

Abstract
We assessed the relative contribution of the mitochondrial respiratory chain and NADPH (nicotinamide adenine dinucleotide phosphate) oxidase to deoxycorticosterone acetate (DOCA)-salt hypertension in mice. The daily mean arterial pressure was monitored by radiotelemetry in DOCA-salt-treated mice given vehicle or the mitochondrial respiratory chain complex I inhibitor rotenone. This treatment produced remarkable attenuation of DOCA-salt hypertension. Similar results were obtained with other inhibitors of mitochondrial function, including 5-hydroxydecanoate (specific for mitochondrial potassium-ATP channels), benzylguanidine (complexes I and III), and the cell-permeable manganese tetrakis (4-benzoic acid) porphyrin (a mimic of mitochondrial superoxide dismutase). In parallel with the blood pressure-lowering effect of rotenone, the DOCA-salt-induced increases in urinary 8-isoprostane excretion and in reactive oxygen species production of isolated kidney mitochondria were both significantly attenuated. Conversely, the DOCA-salt-induced reduction of urinary nitrate/nitrite excretion was significantly elevated. Following DOCA-salt treatment, mice deficient in NADPH oxidase subunits gp91phox or p47phox exhibited a partial attenuation of the hypertensive response at early but not later time points. Thus, the mitochondrial respiratory chain is a major source of oxidative stress in DOCA-salt hypertension, whereas NADPH oxidase may have a relatively minor role during the early stage of hypertension.
Keywords: hypertension, mitochondria, NADPH oxidase, oxidative stress
The association between oxidative stress and hypertension has been well established.1,2 Oxidative stress is found in almost all models of hypertension, and antioxidant treatments lower blood pressure (BP).2 The mechanism of the prohypertensive action of oxidative stress primarily involves superoxide (O2.)-mediated nitric oxide (NO) inactivation that leads to the diminished NO availability in the vasculature and the kidney.3 In addition, a product of the oxidative stress-mediated peroxidation of arachidonic acid, F2-isoprostane, also accounts for enhanced vascular activity through activation of thromboxane receptors.4,5
It is of great significance to identify the source of oxidative stress in animal models of hypertension. The rodent model of deoxycorticosterone acetate (DOCA)-salt hypertension is a widely used animal model of steroid and salt-sensitive hypertension. This model exhibited severe hypertension associated with aortic and renal O2. production as well as renal monocyte/macrophage infiltration.6,7 The source of oxidative stress in this model, as in many other animal models of hypertension, is still incompletely understood. Increased aortic NADPH (nicotinamide adenine dinucleotide phosphate) oxidase activity and decreased aortic copper/zinc superoxide dismutase activity have been reported in the DOCA-salt hypertensive rat.8 Treatment with NADPH oxidase inhibitor, apocynin, in this model, reduced aortic O2. production and induced a varying degree of BP-lowering effects.6,9 This finding calls for a need to define the role of NADPH oxidase subunits and also to identify other sources of oxidative stress than NADPH oxidase. Neither the treatment with the NO synthase inhibitor, N-nitro-l-arginine, nor the xanthine oxidase inhibitor, allopurinol, affected hypertension or aortic O2. production in DOCA-salt rats,6 thus ruling out the contribution from uncoupled endothelial NO synthase and xanthine oxidase. By tail-cuff plethysmography, mice deficient in either p47phox or gp91phox exhibit blunted hypertensive responses to DOCA-salt treatment, providing further support of the involvement of NADPH oxidase.10,11
On the other hand, there is growing evidence supporting mitochondrial respiratory chain as an important source of oxidative stress in normal and pathological states.12–14 Recently, in DOCA-salt rats, endothelin receptor type A antagonism lowered BP and normalized mitochondrial reactive oxygen species (ROS) production without affecting NADPH oxidase activity, representing indirect evidence for the involvement of mitochondrial-derived ROS.15 This study was undertaken to employ telemetry to assess the relative importance of mitochondria respiratory chain versus NADPH oxidase in DOCA-salt-induced hypertension.
RESULTS
Effect of NADPH oxidase deficiency on the progression of hypertension
The baseline mean arterial pressure (MAP) was lower in both p47phox−/− and gp91phox−/Y mice than in their wild-type (WT) controls (WT: 108±2 mm Hg; p47phox−/− 98±2 mm Hg; P<0.05; gp91phox−/Y: 101±3 mm Hg, P<0.05). A 2-week DOCA-salt treatment induced significant increases in MAP in all three strains of mice. Figure 1 shows the BP response as a change from the baseline. Interaction between mouse strain and day of treatment indicated a different pattern of increase in MAP over treatment day between WT and either strain of the null mice (P<0.05, repeated measures analysis of variance). When compared with the WT control, there was a partial attenuation of the increases in both p47phox−/− and gp91phox−/Y mice within the first 2 or 3 days of DOCA-salt treatment but not at later times (Figure 1).
Figure 1 |. Radiotelemetric determination of the hypertensive response to deoxycorticosterone acetate (DOCA)-salt treatment in wild-type (WT; n=8), gp91phox−/Y (n=6), and p47phox−/− (n=6) mice.

Shown is the change of mean arterial pressure (MAP) from the baseline. *P<0.05 vs WT for the corresponding period.
Effect of NADPH oxidase deficiency on systemic oxidative stress
Urinary 8-isoprostane levels were lower in p47phox−/− mice than in WT controls, but comparable in gp91phox−/Y mice and WT controls. In parallel with the level of MAP, urine 8-isoprostane increased in all three strains of mice following DOCA-salt treatment (Figure 2). The increases in urinary 8-isoprostane levels in WT, p47phox−/−, and gp91phox−/Y mice in response to DOCA-salt were 4.6-fold, 4.1-fold, and 4.5-fold (P>0.05), respectively.
Figure 2 |. Determination of 24-h urine output of 8-isoprostane in deoxycorticosterone acetate (DOCA)-salt-treated wild-type (WT; n=8), gp91phox−/Y (n=6), and p47phox−/− (n=6) mice.

Urinary 8-isoprostane was measured by enzyme immunoassay (EIA). ⋇P<0.05 and *P<0.01 vs Control in the same strain; #P<0.01 vs WT DOCA-salt.
Effect of rotenone and other inhibitors on DOCA-salt hypertension
We examined the effect of the mitochondrial respiratory chain complex I inhibitor rotenone on DOCA-salt hypertension in WT mice. Daily MAP was monitored by telemetry. On the second day of DOCA-salt treatment, MAP in the vehicle-treated mice increased from 110±1.3 to 138±2.0 mm Hg, which remained roughly constant throughout the entire experimental period. Rotenone treatment induced an immediate and significant reduction of MAP; MAP decreased from 144±4 to 114±3 mm Hg (n = 6, P<0.01) at the end of the treatment (Figure 3a). To validate the effect of rotenone, we used several other inhibitors that reduce mitochondrial oxidative stress through diverse mechanisms. 5-hydroxydecanoate (5-HD) is a specific mitochondrial ATP-sensitive potassium channel inhibitor that inhibits mitochondrial oxidative stress via reduction of the mitochondrial inner membrane potential.16–18 Benzylguanidine (BG), an analog of the mitochondrial inhibitor m-iodobenzylguanidine, inhibits complexes I and III.19–23 A cell-permeable manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) scavenges mitochondrial ROS, possibly by mimicking mitochondrial superoxide dismutase.24–27 Like rotenone, 5-HD, BG, and MnTBAP all effectively lowered BP in DOCA-salt-treated mice (Figure 3b–d). Interestingly, none of these compounds affected baseline MAP in normotensive mice. MAP values (in mm Hg) before and after a 5-day drug treatment were as follows: before rotenone 100.5±5.2 vs after rotenone 103.5±5.5 (n=4 in each group, P>0.05); before 5-HD 101.0±2.7 vs after 5-HD 99.3±3.3 (n=3 in each group, P>0.05); before BG 98.0±1.7 vs after BG 96.7±0.9 (n=3 in each group, P>0.05); before MnTBAP 100.0±3.4 vs after MnTBAP 101.0±3.6 (n=4 in each group, P>0.05). Table 1 shows sodium intake in various groups. None of the mitochondrial inhibitors had a significant effect on sodium intake.
Figure 3 |. Radiotelemetric monitoring of mean arterial pressure (MAP).

Wild-type mice were treated with deoxycorticosterone acetate (DOCA)-salt and on day 4 they were randomly divided to receive vehicle, (a) rotenone (ROT), (b)5-hydroxydecanoate (5-HD), (c) benzylguanidine (BG), or (d) manganese tetrakis (4-benzoic acid) porphyrin (MnTBAP) for 5–8 days. *P<0.01 vs DOCA-salt alone. #P<0.05 vs the baseline values on day 4. DOCA-salt alone: n=6; ROT: n=6; 5-HD: n = 5; BG: n=3; MnTBAP: n=4.
Table 1 |.
Sodium intake (mmol per 24 h)
| Day 0 | Day 7 | Day 12 | |
|---|---|---|---|
| Control | 0.33±0.02 | 0.41±0.03 | 0.35±0.02 |
| DOCA-salt | 0.43±0.02 | 3.20±0.36 | 3.09±0.41 |
| DOCA-salt + ROT | 0.39±0.01 | 3.01±0.08 | 3.02±0.31 |
| DOCA-salt + BG | 0.32±0.02 | 3.13±0.30 | 3.21±0.28 |
| DOCA-salt + 5-HD | 0.51±0.11 | 3.73±0.27 | 3.72±0.28 |
Abbreviations: BG, benzylguanidine; DOCA, deoxycorticosterone acetate; 5-HD, 5-hydroxydecanoate; ROT, rotenone.
To verify the specificity of the mitochondrial inhibitors, we performed an assay for NADPH oxidase activity in the kidney homogenates from DOCA-salt-treated mice in the presence and absence of the NADPH oxidase inhibitor apocynin or mitochondrial inhibitors such as MnTBAP, rotenone, 5-HD, or BG. The data indicated that apocynin effectively inhibited NADPH oxidase activity, but mitochondrial inhibitors, including MnTBAP, rotenone, 5-HD, and BG, had no effect (Figure 4).
Figure 4 |. Effects of apocynin and mitochondrial inhibitors on nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity.

The kidneys from deoxycorticosterone acetate (DOCA)-salt-treated mice were homogenized and NADPH oxidase activity was assessed in the presence and absence of apocynin or the mitochondrial inhibitors. *P<0.01 vs control, n=6 for each group.
Effect of rotenone on urinary excretion of 8-isoprostane and nitrogen oxide (NOx)
Urine 8-isoprostane and NOx levels were determined by enzyme immunoassay and the Greiss method, respectively. DOCA-salt elevated the urine output of 8-isoprostane from 275.47±44.65 to 1654.97±316.15 pg per 24 h, which was reduced to 826.04±141.24 pg per 24 h after rotenone treatment (Figure 5a). Conversely, DOCA-salt decreased urine NOx output from 466.73±84.14 to 68.30±15.17 pmol/g, which was restored to 38.11±44.53 pmol per 24 h by rotenone treatment (Figure 5b).
Figure 5 |. Effects of rotenone on systemic and mitochondrial oxidative stress in deoxycorticosterone acetate (DOCA)-salt-treated mice.

(a) The 24-h urine output of 8-isoprostane; (b) the 24-h urine output of nitrate/nitrite (NOx); (c) determination of reactive oxygen species (ROS) generation from isolated mitochondria using the enhanced luminal or isoluminol chemiluminescence; and (d) determination of the protein carbonyls from isolated mitochondria. *P<0.01 vs Control; #P<0.01 vs DOCA-salt alone.
Effect of rotenone on mitochondrial ROS production
To evaluate the mitochondrial-derived ROS, we performed two independent assays—luminal- or isoluminol-enhanced chemiluminescence and protein carbonyl assay—on kidney mitochondria isolated from DOCA-salt mice receiving vehicle or rotenone. DOCA-salt treatment induced an ~2.5-fold increase in both mitochondrial ROS production (Figure 5c) and protein carbonyls (Figure 5d), both of which were remarkably inhibited by rotenone. To evaluate the purity of the mitochondrial preparation, we compared the abundance of gp91phox, p47phox, and cytochrome b between the supernatant and the mitochondrial fraction. As expected, mitochondrial protein cytochrome b was detected predominantly in the mitochondrial fraction, whereas cytosolic protein p47phox and membranous protein gp91phox were mostly in the supernatant (Figure 6). This supernatant was prepared at low centrifugation (3500 g) and expected to contain both cytosolic and membranous fractions.
Figure 6 |. Evaluation of the purity of the mitochondrial preparation.

The presence of (a) p47phox, (b) gp91phox, and (c) cytochrome b (cyt b) in the supernatant and the mitochondrial fraction. The pelleted mitochondria and the supernatant were isolated from the mouse kidney, loaded in the same amount, and analyzed for gp91phox, p47phox, and cyt b using immunoblotting. Shown are representatives of 2 to 3 experiments.
Effect of rotenone and NADPH oxidase subunit deficiency on DOCA-salt-induced albuminuria
Proteinuria is an important feature of DOCA-salt-induced renal injury. Therefore, we compared the effects of rotenone and NADPH oxidase subunit deficiency on urinary albumin excretion in DOCA-salt-treated mice. DOCA-salt elevated the urine albumin excretion from 28.24±4.83 to 145.54±15.64 μg per 24 h (P<0.01), which was reduced to 90.80±15.55 μg per 24 h (P<0.01) by rotenone treatment (Figure 7a). In sharp contrast, DOCA-salt-induced albuminuria was not inhibited, but was modestly aggravated in both gp91phox−/Y and p47phox−/− mice (baseline: 26.18±1.87 in WT, 18.79±3.12 in p47phox−/−, and 15.55±2.23 μg per 24 h in gp91phox−/Y; DOCA-salt: 150.67±14.18 in WT, 172.80±12.11 in p47phox−/−, and 196.34±12.14 μg per 24 h in gp91phox−/Y; Figure 7b).
Figure 7 |. Effects of rotenone and gp91phox and p47phox deficiency on deoxycorticosterone acetate (DOCA)-salt-induced albuminuria.
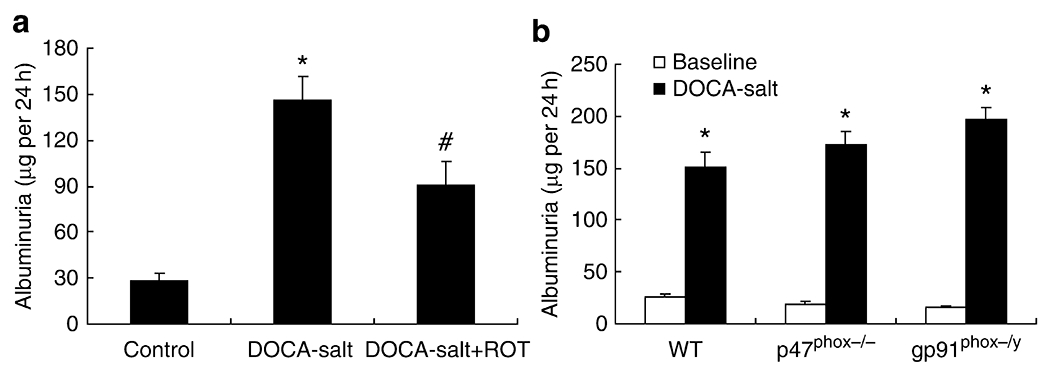
(a) Urinary albumin excretion in control, DOCA-salt treated, or DOCA-salt + rotenone-treated wild-type (WT) mice; (b) urinary albumin excretion in vehicle and rotenone-treated wild type, gp91phox−/Y, and p47phox−/− mice. *P<0.01 vs Control; #P<0.01 vs DOCA-salt alone. (a) n=6 in each group. (b) WT: n=8; gp91phox−/Y: n=6; p47phox−/−: n=6.
The toxicity profile of rotenone
The toxicity of rotenone was assessed by monitoring changes in body weight and performing blood biochemical analysis, tissue histological analysis, and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. Body weight was monitored daily before and during the induction of hypertension. After 2 weeks of DOCA-salt treatment, body weight increased in the vehicle group from 29.02±1.18 to 32.03±1.24 g (P<0.01), whereas the body weight of the rotenone group remained roughly constant (28.87±1.17 vs 28.18±1.73 g, P>0.05). Plasma blood urea nitrogen, creatinine, lactate dehydrogenase, and creatine kinase-MB were determined, and none of these parameters was significantly affected by rotenone treatment (Table 2). Hematoxylin and eosin staining of major internal organs, including brain, heart, liver, and kidney, did not reveal obvious abnormalities associated with rotenone treatment (Figure 8), nor did TUNEL assay on the kidney. The numbers of apoptotic cells in the control, DOCA-salt-alone, and DOCA-salt plus rotenone groups were 1.02±0.18, 1.22±0.19, and 1.27±0.22 per field (P>0.05), respectively. Body weights were determined before and 5 days after treatment with 5-HD, BG, or MnTBAP and were not significantly affected by any of these compounds (before 5-HD 30.025±0.62 vs after 5-HD 29.65±1.3 g, n = 4 in each group, P>0.05; before BG 29.8±0.1.2 vs after BG 29.6±0.55 g, n=3 in each group, P>0.05; before MnTBAP 27.66±1 vs after MnTBAP 27.19±0.75 g, n=4 in each group, P>0.05).
Table 2 |.
Plasma BUN, creatinine, LDH, and CK-MB
| BUN (mg/dl) | Creatinine (mg/dl) | LDH (U/l) | CK-MB (U/l) | |
|---|---|---|---|---|
| Control | 13.67±1.45 | 0.23±0.03 | 1195.33±172.58 | 25.07±2.32 |
| DOCA-salt | 10.67±1.20 | 0.25±0.04 | 1351.00±279.58 | 27.07±5.97 |
| DOCA-salt + ROT | 13.00±1.58 | 0.22±0.03 | 1198.50±129.56 | 26.33±2.79 |
Abbreviations: BUN, blood urea nitrogen; CK-MB, creatine kinase-MB; DOCA, deoxycorticosterone acetate; LDH, lactate dehydrogenase; ROT, rotenone.
Figure 8 |. Effect of rotenone on tissue histology.

Wild-type (WT) mice were treated as in Figure 3 and tissue sections were hematoxylin and eosin (HE) stained. Shown are representative sections of (a–c) brain, (d–f) heart, (g–i) liver, and (j–l) kidney from (a, d, g, l) control, (b, e, h, k) deoxycorticosterone acetate (DOCA)-salt, and (c, f, i, l) DOCA-salt with rotenone-treated mice. Micrographs were taken at × 200 magnification.
Effect of rotenone and apocynin on aldosterone-induced ROS production in cultured vascular smooth muscle cells (VSMCs)
We attempted to use a cell culture model to compare the direct effects of rotenone and the NADPH oxidase inhibitor apocynin on aldosterone-induced ROS production. In cultured VSMCs, exposure for 40 min to aldosterone in the range of 100–1000 nmol/l dose dependently induced ROS production as assessed by 2′,7′-dichlorofluorescein fluorescence (Figure 9a). Based on this observation, 1000 nmol/l was used for subsequent experiments to determine the time course of ROS production and also the effects of rotenone and apocynin. The ROS production in response to aldosterone treatment was detected as early as 10 min and remained elevated over the next 40-min period. The stimulation of ROS production was normalized by 10 μmol/l rotenone at all time points and was partially attenuated by 100 μmol/l apocynin only at 10 min but not at later time points (Figure 9b).
Figure 9 |. Effect of rotenone and apocynin on the aldosterone-induced reactive oxygen species (ROS) generation in vascular smooth muscle cells (VSMCs).
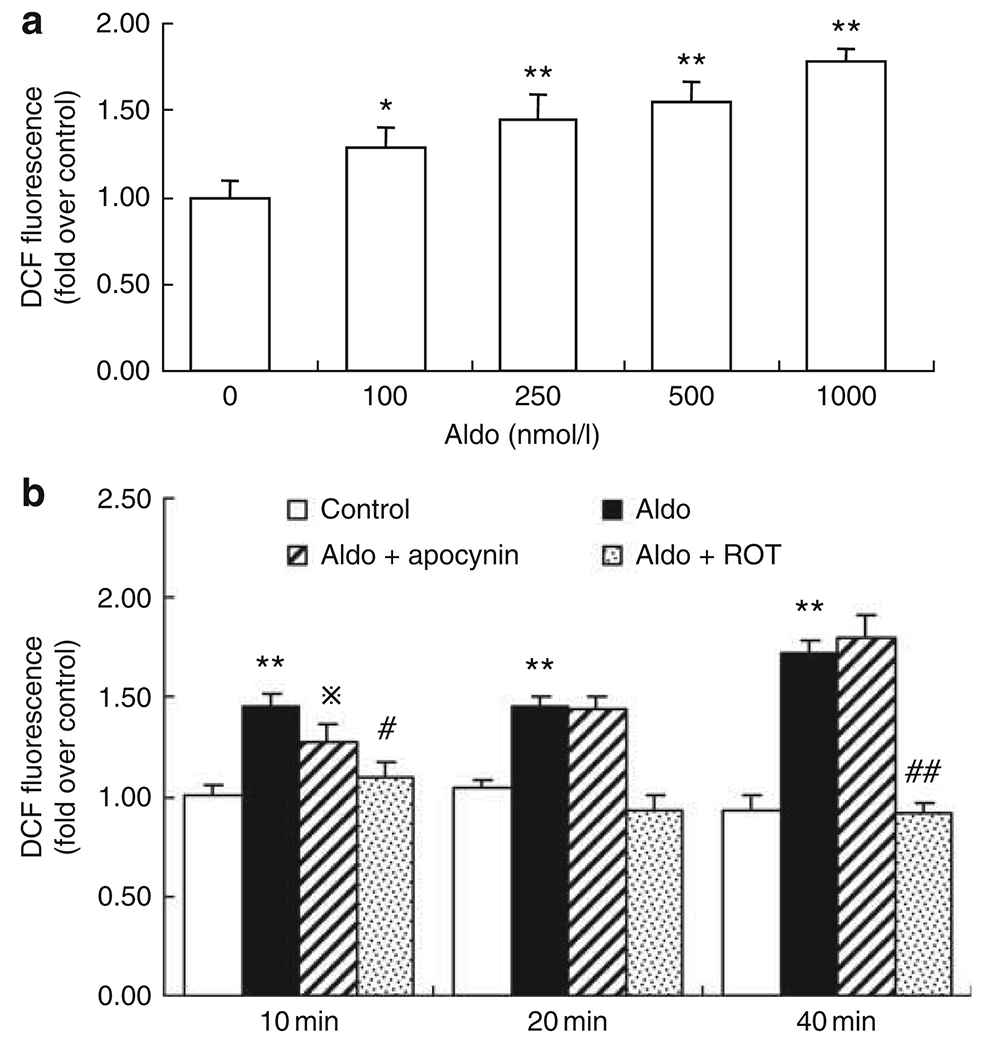
(a) Confluent VSMCs in 96-well plates were treated with the indicated dose (100–1000 nmol/l) of aldosterone for 40 min, or (b) pretreated with apocynin (100 μmol/l) or rotenone (10 μmol/l), and then stimulated by 1000 nmol/l aldosterone for the indicated time in the presence of DCF. Fluorescence was quantified using FLUOstar OPTIMA (BMG Labtech, Cary, NC). *P<0.05, **P<0.01 vs control; ⋇P<0.05, #P<0.01 vs aldo-treated VSMCs. DCF, 2′,7′-dichlorofluorescein.
DISCUSSION
The mitochondrial respiratory chain and NADPH oxidase have emerged as major sources of ROS production.28,29 In the present study, using a radiotelemetric technique, we critically assessed the relative importance of the two types of ROS sources in a mouse model of DOCA-salt hypertension. Treatment with the mitochondrial complex I inhibitor rotenone induced a marked reduction of MAP associated with parallel attenuation of systemic oxidative stress, mitochondrial ROS production, and albuminuria. Several other inhibitors that reduce mitochondrial oxidative stress through diverse mechanisms all produced a BP-lowering effect similar to that of rotenone. In contrast, the hypertensive responses to DOCA-salt treatment in neither p47phox−/− nor gp91phox−/Y mice were significantly different from those of WT controls except at the early phase in the development of hypertension. In cultured VSMCs, rotenone effectively prevented aldosterone-induced ROS production up to 40 min, whereas the NADPH oxidase inhibitor apocynin only inhibited it at 10 min but not at later time points. These results favor the mitochondrial respiratory chain as a major source of oxidative stress in the mouse model of DOCA-salt hypertension, whereas NADPH oxidase may have a relatively minor role.
NADPH oxidase is a multicomponent enzyme comprising the membrane-associated subunits p22phox and gp91phox and the cytosolic subunits p47phox, p67phox, and p40phox. Among these subunits, gp91phox and p47phox are of particular importance, as the former contains the catalytic domain and the latter is necessary for cytosolic subunit translocation and for initiation of NADPH oxidase assembly. In recent years, four homologs of gp91phox (Nox2), Nox1 (Suh et al.30) Nox3, Nox4, and Nox5 (Lambeth29) have been identified, whereas the homologs of p47phox have not yet been found. The involvement of NADPH oxidase in rat models of DOCA-salt hypertension was initially suggested through pharmacological approaches.6,9 Subsequently, mice deficient in p47phox or gp91phox have been shown to be resistant to the development of hypertension following DOCA-salt treatment.10,11 Surprisingly, we found that the hypertensive responses to DOCA-salt treatment in both gp91phox- and p47phox-deficient mice remained largely intact. The exact reason for the disparities is unclear, but it could be related to different techniques used for measurements of BP. In previous studies,10,11 arterial pressure was measured by tail-cuff plethysmography, a noninvasive measure that reports only systolic pressure and requires restraint and tail-cuff inflation that introduces stress. In the present study, however, MAP was monitored by radiotelemetry, which is not associated with the limitations of tail-cuff method. Second, the aforementioned disparities may be related to differences in experimental protocols for generating mouse models of DOCA-salt hypertension. Uninephrectomy was performed and a standard solid chow diet used in the previous two studies, whereas we did not perform uninephrectomy but used high-salt diets (1.5% NaCl in the gelled formulation). The use of the gelled formulation of a high-salt diet in conjunction with saline as the drinking fluid in the present study may make sodium loading more efficient. Third, Landmesser et al.31 only presented the absolute BP values in WT and p47phox−/− mice after DOCA-salt treatment, whereas we showed the change of MAP from the baseline. As p47phox−/− mice had low baseline BP, the magnitude of the change of BP will be more informative. Of note, the involvement of Nox4 cannot be excluded in our models because the activity of Nox4 does not require known cytosolic oxidase proteins or the GTPase Rac.32 Unlike other Nox proteins, Nox4 produces large amounts of hydrogen peroxide constitutively.32
Mitochondria are important cellular organelles for oxidative phosphorylation, the energy production pathway that is associated with the generation of the by-product superoxide anion (O2.). Approximately 0.4–4% of the molecular oxygen (O2) consumed by the mitochondria is reduced by a single electron transfer from the initial steps of the electron transport chain to form O2..33–35 There is a growing appreciation of the mitochondria respiratory chain as an important source of oxidative stress under states of aging, diabetes, and so on.14,36–40 Compared with an overwhelming understanding about NADPH oxidase in hypertension, scant information is available about the role of mitochondria in this disease process. The mitochondrial complex I inhibitor rotenone is widely used to block mitochondrial-derived ROS production in various cell types.41 Using radiotelemetric technique, we provide new evidence that the inhibition of mitochondrial ROS production by rotenone induced a striking BP-lowering effect in the mouse model of DOCA-salt hypertension. This effect is associated with the suppression of systematic oxidative stress and the almost compete normalization of ROS production from isolated kidney mitochondria, and with the restoration of NO levels. These observations support the notion that the BP-lowering effect of rotenone may be related to inhibition of mitochondrial ROS production.
Given the important role of mitochondria in energy production, rotenone is suspected to induce some degree of toxicity. An issue arises as to whether the BP-lowering effect is secondary to rotenone-induced organ damage. Therefore, we performed histological analysis of several internal organs as well as a TUNEL assay on the kidney. These assays did not reveal obvious tissue injuries associated with rotenone treatment. During 2 weeks of DOCA-salt treatment, the body weight of the animals increased ~ 3g (~ 10% body weight), far exceeding natural growth in adulthood, a possible sign of fluid retention. In contrast, body weight gain was completely prevented by rotenone treatment. This phenomenon may reflect lack of fluid retention following rotenone treatment. A study from the National Toxicology Program reported that rotenone treatment for 14 days at doses of ≥ 1200 p.p.m. induced decreases in body weight gain in rats but not in mice, and that this dose range was well tolerated for up to 2 years.42 Overall, the relatively low dose and short duration of rotenone treatment in the present study was unlikely to have produced significant toxicities, which may account for the remarkable BP-lowering effect of the compound.
Considering the specificity issue of rotenone, we used several other inhibitors that interfere with various steps of mitochondrial ROS production. Mitochondrial superoxide production occurs in complexes I and III of the electron transport chain.43 This process is modulated by the mitochondrial inner membrane potential, which is determined by potassium influx through the mitochondrial ATP-sensitive potassium channel.44 A specific mitochondrial ATP-sensitive potassium channel inhibitor, 5-HD, inhibits mitochondrial oxidative stress, whereas the channel openers increase it.16–18 BG is an analog of the mitochondrial inhibitor m-iodobenzylguanidine, which inhibits complexes I and III.19–23 MnTBAP belongs to a class of catalytic antioxidants. A sizable literature shows that MnTBAP is capable of scavenging mitochondrial ROS, possibly by mimicking mitochondrial superoxide dismutase.24–27 Like rotenone, 5-HD, BG, and MnTBAP all effectively lowered BP in DOCA-salt-treated mice. The consistent results obtained with various mitochondrial inhibitors have substantiated the role of mitochondrial ROS in mediating DOCA-salt hypertension.
Another possible explanation for our findings is that different oxidant-producing systems, namely, NADPH oxidase and the mitochondrial respiratory chain, act in concert to sustain ROS production at different stages of the hypertension development. Landmesser et al.11 suggest that the initial activation of NADPH oxidase may lead to greater ROS production later from other sources, such as NO synthase uncoupling. The activation of NADPH oxidase may trigger ROS production from the mitochondrial respiratory chain in addition to NO synthase uncoupling. Along this line, we found that in cultured VSMCs, apocynin partially attenuated the initial rise of aldosterone-induced ROS production. In this cell culture model, rotenone almost completely blocked the ROS production, again supporting the mitochondrial respiratory chain as a dominant source of oxidative stress during aldosterone excess. The two oxidant-producing systems may interact to initiate the ROS generation in response to mineralocorticosteroid excess and the nature of such interaction needs to be explored in future studies.
The present study is limited in the inability to address whether oxidative stress is a cause or a consequence of hypertension. A temporal analysis of oxidative stress levels might be helpful, although it cannot establish causality. This experiment was not performed because of technical difficulty. Instead, this issue has been partially addressed by the in vitro study showing that rotenone rapidly and effectively blocked aldosterone-induced ROS generation. However, the in vitro data are only suggestive of, but do not prove, a cause–effect relationship.
In summary, we used telemetry in the present study to thoroughly examine the relative contribution of the mitochondrial respiratory chain, in comparison with that of NADPH oxidase, to DOCA-salt hypertension in mice. The hypertensive response to DOCA-salt largely remains intact in both gp91phox- and p47phox-deficient mice except at the early phases of the hypertension development. In contrast, pharmacological suppression of mitochondrial ROS levels produces a marked BP-lowering effect. Our results suggest that the mitochondrial respiratory chain may represent a major source of ROS production in DOCA-salt hypertension, whereas NADPH oxidase may have a relatively minor role. Future studies are needed to critically evaluate the relative importance of the two ROS-generating systems in other forms of hypertension. A better understanding of the source of oxidative stress in animal models of hypertension may be helpful for the development of novel antihypertensive therapies.
MATERIALS AND METHODS
Animals
The entire experimental cohorts of male mice deficient in gp91phox (gp91phox−/Y, stock no. 002365) and p47phox (p47phox−/−, stock no. 004742) and WT control (inbred C57BL/6) mice at the age of 3 to 4 months (25–30 g) were obtained from Jackson Laboratories (Bar Harbor, ME). The genotyping of the mutant strains has been validated by the vendor. No breeding was conducted at the University of Utah. gp91phox-deficient mice were originally produced from the laboratory of MC Dinauer,45 and p47phox-deficient mice carry a spontaneous mutation of the Ncf1 gene, which leads to a complete loss of p47phox protein.46 Both strains of mice were backcrossed at least 8 × to the C57BL/6 background at Jackson Laboratories. All mice were housed in an air-conditioned room with a 12-h light/dark cycle. All procedures and protocols were in accordance with the guidelines set by the laboratory animal care committee at the University of Utah.
Animal experimental protocols
Protocol 1:
This protocol was designed to evaluate the role of NADPH oxidase subunits in DOCA-salt hypertension. The radiotelemetric device was implanted into gp91phox−/Y (n=6), and p47phox−/− (n=6) and WT (n=8) mice through catheterization of the carotid artery (model no. TA11PA-C20; DSI, St Paul, MN) as previously described.47 After allowing 1 week for recovery, baseline MAP was collected. A slow-release (21-day) 50 mg DOCA pellet was then implanted subcutaneously through a midscapular incision. Following the surgery, animals received 1.5% NaCl in gelled diets (TestDiet, cat no. 0007551, Richmond, IN) and 1% NaCl in drinking water. The gelled diets were made as previously described.48 Daily MAP was recorded as mean values of 4-h recordings from 0900 to 1300 hours as previously described.47 Mice were placed in metabolic cages (Hatteras Instruments, Cary, NC) for 24-h urine collections before and at the end of experiments.
Protocol 2:
This protocol was designed to evaluate the involvement of mitochondrial oxidative stress in DOCA-salt hypertension in mice. The WT mice were implanted with the radiotelemetric device and treated with DOCA-salt as described above. After stabilization of BP for 4 days, the mice were randomized to receive vehicle, rotenone (600 p.p.m. in a gelled diet), 5-HD (10 mg/kg/day, intraperitoneal), BG (10 mg/kg/day, intraperitoneal), or MnTBAP (15 mg/kg/day, intraperitoneal). All compounds were purchased from Sigma (St Louis, MO) unless otherwise specified.
BP and heart rate measurements
The telemetric device was implanted into mice through catheterization of the carotid artery, according to the manufacturer’s instructions (model no. TA11PA-C20; DSI). Animals were allowed to recover from surgery for at least 1 week before DOCA-salt treatment. Daily MAP and heart rate were monitored every 20 min from 0900 to 1300 hours. The daily value of MAP and heart rate in each animal is the mean value of the 4-h recording.
Blood sample analysis
The plasma samples were diluted 1:50 with distilled water before assays. Plasma blood urea nitrogen, creatinine, lactate dehydrogenase, and creatine kinase isoenzyme MB were measured using an autoanalyzer (Victors 750, Johnson & Johnson, Rochester, NY).
Urine analysis of albumin, 8-isoprostane, and NOx
Urinary concentrations of albumin and 8-isoprostane were determined using enzyme immunoassay kits from Exocell (Philadelphia, PA) and Cayman (Ann Arbor, MI), respectively. Before 8-isoprostane enzyme immunoassay, urine samples were purified using an 8-isoprostane affinity column (Cayman Chemicals model no. 416358). Urinary NOx concentrations were determined using the Griess method (nitrate/nitrite colorimetric assay kit; Cayman Chemicals). For all the assays, samples were run in duplicate and results averaged.
Assays for mitochondrial oxidative stress
Mitochondria from the kidneys of DOCA-salt group and DOCA-salt plus rotenone group were isolated using a kit from Sigma (MITO-ISO1; Sigma Chemical) according to the manufacturer’s protocol. Briefly, whole kidney was homogenized in Extraction Buffer A (50 mmol/l HEPES, 1 m mannitol, 350 mmol/l sucrose, and 5 mmol/l EGTA, pH 7.5) and centrifuged twice at 1200 g for 5 min to pellet unbroken cells, nuclei, and large debris. The aqueous phase was carefully transferred to a new tube and centrifuged twice at 3500 g for 10 min to pellet the mitochondria. The isolated mitochondria were resuspended in storage buffer and protein concentration was determined by Coomassie reagent. The rate of ROS generation by intact mitochondria (0.50–0.60 mg total protein in 1 ml of assay) was detected by enhancer-amplified luminal chemiluminescence using superoxide/reactive oxygen species determination kit (WPI, Sarasota, FL) according to the manufacture’s protocol. Luminescence was measured every 1.8 s for 3 min.
Assay for mitochondrial protein carbonyls
Mitochondrial protein carbonyls were determined by derivatization with 2,4-dinitrophenylhydrazine, according to the report from Levine et al.36 Carbonyl content was measured spectrophotometrically at 366 nm. Results were expressed as nanomoles carbonyl per mg protein.
Assay for NADPH oxidase activity
The homogenates of the kidney from DOCA-salt- treated mice were prepared in Krebs solution using a glass-to-glass homogenizer, and NADPH oxidase activity was measured as previously reported.49 The activity was expressed as arbitrary units per mg protein.
Immunoblotting
The cytosolic and mitochondrial proteins (30 μg each) were denatured in boiling water for 10 min, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The blots were blocked overnight with 5% nonfat dry milk in Tris-buffered saline, followed by incubation for 1 h with mouse anti-gp91phox or anti-p47phox monoclonal antibodies (BD Biosciences, San Jose, CA) at a dilution of 1:1000. After washing with Tris-buffered saline, blots were incubated with a goat anti-horseradish peroxidase-conjugated secondary antibody (1:1000 dilution) and visualized with ECL kits (Amersham, Piscataway, NJ).
Histological analysis
Several internal organs including brain, heart, liver, and kidney were fixed in 3% paraformaldehyde, embedded in paraffin, and then sectioned (2 μm thickness). Sections were stained with hematoxylin/eosin and examined with light microscope. Magnification of micrographs was × 200.
TUNEL assay
Cells undergoing apoptosis in the kidney were identified using an ApopTag In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA). After being dewaxed, the tissue sections were treated with proteinase K (20 μg/ml) and incubated with equilibration buffer in a humidified chamber for 10 min at room temperature, followed by incubation with working-strength TdT enzyme solution in a humidified chamber at 37 °C for 1 h. The reaction was terminated by incubation in working-strength stop/wash buffer for 10 min at room temperature. After being rinsed with phosphate-buffered saline, the tissue sections were incubated with fluorescein isothiocyanate-conjugated antidigoxigenin antibody in a humidified chamber for 30 min at room temperature. As a positive control, slides were treated with DNase (20 Kunitz U/ml) and the slides for the negative control were treated with buffer lacking TdT. The number of TUNEL-positive cells was counted at 20 different fields in each section under × 200 magnification.
Cell culture
The normal human aorta VSMC line (American Type Culture Collection (ATCC) CRL-1999, Manassas, VA) was maintained in F12K Kaighn’s modification media containing 10 mmol/l N-tris [hydroximethyl]methyl-2-aminoethanesulfonic acid, 0.3 mmol/l l-ascorbic acid, 0.001 mmol/l insulin, 0.001% transferrin, 58 nmol/l sodium selenite, 0.003% endothelial growth supplement, 1% antibiotic/antimycotic, 1% l-glutamine, 1% HEPES, 1% non-essential amino acids, 1% sodium pyruvate, and 10% heat-inactivated fetal calf serum. The cells were split once a week as per ATCC recommendation and were maintained for no more than 10 divisions.
Statistical analysis
All data are given as mean ± s.e. For analysis of the hypertensive response in Figure 1, repeated measures analysis of variance was performed to detect an interaction (time × strain). Differences between groups in other figures were assessed by analysis of variance and Student-Newman-Keuls post hoc test for multiple comparisons, with a P-value < 0.05 considered to be significant.
ACKNOWLEDGMENTS
This work was supported by NIH Grants HL079453, DK079162, VA Merit Review (to TY). TY is an Established Investigator from American Heart Association and Research Career Scientist at the Department of Veterans Affairs.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? Am J Physiol Regul Integr Comp Physiol 2005; 289: R913–R935. [DOI] [PubMed] [Google Scholar]
- 2.Wilcox CS. Reactive oxygen species: roles in blood pressure and kidney function. Curr Hypertens Rep 2002; 4: 160–166. [DOI] [PubMed] [Google Scholar]
- 3.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87: 840–844. [DOI] [PubMed] [Google Scholar]
- 4.Salahudeen AK, Reckelhoff JF, Morrow JD et al. F2-isoprostanes and the kidney. Drug News Perspect 1998; 11: 287–290. [DOI] [PubMed] [Google Scholar]
- 5.John GW, Valentin JP. Analysis of the pulmonary hypertensive effects of the isoprostane derivative, 8-iso-PGF2alpha, in the rat. Br J Pharmacol 1997; 122: 899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beswick RA, Dorrance AM, Leite R et al. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension 2001; 38: 1107–1111. [DOI] [PubMed] [Google Scholar]
- 7.Manning RD Jr, Meng S, Tian N. Renal and vascular oxidative stress and salt-sensitivity of arterial pressure. Acta Physiol Scand 2003; 179: 243–250. [DOI] [PubMed] [Google Scholar]
- 8.Wu R, Millette E, Wu L et al. Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J Hypertens 2001; 19: 741–748. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh M, Wang HD, McNeill JR. Role of oxidative stress and nitric oxide in regulation of spontaneous tone in aorta of DOCA-salt hypertensive rats. Br J Pharmacol 2004; 141: 562–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujii A, Nakano D, Katsuragi M et al. Role of gp91phox-containing NADPH oxidase in the deoxycorticosterone acetate-salt-induced hypertension. Eur J Pharmacol 2006; 552: 131–134. [DOI] [PubMed] [Google Scholar]
- 11.Landmesser U, Dikalov S, Price SR et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003; 111: 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramachandran A, Levonen AL, Brookes PS et al. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic Biol Med 2002; 33: 1465–1474. [DOI] [PubMed] [Google Scholar]
- 13.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006; 1757: 509–517. [DOI] [PubMed] [Google Scholar]
- 14.Semenkovich CF. Insulin resistance and atherosclerosis. J Clin Invest 2006; 116: 1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callera GE, Tostes RC, Yogi A et al. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin Sci (Lond) 2006; 110: 243–253. [DOI] [PubMed] [Google Scholar]
- 16.Shinmura K, Tamaki K, Sato T et al. Prostacyclin attenuates oxidative damage of myocytes by opening mitochondrial ATP-sensitive K+ channels via the EP3 receptor. Am J Physiol Heart Circ Physiol 2005; 288: H2093–H2101. [DOI] [PubMed] [Google Scholar]
- 17.Kimura S, Zhang GX, Nishiyama A et al. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 2005; 45: 438–444. [DOI] [PubMed] [Google Scholar]
- 18.Garlid KD, Dos Santos P, Xie ZJ et al. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K(+) channel in cardiac function and cardioprotection. Biochim Biophys Acta 2003; 1606: 1–21. [DOI] [PubMed] [Google Scholar]
- 19.Cornelissen J, Van Kuilenburg AB, Voute PA et al. The effect of the neuroblastoma-seeking agent meta-iodobenzylguanidine (MIBG) on NADH-driven superoxide formation and NADH-driven lipid peroxidation in beef heart submitochondrial particles. Eur J Cancer 1997; 33: 421–424. [DOI] [PubMed] [Google Scholar]
- 20.Cornelissen J, Van Belzen R, Van Gennip AH et al. Specification of the inhibitory action of MIBG on the respiratory chain by EPR scanning. Anticancer Res 1997; 17: 265–268. [PubMed] [Google Scholar]
- 21.Cornelissen J, Wanders RJ, Van den Bogert C et al. Meta-iodobenzylguanidine (MIBG) inhibits malate and succinate driven mitochondrial ATP synthesis in the human neuroblastoma cell line SK-N-BE(2c). Eur J Cancer 1995; 31A: 582–586. [DOI] [PubMed] [Google Scholar]
- 22.Cornelissen J, Wanders RJ, Van Gennip AH et al. Meta-iodobenzylguanidine inhibits complex I and III of the respiratory chain in the human cell line Molt-4. Biochem Pharmacol 1995; 49: 471–477. [DOI] [PubMed] [Google Scholar]
- 23.Kuin A, Aalders M, Lamfers M et al. Potentiation of anti-cancer drug activity at low intratumoral pH induced by the mitochondrial inhibitor miodobenzylguanidine (MIBG) and its analogue benzylguanidine (BG). Br J Cancer 1999; 79: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee BI, Chan PH, Kim GW. Metalloporphyrin-based superoxide dismutase mimic attenuates the nuclear translocation of apoptosis-inducing factor and the subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. Stroke 2005; 36: 2712–2717. [DOI] [PubMed] [Google Scholar]
- 25.Patel MN. Metalloporphyrins improve the survival of Sod2-deficient neurons. Aging Cell 2003; 2: 219–222. [DOI] [PubMed] [Google Scholar]
- 26.Zingarelli B, Day BJ, Crapo JD et al. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br J Pharmacol 1997; 120: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol 1998; 80: 452–457. [DOI] [PubMed] [Google Scholar]
- 28.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002; 84: 131–141. [DOI] [PubMed] [Google Scholar]
- 29.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4: 181–189. [DOI] [PubMed] [Google Scholar]
- 30.Suh YA, Arnold RS, Lassegue B et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999; 401: 79–82. [DOI] [PubMed] [Google Scholar]
- 31.Landmesser U, Cai H, Dikalov S et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002; 40: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martyn KD, Frederick LM, von Loehneysen K et al. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 2006; 18: 69–82. [DOI] [PubMed] [Google Scholar]
- 33.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979; 59: 527–605. [DOI] [PubMed] [Google Scholar]
- 34.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med 1990; 8: 523–539. [DOI] [PubMed] [Google Scholar]
- 35.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 1985; 237: 408–414. [DOI] [PubMed] [Google Scholar]
- 36.Levine RL, Garland D, Oliver CN et al. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 1990; 186: 464–478. [DOI] [PubMed] [Google Scholar]
- 37.Muscari C, Guarnieri C, Biagetti L et al. Influence of age on oxidative damage in mitochondria of ischemic and reperfused rat hearts. Cardioscience 1990; 1: 275–278. [PubMed] [Google Scholar]
- 38.Birch-Machin MA. The role of mitochondria in ageing and carcinogenesis. Clin Exp Dermatol 2006; 31: 548–552. [DOI] [PubMed] [Google Scholar]
- 39.Passos JF, von Zglinicki T, Saretzki G. Mitochondrial dysfunction and cell senescence: cause or consequence? Rejuvenation Res 2006; 9: 64–68. [DOI] [PubMed] [Google Scholar]
- 40.Pamplona R, Barja G. Mitochondrial oxidative stress, aging and caloric restriction: the protein and methionine connection. Biochim Biophys Acta 2006; 1757: 496–508. [DOI] [PubMed] [Google Scholar]
- 41.Yang T, Zhang A, Honeggar M et al. Hypertonic induction of COX-2 in collecting duct cells by reactive oxygen species of mitochondrial origin. J Biol Chem 2005; 280: 34966–34973. [DOI] [PubMed] [Google Scholar]
- 42.National Toxicology Program. NTP toxicology and carcinogenesis studies of rotenone (CAS no. 83-79-4) in F344/N rats and B6C3F1 mice (feed studies). Natl Toxicol Program Tech Rep Ser 1988; 320: 1–158. [PubMed] [Google Scholar]
- 43.Li Y, Zhu H, Trush MA. Detection of mitochondria-derived reactive oxygen species production by the chemilumigenic probes lucigenin and luminol. Biochim Biophys Acta 1999; 1428: 1–12. [DOI] [PubMed] [Google Scholar]
- 44.Holmuhamedov EL, Jovanovic S, Dzeja PP et al. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol 1998; 275: H1567–H1576. [DOI] [PubMed] [Google Scholar]
- 45.Pollock JD, Williams DA, Gifford MA et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 1995; 9: 202–209. [DOI] [PubMed] [Google Scholar]
- 46.Hultqvist M, Olofsson P, Holmberg J et al. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci USA 2004; 101: 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jia Z, Zhang A, Zhang H et al. Deletion of microsomal prostaglandin E synthase-1 increases sensitivity to salt loading and angiotensin II infusion. Circ Res 2006; 99: 1243–1251. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Zhang A, Kohan DE et al. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci USA 2005; 102: 9406–9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia Z, Guo X, Zhang H et al. Microsomal prostaglandin synthase-1-derived prostaglandin E2 protects against angiotensin II-induced hypertension via inhibition of oxidative stress. Hypertension 2008; 52: 952–959. [DOI] [PubMed] [Google Scholar]