

Abstract
Foamy viruses are complex retroviruses whose replication strategy resembles that of conventional retroviruses. However, foamy virus replication also resembles that of hepadnaviruses in many respects. Because hepadnaviruses replicate in an integrase-independent manner, we were interested in investigating the characteristics of human foamy virus (HFV) integration. We have shown that HFV requires a functional integrase protein for infectivity. Our analyses have revealed that in single-cell clones derived from HFV-infected erythroleukemia-derived cells (H92), there were up to 20 proviral copies per host cell genome as determined by Southern blot and fluorescent in situ hybridization analysis. Use of specific probes has also shown that a majority of the proviruses contain the complete tas gene, which encodes the viral transactivator, and are not derived from Δtas cDNAs, which have been shown to arise rapidly in infected cells. To demonstrate that the multiple proviral sequences are due to integration instead of recombination, we have sequenced the junctions between the proviral sequences and the host genome and found that the proviruses have authentic long terminal repeat ends and that each integration is at a different chromosomal site. A virus lacking the Gag nuclear localization signal accumulates fewer proviruses, suggesting that nuclear translocation is important for high proviral load. Since persistently infected H92 clones are not resistant to superinfection, the relative importance of an intracellular versus extracellular mechanism in proviral acquisition has yet to be determined.
Foamy viruses, also known as spumaviruses, comprise one of the genera of the family Retroviridae. Foamy viruses are less well characterized than other family members such as the lentiviruses, but recent investigations of foamy virus replication have revealed many unusual characteristics. In some respects, foamy viruses appear to bridge the gap between retroviruses and hepadnaviruses, although there are features of the foamy virus replication pathway that are distinct from both (reviewed in reference 28). The prototype spumavirus, human foamy virus (HFV), was originally isolated from human nasopharyngeal carcinoma cells (1). The HFV genome contains the canonical gag, pol, and env genes as well as several accessory genes located between the env gene and the 3′ long terminal repeat (LTR) (17, 31, 38). HFV is virtually identical to simian foamy viruses from chimpanzees and is unlikely to be of human origin (21, 22).
Unlike the case for all other retroviruses, foamy virus Pol is expressed from a spliced message and does not contain any gag sequences (12, 29, 56). Nascent HFV Pol contains protease (PR), reverse transcriptase (RT), RNase H (RN), and integrase (IN) domains (31). A single cleavage event of the 127-kDa Pol protein results in two proteins, an approximately 85-kDa protein containing the PR, RT, and RN domains and a 40-kDa protein consisting of only the IN domain (33).
RT activity of foamy virus Pol was first demonstrated in 1971 (36). One unusual characteristic of HFV Pol is that RT activation occurs before or during viral morphogenesis, late in the HFV life cycle (32, 60). One result of this timing is that approximately 25% of nascent particles contain full-length DNA which can serve as an infectious genome (60). A second, less well understood consequence of reverse transcription prior to particle formation is the accumulation of large quantities of extrachromosomal DNA in foamy virus-infected cells (32, 47, 56).
Integration is an obligate, two-step process in all retroviral life cycles (reviewed in reference 7). The IN of HFV is enzymatically active in vitro and shares significant homology with other retroviral integrases (10, 11, 35, 46). The central region of HFV IN contains a conserved D,D-35-E motif which is required for IN function in all retroviral integrases studied (18, 26, 50). Although there is ample evidence for foamy virus integration, when this work was initiated the requirement for integration in the foamy virus life cycle had not been determined (23, 39, 45).
An additional characteristic of HFV is the presence of three Gly-Arg (GR) boxes in the putative nucleocapsid domain of Gag (43). The precise function of the GR boxes remains unclear, but a deletion in GR box I does not bind nucleic acid and does not replicate (57). GR box II contains a nuclear localization sequence (NLS) which is required for Gag localization to the nucleus (43). However, deletion of GR box II results in virus which replicates, although at lower levels (57).
In the present study, we have used a mutation in the conserved catalytic active site of HFV IN and determined that integration is a requirement for HFV replication. We have also demonstrated that human erythroblastoid cells persistently infected with wild-type (wt) HFV accumulate large numbers of independently integrated proviruses. A replication-defective form of HFV which lacks functional tas, termed HFVΔtas, has been implicated in persistent infection both in vitro and in vivo (34, 40–42). We have shown that although large amounts of both full-length HFV and HFVΔtas are present extrachromosomally, full-length HFV is the predominant proviral species in persistently infected H92 cells. Furthermore, we have demonstrated both by fluorescent in situ hybridization (FISH) and by sequencing of viral integration sites that the accumulation of multiple integrations is via de novo integration. Our studies suggest that the GR box II may be required for the high copy number of integrations.
MATERIALS AND METHODS
Cells.
The FAB indicator cell line, which contains β-galactosidase (β-Gal) under control of the HFV LTR and expresses β-Gal only upon transfection or infection with HFV, was previously described (58). Diploid human embryonic lung (HEL) cells (ATCC CCl-137), baby hamster kidney (BHK-21) cells (ATCC CCL-10), and FAB cells were grown in Dulbecco's modified Eagle medium with 5% fetal bovine serum (FBS) and antibiotics. Human erythroleukemia (H92) cells (ATCC TIB-180) were grown in RPMI 1640 supplemented with 10% FBS and antibiotics. Since it is difficult to infect hematopoietic cell lines with free virus, mass cultures of infected H92 cells were obtained by infecting HEL cells with wt HFV or H3RR lacking GR box II. When syncytia appeared, exponentially growing H92 cells were added to the HEL cells and the medium was changed to RPMI 1640–10% FBS. The HEL cells were completely lysed within a week, and the infected H92 cells were diluted before the culture reached stationary phase (about 5 × 105 cells per ml). After several months of growth, a majority of the H92 cells were infected and clonal populations were derived. For single-cell clones of H92 cells infected with wt virus, cells were diluted and plated in the same medium containing 0.36% agar, and single colonies were picked using capillary pipettes. For single-cell clones of H3RR-infected H92 cells, cells were diluted and 100 cells were plated per 96-well microtiter plate. Since H92 cells grow in a semiadherent manner, it was possible to select wells which had only a single colony. In the case of H92 clones infected with wt HFV, all clones were infected with virus. In the case of H3RR clones, about 70% of the clones were infected. In some experiments, the single-cell-derived H92 clones were continuously treated with 3′-azido-3′-deoxythymidine (AZT; 100 μg/ml; Sigma) and samples were taken at the indicated times. Under these conditions, no infectious virus could be detected in the culture medium of AZT-treated cells (60).
Plasmids.
The infectious molecular clone pHFV13 was provided by R. Flugel (30). Plasmid pHFVΔEnv was recently described (3). Plasmid pFOV-7 was provided by A. Rethwilm (44) and used to generate pHFV-rGFP as follows. A 2,774-bp BlpI/SalI fragment from pHFV13 was subcloned into pNEB193 and named pSub5. A 2,052-bp BlpI/XbaI fragment was cloned from pFOV-7 into BlpI/XbaI-digested pSub5 and named pSub5fov. The green fluorescent protein (GFP) from pEGFP1 (Clontech, Palo Alto, Calif.) was amplified using the primers 5′-CTACCCGGGCGCCACCATGGTGAGCAAG-3′ and 5′-GATCCCGGGCTATTACTTGTACAGCTCGTCCATG-3′. This product was digested with SmaI and ligated into SmaI-digested pSub5fov; the resulting plasmid was named pSub5gfp. The minimal Rous sarcoma virus (RSV) promoter was excised from pRSVneo, blunt ended with Klenow enzyme, and cloned into EcoRV-digested pSub5gfp. A 3,474-bp BlpI/SalI fragment from the resulting plasmid, pSub5-rGFP, was subcloned into BlpI/SalI-digested pHFV13, resulting in the infectious clone pHFV-rGFP. The virus encoded by this plasmid expresses GFP from the RSV promoter and a truncated Bet protein from the internal promoter.
Site-directed mutagenesis.
To create a mutation in the putative active site of HFV IN, we used an oligonucleotide-directed mutagenesis kit (MORPH; 5 Prime - 3 Prime, Boulder, Colo.). A 5,475-bp NcoI fragment of pHFV13 which includes the IN domain was subcloned into NcoI-digested pSL1180 (6). The resulting plasmid was used as a template for mutagenesis. A 32-mer, 5′-GGTGATTCACTCTATTCAAGGTGCAGCATT C-3′, was used to mutate codon 936 of the pol gene from Asp (GAT) to Ile (ATT; underlined) at nucleotide positions 5923 and 5924. After confirmation by sequencing, the mutation was introduced into pHFV13 as a 5,475-bp NcoI fragment, and the resulting plasmid was termed pHFV-D9361.
Virus stocks and titer.
Virus stocks were obtained by transfection of FAB cells with proviral DNA constructs, using Lipofectamine (Life Technologies, Inc.) or a modified calcium phosphate method (8). Viruses were titered 48 h posttransfection when used in Western blotting experiments. Otherwise, virus stocks were prepared and titered as previously described (58). Transfection efficiencies were determined by measuring β-Gal production in FAB cells. Cell monolayers were lysed in Ab buffer (20 mM Tris-Cl [pH 7.5], 50 mM NaCl, 0.5% Nonidet P-40, 0.5% sodium dodecyl sulfate [SDS], 0.5% deoxycholate, 0.5% aprotinin), and total protein concentration was measured by optical density at 280 nm. β-Gal activity was measured at 420 nm using o-nitrophenyl-β-d-galactopyranoside (ONPG) (2).
Western blotting.
Western blot analyses were performed using anti-Gag polyclonal rabbit antiserum (3) at 1:2,000 dilution. FAB cells (106) were transfected in triplicate in 10-cm-diameter dishes with viral or mock plasmid constructs, using Lipofectamine (Life Technologies) according to the manufacturer's instructions. At 40 h posttransfection, supernatants were collected, passed through a 0.45-μm-pore-size filter, and then pelleted through 20% sucrose by ultracentrifugation (24,000 rpm, SW28 rotor). Pellets were lysed in Ab buffer, and loading buffer was added; the mixture was boiled and subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% gels. Transfected FAB cell monolayers were lysed in Ab buffer; cell debris was pelleted by low-speed centrifugation; cleared lysates were diluted in loading buffer, boiled, and subjected to SDS-PAGE on 10% gels. H92 cells were pelleted by low-speed centrifugation; cleared lysates were prepared and separated by SDS-PAGE on 10% gels. Proteins were transferred to Immobilon-P membranes (Millipore) and blocked overnight in Wb buffer (1× phosphate-buffered saline, 4% dry nonfat milk, 0.5% Tween 20). Membranes were probed using anti-Gag polyclonal rabbit antiserum (3) at 1:2,000 dilution in Wb buffer for 2 h at 4°C. Membranes were washed three times for 20 min each in Wb buffer; then secondary horseradish peroxidase-conjugated anti-rabbit immunoglobulin (Amersham), was added at 1:8,000 dilution for 1 h. Blots were washed and then visualized using enhanced chemiluminescence (Amersham).
Southern blotting.
Genomic DNA was isolated from uninfected and persistently infected H92 cells by standard techniques (2). Genomic DNA (20 μg) was completely digested with NheI, which cuts once at position 9249 in the HFV genome. Radiolabeled probes were synthesized by random priming using a Prime-It II kit (Stratagene). A bet probe was made using a Qiaex II (Qiagen)-purified 422-bp ClaI/Tht111I fragment of pHSRVΔGPE (58). A Δtas probe was made from a Qiaex II-purified, 203-bp StuI/NsiI fragment of pSub5. A myc probe was made from a 1,280-bp NotI fragment of human c-myc.
FISH.
Metaphase spreads were prepared from persistently infected H92 cells or uninfected H92 cells by standard techniques (4). An EagI/SalI fragment encompassing the entire HFV genome was biotinylated by nick translation using a BioNick labeling kit as instructed by the manufacturer (Life Technologies, Inc.). Human Cot1 DNA was added to the probe to suppress background hybridization. After overnight hybridization at 37°C and washing, the hybridized biotinylated probe was detected with fluorescein isothiocyanate (FITC)-avidin, and the signal was amplified with biotinylated goat antiavidin antibody and subsequent detection with FITC-avidin. Slides were stained with propidium iodide (PI), mounted in antifade solution, and then viewed using a Deltavision microscope. Digital images were collected in both PI and FITC channels and merged.
Sequence analysis of integration sites.
Junctions between viral and host DNA were amplified and sequenced as described elsewhere (51), with minor modifications. Ten micrograms of genomic DNA isolated from persistently infected H92 clones treated with AZT was digested with 30 U of NlaIII, which cuts at position 1115 bp in the viral LTR and randomly in the host genome, leaving 5′-CATG-3′ overhangs. Two hundred picomolar oligonucleotide IPCR-NlaIII (5′-TCATGATCAATGGGACGATCACATG-3′), containing the NlaIII CATG overhang (underlined), was ligated to 5 μg of NlaIII-digested and purified genomic DNA using 20 U of T4 DNA ligase (Roche). Phenol-chloroform extracted, ethanol-precipitated ligation reactions were amplified in a linear PCR with oligonucleotide IPCR1 (5′-GCTTAAGCAGATATAATATTG-3′; positions 411 to 390 in pHFV13). Reactions were performed as instructed by the manufacturer (Perkin-Elmer). Cycling conditions were 50 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min. Linear reaction products were purified using a Qiaex II kit (Qiagen). A standard PCR using IPCR2 (5′-GGCGCGCCATGAGGAGCAGGAGTATTTG-3′) and IPCR3 (5′-GGCGCGCCTCATGATCAATGGGACGATCA-3′) was performed under the following cycling conditions: 94°C for 30 s; 56°C for 30 s, and 72°C for 1 min; (35 cycles). Amplification products were purified and digested with AscI (boldface) then subcloned into AscI-digested pNEB193 and sequenced.
Microscopy and FACS analysis of GFP-infected cells.
Infection of H92 and A3 cells was performed either by coculture with infected HEL cells or via direct infection with cell-free virus. Infection by coculture was performed by seeding 2.5 × 105 HEL cells in 60-mm-diameter dishes and the following day infecting them with 105 infectious units (IU) of cell-free HFV13 or HFV-rGFP. After 3 days significant cytopathic effect was visible, and 2.5 × 105 H92 or A3 suspension cells were added. Cells were passaged for approximately 14 days until no adherent cells were visible. At 3 weeks postinfection, visualization of GFP fluorescence was performed via microscopy and fluorescence-activated cell sorting (FACS). Infection with cell-free virus was performed by infecting 105 H92 or A3 cells with 5 × 104 IU of either HFV or HFV-rGFP in a final volume of 1.5 ml of medium. At 48 h postinfection, cells were visualized by microscopy and analyzed by FACS. All microscopy was performed on a Nikon Eclipse TE300 inverted microscope using a FITC filter set. Digital images were collected using a charge-coupled device camera and the MetaMorph software package (Universal Imaging Corporation). All FACS analysis was done using Becton Dickinson FACSCalibur instrumentation. Live cells were washed once and resuspended in phosphate-buffered saline containing 1% FCS and 1 μg of PI per ml. Postanalysis was performed using the WinMDI software package. All analyses were performed on cells gated for similar size and granularity.
RESULTS
A functional IN protein is required for viral infectivity.
Foamy virus replication is distinct from that of other retroviruses, and resembles that of hepadanaviruses, in several respects (28). Hepadnaviruses accumulate multiple extrachromosomal copies of their genome per cell which serve as templates for replication (48, 49). Foamy viruses also possess extrachromosomal DNA, and their genome appears to be DNA as for hepadnaviruses (32, 47, 60). For these reasons, we were interested in determining if HFV could replicate in an IN-independent manner. A mutation was introduced into the second aspartic acid in the IN D,D-35-E motif. This motif is the catalytic region of all studied retroviral integrases (18, 26, 50). We predicted that the resultant virus, HFV-D936I, would be capable of viral gene expression if transfected into a host cell but would be unable to initiate a subsequent infection because it would fail to integrate into the target cell genome. Transfections were performed in FAB cells, which express β-Gal in the presence of the HFV transactivating protein Tas (58), permitting normalization of transfection efficiencies using the chromogenic substrate ONPG. Transfection of pHFV13, pHFV-D936I, and pHFVΔEnv into FAB cells resulted in expression of similar levels of β-Gal (data not shown). pHFVΔEnv was used as a control because it is replication defective and cannot spread in culture. Expression of β-Gal by these constructs, including pHFV-D936I, indicated that this construct was capable of viral gene expression. This was further supported by Western blotting of cell lysates normalized for β-Gal expression. Using polyclonal anti-Gag antiserum, similar levels of viral Gag expression were observed in all cell lysates (Fig. 1A).
FIG. 1.

Analysis of wt HFV, envelope deletion mutant (HFVΔEnv), or IN active site mutant (pHFV-D936I) virus expression in transfected FAB cells. (A) Whole cell lysates subjected to Western blotting and probed with anti-Gag antiserum. Uncleaved 78-kDa Gag protein is indicated by the solid arrow, and cleaved 74-kDa Gag protein is marked by the open arrow. (B) Cell-free virus supernatants as tested in panel A.
To determine if HFV-D936I was capable of viral replication, lysates from transfected cells were titered on FAB cells as previously described (58). Only wt HFV was capable of initiating a second round of infection in FAB cells, resulting in titers of 4.5 × 104 and 5 × 104 IU/ml as assayed on FAB cells in two independent experiments. HFVΔEnv had a titer of <3 IU/ml in two separate experiments. HFV-D936I had titers of <3, 450, and 920 IU/ml in three independent experiments. HFV-D936I was not able to initiate further rounds of replication in any of the three experiments. To examine whether HFV-D936I could produce normal levels of extracellular virus, Western blotting using Gag antiserum was performed on cell-free virus particles. Both wt HFV and HFV-D936I were able to produce extracellular virus (Fig. 1B). As previously shown, HFVΔEnv does not release viral particles into the supernatant (3). These results show that the IN mutation in HFV-D936I does not affect virus assembly or release. While this work was in progress, similar results were reported (13).
Persistently infected H92 cells accumulate multiple proviruses.
Foamy virus infection of cells in vitro results in accumulation of large amounts of unintegrated viral DNA (32, 47, 56, 60). The copy number of integrated DNA has not been determined, partly because foamy virus infection of many cell types results in cell lysis, making such an analysis difficult. However, we were able to determine proviral copy number in the human erythroleukemia cell line H92, which has been shown to support persistent infection with HFV (59).
HFV-infected H92 cells were diluted and plated in soft agar, and individual colonies picked (see Materials and Methods). These individual clones were then expanded, and genomic DNA was digested with the restriction enzyme NheI, which cuts once in the HFV genome and randomly in the host genome (Fig. 2A). Southern blotting with a radiolabeled HFV probe which hybridizes only to the 3′ fragment of the HFV genome permitted quantitation of individual proviruses. We noted multiple integrations in these cell clones, with some accumulating approximately 20 proviruses (Fig. 2B, clone A3). A large quantity of unintegrated viral DNA arising from reverse transcription occurring late in the replication cycle (32, 60) hindered visualization of bands migrating near 3 and 12 kb (Fig. 2B, left). NheI-digested, unintegrated cDNA is indicated by lower arrows; since complete digestion by NheI was not obtained, linear, unintegrated full-length cDNA is indicated by upper arrows. The smaller band in each pair is the result of a deletion in the tas gene, discussed further below. When AZT was used to inhibit the amount of unintegrated DNA, integrated proviruses migrating near 3 and 12 kb were more clearly visualized (Fig. 2b, right). To ensure that treatment with AZT did not alter viral gene expression, Western blot analysis was done using Gag antiserum on cell lysates from AZT-treated and untreated cells (Fig. 2C). AZT treatment did not alter the amount of Gag protein produced. When DNA from uninfected H92 cells was probed as in Fig. 2B, there were no discernible bands (data not shown and Fig. 5A).
FIG. 2.

Proviral copy number in persistently infected H92 cells. (A) Schematic of the HFV genome showing the location of the unique NheI restriction site. The [32P]dCTP-labeled probe is indicated by a solid rectangle. (B) Southern blot of persistently infected H92 clones either untreated or treated with AZT. Upper arrows indicate uncut unintegrated viral DNA, while lower arrows indicate NheI-digested, unintegrated viral DNA. Solid arrows indicate undigested (upper band) or NheI-digested (lower band) full-length unintegrated viral DNA. Open arrows indicate undigested (upper band) or NheI-digested (lower band) unintegrated HFVΔtas DNA. (C) Western blot analysis using Gag antiserum on culture supernatants from clones A2 and A14 grown with or without AZT.
FIG. 5.
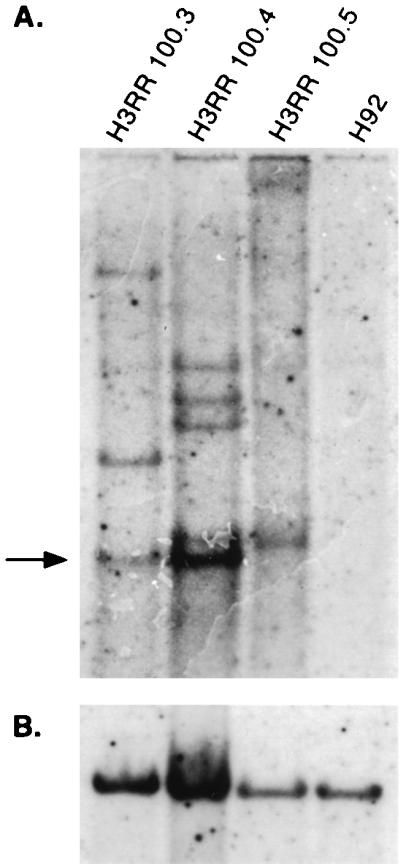
Proviral copy number in H92 cells persistently infected with H3RR virus. (A) Southern blot analysis of H3RR proviral integrations in clones 100.3, 100.4, and 100.5 and in uninfected H92 cells. Arrow indicates unintegrated, NheI-digested viral DNA. (B) The same blot stripped and reprobed with human c-myc, control cDNA.
Multiple integrations in infected clones are predominantly full length.
A defective HFV provirus containing specific 301-bp deletion in the tas gene has been described (42). We previously showed by PCR amplification the presence of large amounts of this 301-bp deletion in the tas gene of persistently infected H92 cells (59). In that study, it was not determined if the PCR template was extrachromosomal or integrated proviral DNA. The tas-deleted form of HFV (HFVΔtas) has been suggested to play a role in chronic infection (40, 41). To address whether HFVΔtas genomes were also present in the genome of H92 clones, Southern blotting was performed. Genomic DNAs from AZT-treated cells were digested with NheI, and the blots were hybridized with a probe within the 301-bp tas deletion (Δtas) and with a probe outside the deletion (bet) (Fig. 3A). Since the 422-bp bet probe recognizes both full-length HFV and HFVΔtas, while the Δtas probe recognizes only full-length viral genomes, HFVΔtas proviruses will produce a band with the bet probe but not with the Δtas probe. Genomic DNA from clones A2 and A5 was compared by Southern blotting using these two probes. The majority of proviruses present in these two clones were of the full-length form. Only a few HFVΔtas proviruses appeared to be present (Fig. 3B). Thus, cDNA from full-length genomes is preferentially integrated in these cells.
FIG. 3.

Identification of HFVΔtas proviruses in persistently infected H92 clones A2 and A5 treated with AZT. (A) The 301-bp deletion lacking in HFVΔtas is indicated by a hatched rectangle. The gray rectangle indicates the 422-bp bet probe, which detects both HFV and HFVΔtas; the black rectangle indicates the Δtas probe, which detects only wt HFV provirus. (B) Southern blot of genomic DNA from clones A2 and A5, using probes described for panel A. The asterisks indicate possible HFVΔtas integrations.
Visualization and sequencing of multiple proviral integrations in persistently infected clones.
Despite the precautions taken to ensure cloning of individual cells infected with HFV, the banding pattern detected by Southern blot analysis (Fig. 2) could result from individual integration events in a number of different cells instead of multiple integrations in a single progenitor cell. To address this issue, we used the FISH technique to directly visualize viral DNA in cells. Metaphase spreads from infected cells probed with a full-length HFV DNA probe showed that the multiple integrations occurred in the same cell (Fig. 4A). Instances where viral sequences are clearly present on both sister chromatids are indicated by arrowheads. Condensed metaphase chromosomes from clones A2 and A5 clearly showed multiple viral sequences within a single nucleus, while uninfected H92 cells only showed random, background staining. Figure 4A also demonstrates that the numbers of integration events visualized by FISH analysis are in agreement with the numbers of integrations present in the same genomic DNA samples when probed by Southern blot (Fig. 2B).
FIG. 4.

Analysis of multiple integrations in persistently infected H92 cells. (A) FISH analysis of uninfected H92 cells and persistently infected H92 clones A2, A3, and A5. Arrows indicate locations where the FITC-labeled HFV DNA probe is clearly visible on both sister chromatids. (B) Sequence of PCR-amplified DNAs at the junction between the HFV provirus 5′ LTR and host cell DNA in HFV-infected clones A3 (3.x) and A14 (14.x).
Because persistent infection results in accumulation of large amounts of unintegrated viral DNA, we wanted to determine whether the multiple viral DNA sequences present in the host genome arose through de novo integration or via gene duplication or recombination events. We PCR amplified and sequenced the cell junctions of four 5′ LTR ends from clone A3 and six 5′ LTR ends from clone A14. We found that in all cases the authentic TG dinucleotide present at the 5′ end of HFV is present at the junction between the LTR and the host DNA (Fig. 4B). Two 3′ LTR junctions from clone H3RR 100.4 were also sequenced and showed the authentic CA dinucleotide present at 3′ end of the HFV genome (data not shown). In addition, each 5′ LTR had different flanking sequences, indicating that each is located at a different cellular site (Fig. 4B).
Nuclear localization of Gag is necessary for high integrated copy number.
We previously demonstrated that a virus with a deletion in GR box II of Gag, termed H3RR, showed greatly reduced nuclear localization of Gag protein but replicated at levels only slightly lower than those for wt HFV (57). One possible role for the NLS is transport of HFV particles into the nucleus for integration. We hypothesized that because H3RR lacks the ability to localize Gag to the nucleus, it would not be able to integrate with the efficiency of wt HFV. Therefore, we generated clones of H3RR-infected H92 cells by limiting dilution and analyzed their genomic DNA for proviral sequences by Southern blotting using the same procedure as for wt virus. The majority of clones contained integrated provirus, indicating that H3RR virus spreads effectively in H92 cells. Three separate H3RR clones, 100.3, 100.4, and 100.5, were further analyzed by Southern blotting using the bet probe described in Fig. 2. The H3RR clones had one to four proviruses integrated into their genomes (Fig. 5A). Interestingly, these clones contained much less unintegrated DNA than wt-infected cells. In fact, clone 100.5 did not contain detectable amounts of unintegrated viral DNA. Also, these clones contained no discernible HFVΔtas genomes either unintegrated or integrated, as determined by Southern blot analysis using the Δtas probe described in Fig. 3A (data not shown). Uninfected H92 cells contained no discernible bands. All lines contained DNA which hybridized to a control myc probe (Fig. 5B).
Persistently infected H92 cells are not resistant to superinfection.
There are at least two possible pathways leading to the accumulation of proviruses in the host genome. One is via integration of intracellular HFV particles which have not budded from the plasma membrane. Another mechanism is repeated superinfection by extracellular virus, a process which is not common but has been documented for other retroviruses (37, 52). To test whether superinfection of infected H92 cells was possible, we constructed a GFP-expressing vector based on the construct pFOV-7 (44) (Fig. 6A). The vector pHFV-rGFP expresses GFP from the RSV minimal promoter, which has been inserted just downstream of tas, which is expressed from the HFV internal promoter. The encoded virus expresses GFP upon infection of BHK cells (Fig. 6B), H92 cells (Fig. 6C), and A3 cells (Fig. 6D). After amplification on HEL cells, the wt HFV titer was 2.0 × 106 IU/ml and the HFV-rGFP titer was 106 IU/ml as assayed on FAB cells (59).
FIG. 6.

Schematic diagram and expression of pHFV-rGFP. (A) Diagram of pHFV-rGFP indicating expression of GFP from the RSV promoter (cross-hatched box) as well as a truncated Bet protein (black boxes). Tas is expressed from the HFV internal promoter (arrow). (B) GFP expression at 48 h post infection in BHK cells after direct infection with cell-free HFV-rGFP at a multiplicity of infection of 1:5. (C) GFP expression in H92 after infection with HFV-rGFP by coculture with infected HEL cells; see Materials and Methods. (D) As for panel C except using clone A3.
We used HFV-rGFP virus to infect clone A3 cells to determine whether they are susceptible to superinfection. In a first experiment, we cocultured HFV-rGFP-infected HEL cells with A3 and H92. After all infected HEL were lysed by the viral infection, we monitored GFP fluorescence by microscopy and FACS. We found by FACS (Fig. 7A) that about 8% of the H92 cells as well as A3 cells were GFP positive. This indicates that in this cell line there is no superinfection interference when virus is introduced by cell-to-cell contact. We further infected both cultures with cell-free HFV-rGFP virus at a multiplicity of infection, as measured on FAB cells, of 0.5 (Fig. 7B). Although the percentage of infected cells was less, about 1% in both cases, there was no difference in infection of the uninfected and already infected populations as measured by FACS. Taken together, these results show that superinfection interference does not occur in persistently infected H92 cells, and therefore we cannot distinguish between intracellular and extracellular pathways for multiple proviral acquisition.
FIG. 7.

FACS analysis of HFV-rGFP-superinfected A3 cells. Vertical axes show cell viability as measured by PI uptake, while horizontal axes show intensity of GFP fluorescence. The percentages of cells which are viable and GFP positive are indicated in the lower right quadrant. (A) H92 cells and A3 cells infected with either HFV13 or HFV-rGFP via coculture; see Materials and Methods. (B) Infection of H92 and A3 cells with cell-free HFV-rGFP.
DISCUSSION
Mutation of the D,D-35-E motif in pHFV13 resulted in virus which was no longer able to replicate (Fig. 1). This indicates that a functional HFV IN protein is required for HFV replication and that this aspect of HFV replication is like that of conventional retroviruses. These results are similar to those recently published (13). Conventional retroviruses undergo reverse transcription early in their life cycle, resulting in integration of the newly synthesized DNA. Late in the life cycle, RNA genomes are packaged into nascent viral particles, which are blocked from reinfecting the same cell. Such infection interference is believed to occur either through downregulation of the cellular receptor or by functional blockage of the cellular receptor. As a result, a pool of integration-competent DNA is present only during the initial infection of the host cell. However, reverse transcription occurs late in the replication cycle of HFV and the infectious genome of HFV appears to be DNA (32, 60), which raises the possibility that integration-competent DNA could be present in HFV-infected cells. To test this possibility, we characterized the proviral integration pattern in hematopoietic cell lines persistently infected with HFV. Our analyses indicate that there are indeed multiple, de novo integrations in clonal cells (Fig. 2B). Our Southern blot data (Fig. 2B and 3B) show bands of unequal intensity. Bands of lesser intensity may indicate that some proviral acquisitions occurred shortly after the initial cloning and therefore are present in only a fraction of the total cell population.
The mechanism of multiple HFV provirus accumulation is unknown, but there are at least two possible pathways. The first mechanism is via extracellular reinfection. Superinfection studies using HFV-rGFP have demonstrated that extracellular reinfection could account for multiple proviral integrations in H92 cells (Fig. 7). In addition, using our HFV-rGFP virus, we were able to infect a BHK-derived cell line, BHKenv, which constitutively expresses foamy virus envelope protein (20) with only a fivefold decrease in efficiency compared to BHK cells (data not shown). Thus, lack of superinfection interference may be a general feature of foamy virus infection. Indeed, the fact that little viral budding or Env expression occurs on the cell surface, but rather is detected in the endoplasmic reticulum, could explain this (19). The second mechanism is acquisition of multiple integrations via an intracellular pathway. This could occur either by de novo integration or by gene duplication or recombination. To eliminate gene duplication or recombination as a mechanism, we demonstrated that the authentic LTR ends were present at multiple cellular locations (Fig. 5B). Therefore, the acquisition of multiple proviruses likely occurs via de novo integration. However, the contributions of extracellular versus intracellular reinfection to the proviral pool cannot be determined by these experiments. Our data are consistent with either mechanism.
An early pathway in hepadnavirus replication is the intracellular recycling of DNA containing core particles from the cytoplasm into the nucleus. As a result of this pathway, an accumulation of covalent closed circular DNA (cccDNA) is observed (54). The mechanism by which core recycling and cccDNA accumulation occurs is not fully understood, but the core protein of hepadnaviruses contains an arginine-rich NLS (55). Transport to the nucleus of hepadnavirus core protein is an important step in cccDNA accumulation (5, 25). Interestingly, the GR box II of HFV, which is dispensable for replication in vitro, is an NLS important for nuclear accumulation of newly synthesized Gag protein (43, 57). In the present study, cells persistently infected with a GR box II mutant virus contained far fewer integrated proviruses. Thus, in both HFV and hepadnaviruses, a mechanism for nuclear localization of core proteins exists, leading to the accumulation of integrated proviruses and cccDNA, respectively. Further evidence supporting a relationship between the replication pathways of foamy viruses and hepadnaviruses comes from analysis of their envelope glycoproteins. In the case of hepadnaviruses, mutation of the envelope protein blocks particle release and results in a drastic increase of cccDNA (27, 48, 49). For HFV, deletion of the envelope glycoprotein blocks particle release and results in accumulation of intracellular viral capsids (3, 16).
Infection of H92 or A3 cells by free virus is rather inefficient (Fig. 7B), but it could account for the proviral copy number that we observe. We are interested in determining whether the putative intracellular pathway contributes to proviral load. By comparing wt HFV to a virus which is released from the cell but which cannot infect subsequent cells due to a block at the level of viral entry, we can directly determine if the putative intracellular pathway contributes to proviral number. In addition, because ΔEnv viruses are not released from the cell, we can determine the effects of viral egress on proviral acquisition. Furthermore, we plan to address the importance of Gag nuclear localization in both of these pathways.
Because little is known about the cellular tropism of HFV, it is not known if multiple integrations occur in vivo. However, if multiple integrations do occur in vivo, there are several implications. Insertional mutagenesis by retroviruses is well documented and generally involves either enhancer insertion, leader insertion, terminator insertion, or direct inactivation (reviewed in references 9 and 15). Multiple integrations in a host cell clearly increases the probability of any of these events occurring. However, tumors have not been associated with foamy virus infection in any infected species. Thus, either cells with stably integrated foamy virus genomes are not abundant or the cells in which provirus exists are not rapidly proliferating, which is a prerequisite for tumor induction by other retroviruses. It will be interesting to isolate target cells of infected animals and determine the proviral copy number.
Persistence is a characteristic of foamy virus infection in vivo, but in vitro infection is generally characterized by rapid appearance of cytopathic effects including syncytium formation, cytoplasmic vacuolation, and ultimately cell lysis (24). If lytically infected cells are kept in culture, the few cells which survive the initial infection can be recovered. They are characterized by the presence of defective HFVΔtas proviruses and the absence of virus production (34, 40). However, in persistently infected H92 cells which continually produce relatively high levels of infectious virus, there are very few HFVΔtas proviruses (Fig. 4). This finding was unexpected, since there is a large amount of HFVΔtas cDNA in the infected cells (Fig. 2B, open arrows). There is no obvious reason for why the integrations are primarily from full-length cDNAs. Both the wt and HFVΔtas genomes are present in virions from infected H92 cells (data not shown) and should contain all of the cis-acting elements necessary for packaging, reverse transcription, and integration (14, 53). It is possible that there is an undescribed element in tas which is required for efficient integration. Our results indicate that in H92 cells, integration of HFVΔtas does not appear to play a central role in persistence. Therefore, there are likely other factors which permit cells such as H92 to continually produce infectious virus without suffering the cytopathic effects observed in other cell types. A large proportion of natural hosts are infected with foamy viruses (24). However, infection occurs in the absence of high levels of viral replication. In vivo, more than one mechanism may be involved in persistence. HFVΔtas may be important in attenuating infection in cells which are susceptible to cell lysis, while a reservoir of cells persistently infected with intact genomes which can produce replication competent virus may allow for horizontal transmission.
ACKNOWLEDGMENTS
This investigation was supported by NIH grants R01 CA18282 and P01 HL53762 to M.L.L. C.D.M. was supported by grants T32 GM07270 and T32 CA80416, The Hearst Foundation, and the University of Washington Graduate School. K.E.C. was supported by grant T32 CA09229.
We thank Michael Emerman for critical review of the manuscript, David Baldwin for continued discussion regarding this research, Paul Lampe for use of the fluorescence microscope, Barbara Trask for assistance with FISH analysis, Axel Rethwilm (University of Dresden) for the construct pFOV-7, and Matt Query for technical assistance.
REFERENCES
- 1.Achong B G, Mansell P W A, Epstein M A, Clifford P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J Natl Cancer Inst. 1971;46:299–307. [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. Vol. 1. New York, N.Y: John Wiley & Sons, Inc.; 1998. [Google Scholar]
- 3.Baldwin D N, Linial M L. The roles of Pol and Env in the assembly pathway of human foamy virus. J Virol. 1998;72:3658–3665. doi: 10.1128/jvi.72.5.3658-3665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birren B W, Tachi-iri Y, Kim U J, Nguyen M, Shizuya H, Korenberg J R, Simon M I. A human chromosome 22 fosmid resource: mapping and analysis of 96 clones. Genomics. 1996;34:97–106. doi: 10.1006/geno.1996.0246. [DOI] [PubMed] [Google Scholar]
- 5.Bock C T, Schwinn S, Schroder C H, Velhagen I, Zentgraf H. Localization of hepatitis B virus core protein and viral DNA at the nuclear membrane. Virus Genes. 1996;12:53–63. doi: 10.1007/BF00370001. [DOI] [PubMed] [Google Scholar]
- 6.Brosius J. Superpolylinkers in cloning and expression vectors. DNA. 1989;8:759–777. doi: 10.1089/dna.1989.8.759. [DOI] [PubMed] [Google Scholar]
- 7.Brown P O. Integration of retroviral DNA. Curr Top Microbiol Immunol. 1990;157:19–48. doi: 10.1007/978-3-642-75218-6_2. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffin J M. Retroviridae: the viruses and their replication. In: Fields B N, editor. Fields virology. 3rd ed. Vol. 2. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1814–1822. [Google Scholar]
- 10.Dias H W, Aboud M, Flugel R M. Analysis of the phylogenetic placement of different spumaretroviral genes reveals complex pattern of foamy virus evolution. Virus Genes. 1995;11:183–190. doi: 10.1007/BF01728657. [DOI] [PubMed] [Google Scholar]
- 11.Engelman A, Craigie R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J Virol. 1992;66:6361–6369. doi: 10.1128/jvi.66.11.6361-6369.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enssle J, Jordan I, Mauer B, Rethwilm A. Foamy virus reverse transcriptase is expressed independently from the Gag protein. Proc Natl Acad Sci USA. 1996;93:4137–4141. doi: 10.1073/pnas.93.9.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enssle J, Moebes A, Heinkelein M, Panhuysen M, Mauer B, Schweizer M, Neumann-Haefelin D, Rethwilm A. An active foamy virus integrase is required for virus replication. J Gen Virol. 1999;80:1445–1452. doi: 10.1099/0022-1317-80-6-1445. [DOI] [PubMed] [Google Scholar]
- 14.Erlwein O, Bieniasz P D, McClure M O. Sequences in pol are required for transfer of human foamy virus-based vectors. J Virol. 1998;72:5510–5516. doi: 10.1128/jvi.72.7.5510-5516.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan H. Retroviruses and their role in cancer. In: Levy J A, editor. Retroviridae. Vol. 3. New York, N.Y: Plenum Press; 1994. pp. 313–362. [Google Scholar]
- 16.Fischer N, Heinkelein M, Lindemann D, Enssle J, Baum C, Werder E, Zentgraf H, Muller J G, Rethwilm A. Foamy virus particle formation. J Virol. 1998;72:1610–1615. doi: 10.1128/jvi.72.2.1610-1615.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flugel R M, Rethwilm A, Maurer B, Darai G. Nucleotide sequence analysis of the env gene and its flanking regions of the human spumaretrovirus reveals two novel genes. EMBO J. 1987;6:2077–2084. doi: 10.1002/j.1460-2075.1987.tb02473.x. . (Erratum, 9:3806, 1990.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerton J L, Ohgi S, Olsen M, DeRisi J, Brown P O. Effects of mutations in residues near the active site of human immunodeficiency virus type 1 integrase on specific enzyme-substrate interactions. J Virol. 1998;72:5046–5055. doi: 10.1128/jvi.72.6.5046-5055.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goepfert P A, Shaw K L, Ritter G, Jr, Mulligan M J. A sorting motif localizes the foamy virus glycoprotein to the endoplasmic reticulum. J Virol. 1997;71:778–784. doi: 10.1128/jvi.71.1.778-784.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herchenroder O, Moosmayer D, Bock M, Pietschmann T, Rethwilm A, Bieniasz P D, McClure M O, Weis R, Schneider J. Specific binding of recombinant foamy virus envelope protein to host cells correlates with susceptibility to infection. Virology. 1999;255:228–236. doi: 10.1006/viro.1998.9570. [DOI] [PubMed] [Google Scholar]
- 21.Herchenroder O, Renne R, Loncar D, Cobb E K, Murthy K K, Schneider J, Mergia A, Luciw P A. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFV cpz): high homology to human foamy virus (HFV) Virology. 1994;201:187–199. doi: 10.1006/viro.1994.1285. [DOI] [PubMed] [Google Scholar]
- 22.Herchenroder O, Turek R, Neumann-Haefelin D, Rethwilm A, Schneider J. Infectious proviral clones of chimpanzee foamy virus (SFV cpz) generated by long PCR reveal close functional relatedness to human foamy virus. Virology. 1995;214:685–689. doi: 10.1006/viro.1995.0086. [DOI] [PubMed] [Google Scholar]
- 23.Hirata R K, Miller A D, Andrews R G, Russell D W. Transduction of hematopoietic cells by foamy virus vectors. Blood. 1996;88:3654–3661. [PubMed] [Google Scholar]
- 24.Hooks J J, Gibbs C., Jr The foamy viruses. Bacteriol Rev. 1975;39:169–185. doi: 10.1128/br.39.3.169-185.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kann M, Bischof A, Gerlich W H. In vitro model for the nuclear transport of the hepadnavirus genome. J Virol. 1997;71:1310–1316. doi: 10.1128/jvi.71.2.1310-1316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leavitt A D, Robles G, Alesandro N, Varmus H E. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol. 1996;70:721–728. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenhoff R J, Summers J. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J Virol. 1994;68:4565–4571. doi: 10.1128/jvi.68.7.4565-4571.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linial M L. Foamy viruses are unconventional retroviruses. J Virol. 1999;73:1747–1755. doi: 10.1128/jvi.73.3.1747-1755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lochelt M, Flugel R M. The human foamy virus pol gene is expressed as a Pro-Pol polyprotein and not as a Gag-Pol fusion protein. J Virol. 1996;70:1033–1040. doi: 10.1128/jvi.70.2.1033-1040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lochelt M, Zentgraf H, Flugel R M. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology. 1991;184:43–54. doi: 10.1016/0042-6822(91)90820-2. [DOI] [PubMed] [Google Scholar]
- 31.Maurer B, Bannert H, Darai G, Flugel R M. Analysis of the primary structure of the long terminal repeat and the gag and pol genes of the human spumaretrovirus. J Virol. 1988;62:1590–1597. doi: 10.1128/jvi.62.5.1590-1597.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moebes A, Enssle J, Bieniasz P D, Heinkelein M, Lindemann D, Bock M, McClure M O, Rethwilm A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J Virol. 1997;71:7305–7311. doi: 10.1128/jvi.71.10.7305-7311.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Netzer K O, Schliephake A, Maurer B, Watanabe R, Aguzzi A, Rethwilm A. Identification of pol-related gene products of human foamy virus. Virology. 1993;192:336–338. doi: 10.1006/viro.1993.1039. [DOI] [PubMed] [Google Scholar]
- 34.Neves M, Peries J, Saib A. Study of human foamy virus proviral integration in chronically infected murine cells. Res Virol. 1998;149:393–401. doi: 10.1016/s0923-2516(99)80007-7. [DOI] [PubMed] [Google Scholar]
- 35.Pahl A, Flugel R M. Endonucleolytic cleavages and DNA-joining activities of the integration protein of human foamy virus. J Virol. 1993;67:5426–5434. doi: 10.1128/jvi.67.9.5426-5434.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parks W P, Todaro G J, Scolnick E M, Aaronson S A. RNA dependent DNA polymerase in primate syncytium-forming (foamy) viruses. Nature. 1971;229:258–260. doi: 10.1038/229258a0. [DOI] [PubMed] [Google Scholar]
- 37.Pauza C D, Galindo J E, Richman D D. Reinfection results in accumulation of unintegrated viral DNA in cytopathic and persistent human immunodeficiency virus type 1 infection of CEM cells. J Exp Med. 1990;172:1035–1042. doi: 10.1084/jem.172.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rethwilm A, Darai G, Rosen A, Maurer B, Flugel R M. Molecular cloning of the genome of human spumaretrovirus. Gene. 1987;59:19–28. doi: 10.1016/0378-1119(87)90262-9. [DOI] [PubMed] [Google Scholar]
- 39.Rhodes-Feuillette A, Mahouy G, Lasneret J, Flandrin G, Peries J. Characterization of a human lymphoblastoid cell line permanently modified by simian foamy virus type 10. J Med Primatol. 1987;16:277–289. [PubMed] [Google Scholar]
- 40.Saib A, Koken M H, van der Spek P, Peries J, de The H. Involvement of a spliced and defective human foamy virus in the establishment of chronic infection. J Virol. 1995;69:5261–5268. doi: 10.1128/jvi.69.9.5261-5268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saib A, Neves M, Giron M L, Guillemin M C, Valla J, Peries J, Canivet M. Long-term persistent infection of domestic rabbits by the human foamy virus. Virology. 1997;228:263–268. doi: 10.1006/viro.1996.8383. [DOI] [PubMed] [Google Scholar]
- 42.Saib A, Peries J, de The H. A defective human foamy provirus generated by pregenome splicing. EMBO J. 1993;12:4439–4444. doi: 10.1002/j.1460-2075.1993.tb06129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schliephake A W, Rethwilm A. Nuclear localization of foamy virus Gag precursor protein. J Virol. 1994;68:4946–4954. doi: 10.1128/jvi.68.8.4946-4954.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt M, Rethwilm A. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology. 1995;210:167–178. doi: 10.1006/viro.1995.1328. [DOI] [PubMed] [Google Scholar]
- 45.Schweizer M, Fleps U, Jackle A, Renne R, Turek R, Neumann-Haefelin D. Simian foamy virus type 3 (SFV-3) in latently infected Vero cells: reactivation by demethylation of proviral DNA. Virology. 1993;192:663–666. doi: 10.1006/viro.1993.1084. [DOI] [PubMed] [Google Scholar]
- 46.Schweizer M, Neumann-Haefelin D. Phylogenetic analysis of primate foamy viruses by comparison of pol sequences. Virology. 1995;207:577–582. doi: 10.1006/viro.1995.1120. [DOI] [PubMed] [Google Scholar]
- 47.Schweizer M, Renne R, Neumann-Haefelin D. Structural analysis of proviral DNA in simian foamy virus (LK-3)-infected cells. Arch Virol. 1989;109:103–114. doi: 10.1007/BF01310521. [DOI] [PubMed] [Google Scholar]
- 48.Summers J, Smith P, Horwich A. Hepadnavirus envelope proteins regulate closed circular DNA amplification. J Virol. 1990;64:2819–2824. doi: 10.1128/jvi.64.6.2819-2824.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Summers J, Smith P, Huang M, Yu M. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol. 1991;65:1310–1317. doi: 10.1128/jvi.65.3.1310-1317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Ent F M, Vos A, Plasterk R H. Mutational scan of the human immunodeficiency virus type 2 integrase protein. J Virol. 1998;72:3916–3924. doi: 10.1128/jvi.72.5.3916-3924.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wattel E, Vartanian J, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. 1995;69:2863–2868. doi: 10.1128/jvi.69.5.2863-2868.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weller S K, Joy A E, Temin H M. Correlation between cell killing and massive second-round superinfection by members of some subgroups of avian leukosis virus. J Virol. 1980;33:494–506. doi: 10.1128/jvi.33.1.494-506.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu M, Chari S, Yanchis T, Mergia A. cis-acting sequences required for simian foamy virus type 1 vectors. J Virol. 1998;72:3451–3454. doi: 10.1128/jvi.72.4.3451-3454.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu T T, Coates L, Aldrich C E, Summers J, Mason W S. In hepatocytes infected with hepatitis B virus, the template from viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 55.Yeh C T, Liaw Y F, Ou J H. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J Virol. 1990;64:6141–6147. doi: 10.1128/jvi.64.12.6141-6147.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu S F, Baldwin D N, Gwynn S R, Yendapalli S, Linial M L. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science. 1996;271:1579–1582. doi: 10.1126/science.271.5255.1579. [DOI] [PubMed] [Google Scholar]
- 57.Yu S F, Edelmann K, Strong R K, Moebes A, Rethwilm A, Linial M L. The carboxyl terminus of the human foamy virus Gag protein contains separable nucleic acid binding and nuclear transport domains. J Virol. 1996;70:8255–8262. doi: 10.1128/jvi.70.12.8255-8262.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu S F, Linial M L. Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J Virol. 1993;67:6618–6624. doi: 10.1128/jvi.67.11.6618-6624.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu S F, Stone J, Linial M L. Productive persistent infection of hematopoietic cells by human foamy virus. J Virol. 1996;70:1250–1254. doi: 10.1128/jvi.70.2.1250-1254.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu S F, Sullivan M D, Linial M L. Evidence that the human foamy virus genome is DNA. J Virol. 1999;73:1565–1572. doi: 10.1128/jvi.73.2.1565-1572.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]