

Abstract
EBNA2 is essential for Epstein-Barr virus (EBV) immortalization of B lymphocytes. EBNA2 functions as a transcriptional activator and targets responsive promoters through interaction with the cellular DNA binding protein CBF1. We have examined the mechanism whereby EBNA2 overcomes CBF1-mediated transcriptional repression. A yeast two-hybrid screen performed using CBF1 as the bait identified a protein, SKIP, which had not previously been recognized as a CBF1-associated protein. Protein-protein interaction assays demonstrated contacts between SKIP and the SMRT, CIR, Sin3A, and HDAC2 proteins of the CBF1 corepressor complex. Interestingly, EBNA2 also interacted with SKIP in glutathione S-transferase affinity and mammalian two-hybrid assays and colocalized with SKIP in immunofluorescence assays. Interaction with SKIP was not affected by mutation of EBNA2 conserved region 6, the CBF1 interaction region, but was abolished by mutation of conserved region 5. Mutation of conserved region 5 also severely impaired EBNA2 activation of a reporter containing CBF1 binding sites. Thus, interaction with both CBF1 and SKIP is necessary for efficient promoter activation by EBNA2. A model is presented in which EBNA2 competes with the SMRT-corepressor complex for contacts on SKIP and CBF1.
The Epstein-Barr virus (EBV)-encoded latency protein EBNA2 is a nuclear transcriptional activator that is essential for EBV-induced B-cell transformation (27). EBNA2 contributes to immortalization by regulating EBV latency gene expression and by activating expression of cellular genes. EBNA2 regulates expression of the EBV latency Cp promoter that drives expression of the EBNA family of nuclear proteins in type III latency and also contributes to the positive regulation of the LMP2A, LMP2B, and LMP1 promoters (21, 36, 46, 51, 62). EBNA2 does not bind to DNA directly but rather is targeted to responsive promoters through interactions with cellular DNA binding proteins. Targeting through Pu.1 (Spi1) has been described for the LMP1 promoter (22, 30), while CBF1 (RBP-Jκ, RBP-2N, Jκ) has been identified as the targeting partner for the viral Cp, LMP2A, and divergent LMP1 and LMP2B promoters (9, 14, 35, 54, 63). EBNA2 regulates its own expression, and deletion of the region containing the CBF1 binding site from Cp biases promoter usage toward Wp (60). A number of cellular genes, such as those encoding CD23, interleukins, and beta interferon that respond to EBNA2 also have CBF1 binding sites in their promoters (24, 28, 34, 56). However, there are some responsive genes, such as the cyclin D2 gene (26, 43), for which the mechanism of activation is unknown. These genes may be activated as part of a downstream response cascade, or additional EBNA2 targeting mechanisms may exist. Optimal activation by EBNA2 also requires cooperation with other transcription factors. The Cp EBNA2-responsive element contains a CBF2 binding site adjacent to the CBF1 binding site, and the CBF2 site contributes to EBNA2 responsiveness (7, 21, 35, 40). The LMP1 promoter is complexly regulated with both CBF1 and PU.1 targeting sites and multiple transcription factor binding sites that enhance responsiveness (22, 31, 44). The EBNA2-responsive elements are conserved in the Cp and LMP1 promoters of other primate lymphocryptoviruses (8, 39).
CBF1 is an evolutionarily conserved protein that binds to the motif GTGGGAA (34, 52). CBF1 represses transcription in part by tethering a histone deacetylase (HDAC) corepressor complex to the promoter (6, 16, 25, 53). The corepressor proteins SMRT, CIR, SAP30, HDAC1, and HDAC2 have been shown to be components of this complex (19, 25). Repression is believed to be a consequence of histone deacetylation which leads to chromatin remodeling and loss of transcription factor access to the nucleosome-associated promoter sequences. EBNA2 activates expression by binding to the repression domain of CBF1 to relieve repression and bringing a transcriptional activation domain to the promoter (16). Changes in promoter conformation that contribute to activation may also be mediated through EBNA2 interaction with the SNF-SWI complex (57). The EBNA3 proteins also bind to CBF1 (3, 40, 61). Their interaction abolishes CBF1 DNA binding activity (23, 55, 61) and thus modulates the effects of EBNA2.
An important insight into the role of EBNA2 in B-cell immortalization derived from the realization that CBF1 is also the intranuclear target of Notch signaling (17, 20, 47). Notch is a cell surface receptor that when activated by ligand influences a broad spectrum of developmental processes (1). Although the mechanism by which Notch signaling activates downstream target genes is not completely understood, a current model involves ligand binding inducing a proteolytic cleavage event that releases the intracellular domain of Notch, NotchIC, which then translocates to the nucleus (32, 42, 45). NotchIC binds to CBF1 and has a remarkably similar mechanism of action in that it also binds to the CBF1 repression domain to relieve repression and further activates transcription through an endogenous transcriptional activation domain (17, 18). The commonality of the EBNA2 and NotchIC interactions with CBF1 suggested that the early steps in EBV immortalization may mimic an aspect of Notch signaling. Further, several CBF1-regulated genes have been shown to respond to both NotchIC and EBNA2, and EBNA2 has been found to share with NotchIC the ability to block muscle cell differentiation (15, 41).
Comparisons of the EBV EBNA2 amino acid sequence with that of baboon herpesvirus papio revealed a series of nine conserved regions (CR) within EBNA2 (36). Subsequent analyses identified the most carboxy-terminal CR as a strong nuclear localization signal (36) and the adjacent CR as the transcriptional activation domain that interacts with components of the cellular basal transcription complex (4, 36, 48–50). CR6 proved to be the CBF1 targeting domain. Mutation of two tryptophan residues in this region abolished CBF1 interaction (33, 35, 58). This mutation also abolished the ability of EBNA2 to activate reporters carrying CBF1 binding sites (16, 35) and when transferred into the EBNA2 open reading frame within the EBV genome resulted in a virus variant that was nonimmortalizing (58). Mutation of the adjacent region, CR5, resulted in an EBNA2 that retained CBF1 interaction but had a diminished ability to activate a Cp reporter (33). Deletion of the CR5 region from EBNA2 in the context of the EBV genome also resulted in a nonimmortalizing mutant EBV (10).
While it was clear that CR5 made an important contribution to EBNA2 transactivation function, the nature of that contribution was not known. In seeking to better understand the composition of the CBF1 targeting complex, we used a yeast two-hybrid screen to identify CBF1-interacting proteins. We describe the identification of SKIP (Ski-interacting protein) as a component of the CBF1 corepressor complex. SKIP is a nuclear protein with a broad tissue distribution and was originally identified as an interacting partner of the avian retroviral oncoprotein v-Ski (5). We demonstrate that SKIP also interacts with EBNA2 and that it is CR5 that mediates the contacts between EBNA2 and SKIP. The behavior of the CR5 and CR6 EBNA2 mutants suggests that contacts on both CBF1 and SKIP are required for effective EBNA2 targeting to DNA.
MATERIALS AND METHODS
Plasmids.
SKIP cDNA was isolated from a B-cell library (Clontech) in a yeast two-hybrid screen with Gal4DBD (Gal4 DNA binding domain [DBD])-CBF1 as the bait protein. The SKIP sequence is identical to that described in accession no. U51432 and one base different from that of NcoA-62 (accession no. AF045184). Proteins were expressed in yeast as Gal4ACT (Gal4 activation domain [ACT]) fusions in the vector pACTII or as DBD fusions in the vector pAS1-CYH2 (SKIP-ACT, pJH177; DBD-SKIP, pJH313). DBD-CIR (pJH491), ACT-CIR (pJH178), and ACT-mHDAC2(286-489) have been previously described (19). Bacterially expressed glutathione S-transferase (GST) fusions were generated in the pGEX2T (Promega)-derived plasmid pGH413 [GST-CBF1(1-500), pJH163; GST-SKIP(1-536), pJH286-2].
The Gal4 fusions used in the mammalian two-hybrid assays were generated in pGH250, which has a simian virus 40 promoter [Gal4-CBF1(1-500), pJH93; Gal4-SKIP(1-536), pJH274]. Gal4-mHDAC2 was obtained from W.-M. Yang (59). The SG5 vector (Stratagene) was modified to incorporate either Flag (pJH253), hemagglutinin (HA) (pHYC66), or Myc (pJH363) epitopes. These vectors were used to generate Flag-CBF1 (pJH282), Flag-SKIP (pJH281), CIR-Flag (pJH518), Myc-CBF1 (pMF1), and HA-SKIP (pJH277). SG5-SKIP-Rta (pJH511) expresses SKIP fused to the activation domain (amino acids 520 to 605) of the EBV R transactivator (Rta) (11). Flag-SMRT (pCMX-PL2-SMRT-Flag) (25) was obtained from R. Evans, and Myc-mSin3A (29) was obtained from C. Laherty and R. Eisenman. Expression vectors for wild-type EBNA2 (wtEBNA2) (pPDL151), EBNA2(WW323SR) (pPDL152), EBNA2(II307SR) (pPDL159), and EBNA2(PI326SR) (pPDL196) have been described elsewhere (33), as have the reporter plasmids 5xGal4TK (thymidine kinase)-CAT (chloramphenicol acetyltransferase), TK-Luciferase, and 4xCp-CAT (17, 36).
Yeast assays.
The yeast two-hybrid screen and yeast assays for SKIP interactions were performed in Saccharomyces cerevisiae Y190 as previously described (19). β-Galactosidase activity was measured from three independent cotransformants using 2-nitrophenyl β-d-galactopyranoside as the substrate. The amount of 2-nitrophenol liberated after 2 to 4 h of incubation was measured by absorbance at 420 nm.
CAT assays.
HeLa cells were maintained in Dulbecco modified Eagle medium plus 10% fetal calf serum and plated at 1.2 × 105 cells per well in six-well plates (Nunc) 1 day prior to transfection. Cells were transfected by the calcium phosphate procedure and received 0.8 μg of 5xGal4TK-CAT or 4xCp-CAT reporter, 0.5 μg of Gal4 vector or Gal4 fusion plasmid, 0.5 μg of EBNA2 effector plasmid, and 1 μg of TK-Luciferase as an internal control for transfection efficiency. The total DNA was kept constant for each sample by using vector plasmid. Each experiment was repeated at least two times. CAT and luciferase assays were performed as previously described (17).
Immunofluorescence assays.
EBNA2, Flag-HDAC, and HA-SKIP plasmids (0.8 μg of each) were transfected by the calcium phosphate procedure into Vero cells seeded in two-well LabTek slides (Nunc) at 0.8 × 105 cells per well and grown in Dulbecco modified Eagle medium plus 10% fetal calf serum. Two days after transfection, cells were washed and fixed in 1% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature. Fixed cells were washed and permeabilized in 0.2% Triton X-100 in PBS for 20 min on ice. After washing, the cells were incubated with primary antibodies for 1 h at 37°C. Mouse anti-EBNA2 monoclonal antibody (1:200) was obtained from Dako Corp., and rabbit anti-SKIP antibody (1:500) was generated using the peptide Y-H-G-G-S-K-R-P-S-D-S-S-R-P-K-E-S-C as the immunogen. The first amino acid (Y) and the last two amino acids (S-C) were added for stability and ease of conjugation with carrier protein. Secondary antibodies, fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit antibody (1:100) and rhodamine-conjugated goat anti-mouse antibody (1:100) (Chemicon), were incubated for 0.5 h at 37°C. The slides were washed and mounted with Mowiol solution (Calbiochem), and the images were captured using a Leitz fluorescence microscope and Image Pro software (Media Cybernetic, MD).
Immunoprecipitation and Western blotting.
293T cells seeded at 106 per 10-cm-diameter culture dish were transfected with 8 μg of expression plasmid by the calcium phosphate method. Two days after transfection, the cells were washed and lysed in 2.5 ml of ice-cold lysis buffer (0.1% sodium dodecyl sulfate [SDS], 1% deoxycholic acid, 0.5% NP-40, 0.2 mM phenylmethylsulfonyl fluoride, and 2 μg of aprotinin per ml in PBS). The cell suspension was passed five times through a 20-gauge syringe needle, and the extract was clarified by centrifugation for 10 min at 15,000 rpm. Anti-Flag or anti-Myc mouse monoclonal antibody, (Sigma), anti-EBNA2 monoclonal antibody (Dako), and anti-CBF1 or anti-SKIP rabbit polyclonal antibody were mixed with protein A-Sepharose 4B (20 μl; Pharmacia) in 60 μl of lysis buffer and incubated at 4°C for 2 h. The beads were blocked with 3% skim milk in lysis buffer for 15 min and washed three times in lysis buffer. One milliliter of cell extract was added to the beads and incubated for 2 h at 4°C. The beads were then washed six times with lysis buffer and mixed with 35 μl of sample buffer. Samples (5 to 25 μl) were subjected to electrophoresis using a 9% denaturing polyacrylamide gel. The amount of sample loaded in the control lanes (direct immunoprecipitate) was one-quarter of the amount used for the coimmunoprecipitated sample. Western blot analysis was performed using peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G secondary antibodies and the Amersham enhanced chemiluminescence system. Rabbit anti-CBF1 polyclonal antisera were generated using the peptide Y-P-G-K-F-G-E-R-P-P-P-K-R-L-T-R-S-C as immunogen. Molecular mass standards were purchased from GibcoBRL.
GST-protein affinity assays.
293T cells were transfected in 10-cm-diameter dishes with 12 μg of each plasmid. Extracts were prepared 2 days after transfection by washing the cells with PBS followed by lysis in ice-cold lysis buffer (0.2% NP-40, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 5% glycerol, 0.2 mM phenylmethylsulfonyl fluoride, and 2 μg of aprotinin per ml in Tris-HCl [pH 7.4]). The suspension was sonicated for 15 s on ice and clarified by centrifugation for 10 min at 15,000 rpm.
Extracts from bacterial cells induced to express GST-CBF1 or GST-SKIP proteins were prepared by standard procedures. These extracts were incubated for 2 h at 4°C with 20 μl of glutathione-Sepharose 4B beads (Pharmacia). After three washes in lysis buffer, the bound GST fusion proteins were incubated for 2 h at 4°C with transfected 293T cell extract. The beads were then washed six times in lysis buffer and added to 30 μl of sample buffer. Samples were electrophoresed through SDS–9% polyacrylamide gels; the separated proteins were transferred to a nitrocellulose membrane and detected by Western blotting as described above.
RESULTS
SKIP interacts with CBF1.
SKIP was identified as a CBF1-interacting protein in a yeast-two hybrid screen. To demonstrate interaction between SKIP and CBF1 in mammalian cells, GST affinity and immunoprecipitation assays were performed using extracts of 293T cells transfected with expression vectors for epitope-tagged SKIP and CBF1. In a GST affinity assay (Fig. 1A), extract from cells transfected with Myc-CBF1 was incubated with control GST protein or with GST-SKIP, and the bound proteins were subjected to Western blot analysis using anti-Myc antibody to detect Myc-CBF1. Myc-CBF1 did not bind to the control protein GST (Fig. 1A, lane 1), but interaction was detected using GST-SKIP (Fig. 1A, lane 2). This interaction was confirmed by coimmunoprecipitation from cells cotransfected with Flag-SKIP and Myc-CBF1. Immunoprecipitated proteins were analyzed by Western blotting with anti-Flag antibody (Fig. 1B). Flag-SKIP was detected as a coprecipitating protein with Myc-CBF1 in immunoprecipitates generated with rabbit anti-Myc antiserum (lane 1) but not in immunoprecipitates using preimmune rabbit antiserum (lane 2). The identity of Flag-SKIP was confirmed by direct precipitation from the cell extract using mouse anti-Flag antibody (lane 3). Flag-SKIP was not directly precipitated by an irrelevant anti-Zta mouse monoclonal antibody (lane 4).
FIG. 1.

SKIP interaction with CBF1. (A) GST affinity assay using extract from 293T cells expressing Myc-CBF1. Bound protein was detected by Western blotting using anti-Myc antibody. Extract was incubated with control GST beads (lane 1) or GST-SKIP (lane 2). Lane 3 was loaded with 10 μl of transfected cell extract. (B) Lysate from cells cotransfected with Myc-CBF1 and Flag-SKIP was subjected to immunoprecipitation, and Western blots of the immunoprecipitated proteins were probed with anti-Flag antibody to detect Flag-SKIP. Flag-SKIP coprecipitated with Myc-CBF1 in precipitates formed with rabbit anti-Myc antibody (lane 1). Flag-SKIP was not precipitated by control preimmune rabbit antiserum (lane 2). As a positive control, Flag-SKIP was directly immunoprecipitated by anti-Flag monoclonal antibody (lane 3). Flag-SKIP was not observed in immunoprecipitates generated with an irrelevant monoclonal antibody (anti-Zta; lane 4). Fourfold more extract was used in the coprecipitation than in the direct precipitation. The vertical bar indicates the position of the immunoglobulin heavy chain.
SKIP interacts with members of the CBF1 corepressor complex.
To better understand SKIP function, we sought to determine whether SKIP was a component of the CBF1 corepressor complex. We had previously isolated a novel member of the CBF1 corepressor complex, CIR, which is involved in interactions with SAP30 and HDAC (19). Interaction between SKIP and CIR was examined in coimmunoprecipitation and yeast two-hybrid assays. Immunoprecipitation assays were performed on extracts of 293T cells that had been transfected with CIR-Flag and SKIP expression vectors. Immunoprecipitated proteins were analyzed by Western blotting using anti-Flag antibody (Fig. 2A). CIR-Flag coimmunoprecipitated with SKIP in immunoprecipitates obtained using anti-SKIP rabbit antibody (Fig. 2A, lane 1). Direct immunoprecipitation of CIR-Flag by anti-Flag antibody is shown in lane 2. The SKIP-CIR interaction was also demonstrable in a yeast two-hybrid assay in which yeast cells were cotransformed with Gal4DBD or Gal4ACT fusion proteins, and interaction between the fusion proteins was measured by induction of β-galactosidase enzyme activity (Fig. 2B). The DBD-SKIP and ACT-empty vector combination (lane 1) formed the negative control and β-galactosidase induction in yeast cotransformed with the known interactors, EBNA2 and CBF1, is shown in lane 2. β-Galactosidase activity was induced in yeast cotransformed with CIR and SKIP fusion vectors with SKIP as the ACT fusion partner (lane 3) or as the DBD fusion partner (lane 4).
FIG. 2.

SKIP interacts with the CBF1 corepressor proteins CIR and HDAC. SKIP-corepressor interactions were analyzed using coimmunoprecipitation, yeast two-hybrid, and mammalian two-hybrid assays. (A) Coimmunoprecipitation assay using extracts of 293T cells cotransfected with CIR-Flag and SKIP. Western blots of the immunoprecipitated proteins were probed with anti-Flag antibody to detect CIR-Flag. Incubation with rabbit anti-SKIP antibody coprecipitated CIR-Flag (lane 1). CIR-Flag was directly precipitated by mouse anti-Flag antibody (lane 2). The amount of extract used in the direct precipitation was one-fourth of that used in the coprecipitation. Lane 3 was loaded with 10 μl of transfected cell extract. (B) SKIP interacts with CIR and HDAC in a yeast two-hybrid assay in which interaction is measured by induction of β-galactosidase activity. Yeast cells were cotransformed with Gal4DBD-SKIP plus Gal4ACT vector (negative control; lane 1), Gal4DBD-CBF1 plus EBNA2(252-425) (positive control; lane 2), Gal4DBD-CIR plus Gal4ACT-SKIP (lane 3), Gal4DBD-SKIP plus Gal4ACT-CIR (lane 4), or Gal4DBD-SKIP plus Gal4ACT-HDAC2 (lane 5). The results shown are an average of three experiments with the standard deviation indicated. (C) Mammalian two-hybrid assay in which Gal4-HDAC2 is targeted to a 5xGal4TK-CAT reporter and the ability of a SKIP activation domain fusion, SKIP-Rta, to activate reporter expression is used as a measure of SKIP-HDAC interaction. HeLa cells were cotransfected with 5xGal4TK-CAT reporter alone or with Gal4-HDAC plus increasing amounts (0, 0.5, 1.0, and 1.5 μg) of SKIP-Rta. For comparison, the 5xGal4TK-CAT reporter was cotransfected with Gal4-CBF1, which represses reporter expression, and Gal4-CBF1 plus EBNA2, which activates the reporter through tethering to CBF1.
HDAC is a key member of the corepressor complex. Interaction between SKIP and HDAC2 was sought in yeast and mammalian two-hybrid assays. As shown in Fig. 2B (lane 5), SKIP-HDAC2 interaction could also be detected in the yeast assay. The mammalian two-hybrid assay was performed in HeLa cells that were cotransfected with a 5xGal4TK-CAT reporter, TK-Luciferase control, Gal4-HDAC, and increasing amounts of a plasmid expressing SKIP-Rta, a chimeric protein in which SKIP is fused to the transcriptional activation domain of the EBV Rta lytic transactivator. SKIP-Rta activated CAT expression, indicating that there was interaction between SKIP and the promoter-bound Gal4-HDAC2 to bring the Rta activation domain to the promoter (Fig. 2C). SKIP-Rta had no effect when cotransfected with a vector expressing only the Gal4 fusion partner (data not shown). Gal4-CBF1 and Gal4-CBF1 plus EBNA2 were included for comparison (Fig. 2C). Gal4-CBF1 represses 5xGal4TK-CAT reporter expression, and EBNA2 interaction with promoter-bound Gal4-CBF1 activates reporter expression.
Sin3A and SMRT are also constituents of HDAC-associated corepressor complexes (12, 13, 29, 37). SMRT has been demonstrated to be a component of the CBF1-associated corepressor complex (25), but the presence of Sin3A in this complex has not previously been addressed. We tested whether interactions could be demonstrated between mSin3A and CBF1, mSin3A and SKIP, and SMRT and SKIP in GST affinity and immunoprecipitation assays. In a GST affinity assay (Fig. 3A), GST, GST-CBF1, and GST-SKIP were incubated with extract from 293T cells transfected with Myc-mSin3A, and the bound proteins were analyzed on a Western blot probed with anti-Myc antibody. Myc-mSin3A did not interact with GST protein (lane 1). However, Myc-mSin3A bound both GST-CBF1 (lane 2) and GST-SKIP (lane 3). Cell extract is shown in lane 4, and Myc-mSin3A directly precipitated from the extract with anti-Myc antibody is shown in lane 5. Evidence was also obtained for interaction between SMRT and SKIP. Flag-SMRT coimmunoprecipitated with SKIP in assays performed on extracts of 293T cells cotransfected with SKIP and Flag-SMRT (Fig. 3B). Immunoprecipitated proteins were analyzed on a Western blot probed with anti-Flag antibody. Flag-SMRT was not precipitated by preimmune rabbit antibody (lane 1) but was directly precipitated by mouse anti-Flag antibody (lane 3) and also coprecipitated with SKIP in immunoprecipitates formed with anti-SKIP rabbit antibody (lane 4).
FIG. 3.

SKIP also interacts with the corepressor proteins Sin3A and SMRT. (A) GST affinity assay in which an extract of Myc-mSin3A-transfected 293T cells was applied to GST-SKIP, GST-CBF1, or control GST beads and a Western blot of the bound proteins was probed with anti-Myc monoclonal antibody. Myc-mSin3A did not bind to GST alone (lane 1) but bound to both GST-CBF1 (lane 2) and GST-SKIP (lane 3). Transfected cell extract (10 μl) (lane 4) and Myc-mSin3A directly immunoprecipitated with anti-Myc monoclonal antibody (lane 5) served as positive controls. (B) Coimmunoprecipitation of Flag-SMRT and SKIP from extracts of cotransfected 293T cells. Flag-SMRT was detected on a Western blot using mouse anti-Flag antibody. Lane 1, precipitation with by preimmune rabbit antibody; lane 2, transfected cell extract (10 μl); lane 3, direct precipitation of Flag-SMRT with anti-Flag antibody; lane 4, coprecipitation of Flag-SMRT with SKIP from extracts incubated with anti-SKIP rabbit antibody. The amount of extract used in the direct precipitation was one-fourth of that used in the coprecipitation.
Taken together, these protein-protein interaction assays indicate that SKIP is associated with the CBF1 corepressor complex. Some of the interactions observed are relatively weak, and it is likely that SKIP makes direct contacts with only a subset of corepressor proteins and that other members of the corepressor complex are contacted indirectly.
SKIP colocalizes with EBNA2 in transfected cells.
We next sought to determine whether SKIP had any role in EBNA2 activation of CBF1-repressed promoters. First, we compared the physical distribution of SKIP and EBNA2 within the cell. In both transfected and EBV-infected cells, EBNA2 is detected in indirect immunofluorescence assays as characteristic punctate spots within the nucleus. In cotransfected Vero cells, SKIP colocalized with EBNA2, and the colocalization was particularly dramatic when the cells were triply transfected with EBNA2, SKIP, and HDAC, as illustrated in Fig. 4. In this assay, EBNA2 was detected with an anti-EBNA2 monoclonal antibody and a rhodamine-conjugated secondary antibody, while SKIP was detected with an anti-SKIP rabbit antibody and FITC-conjugated secondary antibody. The merged image further substantiates colocalization of EBNA2 and SKIP.
FIG. 4.
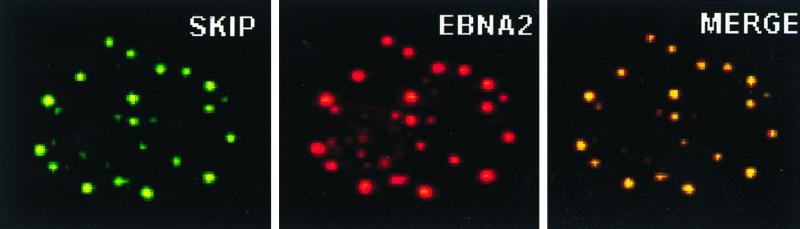
Intranuclear colocalization of SKIP and EBNA2 in the presence of HDAC. Immunofluorescence assay in Vero cells cotransfected with EBNA2, HA-SKIP, and HDAC shows that EBNA2 (red) and SKIP (green) each gives a punctate staining pattern that colocalizes in the merged image (yellow). Primary antibodies were anti-EBNA2 mouse antibody and rabbit anti-SKIP antibody. Secondary antibodies were FITC-conjugated donkey anti-rabbit (green) and rhodamine-conjugated goat anti-mouse (red).
EBNA2 interacts with SKIP and CBF1.
EBNA2 is known to interact with CBF1 (9, 14, 16, 54, 63). A coimmunoprecipitation assay using extracts from 293T cells cotransfected with plasmids expressing EBNA2 and Flag-CBF1 is shown to illustrate this point (Fig. 5A). The Western blot of the precipitated proteins was probed with an anti-EBNA2 monoclonal antibody. EBNA2 coprecipitated with Flag-CBF1 in the anti-Flag immunoprecipitate (lane 1). Direct precipitation of EBNA2 by the anti-EBNA2 monoclonal antibody is shown in lanes 2 and 6. Cell extract was loaded in lanes 3 and 7. The specificity of the immunoprecipitation was confirmed by the absence of EBNA2 in precipitates generated with preimmune rabbit antiserum (lane 4) or irrelevant mouse monoclonal antibody (anti-CD23 [Dako]; lane 5). A GST affinity assay using extracts of 293T cells transfected with an EBNA2 expression plasmid was performed to evaluate SKIP-EBNA2 interaction (Fig. 5B). Bound protein was detected by Western blot analysis using an anti-EBNA2 monoclonal antibody. EBNA2 did not bind to GST protein (lane 1). However, binding of EBNA2 to GST-SKIP was detected (lane 2).
FIG. 5.

EBNA2 interacts with SKIP in addition to CBF1. (A) Immunoprecipitation assay using extracts from 293T cells transfected with EBNA2 plus Flag-CBF1 to show interaction between EBNA2 and CBF1. EBNA2 was detected by Western blot analysis using anti-EBNA2 mouse monoclonal antibody. Lane 1, rabbit anti-CBF1 antibody-coprecipitated EBNA2; lanes 2 and 6, direct immunoprecipitation of EBNA2 by anti-EBNA2 mouse monoclonal antibody; lanes 3 and 7, transfected cell extract (10 μl); lane 4, precipitation with preimmune rabbit antiserum; lane 5, precipitation with irrelevant mouse monoclonal antibody (anti-CD23). The amount of extract used in the direct precipitation was one-fourth of that used in the coprecipitation. (B) GST affinity assay in which extracts from 293T cells transfected with EBNA2 were incubated with GST (lane 1) or GST-SKIP (lane 2). Transfected cell extract (10 μl) was loaded in lane 3.
EBNA2 interaction with both CBF1 and SKIP is blocked by SMRT.
The interactions between SKIP and members of the CBF1 corepressor complex suggested that SKIP was a constituent of the corepressor complex. On the other hand, interaction between SKIP and EBNA2 implies that SKIP is also present in the CBF1-EBNA2 transcriptional activation complex. To better understand the contribution of SKIP, we compared the effects of cotransfection of SKIP versus cotransfection of SMRT on EBNA2 activation mediated by CBF1. HeLa cells were cotransfected with a 5xGal4TK-CAT reporter, TK-Luciferase control, and Gal4-CBF1 alone or in the presence of EBNA2 (Fig. 6A). As expected, Gal4-CBF1 repressed expression from 5xGal4TK-CAT and addition of EBNA2 led to reporter activation. EBNA2 was unable to activate the reporter in the presence of Gal4 alone, indicating that promoter targeting through the CBF1 partner in Gal4-CBF1 was required for activation (not shown). Addition of increasing amounts of SKIP had a mild stimulatory effect on EBNA2 activation. In marked contrast, cotransfection of increasing amounts of SMRT completely abolished EBNA2 activation (Fig. 6A). This result is consistent with competition between SMRT and EBNA2 for binding to CBF1.
FIG. 6.

SMRT competes for EBNA2 binding to both CBF1 and SKIP. In mammalian two-hybrid assays, SKIP facilitates EBNA2 interaction with Gal4-CBF1 while SMRT interferes with the ability of EBNA2 to bind to (A) Gal4-CBF1 or (B) Gal4-SKIP and activate expression from a 5xGal4TK-CAT reporter. (A) HeLa cells were cotransfected with 5xGal4TK-CAT reporter, TK-Luciferase control, Gal4-CBF1 alone or in the presence of EBNA2, and increasing amounts of either SKIP or SMRT (0.1, 0.5, and 2 μg) as indicated. SKIP facilitated the EBNA2-CBF1 interaction, while SMRT abolished activation of the reporter by EBNA2. (B) HeLa cells were cotransfected with 5xGal4TK-CAT reporter, TK-Luciferase control, Gal4-SKIP alone or in the presence of EBNA2, and increasing amounts of SMRT (0.1, 0.5, and 2 μg) as indicated. SMRT also abolished reporter activation by SKIP-tethered EBNA2.
We next asked whether SMRT affected interactions between EBNA2 and SKIP. A similar assay was performed using the Gal4TK-CAT reporter, Gal4-SKIP, and EBNA2 transfected in the presence or absence of increasing amounts of an SMRT expression plasmid (Fig. 6B). Cotransfection of Gal4-SKIP repressed expression from the 5xGal4TK-CAT reporter, and addition of EBNA2 led to reporter activation, consistent with interaction between EBNA2 and the DNA-tethered Gal4-SKIP. Again, addition of SMRT abolished EBNA2 activation of the 5xGal4TK-CAT reporter. Thus, SMRT competes with EBNA2 for binding to both CBF1 and SKIP.
The EBNA2 domain that interacts with SKIP is distinct from the CBF1 interaction domain.
The observation that SMRT competed with EBNA2 for binding to CBF1 as well as to SKIP raised the possibility that the apparent SKIP-EBNA2 interaction might be an indirect interaction mediated through CBF1; i.e., EBNA2 interacts with CBF1 and CBF1 interacts with SKIP, but EBNA2 does not contact SKIP itself. To distinguish a SKIP-EBNA2 interaction from a SKIP-CBF1-EBNA2 interaction, mammalian two-hybrid assays were performed using a CR6 EBNA2 mutant, E2(WW323SR), which has previously been shown to have lost the ability to interact with CBF1 (16, 33, 35). HeLa cells were cotransfected with the 5xGal4TK-CAT reporter and Gal4-CBF1 or Gal4-SKIP alone or in the presence of wtEBNA2 or the CR6 mutant E2(WW323SR) (Fig. 7). As previously shown, both Gal4-CBF1 and Gal4-SKIP repressed expression of 5xGal4TK-CAT, while addition of wtEBNA2 activated reporter expression through tethering of EBNA2 to the DNA-bound Gal4-CBF1 and Gal4-SKIP. The non-CBF1-interacting mutant E2(WW323SR) did not alter the level of 5xGal4TK-CAT expression seen in the presence of Gal4-CBF1. However, this mutant EBNA2 was as effective as wtEBNA2 in activating the Gal4-SKIP bound reporter (Fig. 7). The behavior of the E2(WW323SR) mutant indicates that the region of EBNA2 that interacts with SKIP is distinct from the region that interacts with CBF1 and hence that EBNA2 interacts with SKIP independently of its interaction with CBF1.
FIG. 7.

EBNA2 interaction with SKIP is not mediated by the CBF1 interaction domain. Mammalian two-hybrid assay shows that the EBNA2(WW323SR) mutant that does not interact with Gal4-CBF1 retains interaction with Gal4-SKIP. HeLa cells were transfected with a 5xGal4TK-CAT reporter, TK-Luciferase control, Gal4-CBF1 or Gal4-SKIP, and wtEBNA2 (wtE2) or mutant EBNA2 [E2(WW323SR)]. The amount of transfected DNA was equalized using vector DNA. Both Gal4-CBF1 and Gal4-SKIP repress CAT reporter expression. Activation of expression by EBNA2 is indicative of interaction between EBNA2 and Gal4-CBF1 or Gal4-SKIP. E2(WW323SR) activates expression in the presence of Gal4-SKIP but not in the presence of Gal4-CBF1.
Interaction with SKIP is necessary for efficient EBNA2 activation of a CBF1-repressed promoter.
We had previously observed that mutation of CR5 of EBNA2 did not prevent binding of EBNA2 to CBF1 but did affect EBNA2 function (33). The effect of the CR5 mutation II307SR on transactivation of a Cp reporter (4xCp-CAT) is illustrated in Fig. 8A. HeLa cells were transfected with the 4xCp-CAT reporter alone or in the presence of wtEBNA2 or the E2(II307SR) mutant, and CAT activity was assayed in extracts harvested 2 days after transfection. 4xCp-CAT was efficiently activated by wtEBNA2, but E2(II307SR) was an ineffective activator. E2(II307SR) is expressed in transfected cells at levels similar to those for wtEBNA2 (33). The ability of E2(II307SR) to bind to SKIP was assessed in a mammalian two-hybrid assay (Fig. 8B). HeLa cells were transfected with the 5xGal4TK-CAT reporter, Gal4-SKIP, and either E2(II307SR) or a second CR6 mutant, E2(PI326SR) (33). Gal4-SKIP repressed expression from the 5xGal4TK-CAT reporter. Addition of the CR6 mutant E2(PI326SR) led to activation of CAT expression, indicating interaction between E2(PI326SR) and the reporter bound Gal4-SKIP. However, cotransfection of an expression plasmid for the CR5 mutant E2(II307SR) had no activating effect, and Gal4-SKIP continued to repress expression from 5xGal4TK-CAT. This result is consistent with mutation of EBNA2 CR5 leading to loss of interaction with SKIP. The relative locations of the CBF1 and SKIP interaction domains and of the mutations introduced into CR5 and CR6 are summarized in Fig. 9A. The combined results also indicate that efficient activation of a CBF1-repressed promoter by EBNA2 requires that EBNA2 contact not only CBF1 but also SKIP.
FIG. 8.

SKIP interaction is necessary for effective EBNA2 targeting of promoter-bound CBF1. Transient expression and mammalian two-hybrid assays linking the inability of EBNA2(II307SR) to efficiently activate 4xCp-CAT with an inability to interact with SKIP. (A) CAT assay performed using extracts of HeLa cells transfected with a 4xCp-CAT reporter alone or in the presence of either wtEBNA2 (wtE2) or the EBNA2 mutant E2(II307SR). (B) Mammalian two-hybrid assay performed in HeLa cells transfected with a 5xGal4TK-CAT reporter, Gal4-SKIP as indicated, and either the CR5 EBNA2 mutant E2(II307SR) or the CR6 mutant E2(PI326SR). Gal4-SKIP represses expression from 5xGal4TK-CAT. This repression is overcome by the E2(PI326SR) mutant but not by the E2(II307SR) mutant, indicating that mutation in CR5 ablates interaction of EBNA2 with SKIP, whereas a CR6 mutant retains SKIP interaction.
FIG. 9.

(A) Schematic representation of EBNA2 illustrating the relative locations of characterized functional domains and of the mutants used in this study. The amino acid numbers are indicated. CR5, CR6 (33), and a nuclear localization signal (NLS) (36) are indicated. (B) Model for EBNA2 activation of CBF1-repressed promoters. CBF1 binds to the DNA sequence GTGGGAA in responsive promoters. SKIP is bound to CBF1. SMRT contacts both SKIP and CBF1. SMRT is a component of a corepressor complex that includes Sin3A, SAP30, CIR, HDAC1, and HDAC2 (19, 25), and potentially other Sin3-associated proteins (indicated by x). This complex mediates promoter repression through chromatin remodeling. EBNA2 competes with SMRT for contacts on both SKIP and CBF1. Displacement of the SMRT-corepressor complex relieves repression, and introduction of the EBNA2 activation domain induces transcriptional activation. EBNA2 mutants in CR5 and CR6 lose the ability to interact with SKIP and CBF1, respectively. Loss of either interaction impairs the ability of EBNA2 to activate CBF1-repressed promoters.
The observations that (i) SKIP is associated with the CBF1 corepressor complex, (ii) there is competition between SMRT and EBNA2 for contacts on both CBF1 and SKIP, and (iii) EBNA2 contacts with SKIP are important for functional activation lead to the model of EBNA2 activation of CBF1-repressed promoters presented in Fig. 9B.
DISCUSSION
DNA-bound CBF1 acts as a transcriptional repressor. A recurring theme in transcriptional repression is the recruitment of an HDAC-containing corepressor complex that is tethered to the promoter through contacts with a DNA binding protein. Deacetylation of lysine residues at the N terminus of the core histones is believed to strengthen histone binding to DNA, with the resulting changes in chromatin structure limiting access of the transcriptional machinery to the promoter (2). CBF1 associates with such a corepressor complex. Interactions with the corepressor proteins SMRT, HDAC1, HDAC2, SAP30, and CIR have been described (19, 25), and in the present study we were also able to demonstrate interaction between CBF1 and Sin3A. The biological significance of the corepressor interactions was originally substantiated in experiments demonstrating that a CBF1 mutant, CBF1(EEF233AAA), that had lost repression activity was also unable to interact with either SMRT or CIR (19, 25).
We have now identified SKIP as a CBF1-interacting protein and presented evidence for the presence of SKIP in the CBF1 repression complex by demonstrating interactions between SKIP and other members of the complex, namely, SMRT, Sin3A, CIR, and HDAC2. A yeast two-hybrid screen previously identified SKIP as a Ski-interacting protein (5). c-Ski binds both Sin3A and N-CoR, a corepressor protein related to SMRT, and has recently been shown to be a component of the thyroid hormone receptor corepressor complex and the Mad corepressor complex (38). These observations reinforce the point that the CBF1 corepressor complex contains many of the same protein constituents as the Mad and nuclear hormone receptor corepressor complexes.
DNA-bound CBF1 is converted from a transcriptional repressor to an activator in two known circumstances: in the presence of EBNA2 and in the presence of the activated form of cellular Notch, NotchIC (16, 17). The identification of SKIP as a CBF1-interacting protein provides additional insight into the way in which EBNA2 brings about this conversion. SKIP appears to serve as a tethering point both for the SMRT corepressor complex and for EBNA2. In contrast to SMRT, SKIP does not compete EBNA2 from CBF1 but rather appears to strengthen the CBF1-EBNA2 interaction. Further, the interaction of SKIP with SMRT is mutually exclusive of the SKIP-EBNA2 interaction. SMRT competes EBNA2 off SKIP just as it competes with EBNA2 for binding to CBF1. Thus, a model evolves in which the conversion from transcriptional repression to activation involves both CBF1 and SKIP exchanging partners from the SMRT-corepressor complex to the EBNA2 transactivation complex. This model is also compatible with the previous observation that EBNA2 had two separable effects on CBF1 repressed promoters, relief of repression and activation. An EBNA2 mutant deleted for the transcriptional activation domain remained capable of relieving CBF1-mediated repression (16). This relief of repression can now be correlated with displacement of the SMRT-corepressor complex from SKIP and CBF1 by EBNA2.
EBNA2 amino acids 307 and 308 form part of a 14-amino-acid motif, CR5, that is highly conserved between the EBNA2 proteins of EBV and baboon herpesvirus papio (36). Mutation of CR5 had previously been shown to impair EBNA2 transactivation function in transient expression assays (33), and deletion of the entire motif from EBNA2 resulted in an EBV variant that was unable to transform B cells in an in vitro outgrowth assay (10). While it was recognized that CR5 was important for EBNA2 function, the role played by the CR5 motif was unclear. We have now correlated the inability of the CR5 mutant E2(II307) to efficiently activate reporters containing CBF1 binding sites with an inability of this mutant to bind to SKIP. The adjacent conserved region in EBNA2, CR6, contains two tryptophan residues. Mutation of these residues abolishes EBNA2 interaction with CBF1 along with EBNA2 biological function (35, 38). Since the E2(II307) mutant continues to interact effectively with CBF1 (33), the behavior of the CR5 mutant substantiates a model in which contacts with both CBF1 and SKIP are needed for effective displacement of the corepressor complex by EBNA2.
A significant contribution of EBNA2 to immortalization is attributable to EBNA2-mediated activation of CBF1-repressed cell genes. Activation of genes required for progression into G1 may be particularly relevant (26, 43). The ability of EBNA2 to bind to SKIP and the presence of Ski, and therefore presumably SKIP, in the Mad and nuclear hormone receptor complexes also raises the potential of alternative ways in which EBNA2 might affect cellular gene expression. Sequestering of SKIP by EBNA2 might affect the functioning of transcription complexes in which Ski and SKIP normally participate, and there is also the possibility that EBNA2 might, in some circumstances, be capable of targeting to promoters through non-CBF1 complexes containing SKIP.
ACKNOWLEDGMENTS
We are grateful to R. Evans, C. Laherty, and W.-M. Yang for gifts of SMRT, mSin3A, and HDAC2 plasmids. We thank M. Chiu for technical assistance and F. Chang for help with manuscript preparation.
This work was supported by National Institutes of Health grant RO1 CA42245 to S.D.H.
REFERENCES
- 1.Artavanis-Tsakonas S, Rand M D, Lake R J. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 2.Ayer D E. Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999;9:193–198. doi: 10.1016/s0962-8924(99)01536-6. [DOI] [PubMed] [Google Scholar]
- 3.Bain M, Watson R J, Farrell P J, Allday M J. Epstein-Barr virus nuclear antigen 3C is a powerful repressor of transcription when tethered to DNA. J Virol. 1996;70:2481–2489. doi: 10.1128/jvi.70.4.2481-2489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen J I, Kieff E. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J Virol. 1991;65:5880–5885. doi: 10.1128/jvi.65.11.5880-5885.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahl R, Wani B, Hayman M J. The Ski oncoprotein interacts with Skip, the human homolog of Drosophila Bx42. Oncogene. 1998;16:1579–1586. doi: 10.1038/sj.onc.1201687. [DOI] [PubMed] [Google Scholar]
- 6.Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T, Vales L D. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol Cell Biol. 1994;14:3310–3319. doi: 10.1128/mcb.14.5.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuentes-Panana E M, Ling P D. Characterization of the CBF2 binding site within the Epstein-Barr virus latency C promoter and its role in modulating EBNA2-mediated transactivation. J Virol. 1998;72:693–700. doi: 10.1128/jvi.72.1.693-700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuentes-Panana E M, Swaminathan S, Ling P D. Transcriptional activation signals found in the Epstein-Barr virus (EBV) latency C promoter are conserved in the latency C promoter sequences from baboon and rhesus monkey EBV-like lymphocryptoviruses (cercopithicine herpesviruses 12 and 15) J Virol. 1999;73:826–833. doi: 10.1128/jvi.73.1.826-833.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grossman S R, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the Jk recombination signal binding protein. Proc Natl Acad Sci USA. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harada S, Yalamanchili R, Kieff E. Residues 231 to 280 of the Epstein-Barr virus nuclear protein 2 are not essential for primary B-lymphocyte growth transformation. J Virol. 1998;72:9948–9954. doi: 10.1128/jvi.72.12.9948-9954.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardwick J M, Tse L, Applegren N, Nicholas J, Veliuona M A. The Epstein-Barr virus R transactivator (Rta) contains a complex, potent activation domain with properties different from those of VP16. J Virol. 1992;66:5500–5508. doi: 10.1128/jvi.66.9.5500-5508.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hassig C A, Fleischer T C, Billin A N, Schreiber S L, Ayer D E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- 13.Heinzel T, Lavinsky R M, Mullen T M, Soderstrom M, Laherty C D, Torchia J, Yang W M, Brard G, Ngo S D, Davie J R, Seto E, Eisenman R N, Rose D W, Glass C K, Rosenfeld M G. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 14.Henkel T, Ling P D, Hayward S D, Peterson M G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein Jk. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- 15.Hofelmayr H, Strobl L J, Stein C, Laux G, Marschall G, Bornkamm G W, Zimber-Strobl U. Activated mouse Notch1 transactivates Epstein-Barr virus nuclear antigen 2-regulated viral promoters. J Virol. 1999;73:2770–2780. doi: 10.1128/jvi.73.4.2770-2780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh J J-D, Hayward S D. Masking of the CBF1/RBPJk transcriptional repression domain by Epstein-Barr virus EBNA2. Science. 1995;268:560–563. doi: 10.1126/science.7725102. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh J J-D, Henkel T, Salmon P, Robey E, Peterson M G, Hayward S D. Truncated mammalian Notch1 activates CBF1/RBPJκ-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh J J-D, Nofziger D E, Weinmaster G, Hayward S D. Epstein-Barr virus immortalization: Notch2 interacts with CBF1 and blocks differentiation. J Virol. 1997;71:1938–1945. doi: 10.1128/jvi.71.3.1938-1945.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsieh J J-D, Zhou S, Chen L, Young D B, Hayward S D. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc Natl Acad Sci USA. 1999;96:23–28. doi: 10.1073/pnas.96.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarriault S, Brou C, Logeat F, Schroeter E H, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 21.Jin X W, Speck S H. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J Virol. 1992;66:2846–2852. doi: 10.1128/jvi.66.5.2846-2852.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman S R. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by Jκ and PU.1. J Virol. 1995;69:253–262. doi: 10.1128/jvi.69.1.253-262.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johannsen E, Miller C L, Grossman S R, Kieff E. EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJκ in Epstein-Barr virus-transformed B lymphocytes. J Virol. 1996;70:4179–4183. doi: 10.1128/jvi.70.6.4179-4183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanda K, Kempkes B, Bornkamm G W, von Gabain A, Decker T. The Epstein-Barr virus nuclear antigen 2 (EBNA2), a protein required for B lymphocyte immortalization, induces the synthesis of type I interferon in Burkitt's lymphoma cell lines. Biol Chem. 1999;380:213–221. doi: 10.1515/BC.1999.029. [DOI] [PubMed] [Google Scholar]
- 25.Kao H-Y, Ordentlich P, Koyano-Nakagawa N, Tang Z, Downes M, Kintner C R, Evans R M, Kadesch T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998;12:2269–2277. doi: 10.1101/gad.12.15.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart J W, Kremmer E, Delecluse H-J, Rottenberger C, Bornkamm G W, Hammerschmidt W. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 1995;14:88–96. doi: 10.1002/j.1460-2075.1995.tb06978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kieff E. Epstein-Barr virus and its replication. In: Field B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 2. New York, N.Y: Raven Press; 1996. pp. 2343–2396. [Google Scholar]
- 28.Krauer K G, Belzer D K, Liaskou D, Buck M, Cross S, Honjo T, Sculley T. Regulation of interleukin-1 beta transcription by Epstein-Barr virus involves a number of latent proteins via their interaction with RBP. Virology. 1998;252:418–430. doi: 10.1006/viro.1998.9441. [DOI] [PubMed] [Google Scholar]
- 29.Laherty C D, Yang W M, Sun J M, Davie J R, Seto E, Eisenman R N. Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 30.Laux G, Adam B, Strobl L J, Moreau-Gachelin F. The Spi-1/PU.1 and Spi-B ets family transcription factors and the recombination signal binding protein RBP-J kappa interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J. 1994;13:5624–5632. doi: 10.1002/j.1460-2075.1994.tb06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laux G, Dugrillon F, Eckert C, Adam B, Zimber S U, Bornkamm G W. Identification and characterization of an Epstein-Barr virus nuclear antigen 2-responsive cis element in the bidirectional promoter region of latent membrane protein and terminal protein 2 genes. J Virol. 1994;68:6947–6958. doi: 10.1128/jvi.68.11.6947-6958.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lecourtois M, Schweisguth F. Indirect evidence for Delta-dependent intracellular processing of Notch in Drosophila embryos. Curr Biol. 1998;8:771–774. doi: 10.1016/s0960-9822(98)70300-8. [DOI] [PubMed] [Google Scholar]
- 33.Ling P D, Hayward S D. Contribution of conserved amino acids in mediating interaction between EBNA2 and CBF1/RBPJκ. J Virol. 1995;69:1944–1950. doi: 10.1128/jvi.69.3.1944-1950.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling P D, Hsieh J J-D, Ruf I K, Rawlins D R, Hayward S D. EBNA-2 upregulation of Epstein-Barr virus latency promoters and the cellular CD23 promoter utilizes a common targeting intermediate, CBF1. J Virol. 1994;68:5375–5383. doi: 10.1128/jvi.68.9.5375-5383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ling P D, Rawlins D R, Hayward S D. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc Natl Acad Sci USA. 1993;90:9237–9241. doi: 10.1073/pnas.90.20.9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ling P D, Ryon J J, Hayward S D. EBNA-2 of herpesvirus papio diverges significantly from the type A and type B EBNA-2 proteins of Epstein-Barr virus but retains an efficient transactivation domain with a conserved hydrophobic motif. J Virol. 1993;67:2990–3003. doi: 10.1128/jvi.67.6.2990-3003.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagy L, Kao H Y, Chakravarti D, Lin R J, Hassig C A, Ayer D E, Schreiber S L, Evans R M. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 38.Nomura T, Khan M M, Kaul S C, Dong H-D, Wadhwa R, Colmenares C, Kohno I, Ishii S. Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 1999;13:412–423. doi: 10.1101/gad.13.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivailler P, Quink C, Wang F. Strong selective pressure for evolution of an Epstein-Barr virus LMP2B homologue in the rhesus lymphocryptovirus. J Virol. 1999;73:8867–8872. doi: 10.1128/jvi.73.10.8867-8872.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson K D, Hayward S D, Ling P D, Samid D, Ambinder R F. Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol Cell Biol. 1995;15:6150–6159. doi: 10.1128/mcb.15.11.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakai T, Taniguchi Y, Tamura K, Minoguchi S, Fukuhara T, Strobl L J, Zimber-Strobl U, Bornkamm G W, Honjo T. Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2. J Virol. 1998;72:6034–6039. doi: 10.1128/jvi.72.7.6034-6039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroeter E H, Kisslinger J A, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 43.Sinclair A J, Palmero I, Peters G, Farrell P J. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J. 1994;13:3321–3328. doi: 10.1002/j.1460-2075.1994.tb06634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sjoblom A, Yang W, Palmqvist L, Jansson A, Rymo L. An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J Virol. 1998;72:1365–1376. doi: 10.1128/jvi.72.2.1365-1376.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- 46.Sung N S, Kenney S, Gutsch D, Pagano J S. EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J Virol. 1991;65:2164–2169. doi: 10.1128/jvi.65.5.2164-2169.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jk/Su(H) Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 48.Tong X, Drapkin R, Reinberg D, Kieff E. The 62- and 80-kDa subunits of transcription factor II H mediate the interaction with Epstein-Barr virus nuclear protein 2. Proc Natl Acad Sci USA. 1995;92:3259–3263. doi: 10.1073/pnas.92.8.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong X, Drapkin R, Yalamanchili R, Mosialos G, Kieff E. The Epstein-Barr virus nuclear protein 2 acid domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol Cell Biol. 1995;15:4735–4744. doi: 10.1128/mcb.15.9.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tong X, Wang F, Thut C J, Kieff E. The Epstein-Barr virus nuclear protein 2 acidic domain can interact with TFIIB, TAF40, and RPA70 but not with TATA-binding protein. J Virol. 1995;69:585–588. doi: 10.1128/jvi.69.1.585-588.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsang S-F, Wang F, Izumi K M, Kieff E. Delineation of the cis-acting element mediating EBNA-2 transactivation of latent infection membrane protein expression. J Virol. 1991;65:6765–6771. doi: 10.1128/jvi.65.12.6765-6771.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994;22:965–971. doi: 10.1093/nar/22.6.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waltzer L, Bourillot P Y, Sergeant A, Manet E. RBP-J-kappa repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transcription factor EBNA2. Nucleic Acids Res. 1995;23:4939–4945. doi: 10.1093/nar/23.24.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. The human J kappa recombination signal sequence binding protein (RBP-J kappa) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J. 1994;13:5633–5638. doi: 10.1002/j.1460-2075.1994.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waltzer L, Perricaudet M, Sergeant A, Manet E. Epstein-Barr virus EBNA3A and EBNA3C proteins both repress RBP-Jκ–EBNA2-activated transcription by inhibiting the binding of RBP-Jκ to DNA. J Virol. 1996;70:5909–5915. doi: 10.1128/jvi.70.9.5909-5915.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang F, Kikutani H, Tsang S, Kishimoto T, Kieff E. Epstein-Barr virus nuclear protein 2 transactivates a cis-acting CD23 DNA element. J Virol. 1991;65:4101–4106. doi: 10.1128/jvi.65.8.4101-4106.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu D Y, Kalpana G V, Goff S P, Schubach W H. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J Virol. 1996;70:6020–6028. doi: 10.1128/jvi.70.9.6020-6028.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yalamanchili R, Tong X, Grossman S, Johannsen E, Mosialos G, Kieff E. Genetic and biochemical evidence that EBNA 2 interaction with a 63-kDa cellular GTG-binding protein is essential for B lymphocyte growth transformation by EBV. Virology. 1994;204:634–641. doi: 10.1006/viro.1994.1578. [DOI] [PubMed] [Google Scholar]
- 59.Yang W-M, Yao Y-L, Sun J-M, Davie J R, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 60.Yoo L I, Mooney M, Puglielli M T, Speck S H. B-cell lines immortalized with an Epstein-Barr virus mutant lacking the Cp EBNA2 enhancer are biased toward utilization of the oriP-proximal EBNA gene promoter Wp1. J Virol. 1997;71:9134–9142. doi: 10.1128/jvi.71.12.9134-9142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao B, Marshall D R, Sample C E. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of Jκ. J Virol. 1996;70:4228–4236. doi: 10.1128/jvi.70.7.4228-4236.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimber-Strobl U, Kremmer E, Grasser F, Marschall G, Laux G, Bornkamm G W. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. 1993;12:167–175. doi: 10.1002/j.1460-2075.1993.tb05642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimber-Strobl U, Strobl L J, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm G W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994;13:4973–4982. doi: 10.1002/j.1460-2075.1994.tb06824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]