

Earth probably harbours a million times more virus particles than there are stars in the observable Universe. These viruses could hold solutions to many of humanity’s current problems.
Phage therapy could someday be used to treat diseases caused by multidrug-resistant bacteria, for instance. Enzymes encoded by new viruses could help researchers to develop pharmaceuticals. Or viruses that kill algal cells could be used to control harmful blooms.
Tapping into the benefits — and threats — requires describing and cataloguing viruses and mapping their evolutionary relationships.
But, so far, just 4,958 virus species have been formally described. Comparative analyses of the genomes of these and numerous unclassified viruses show that the current taxonomy is vastly incomplete and, in places, even wrong.
Last month a momentous step was announced by the International Committee on Taxonomy of Viruses (ICTV), which authorizes and organizes the classification and naming of virus species. (Five of us have roles in this organization: J.H.K., M.K., Y.-Z.Z., P.M. and V.V.D.) The committee endorsed the establishment of taxa above the rank of order1. It introduced the first-ever virus phylum, Negarnaviricota, for single-stranded negative-sense RNA viruses (see ‘Branching out’). Similar high-rank taxa could be established for other major virus groups.
BRANCHING OUT.
A first for viruses
Higher taxonomic ranks of RNA viruses can be established on the basis of a phylogeny of a universal marker. (In a similar way, the taxonomy of cellular organisms is based largely on the phylogenetic trees of universally conserved genes, such as those for ribosomal RNA.)
For RNA viruses, this marker is the RNA-dependent RNA polymerase (RdRp), the only protein shared by all bona fide RNA viruses. In the RdRp tree6, all negative-sense RNA viruses form a single branch, justifying the phylum Negarnaviricota for single-stranded, negative-sense RNA viruses. This single branch splits into two subbranches, the subphyla Haploviricotina and Polyploviricotina (see ‘Classifying RNA viruses’ and supplementary information).
The need for two subphyla is also supported by another major molecular marker. Haploviricotines synthesize a cap structure on virus messenger RNAs that is required for protein synthesis using a virus enzyme. Polyploviricotines, by contrast, ‘snatch’ this cap from host mRNAs. J.H.K. et al.
CLASSIFYING RNA VIRUSES.
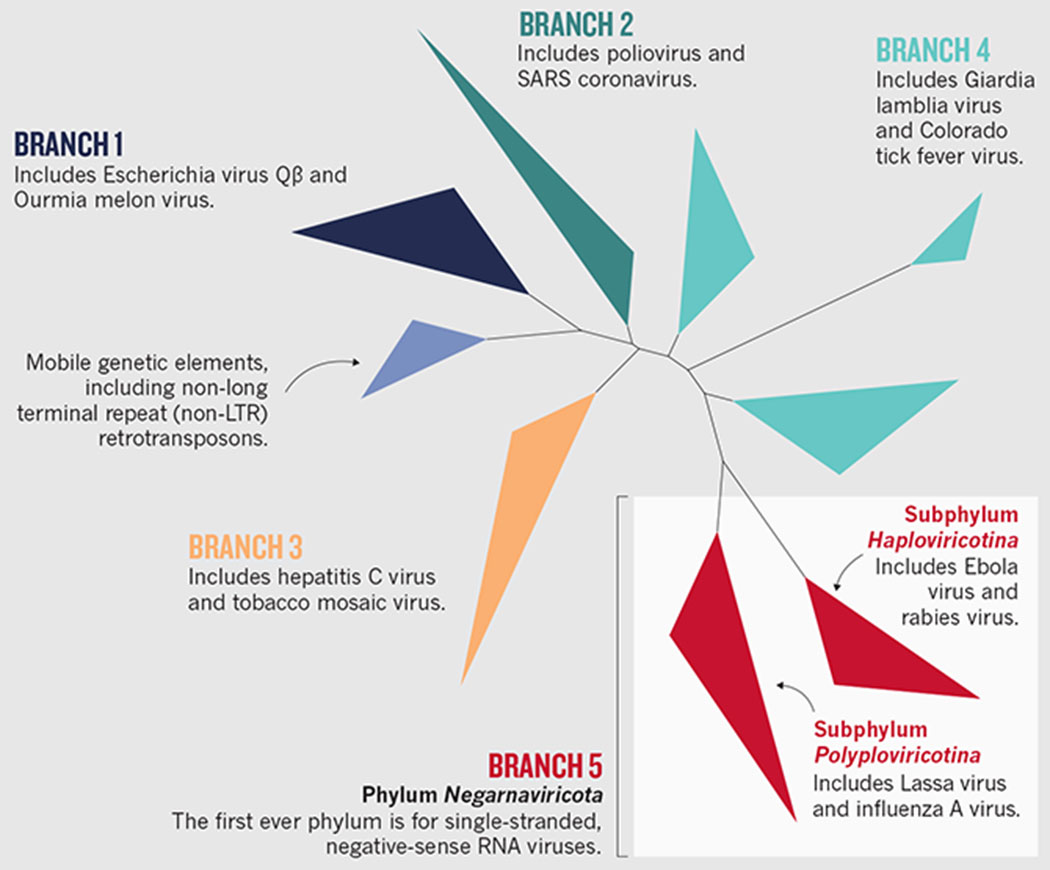
Phylogenetic tree determined using RNA-dependent RNA polymerases.
Comprehensive classification of the entire virosphere now seems within reach. But major changes are needed to make that happen.
HIDDEN DIVERSITY
There are probably billions of distinct viruses. Viruses depend on host cells to replicate, as do other ‘selfish’ genetic elements, such as viroids and plasmids. The same host (say, humans or the Escherichia coli bacterium) can be infected by many, very different viruses. However, most of the viruses that have been described infect only a few hosts. In fact, bacterial viruses (bacteriophages) often infect members of just one species, so are sometimes used to identify the bacteria in a sample. In other words, the diversity of viruses is probably at least as great as that of their available hosts.
Only around 5,500 species of mammal and about 391,000 species of vascular plant have been established. But the smaller the organism, the higher the numbers typically get. There are 1 million to 10 million species of arthropod and probably more than 5.1 million distinct fungi. And all bets are off when it comes to prokaryotes (bacteria and archaea). Estimates differ widely, but an analysis published earlier this month suggests that there could be as many as 1.6 million distinct microbes on Earth2. Each of these microbes is almost certainly associated with at least one virus.
During one of the Tara Oceans expeditions (between 2009 and 2013), investigators identified 5,476 populations of double-stranded DNA viruses in 43 samples taken from 26 stations. And only 39 of these populations were found to closely resemble lineages recognized by the ICTV3. Only last year, a study of marine microbial metagenomes revealed more phylogenetic diversity in one proposed family of large DNA viruses (‘Megaviridae’) than currently exists in bacteria and archaea, combined4.
In short, the planet hosts massive virus diversity.
UNTAPPED POTENTIAL
“So what?” some might ask. Only a few hundred viruses are known to cause animal or plant diseases, and although more pathogenic viruses will certainly be discovered, they will probably number only in the hundreds.
In our view, limiting analyses only to potentially pathogenic viruses is short-sighted and ultimately negligent. Most antibiotics in clinical use were discovered from harmless soil bacteria. A key component of commercial detergents — heat-resistant proteases — was discovered in deep-sea prokaryotes not known to harm any eukaryotes. Most influential biotechnological breakthroughs of the twentieth and twenty-first centuries are based on enzymes from seemingly ‘unimportant’ bacteria. Examples include: the restriction enzymes that launched genetic engineering in the 1970s; the polymerase chain reaction (PCR) used to make many copies of a DNA segment; and the CRISPR technology now revolutionizing genome engineering. Indeed, CRISPR–Cas systems were discovered through the study of prokaryotic immune responses against viruses that no clinician could name.
It is quite possible that the proteases and polymerases encoded by the viruses of deep-sea organisms will offer researchers qualities different from those they have access to today. And bacterial and archaeal viruses encode an enormous diversity of proteins that could enable geneticists to fine-tune their use of CRISPR to engineer organisms.
We cannot know what the trove of ‘unimportant’ viruses could possibly amount to until we have examined them.
What’s more, virologists are discovering that the phenomenon of switching hosts is not as random and rare as was thought. Given the growing threat of emerging virus diseases in the face of human-induced changes such as population growth, urbanization and climate change, describing and cataloguing the viromes of healthy and sick humans is crucial. Cataloguing should include all the viruses in human tissues; those in the bacteria, protists, fungi and worms that inhabit human bodies; those in our close associates — pets, pests and farm animals; and the viruses in the microfauna that inhabit these animals.
Ultimately, we must decipher patterns in the virosphere — for instance, how viruses interact with each other and with microbes, as well as the triggers that cause them to jump hosts. Then, we might be able to predict the emergence of the next HIV-1, Ebola virus, or influenza A virus that is capable of causing another deadly pandemic. Antiviral therapy could even be feasible for a person infected with a new virus, if researchers have a deep understanding of the virus’s closest relatives. (A similar approach is now routinely used to treat bacterial infections.)
Last but not least, comprehensive classification is essential for understanding how viruses and their cellular hosts have shaped each other, as well as how life on Earth began and has evolved.
MANY CHALLENGES
Classifying millions of viruses into hierarchically organized taxa requires a huge, community-wide effort and a broad array of researchers — including virologists, microbiologists, ecologists, veterinary surgeons and zoologists. These scientists will need incentives. Changes should also be made to the classification process itself.
Currently, for a newly discovered virus or group of viruses to be classified, researchers need to submit a proposal detailing the rationale to the ICTV. The proposals then undergo a multi-stage review process that involves, among others, ICTV study groups (experts on the virus group in question), subcommittees and the executive committee.
If the executive committee approves the proposal, it is published on the ICTV website (http://ictv.global), and members can elect to ratify it through electronic vote. Newly approved classifications are announced at least once a year.
Any researcher can submit a proposal, at https://talk.ictvonline.org. Yet, for various reasons, many virus discoverers think that their role in the process of classification ends when they publish their work in a scientific journal. The rest — the actual cataloguing — is left to the ICTV.
The ICTV runs on a shoestring budget; regular donations of around US$12,000 per year come from a few microbiology societies, including the International Union of Microbiological Societies, the American Society for Virology, the Microbiology Society and, occasionally, organizations such as the UK Wellcome Trust. The ICTV functions almost exclusively through the efforts of a few hundred volunteers who advance virus taxonomy using their free time and, very often, personal funds. Thus, the classification of viruses is somewhat neglected — especially for ones that do not obviously fall under the purview of the existing study groups or are discovered through metagenomics surveys.
In 2017, the ICTV greatly eased the process by allowing viruses to be classified on the basis of genome-sequence information alone5. Before this, researchers had to grow and study them in the laboratory — a process that is feasible only for some virus groups. The establishment of the first taxon above the rank of order, the Negarnaviricota phylum, is another major step forward by the ICTV.
But more changes are needed.
CALL FOR HELP
First, the ICTV needs dedicated and continuous funding, perhaps from an organization such as the Bill & Melinda Gates Foundation or the Gordon and Betty Moore Foundation. Such investment is needed to create a centralized, online platform for classification. This platform could incorporate cutting-edge taxonomic methodologies and increase the efficiency of submitting proposals that are based on massive amounts of data. Funds are also needed to help curate and maintain the taxonomy database and for the development of analytical tools such as algorithms that automatically classify virus genomes.
Second, schemes are needed to motivate virologists and others to get more involved in taxonomy. Proposals to the ICTV could count as formal publications in their own right. Or publishers could make submitting a proposal to the ICTV a condition of publishing papers on newly described viruses (much of the information required for both is the same). In principle, the classification of viruses could become routine, much the same as the deposition of gene-sequence information to public databases such as GenBank has become commonplace in genomics.
If more virologists joined ICTV study groups, and all discoverers submitted taxonomic proposals and manuscripts in tandem, the specialists on the new viruses would routinely join the ICTV volunteers in preparing taxonomic proposals. That alone would greatly improve classification efforts.
We call on funders to give more priority to projects based on evolutionary biology and discovery, so that more virologists can harness the rapidly developing analytical tools and get involved in taxonomy.
Virus classification is a straightforward way to contribute today to solving the global problems of tomorrow. ■
Supplementary Material
Viruses that infect humans, such as Ebola, are a tiny fraction of what needs to be characterized.
A researcher observes the life cycle of Pithovirus, a giant virus that infects amoebae.
Footnotes
Supplementary information accompanies this article: see go.nature.com/2tk6fpj.
Contributor Information
Jens H. Kuhn, Integrated Research Facility at Fort Detrick, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Frederick, Maryland, USA.
Yuri I. Wolf, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA.
Mart Krupovic, Department of Microbiology, Pasteur Institute, Paris, France.
Yong-Zhen Zhang, Shanghai Public Health Clinical Center & Institutes of Biomedical Sciences, Fudan University, Shanghai, China; Chinese Center for Disease Control and Prevention, Beijing, China.
Piet Maes, Zoonotic Infectious Diseases Unit, Catholic University of Leuven (KU Leuven), Leuven, Belgium.
Valerian V. Dolja, Department of Botany and Plant Pathology, Oregon State University, Corvallis, Oregon, USA.
Eugene V. Koonin, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA.
References
- 1.Siddell SG et al. Arch Virol. 10.1007/s00705-018-04136-2 (2019). [DOI] [Google Scholar]
- 2.Louca S, Mazel F, Doebeloi M & Parfrey LW PLoS Biol. 17, e3000106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brum JR et al. Science 348, 1261498 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Mihara T. et al. Microbes Environ. 33, 162–171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simmonds P et al. Nature Rev. Microbiol 15, 161–168 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Wolf YI et al. MBio 9, e02329–18 (2018).30482837 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.