and p53 target genes except for Noxa. Bcl‐2 family proteins regulate apoptosis and cell cycle control.( 31 ) Thus, we assessed the effect of QS‐ZYX‐1‐61 on expression of Bcl‐2 family proteins and other anti‐apoptotic proteins. QS‐ZYX‐1‐61 had no significant effect on the expression of anti‐apoptotic proteins (Bcl‐2, Bcl‐XL) or on the expression of a pro‐apoptotic protein (Bax) (Fig. 4B). Survivin is a member of the inhibitor of apoptosis protein family and interacts with caspases to block apoptosis.( 32 ) Our results indicate that QS‐ZYX‐1‐61 downregulates survivin within 6 h (Fig. 4B). We further examined the effect of QS‐ZYX‐1‐61 by studying the effect of siRNA‐mediated knockdown of p53 on rescue from QS‐ZYX‐1‐61‐mediated apoptosis. Figure 4(C,D)shows that knockdown of p53 rendered A549 cells resistant to QS‐ZYX‐1‐61‐mediated apoptotic death (PARP cleavage and caspase‐3 activation) and cell growth inhibition. These results suggest that p53 has an important role in QS‐ZYX‐1‐61‐induced apoptosis in A549 cells.
Figure 4.

Effect of QS‐ZYX‐1‐61 on the biomarkers associated with apoptosis in A549 non‐small‐cell lung cancer cells. (A) QS‐ZYX‐1‐61 induces p53 activation. Cells were treated with drugs for indicated times, and the cell lysates were subjected to Western blot analysis. (B) Change in expression of anti‐apoptotic and pro‐apoptotic Bcl‐2 family proteins, and member of the inhibitor of apoptosis protein family, survivin, in A549 cells following QS‐ZYX‐1‐61 and VP‐16 treatment. Cells were treated for indicated times, and the cell lysates were subjected to immunoblotting using indicated antibodies. (C,D) Knockdown of p53 protects A549 cells from QS‐ZYX‐1‐61‐induced apoptotic cell death (C) and cell growth inhibition (D). A549 cells were transfected with control siRNA or siRNA against p53, and those cells were treated with DMSO or QS‐ZYX‐1‐61 (1 μM) for 24 h (for immunoblot analyses) and 48 h (for MTT assay). CTL, control. **P < 0.01; ***P < 0.001.
QS‐ZYX‐1‐61 triggers DSBs and DNA damage checkpoints. The comet assay is a rapid, simple, and sensitive method for measuring DNA strand breaks. VP‐16, a topo II‐targeting anticancer drug that efficiently induces topo II‐mediated DNA damage,( 33 ) may be considered as a positive control. Figure 5(A) shows that QS‐ZYX‐1‐61, like VP‐16, induces chromosomal DNA strand breaks in a concentration‐dependent manner. Activation of nuclear kinase ATM is one of the earliest signs of DNA damage( 34 ) and activated ATM kinase is known to phosphorylate and activate numerous substrates.( 24 ) Histone H2AX, one of its substrates, is phosporylated at Ser‐139.( 22 ) A previous study indicated that phosphorylation of histone H2AX (termed γH2AX) was a biomarker for drug‐induced DNA damage at DSB sites.( 35 , 36 ) Thus, we assessed the effect of QS‐ZYX‐1‐61 on the level of γH2AX and DNA damage checkpoint kinases in A549 cells. Figure 5(B) shows that treatment of A549 cells with QS‐ZYX‐1‐61 or VP‐16 increased the levels of phospho‐ATM, phospho‐Chk2, and γH2AX. The results also indicate that QS‐ZYX‐1‐61 is more potent than VP‐16 (lanes 9 and 10), and that both agents induce activation of ATM kinase and its downstream protein targets. Taken together, our data indicate that QS‐ZYX‐1‐61 is a potent DNA damaging agent that triggers the DNA damage checkpoint signaling pathway, involving ATM, Chk2, p53, and γH2AX.
Figure 5.
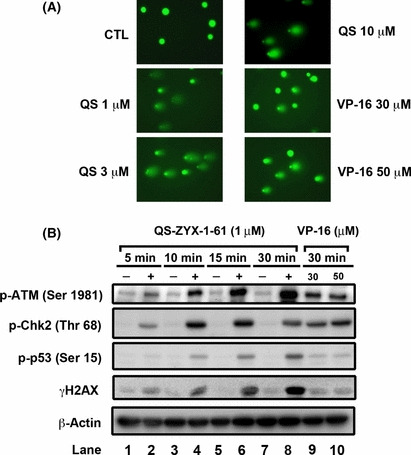
QS‐ZYX‐1‐61 (QS) induces double strand breaks and DNA checkpoint pathway in a time‐dependent manner. (A) Chromosomal DNA strand breaks were detected by comet assay. Non‐small‐cell lung cancer cells were treated with QS‐ZYX‐1‐61 or VP‐16 at various concentrations for 30 min. After treatment, comet assay was carried out to analyze the chromosome DNA integrity in A549 cells. CTL, control. (B) QS‐ZYX‐1‐61 activates DNA damage signaling. Cells were treated with QS‐ZYX‐1‐61 (1 μM) or VP‐16 (30 and 50 μM) in 10% FBS‐containing RPMI‐1640 for 30 min. Western blot analysis of the expression levels of indicated proteins in A549 cells.
QS‐ZYX‐1‐61 inhibits topo II activity and induces reversible cleavable complexes. Next, we used a commercial assay for in vitro measurement of changes in supercoiled DNA following QS‐ZYX‐1‐61 treatment. Figure 6(A) shows that QS‐ZYX‐1‐61 inhibited the DNA relaxation activity of topo II. VP‐16 is a known topo II inhibitor and is considered a topo II poison because it induces and stabilizes DNA cleavable complexes.( 20 ) The results of our band‐depletion assay indicated that QS‐ZYX‐1‐61 decreased free topo IIα and IIβ expression in a concentration‐dependent (Fig. 6B, left panel) and time‐dependent (Fig. 6B, right panel) manner because of “trapping” of topo IIα and IIβ to DNA into protein–DNA complexes. However, QS‐ZYX‐1‐61 had no effect on topo I–DNA complexes in A549 cells (Fig. 6C). The topo I poison camptothecin was used as the positive control.( 37 ) Further experiments indicated that these complexes were reversible, as indicated by restoration of the original condition after replacement of the medium with fresh conditioned medium (Fig. 6D). Based on these findings, we conclude that QS‐ZYX‐1‐61 and VP‐16 reduce the intensity of the topo II protein band, and QS‐ZYX‐1‐61 was more effective than VP‐16 (Fig. 6B).
Figure 6.
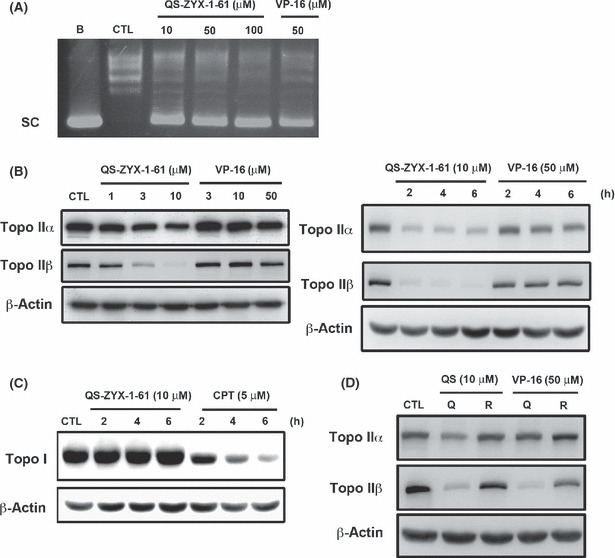
Effect of QS‐ZYX‐1‐61 on topoisomerase II (topo II). (A) DNA relaxation assay. The results showed that QS‐ZYX‐1‐61 inhibited the ability of topo II to relax supercoiled DNA. (B) Drugs induced topo II–DNA cleavable complex in A549 cells in a concentration‐dependent (left panel) and time‐dependent (right panel) manner by band‐depletion assay. Cells were treated with indicated drugs for 1 h. Cell lysates were immunoblotted with indicated antibodies. Reduction of topo IIα and IIβ bands is indicative of the formation of topo II–DNA covalent complexes. (C) Effect of QS‐ZYX‐1‐61 on trapping topo I–DNA cleavable complex. Cells were treated with QS‐ZYX‐1‐61 and camptothecin (CPT) at indicated concentrations for indicated times. Cell lysates were immunoblotted with anti‐topo I and β‐actin. (D) The formation of reversible topo IIα and IIβ cleavable complexes was investigated by removing drug treatment with 10 μM QS‐ZYX‐1‐61 (QS) or 50 μM VP‐16 and adding fresh medium (R) for 1 h after 1 h treatment with QS‐ZYX‐1‐61 (Q) and VP16.
Topoisomerase IIβ is required for activation of p53 and induction of apoptosis. To validate whether topo II was required for the activation of ATM and downstream signaling, we examined the effect of shRNA‐mediated knockdown of topo II isoforms (topo IIα and IIβ) on rescuing QS‐ZYX‐1‐61‐mediated apoptotic death in A549 cells. We transfected A549 cells with plasmids encoding shRNAs against individual topo II isoforms, then carried out clonal selection. This shRNA knockdown was highly specific, as indicated by the absence of cross‐silencing of other topo II isoforms (α and β) (Fig. 7A). The results indicate that QS‐ZYX‐1‐61 retains its ability to trap the topo II–DNA cleavable complex in the presence of topo IIα and IIβ deficiency (Fig. 7B). In addition, the knockdown of topo IIβ led A549 cells to reduce the activation of QS‐ZYX‐1‐61‐mediated checkpoint pathways (p‐ATM, p‐p53) and DNA DSBs (Fig. 7C), whereas the effect on topo IIα was negligible. As there exists a mechanistic link between checkpoint pathway activation and the topo II inhibition, we further examined whether topo II inhibition was the underlying mechanism of QS‐ZYX‐1‐61‐induced cell death. Figure 7(D) shows that topo IIα and IIβ knockdown largely abolished caspase‐3 activation and PARP cleavage, but that topo IIβ had a greater effect on this protection. Moreover, blockade of topo IIα and IIβ expression also protected A549 cells from QS‐ZYX‐1‐61‐induced cell growth inhibition (Fig. 7E,F). Overall, these results suggest that topo IIβ plays a more crucial role than topo IIα in QS‐ZYX‐1‐61‐induced activation of DNA damage signaling and apoptotic cell death.
Figure 7.
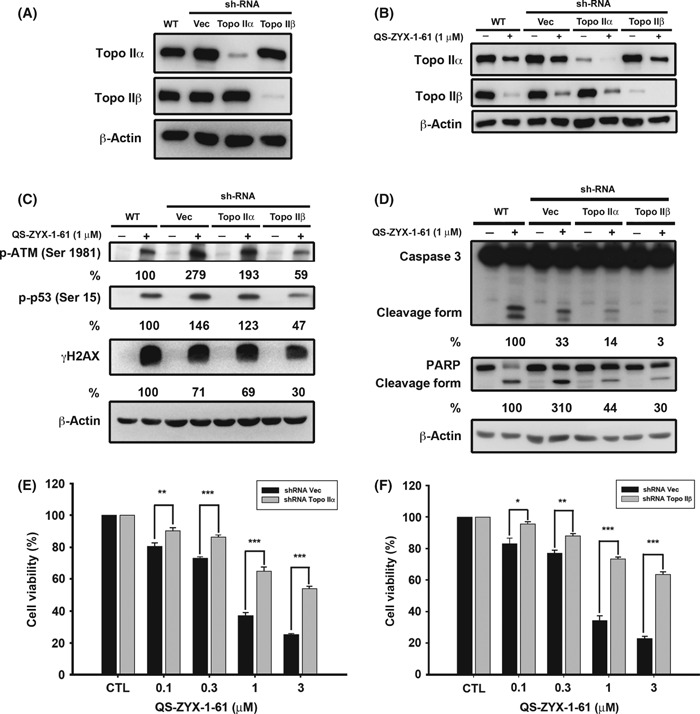
Effects of knockdown of topoisomerase (topo) IIα and IIβ on QS‐ZYX‐1‐61‐induced DNA damage response, caspase‐3, poly‐ADP‐ribose polymerase (PARP) activation, and cell growth inhibition. (A) Western blot analysis of the expression levels of topo IIα and IIβ in A549 non‐small‐cell lung cancer cells subjected to different treatments, as indicated. (B) Effects of drug‐trapped topo II–DNA cleavable complex. The shRNA‐mediated knockdown of topo IIα and topo IIβ was applied to further confirm the effect of QS‐ZYX‐1‐61 on trapping topo II–DNA complex in A549 cells by band depletion assay. (C) Knockdown of topo IIα and IIβ could protect A549 cells from QS‐ZYX‐1‐61‐induced DNA damage response. Western blot analysis of the expression levels of indicated proteins in A549 cells subjected to QS‐ZYX‐1‐61 for 30 min. (D) Knockdown of topo IIβ protects A549 cells against QS‐ZYX‐1‐61‐induced apoptosis. Western blot analysis of the expression levels of caspase‐3, PARP, and β‐actin in A549 cells subjected to QS‐ZYX‐1‐61 for 24 h. The values in percentages, the average of two independent experiments, denote the relative intensities of protein bands of drug‐treated samples (shRNA infected groups) to that of the drug‐treated control (WT) after normalization to β‐actin. (E,F) Effect of topo IIα (E) and IIβ (F) knockdown on suppressing QS‐ZYX‐1‐61‐induced cell death. Cells were transfected with control (CTL) or plasmids encoding shRNA against topo IIα or IIβ, and those stable clones were treated with QS‐ZYX‐1‐61 as indicated. Cell viability was measured by MTT assay. Vec, control vector. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Etoposide is currently approved for treatment of a wide variety of cancers, including small‐cell lung cancer,( 19 , 38 ) NSCLC,( 19 , 27 ) testicular cancer,( 19 ) and lymphomas.( 19 , 39 ) Despite the crucial clinical role of etoposide, there are problems in its use in the treatment for human cancers,( 40 , 41 ) motivating the search for additional anticancer agents. The present study indicates that QS‐ZYX‐1‐61, an analogue of etoposide, suppressed the proliferation of A549 cells at least in part by stimulation of checkpoint kinases, leading to p53 activation, then caspase‐3‐dependent apoptosis. Our results also indicate that QS‐ZYX‐1‐61 has 100‐fold greater potency than VP‐16 in A549 cells. Furthermore, our lactate dehydrogenase cytotoxicity assay confirmed that QS‐ZYX‐1‐61 inhibited cell growth and stimulated apoptosis without induction of necrosis (Fig. S1).
Previous research of multiple types of cancer indicated that the level of survivin expression correlates with poor prognosis and predicts response to diverse anticancer therapies,( 42 ) and that reducing the expression of survivin sensitizes cells to lung cancer treatment.( 43 , 44 ) Our results show that QS‐ZYX‐1‐61 downregulates the expression of the anti‐apoptotic protein, survivin, but had no effect on the protein levels of Bax, Bcl‐2, and Bcl‐XL. Several mechanisms have been proposed to explain the effect of DNA damaging agents on the sensitization or chemosensitization of cancer cells.( 45 , 46 ) In addition, some studies have shown that p53 is stabilized in response to DNA damaging agents.( 47 , 48 ) Our results indicate that QS‐ZYX‐1‐61 causes DDR, leading to accumulation of p53 protein, increased expression of p53 target genes (p21 and puma), and induction of apoptosis signaling pathways. Moreover, we found that p53 silencing by p53 siRNA protects cells from the effect of QS‐ZYX‐1‐61 on p21 upregulation, survivin downregulation, PARP cleavage, and caspase‐3 activation (Fig. 4C). It has been reported that loss of p53 function in cancer cells might lead to induction of survivin and resistance to DNA damaging agents.( 49 ) Based on previous findings and our current results, p53 plays a crucial role in the QS‐ZYX‐1‐61‐mediated apoptosis of A549 cells.
DNA damage responses, generated by increased levels of topo II–DNA covalent complexes, are powerful activators and further engage a signal amplification cascade.( 50 ) Cells can undergo p53‐dependent and p53‐independent signaling in response to DNA damage.( 51 ) The relationship between p53 status and response to topo II inhibitor raised many questions and led to diverse conclusions.( 52 ) Previous studies have shown that wild‐type p53 accelerates cell death induced by DNA damaging agents in both normal and cancer cells.( 53 ) Moreover, various chemotherapeutic agents can induce cell death in tumor cells through p53‐independent pathways,( 54 , 55 ) suggesting that those cells do not loss their ability to undergo apoptosis completely and they can activate p53‐independent apoptosis through a backup system. Our results showed that QS‐ZYX‐1‐61 can cause cell death efficiently in wild‐type p53 (A549), whereas higher IC50 levels of the drug were shown in p53‐mutant (NCI‐H226), and p53‐null (H1299) cell lines (Fig. 2A). We also carried out siRNA‐mediated knockdown of topo IIα and IIβ in H1299 and NCI‐H226 cells and found either topo IIα or IIβ‐knockdown cells can reduce growth inhibition induced by QS‐ZYX‐1‐61 (Fig. S2). In addition, the differential effect of QS‐ZYX‐1‐61 was also observed in prostate cancer cells (DU145, PC3, and LNCaP) with different p53 status (Fig. S3). Such findings suggested that the direct function of QS‐ZYX‐1‐61 in targeting topo II seems to be the crucial role in determining cellular fate, and wild‐type p53 protein acts as a downstream effector in cells to facilitate the cell death triggered by QS‐ZYX‐1‐61.
QS‐ZYX‐1‐61 has the ability to interfere with topo II by induction of reversible cleavage complexes in a reaction, therefore, we propose that QS‐ZYX‐1‐61 might exert its anticancer activity through topo II. Surprisingly, we also found that QS‐ZYX‐1‐61‐induced apoptotic markers are reduced more in topo IIβ‐deficient A549 cells than in topo IIα‐deficient A549 cells. Previous studies have reported that topo IIβ (rather than topo IIα) cleavage complexes are rapidly converted into DNA DSBs through proteasomal degradation, suggesting a preferential role for topo IIβ in DNA DSB‐mediated apoptosis.( 56 , 57 )
In conclusion, our results indicate that QS‐ZYX‐1‐61 mediates apoptosis through topo II in A549 cells and exhibits strong antitumor activities in NSCLC cells. These antitumor activities include trapping of topo II to the topo II–DNA complex, activation of DNA damage pathways, and post‐translational upregulation of the apoptotic regulator, p53. Taken together, our results provide compelling evidence that QS‐ZYX‐1‐61 has great potential as an individual agent or in combination with other drugs in the treatment of NSCLC.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Lactate dehydrogenase assay of QS‐ZYX‐1‐61‐treated A549 non‐small‐cell lung cancer cells.
Fig. S2. Effect of topoisomerase (topo) IIα and IIβ knockdown on suppressing QS‐ZYX‐1‐61‐induced cell growth inhibition in NCI‐H226 and H1299 cells.
Fig. S3. Effect of QS‐ZYX‐1‐61 on cell viability in prostate cancer cells.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We would like to give our thanks to Ting‐Hsiang Huang for technical assistance. This work was supported by the National Science Council (NSC 98‐2320‐B‐002‐009‐MY3).
References
- 1. Hurley LH. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2002; 2: 188–200. [DOI] [PubMed] [Google Scholar]
- 2. Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009; 9: 338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neidle S, Thurston DE. Chemical approaches to the discovery and development of cancer therapies. Nat Rev Cancer 2005; 5: 285–96. [DOI] [PubMed] [Google Scholar]
- 4. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8: 193–204. [DOI] [PubMed] [Google Scholar]
- 5. Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer 2004; 4: 216–25. [DOI] [PubMed] [Google Scholar]
- 6. Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 2009; 9: 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen AY, Liu LF. DNA topoisomerases: essential enzymes and lethal targets. Annu Rev Pharmacol Toxicol 1994; 34: 191–218. [DOI] [PubMed] [Google Scholar]
- 8. Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem 1989; 58: 351–75. [DOI] [PubMed] [Google Scholar]
- 9. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002; 3: 430–40. [DOI] [PubMed] [Google Scholar]
- 10. Li TK, Liu LF. Tumor cell death induced by topoisomerase‐targeting drugs. Annu Rev Pharmacol Toxicol 2001; 41: 53–77. [DOI] [PubMed] [Google Scholar]
- 11. Felix CA, Kolaris CP, Osheroff N. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst) 2006; 5: 1093–108. [DOI] [PubMed] [Google Scholar]
- 12. McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 2007; 623: 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hasinoff BB, Abram ME, Barnabe N, Khelifa T, Allan WP, Yalowich JC. The catalytic DNA topoisomerase II inhibitor dexrazoxane (ICRF‐187) induces differentiation and apoptosis in human leukemia K562 cells. Mol Pharmacol 2001; 59: 453–61. [DOI] [PubMed] [Google Scholar]
- 14. Andoh T, Ishida R. Catalytic inhibitors of DNA topoisomerase II. Biochim Biophys Acta 1998; 1400: 155–71. [DOI] [PubMed] [Google Scholar]
- 15. Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive‐stage small cell lung carcinoma: current status and future prospects. Eur Respir J 2010; 35: 202–15. [DOI] [PubMed] [Google Scholar]
- 16. Kuruvilla J. Standard therapy of advanced Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program 2009; 16: 497–506. [DOI] [PubMed] [Google Scholar]
- 17. Isoyama Y, Shioyama Y, Nomoto S et al. Carboplatin and etoposide combined with radiotherapy for limited‐stage small‐cell esophageal carcinoma: three cases and review of the literature. Jpn J Radiol 2010; 28: 181–7. [DOI] [PubMed] [Google Scholar]
- 18. Kosmas C, Daladimos T, Athanasopoulos A et al. Double high‐dose chemotherapy and stem cell transplantation in adult Wilms’ tumor. Future Oncol 2010; 6: 1803–9. [DOI] [PubMed] [Google Scholar]
- 19. Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer 1998; 34: 1514–21. [DOI] [PubMed] [Google Scholar]
- 20. Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett 2007; 252: 9–18. [DOI] [PubMed] [Google Scholar]
- 21. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26: 7773–9. [DOI] [PubMed] [Google Scholar]
- 22. Bekker‐Jensen S, Mailand N. Assembly and function of DNA double‐strand break repair foci in mammalian cells. DNA Repair (Amst) 2010; 9: 1219–28. [DOI] [PubMed] [Google Scholar]
- 23. Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 2004; 5: 792–804. [DOI] [PubMed] [Google Scholar]
- 24. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003; 3: 155–68. [DOI] [PubMed] [Google Scholar]
- 25. Evans T. Chemotherapy in advanced non‐small cell lung cancer. Semin Respir Crit Care Med 2005; 26: 304–13. [DOI] [PubMed] [Google Scholar]
- 26. Raez LE, Lilenbaum R. Chemotherapy for advanced non‐small‐cell lung cancer. Clin Adv Hematol Oncol 2004; 2: 173–8. [PubMed] [Google Scholar]
- 27. Goksel T, Hatipoglu ON, Ozturk C et al. A prospective, multicentre clinical trial comparing cisplatin plus gemcitabine with cisplatin plus etoposide in patients with locally advanced and metastatic non‐small cell lung cancer. Respirology 2005; 10: 456–63. [DOI] [PubMed] [Google Scholar]
- 28. Xiao Z, Bastow KF, Vance JR et al. Antitumor agents. 234. Design, synthesis, and biological evaluation of novel 4beta‐[(4’‘‐benzamido)‐amino]‐4’‐o‐demethyl‐epipodophyllotoxin derivatives. J Med Chem 2004; 47: 5140–8. [DOI] [PubMed] [Google Scholar]
- 29. Xiao Z, Bastow KF, Vance JR, Lee KH. Antitumor agents. Part 227: studies on novel 4’‐O‐demethyl‐epipodophyllotoxins as antitumor agents targeting topoisomerase II. Bioorg Med Chem 2004; 12: 3339–44. [DOI] [PubMed] [Google Scholar]
- 30. Chen TH, Pan SL, Guh JH et al. Moscatilin induces apoptosis in human colorectal cancer cells: a crucial role of c‐Jun NH2‐terminal protein kinase activation caused by tubulin depolymerization and DNA damage. Clin Cancer Res 2008; 14: 4250–8. [DOI] [PubMed] [Google Scholar]
- 31. Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ 2006; 13: 1351–9. [DOI] [PubMed] [Google Scholar]
- 32. Altieri DC. Survivin, cancer networks and pathway‐directed drug discovery. Nat Rev Cancer 2008; 8: 61–70. [DOI] [PubMed] [Google Scholar]
- 33. Fan JR, Peng AL, Chen HC, Lo SC, Huang TH, Li TK. Cellular processing pathways contribute to the activation of etoposide‐induced DNA damage responses. DNA Repair (Amst) 2008; 7: 452–63. [DOI] [PubMed] [Google Scholar]
- 34. Kurz EU, Lees‐Miller SP. DNA damage‐induced activation of ATM and ATM‐dependent signaling pathways. DNA Repair (Amst) 2004; 3: 889–900. [DOI] [PubMed] [Google Scholar]
- 35. Chen CS, Wang YC, Yang HC et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double‐strand breaks by targeting Ku70 acetylation. Cancer Res 2007; 67: 5318–27. [DOI] [PubMed] [Google Scholar]
- 36. Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double‐strand breaks. Cancer Res 2003; 63: 4347–50. [PubMed] [Google Scholar]
- 37. Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr, Stewart L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci USA 2002; 99: 15387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ignatiadis M, Mavroudis D, Veslemes M et al. Sequential versus alternating administration of cisplatin/etoposide and topotecan as first‐line treatment in extensive‐stage small‐cell lung cancer: preliminary results of a Phase III Trial of the Hellenic Oncology Research Group. Clin Lung Cancer 2005; 7: 183–9. [DOI] [PubMed] [Google Scholar]
- 39. Prochazka V, Faber E, Raida L et al. Long‐term outcome of patients with peripheral T‐cell lymphoma treated with first‐line intensive chemotherapy followed by autologous stem cell transplantation. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2011; 155: 63–9. [DOI] [PubMed] [Google Scholar]
- 40. Yoshino M, Okazaki N, Yoshida T et al. A phase II study of etoposide in patients with hepatocellular carcinoma by the Tokyo Liver Cancer Chemotherapy Study Group. Jpn J Clin Oncol 1989; 19: 120–2. [PubMed] [Google Scholar]
- 41. Ekstrom K, Hoffman K, Linne T, Eriksson B, Glimelius B. Single‐dose etoposide in advanced pancreatic and biliary cancer, a phase II study. Oncol Rep 1998; 5: 931–4. [DOI] [PubMed] [Google Scholar]
- 42. Duffy MJ, O’Donovan N, Brennan DJ, Gallagher WM, Ryan BM. Survivin: a promising tumor biomarker. Cancer Lett 2007; 249: 49–60. [DOI] [PubMed] [Google Scholar]
- 43. Yonesaka K, Tamura K, Kurata T et al. Small interfering RNA targeting survivin sensitizes lung cancer cell with mutant p53 to adriamycin. Int J Cancer 2006; 118: 812–20. [DOI] [PubMed] [Google Scholar]
- 44. Chen W, Wang X, Zhuang J, Zhang L, Lin Y. Induction of death receptor 5 and suppression of survivin contribute to sensitization of TRAIL‐induced cytotoxicity by quercetin in non‐small cell lung cancer cells. Carcinogenesis 2007; 28: 2114–21. [DOI] [PubMed] [Google Scholar]
- 45. Jacob S, Aguado M, Fallik D, Praz F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res 2001; 61: 6555–62. [PubMed] [Google Scholar]
- 46. Kim JH, Chae M, Kim WK et al. Salinomycin sensitizes cancer cells to the effects of doxorubicin and etoposide treatment by increasing DNA damage and reducing p21 protein. Br J Pharmacol 2011; 162: 773–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010; 9: 472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 2003; 22: 6068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou M, Gu L, Li F, Zhu Y, Woods WG, Findley HW. DNA damage induces a novel p53‐survivin signaling pathway regulating cell cycle and apoptosis in acute lymphoblastic leukemia cells. J Pharmacol Exp Ther 2002; 303: 124–31. [DOI] [PubMed] [Google Scholar]
- 50. d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA‐damage response. Nat Rev Cancer 2008; 8: 512–22. [DOI] [PubMed] [Google Scholar]
- 51. Norbury CJ, Zhivotovsky B. DNA damage‐induced apoptosis. Oncogene 2004; 23: 2797–808. [DOI] [PubMed] [Google Scholar]
- 52. Valkov NI, Sullivan DM. Tumor p53 status and response to topoisomerase II inhibitors. Drug Resist Updat 2003; 6: 27–39. [DOI] [PubMed] [Google Scholar]
- 53. Blagosklonny MV, El‐Deiry WS. Acute overexpression of wt p53 facilitates anticancer drug‐induced death of cancer and normal cells. Int J Cancer 1998; 75: 933–40. [DOI] [PubMed] [Google Scholar]
- 54. Park JH, Kim TH. Release of cytochrome c from isolated mitochondria by etoposide. J Biochem Mol Biol 2005; 38: 619–23. [DOI] [PubMed] [Google Scholar]
- 55. Roos WP, Batista LF, Naumann SC et al. Apoptosis in malignant glioma cells triggered by the temozolomide‐induced DNA lesion O6‐methylguanine. Oncogene 2007; 26: 186–97. [DOI] [PubMed] [Google Scholar]
- 56. Harker WG, Slade DL, Parr RL, Feldhoff PW, Sullivan DM, Holguin MH. Alterations in the topoisomerase II alpha gene, messenger RNA, and subcellular protein distribution as well as reduced expression of the DNA topoisomerase II beta enzyme in a mitoxantrone‐resistant HL‐60 human leukemia cell line. Cancer Res 1995; 55: 1707–16. [PubMed] [Google Scholar]
- 57. Sunter NJ, Cowell IG, Willmore E, Watters GP, Austin CA. Role of topoisomerase IIbeta in DNA damage response following ir and etoposide. J Nucleic Acids 2010; 2010: 710589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Lactate dehydrogenase assay of QS‐ZYX‐1‐61‐treated A549 non‐small‐cell lung cancer cells.
Fig. S2. Effect of topoisomerase (topo) IIα and IIβ knockdown on suppressing QS‐ZYX‐1‐61‐induced cell growth inhibition in NCI‐H226 and H1299 cells.
Fig. S3. Effect of QS‐ZYX‐1‐61 on cell viability in prostate cancer cells.
Supporting info item
Supporting info item
Supporting info item