

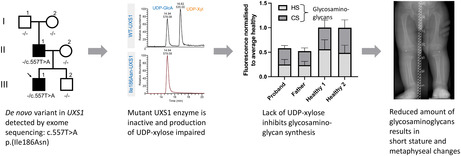
Abstract
Background
Serine residues in the protein backbone of heavily glycosylated proteoglycans are bound to glycosaminoglycans through a tetrasaccharide linker. UXS1 encodes UDP‐glucuronate decarboxylase 1, which catalyzes synthesis of UDP‐xylose, the donor of the first building block in the linker. Defects in other enzymes involved in formation of the tetrasaccharide linker cause so‐called linkeropathies, characterized by short stature, radio‐ulnar synostosis, decreased bone density, congenital contractures, dislocations, and more.
Methods
Whole exome sequencing was performed in a father and son who presented with a mild skeletal dysplasia, as well as the father's unaffected parents. Wild‐type and mutant UXS1 were recombinantly expressed in Escherichia coli and purified. Enzyme activity was evaluated by LC–MS/MS. In vivo effects were studied using HeparinRed assay and metabolomics.
Results
The son had short long bones, normal epiphysis, and subtle metaphyseal changes especially in his legs. The likely pathogenic heterozygous variant NM_001253875.1(UXS1):c.557T>A p.(Ile186Asn) detected in the son was de novo in the father. Purified Ile186Asn‐UXS1, in contrast to the wild‐type, was not able to convert UDP‐glucuronic acid to UDP‐xylose. Plasma glycosaminoglycan levels were decreased in both son and father.
Conclusion
This is the first report linking UXS1 to short‐limbed short stature in humans.
Keywords: medical genetics, monogenic disorder, skeletal dysplasia
This is the first described family where a likely pathogenic variant in UXS1 causes short stature and metaphyseal changes through impaired glycosaminoglycan biosynthesis.
1. INTRODUCTION
Proteoglycans are heavily glycosylated proteins abundant in cartilage and bone extracellular matrix. Together with other components of the extracellular matrix, they control many cellular events such as differentiation, migration, and adhesion. Proteoglycans consist of a protein backbone with covalently attached glycosaminoglycan sugar chains, such as chondroitin sulfate and heparan sulfate.
The UXS1 gene (HGNC:17729, OMIM: 609749) encodes the enzyme UDP‐glucuronate decarboxylase 1, which is evolutionary highly conserved with 75%–80% amino acid sequence identity and 90% similarity between plants and mammals (Moriarity et al., 2002). UXS1 is located in the perinuclear Golgi apparatus where it catalyzes the synthesis of UDP‐xylose (UDP‐Xyl) from UDP‐glucuronic acid (UDP‐GlcA) (Figure 1).
FIGURE 1.
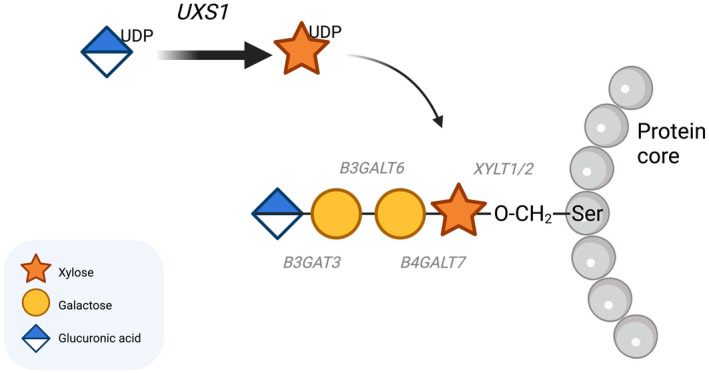
Tetrasaccharide linker biosynthesis. UXS1 catalyzes the synthesis of UDP‐xylose from UDP‐glucuronic acid, and the glycosyltransferases XYLT1 and XYLT2 transfer the xylosyl group to a serine residue on the core protein. B4GALT7, B3GALT6 and B3GAT3 then add a β1,4‐linked galactose, a β1,3‐linked galactose and a β1,3‐linked glucuronic acid, respectively.
The glycosyltransferases XYLT1 (OMIM: 608124) and XYLT2 (OMIM: 608125) transfer a xylosyl group from UDP‐Xyl to a serine residue on the protein backbone. This is the first step in generating the tetrasaccharide linker on which glycosaminoglycans are biosynthesised. Subsequent steps are catalyzed by enzymes encoded by B4GALT7 (OMIM: 604327), B3GALT6 (OMIM: 615291), and B3GAT3 (OMIM 606374), which add a β1,4‐linked galactose, a β1,3‐linked galactose, and a β1,3‐linked glucuronic acid, respectively.
Abolished or reduced activity of the enzymes producing the tetrasaccharide linker cause rare disorders referred to as linkeropathies, characterized by short stature, joint laxity and dislocations, prominent eyes and forehead, pectus abnormalities, foot abnormalities, broad tips of digits, hypotonia, and developmental delay (Jones et al., 2015).
Zebrafish uxs1 knockouts have demonstrated that uxs1 activity is necessary for extracellular skeletal matrix production and organization (Eames et al., 2010).
Skeletal dysplasias are a clinically and molecularly heterogenous group of conditions, some of which have significant extra‐skeletal features. A molecular genetic diagnosis may have consequences for treatment options and is the best basis for genetic counseling. Several skeletal dysplasias are caused by glycosaminoglycan/proteoglycan dysfunction (Unger et al., 2023).
Here, we present a novel autosomal dominant form of short‐limbed short stature due to an inactivating variant in UXS1 and describe two affected first‐degree relatives.
2. METHODS
2.1. Whole exome sequencing
DNA was extracted from EDTA blood and Nextera Rapid Capture Exome (Illumina, California, USA) sample prep, enrichment, sequencing, and bioinformatics were carried out as previously described (Smeland et al., 2023). Samples from individuals I‐1, I‐2, II‐1 (father), II‐2, and III‐1 (proband) were exome sequenced. III‐2 was Sanger sequenced to determine UXS1 status.
2.2. In silico structural analysis
Structural analysis of the Ile186Asn mutant was based on the previously determined crystal structure of human UXS1 in complex with NAD+ and UDP at 1.21 Å (PDB entry 2B69) (Eixelsberger et al., 2012).
2.3. Recombinant UXS1 expression and purification
The coding region of a truncated form of human UXS1 (residues 85–402) lacking the N‐terminal transmembrane domain was synthesized with codon usage optimized for expression in Escherichia coli (E.coli, General Biosystems). It was then inserted into the NcoI and BamH1 sites of the pET‐derived pETM‐11 vector (EMBL collection), which added an N‐terminal hexahistidine (His) tag and a tobacco etch virus (TEV) protease cleavage site to the protein. The expression vector was transformed into One Shot™ BL21(DE3) Chemically Competent E. coli expression cells.
Cells were grown in LB medium with 50 μg/mL of kanamycin at 37°C until they reached an OD600 of ~1.0 when expression was induced by adding isopropyl‐β‐d‐thiogalactopyranoside to a final concentration of 0.25 mM. Induced cells were incubated for an additional 2 h at 37°C before being harvested by centrifugation at 6000g. Cell pellets were resuspended in lysis buffer (50 mM Tris–HCl pH 8.6 and 500 mM NaCl) and lysed by sonication. Cell‐free protein extract was prepared by centrifugation, and applied to Ni‐NTA resin, equilibrated in a lysis buffer, for approximately 1 h. The mixture was washed with a lysis buffer, and the bound protein was released from the resin using a lysis buffer with 300 mM imidazole. The purified protein was dialysed into a buffer containing 20 mM Tris–HCl pH 8.6, 150 mM NaCl. Following dialysis, the protein was concentrated to approximately 1.5 mg/mL using Amicon Ultra Centrifugal Filters. Subsequently, glycerol was added to a final concentration of 50% (v/v) before storing at −20°C.
2.4. Enzymatic assay of UXS1 using liquid‐chromatography with electrospray ionization tandem mass spectrometry (LC‐ESI‐MS/MS)
The reaction mixture (50 ul) contained 10 μL 12.5 mM NAD+ (Sigma‐Aldrich), 1 μL 1 mM UDP‐glucuronic acid (UDP‐GlcA) (Sigma‐Aldrich), and an appropriate amount of enzyme diluted with phosphate‐buffered saline (PBS) buffer. The reaction mixture was incubated at 37°C overnight. Nucleotide sugars were purified using Hypercarb PGC slurry. Hypercarb PGC suspended in methanol was packed in a C18 Ziptip to a final bed volume of 30 μL. The tip was washed with 3 × 25 μL 90% acetonitrile (MeCN) in 0.5% trifluoroacetic acid (TFA) and 3 × 25 μL 0.5% TFA. After loading the sample, the tip was washed with 3 × 25 μL 0.5% TFA. Nucleotide sugars were eluted with 3 × 25 μL 65% MeCN in 0.5% TFA. Samples were dried in SpeedVac, resuspended in 400 μL of water, and 2 μL of each sample were analyzed by liquid‐chromatography with electrospray ionization tandem mass spectrometry (LC‐ESI‐MS/MS).
Nucleotide sugars were separated on a column (10 cm × 250 μm) packed in‐house with 5 μm porous graphite particles (Hypercarb, Thermo‐Hypersil, Runcorn, UK), then injected onto the column and eluted with an acetonitrile gradient (Buffer A, 10 mM ammonium bicarbonate; Buffer B, 10 mM ammonium bicarbonate in 80% MeCN). The gradient (0%–45% Buffer B) was eluted for 46 min, followed by a wash step with 100% Buffer B and equilibrated with Buffer A for the next 24 min. A 40 cm × 50 μm i.d. fused silica capillary was used as a transfer line to the ion source.
Samples were analyzed in negative ion mode on a LTQ linear ion trap mass spectrometer (Thermo Electron, San José, CA), with an IonMax standard ESI source equipped with a stainless steel needle kept at −3.5 kV. Compressed air was used as nebulizer gas. The heated capillary was kept at 270°C, and the capillary voltage was −50 kV. Full scan (m/z 380–2000, two microscan, maximum 100 ms, and target value of 30,000) was performed, followed by data‐dependent MS2 scans (two microscans, maximum 100 ms, and target value of 10,000) with normalized collision energy of 35%, isolation window of 2.5 units, activation q = 0.25, and activation time 30 ms. The threshold for MS2 was set to 300 counts. Data acquisition and processing were conducted with Xcalibur software (Version 2.0.7).
2.5. Metabolomics
Metabolites were extracted from dried blood spot samples and metabolomics untargeted MS analyses were performed according to a procedure described previously (Skogvold et al., 2021). Water was prepared in the same way as a blank sample. Samples were also analyzed using data‐dependent MS/MS mode (top 5, resolution: 17,500 FWHM (at m/z 200), AGC target value: 5e5 ion counts, maximum injection time: 100 ms and dynamic exclusion time: 10 s). All samples were injected five times in random order. A pooled quality control sample was prepared by mixing equal volume of the three prepared samples and injected between every fifth sample injection.
FreeStyle 1.8 software was used to observe extracted ion chromatograms and mass spectra, and the workflow template “Untargeted Metabolomics with Statistics Detect Unknowns with ID Using Online Databases and mzLogic” in Compound Discoverer 3.3 was used for data processing and statistical analyses. Using a mzVault based in‐house library, containing retention time and MS/MS spectra of about 500 compounds, metabolite identification was accomplished. All software was from Thermo Scientific (Waltham, MA, USA).
All metabolites discussed in the current report, UDP‐glucose, glucuronate, glucose, glucose‐1‐phosphate, glucose‐6‐phosphate, arabinose, and choline, were present in the in‐house library.
2.6. GAG measurements
Plasma GAGs were measured using HeparinRed assay (RedProbes) following the manufacturer's instructions. To determine heparan sulfate (HS) and chondroitin sulfate (CS) levels, 2 mU/mL of heparinases I, II, III (Iduron) or chondroitinase ABC (Sigma) enzymes were incubated at 37°C overnight in enzyme digestion buffer (100 mM sodium acetate, 10 mM calcium acetate, pH 7 for HS or 50 mM tris, 50 mM sodium acetate, and pH 8 for CS) spiked into 60 μL plasma. 20 μL of digested aliquots were transferred to a fresh 96 well plate and 80 μL HeparinRed reagent added. To remove non‐GAG‐related fluorescence, the reduced fluorescence value after enzymatic digestion was subtracted from total fluorescence to provide the attributed fluorescence for each GAG type. These values were combined to generate total secreted HS/CS and to calculate the contribution of each GAG type in the sample. Data were obtained in triplicate technical replicates, averaged and standard deviation calculated. Data were normalized to the value of 1.00 using the average of the 2 healthy control samples.
3. RESULTS
3.1. Clinical course
The proband came to medical attention at age 10 weeks because of reduced height velocity and suspicion of an unknown inherited skeletal dysplasia since his father, who was 34 years old at the time, had short stature of unknown cause (Figures 2 and 3). Intrauterine ultrasound of the proband had shown short extremities. His length and weight at birth were within the normal range (51 cm, 50th centile and 4230 g, 89th centile (Juliusson et al., 2009)). At 6 months of age, his height was at the 2nd centile and at 2.5 years it was below the 1st centile (Figure 2). He had normal psychomotor development.
FIGURE 2.
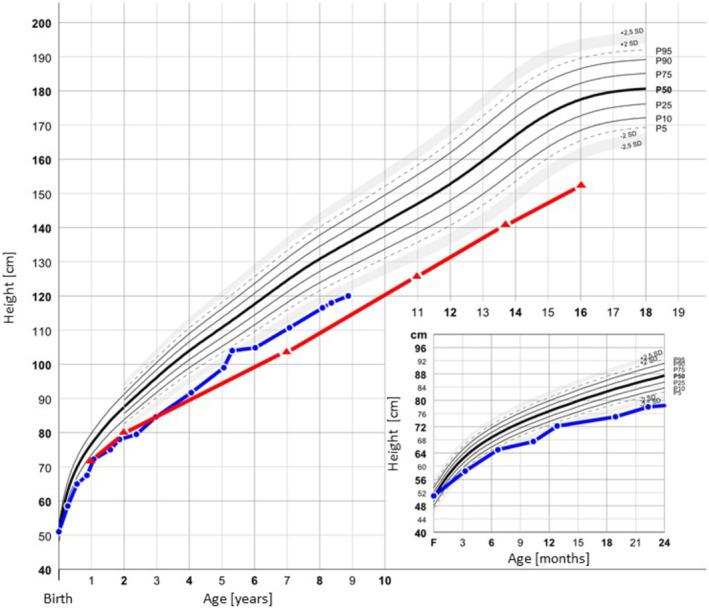
Growth charts. Growth charts for III‐1 (blue) and II‐1 (red). Median growth percentiles (P) according to Júlíusson et al. are shown in black (Juliusson et al., 2009).
FIGURE 3.
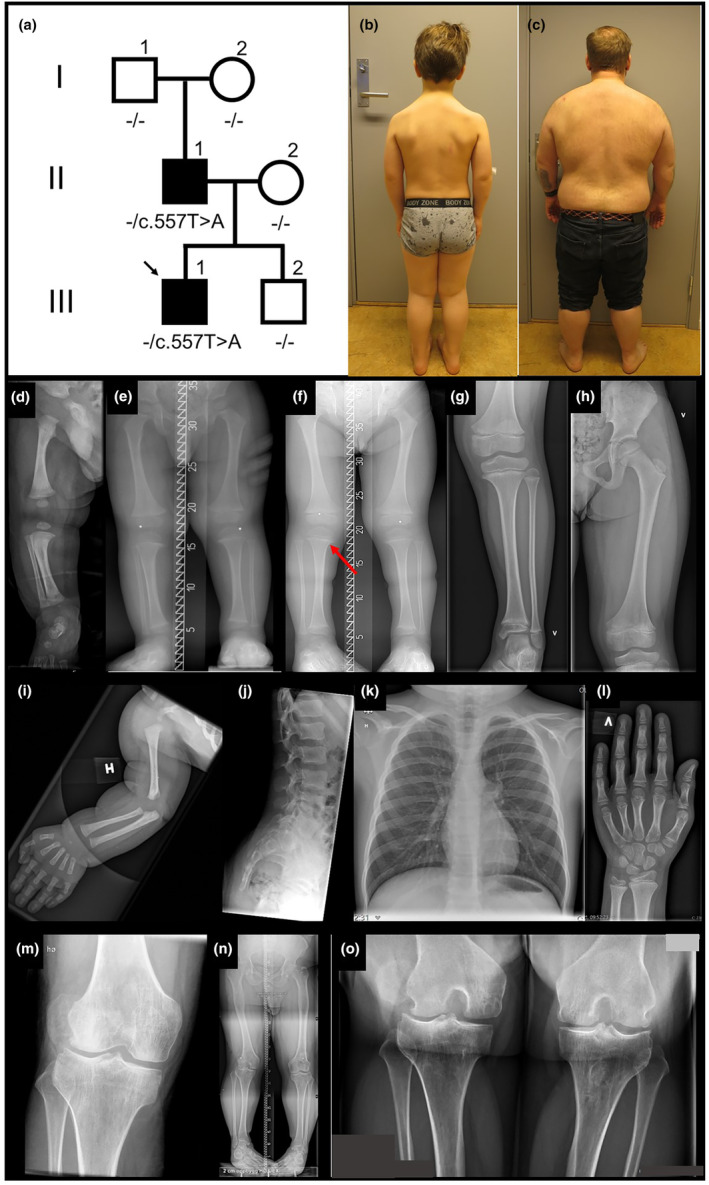
Pedigree, photos and x‐rays. (a) Pedigree, (b) III‐1 photo at age eight years one month, (c) II‐1 photo at age 41 years nine months, (d–l) III‐1 x‐rays, (d) and (i) age one month, (e) age two years, (f) age three and one half years, (g), (h), (j) and (k) age eight years, (l) age eight and one half years. Short long bones clearly apparent at age three and one half years (f), a slight medial beaking of the metaphysis in the proximal tibiae (f, arrow). Flattening of the vertebrae is considered within normal limits for a child (j). (m–o) II‐1 knees at age 40 years show broad metaphyses with bulbous joints.
At age one year, the proband had short extremities. A skeletal survey revealed short long bones and accelerated skeletal maturation with skeletal age approximately two years at chronological age 15 months. There was slight metaphyseal beaking medially in the proximal tibiae and subtle changes in the other metaphyses, as seen in Figure 3. His spine was normal without narrowing of the inferior lumbar interpedicular distance. Cranial and pelvic radiographs were unremarkable. Radiographs at age eight years showed short long bones with slight metaphyseal changes, most evident in the proximal tibiae. There were no dysplastic changes in the pelvis or spine. The slight flattening of the vertebrae seen in Figure 3j is considered within normal limits for an eight‐year‐old child.
Skeletal maturation at eight and a half years was in keeping with chronological age. Measurements of the proband's limb length on radiographs at age 21 days, 12 months (not shown), and age eight years showed rhizo‐mesomelic shortening (Table 1). Testing for growth hormone (GH) deficiency was negative, and serum vitamin D and calcium levels have been normal.
TABLE 1.
Clinical features.
| III‐1 (proband) | II‐1 (father) | |
|---|---|---|
| Age at latest examination | 8 years 1 month | 41 years 9 months |
| Height | 116.5 cm (−2.72 SD) (Juliusson et al., 2009) | 153 cm (−3.47 SD) (Juliusson et al., 2009) |
| Arm span | 113.5 cm | 157.5 cm |
| Sitting height (upper segment) | 68.7 cm, (Between 0 SD and −1 SD (Fredriks et al., 2005)) | 94.8 cm, (0 SD for 21 year olds (Fredriks et al., 2005)) |
| Sitting height/height ratio |
68.7/116.5 = 0.59 (>+2.5 SD (Fredriks et al., 2005)) |
94.8/153 = 0.62 (>+2.5 SD for 21 year olds (Fredriks et al., 2005)) |
| Weight | 27.3 kg (−0.11 SD) (Juliusson et al., 2009) | 109 kg (+2.36 SD) |
| Birth weight, length | 4230 g (1.13 SD) (Juliusson et al., 2009), 51 cm (0.13 SD) (Juliusson et al., 2009) | NA |
| Occipto‐frontal circumference | 52.5 cm (−0.35 SD) (Juliusson et al., 2009) | 60.5 cm, 0.5 cm above the 97.5th centile (Waaler, 1983) |
| Inner canthal distance | 2.5 cm (−1 SD to −2 SD (Hall, 2007)) |
2.8 cm (~−1 SD for 16 year olds (Hall, 2007)) |
| Hyperextensible interphalangeal joints (fingers and toes) | + |
+ |
| Joint luxations | − | Both patellas after initiating growth hormone treatment at age 11 years |
| Gross motor delay | ‐ Walked at ~1 year | NA |
| Proptosis/prominent eyes | − | − |
| Short neck | − | + |
| Webbing | + | + |
| Sloping shoulders | (+) | + |
| Prominent forehead | − | − |
| Low‐set ears | − | − |
| Downturned palpebral fissures | − | − |
| Microstomia | − | − |
| Atrophic scars | − | + |
| Micrognathia | − | − |
| Low nasal bridge | − | − |
| Pectus excavatum/carninatum | − | − |
| Broad fingers/toes | − | − |
| Sandal gap | + | + |
| Broad feet | − | + |
| Hypotonia | − | − |
| Fractures | − | − |
| Radio‐ulnar synostosis | − | − |
| Congenital heart defect | − | − |
| Blue sclerae | − | − |
| Glaucoma | Not examined | Not examined |
| Reduced bone density | Not examined | Not examined |
|
Humerus length [mm] (AVG ± 2 SD) |
21 days: 58 (63–79) 12 months: 90 (94–116) 8 years: 183 (199–246) |
40 years: NA |
|
Radius length [mm] (AVG ± 2 SD) |
21 days: 49 (54–64) 12 months: 69 (76–93) 8 years: 130 (149–186) |
40 years: NA |
|
Ulna length [mm] (AVG ± 2 SD) |
21 days: 57 (61–72) 12 months: 82 (85–103) 8 years: 147 (163–201) |
40 years: NA |
|
Femur length [mm] (AVG ± 2 SD) |
21 days: 64 (72–96) 12 months: 111 (128–149) 8 years: 258 (294–351) |
40 years: 440* (481–574) |
|
Tibia length [mm] (AVG ± 2 SD) |
21 days: 58 (60–83) 12 months: 86 (99–122) 8 years: 206 (230–296) |
40 years: 295* (390–496) |
|
Fibula length [mm] (AVG ± 2 SD) |
21 days: 56 (56–80) 12 months: 92 (96–117) 8 years: 210 (228–293) |
40 years: 320* (379–475) |
Note: Clinical features of the proband and father.
Abbreviations: NA, not available; SD, standard deviation.
After femoral lengthening.
At age eight and a half years, the proband is an active boy who participates in sports and attends regular school. He does not complain of joint pain and has not experienced patellar luxations. His vision and hearing are normal. Both the proband and his father have webbing of the neck and sloping shoulders, the latter more pronounced in the father.
The father's height at age one year was at the 1st centile and lower by age two years (Figure 2). He was thought to have hypochondroplasia and treated with GH from 11 to 13 years of age. Why treatment was stopped after two years is not clear. His self‐reported predicted final height pretreatment was 148 cm, at discontinuation of GH his height was 141 cm. His height at completed growth was 153 cm, −3.47 SD (Juliusson et al., 2009). At age 14 years, he had surgery to correct a 3 mm underbite. He underwent femoral osteotomies for leg lengthening at ages 18 and 21 years, and his adult height is 162.5 cm. As a child, teenager, and young adult, he did not have joint pain, or problems related to his spine. After GH treatment, he has had repeated bilateral patellar luxations, resulting in two left knee surgeries. In adulthood, increasing pain in his right knee due to secondary arthrosis led to a total knee replacement at age 40 years. He has normal vision, but reduced hearing in one ear after recurrent otitis media in childhood. He has hypermobile interphalangeal finger and toe joints (as does his son), in addition to sandal gap bilaterally. The father's radiological investigations prior to adulthood are not available. The diagnosis of hypochondroplasia in childhood was not confirmed molecularly.
The father's siblings have not received GH treatment and have adult heights between 167 and 177 cm. The paternal grandfather (I‐1) is 174 cm, and the paternal grandmother (I‐2) is 173 cm.
3.2. Genetics
Sequencing of the proband's DNA with a skeletal dysplasia gene panel and MLPA for SHOX were normal. Whole exome sequencing of the proband, affected father and unaffected paternal grandparents revealed a heterozygous variant of interest in UXS1: NM_001253875.2:c.557T>A p.(Ile186Asn) GRCh37 Chr 2:106746150A>T, not previously observed in‐house or in gnomAD v4.0.0. The variant was confirmed de novo in the father and inherited in the proband by Sanger sequencing (Figure 4). In silico prediction suggested that the variant was pathogenic (REVEL score 0.886). Neither contacting other laboratories directly nor registration in GeneMatcher, led to identification of additional individuals with potentially deleterious variants in UXS1. According to the guidelines published by American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015), we classified the variant as likely pathogenic based on the following criteria: PS2P (the variant is de novo), PS3M (evidence from functional studies, see sections UXS1 enzymatic activity and GAG measurements below), PM2 (the variant is absent from gnomAD), and PP3 (computational evidence supporting a deleterious effect).
FIGURE 4.
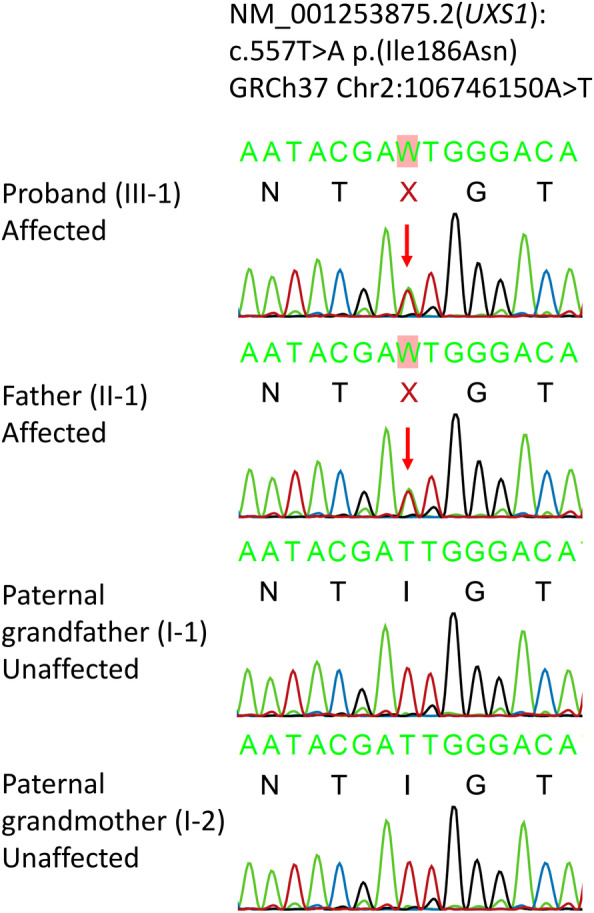
Sanger sequencing. Sanger sequencing electropherograms show the heterozygous variant NM_001253875.2(UXS1): c.557T>A p.(Ile186Asn) in the proband and father. The unaffected paternal grandparents are wild‐type.
3.3. Structural analysis of Ile186Asn‐UXS1 protein
UXS1 consists of two domains, a large NAD+‐binding domain and a smaller UDP‐GlcA binding domain. The enzyme's active site has six highly conserved residues, which lie in a cavity formed between the two domains (Figure 5). It has previously been shown that the active enzyme is a homodimer with a dimer interface composed of helices α5 and α7 forming a helical bundle (Figure 5) (Eixelsberger et al., 2012). Residue Ile186 is located in α5 within the dimer interface where it makes intermolecular hydrophobic contact with Ile186 in the other subunit. Changing hydrophobic isoleucine to polar asparagine at this position is likely to disturb hydrophobic interactions and modify the intermolecular forces between the monomers, ultimately impacting enzymatic activity.
FIGURE 5.
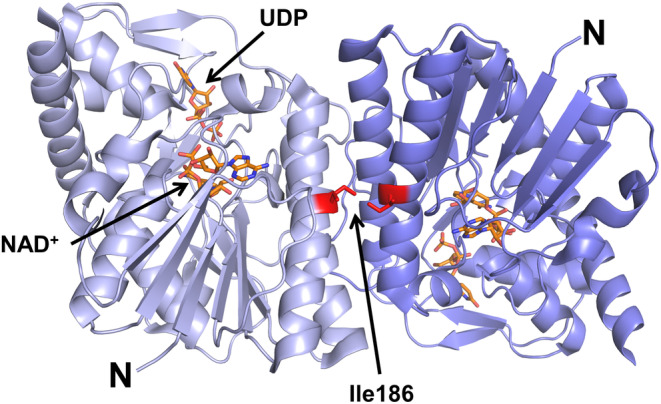
Three dimensional structure of UXS1. Cartoon representation of the hUXS1 crystallographic dimer based on the 1.26 Å crystal structure of hUXS1 bound with NAD+ and UDP (PDB ID: 2B69). The two UXS1 monomers are shown in light and dark blue. NAD+ and UDP are shown as orange sticks. Ile186 is located in the dimer interface where it makes intermolecular hydrophobic contact with Ile186 in the other subunit.
3.4. UXS1 enzymatic activity
To determine whether Ile186Asn affects enzymatic activity, the cytoplasmic domain of WT‐UXS1 and Ile186Asn‐UXS1 were overexpressed and purified from E. coli. The two versions of the enzyme were incubated overnight with UDP‐GlcA and NAD+ before the nucleotide sugar substrate (UDP‐GlcA) and product (UDP‐Xyl) were analyzed by LC–MS/MS in negative mode (Figure 6). Two peaks eluting at 14.94 and 16.82 min were observed in WT‐UXS1, but only the first peak was present in Ile186Asn‐UXS‐1 (Figure 6a). The MS/MS spectrum of the first peak with precursor ions at m/z 579 was consistent with UDP‐GlcA, while the second with precursor ions at m/z 535 was consistent with UDP‐Xyl (Figure 6b). The MS/MS ion fragments at m/z 235, 323, and 403 are assigned [UDP‐uridine‐H−]−, [UMP‐H−]−, and [UDP‐H−]−, respectively. These data suggest that Ile186Asn‐UXS1 does not synthesize UDP‐Xyl. For WT enzyme, biosynthesis of UPD‐Xyl increased with increasing amount of enzyme (Figure 6c) and produced higher activity in glycerol stock than in aqueous solution (data not shown). Allthough Ile186Asn‐UXS1 fails to convert UDP‐GlcA to UDP‐Xyl, it nonetheless reduces the level of UPD‐GlcA (Figure 6c).
FIGURE 6.
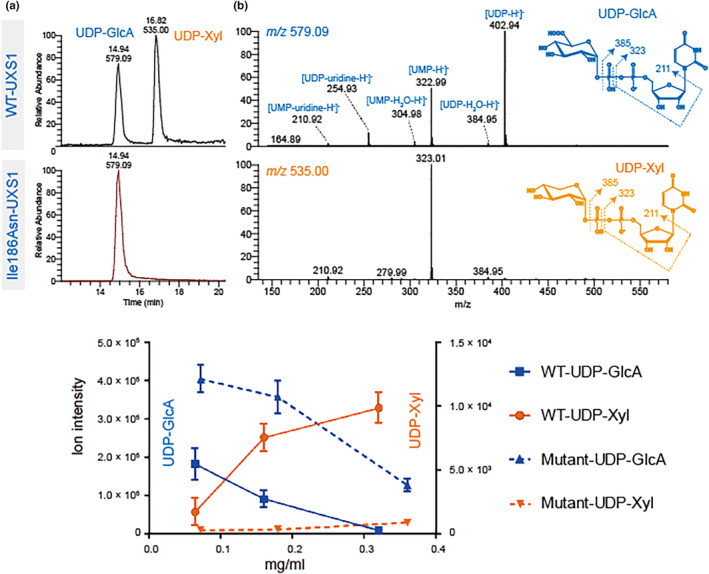
Activity of recombinant Ile186Asn‐UXS1 and WT‐UXS1. Purified recombinant enzymes were incubated with UDP‐GlcA in the presence of NAD+. Analysis of nucleotide sugars was carried out by PGC‐LC‐ESI‐MS/MS. (a) Extracted ion chromatogram at m/z 535 and 579, corresponding to deprotonated UDP‐Xyl and UDP‐GlcA, respectively. Only WT‐UXS1 converts UDP‐GlcA to UDP‐Xyl. (b) MS/MS spectrum of precursor ions at m/z 579.09 (upper) and 535.00 (lower). Fragmentation ions at m/z 211, 323, and 403 are consistent with predicted fragmentation of a UDP‐sugar into [UDP‐H−]−, [UMP‐H−]−, and [UDP‐uridine‐H−]−, respectively. (c) Ion intensity of ions at m/z 535 and 579. The decrease of UDP‐GlcA is commensurate with increased UDP‐Xyl reflecting the activity of WT enzyme. With mutant enzyme, only a decrease of UDP‐GlcA was observed.
3.5. Metabolomics
After collection of the global metabolomes from dried blood spots and validated signal correction, metabolites in the biochemical pathway of proteoglycan synthesis were examined. Metabolites directly upstream and downstream of the altered enzyme (Figure 7) could not be detected using the metabolomics method. Examining compounds in connected pathways, however, we noted that the proband and father had on average 80% higher levels of glucuronate than the unaffected brother (III‐2, Figure 7b). For related metabolic compounds such as glucose, glucose‐1‐phosphate, glucose‐6‐phosphate, arabinose, and UDP‐glucose, there were less than 30% differences in amounts between affected father/proband and the unaffected brother.
FIGURE 7.
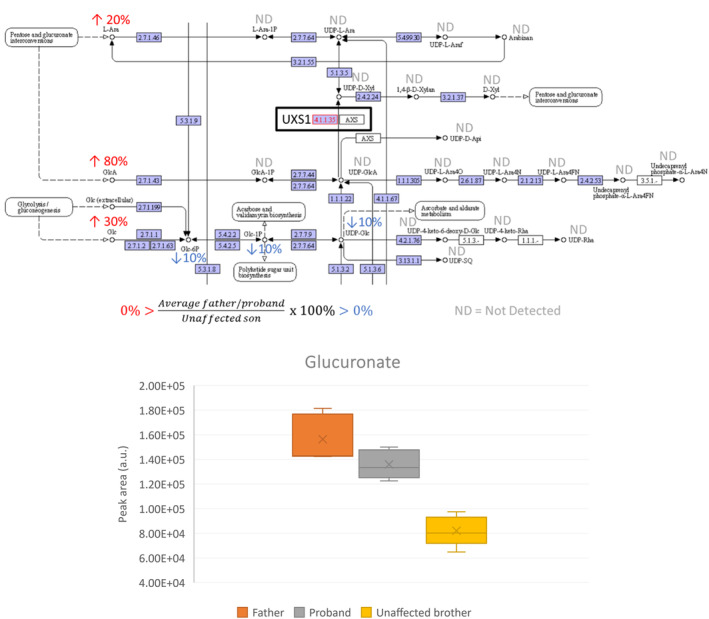
Focused metabolomics of proteoglycan synthesis pathway. (a) Part of amino sugar and nucleotide sugar metabolism in KEGG database (Kanehisa Laboratories, n.d.). % Peak area of compound in samples from father and proband compared to unaffected brother. ND = not detected by the metabolomics method. (b) Glucuronate peak area in samples of father, proband and unaffected brother (N = 5). Father and proband had an ~80% higher level than the unaffected brother (90% and 70%, respectively).
Next, a discovery step was performed by comparing the global metabolomes from the proband and affected father, with the unaffected brother. Choline and a yet unidentified component with m/z 129.06596 and putative chemical formula C5H8N2O2 (neutral mass) were significantly increased in amount in the affected father/proband compared to the unaffected brother (data not shown). However, both of these compounds were in the normal range for all three when compared to a healthy population (N = 50, data collected in earlier projects).
3.6. GAG measurements
Finally, secreted GAGs were measured by HeparinRed assay on plasma from the proband, father and two healthy controls (Figure 8). Overall plasma GAG levels were decreased in both the proband (0.58) and father (0.52) compared to healthy controls (1.00), and affected both HS and CS (0.25, 0.24, and 0.33, 0.28 for proband, father for HS and CS, respectively).
FIGURE 8.
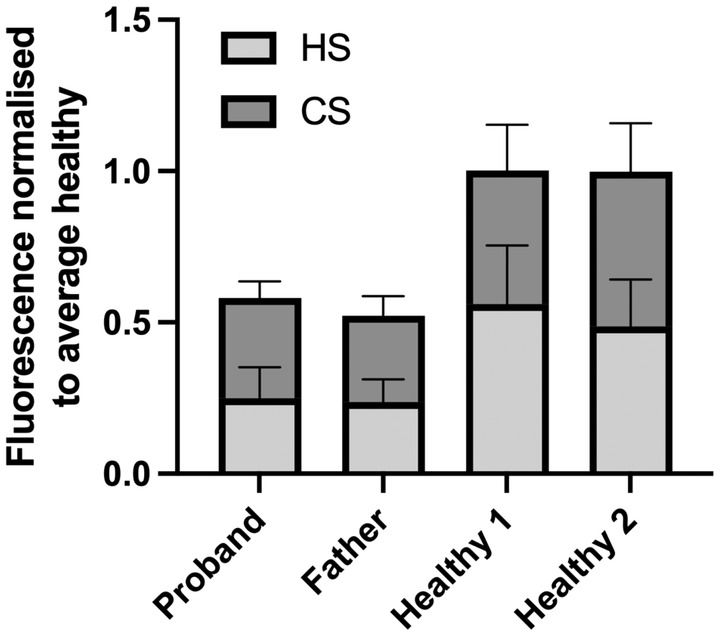
GAG secretion. Average plasma heparan sulfate (HS) and chondroitin sulfate (CS) levels in proband, father and two healthy individuals. Data normalized (Moriarity et al., 2002) to average of healthy controls. Error bars represent standard deviation of three technical replicates.
4. DISCUSSION
Linkeropathies are a clinically heterogeneous group of connective tissue disorders caused by defects in the biosynthesis of proteoglycans (Jones et al., 2015). Here, we describe a novel linkeropathy, a type of short‐limbed short stature with mild radiographic changes, presumably caused by a likely pathogenic, heterozygous variant in UXS1. UXS1 catalyzes the formation of UDP‐xylose necessary for the first step in formation of the tetrasaccharide linker in proteoglycan biosynthesis of heparan sulfate/heparin and chondroitin‐/dermatan sulfate. UDP‐xylose is rarely involved in other catabolic pathways in humans, and its effect on proteoglycans is likely to be quite specific.
Twenty‐six unique UXS1 loss‐of‐function (LoF) variants are registered in gnomAD (expected 44.8, observed 26, pLI = 0), making LoF an unlikely mechanism for a rare autosomal dominant skeletal dysplasia. Taken together with the report of a single heterozygous deletion classified as likely benign in ClinVar (National Center for Biotechnology Information, n.d.), we suggest that Ile186Asn disrupts enzyme activity through a possible dominant negative effect, as is often the case in disorders due to aberrations in homomeric complexes (Gerasimavicius et al., 2022). Allthough Ile186Asn is absent from gnomAD, a different missense variant affecting the same codon, NM_001253875.2:c.557T>C p.(Ile186Thr), is present in four alleles. Given the relatively mild clinical phenotype we describe herein, it is reasonable to assume that data from other individuals with a UXS1‐associated linkeropathy may be present in gnomAD. In keeping with this hypothesis, is the presence of eight alleles with the common pathogenic variant, NM_000142.4 (FGFR3):c.1138G>A p.(Gly380Arg), which causes hypochondroplasia, another relatively mild skeletal dysplasia.
Ile186Asn is located in the dimer interface, potentially disrupting interactions necessary for dimer formation. Disruption of this interface mediated by the previously published zebrafish mutant (equivalent to human Arg236His) leads to deactivation of UXS1 (Eames et al., 2010). Ile186 is highly conserved, and a REVEL score of 0.886 also supports pathogenicity of Ile186Asn.
4.1. In vitro assays
Compared to purified, recombinant WT UXS1, Ile186Asn‐UXS1 is inactive (Figure 6). However, the level of UDP‐GlcA decreased with increasing amounts of Ile186Asn‐UXS1 enzyme (as well as extended incubation, data not shown). The reason for this decrease is unknown.
The uxs1 mutant zebrafish has demonstrated that Uxs1 activity is essential for production and organization of the extracellular skeletal matrix (Eames et al., 2010). Uxs1 plays a role in several aspects of skeletal morphogenesis including chondrocyte intercalating and stacking and cartilage elongation. The shorter cartilage elements in the mutant fish (Eames et al., 2010) are in keeping with the short long bones in the affected individuals we describe.
Global metabolomic analysis of dried blood spots revealed some differences between the two affected individuals and an unaffected family member. However, levels were in the normal range compared to a healthy population. The focused analysis of metabolites in the biochemical pathway of proteoglycan synthesis showed increased amounts of glucuronate (GlcA) a few enzymatic steps upstream of UXS1 (Figure 7). This is in line with reduced UXS1 activity, since decreased ability to convert UDP‐GlcA to UDP‐Xyl would lead to an increase in substrate. Unfortunately, metabolites directly upstream or downstream could not be detected with our assays.
The proband and father showed reduced, but detectable, GAG levels in plasma; consistent with the expectation that a heterozygous variant with a dominant negative effect would likely result in residual UXS1 activity.
4.2. Phenotype
Phenotypic features present in both the proband and father include short stature with short long bones, hypermobile interphalangeal joints in fingers and toes, sandal gap, webbing of the neck, and sloping shoulders (Figures 2 and 3). The father's recurrent patellar luxations may be a feature of the disorder, but could also be related to previous leg lengthening surgery. In the most recent nosology of skeletal dysplasias (Unger et al., 2023), linkeropathies are classified as dysplasias with multiple dislocations. Linkeropathies exhibit a broad spectrum of phenotypic features, including a variety of skeletal anomalies (Taylan & Makitie, 2016). However, short stature, joint laxity, joint dislocations, and sandal gap are shared by all (Jones et al., 2015). The phenotype in this family is compatible with, but not diagnostic of, a linkeropathy. The slight metaphyseal changes present in the proband are similar to what may be seen in linkeropathies caused by pathogenic variants in B3GAT3, B4GALT7, B3GALT6 and XYLT1 (Ritelli et al., 2019).
The father has a head circumference slightly above the 97.5th centile, as do several individuals in his family, including first‐ and second‐degree relatives of average stature. Therefore, we suspect that his head size is unrelated to the UXS1 variant.
4.3. Differential diagnoses
Conditions included in the metaphyseal dysplasia group in the current nosology could be relevant differential diagnoses although metaphyseal involvement in our family is slight. For example, Schmid type of metaphyseal dysplasia (Unger et al., 2023) might be considered, although this disorder typically presents with bowed limbs (Al Kaissi et al., 2018) in addition to short stature.
Cartilage‐hair hypoplasia (CHH) also affects the metaphyses. In CHH, short stature is usually apparent prenatally or at birth, which was not unequivocally the case in our family. Additionally, normal hair growth makes CHH unlikely.
Identifying the likely pathogenic variant in UXS1, alleviates concerns about the development of serious extra‐skeletal features, such as immunodeficiency in CHH and pancreatic insufficiency in Shwachman‐Diamond syndrome.
4.4. Growth hormone treatment
The father received GH for two years from age 11 years. His predicted final height pre‐treatment was 148 cm. His final height exceeded this by 5 cm; this increase is within the margin of error for height prediction and cannot be attributed to GH treatment.
The father underwent surgery for mandibular prognatism after having received GH. Mandibular prognathism, not previously described as a feature of linkeropathies, has been described in conjunction with GH treatment (Kjellberg & Wikland, 2007; Pan et al., 2013). At age eight and a half years, the proband has an unremarkable mandible.
In the absence of GH resistance, and given his normal GH level, as well as the likely pathogenic UXS1‐variant, we have not offered GH treatment to the proband.
5. CONCLUSION
We describe a novel short‐limbed short stature condition in the linkeropathy category likely caused by a monoallelic variant in UXS1. Given the broad phenotype of linkeropathies, and the nonspecific clinical and radiological features in the family we describe, we suggest including UXS1 in gene panels for short stature and skeletal dysplasias. Further case reports are required to validate the phenotypic effects of deleterious variants in UXS1.
AUTHOR CONTRIBUTIONS
All authors of this manuscript fulfill the Vancouver criteria for authorship. CFR conducted clinical investigations and genetic counseling. PHB expressed and purified proteins and did structural analysis. CJ performed chromatography and enzymatic assays. NK contributed in planning and design of enzymatic assays and interpretation of data. MLMH contributed in planning, design, and performance of the GAG secretion assay and interpretation of data. EM conducted radiographic examinations. BW, ESS, and KBPE performed metabolomics studies. KT, TP, and ØLH performed exome sequencing and variant interpretation. ØLH planned the study. CFR, TP, and ØLH drafted the manuscript. All authors took part in the writing process and revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
ETHICAL COMPLIANCE
The study was approved by The Norwegian Regional Committee for Medical and Health Research Ethics (2019/616) and complies with the Declaration of Helsinki. The proband's parents provided informed, written consent on behalf of themselves and their children under 16 years of age. Initial investigations were performed in a diagnostic setting. Parental/guardian consent for publication was obtained.
ACKNOWLEDGMENTS
We are grateful to the family for their participation in this work. PHB was supported by the Southeastern Norway Regional Health Authorities Technology Platform for Structural Biology (grant 2015095). MLMH received funding from MSCA Postdoctoral Fellowship (GlycoMap grant 101107665). We also thank SciLifeLab and BioMS, funded by the Swedish research council, for providing financial support to the Proteomics Core Facility, Sahlgrenska Academy (CJ). NK received funding from Oslo Metropolitan University and University of Gothenburg. Figure 1 was created using BioRender.com.
Rustad, C. F. , Backe, P. H. , Jin, C. , Merckoll, E. , Tveten, K. , Maciej‐Hulme, M. L. , Karlsson, N. , Prescott, T. , Sand, E. S. , Woldseth, B. , Elgstøen, K. B. P. , & Holla, Ø. L. (2024). A monoallelic UXS1 variant associated with short‐limbed short stature. Molecular Genetics & Genomic Medicine, 12, e2472. 10.1002/mgg3.2472
Cecilie F. Rustad and Paul Hoff Backe contributed equally to this study.
DATA AVAILABILITY STATEMENT
Data are available in a public, open access repository: Glycopost (https://glycopost.glycosmos.org/preview/23867747464d0c06bb6b1b).
REFERENCES
- Al Kaissi, A. , Ghachem, M. B. , Nabil, N. M. , Kenis, V. , Melchenko, E. , Morenko, E. , Grill, F. , Ganger, R. , & Kircher, S. G. (2018). Schmid's type of metaphyseal chondrodysplasia: Diagnosis and management. Orthopaedic Surgery, 10(3), 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eames, B. F. , Singer, A. , Smith, G. A. , Wood, Z. A. , Yan, Y. L. , He, X. , Polizzi, S. J. , Catchen, J. M. , Rodriguez‐Mari, A. , Linbo, T. , Raible, D. W. , & Postlethwait, J. H. (2010). UDP xylose synthase 1 is required for morphogenesis and histogenesis of the craniofacial skeleton. Developmental Biology, 341(2), 400–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eixelsberger, T. , Sykora, S. , Egger, S. , Brunsteiner, M. , Kavanagh, K. L. , Oppermann, U. , Brecker, L. , & Nidetzky, B. (2012). Structure and mechanism of human UDP‐xylose synthase: Evidence for a promoting role of sugar ring distortion in a three‐step catalytic conversion of UDP‐glucuronic acid. The Journal of Biological Chemistry, 287(37), 31349–31358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriks, A. M. , van Buuren, S. , van Heel, W. J. , Dijkman‐Neerincx, R. H. , Verloove‐Vanhorick, S. P. , & Wit, J. M. (2005). Nationwide age references for sitting height, leg length, and sitting height/height ratio, and their diagnostic value for disproportionate growth disorders. Archives of Disease in Childhood, 90(8), 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimavicius, L. , Livesey, B. J. , & Marsh, J. A. (2022). Loss‐of‐function, gain‐of‐function and dominant‐negative mutations have profoundly different effects on protein structure. Nature Communications, 13(1), 3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. G. (2007). Handbook of physical measurements (2nd ed.). Oxford University Press. [Google Scholar]
- Jones, K. L. , Schwarze, U. , Adam, M. P. , Byers, P. H. , & Mefford, H. C. (2015). A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. American Journal of Medical Genetics. Part A, 167A(11), 2691–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliusson, P. B. , Roelants, M. , Eide, G. E. , Moster, D. , Juul, A. , Hauspie, R. , Waaler, P. E. , & Bjerknes, R. (2009). [Growth references for Norwegian children]. Tidsskrift for den Norske Lægeforening, 129(4), 281–286. [DOI] [PubMed] [Google Scholar]
- Kanehisa Laboratories KEGG PATHWAY Database; [map00520]. https://www.genome.jp/pathway/map00520+C00191
- Kjellberg, H. , & Wikland, K. A. (2007). A longitudinal study of craniofacial growth in idiopathic short stature and growth hormone‐deficient boys treated with growth hormone. European Journal of Orthodontics, 29(3), 243–250. [DOI] [PubMed] [Google Scholar]
- Moriarity, J. L. , Hurt, K. J. , Resnick, A. C. , Storm, P. B. , Laroy, W. , Schnaar, R. L. , & Snyder, S. H. (2002). UDP‐glucuronate decarboxylase, a key enzyme in proteoglycan synthesis: Cloning, characterization, and localization. The Journal of Biological Chemistry, 277(19), 16968–16975. [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information ClinVar; [VCV000443673.2]. https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000443673.2
- Pan, C. Y. , Lan, T. H. , Chou, S. T. , Tseng, Y. C. , Chang, J. Z. , & Chang, H. P. (2013). Orthodontic treatment for a mandibular prognathic girl of short stature under growth hormone therapy. Journal of the Formosan Medical Association, 112(12), 801–806. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritelli, M. , Cinquina, V. , Giacopuzzi, E. , Venturini, M. , Chiarelli, N. , & Colombi, M. (2019). Further defining the phenotypic spectrum of B3GAT3 mutations and literature review on linkeropathy syndromes. Genes (Basel), 10(9), 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skogvold, H. B. , Sandas, E. M. , Osteby, A. , Lokken, C. , Rootwelt, H. , Ronning, P. O. , Rootwelt, H. , & Elgstøen, K. B. P. (2021). Bridging the polar and hydrophobic metabolome in single‐run untargeted liquid chromatography‐mass spectrometry dried blood spot metabolomics for clinical purposes. Journal of Proteome Research, 20(8), 4010–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeland, M. F. , Brouillard, P. , Prescott, T. , Boon, L. M. , Hvingel, B. , Nordbakken, C. V. , Nystad, M. , Holla, Ø. L. , & Vikkula, M. (2023). Biallelic ANGPT2 loss‐of‐function causes severe early‐onset non‐immune hydrops fetalis. Journal of Medical Genetics, 60(1), 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylan, F. , & Makitie, O. (2016). Abnormal proteoglycan synthesis due to gene defects causes skeletal diseases with overlapping phenotypes. Hormone and Metabolic Research, 48(11), 745–754. [DOI] [PubMed] [Google Scholar]
- Unger, S. , Ferreira, C. R. , Mortier, G. R. , Ali, H. , Bertola, D. R. , Calder, A. , Cohn, D. H. , Cormier‐Daire, V. , Girisha, K. M. , Hall, C. , Krakow, D. , Makitie, O. , Mundlos, S. , Nishimura, G. , Robertson, S. P. , Savarirayan, R. , Sillence, D. , Simon, M. , Sutton, V. R. , … Superti‐Furga, A. (2023). Nosology of genetic skeletal disorders: 2023 revision. American Journal of Medical Genetics. Part A, 191(5), 1164–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaler, P. E. (1983). Anthropometric studies in Norwegian children. Acta Paediatrica Scandinavica. Supplement, 308, 1–41. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available in a public, open access repository: Glycopost (https://glycopost.glycosmos.org/preview/23867747464d0c06bb6b1b).