

Abstract
Field emission scanning electron microscopy (FESEM) is a well-established technique for acquiring three-dimensional surface images of nuclear pore complexes (NPCs). We present an optimized protocol for the exposure of mammalian cell nuclei and direct surface imaging of nuclear envelopes by FESEM, allowing for a detailed morphological comparison of individual NPCs, without the need for averaging techniques. This provides a unique high resolution tool for studying the effects of cellular stress, specific genetic manipulations and inherited diseases on the ultrastructure of human NPCs.
Keywords: Scanning electron microscopy, Nuclear pore complex, Nuclear envelope, Mammalian cell culture, Auxin-inducible degron, Central channel particles
1. Introduction
High-resolution scanning electron microscopy of nuclear envelopes and NPCs was pioneered in the 1990s by Martin Goldberg and Terry Allen, working primarily with the giant nuclei of amphibian oocytes [1–3]. Imaging mammalian NPCs is more challenging, because it requires unobstructed views of the outer surface of nuclei and this should ideally be achieved without the use of detergents, to preserve the native membrane-embedded structure. We have previously developed a method for nuclear exposure based on repeated hypotonic treatment of adherent cultured cells, in the absence of detergents [4, 5]. This method has been adjusted for use with various human cells, including immortalized cell lines, patient-derived primary fibroblasts and even induced pluripotent stem cell (iPSC)-derived neurons [6–10]. Improved ultrastructural analysis is achieved by the standardized use of iridium coating, depositing a delicate grain-less conductive metal layer over the samples. This provides improved resolution and finer details in the FESEM images of individual NPCs. Importantly, when large expanses of the nuclear envelope can reproducibly be imaged by this method in a given human cell line, it becomes possible to assess the distribution of NPCs, to identify unique structural subtypes and to test the effect of specific treatments on NPC structure.
Here, we present an optimized procedure, with practical notes that may be helpful in adapting the protocol to additional experimental systems. Two examples demonstrate the type of qualitative and quantitative analysis that can be obtained from a large number of high resolution images of NPCs or NPC remnants (see below).
The first example focuses on central NPC channel particles, also known as “plugs,” that we have previously linked with a rare inherited disease: acute febrile encephalopathy [7]. The disease is caused by genetic mutations destabilizing Nup214 on the cytoplasmic façade of the NPC and patients suffer recurring febrile (high temperature) episodes resulting in seizures, neurodevelopmental regression and cerebellar atrophy. We have demonstrated that the phenotype at the intracellular level in patient fibroblasts is a dramatic increase in the presence of central plugs/particles in NPC channels. Our next goal is to examine the effect of heat shock on central channel particles in an attempt to understand the mechanism of pathogenesis. Figure 1 demonstrates this type of analysis, performed in human fibroblasts derived from a healthy individual, including the categorizing of 3 types of channels in individual NPCs and images acquired at different magnifications. Note that the extraordinarily flat nuclei of fibroblasts provide large expanses of relatively flat and exposed nuclear envelope and a multitude of visible NPCs.
Fig. 1.
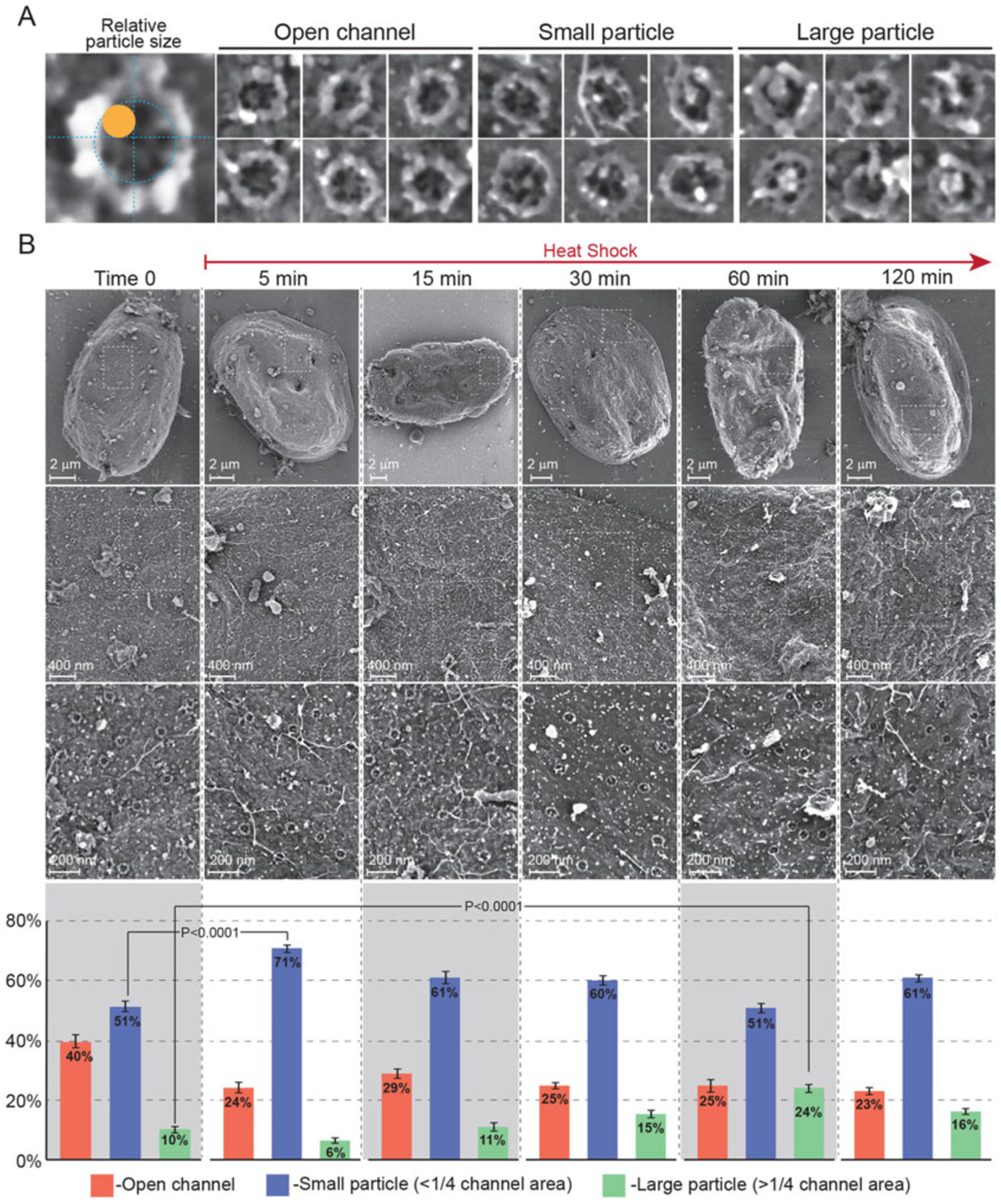
FESEM analysis of central channel plugs in the NPCs of fibroblasts exposed to heat shock stress. Primary human skin fibroblasts derived from a healthy individual were exposed to continuous heat shock at 43 °C. Samples were removed at various time-points between 0 and 120 min of heat shock and prepared for direct surface imaging of exposed nuclei by FESEM as described in the text. (a) Individual NPCs were scored according to the example shown on the left, as having an open central channel, a channel occupied by a small particle (<¼ of projected channel area) or a channel occupied by a large particle (>¼ channel area). (b) Different magnifications of representative nuclei from time 0 and 5 different heat shock time-points. Whole nuclei are shown in the top row and areas marked by dashed rectangles are enlarged in subsequent rows. Bar charts below each time-point show a quantitative analysis of the three categories of NPC channels, compiled from three independent experiments. Only two statistically significant comparisons, for small and large particles, are shown for clarity. Error bars indicate SEM
The second example concerns the use of a rapid Auxin-Inducible Degron (AID) system to target individual nucleoporins for degradation during interphase, in preexisting NPCs. The extended study includes a systematic comparison of many nucleoporins showing varying effects on distinct structural modules of the NPC [11]. Here, we show one example of AID-tagged Nup93, which was found to be a linchpin subunit with critical connections affecting the entire structure of the NPC. The resultant structures are defined as NPC remnants and can be classified into different subtypes (see gallery at the bottom of Fig. 2). Of note, is the drastically different morphology of exposed DLD-1 cell nuclei used in this study, as compared to the primary fibroblasts shown in Fig. 1. DLD-1 nuclei are characterized by steep angles and abundant obscuring debris, revealing minimal useable areas on the top surface, while extended flat areas of nuclear envelope are often found on the outskirts of the nucleus (Fig. 2).
Fig. 2.
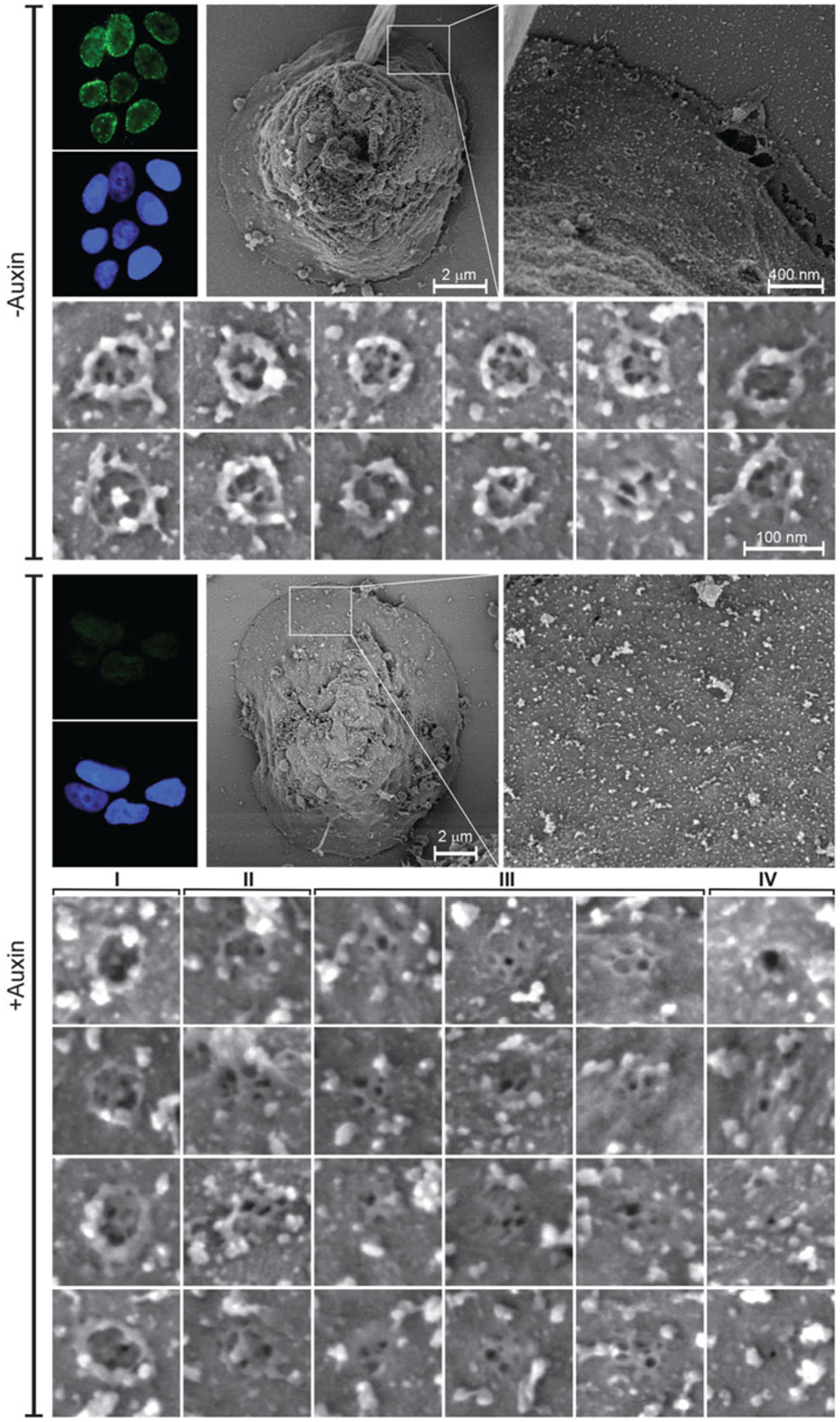
FESEM analysis of the effect of targeted degradation of Nup93 on NPC architecture. Direct surface imaging of exposed nuclei was performed on control (top) and auxin-treated (bottom) NUP93::NeonGreen-AID DLD-1 cells. See ref. (11) for further details on the experimental system. Complete degradation of the targeted nucleoporin was verified by fluorescence microscopy, as shown on the top left panel of each part (−Auxin; +Auxin). Images show a whole nucleus, an enlarged selected area and galleries of individual NPCs or NPC remnants. The +Auxin gallery at the bottom shows the classification of four different subtypes of NPC remnants, with subtype I being the rarest form and subtype III the most common
Overall, the detailed method described below provides a generalized protocol, that can be adapted to different adherent cell-culture lines. Once the conditions for reproducibly obtaining large expanses of exposed nuclear envelope have been determined, it becomes possible to compare different treatments or sources of cells. This includes exposure to cellular stress, genetic manipulation such as the AID targeting of nucleoporins, or specific genetic backgrounds in cells derived from human patients. It remains to be seen whether the large number of high resolution images of NPCs or NPC remnants accumulated by this strategy can also be used in something akin to single particle analysis, to obtain additional structural insight.
2. Materials
2.1. Buffers
Phosphate-buffered saline (PBS): 2.67 mM KCl, 1.47 mM KH2PO4, 138 mM NaCl, 8.1 mM Na2HPO4, pH 7.4. Sterilize and store at 4 °C.
Hypotonic buffer: 15 mM Tris–HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2. Prepare 50 mL of the buffer from separate stock solutions and ultrapure water, filter through a 0.22 !m filter, divide into 5 mL aliquots, and freeze at −20 °C.
5 x Buffer F: 400 mM PIPES KOH, pH 6.8, 5 mM MgCl2, 750 mM sucrose.
Sodium cacodylate buffer, 0.2 M, pH 7.4. Store at 4 °C.
2.2. Materials and Reagents for Sample Preparation
Poly-L-lysine hydrobromide (e.g., P1524, Sigma). Dissolve at 0.1 mg/mL in sterile ultrapure water, filter through a 0.22 μm filter, and divide into 120 μL aliquots. Store at −20 °C.
4″ Silicon wafer in 5 × 5 mm chips (e.g., #16008, Ted Pella).
Glutaraldehyde, 8% EM grade.
Paraformaldehyde, 16% EM grade.
Modified Karnovsky’s fixation solution: 2% paraformaldehyde, 2% glutaraldehyde in buffer F. Dilute in ultrapure water and filter through a 0.22 μm filter.
Osmium tetroxide, 2% (e.g., #19152, Electron Microscopy Sciences). Dilute 1:1 with cacodylate buffer immediately before use. Protect solution from light.
Graded ethanol series: 3.25%, 7.5%, 15%, 30%, 50%, 70%, 90%, 95%, and 100% ethanol. Prepare from absolute, molecular biology-grade ethanol and ultrapure water, filter through a 0.22 μm filter and store at −20 °C.
PELCO conductive silver 187 (e.g., #16045, Ted Pella).
Aluminum SEM specimen mounting stubs.
Carbon adhesive tabs (e.g., #77825–12, Electron Microscopy Sciences).
Diamond pen.
2.3. Equipment
Refrigerated benchtop centrifuge with fixed-angle and swing-bucket rotors (e.g., 5810R, Eppendorf).
Critical point dryer (e.g., K850 CPD, Quorum Technologies) attached to a high purity liquid CO2 cylinder.
Sputter-coating apparatus (e.g., Q150R S, Quorum Technologies) equipped with an iridium target.
High resolution field emission scanning electron microscope (e.g., Merlin, Zeiss).
3. Methods
3.1. Experimental Set-up
All the items and working solutions needed for fixation and sample preparation for FESEM should be prepared on the day of the experiment and before starting the hypotonic treatment for nuclear exposure (see Note 1). When additional treatment of cell cultures is part of the experimental plan (e.g., exposure to stress conditions or auxin-induced degradation of proteins), these steps should be integrated into the time-line, preferably within the same day.
Prepare polylysine-coated silicon chips and first mark individual chips by engraving numbers or letters in one corner with a diamond pen. Plan on using four 5 × 5 mm silicon chips for one million cells. This number may vary for different cell types, because of cellular dimensions and the efficiency of nuclear exposure and recovery.
Spread a drop of 20 μL of 0.1 mg/mL polylysine over the surface of each chip (shiny side up) and incubate for 20 min at room temperature. Wash the chip in filtered ultrapure water and air-dry.
We recommend the use of improvised low-speed centrifugation devices or “microtube chambers,” as described in Fig. 1 of Chapter 27, this volume, and in our previous publications [4, 5]. This improves the yield of cells and nuclei in the recovery stage, after the hypotonic treatment, ensuring they are spread over the entire surface of the silicon chips.
3.2. Hypotonic Treatment and Preparation for FESEM
Grow cells under standard conditions, detach by trypsinization, and pellet by centrifugation for 5 min at 1000 × g, 4 °C.
Resuspend, count and transfer ~250,000 cells destined for each silicon chip into a separate Eppendorf tube.
Wash cells in cold PBS, pellet by centrifugation for 5 min at 1000 × g, 4 °C.
Gently resuspend the cells in 0.5 mL of cold hypotonic buffer in each tube. Incubate on ice for 5 min.
Pass the cells twice through a 1 mL syringe with a 21-gauge needle and recover the samples (containing a mixture of intact cells and partially exposed nuclei) by centrifugation for 3 min at 1500 × g, 4 °C in a fixed-angle rotor (see Note 2).
Resuspend each pellet in 1 mL of PBS/10% glycerol, move into a microtube chamber (see Subheading 3.1, step 3) and centrifuge directly onto the polylysine-coated silicon chip for 10 min at 800 × g, 4 °C in a swing-bucket rotor. The improvised microtube chambers should be placed in 14 mL centrifuge tubes stuffed with paper up to the 10.5 mL mark (see Note 3).
From this point on, all steps should be carried out at room temperature, in a 24-well cell culture plate, taking extreme caution to avoid any drying. We recommend the simultaneous use of two micropipettes: one for removing liquid from the well and the other for the swift addition of a new solution [4].
Use high-precision tweezers to transfer the silicon chips into a 24-well cell culture plate filled with PBS. Keep track of chip numbering.
Wash chips 2 × 2 min in 1× Buffer F, 1 mL solution per well.
Fix in modified Karnovsky’s fixation solution for 20 min.
Wash 3 × 2 min in 0.1 M cacodylate buffer pH 7.4.
Incubate for 10 min in secondary fixative containing 1% OsO4 in 0.1 M cacodylate buffer pH 7.4; protect from light.
Wash 3 × 2 min in ultrapure water.
Carefully start dehydration through a graded ethanol series: 3.25%, 7.5%, 15%; 1 × 2 min incubation for each step. Continue from 30% through 50%, 70%, 90%, and 95% ethanol; 2 × 2 min incubation for each step.
Conclude dehydration by washing and exchanging 100% ethanol 3 × 2 min (see Note 4).
Transfer the silicon chips to a critical point drying apparatus (preferably to a semiautomatic model) filled with 100% ethanol. Exchange into high-purity liquid CO2 and subsequently from liquid to gas, according to the manufacturer’s instructions (see Note 5).
From this point on, store the dried samples under vacuum, or under nitrogen or argon gas, in a desiccator. This is important for minimizing damage by oxidation and rehydration.
Mount individual silicon chips on aluminum stubs using conductive silver paint or double-sided carbon adhesive tabs.
Dry the chips under a gentle flow of argon, in a desiccator, for 30 min.
Transfer the samples to a sputter-coating apparatus and coat with a ~1 nm thick layer of iridium (see Note 6).
3.3. High Resolution Imaging by FESEM
FESEM imaging should be performed by an experienced professional operator on the appropriate instrument. Sample analysis is time consuming and requires patience. It is only at this stage, that a successful procedure can be confirmed by the identification of exposed nuclei and NPCs. We recommend examining large areas of each silicon chip at a magnification of 4000 to 5000× and zooming in to magnifications of 20,000 to 30,000× to verify the detection of genuine nuclei. Most of the objects scattered over the surface of the chip will be intact cells, cellular debris and partially obscured nuclei. The first objective is therefore identifying intact, undamaged nuclei. This is usually based on the identification of multiple NPCs, with their distinctive appearance, but it can become more challenging when the abundance of mature NPCs drops, or when they are replaced by NPC remnants (see below).
We can only offer some general tips and guidelines for this part of the procedure and stress that every new type of sample should be analyzed with careful attention. Conclusions should be based on biological, rather than simple technical, repeats.
In most cell types, exposed nuclei tend to be more round or oval shaped, but not necessarily smaller than intact cells, when viewed at low magnification.
Likewise, nuclei tend to have less irregularities on their surface, as compared to cells.
Cells and nuclei also differ in electron emission efficiency. In most cases, exposed nuclei emit more signal and look brighter than whole cells, although we have also observed rare darker nuclei within the specimens.
Finding many exposed nuclei on a single silicon chip is not necessarily a good sign, because it can be the result of an exaggerated hypotonic treatment or mechanical disruption. Carefully screen the sample for signs of torn or damaged nuclear membranes and adjust the treatment, if necessary (see Notes 2 and 3).
3.4. Image Analysis
When large numbers of high resolution images become available, it is possible to move on to image analysis and consider a comparison of individual NPCs. Again, it is important to stress that conclusions should not be reached based on single experiments and it is important to perform biological repeats and collect many images from multiple nuclei.
Figure 1 demonstrates the analysis of central channel plug prevalence following heat shock in primary fibroblasts obtained from a healthy individual. The top row in panel B shows whole nuclei at a low magnification, with subsequent rows “zoomed-in” to higher magnifications and demonstrating the abundance of easily recognizable NPCs. In this case, our preliminary analysis of the images suggested that central plugs/particles of different sizes were accumulating during heat shock exposure. The whole set of individual NPC images was next divided into 3 categories: open channels, channels occupied by “small” particles (up to ¼ of the projected cross-sectional area of the central channel), or channels occupied by “large” particles (>¼ of the projected area; see Fig. 1, panel A). A quantitative analysis of these categories (bar chart at the bottom of panel B) indeed shows a sharp but transient increase in small particles within 5 min of heat shock, followed by a slower accumulation of large particles most clearly evident at 1 h of exposure to heat shock.
The second example, shown in Fig. 2, involves a very different type of nuclear morphology, typical of the DLD-1 cell line used for auxin-induced degradation (see Chapter 9, this volume, for details of the auxin inducible degron system). In this case, only relatively small areas of flat and exposed nuclear envelope are available and data needs to be combined and collected from a larger number of nuclei. Fortunately, the degradation of the target can be followed by a fluorescent marker and the results are very reproducible. The FESEM images in Fig. 2 demonstrate the characteristic features of mature NPCs in the -Auxin control, while the abundance of NPC-related structures drops abruptly in the +Auxin sample and these can be classified into several categories (see gallery at the bottom of Fig. 2). Most of these NPC remnants do not display a wide channel or the full rotational symmetry of an intact scaffold, but contain smaller structural elements reminiscent of the nuclear basket. A potential sequence starting from subtype I and ending in subtype IV can be envisioned for the collapse of NPC structure following the degradation of AID-tagged Nup93. Thus, a careful comparison of individual high-resolution FESEM images can prompt new questions for a follow-up study in this case.
4. Notes
Purchase paraformaldehyde, glutaraldehyde, and osmium tetroxide in single-use, sealed ampoules. Prepare the working solutions for Karnovsky’s fixative and the secondary fixative with 0.22 μm filtered buffers shortly before use.
This is the most sensitive and critical step in the entire protocol. Finding the optimal conditions for the exposure of intact nuclei is clearly a matter of trial and error, as well as patient inspection of the results by FESEM. It is advisable to prepare several chips for each condition so that you can slightly vary the pressure applied to the plunger of the syringe or change the number of passages through the syringe. As a rule of thumb for the required pressure: train yourself by filling 0.5 mL of water in a 1 mL syringe mounted with a 21-gauge needle, point it upward in a 45° angle and find the pressure needed to shoot the liquid to a distance of 2.5–3 m. Avoid air bubbles in the solution.
For additional notes on the adjustment and application of this protocol to different types of adherent cell lines, see previous reference (5). However, this is ultimately a question of trial and error. We note that in one unusual case, we found that adjusting the hypotonic buffer to 80 mM NaCl, resulted in optimal exposure of nuclei from iPSC-derived neurons [6].
Take care to avoid any drying throughout steps 7–15 of the procedure. One to three silicon chips can be treated together in a single well of a 24-well culture plate, but keep track of chip numbering and avoid large bursts of liquid that might overturn the chips.
Successful critical point drying is essential for avoiding the collapse and deformation of biological structures during sample preparation. If this step fails, all the samples in an experiment are likely to be severely damaged. By contrast, if one or more of the silicon chips are transiently dried (even for a few seconds) during handling, there are likely to be extreme differences between different chips in the same experiment.
Coating samples with iridium is superior to any other type of metal coating we have tried. It provides a stable, virtually grain-less conductive coating layer over the samples and allows more details of NPC ultrastructure to be discerned.
Acknowledgments
This work was supported by research grants from the Israel Science Foundation (958/15) and the Christians for Israel Chair in Medical Research to A.H.
References
- 1.Goldberg MW, Allen TD (1992) High resolution scanning electron microscopy of the nuclear envelope: demonstration of a new, regular, fibrous lattice attached to the baskets of the nucleoplasmic face of the nuclear pores. J Cell Biol 119:1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg MW, Allen TD (1993) The nuclear pore complex: three-dimensional surface structure revealed by field emission, in-lens scanning electron microscopy, with underlying structure uncovered by proteolysis. J Cell Sci 106(Pt 1): 261–274 [DOI] [PubMed] [Google Scholar]
- 3.Goldberg MW, Allen TD (1995) Structural and functional organization of the nuclear envelope. Curr Opin Cell Biol 7:301–309 [DOI] [PubMed] [Google Scholar]
- 4.Fichtman B, Shaulov L, Harel A (2014) Imaging metazoan nuclear pore complexes by field emission scanning electron microscopy. Methods Cell Biol 122:41–58 [DOI] [PubMed] [Google Scholar]
- 5.Shaulov L, Fichtman B, Harel A (2014) High-resolution scanning electron microscopy for the imaging of nuclear pore complexes and ran-mediated transport. Methods Mol Biol 1120:253–261 [DOI] [PubMed] [Google Scholar]
- 6.Coyne AN, Zaepfel BL, Hayes L, Fitchman B, Salzberg Y, Luo EC, Bowen K, Trost H, Aigner S, Rigo F et al. (2020) G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron 107:1124–1140.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fichtman B, Harel T, Biran N, Zagairy F, Applegate CD, Salzberg Y, Gilboa T, Salah S, Shaag A, Simanovsky N et al. (2019) Pathogenic variants in NUP214 cause “plugged” nuclear pore channels and acute febrile encephalopathy. Am J Hum Genet 105:48–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fichtman B, Zagairy F, Biran N, Barsheshet Y, Chervinsky E, Ben Neriah Z, Shaag A, Assa M, Elpeleg O, Harel A et al. (2019) Combined loss of LAP1B and LAP1C results in an early onset multisystemic nuclear envelopathy. Nat Commun 10:605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreira TG, Zhang L, Shaulov L, Harel A, Kuss SK, Williams J, Shelton J, Somatilaka B, Seemann J, Yang J et al. (2015) Sec13 regulates expression of specific immune factors involved in inflammation in vivo. Sci Rep 5:17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravindran E, Juhlen R, Vieira-Vieira CH, Ha T, Salzberg Y, Fichtman B, Luise-Becker L, Martins N, Picker-Minh S, Bessa P et al. (2021) Expanding the phenotype of NUP85 mutations beyond nephrotic syndrome to primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders. Hum Mol Genet 30(22):2068–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regmi SG, Lee H, Kaufhold R, Fichtman B, Chen S, Aksenova V, Turcotte E, Harel A, Arnaoutov A, Dasso M (2020) The nuclear pore complex consists of two independent scaffolds. bioRxiv 2020.2011.2013.381947 [Google Scholar]