

Abstract
Programmed cell death-1 (PD-1) is a potent immune checkpoint receptor on T lymphocytes. Upon engagement by its ligands, PD-L1 or PD-L2, PD-1 inhibits T cell activation and can promote immune tolerance. Antagonism of PD-1 signaling has proven effective in cancer immunotherapy, and conversely, agonists of the receptor may have a role in treating autoimmune disease. Some immune receptors function as dimers, but PD-1 has been considered monomeric. Here, we show that PD-1 and its ligands form dimers as a consequence of transmembrane domain interactions and that propensity for dimerization correlates with the ability of PD-1 to inhibit immune responses, antitumor immunity, cytotoxic T cell function, and autoimmune tissue destruction. These observations contribute to our understanding of the PD-1 axis and how it can potentially be manipulated for improved treatment of cancer and autoimmune diseases.
INTRODUCTION
T cell activation is tightly regulated to allow for an adaptive immune response (1). If overly weighted toward tolerance, host defense against pathogens and cancer immune surveillance are compromised (2). Conversely, excessive T cell activation underlies many autoimmune disorders (3). T cell costimulatory receptors, such as CD28 and inducible T cell co-stimulator (ICOS), as well as co-inhibitory/immune checkpoint receptors, such as cytotoxic T lymphocyte associated protein 4 (CTLA-4) and programmed cell death-1 (PD-1), modulate T cell receptor (TCR) activation to provide this balance (4). The ligands for these receptors are members of the B7 family of transmembrane proteins and include CD80, CD86, ICOSL, programmed death-ligand 1 (PD-L1), and PD-L2 (5). Both PD-L1 and PD-L2 ligands bind to PD-1 (6, 7), but whereas PD-L1 is ubiquitously expressed, PD-L2 is restricted to antigen-presenting cells (APCs) (8). Recent breakthroughs in cancer immunotherapy have centered around monoclonal antibodies (mAbs) that block the interaction of PD-1 or CTLA-4 with their cognate ligands (9). Conversely, a CTLA-4 receptor mimic is in use to treat some autoimmune disorders (10). Clinically useful PD-1 agonist mAbs are emerging and hold great promise in the treatment of T cell–mediated autoimmune diseases (11).
Members of the CD28 family of coreceptors interact primarily with B7 family ligands in trans (from the surface of one cell to another). However, members of these protein families also have consequential relationships in cis (in the plane of the membrane of a single cell). CD28, ICOS, and CTLA-4 are constitutive homodimers as a consequence of a disulfide linkage in their membrane proximal extracellular stalk regions (12–14). PD-1 lacks this cysteine, and crystal structures of its extracellular domain (ECD) have suggested that it is monomeric (15). Analytical ultracentrifugation and crystallographic analysis of the CD80 and ICOSL ECDs indicate that each of these B7 ligands dynamically homodimerizes on the cell surface through their ECDs (16, 17). In contrast, the PD ligands are thought to be distributed in monomeric form within the plasma membrane (6, 18, 19). Nevertheless, PD-L1 on T cells has been shown to undergo regulatory cis interactions with PD-1 and CD80. The PD-1/PD-L1 cis interaction prevents PD-1 from binding PD-L1 in trans (20). In addition, a PD-L1/CD80 cis interaction is reported to prevent PD-L1 from binding PD-1, which attenuates the CD80 trans interaction with CTLA-4 but not CD28 (21, 22). Homotypic cis interactions in the PD-1 and its ligands form dimers as a consequence of transmembrane domain (TMD) interactions and that propensity for dimerization correlates with the PD-1 ability to inhibit immune responses, antitumor immunity, cytotoxic T cell function, and autoimmune tissue destruction.
RESULTS
PD-1 and its ligands dimerized in cis
To determine whether PD-1 and its ligands dimerize in cis, we used flow cytometric analysis of fluorescence resonance energy transfer (flow-FRET) (fig. S1A) (23). To validate this approach, we also examined a nominal monomer, CD86 (16, 24, 25); a dynamic dimer, CD80 (26, 27); a constitutive dimer, CD8α (28); and non-interacting CD80 and CD86. No FRET was observed between CD80 and CD86, confirming lack of interaction, but when B7 proteins were analyzed for homodimerization, FRET increased with their reported propensities for homodimerization: CD86 < CD80 < CD8α (Fig. 1 and fig. S1, B and C). Notably, PD ligands showed both homo- and heterodimerization in the range of CD80, and PD-1 showed robust homodimerization approaching that of the constitutive CD8α control dimer (Fig. 1, B, D, E, and F).
Fig. 1. PD-1 and its ligands dimerize in cis.
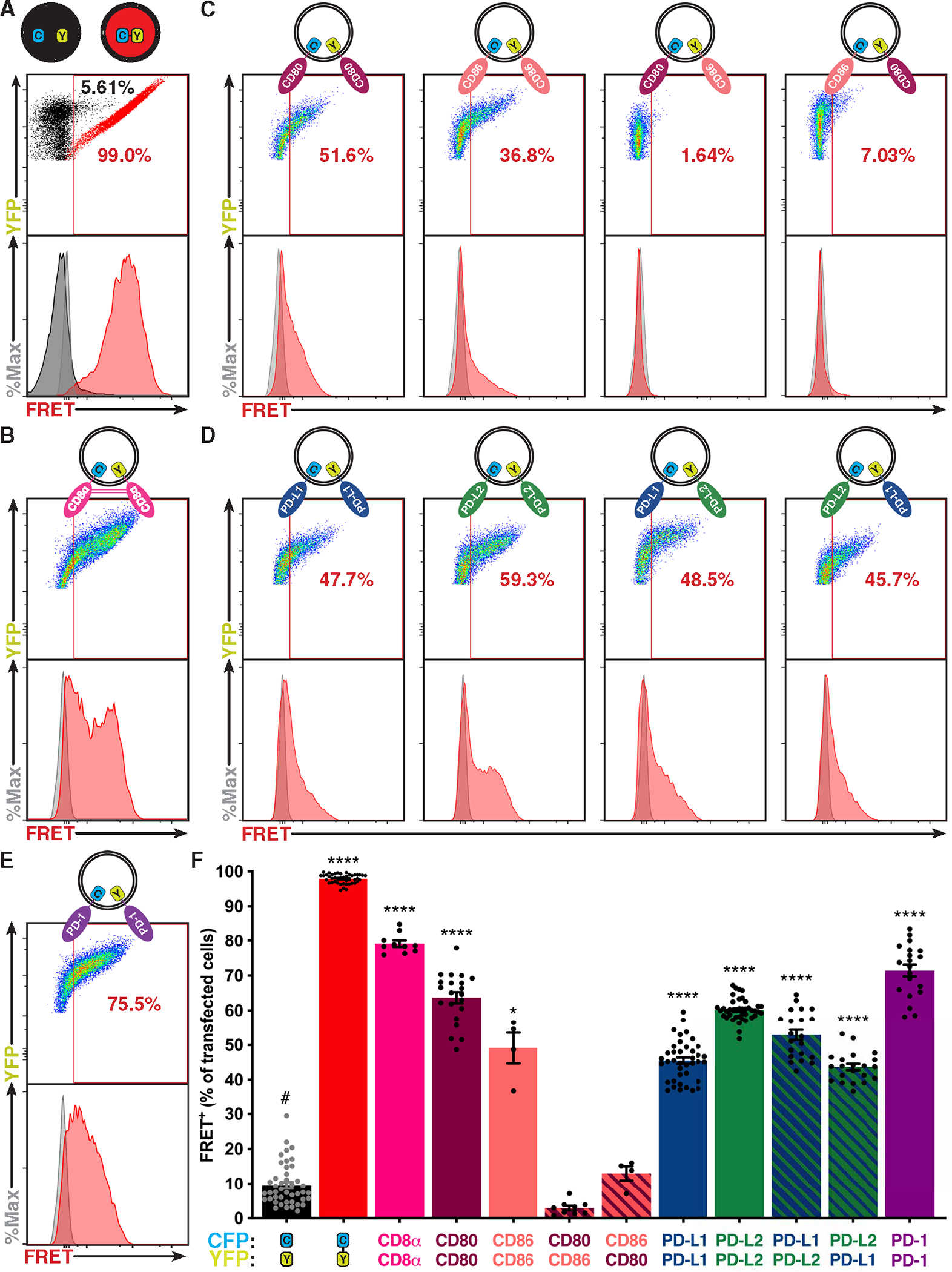
(A to E) Flow-FRET analysis of immune receptors expressed in CHO cells as CFP or YFP fusion proteins as depicted above each panel. FRET was determined with gates described in fig. S1A and shown as a representative scatterplot (YFP versus FRET) and histogram (events versus FRET). (A) Controls for minimum (CFP and YFP coexpressed, black) and maximum (CFP fused to YFP, red) flow-FRET. (B) CD8α flow-FRET pair. (C) CD80 and CD86 homo– and hetero–flow-FRET pairs for validation. (D) PD-L1 and PD-L2 homo– and hetero–flow-FRET pairs. (E) PD-1 flow-FRET. (F) Cumulative flow-FRET data (n ≥ 3) displayed as the percent of FRET+ transfected cells ± SEM. Significance shown as paired t tests versus negative control (#): *P < 0.05 and ****P < 0.0001.
To validate PD-1 homodimerization, we turned to two sensitive measures of receptor proximity, fluorescence lifetime imaging microscopy (FLIM) and fluorescence cross-correlation spectroscopy (FCCS). FRET efficiency was calculated from the fluorescence lifetime of the donor, a method that is independent of relative fluorophore concentration (29). FLIM FRET measurements confirmed PD-1 dimerization (Fig. 2A). FCCS, a form of fluorescence fluctuation spectroscopy independent of FRET (30), further validated this finding (Fig. 2B). These orthogonal approaches strongly suggest that the PD-1 receptor dimerizes in cis, despite lacking a constitutive disulfide link.
Fig. 2. Fluorescence lifetime imaging and cross-correlation spectroscopy demonstrates PD-1 dimerization.
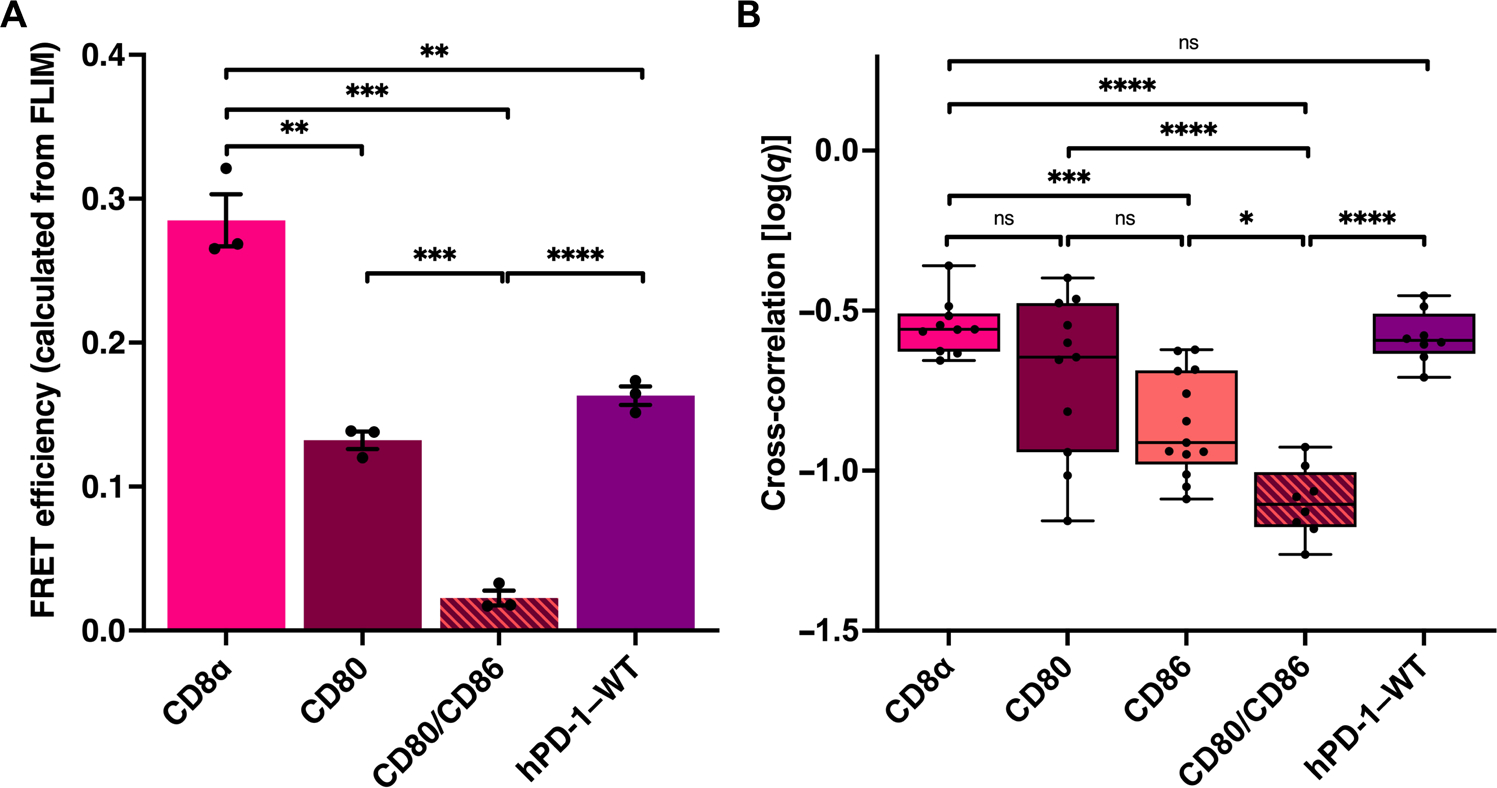
(A) FRET efficiency calculated from FLIM of the indicated mEGFP, mCherry2 fusion protein pairs expressed in baby hamster kidney cells (see fig. S6). (B) FCCS measurements of the indicated mEGFP, mCherry2 fusion protein pairs expressed in HEK293T cells (see fig. S7). Significance tested by unpaired t tests: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns, not significant.
To measure dimerization of endogenous receptors and ligands, we purified monovalent antigen-binding fragments (Fabs) against CD4, PD-L1, and PD-1 and conjugated them to Alexa Fluor (AF) 555 and/or AF 647, an optimal FRET pair (fig. S2). CD4 was chosen as a negative control because of its monomeric surface distribution (31). A dual-labeled Fab served as a positive control (fig. S2). Using these reagents in flow-FRET analysis, we observed homodimerization of endogenous PD-L1 on a Hodgkin lymphoma cell line, HDLM-2 (fig. S2, B and C) (32). We also observed homodimerization of endogenous PD-1 in stimulated Jurkat T cells (fig. S2, D and E).
TMDs drive dimerization of PD-1 and its ligands
Dimerization of transmembrane proteins can be mediated by interactions of the ECD, TMD, or both (33–35). The structure of the dimer interface between homo-interacting CD80 ECDs is known at atomic resolution (16). Flow-FRET analysis confirmed that the ECD, but not the TMD, mediates homodimerization of CD80 (Fig. 3A). By contrast, the TMDs of both PD-1 and its ligands were sufficient to drive dimerization (Fig. 3B).
Fig. 3. TMDs drive dimerization of PD-1 and its ligands.
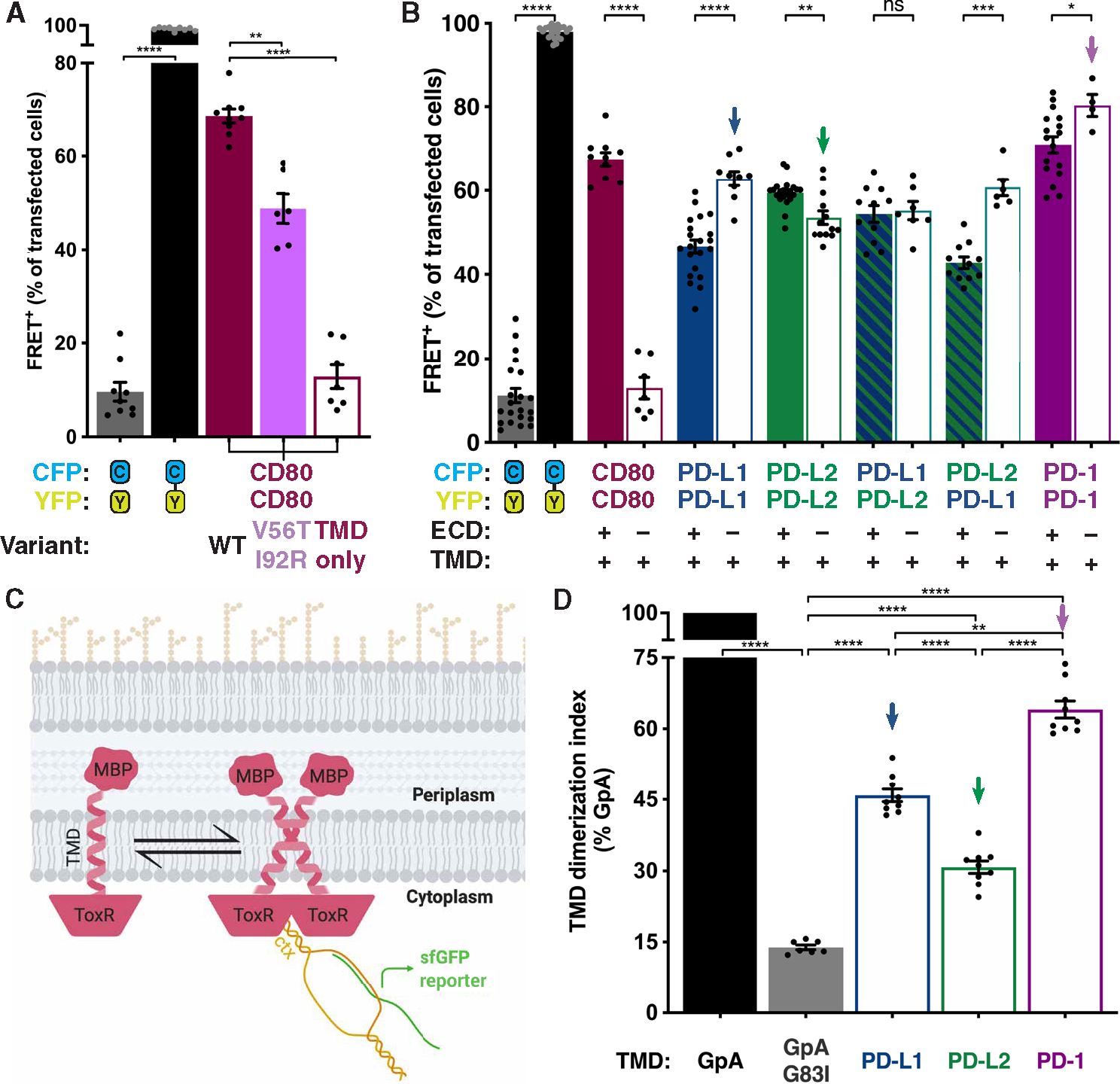
(A and B) Flow-FRET analysis of immune receptors as in Fig. 1. (A) Flow-FRET analysis of CD80: WT, V56T + I92R ECD interface mutations and TMD only to validate assay for ECD-mediated dimerization. (B) Flow-FRET analysis of CD80, PD-L1, PD-L2, and PD-1 TMDs ± ECDs. (C) Schematic of bacterial TOXGREEN TMD dimerization assay (created with BioRender). (D) TOXGREEN analysis of PD-L1, PD-L2, and PD-1 TMD dimerization propensity relative to that of GpA ± G83I mutation that abolished dimerization. Downward arrows in (B) and (D) indicate corresponding conditions. Significance shown as paired t tests: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The isolated TMD of PD-L1 gave a significantly stronger flow-FRET homodimerization signal than did the ECD + TMD protein, suggesting that the PD-L1 ECD acts to weaken homodimerization (Fig. 3B). The same phenomenon was observed for PD-1, albeit with less magnitude, but not PD-L2. We mapped the PD-L1 residues responsible for weakening the cis interaction to a five–amino acid stretch (PD-L1 residues 170 to 174) in its membrane proximal immunoglobulin constant domain (fig. S3). This region was lost in PD-L2 between bird and mammal evolutionary divergence (fig. S3, A and B), suggesting that modulating the efficiency of dimerization may have been part of the functional refinement of PD-L2 in mammals (7, 36).
TMD-mediated homodimerization is a paradigm that is well documented in receptor tyrosine kinases (RTKs). The TMDs of the 58 members of the human RTK family mediate a wide range of degrees of homodimerization, a property that has been quantified with the bacterial TOXR transcription factor TMD dimerization (TOXGREEN) assay (Fig. 3C) (33, 37). In the TOXGREEN assay, a TMD of interest is expressed between a periplasmic maltose binding protein and a cytoplasmic TOXR dimerization–dependent transcription factor. The TOXR transcription factor drives expression of super-fold green fluorescent protein (sfGFP) only when TOXR is in the dimeric state such that sfGFP fluorescence intensity is a measure of dimerization propensity. TOXGREEN analysis confirmed TMD-mediated dimerization of the PD-1 axis with an order of efficiency of PD-1–TMD > PD-L1–TMD > PD-L2–TMD (Fig. 3D), concordant with the flow-FRET analysis (Fig. 3B).
Human and murine PD-1 TMDs dimerize using N-terminal contacts
Gly-XXX-Gly or small-XXX-small is a recurrent amino acid motif that has been shown to drive a subset of TMD helix interactions (38–40). Although the human PD-1 (hPD-1) TMD has two tandem Gly-XXX-Gly motifs and one small-XXX-small motif that is conserved, mutation of these sequences did not disrupt dimerization (fig. S4). We therefore used an unbiased approach to elucidate the molecular basis for the PD-1 TMD homotypic interaction. We performed a tryptophan (Trp) scan (41, 42) through the 23 amino acids of the hPD-1 TMD using the TOXGREEN assay as a readout of dimerization (Fig. 4A). Trp has a bulky side chain that can disrupt helical interactions that require close proximity or enhance interactions of more widely spaced helices by establishing π-stacking interactions. We observed that introduction of Trp in the PD-1 TMD near the helical N terminus (V173W) reduced dimerization, whereas Trp substitutions near the C terminus (L184W) strengthened dimerization (Fig. 4, A and B, and fig. S5, A and B). These data confirm that the TMD helix mediates the homotypic interaction and suggest that, as has been established for glycophorin A (GpA) dimerization (43), the PD-1 TMD helices adopt an “A” shaped conformation in the plane of the membrane (fig. S5B).
Fig. 4. hPD-1 and mPD-1 TMDs dimerize using N-terminal contacts.
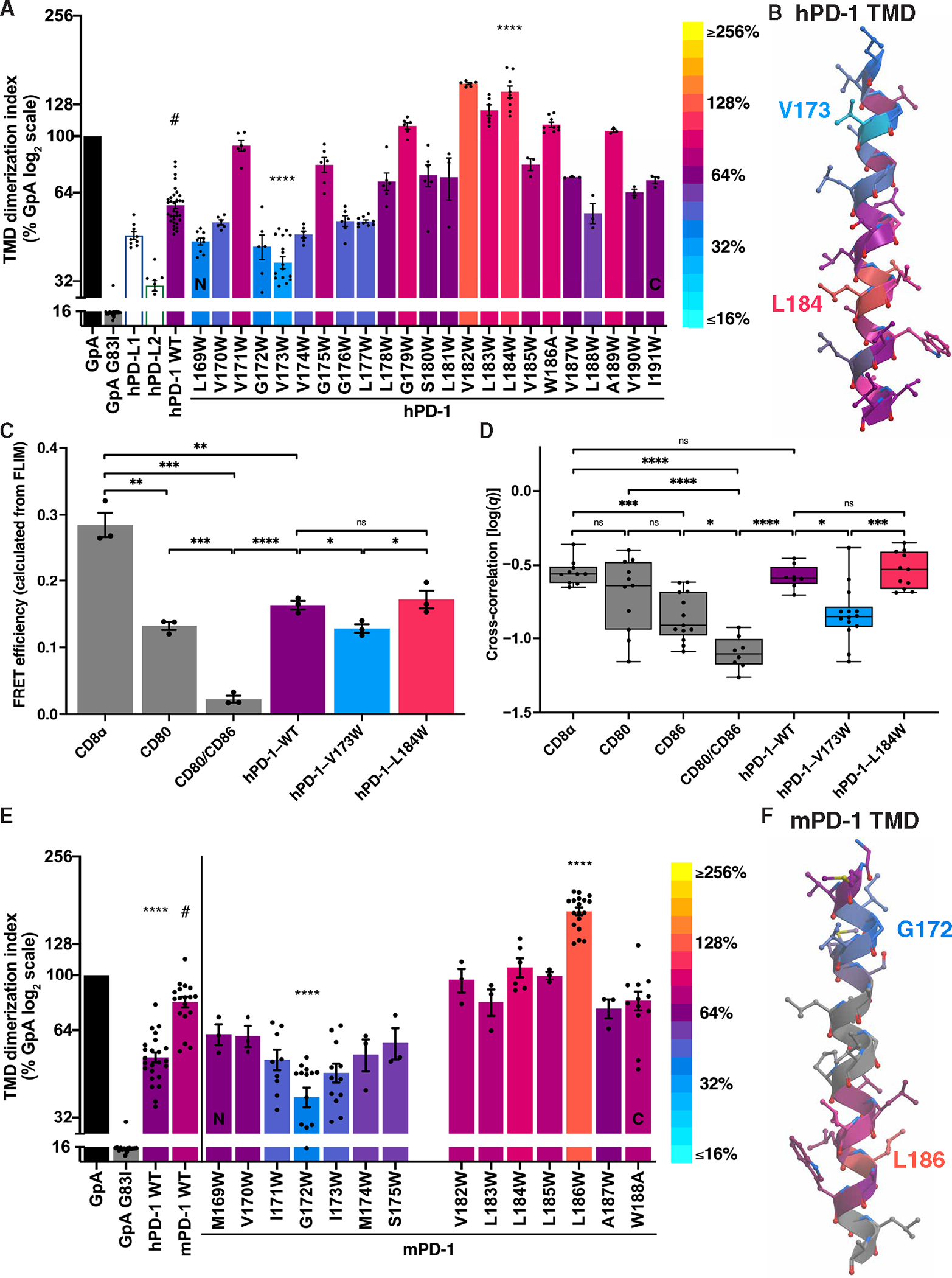
(A) The effects on dimerization as determined by TOXGREEN of Trp substitutions at the indicated positions in the hPD-1 TMD. Bars were assigned a color on the basis of the log2 scaled look-up table (right). (B) Helical model of the hPD-1 TMD color-coded as in (A) with the V173hPD-1 low and L184hPD-1 high Trp substitutions labeled. (C) FRET efficiency calculated from FLIM of the indicated pairs of mEGFP- and mCherry2-tagged constructs expressed in baby hamster kidney cells. (D) FCCS measurements of mEGFP- and mCherry2-tagged constructs expressed in HEK293T cells pooled across four biological replicates. (E and F) Analysis and color-coding of the mPD-1 TMD as in (A) and (B) revealing G172mPD-1 low and L186mPD-1 high Trp substitutions. Significance in (A), (C), and (E) was determined by unpaired t tests; significance in (D) was determined by ANOVA test with Tukey’s correction for multiple comparisons: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To validate the low and high dimerization mutants in a mammalian cell expression system, we turned to FLIM-FRET and FCCS. FLIM-FRET revealed that the hPD-1–V173W variant dimerizes less than its wild-type (WT) and L184W counterparts (Fig. 4C and fig. S6, A and B). FCCS corroborated these findings with a method that does not rely on FRET (Fig. 4D and fig. S7, A and B). Thus, TMD dimerization perturbations detected with the bacterial TOXGREEN assay correlate with dimerization tendency for receptors expressed in mammalian cells.
If TMD-driven dimerization plays a functional role in PD-1 signaling, then it is likely conserved among species. However, the murine PD-1 (mPD-1) TMD sequence differs from human (figs. S4A and S5E). This divergence is similar to that seen among RTKs and reflects the sequence promiscuity of TMDs that can support dimerization (33). The mPD-1 TMD supported dimerization more efficiently than the hPD-1 TMD (Fig. 4E). The TMD of mPD-1 predicted by hydropathy analysis is 25 rather than 23 amino acids in length because of two additional leucine (2Leu) residues, and it contains a proline, likely providing a kink in the murine helix (figs. S4A and S5E). We performed a Trp scan of the mPD-1–TMD (Fig. 4, E and F, and fig. S5, C to E). Our results suggest that, like the hPD-1 TMD, the mPD-1 TMD forms an analogous A-shaped dimeric interface, but the interacting helical face is shifted ~130° from that of the hPD-1 TMD (fig. S5, A to D). Both TMD helices contain a native Trp residue near the C terminus; however, because of the 2Leu deletion in hPD-1 that evolved with primates (fig. S4A), this Trp likely resides on an opposite helical face (Fig. 4, B and F, and fig. S5, A and C). Consistent with this prediction, substitutions of Ala for these Trps affected only the human interaction (fig. S5F). Addition of 2Leu into the hPD-1 TMD or removal of 2Leu from mPD-1 TMD had opposite effects on dimerization (fig. S5G), consistent with the native Trp disposed on opposite sides of the helix. These data reveal that the PD-1 TMDs of both human and mouse support dimerization with topologically similar regions of interaction. They also revealed substitutions that could be exploited to produce low (hPD-1–V173W and mPD-1–G172W) or high (hPD-1–L184W and mPD-1–L186W) dimerizing mutants of PD-1 that could be applied to functional analyses (fig. S5H).
PD-1 dimerization facilitates T cell inhibition
PD-1 signals at the immunological synapse (IS) in part by recruiting the Src homology 2 (SH2) domain containing protein tyrosine phosphatase (PTP) SHP2 (44). To determine the role of PD-1 dimerization in this process, we used Jurkat T cells and a supported, planar lipid bilayer that incorporated intercellular adhesion molecule–1 (ICAM-1), anti-CD3 Fab, and PD-L1 to mimic an APC. We generated a PD-1 null Jurkat T cell clone (2E4) using CRISPR-Cas9 and rescued PD-1 expression by lentiviral transduction with GFP-tagged hPD-1 that incorporated the WT TMD, the low dimerization V173W mutant, or the high dimerization L184W mutant (fig. S8, A to F). The three hPD-1–GFP variants displayed the same plasma membrane/Golgi apparatus pattern of distribution typical of a type I transmembrane protein as well as equivalent surface expression (fig. S8, D to H). The hPD-1–V173W low dimerization mutant recruited less, and the hPD-1–L184W high dimerization mutant recruited more, PD-L1 and SHP2 to the synapse compared with WT, and low dimerization of PD-1 was associated with enhanced tyrosine phosphorylation (pTyr) (Fig. 5, A to C, and fig. S8I). Further, the Jurkat T cells rescued with hPD-1–V173W (low dimerization) displayed impaired inhibition as determined by increased CD69 positivity of cells stimulated with plate-bound anti-CD3 in the presence of immobilized PD-L1 (Fig. 5D). These data support a model whereby PD-1 TMD–driven dimerization enhances T cell inhibition by limiting signaling in the IS.
Fig. 5. hPD-1 TMD dimerization affects T cell inhibition.
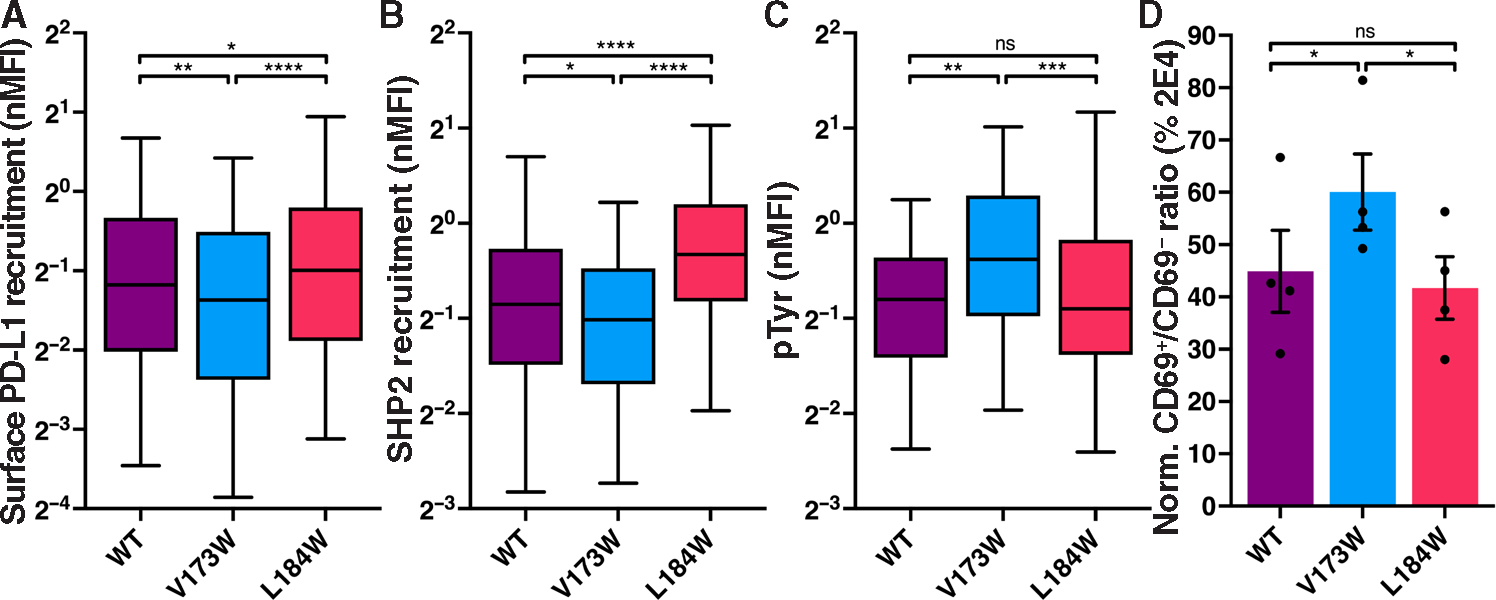
(A to C) The cells shown in fig. S8 (D to F) were applied to a supported lipid bilayer incorporating ICAM-1, anti-CD3, and PD-L1, and the IS was analyzed by TIRF microscopy. Normalized recruitment of PD-L1, SHP2, or pTyr to the IS is depicted as box and whisker plots. Mean (solid line) and 5 to 5% range (whiskers) are indicated (n ≥ 119). Significance was determined by Mann-Whitney test: *P < 0.05, ***P < 0.001, and ****P < 0.0001. (D) Jurkat cell lines were activated with immobilized anti-CD3 and soluble anti-CD28 in the presence of immobilized PD-L1 and subsequently stained for CD69 expression. Data are displayed as a ratio of CD69+:CD69− cells normalized to the CD69 stain ratio of 2E4 PD-1–KO cells. nMNFI, normalized mean fluorescence intensity.
To determine the functional consequences of PD-1 dimerization ex vivo and in vivo, we bred PD-1 null (PD-1−/−) mice with OT1 transgenic TCR mice to generate mice reactive to ovalbumin peptide (pOVA) and deficient in PD-1. CD8+ T cells from these OT1, PD-1−/− mice were isolated and retrovirally transduced with PD-1 variants to rescue PD-1 function (Fig. 6, A to C). IS formation was studied with a supported lipid bilayer containing H-2Kb-pOVA, ICAM-1, and PD-L1. Rescue with WT PD-1 markedly decreased the release of granzyme B into the IS, an effect that was diminished by the low dimerization mutant, mPD-1–G172W, and preserved by the high dimerization mutant, mPD-1–L186W (fig. S9). Endogenous PD-1 surface expression on activated OT1 T cells was similar to surface expression of the transduced gene in PD-1−/− OT1 CD8 cells rescued with the PD-1 variants (fig. S9C). Thus, ex vivo analysis of these PD-1 variant rescued OT1 cells demonstrates that PD-1 suppresses a critical component of CD8 T cell cytotoxicity and that this effector function is sensitive to perturbations of the PD-1 oligomeric state.
Fig. 6. PD-1 dimerization limits cytotoxic T cell antitumor activity in vivo.
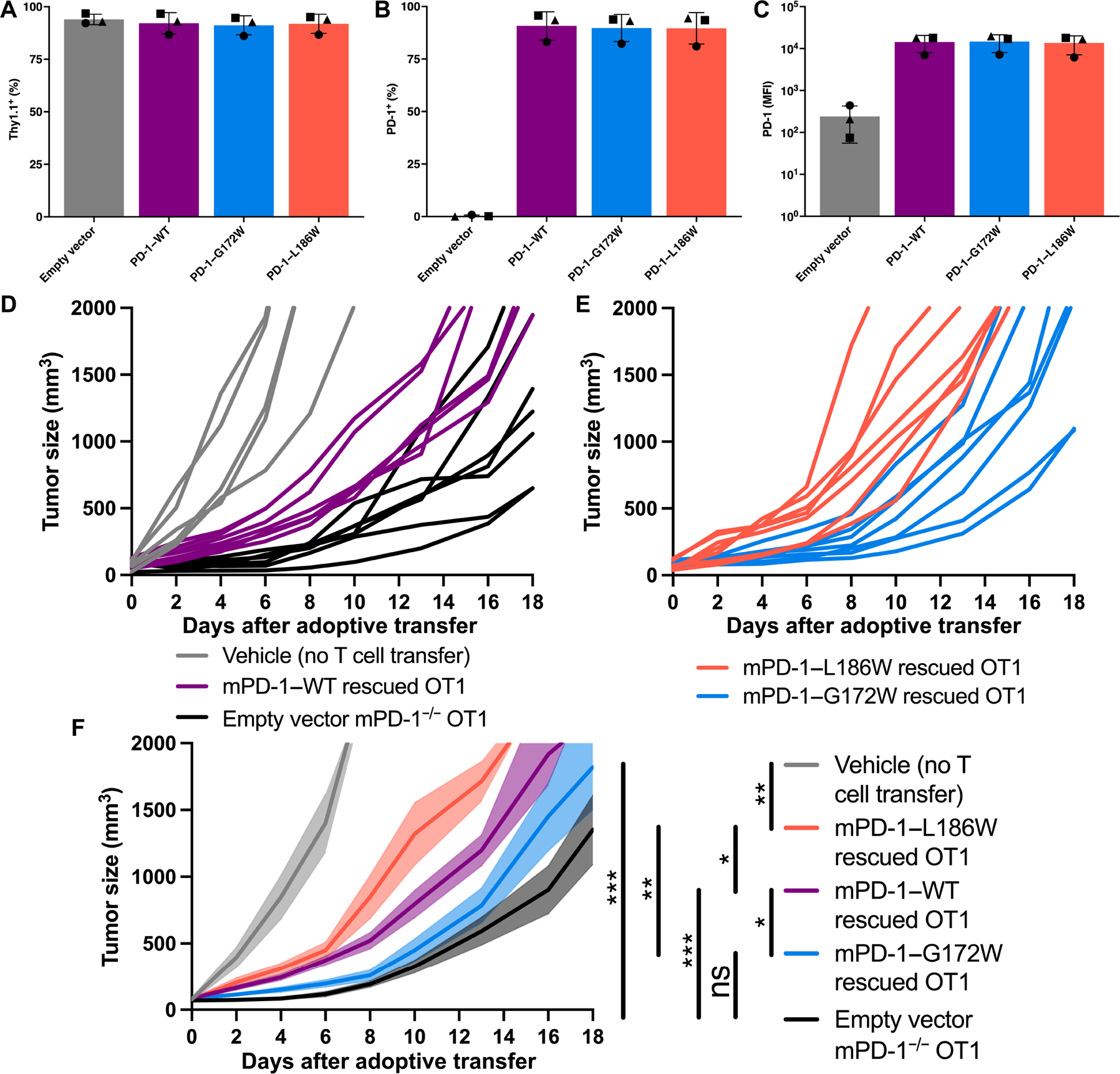
(A to C) Thy1.1 and PD-1 expression on reconstituted PD-1−/− OT1 CD8+ T cells after RV transduction with the indicated vector. (D to F) B16-OVA tumor growth in WT C57BL/6 mice after adoptive transfer with OT1 T cells retrovirally transduced as in (A) to (C). (D) Spider chart of tumor growth in individual mice with no T cell transfer (gray), PD-1–WT reconstituted OT1 cells (purple), or PD-1−/− EV transduced OT1 cells (black). Spider chart of tumor growth with PD-1–G172W reconstituted OT1 cells (blue) or PD-1–L186W reconstituted OT1 cells (orange). (F) Cumulative B16-OVA tumor growth as in (D) and (E) with SEM indicated by transparent shading. Representative experiment with five mice in the vehicle group and seven mice in the EV, mPD-1–WT, mPD-1–G172W, and mPD-1–L186W groups. Significance was tested by two-way ANOVA: *P < 0.05, **P < 0.01, and ***P < 0.001.
To explore the functional impact of PD-1 TMD dimerization in vivo, we used a tumor model known to be responsive to anti–PD-1 therapy (45). We adoptively transferred retrovirally PD-1–rescued OT1 CD8+ cells into WT recipient mice bearing OVA-expressing melanoma tumors (B16-OVA) (Fig. 6, D–F). Transfer of PD-1−/− OT1 cells transduced with an empty vector (EV) markedly delayed tumor progression compared with animals without transferred T cells (Fig. 6, D and F), consistent with antitumor action of CD8+ effector cells lacking a critical inhibitory checkpoint. Transfer of OT1 cells rescued with mPD-1–WT was less effective at delaying tumor growth than the PD-1−/− cells demonstrating the inhibitory effect of PD-1 on cytotoxic T cells (Fig. 6, D and F). Transfer of OT1 cells rescued with the low dimerization variant, mPD-1–G172W, delayed tumor progression to a greater degree than the PD-1–WT OT1 cells (Fig. 6, E and F), consistent with diminished inhibitory function. Conversely, transfer of OT1 cells rescued with the enhanced dimerization variant, mPD-1–L186W, was less effective at delaying tumor growth (Fig. 6, E and F). Thus, the capacity of PD-1 to limit antitumor cytotoxic T cells in this model correlates directly with its capacity to homodimerize.
To explore the impact of PD-1 TMD dimerization in autoimmunity, we turned to a type 1 diabetes (T1D) mouse model. PD-1 is critical in maintaining immune tolerance of pancreatic islets (46, 47). One demonstration of the role of PD-1 in human autoimmunity can be found in a recent case report where two Turkish boys who are the products of a consanguineous marriage acquired germline PD-1–null mutations and developed T1D before 7 months of age, before both boys succumbed to other manifestations of autoimmune disease (48). To model this disease process, we used mice transgenic for a gene encoding a fusion of OVA with a transmembrane segment under the control of the rat insulin promotor (RIP-mOVA) (49). Upon adoptive transfer of PD-1−/− OT1 cells, all RIP-mOVA mice developed diabetes, whereas littermates that did not receive an OT1 cell adoptive transfer remained T1D-free (Fig. 7). Whereas some PD-1–WT and PD-1–L186W recipient RIP-mOVA mice develop delayed onset T1D, all mice that received PD-1–G172W rescued OT1 cells developed rapid onset T1D equivalent to PD-1–null OT1 recipient mice (Fig. 7). Thus, as was observed in the B16-OVA tumor model, cytotoxic T cell–mediated autoimmune T1D was limited by PD-1, and the efficiency of this activity correlated directly with PD-1 homodimerization potential.
Fig. 7. PD-1 dimerization regulates islet cell–specific cytotoxic T cells that induce T1D in vivo.
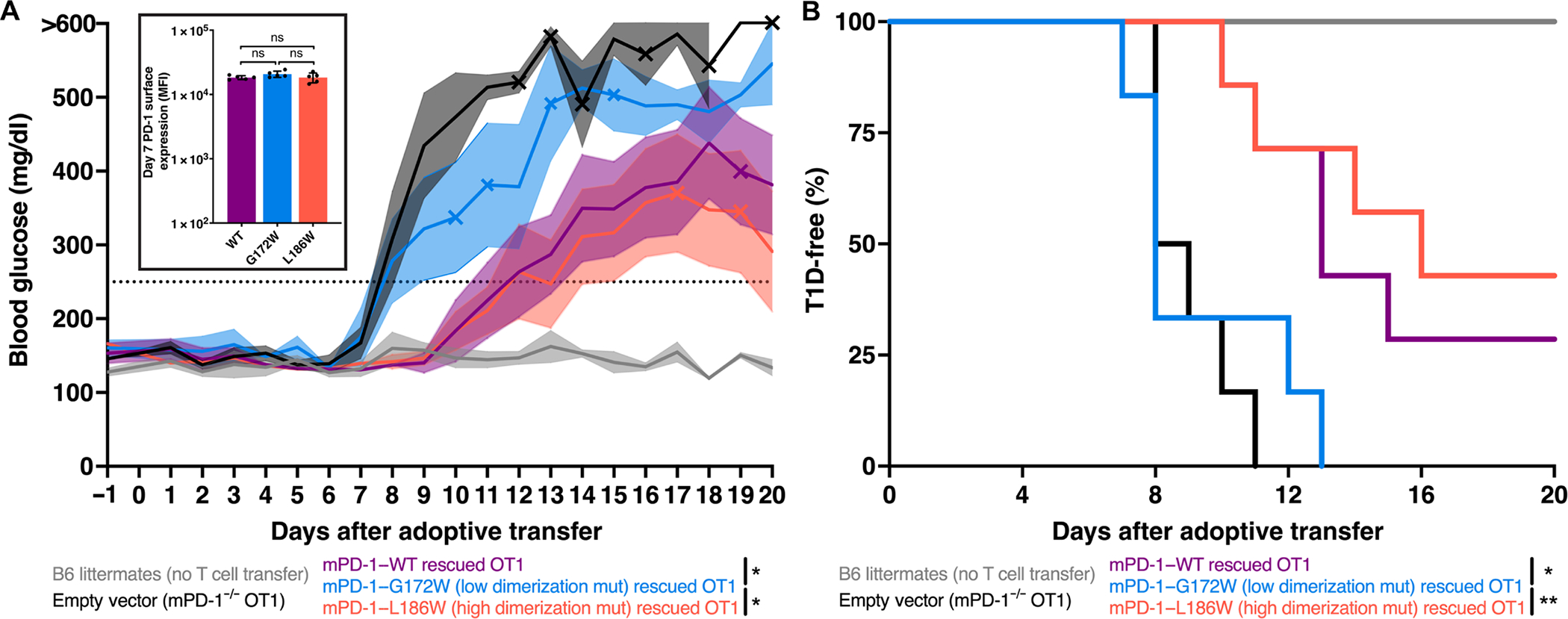
(A) Blood glucose measurements after adoptive transfer of indicated OT1 cells into recipient RIP-mOVA mice with SEM indicated by transparent shading. Mice were considered diabetic after two consecutive readings above 250 mg/dl (dotted line). X symbol indicates deaths. Significance was tested by two-way ANOVA on data before day 11: *P < 0.05. Insert: PD-1 surface expression on day 7 after adoptive transfer. (B) Kaplan-Meyer plot of diabetes-free mice from (A). Representative experiment with four mice in the littermates group, six mice in the EV and mPD-1–G172W groups, and seven mice in the mPD-1–WT and mPD-1–L186W groups. Significance was tested by Gehan-Breslow-Wilcoxon test: *P < 0.05 and **P < 0.01.
DISCUSSION
Our results demonstrate that, like other T cell coreceptors and ligands, PD-1 and its ligands dimerize in cis (fig. S10). Unlike B7 family paralogs that dimerize via their ECDs, PD-1 axis dimerization is driven by TMD interactions (fig. S10) in a fashion analogous to RTKs. In the case of the PD ligands, we observed both homo- and heterodimerization. The relative propensities of the PD ligands to homo- or heterodimerize must be considered in the context of their relative expression on APCs. Because PD-L1 is typically present in 7- to 15-fold excess relative to PD-L2 on APCs (6), PD-L1/PD-L2 heterodimers will be more abundant and are more likely to play a physiologic role than PD-L2 homodimers. However, despite lower expression levels, PD-L2 homodimerization may play a role when high CD80 expression sequesters PD-L1 via the PD-L1/CD80 cis interaction (21, 22), allowing PD-L2 to become a more predominant PD-1 ligand within the IS. Furthermore, the loss of a domain that modulates PD-L2 homodimerization upon the evolutionary divergence of mammals is consistent with a critical role of dimerization in PD ligand function.
In our study, manipulation of PD-1 TMD dimerization revealed a structure-function relationship whereby PD-1 dimerization potentiates its function. In support of this model, a secreted, disulfide-linked dimeric PD-L1 splice variant was recently shown to engage PD-1 and inhibit T cells (50). In addition, it was recently reported that a single SHP2 molecule can bridge two PD-1 cytoplasmic tails with its two SH2 domains (51), a conformation that would be facilitated by upstream, intrinsic dimerization of the receptor. Last, membrane proximal anti–PD-1 mAbs impart PD-1 agonism (52), a finding for which this work provides mechanistic support. Peresolimab, a PD-1 agonist mAb, was recently reported in a phase 2 clinical trial to be effective in treating rheumatoid arthritis (11). However, the mechanism whereby peresolimab acts as an agonist was not reported. Our characterization of PD-1 TMD dimerization may help inform evolving strategies for developing both agonists and antagonists.
In contrast to other coreceptors like CD28, ICOS, and CTLA-4 that are constitutive dimers established by disulfide bonds, PD-1 dimerization is driven by noncovalent association of its TMD. The more dynamic dimerization of the PD-1 axis described here affords an additional mode of regulation. Chimeric antigen receptor (CAR) T cells have been engineered to recombine extracellular and cytoplasmic domains so as to weaponize antitumor T cells (53). TMD manipulation in CAR T cells may afford a useful way to fine-tune thresholds for positive or negative regulators. Our data suggest that manipulation of the oligomeric state of the PD-1 axis may offer an additional modality in the effort to enhance cancer immunotherapy or diminish autoimmunity.
Whereas our model, informed by mutational analysis, suggests that PD-1 dimerizes with a TMD A shape interaction akin to GpA, our data cannot rule out higher-order oligomeric interactions. Further investigation is needed to determine whether PD-1 clustering results in lattice formation or allows for two-dimensional phase separation. Moreover, because PD-1 function is contingent on proximity to TCR microclusters (44), a more complete understanding of the broader cis-interaction network within the microcluster will help contextualize the role of PD-1 oligomerization.
MATERIALS AND METHODS
Study design
The study sought to determine whether there exists homotypic dimerization within the PD-1 axis, which domain of PD-1 drives receptor dimerization, and what functional impact PD-1 dimerization has on T cell inhibition. Complementary biochemical techniques were used to examine PD-1 dimerization including Flow-FRET, FLIM-FRET, FCCS, and TOXGREEN. Fluorophore-conjugated Fabs were used to validate dimerization on endogenous receptors. PD-1−/− Jurkat T cells were rescued with high and low dimerization PD-1 mutants and studied on a supported lipid bilayer to study dimerization effects on proximal PD-1 signaling. B6 mice with B16 melanoma tumors were adoptively transferred with PD-1−/− OT1 cells rescued with high and low dimerization PD-1 mutants to measure the functional impact of PD-1 dimerization on antitumor immunity and cytotoxic T cell function. Last, these PD-1−/− OT1 cells rescued with high and low dimerization PD-1 mutants were adoptively transferred into RIP-mOVA T1D model mice to measure the impact of PD-1 dimerization on autoimmune tissue destruction.
Cell lines
The parental Jurkat T cell line was acquired from American Type Culture Collection. The Hodgkin lymphoma cell line, HDLM-2, was provided by C. Diefenbach [New York University (NYU)]. These cells were grown in RPMI medium complete with 10% fetal bovine serum (FBS). The B16-OVA melanoma line was gifted by S. Feske and grown in Dulbecco’s modified Eagle’s medium (DMEM) + 10% FBS.
Mice
A PD-1−/− female was purchased from the Jackson Laboratory (028276, B6.Cg-Pdcd1tm1.1Shr/J). OT1+, UBC-GFP+ male mice were provided by S. Schwab (NYU). These mice were bred, and subsequent pups were back-crossed to the PD-1−/− female to generate PD-1−/−, OT1+, GFP+ mice. OT1 cells from pups lacking the GFP transgene were used in total internal reflection fluorescence microscopy (TIRFM) experiments. A hemizygous RIP-mOVA male was purchased from the Jackson Laboratory [005431, C57BL/6-Tg(Ins2-TFRC/OVA)296Wehi/WehiJ] and crossed to six WT black females. Female pups confirmed to be hemizygous for the RIP-mOVA transgene were used in T1D experiments.
Flow cytometric analysis of FRET
CD80, PD-L1, PD-L2, and PD-1 ECDs from protein expression plasmids were extended with primers at their 3′ ends to add respective TMDs by two sequential primer-overhang extension polymerase chain reactions (PCRs). ECDs and TMDs were then cloned into cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) N1 vectors using Xho I and Hind III restriction sites. CD86 was similarly cloned after PCR amplification from a cDNA plasmid template (Addgene plasmid #98284). Full-length CD8α cDNA was cloned into CFP and YFP N1 plasmids into the Eco RI and Bam HI restriction sites. ECD removal of CD80, PD-L1, PD-L2, and PD-1 plasmids was performed by overlapping sequential PCR, followed by cloning into CFP and YFP N1 vectors as described above. In each case, the signal peptide was maintained, and six stalk amino acids attached to the N terminus of the TMD were maintained.
Suspension ExpiCHO-S cells (Thermo Fisher Scientific) were transfected with CFP and YFP expression plasmids using the Lipofectamine 3000 (Thermo Fisher Scientific) reagent in a 24-well plate using the ExpiCHO serum-free expression medium (Thermo Fisher Scientific). After a 40-hour incubation at 37°C in 5% CO2, cells were analyzed in a BD LSRII flow cytometer. Blue (488 nm) and violet (407 nm) lasers were used for excitation. YFP fluorescence was measured using a 530/30 detector off of the blue laser. CFP fluorescence was measured using a 450/50 detector off of the violet laser. FRET was measured using the 530/30 detector off of the violet laser. The gating strategy is described in fig. S1. Data were analyzed using FlowJo 10 software.
Fluorescence lifetime imaging microscopy–FRET
CD8a, CD80, CD86, PD-1, PD-1–V173W, and PD-1–L184W ECDs were cloned into the monomeric enhanced green fluorescent protein (mEGFP)–N1 and mCherry2-N1 plasmids (Addgene plasmids #54767 and #54517) using the Xho I and Hind III restriction sites. Approximately 1 × 106 baby hamster kidney cells were seeded on glass coverslips. On the following day, the cells were transiently transfected with 0.8 μg of mEGFP-tagged plasmid and 0.8 μg of EV pC1 or mCherry2-tagged plasmid. On the third day, cells were washed with phosphate-buffered saline (PBS), fixed using 4% deionized water, and mounted on glass slides. The mounting glass coverslips were baked at 37°C for 1 hour before imaging. The cells were imaged using a Nikon TiE wide-field fluorescence microscope with a 60× Plan-Apo/1.40 numerical aperture oil (NA) immersion objective. mEGFP was excited via a high-power light-emitting diode (3 watt, 497 nm at 40 MHz) of a Lambert Instruments (Roden, the Netherlands) LIFA fluorescence lifetime imaging module attached to the Nikon TiE microscope. Frequency-domain fluorescence life-time measurements were collected and calculated via the Lambert software. For each condition, three individual experiments were performed, and a total of at least 60 cells were imaged for FLIM measurements. FRET efficiency was calculated as 1 – [experimental lifetime/(lifetime of mEGFP component with EV)]. Data are shown as mean fluorescence lifetime values in nanoseconds, as well as FRET efficiency, ±SEM. Student’s t tests were performed to evaluate statistical significance.
Fluorescence cross-correlation spectroscopy
Human embryonic kidney (HEK) 293T cells were seeded in eight-well chambers on glass coverslips (ibidi μ-Slide, #80807) and transfected using GeneJuice transfection reagent (Novagen). Cells were imaged 24 hours posttransfection on a Zeiss LSM 780 confocal microscope fitted with a 60× 1.2 NA objective and using 488- and 594-nm lasers to excite mEGFP and mCherry2, respectively. Scanning FCCS was performed by acquiring 100,000 frames of a 52-pixel line scan at the basal cell surface at a pixel dwell time of 3.94 μs. Intensity traces were correlated and fitted using FoCuS software (54), and statistical analyses were performed with GraphPad Prism v9.0.2. The cross-correlation quotient (q) was calculated as the ratio between the amplitude of the cross-correlation curve and the minimal autocorrelation amplitude. Normality was assessed using a Kolmogorov-Smirnov test, and statistical comparison was performed using an analysis of variance (ANOVA) test with Tukey’s correction for multiple comparisons.
TOXR transcription factor TMD dimerization
The TOXGREEN assay for TMD dimerization in Escherichia coli was performed at stationary phase as described (37). Briefly, TMDs were PCR-amplified with Nhe I and Bam HI flanking restriction sites and cloned into the pccGFPKAN plasmid (Addgene plasmid #73649). MM39 bacteria (Addgene bacterial strain #42894) were transformed with these plasmids as well as with pccGFP-GpA (Addgene plasmid #73651), pccGFP-GpA-G83I (Addgene plasmid #73650), and pccGFPKAN as the no-transmembrane (TM) control. Transformed colonies were grown in 3-ml LB medium for 16 hours at 37°C with shaking. Three hundred microliters of culture was loaded into a 96-well black-walled clear-bottom plate in triplicate. Plates were read using a fluorescence multiwell plate reader (EnVision Multilabel Plate Reader; PerkinElmer) with an excitation wavelength of 485 nm and fluorescence emission at 512 nm along with an optical density reading at 600 nm for normalization. Results were further normalized by subtracting the average of the no TM control followed by normalization to the GpA-positive control.
AF-conjugated Fab FRET
OKT4 (anti-CD4 mAb) and pembrolizumab (anti–PD-1 mAb) were digested with papain, and Fab fragments were purified by sequential protein A Fc removal and high-performance liquid chromatography gel filtration. OKT4 is known to disrupt potential CD4 oligomerization and thus was chosen as a negative control (55). The anti–PD-L1 Fab (from BMS-936559 mAb sequence) was expressed in suspension HEK293F cells using a polyethylenimine (PEI) lipid–based transfection as described previously (7). Purified Fab fragments were conjugated to AF 555 and AF 647 via primary amine labeling according to the manufacturer’s instructions (Invitrogen Molecular Probes). T cells were stained with the labeled Fabs for 20 min on ice, protected from light. After washing, the cells were analyzed in a BD LSRII flow cytometer. AF 555 fluorescence was measured using a 586/15 filter off of the 561-nm laser. AF 647 was detected with a 670/30 filter off of the 633-nm laser. FRET was measured with a 670/20 filter off of the 561-nm laser. Data were analyzed using FlowJo 10 software.
Evolutionary sequences and synteny analysis
Blast searches were performed by searching the various genome sequencing projects with the Blast-T program with multiple starting queries using the National Center for Biotechnology Information (http://ncbi.nlm.nih.gov) and the Ensembl (http://ensembl.org).
Lentivirus production
The PD-1–EGFP–pHR lentivirus vector was provided by Hui et al. (56). The PD-1 insert was recreated by overlapping PCRs to create the PD-1–G173W and PD-1–L184W TMD variants. Lentivirus was generated by transfecting the above pHR plasmids along with psPAX2 and pMD.2G packaging plasmids into 293T cells using Lipofectamine 3000 transfection reagents. On days 2 and 3 after transfection lentivirus, particles were harvested by filtering the viral supernatant (0.45 μM). Lentivirus was frozen at −80°C.
CRISPR-Cas9 knockout of PDCD1 in Jurkat T cells and subsequent PD-1 lentiviral rescue
Parental Jurkat T cells were electroporated (AMAXA) with a combination of two PDCD1 CRISPR RNA guides (2: CACGAAGCTCTCCGATGTGT and 3: GCGTGACTTCCACATGAGCG) in PX458 (GenScript). PX458 is a pSpCas9(BB)-2A-GFP plasmid. Thus, electroporated cells are rendered transiently GFP+. One to 2 days after electroporation, the GFP+ electroporated Jurkat cells were single-cell–sorted into 96-well plates. Screening for the loss of PD-1 surface expression in clones after anti-CD3/28 overnight T cell stimulation identified the 2E4 clone. PD-1 was rescued in 2E4 cells using the lentiviral particles described above. Approximately 1 week after transduction, the transduced cells were sorted for GFP. PD-1 surface expression for the three PD-1 variants was verified by microscopy and flow cytometry surface staining.
Planar-supported lipid bilayer preparation
Small unilamellar vesicle mixtures were injected into flow chambers formed by sealing sterilized glass coverslips to adhesive-backed plastic manifolds with six flow channels (StickySlide VI 0.4, Ibidi). After 20 min, the channels were flushed with Hemo bioscience (HBS)–bovine serum albumin (BSA) [20 mM Hepes (pH 7.2), 137 mM NaCl, 5 mM KCl, 700 μM Na2HPO4, 6 mM d-glucose, 10 mM CaCl2, 10 mM MgCl2, and 0.1% BSA (w/v)] without introducing air bubbles to remove excess small unilamellar vesicles (SUVs). After blocking for 20 min with 5% BSA, His-tagged and/or biotinylated proteins were incubated on bilayers for an additional 20 min. Protein concentrations required to achieve desired densities on bilayers were calculated from calibration curves constructed from flow cytometric measurements of bead-supported lipid bilayers compared with reference beads containing known numbers of the appropriate fluorescent dyes (Bangs Laboratories).
Jurkat or OT1 cells were incubated at 37°C on planar-supported lipid bilayers containing ICAM-1-AF405, UCHT1-Fab or monobiotinylated pMHC [H-2Kb SIINFEKL, Medical & Biological Laboratories (MBL)], and PD-L1-AF647 for 15 min (Jurkat) or 30 min (OT1). The cells were subsequently fixed with 4% paraformaldehyde in PIPES HEPES EGTA magnesium (PHEM) buffer [10 mM EGTA, 2 mM MgCl2, 60 mM Pipes, and 25 mM Hepes (pH 7.0)], washed three times with PBS, and permeabilized with 0.1% Triton X-100. They were then washed again three times with PBS, blocked with 6% BSA for 45 min, and stained with primary antibodies overnight. The washing step was repeated before the cells were incubated with secondary antibodies for 45 min for Jurkat cells. OT1 cells were stained once with anti–granzyme B–fluorescein isothiocyanate (FITC) overnight. After a final washing step, the cells were imaged by TIRFM. Individual cells attached to the supported lipid bilayer (SLB) were automatically segmented from each TIRF micrograph using FIJI software (version 2.3.0/1.53f) based on either an interference reflection microscopy image (Jurkat cells) or a bright-field image (OT-1 cells). The mean fluorescence intensity of proteins of interest within the segmented areas was then quantified and defined as protein recruitment to the IS. Jurkat TIRFM data were normalized to PD-1–GFP expression. Antibodies used were as follows: AF 488 anti-phosphotyrosine (PY20, BioLegend), FITC anti-human/mouse granzyme B (BioLegend), and SH-PTP2 (B-1) (Santa Cruz Biotechnology).
Total internal reflection fluorescence microscopy
TIRFM of the Jurkat cells was performed on an Olympus IX83 inverted microscope equipped with a four-line (405-, 488-, 561-, and 640-nm laser) illumination system. The system was fitted with an Olympus UApON 150 × 1.45 NA objective and a Photometrics Evolve delta electron-multiplying charge-coupled device (EMCCD) camera to provide Nyquist sampling. TIRFM of the OT-1 cells was performed on a Deltavision OMX V4 system (Applied Precision, GE Healthcare). The microscope was equipped with a 60× ApoN NA 1.49 objective (Olympus), three cooled scientific complementary metal–oxide–semiconductor (sCMOS) cameras (PCO), and 405-nm (100-mW diode), 488-nm [100-mW optically pumped semiconductor laser (OPSL)], and 568- and 642-nm (110-mW diode) lasers. The system was controlled using the OMX Acquisition control software running under a Windows 7 operating system. Image registration was performed using the SoftWoRx software package (Applied Precision, GE Healthcare). Quantification of fluorescence intensity was performed with ImageJ software (National Institutes of Health).
Jurkat plate–bound activation/inhibition
Plates (48-well) were coated overnight at 4°C with anti-human CD3 mAb (10 μg/ml; UCHT1 Ultra-LEAF purified, BioLegend) and PD-L1 monomer protein (1.5 μg/ml) (7) in 200 μl per well. The next day, wells were washed once with medium, followed by addition of 5 × 105 Jurkat T cells along with soluble anti-human CD28 mAb (2 μg/ml; CD28.2 Ultra-LEAF–purified, BioLegend) in a 500-μl volume of T cell media (TCM) per well. After an 18- to 24-hour 37°C incubation, the cells were harvested, washed, and stained for the CD69 activation marker using the FN50-PE (BioLegend). A ratio of CD69+/CD69− cells was generated using FlowJo software.
Retrovirus production
mPD-1, mPD-1–G172W, and mPD-1–L186W open reading frames were gene-synthesized using mouse optimized codons (GeneScript) and cloned into the MSCV-IRES-Thy1.1 retroviral (RV) vector using the Sal I and Bam HI restriction sites. Plat-E cells were used to prepare ecotropic RV particles for T cell transduction using Lipofectamine 3000 transfection reagents. Briefly, 70 to 80% confluent Plat-E cells grown in DMEM + 10% FBS were transfected. After a 24-hour incubation, medium was changed to RPMI + 10% FBS. On days 2 and 3 after transfection, RV particles were harvested by filtering the viral supernatant (0.45 μM). RV was used fresh or frozen at −80°C.
Ex vivo culture of CD8+ T cells and RV transduction
OT1 T cells were isolated from mouse spleen and lymph nodes and purified by positive selection with magnetic-activated cell sorting anti-CD8 magnetic beads according to the manufacturer’s protocol (Miltenyi Biotec). For T cell activation, wells were precoated with goat anti-hamster immunoglobulin G at 1:40 dilution (1 mg/ml; MP Biomedicals). T cells were resuspended in TCM: RPMI supplemented with 10% FBS, 4 mM glutamine, 50 μM β-mercaptoethanol, and 20 mM Hepes. For activation, TCM was further supplemented with anti-CD3 (1 μg/ml, Bio X Cell) and anti-CD28 (1 μg/ml, Bio X Cell). After overnight activation, T cells were transduced by spin infection with RV particles with polybrene (10 μg/ml; Sigma-Aldrich). Spin infection was performed by centrifugation at 850g at 32°C for 1.5 hours. T cells were kept in TCM supplemented with human interleukin-2 (100 U/ml; PeproTech) for 2 days before use.
B16-OVA tumor OT1 adoptive transfer
B16-OVA cells (2 × 105) were injected into six 8- to 9-week-old WT black mice on day −10. On day zero, 5 × 106 retrovirally transduced GFP+, OT1 T cells were injected intravenously. Tumor growth was measured for 18 days after adoptive transfer or until death/tumor volume exceeded 2000 mm3.
RIP-mOVA T1D model
OT1 cells (1 × 106) were adoptively transferred via tail vein injection into hemizygous RIP-mOVA mice 7 to 12 weeks of age. Blood glucose measurements were taken daily from the tail tip using the AimStrip Plus glucose meter. Mice were considered diabetic if blood glucose readings were above 250 mg/dl for >2 days.
Statistics
Means are shown with error bars indicating SEM. Statistical analyses were performed using GraphPad v.8.0. Unpaired versus paired Student’s t tests are indicated in the figure legends. Two-way ANOVA analysis was used to compare the tumor growth curves and blood glucose serial measurements. The Mann-Whitney test was used to evaluate the TIRFM experiments. Significance is indicated as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Funding:
This work was supported in part by NIH grants T32AR069515 (E.A.P.), T32GM007308 (E.A.P.), R37CA273333–01 (J.W.), R01CA269898 (J.W.), and R01AI125640 (A.M.M.). The work was also supported by an Institutional Fund from NYU Grossman School of Medicine (X.-P.K.), Wellcome and Kennedy Trust for Rheumatology Research Principal Research Fellowship 100262Z/12/Z (M.L.D.), a young investigator award from the Melanoma Research Alliance (J.W.), a pilot award from the NYU Colton Center for Autoimmunity (J.W.), the V Foundation (J.W.), the Mark Foundation ASPIRE award (J.W.), the Research Council of Norway in conjunction with Marie Sklodowska-Curie Actions 275466 (A.K.), and a Wellcome Trust PhD Studentship in Science 108869/Z/15/Z (A.M.M.). The authors acknowledge support from the Wolfson Imaging Centre at the MRC Weatherall Institute of Molecular Medicine, University of Oxford and the Research Council of Norway in conjunction with Marie Sklodowska-Curie Actions 275466 (A.K.).
Footnotes
Competing interests: J.W. is a consultant for Rootpath Genomics, Bristol Myers Squibb (Relatlimab Advisory Council), and Hanmi Pharmaceutical Co. The other authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Materials generated as part of this study can be shared upon the completion of standard material transfer agreements with NYU and/or the University of Oxford.
REFERENCES AND NOTES
- 1.Sharma P, Allison JP, Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 161, 205–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wherry EJ, T cell exhaustion. Nat. Immunol. 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Tai Y, Wang Q, Korner H, Zhang L, Wei W, Molecular mechanisms of T cells activation by dendritic cells in autoimmune diseases. Front. Pharmacol. 9, 642 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharpe AH, Freeman GJ, The B7-CD28 superfamily. Nat. Rev. Immunol. 2, 116–126 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Greenwald RJ, Freeman GJ, Sharpe AH, The B7 family revisited. Annu. Rev. Immunol. 23, 515–548 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, Huo J, Yu C, Evans EJ, Leslie AJ, Griffiths M, Stubberfield C, Griffin R, Henry AJ, Jansson A, Ladbury JE, Ikemizu S, Carr MD, Davis SJ, Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 288, 11771–11785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philips EA, Garcia-Espana A, Tocheva AS, Ahearn IM, Adam KR, Pan R, Mor A, Kong XP, The structural features that distinguish PD-L2 from PD-L1 emerged in placental mammals. J. Biol. Chem. 295, 4372–4380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keir ME, Butte MJ, Freeman GJ, Sharpe AH, PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribas A, Wolchok JD, Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dall’Era M, Davis J, CTLA4Ig: A novel inhibitor of costimulation. Lupus 13, 372–376 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Tuttle J, Drescher E, Simon-Campos JA, Emery P, Greenwald M, Kivitz A, Rha H, Yachi P, Kiley C, Nirula A, A phase 2 trial of peresolimab for adults with rheumatoid arthritis. N. Engl. J. Med. 388, 1853–1862 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Greene JL, Leytze GM, Emswiler J, Peach R, Bajorath J, Cosand W, Linsley PS, Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J. Biol. Chem. 271, 26762–26771 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Linsley PS, Nadler SG, Bajorath J, Peach R, Leung HT, Rogers J, Bradshaw J, Stebbins M, Leytze G, Brady W, Malacko AR, Marquardt H, Shaw SY, Binding stoichiometry of the cytotoxic T lymphocyte-associated molecule-4 (CTLA-4). A disulfide-linked homodimer binds two CD86 molecules. J. Biol. Chem. 270, 15417–15424 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA, ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 397, 263–266 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, Nathenson SG, Almo SC, Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–347 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Ikemizu S, Gilbert RJ, Fennelly JA, Collins AV, Harlos K, Jones EY, Stuart DI, Davis SJ, Structure and dimerization of a soluble form of B7–1. Immunity 12, 51–60 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Chattopadhyay K, Bhatia S, Fiser A, Almo SC, Nathenson SG, Structural basis of inducible costimulator ligand costimulatory function: Determination of the cell surface oligomeric state and functional mapping of the receptor binding site of the protein. J. Immunol. 177, 3920–3929 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Zak KM, Kitel R, Przetocka S, Golik P, Guzik K, Musielak B, Domling A, Dubin G, Holak TA, Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 23, 2341–2348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC, Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. U.S.A. 105, 10483–10488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Harrison DL, Song Y, Ji J, Huang J, Hui E, Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 24, 379–390.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, Xiao C, Bonorino C, Lu LF, Bui JD, Hui E, PD-L1:CD80 Cis-heterodimer triggers the Co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 51, 1059–1073.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, Okazaki T, Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Siegel RM, Chan FK, Zacharias DA, Swofford R, Holmes KL, Tsien RY, Lenardo MJ, Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci STKE 2000, pl1 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Bhatia S, Edidin M, Almo SC, Nathenson SG, Different cell surface oligomeric states of B7–1 and B7–2: Implications for signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 15569–15574 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatia S, Edidin M, Almo SC, Nathenson SG, B7–1 and B7–2: Similar costimulatory ligands with different biochemical, oligomeric and signaling properties. Immunol. Lett. 104, 70–75 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC, Structural basis for co-stimulation by the human CTLA-4/B7–2 complex. Nature 410, 604–608 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Schwartz JC, Almo SC, Nathenson SG, Crystal structure of the receptor-binding domain of human B7–2: Insights into organization and signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 2586–2591 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacDonald HR, Schreyer M, Howe RC, Bron C, Selective expression of CD8α (Ly-2) subunit on activated thymic γ/δ cells. Eur. J. Immunol. 20, 927–930 (1990). [DOI] [PubMed] [Google Scholar]
- 29.Datta R, Heaster TM, Sharick JT, Gillette AA, Skala MC, Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 25, 1–43 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieger JW, Singh AP, Bag N, Garbe CS, Saunders TE, Langowski J, Wohland T, Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. Nat. Protoc. 10, 1948–1974 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Lange G, Lewis SJ, Murshudov GN, Dodson GG, Moody PC, Turkenburg JP, Barclay AN, Brady RL, Crystal structure of an extracellular fragment of the rat CD4 receptor containing domains 3 and 4. Structure 2, 469–481 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA, Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116, 3268–3277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finger C, Escher C, Schneider D, The single transmembrane domains of human receptor tyrosine kinases encode self-interactions. Sci. Signal. 2, ra56 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Lemmon MA, Schlessinger J, Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter D, Moraga I, Winkelmann H, Birkholz O, Wilmes S, Schulte M, Kraich M, Kenneweg H, Beutel O, Selenschik P, Paterok D, Gavutis M, Schmidt T, Garcia KC, Muller TD, Piehler J, Ligand-induced type II interleukin-4 receptor dimers are sustained by rapid re-association within plasma membrane microcompartments. Nat. Commun. 8, 15976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dustin ML, Zenclussen AC, A checkpoint cliffhanger at the dawn of placental mammals. J. Biol. Chem. 295, 4381–4382 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong CR, Senes A, Screening for transmembrane association in divisome proteins using TOXGREEN, a high-throughput variant of the TOXCAT assay. Biochim. Biophys. Acta 1858, 2573–2583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russ WP, Engelman DM, The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 296, 911–919 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Lemmon MA, Flanagan JM, Hunt JF, Adair BD, Bormann BJ, Dempsey CE, Engelman DM, Glycophorin A dimerization is driven by specific interactions between transmembrane alpha-helices. J. Biol. Chem. 267, 7683–7689 (1992). [PubMed] [Google Scholar]
- 40.Brosig B, Langosch D, The dimerization motif of the glycophorin A transmembrane segment in membranes: Importance of glycine residues. Protein Sci. 7, 1052–1056 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monks SA, Needleman DJ, Miller C, Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 113, 415–423 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharp LL, Zhou J, Blair DF, Features of MotA proton channel structure revealed by tryptophan-scanning mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 92, 7946–7950 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacKenzie KR, Prestegard JH, Engelman DM, A transmembrane helix dimer: Structure and implications. Science 276, 131–133 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T, Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curran MA, Montalvo W, Yagita H, Allison JP, PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin-Orozco N, Wang YH, Yagita H, Dong C, Cutting Edge: Programmed death (PD) ligand-1/PD-1 interaction is required for CD8+ T cell tolerance to tissue antigens. J. Immunol. 177, 8291–8295 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Keir ME, Freeman GJ, Sharpe AH, PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 179, 5064–5070 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, Ali FA, Rahman M, Delmonte OM, Chrabieh M, Zhang P, Gruber C, Pelham SJ, Spaan AN, Rosain J, Lei WT, Drutman S, Hellmann MD, Callahan MK, Adamow M, Wong P, Wolchok JD, Rao G, Ma CS, Nakajima Y, Yaguchi T, Chamoto K, Williams SC, Emile JF, Rozenberg F, Glickman MS, Rapaport F, Kerner G, Allington G, Tezcan I, Cagdas D, Hosnut FO, Dogu F, Ikinciogullari A, Rao VK, Kainulainen L, Beziat V, Bustamante J, Vilarinho S, Lifton RP, Boisson B, Abel L, Bogunovic D, Marr N, Notarangelo LD, Tangye SG, Honjo T, Gros P, Boisson-Dupuis S, Casanova JL, Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat. Med. 27, 1646–1654 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H, Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 184, 923–930 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, Eisenhaure TM, Pendergraft III WF, Hua P, Pham HC, Bu X, Zhu B, Hacohen N, Fritsch EF, Boussiotis VA, Wu CJ, Freeman GJ, A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol. Immunother. 68, 421–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patsoukis N, Duke-Cohan JS, Chaudhri A, Aksoylar HI, Wang Q; Asia Council A Berg GJ. Freeman VA. Boussiotis, Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 3, 128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki K, Tajima M, Tokumaru Y, Oshiro Y, Nagata S, Kamada H, Kihara M, Nakano K, Honjo T, Ohta A, Anti-PD-1 antibodies recognizing the membrane-proximal region are PD-1 agonists that can down-regulate inflammatory diseases. Sci. Immunol. 8, eadd4947 (2023). [DOI] [PubMed] [Google Scholar]
- 53.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC, CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Waithe D, Schneider F, Chojnacki J, Clausen MP, Shrestha D, de la Serna JB, Eggeling C, Optimized processing and analysis of conventional confocal microscopy generated scanning FCS data. Methods 140–141, 62–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lynch GW, Sloane AJ, Raso V, Lai A, Cunningham AL, Direct evidence for native CD4 oligomers in lymphoid and monocytoid cells. Eur. J. Immunol. 29, 2590–2602 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD, T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Materials generated as part of this study can be shared upon the completion of standard material transfer agreements with NYU and/or the University of Oxford.