

Abstract
Adoptive cell therapy with lymphocytes that have been genetically engineered to express tumor‐reactive T‐cell receptors (TCR) is a promising approach for cancer immunotherapy. We have been exploring the development of TCR gene therapy targeting cancer/testis antigens, including melanoma‐associated antigen (MAGE) family antigens, that are ideal targets for adoptive T‐cell therapy. The efficacy of TCR gene therapy targeting MAGE family antigens, however, has not yet been evaluated in vivo. Here, we demonstrate the in vivo antitumor activity in immunodeficient non‐obese diabetic/SCID/γcnull (NOG) mice of human lymphocytes genetically engineered to express TCR specific for the MAGE‐A4 antigen. Polyclonal T cells derived from human peripheral blood mononuclear cells were transduced with the αβ TCR genes specific for MAGE‐A4, then adoptively transferred into NOG mice inoculated with MAGE‐A4 expressing human tumor cell lines. The transferred T cells maintained their effector function in vivo, infiltrated into tumors, and inhibited tumor growth in an antigen‐specific manner. The combination of adoptive cell therapy with antigen peptide vaccination enhanced antitumor activity, with improved multifunctionality of the transferred cells. These data suggest that TCR gene therapy with MAGE‐A4‐specific TCR is a promising strategy to treat patients with MAGE‐A4‐expressing tumors; in addition, the acquisition of multifunctionality in vivo is an important factor to predict the quality of the T‐cell response during adoptive therapy with human lymphocytes. (Cancer Sci 2012; 103: 17–25)
T‐cell receptor (TCR) gene transfer using retroviral vectors has been shown to be an attractive strategy to redirect the antigen specificity of polyclonal T cells to create tumor‐ or pathogen‐specific lymphocytes.( 1 , 2 , 3 , 4 , 5 , 6 ) This approach is a promising method for the treatment of patients with malignancies that might overcome the limitations of current adoptive T‐cell therapies that have been hampered by difficulties in the isolation and expansion of pre‐existing, antigen‐specific lymphocytes in patients.( 7 , 8 , 9 , 10 ) For the treatment of metastatic melanoma, clinical trials using autologous lymphocytes that have been retrovirally transduced with melanoma/melanocyte antigen‐specific TCR have reported objective cancer regression.( 11 , 12 ) These reports suggest that adoptive cell therapy using TCR gene‐modified lymphocytes is a promising approach to immunotherapy in cancer patients; such reports have encouraged the development of novel TCR gene therapy‐based approaches.
On‐target adverse events, however, have been reported for TCR gene therapies targeting melanocyte differentiation antigens, such as melanoma antigen recognized by T‐cells (MART)‐1 or gp100. Normal tissues in which melanocytic cells exist, such as the skin, eyes, and inner ears, exhibited severe histological destruction, especially when high‐avidity TCR were used.( 12 ) Gene‐modified T cells targeting carcinoembryonic antigen also induced a severe transient inflammatory colitis that served as a dose‐limiting toxicity for all three patients enrolled.( 13 ) Case reports exploring the severe adverse events seen in patients receiving T cells transduced with chimeric antigen receptors bearing the variable regions of human epidermal growth factor receptor type 2 (HER2)/neu‐ or CD19‐reactive antibodies have suggested that these adverse events might be related to the release of cytokines from transferred cells.( 14 , 15 ) These observations highlight the potential risk in the usage of receptor genes that render T cells reactive to both tumor cells and a subset of normal cells.
Cancer/testis antigens are particularly attractive targets for immunotherapy, because of their unique expression profiles. While these antigens are highly expressed on adult male germ cells or placenta, they are typically completely absent from other normal adult tissues, and demonstrate aberrant expression in a variety of malignant neoplasms.( 16 , 17 ) As adult male germ cells do not express MHC class I, CD8+ effector cells theoretically ignore these cells.( 18 ) MAGE‐A, ‐B, and ‐C genes exhibit such an expression pattern, and their immunogenicity as targets for cancer immunotherapy has been well studied.( 19 , 20 , 21 ) MAGE‐A4 expression was reported in 56.6% of serous carcinoma of the ovary, 61.4% of melanoma, 28.4% of non‐small cell lung carcinoma, 20% of hepatocellular carcinoma, 22.3% of colorectal carcinoma, 90.2% of esophageal squamous cell carcinoma, and 6.7% of esophageal adenocarcinoma.( 22 , 23 , 24 , 25 , 26 , 27 , 28 ) These results suggest that TCR gene therapy targeting the MAGE family of antigens, including MAGE‐A4, represents a promising treatment for malignancies that minimizes the risk of severe on‐target toxicity. The feasibility of TCR gene therapy targeting MAGE family antigens in vivo, however, has not previously been evaluated.
In the present study, we isolated rearranged TCRαβ genes from a human CD8+ T‐cell clone that recognizes a MAGE‐A4‐derived peptide, MAGE‐A4143–151, in the context of HLA‐A*2402.( 29 ) Polyclonal human lymphocytes that were retrovirally transduced with these TCR genes demonstrated stable transgene expression and specific cytotoxicity against MAGE‐A4‐expressing tumor cells in vitro.( 30 , 31 ) These results prompted us to confirm the efficacy of the TCR gene‐modified T cells in vivo prior to clinical evaluation.
In this study, we investigated if human lymphocytes genetically engineered to express this MAGE‐A4‐specific TCR could inhibit the growth of MAGE‐A4‐expressing tumors when adoptively transferred into immunodeficient non‐obese diabetic/SCID/γcnull (NOG) mice. We evaluated the in vivo function of the transferred cells, as well as their migration to the tumor site, and the resultant antitumor effect. We addressed if the combination of adoptive cell therapy and vaccination with peptide antigen could influence the antitumor activity of transferred cells.
Materials and Methods
Peripheral blood mononuclear cells. Peripheral blood mononuclear cells (PBMC) were isolated from healthy donors who provided informed consent. Peripheral blood mononuclear cells were cultured in GT‐T503 media (Takara Bio, Otsu, Japan) supplemented with 1% autologous plasma, 0.2% human serum albumin (HSA; Sigma‐Aldrich, St. Louis, MO, USA), 2.5 mg/mL fungizone (Bristol‐Myers Squibb, New York, NY, USA), and 600 IU/mL interleukin‐2. This study was approved by the ethics review committees of Mie University Graduate School of Medicine (Tsu, Japan) and Takara Bio.
Mice. Studies were conducted using 8‐week‐old female NOG mice (Central Institute for Experimental Animals, Kawasaki, Japan) that had been established as described previously.( 32 ) Mice were maintained at the Animal Center of Mie University Graduate School of Medicine. All experimental protocols were approved by the Ethics Review Committee for Animal Experimentation (of Mie University Graduate School of Medicine).
Cell lines. The KE4 (MAGE‐A4+HLA‐A*2402+ human esophageal carcinoma), QG56 (MAGE‐A4+HLA‐A*2402− human lung carcinoma), and T2‐A*2402 (human T, B hybridoma transfected with HLA‐A*2402 cDNA)( 29 ) cell lines were maintained in RPMI‐1640 media (Sigma‐Aldrich) supplemented with 10% FCS, penicillin (100 U/mL), and streptomycin (100 mg/mL).
Retroviral transduction. A retroviral vector encoding MAGE‐A4‐specific TCRα (TRAV8‐1) and TCRβ (TRBV7‐9) genes (MS‐bPa retroviral vector) was described previously.( 30 ) Peripheral blood mononuclear cells were stimulated with 30 ng/mL OKT‐3 (Janssen Pharmaceutical, Titusville, NJ, USA) and 600 IU/mL interleukin‐2 prior to transduction with MS‐bPa particles. Briefly, retroviral solutions were preloaded onto RetroNectin‐coated plates and centrifuged at 2000 g for 2 h, then rinsed with PBS, according to the RetroNectin (Takara Bio)‐bound virus infection method. Cells were then applied onto preloaded plates; PBMC transduced with the MS‐bPa retroviral vector were designated as gene‐modified cells. Control PBMC were treated similarly, except that MS‐bPa was omitted from the cultures; these specimens were designated as unmodified cells.
Tumor challenge. KE4 tumor cells (2.5 × 106 in 0.2 mL PBS) were subcutaneously inoculated into the right flanks of mice. In the indicated experiments, QG56 tumor cells (2.5 × 106 in 0.2 mL PBS) were subcutaneously inoculated in a similar manner. Tumor size was determined by the product of perpendicular diameters measured with calipers. The mice were killed before the mean diameter of the tumor reached 20 mm, according to institutional guidelines. The statistical significance of the difference between groups in tumor growth was evaluated at the last time point.
Adoptive cell transfer. After two washes in saline containing 1% human serum albumin (HSA), gene‐modified or unmodified cells (1 × 108) were suspended in 0.3 mL saline and intravenously injected into a lateral tail vein of the NOG mice. Prior to injection, gene‐modified cells were analyzed for staining with MAGE‐A4143–151/HLA‐A*2402 tetramer and antihuman CD8 mAb to calculate the proportion of tetramer+CD8+ T cells infused. When indicated, HLA‐A*2402‐positive PBMC were pulsed with 1 μM MAGE‐A4141–153 peptide and co‐administered intravenously as a peptide vaccination.
In vitro stimulation and staining of cells. Cells were incubated for 2 h at 37°C with irradiated (45 Gy) stimulator T2‐A*2402 cells, which had been pulsed with 1 μM MAGE‐A4141–153 or HER263–71 (an irrelevant peptide with HLA‐A*2402 binding activity) peptide, at an effector/stimulator ratio of four in the presence of 0.1 mg/mL phycoerythrin (PE)‐conjugated anti‐CD107a (BD Bioscience, San Diego, CA, USA). We then incubated samples for an additional 6 h in 1 mL/mL GolgiStop (BD Bioscience). The cells were then stained with FITC‐conjugated anti‐CD8 (BD Bioscience) mAb. After permeabilization and fixation using a Cytofix/Cytoperm kit (BD Bioscience) according to the manufacturer’s instructions, the cells were stained intracellularly with allophycocyanin (APC)‐conjugated anti‐γ‐interferon (IFN‐γ) (BD Bioscience) and PE–Cy7‐conjugated antitumor necrosis factor (TNF) (BD Bioscience) mAb.
Flow cytometric analysis. PE‐conjugated MAGE‐A4143–151/HLA‐A*2402 tetramer (provided by the Ludwig Institute for Cancer Research, New York, NY, USA) and FITC‐conjugated antihuman CD4 (BD Bioscience), human CD8 (BD Bioscience), and PerCP–Cy5.5‐conjugated antihuman CD3 (BD Bioscience) mAb were used to detect transduced TCR in specific cell populations. Polychromatic analyses were performed as previously described.( 33 ) Cell staining data were acquired using a FACS CantoI flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA), and analyzed using FACSDiva (Becton Dickinson) and FlowJ (Tree Star, Ashland, OR, USA) software.
Immunohistochemical analysis. Formalin‐fixed and paraffin‐embedded specimens were used. After deparaffinization, tissue sections were pretreated with antigen retrieval solution (DAKO high pH solution, DAKO, Glostrum, Denmark) at 95°C for 20 min. As a primary antibody, antihuman CD8 (clone C8/144B; DAKO) was used. Dextran polymer method with EnVision plus (DAKO) was adopted for secondary detection. 3,3′‐Diaminobenzidine was used as chromogen, and hematoxylin counterstain was performed. Infiltrated CD8‐positive tumor infiltrating lymphocytes (TIL) were counted in the selected 10 independent areas with most abundant TIL infiltration. Tumor‐infiltrated, CD8‐positive cells per high power field (0.0625 mm2) were counted using an ocular grid at ×400 magnification. Three independent counts were performed by a board‐certified pathologist (E.S) with no knowledge of the earlier results. The average TIL counts of 10 fields was used for the statistical analyses.
Statistical analyses. Data were expressed as mean ± SD. Differences between groups were examined for statistical significance using the Student’s t‐test. A P‐value less than 0.01 denoted a statistically‐significant difference.
Results
Adoptive transfer of MAGE‐A4‐specific, TCR‐transduced lymphocytes inhibits tumor progression in a dose‐dependent and antigen‐specific manner. We previously reported the successful retroviral transduction of TCRαβ genes recognizing the MAGE‐A4143–151 peptide in an HLA‐A*2402‐restricted manner into polyclonally‐activated human CD8+ T cells. The TCRαβ‐transduced CD8+ T cells exhibited IFN‐γ production and cytotoxic activity against both peptide‐loaded T2‐A*2402 cells and human tumor cell lines, such as KE4, that express both MAGE‐A4 and HLA‐A*2402.( 30 ) To confirm the efficacy of these gene‐modified T cells in vivo prior to clinical evaluation, we examined the antitumor efficacy of adoptive cell therapy with MAGE‐A4‐specific TCR gene‐modified lymphocytes into NOG mice. We anticipated that a clinical trial to evaluate this therapy would involve the transduction of polyclonally‐activated PBMC with TCR genes, followed by the transfer of these cells into patients without purification of the CD8+ T‐cell subset. To mimic these conditions, the NOG mice received TCR gene‐modified lymphocytes without further purification. The TCR gene‐modified and unmodified cells used for the transfer experiments were stained with anti‐CD8 mAb and a MAGE‐A4143–151/HLA‐A*2402 tetramer that specifically detected the transduced TCR (Fig. 1A). As we reported previously, this TCR bound the tetramer in a CD8 molecule‐dependent manner.( 34 ) These T cells were tested for specific reactivity against antigen peptide presented on HLA‐A*2402 (Fig. 1B,C).
Figure 1.
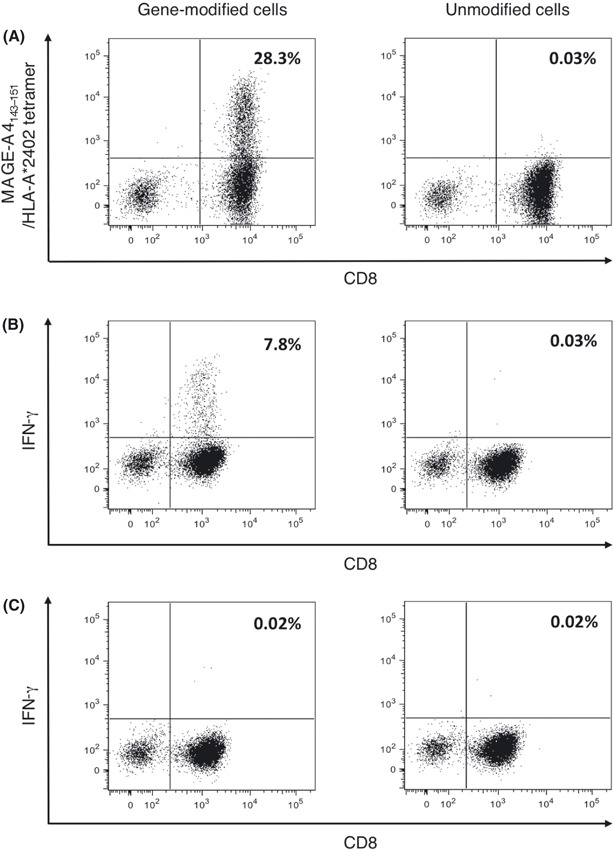
Transduction of melanoma‐associated antigen (MAGE)‐A4‐specific T‐cell receptor (TCR) in human lymphocytes. Peripheral blood mononuclear cells from healthy donors were stimulated with anti‐CD3 mAb and interleukin‐2. Cells were cultured with or without retroviral vector encoding MAGE‐A4‐specific TCR, designated gene‐modified or unmodified cells, respectively. (A) Representative staining for gene‐modified and unmodified cells with MAGE‐A4143–151/HLA‐A*2402 tetramer and antihuman CD8 mAb are shown. (B,C) Gene‐modified and unmodified cells were stimulated with T2‐A*2402 cells pulsed with the MAGE‐A4143–151 peptide (B) or HLA‐A*2402‐binding irrelevant control peptide (C). Representative specific intracellular interferon (IFN)‐γ staining is displayed. Numerical value indicates the percentage of the tetramer+ cells or IFN‐g+ cells among CD8+ cells.
Before transfer, we stained the cells with the MAGE‐A4143–151/HLA‐A*2402 tetramer to calculate the number of tetramer+CD8+ cells. The growth of implanted MAGE‐A4+HLA‐A*2402+ KE4 tumor cells was significantly inhibited when 9 × 106 of tetramer+CD8+ cells were intravenously injected into NOG mice on day 0 (Fig. 2A). The inhibition of KE4 growth required specific recognition of the MAGE‐A4141–153/HLA‐A*2402 complex by the TCR, because unmodified cells derived from the same donor did not alter KE4 growth. In this experiment, 1 × 108 gene‐modified or unmodified lymphocytes derived from the same donor were administered to mice. Although the CD4/CD8 ratio of the in vitro expanded lymphocytes depends on the donor, gene‐modified and unmodified cells derived from the same donor demonstrated similar phenotypes, determined by the expression of cell surface markers, including CD3, CD4, CD8, CD45RA, CD45RO, CD62L, CCR7, CD152, CD25, CD27, and CD28 (data not shown). The growth of the QG56 tumors, which expressed MAGE‐A4, but lacked HLA‐A*2402, was indistinguishable in mice receiving either gene‐modified or unmodified cells (Fig. 2D). Only a modest inhibition of KE4 growth was seen when mice received only 3 × 106 of tetramer+CD8+ cells (Fig. 2B), while no effect was seen upon administration of 1 × 106 of tetramer+CD8+ cells (Fig. 2C).
Figure 2.
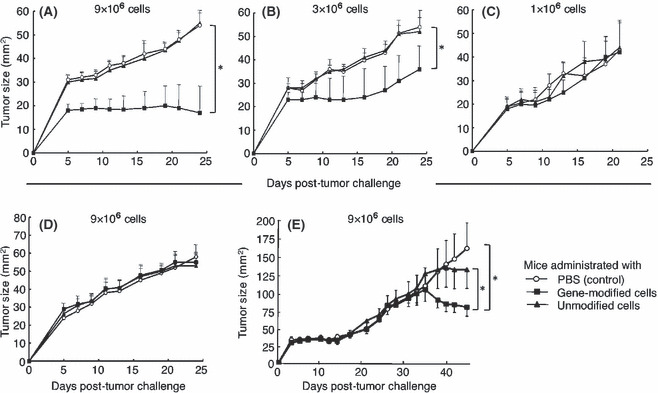
Adoptive transfer of lymphocytes genetically engineered to express MAGE‐A4‐specific T‐cell receptor inhibits human tumor progression in non‐obese diabetic/SCID/γcnull mice. Non‐obese diabetic/SCID/γcnull mice (n = 4 per group) were subcutaneously inoculated with 2.5 × 106 KE4 (A–C) or QG56 (D) tumor cells, and intravenously administered ∼1 × 108 gene‐modified (■) or unmodified (▴) cells or PBS alone (control, ○) on day 0. Total of 9 × 106 (A,D), 3 × 106 (B), or 1 × 106 (C) tetramer+CD8+ cells were confirmed to be adoptively transferred; we subsequently monitored tumor growth over time. (E) Non‐obese, diabetic/SCID/γcnull mice (n = 4 per group) received the treatment 3 days after the subcutaneous inoculation of 2.5 × 106 KE4. Total of 9 × 106 tetramer+CD8+ cells were transferred. Mean tumor size for each group is represented as the average + SD of four mice. Results are representative of three independent experiments. Differences between groups were examined for statistical significance using the Student’s t‐test. *P < 0.01. Numerical value indicates the number of tetramer+CD8+ cells administrated.
We addressed the effect of the adoptive transfer of the gene‐modified cells into the mice with established tumors. We adoptively transferred TCR‐engineered T cells into NOG mice that were inoculated with KE4 tumor cells 3 days earlier. On the day of adoptive T‐cell transfer, we observed the establishment of a KE4 tumor mass in the mice. As shown in Figure 2(E), the administration of gene‐modified cells significantly inhibited the growth of KE4 tumors, although the effect was limited and appeared later compared to the treatment on day 0. Taken together, the adoptive transfer of MAGE‐A4‐specific TCR gene‐modified lymphocytes inhibited human tumor growth in NOG mice in a dose‐dependent and TCR‐specific manner.
Adoptively‐transferred human lymphocytes persist in NOG mice. We monitored the persistence of transferred human lymphocytes in peripheral blood by staining Ficoll‐purified PBMC from NOG mice with mAb specific for human CD8 and CD4. Human CD8+ T cells persisted in NOG mice for more than 40 days after transfer (Fig. 3A). The transferred human CD8+ cells comprised between 10% and 30% of the total peripheral mononuclear cells in NOG mice at almost all time points following transfer of 1 × 108 human lymphocytes. In these experiments, approximately 9 × 106 of the transferred 1 × 108 gene‐modified cells were tetramer+CD8+. The percentage of specifically staining cells in the total peripheral mononuclear cell population was significantly less when mice received 5 × 107 human lymphocytes (Fig. 3B). There was no significant difference in transferred cell survival or percentages between mice receiving gene‐modified and unmodified cells (Fig. 3A,B). Human CD4+ cells comprised less than 10% of all lymphocytes for the first 2 weeks following transfer, but a rapid increase in this population was evident after day 21(Fig. 3C,D). This observation was consistent with reports suggesting that CD4+ T cells play a dominant role in the induction of graft‐versus‐host (GVH) reactions in hosts receiving transfusions.( 35 , 36 ) The NOG mice receiving human lymphocyte transfers demonstrated significant weight loss after day 21, a sign of GVH reactions (Fig. 3E).
Figure 3.
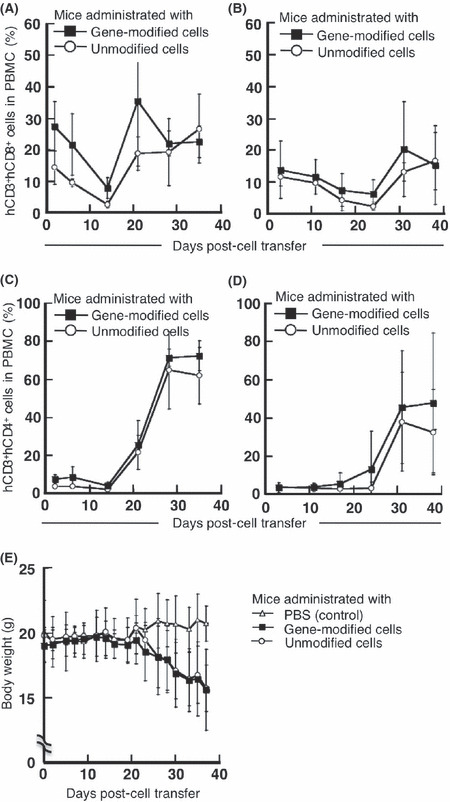
Persistence of adoptively transferred human lymphocytes in non‐obese, diabetic/SCID/γcnull (NOG) mice. Non‐obese, diabetic/SCID/γcnull mice (n = 4 per group) were subcutaneously inoculated with 2.5 × 106 KE4 tumor cells, then intravenously administered 1 × 108 (A,C) or 5 × 107 (B,D) gene‐modified (■) or unmodified (○) cells on day 0. Mononuclear cells were purified from peripheral blood collected from mice on the indicated days. We evaluated the proportion of human CD3+CD8+ (A,B) or CD3+CD4+ (C,D) cells among the mononuclear cell population. (E) We also monitored the body weight of NOG mice administered 1 × 108 gene‐modified (■) or unmodified (○) cells or PBS (control, △) over time. Results are representative of three independent experiments. PBMC, peripheral blood mononuclear cells.
Transferred TCR gene‐modified T cells retain their ability to recognize specific antigens in NOG mice. Lymphocytes harvested from the peripheral blood of NOG mice administered TCR gene‐modified lymphocytes were tested for their antigen‐specific reactivity by intracellular cytokine staining with anti‐IFN‐γ mAb after incubation with peptide‐loaded T2‐A*2402 cells. Antigen‐specific IFN‐γ secretion was detectable by peripheral blood CD8+ cells isolated from mice throughout the 40‐day period after adoptive transfer with either 1 × 108 (Fig. 4A) or 5 × 107 (Fig. 4B) gene‐modified cells. No reactivity of these lymphocytes was seen against T2‐A*2402 cells without loaded peptide (data not shown). Cells from mice that received unmodified lymphocytes did not demonstrate a specific response (Fig. 4A,B). These results indicate that transferred TCR gene‐modified cells remained functional in vivo, recognizing the MAGE‐A4141–153 peptide in the context of HLA‐A*2402. When 5 × 107 cells were transferred, these cells expanded more rapidly in the early phase compared to the group with 1 × 108 cells transferred. We speculate that the adoptive transfer of a lower number of antigen‐specific T cells might induce these cells to expand more rapidly in vivo in the early expansion phase. At the later time points, more antigen‐specific cells persisted in mice receiving 1 × 108 cells.
Figure 4.
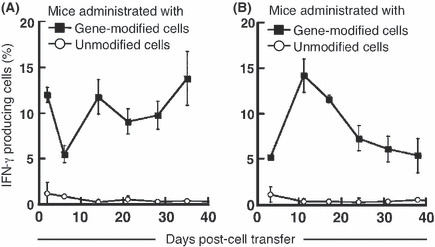
Lymphocytes genetically engineered to express MAGE‐A4‐specific T‐cell receptor‐maintained specific reactivity after in vivo passage. Non‐obese, diabetic/SCID/γcnull mice (n = 4 per group) were subcutaneously inoculated with 2.5 × 106 KE4 tumor cells, then intravenously administered 1 × 108 (A) or 5 × 107 (B) gene‐modified (■) or unmodified (○) cells on day 0. Mononuclear cells were purified from peripheral blood collected from mice on the indicated days. Intracellular γ‐interferon (IFN‐γ) production by these cells was assessed after being stimulated with 1 μM MAGE‐A4141–153 peptide for 6 h. Data are shown as the percentage of IFN‐γ‐producing cells within the total human CD8+ cell population. Results are representative of three independent experiments.
Intratumor infiltration of transferred human CD8+ T cells. To confirm the infiltration of transferred cells into tumor tissue, we examined implanted KE4 and QG56 tumors by immunohistochemical analysis. As antibodies specifically recognizing the transferred TCR (TCRα V8‐1 or TCRβ V7‐9) are not available, we stained tumor specimens with a mAb against human CD8. Significant infiltration of human CD8+ cells was detectable in KE4 tumors harvested from mice as early as 2 weeks after the transfer of gene‐modified cells (Fig. 5A,B). CD8+ cell infiltration in KE4 tumor specimens in the mice that received gene‐modified cells was slightly better than in the mice that received unmodified lymphocytes. However, the difference was not statistically significant (Fig. 5A,B). A similar degree of infiltration was also observed in QG56 tumors. These data were consistent with previous reports analyzing the migration of tumor‐specific T cells by two‐photon laser microscopy that indicated tumor‐specific T cells accumulate in both antigen‐positive and ‐negative tumor tissues to comparable extents, but at different migratory velocities, according to tumor antigen expression.( 37 ) The KE4 tumors in mice that did not receive human lymphocytes lacked any positive staining (Fig. 5B).
Figure 5.
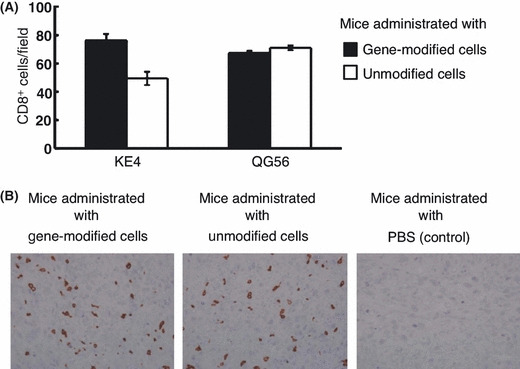
Adoptively‐transferred human CD8+ T cells infiltrate into tumor tissues. Tumor specimens were harvested from non‐obese, diabetic/SCID/γcnull mice 14 days after subcutaneous inoculation with 2.5 × 106 KE4 or QG56 tumor cells, and intravenous administration of 1 × 108 gene‐modified or unmodified cells or PBS (control). We stained formalin‐embedded tumor specimens with an antihuman CD8 monoclonal antibody, clone C8/144B. Average CD8+ TIL counts ± SD in KE4 or QG56 (A) and the representative images from KE4 tissue sections (B) are shown.
Combination of TCR gene therapy and peptide vaccine enhances antitumor efficacy. In animal models of adoptive cell therapy examining the effects against murine tumors with tumor‐specific CD8+ T cells, in vivo vaccinations using agents, such as antigen‐peptide or antigen‐encoding viruses, can increase the antitumor efficacy of adoptive cell therapy.( 9 , 38 ) Therefore, we explored if a peptide vaccination in conjunction with TCR gene‐modified cell transfer could increase the inhibition of tumor growth seen in this model. As the administration of 1 × 106 tetramer+CD8+ cells alone was incapable of inducing tumor growth inhibition in this model (Fig. 2C), we examined if the combination of an in vivo peptide vaccination with cell transfer under these conditions could enhance tumor inhibition. As NOG mice do not possess endogenous antigen‐presenting cells capable of presenting peptide in an HLA‐A*2402‐restricted manner, we used HLA‐A*2402‐positive human PBMC pulsed with the MAGE‐A4143–151 peptide. Tumor‐inoculated NOG mice receiving gene‐modified cells were also administered peptide‐loaded HLA‐A*2402‐positive PBMC derived from the same donor on days 2 and 8 of the tumor challenge. KE4 tumor growth was significantly inhibited in the mice receiving a combination of cell therapy and peptide vaccination in comparison to mice treated by cell therapy alone (Fig. 6A). The peptide vaccination did not alter KE4 growth when combined with the transfer of unmodified cells. The growth of the HLA‐A*2402‐negative QG56 tumor was identical in both groups (Fig. 6B).
Figure 6.
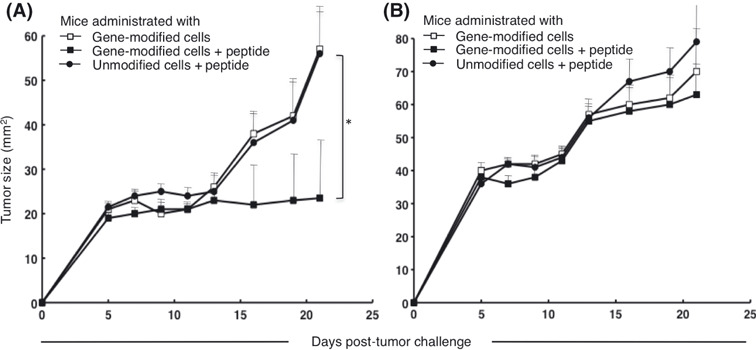
Peptide vaccination enhanced the antitumor efficacy of adoptive therapy using T‐cell receptor, gene‐modified cells. Non‐obese, diabetic/SCID/γcnull mice (n = 4 per group) were subcutaneously inoculated with 2.5 × 106 KE4 (A) or QG56 (B) tumor cells, and intravenously administered 1 × 108 gene‐modified (□) or unmodified (•) cells on day 0. Gene‐modified population included 1 × 106 tetramer+CD8+ cells. We pulsed 4 × 107 peripheral blood mononuclear cells derived from the same donor (HLA‐A*2402 positive) with 1 μM MAGE‐A4141–153 peptide, and intravenously administered these cells into the animals on days 1 and 8 (■ and •). Results are representative of three independent experiments.
Increased multifunctionality in adoptively‐transferred cells when inoculated with peptide vaccine. We previously reported that the multifunctionality of effector cytotoxic T cells (CTL) is a critical determinant of the quality of the T‐cell response and the resultant immunological control of tumor.( 33 , 39 ) We therefore compared the multifunctionality of transferred cells from NOG mice treated with TCR gene‐modified cells and peptide vaccination with that from mice treated by TCR gene cell therapy alone. We assessed IFN‐γ and TNF‐α production and CD107a mobilization by CD8+ T cells at the single‐cell level in specimens harvested from mice on days 2, 7, and 14 after transfer. We selected these functional measures because multifunctionality assessed by these factors defines a sensitive correlate of the immunological control of tumors.( 33 , 39 )
The mice received human lymphocytes with or without peptide vaccination; isolated peripheral blood specimens were tested for their antigen‐specific reactivity of component CD8+ T cells at the indicated time points. On day 2 or 7 after adoptive transfer, we were barely able to detect cells with two or three functions in mice receiving gene‐modified cells without peptide vaccination (Fig. 7); cells with three functions comprised 3.7% of all CD8+ T cells, while bifunctional cells comprised 2.4% on day 14. In contrast, mice receiving combination therapy with gene‐modified cells and peptide vaccination exhibited a population of cells with three and two functions of 1.4% and 2% of the total CD8+ cells, respectively, as early as day 2. Therefore, multifunctional effector CD8+ T cells appear earlier in mice receiving combination therapy in comparison to those receiving cell therapy alone. On day 7, trifunctional and bifunctional cells in mice receiving combination therapy comprised 1.7% and 4.8% of all cells, respectively. The cells with three or two functions were retained as part of the peripheral mononuclear cell population in these animals on day 14.
Figure 7.
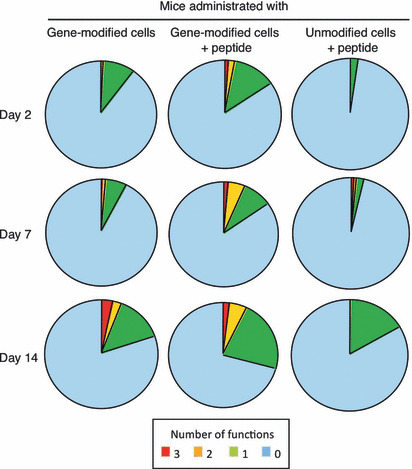
Peptide vaccination increased the multifunctionality of transferred gene‐modified cells. Mice were subcutaneously inoculated with 2.5 × 106 KE4 tumor cells, then intravenously administered 1 × 108 gene‐modified or unmodified cells with or without peptide vaccination. Two, 7, and 14 days after transfer, we collected peripheral blood from mice. After purifying the mononuclear cells in these samples, we evaluated their multifunctionality by measuring γ‐interferon (IFN‐γ) and tumor necrosis factor‐α (TNF‐α) production and CD107a mobilization. Data are summarized in the pie chart, where each wedge represents the frequency of human CD8+ cells expressing all three functions (3), any two functions (2), a single function (1), or no function (0). Results are representative of three independent experiments.
Discussion
Successful clinical responses using adoptive cell therapy with tumor‐reactive T cells in patients with advanced melanoma have encouraged the development of genetic engineering approaches using patient lymphocytes; these studies aim to extend the range of tumor types that can be treated with this technique and to improve the quality of the lymphocytes employed.( 40 , 41 , 42 ) In a recent clinical trial for metastatic synovial cell sarcoma and melanoma, patients were administered autologous lymphocytes genetically engineered to express a high‐avidity TCR against NY‐ESO‐1; objective clinical responses were observed in four (60%) of six patients with synovial cell sarcoma, and five (45%) of 11 patients with melanoma.( 43 ) In this trial, the transferred TCR contained two amino‐acid substitutions in the third complementary determining region of the native TCRα chain that conferred CD8+ T cells with an enhanced avidity. No on‐target toxicities were seen in this trial, in contrast to previous observations of vigorous on‐target toxicity in patients receiving lymphocytes engineered to express melanocyte differentiation antigen‐specific TCR. Genetic engineering also offers the means to endow T cells with enhanced function, as well as resistance to tumor‐mediated immunosuppression through the addition of genes encoding homeostatic or pro‐inflammatory cytokines,( 44 , 45 ) chemokine receptors,( 46 ) anti‐apoptotic molecules,( 47 ) and costimulatory molecules,( 48 , 49 ) as well as the silencing of co‐inhibitory molecules,( 50 ) although these modifications await clinical evaluation. As increased effector function and/or in vivo persistence of cells bearing these modifications might increase on‐target toxicity during therapy, the selection of appropriate target antigens is critical to induce favorable antitumor effects and avoid severe adverse events.
The establishment of an animal model suitable for evaluating the in vivo efficacy and safety of human adoptive cell therapy is an important challenge to facilitate the development of these therapies and prevent toxicity. Non‐obese diabetic/SCID/γcnull‐immunodeficient mice that lack T, B, and natural killer cells, and demonstrate impaired dendritic cell activity, are a helpful animal model to evaluate the in vivo activity of human hematopoietic cells.( 32 ) The NOG mouse model, however, still has limitations, including a homeostatic expansion effect on infused T cells, an allo‐reactive response between infused effector cells and transplanted target cells, and potential GVH reactions. In this study, mice receiving human lymphocytes exhibited severe weight loss, consistent with GVH reaction, which worsened after day 21. Therefore, antitumor efficacy in this model is best evaluated before day 21. Future studies will need to evaluate if the homeostatic proliferation of infused cells and/or a suboptimal allo‐reactivity influenced the treatment effect seen in this model. The lack of an effect by unmodified cells (Fig. 2) and the increased efficacy upon co‐administration of an antigen‐peptide vaccine (Fig. 6), however, strongly suggest that the observed antitumor effect was achieved in a MAGE‐A4‐specific, TCR‐mediated manner. The future development of improved humanized mice will help to better evaluate the optimization of human immunotherapy.
Multifunctionality is the ability of T cells to exhibit multiple functions, including the simultaneous secretion of multiple cytokines, chemokines, or cytotoxic granules at the single‐cell level.( 51 ) The importance of T‐cell multifunctionality has been reported in multiple animal infection models( 52 , 53 ) and in humans infected with HIV, cytomegalovirus, hepatitis B virus, or tuberculosis.( 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 ) We reported the importance of effector T‐cell multifunctionality in antitumor immune response. Specifically, the appearance of multifunctional CD8+ effector cytotoxic T cells in vivo is a critical determinant of effective immunological control of tumors. Regulatory T cells were found to play a role in the inhibition of transferred tumor antigen‐specific T‐cell multifunctionality.( 33 , 39 ) In the present study, effector T‐cell multifunctionality appeared to correlate with the quality of T‐cell responses in adoptive T‐cell therapy utilizing genetically‐engineered human lymphocytes (6, 7). The peptide vaccination did not significantly change the percentage of human CD3+CD8+ cells in the PBMC of NOG mice (data not shown). The TCR‐transduction efficiency in this study was not very high in general. We found that the combination of vaccination with the adoptive transfer of antigen‐specific T cells increased effector T‐cell multifunctionality and made the antitumor effect visible, even with a low number of specific TCR‐transduced T cells transferred. The unmodified cells with background reactivity were the IFN‐γ single producers. We speculate that these cells are positive for IFN‐γ because of their non‐specific activation due to GVH reaction.
To our knowledge, this study represents the first demonstration in vivo of an antitumor effect following the adoptive transfer of human lymphocytes genetically engineered to express a TCR specific for MAGE family antigen. The retroviral vector used in this report is currently under evaluation in a phase I clinical trial designed to treat patients with MAGE‐A4‐expressing esophageal cancer.
In summary, our data suggest that adoptive cell therapy with human lymphocytes engineered to express MAGE‐A4‐specific TCR through retroviral transduction is a promising strategy to treat patients with MAGE‐A4‐expressing tumors. Combination therapy with gene‐modified cell‐adoptive transfer and in vivo vaccination might improve antitumor efficacy, even with low numbers of transferred tumor‐reactive T cells. These data support the rationale to explore clinical trials utilizing gene‐modified lymphocytes prepared using the vector described in this report.
Disclosure Statement
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Morgan RA, Dudley ME, Yu YY et al. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol 2003; 171: 3287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rubinstein MP, Kadima AN, Salem ML et al. Transfer of TCR genes into mature T cells is accompanied by the maintenance of parental T cell avidity. J Immunol 2003; 170: 1209–17. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY‐ESO‐1 antigen‐specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol 2005; 174: 4415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hughes MS, Yu YY, Dudley ME et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T‐cell effector functions. Hum Gene Ther 2005; 16: 457–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coccoris M, Swart E, de Witte MA et al. Long‐term functionality of TCR‐transduced T cells in vivo . J Immunol 2008; 180: 6536–43. [DOI] [PubMed] [Google Scholar]
- 6. Abad JD, Wrzensinski C, Overwijk W et al. T‐cell receptor gene therapy of established tumors in a murine melanoma model. J Immunother 2008; 31: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer 2003; 3: 35–45. [DOI] [PubMed] [Google Scholar]
- 8. Murphy A, Westwood JA, Teng MW, Moeller M, Darcy PK, Kershaw MH. Gene modification strategies to induce tumor immunity. Immunity 2005; 22: 403–14. [DOI] [PubMed] [Google Scholar]
- 9. June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest 2007; 117: 1466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan RA, Dudley ME, Wunderlich JR et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314(5796): 126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson LA, Morgan RA, Dudley ME et al. Gene therapy with human and mouse T‐cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009; 114: 535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parkhurst MR, Yang JC, Langan RC et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19: 620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18: 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 2010; 18: 666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boon T, Old LJ. Cancer tumor antigens. Curr Opin Immunol 1997; 9: 681–3. [DOI] [PubMed] [Google Scholar]
- 17. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 2002; 188: 22–32. [DOI] [PubMed] [Google Scholar]
- 18. Uyttenhove C, Godfraind C, Lethe B et al. The expression of mouse gene P1A in testis does not prevent safe induction of cytolytic T cells against a P1A‐encoded tumor antigen. Int J Cancer 1997; 70: 349–56. [DOI] [PubMed] [Google Scholar]
- 19. van der Bruggen P, Traversari C, Chomez P et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254(5038): 1643–7. [DOI] [PubMed] [Google Scholar]
- 20. De Plaen E, Arden K, Traversari C et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics 1994; 40: 360–9. [DOI] [PubMed] [Google Scholar]
- 21. Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res 2001; 61: 5544–51. [PubMed] [Google Scholar]
- 22. Yakirevich E, Sabo E, Lavie O, Mazareb S, Spagnoli GC, Resnick MB. Expression of the MAGE‐A4 and NY‐ESO‐1 cancer‐testis antigens in serous ovarian neoplasms. Clin Cancer Res 2003; 9: 6453–60. [PubMed] [Google Scholar]
- 23. Peng JR, Chen HS, Mou DC et al. Expression of cancer/testis (CT) antigens in Chinese hepatocellular carcinoma and its correlation with clinical parameters. Cancer Lett 2005; 219: 223–32. [DOI] [PubMed] [Google Scholar]
- 24. Li M, Yuan YH, Han Y et al. Expression profile of cancer‐testis genes in 121 human colorectal cancer tissue and adjacent normal tissue. Clin Cancer Res 2005; 11: 1809–14. [DOI] [PubMed] [Google Scholar]
- 25. Lin J, Lin L, Thomas DG et al. Melanoma‐associated antigens in esophageal adenocarcinoma: identification of novel MAGE‐A10 splice variants. Clin Cancer Res 2004; 10: 5708–16. [DOI] [PubMed] [Google Scholar]
- 26. Tajima K, Obata Y, Tamaki H et al. Expression of cancer/testis (CT) antigens in lung cancer. Lung Cancer 2003; 42: 23–33. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida N, Abe H, Ohkuri T et al. Expression of the MAGE‐A4 and NY‐ESO‐1 cancer‐testis antigens and T cell infiltration in non‐small cell lung carcinoma and their prognostic significance. Int J Oncol 2006; 28: 1089–98. [PubMed] [Google Scholar]
- 28. Forghanifard MM, Gholamin M, Farshchian M et al. Cancer‐testis gene expression profiling in esophageal squamous cell carcinoma: identification of specific tumor marker and potential targets for immunotherapy. Cancer Biol Ther 2011; 12: 191–7. [DOI] [PubMed] [Google Scholar]
- 29. Miyahara Y, Naota H, Wang L et al. Determination of cellularly processed HLA‐A2402‐restricted novel CTL epitopes derived from two cancer germ line genes, MAGE‐A4 and SAGE. Clin Cancer Res 2005; 11: 5581–9. [DOI] [PubMed] [Google Scholar]
- 30. Hiasa A, Hirayama M, Nishikawa H et al. Long‐term phenotypic, functional and genetic stability of cancer‐specific T‐cell receptor (TCR) alphabeta genes transduced to CD8+ T cells. Gene Ther 2008; 15: 695–9. [DOI] [PubMed] [Google Scholar]
- 31. Okamoto S, Mineno J, Ikeda H et al. Improved expression and reactivity of transduced tumor‐specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res 2009; 69: 9003–11. [DOI] [PubMed] [Google Scholar]
- 32. Ito M, Hiramatsu H, Kobayashi K et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002; 100: 3175–82. [DOI] [PubMed] [Google Scholar]
- 33. Imai N, Ikeda H, Tawara I, Shiku H. Tumor progression inhibits the induction of multifunctionality in adoptively transferred tumor‐specific CD8+ T cells. Eur J Immunol 2009; 39: 241–53. [DOI] [PubMed] [Google Scholar]
- 34. Hiasa A, Nishikawa H, Hirayama M et al. Rapid alphabeta TCR‐mediated responses in gammadelta T cells transduced with cancer‐specific TCR genes. Gene Ther 2009; 16: 620–8. [DOI] [PubMed] [Google Scholar]
- 35. Bendle GM, Linnemann C, Hooijkaas AI et al. Lethal graft‐versus‐host disease in mouse models of T cell receptor gene therapy. Nat Med 2010; 16: 565–70. 1p following 70. [DOI] [PubMed] [Google Scholar]
- 36. Schroeder ML. Transfusion‐associated graft‐versus‐host disease. Br J Haematol 2002; 117: 275–87. [DOI] [PubMed] [Google Scholar]
- 37. Mrass P, Takano H, Ng LG et al. Random migration precedes stable target cell interactions of tumor‐infiltrating T cells. J Exp Med 2006; 203: 2749–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Overwijk WW, Theoret MR, Finkelstein SE et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self‐reactive CD8+ T cells. J Exp Med 2003; 198: 569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imai N, Ikeda H, Tawara I et al. Glucocorticoid‐induced tumor necrosis factor receptor stimulation enhances the multifunctionality of adoptively transferred tumor antigen‐specific CD8+ T cells with tumor regression. Cancer Sci 2009; 100: 1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dudley ME, Wunderlich JR, Robbins PF et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dudley ME, Wunderlich JR, Yang JC et al. Adoptive cell transfer therapy following non‐myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005; 23: 2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dudley ME, Yang JC, Sherry R et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 2008; 26: 5233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robbins PF, Morgan RA, Feldman SA et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY‐ESO‐1. J Clin Oncol 2011; 29: 917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hsu C, Hughes MS, Zheng Z, Bray RB, Rosenberg SA, Morgan RA. Primary human T lymphocytes engineered with a codon‐optimized IL‐15 gene resist cytokine withdrawal‐induced apoptosis and persist long‐term in the absence of exogenous cytokine. J Immunol 2005; 175: 7226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu K, Rosenberg SA. Interleukin‐2‐independent proliferation of human melanoma‐reactive T lymphocytes transduced with an exogenous IL‐2 gene is stimulation dependent. J Immunother 2003; 26: 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kershaw MH, Wang G, Westwood JA et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther 2002; 13: 1971–80. [DOI] [PubMed] [Google Scholar]
- 47. Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl‐2 overexpression enhances tumor‐specific T‐cell survival. Cancer Res 2005; 65: 2001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28‐ CD8+ memory effector T cells reconstitutes antigen‐induced IL‐2 production. J Exp Med 2003; 198: 947–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stephan MT, Ponomarev V, Brentjens RJ et al. T cell‐encoded CD80 and 4‐1BBL induce auto‐ and transcostimulation, resulting in potent tumor rejection. Nat Med 2007; 13: 1440–9. [DOI] [PubMed] [Google Scholar]
- 50. Borkner L, Kaiser A, van de Kasteele W et al. RNA interference targeting programmed death receptor‐1 improves immune functions of tumor‐specific T cells. Cancer Immunol Immunother 2010; 59: 1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen‐colour flow cytometry: unravelling the immune system. Nat Rev Immunol 2004; 4: 648–55. [DOI] [PubMed] [Google Scholar]
- 52. Chan KS, Kaur A. Flow cytometric detection of degranulation reveals phenotypic heterogeneity of degranulating CMV‐specific CD8+ T lymphocytes in rhesus macaques. J Immunol Methods 2007; 325: 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darrah PA, Patel DT, De Luca PM et al. Multifunctional TH1 cells define a correlate of vaccine‐mediated protection against Leishmania major. Nat Med 2007; 13: 843–50. [DOI] [PubMed] [Google Scholar]
- 54. De Rosa SC, Lu FX, Yu J et al. Vaccination in humans generates broad T cell cytokine responses. J Immunol 2004; 173: 5372–80. [DOI] [PubMed] [Google Scholar]
- 55. Casazza JP, Betts MR, Price DA et al. Acquisition of direct antiviral effector functions by CMV‐specific CD4+ T lymphocytes with cellular maturation. J Exp Med 2006; 203: 2865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Betts MR, Nason MC, West SM et al. HIV nonprogressors preferentially maintain highly functional HIV‐specific CD8+ T cells. Blood 2006; 107: 4781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Precopio ML, Betts MR, Parrino J et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med 2007; 204: 1405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beveridge NE, Price DA, Casazza JP et al. Immunisation with BCG and recombinant MVA85A induces long‐lasting, polyfunctional mycobacterium tuberculosis‐specific CD4+ memory T lymphocyte populations. Eur J Immunol 2007; 37: 3089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Almeida JR, Price DA, Papagno L et al. Superior control of HIV‐1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 2007; 204: 2473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Duvall MG, Precopio ML, Ambrozak DA et al. Polyfunctional T cell responses are a hallmark of HIV‐2 infection. Eur J Immunol 2008; 38: 350–63. [DOI] [PMC free article] [PubMed] [Google Scholar]