

Abstract
RNA polymerases (RNAPs) accomplish the first step of gene expression in all living organisms. However, the sequence divergence between bacterial and human RNAPs makes the bacterial RNAP a promising target for antibiotic development. The most clinically important and extensively studied class of antibiotics known to inhibit bacterial RNAP are the rifamycins. For example, rifamycins are a vital element of the current combination therapy for treatment of tuberculosis. Here, we provide an overview of the history of the discovery of rifamycins, their mechanisms of action, the mechanisms of bacterial resistance against them, and progress in their further development.
INTRODUCTION
While a handful of natural and synthetic compounds are known to target bacterial RNA polymerases (RNAPs), only two of them have managed to make it into clinical use: rifamycins and lipiarmycin (1). The most clinically important and extensively studied class of antibiotics known to inhibit bacterial RNA polymerase is the rifamycins (RIFs). RIFs are a vital element of the current combination therapy for treatment of tuberculosis (TB), an infection caused by Mycobacterium tuberculosis (2). The mechanism of RNAP inhibition by RIFs is believed to be identical for various bacterial RNAPs, and most of the studies have been performed on Escherichia coli RNAP as a common model system (3).
HISTORY OF RIFAMYCINS
Ansamycins are a group of naturally derived antibiotics which are named after their characteristic basket-like structures, where an aliphatic ansa-chain (ansa = handle in latin) spans an aromatic naphthalenic (e.g., RIF) or benzenic moiety (e.g., geldanamycin) from its nonadjacent positions (4). RIFs, the first antibiotics found to inhibit bacterial RNAPs, were originally isolated from Amycolatopsis mediterranei in 1959 (5–7). Rifamycin B (RIF B) was the first of a kind that was stable enough to be purified from a mixture of RIFs (8). Although RIF B has poor antimicrobial activity (likely due to its inability to penetrate the bacterial cell envelope), it is reversibly converted to RIF O in aqueous oxygenated solutions. RIF O is then hydrolyzed to RIF S, losing a glycolic acid residue. The reduction of RIF S results in RIF SV (Fig. 1) (9, 10). It is believed that the observed activity of RIF B is due at least in part to the transformation into active products (RIF O, S, and SV) (11).
Figure 1.
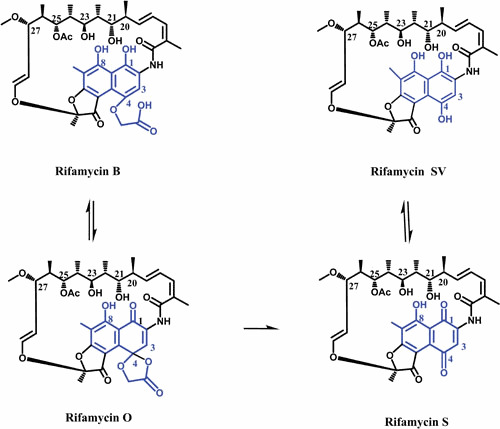
The chemical pathway for conversion of RIF B to RIF SV. The ansa chain and naphthalene moiety of molecules are shown in black and blue, respectively.
RIF SV was the first ansamycin that found clinical application. RIF SV has potent activity, especially against a spectrum of Gram-positive bacteria and a clinically significant pathogen, M. tuberculosis. However, RIF SV has the limitation of oral administration due to low gastrointestinal absorbance (12). Therefore, comprehensive semisynthetic RIF programs aimed at finding a compound with enhanced oral absorption, prolonged existence in blood, and better activity against both Gram-positive and Gram-negative bacteria. Rifampicin (RMP) was the result of preparation and screening of several hundreds of semisynthetic RIFs (13). Since the introduction of RMP, it has remained the first-line treatment for mycobacterial infections, including TB.
RIFAMYCINS IN CLINIC
TB is the ninth leading cause of death and the main cause of death by a single infectious agent worldwide. In 2018, 10 million people fell ill with TB, and about 1.2 million (HIV-negative patients) died because of TB. The current treatment for TB is 6-month combination therapy with RIF, isoniazid, ethambutol, and pyrazinamide. Drug-resistant TB is a serious threat, exacerbated by the fact that as recently as 2018, approximately half a million new cases of RMP-resistant (RMPr) TB were recorded, 78% of which were multidrug-resistant TB (MDR-TB; resistance to RMP and isoniazid). The RMPr-TB and MDR-TB require longer treatments, which typically last for 20 months. The role of RIFs as the main first-line anti-TB drug is so critical that the WHO recommended the Xpert MTB/RIF assay for simultaneous detection of TB and RMP resistance (14). RMP is also one of the relatively few anti-TB drugs with sterilizing activity, the capacity to kill the mycobacteria that remain after the initial phase of treatment (e.g., the population of bacteria that undergo sporadic metabolism) (15). The sterilizing activity of RIFs enables further shortening of the treatment if higher doses are tolerated. Higher doses of RMP (up to 35 mg/kg) are in trials to reduce the treatment period for drug-susceptible TB (14). Five different RIFs are currently marketed in various countries: RMP, rifapentine (RPT), and rifabutin (RBT) for the systemic treatment of mycobacterial infections, rifaximin (RXM) only for travelers’ diarrhea), and RIF SV (with limited availability) (16).
MECHANISM OF ACTION OF RIFs
The antibacterial activity of RIFs is due to inhibition of DNA-dependent RNA synthesis (17). This inhibition is a result of strong binding of RIFs to prokaryotic RNAP, ranking them as the most potent inhibitors of bacterial RNAP (the 50% effective concentration [EC50RMP] for E. coli RNAP is ∼20 nM). The binding constant of RMP for eukaryotic RNAP is at least 100 times higher than its binding constant for prokaryotic enzymes. Inhibition of RNAP is the common mechanism of action (MOA) among all structurally related RIFs with antibacterial activity (15, 18, 19). The antimicrobial activity differences of RIFs in Gram-positive and Gram-negative bacteria are not related to their binding site on RNAP but are due to other factors like efflux pumps in E. coli (20). The binding of RIFs to RNAP was initially verified by mapping almost all RMPr mutations to the rpoB gene encoding the β subunit of RNAP (21).
Studies on the MOA of RIFs revealed that they inhibit RNA synthesis at the very early stages of transcription (15, 22, 23) and that RMP is no longer active when RNA synthesis progresses beyond an early stage (24). It was also determined that RMP binds to core E. coli RNAP and does not need the σ-subunit for its binding (25). McClure and Cech discovered that RMP inhibition induces the release of dinucleotide from E. coli RNAP if transcription is started with nucleoside triphosphate, whereas trinucleotide is released from transcription complexes started with smaller nucleoside mono- or diphosphate. Based on this observation, they proposed that RIFs sterically block the extension of nascent RNA at transcription initiation (26). The well-studied RMP-E. coli RNAP model was used as a prototype for analyzing the RMP binding to other bacterial RNAPs, mostly through investigation of the RMP-resistant mutants (3, 27–30). Resolving the crystal structure of RMP bound to RNAP was a breakthrough in the investigation of RIF’s MOA. According to the structure, the RIF-binding pocket is located in the β subunit of Thermus aquaticus RNAP within the DNA/RNA binding channel in 12-Å proximity to the Mg2+ ion at the active site (3), which is consistent with previous biochemical observations with E. coli RNAP (31, 32). Based on the crystal structure, binding of RMP to RNAP blocks the formation of the second or third phosphodiester bond (Fig. 2) (3).
Figure 2.

Mechanisms of action of different RIFs. A RIF (with or without groups at C-3/C-4 or KglA) bound at the RIF-binding pocket either sterically blocks progression of the growing RNA chain, resulting in abortive synthesis (left), or inhibits the first phosphodiester bond formation by interfering with initiating NTP or with σ region 3.2 that stabilizes the template DNA.
The steric model alone did not explain the differences observed in RIFs. For example, a semisynthetic RIF, rifalazil (RLZ), and RMP are not completely cross-resistant, or RLZ and RBT can develop different resistant mutations compared to RMP (33). Based on the crystal structures of Thermus thermophilus RNAP holoenzymes in complex with RBT and RPT, which lacked catalytic Mg2+ from the active center, Artsimovitch et al. proposed an allosteric inhibition model in which instead of or in addition to the steric model, RIFs allosterically reduce the affinity of catalytic Mg2+ ion to the RNAP active center (19, 34).
Later biochemical and structural studies contradicted the allosteric model of RIF inhibition. Feklistov et al. showed that RIFs do not affect the affinity of Mg2+ to RNAP, and a high concentration of Mg2+ does not confer resistance to RIFs (35), as was proposed for the allosteric mechanism. The crystal structures of the E. coli RNAP σ70 holoenzyme in complex with RMP as well as two RIF derivatives which similarly to RBT inhibit the first phosphodiester bond displayed the binding of RIFs to a RIF-binding pocket without inducing the loss of Mg2+ from the active center (36). Accordingly, the authors suggested that a potential interaction of RIF “tails” with a loop-like domain of σ subunit, σ region 3.2, is responsible for inhibiting the formation of the first phosphodiester bond by some RIF derivatives (36). The σ region 3.2 stretches toward the RNAP active center and plays an important role in transcription initiation via direct interaction with template DNA and stabilizes the initiating nucleoside triphosphates (NTPs) in the RNAP active site (37). Therefore, disengagement of the σ region 3.2 from its position by RIFs may reposition the template DNA relative to the active site and impair the binding of, first, NTP, which consequently may inhibit the formation of the first phosphodiester bond (36–39).
Recently, crystal structures of M. tuberculosis RNAP containing variable synthetic RNA oligomers (2-, 3-, and 4-RNA nucleotide RNA) in complex with RMP demonstrated that it does not expel the catalytic Mg2+ from RNAP (40). The recent studies of kanglemycin A (KglA), a rifamycin molecule with no C-3/C-4 side groups, showed that it does not displace the Mg2+ from the RNAP active center while inhibiting the first phosphodiester bond via a steric and/or electrostatic clash of the additional acid moiety of the ansa bridge with γ and/or β phosphates of the initiating NTP (Fig. 2) (41, 42). Overall, the discrepancy in the MOA of different RIFs could be explained by direct interference of large groups of some RIF molecules (e.g., RBT) with the initiating dinucleotide or other proximal residues such as the σ region 3.2 (rather than allosterically with Mg2+ of the active center), which also would appear as inhibition of synthesis of the first phosphodiester bond (Fig. 2). It should be mentioned that some discrepancies in interpretation of biochemical results may also be caused by the usage of different initiation substrates in different studies, in particular, NTP versus NpN dinucleotide primers (lacking triphosphate moiety at their 5′ end), which have different sizes and charge distributions, which, in turn, may strongly affect their interactions with various RIFs.
The sensitivity of different bacterial RNAPs to RIFs is influenced not only by the binding region (RIF-pocket) but also by other regions which indirectly change the conformation of the binding site (43). The M. tuberculosis and Mycobacterium avium RNAPs have been shown to be more sensitive than E. coli RNAP to RMP (44, 45). As the RIF-binding pocket is highly conserved among all bacteria, such differences are thought to be due to structural differences in the nonconserved regions surrounding the RIF-binding pocket of RNAPs. For instance, a chimeric E. coli RNA polymerase carrying β-subunit regions I and II of M. tuberculosis enzyme was as sensitive as wild-type E. coli enzyme in response to RMP, suggesting that regions outside the RIF pocket can determine sensitivity to RIFs (43). Similarly, T. aquaticus RNAP is about 100-fold less sensitive to RMP compared to E. coli RNAP (42, 46), while amino acids of T. aquaticus RNAP that directly interact with RIF are identical to those in E. coli RNAP. Therefore, the difference is also thought to be due to changes in the regions outside the RIF-binding pocket (3, 47).
STRUCTURE-ACTIVITY RELATIONSHIP OF RIFs
Almost all RIFs form four critical hydrogen bonds with RNAP residues through oxygens at C-1 and C-8 on the naphthalenic ring and C-21 and C-23 hydroxyl groups. A particular spatial arrangement of the above four oxygens is important for the binding. Oxygens at C-1 and C-8 make hydrogen bonds with RNAP residues S531/Q513 and R529, respectively, while hydroxyl oxygen at C-21 makes hydrogen or van der Waals interactions with three residues of RNAP: H526, D516, and F514 (E. coli numbering is used here and throughout the paper). The hydroxyl oxygen at C-23 together with carbonyl oxygen of C-25 make hydrogen bonds to F514 (3, 48). In KglA, because of different orientation of the ansa chain compared with other RIFs, the hydroxyl group at C-23 does not form a hydrogen bond with βF514. Instead, the unique sugar side chain of KglA at C-27 makes contact with βR143 (Fig. 3) (41). Moreover, hydrophobic interactions between RIFs and RNAP residues E565, I572, G534, L533, L511, and Q510 contribute to the binding of RMP (3).
Figure 3.
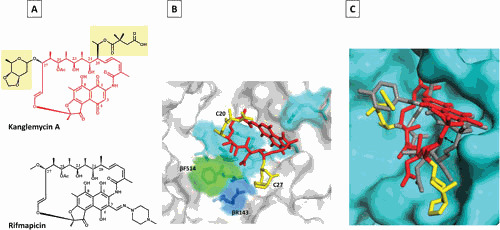
Mode of KglA binding to RNAP compared with RMP. (A) Chemical structures of KglA (in red with C-20 and C-27 side chains highlighted in yellow) and RMP (black). (B) A close-up view of KglA in the RIF-binding pocket of T. thermophilus RNAP (PDB: 6CUU). KglA is shown as a stick model (red) with its deoxysugar and succinate groups shown in yellow. RNAP is shown as a transparent surface model (gray), and RNAP β residues, which form the RIF-binding pocket, are shown as stick models. KglA binds to the same residues that RMP binds (cyan) to, with the exception of βF514 (green). KglA makes additional binding with βR143 (blue). (C) A side view of KglA in the RIF-binding pocket shown in panel B (PDB: 1YNN and 6CUU). The RNAP β subunit is shown in cyan. KglA (red and yellow) is overlaid on RMP (gray). Compared with RMP, KglA maintains a larger distance from the RIF-binding pocket (depicted by the two-headed arrow) (41).
MODIFICATIONS OF THE RIF ANSA BRIDGE
Early studies showed that even minor modifications of the ansa bridge, such as acetylation of C-21 or C-23 alcohols, led to activity loss of RIFs (15). Other examples of unsuccessful modifications in the ansa bridge include the epoxy side chain derivatives (e.g., 18,19-epoxy RIF S), RIF SV analogues with a keto group at C-21 instead of hydroxyl and an extra hydroxyl at C-20 (49), derivatives with a cyclic 3-14 linkage to the nitrogen of the macrocyclic ring, and RIFs with open rings, such as RIF W (50). Very few derivatives of RIFs with ansa bridge modifications have been shown to retain their activity due to only minor conformational changes of the ansa bridge. The hydroxylated C-25 derivative of RIF is one of the modifications of the side chain which is tolerated (49, 51). The 24-desmethyl rifampicin has also been claimed to have the same or enhanced activity against some bacteria compared to RMP (52). The considerable potential of RIF ansa bridge modifications to improve their activity was revealed after the discovery of the KglA MOA (41, 42). KglA, a rifamycin molecule with unusual ansa bridge modifications (C-27 β-O-3,4-O,O′-methylene digitoxose and C-20 2,2-dimethyl succinic acid) is more effective against RMPr RNAPs and bacteria compared with the commonly used RMP. KglA binds to the RIF-binding pocket, but the unique ansa bridge groups of KglA establish additional contacts with the RIF-binding pocket and influence the overall binding mode of KglA in the pocket (Fig. 3) (41). The KglA scaffold provides a rationale for the further development of new RIFs with activity against the RMPr bacteria.
MODIFICATIONS OF THE NAPHTHAQUINONE RING
Modifications at Position C-4 of RIFs
Generally, modifications in the naphthalene ring of RIFs are well tolerated. The first observation indicating the possibility of modifications in the naphthalene ring was the equal RNAP activity of RIF S, RIF B, and RIF SV (15, 53). RIF B, a potent inhibitor of bacterial RNAP, shows poor penetration through the bacterial cell wall due to free carboxylic acid at C-4. Structure-activity relationship studies noted that RIFs with free carboxyl groups at C-3 or C-4 have reduced ability to penetrate the cell wall and therefore possess reduced activity against bacteria (54). The first successful modifications at C-4 were the amide and hydrazide analogues of Rif B with enhanced in vitro and in vivo activity (55). The first RIF B derivative in clinical use was a diethylamide analogue (rifamide), though it was replaced by RMP because of its poor pharmacokinetics (13, 56).
Modifications at Position C-3 of RIFs
Most of the initial successful structure-activity relationship studies concentrated on C-3 modifications of RIFs. A range of substitutions at the C-3 position of RIF S or SV have been shown to maintain their activity against bacteria (57–59). Dampier and Whitlock showed that electronegative groups at C-3 enhance the activity of RIFs, while the electron-donating groups at C-3 reduce the activity against RNAP (60). Early works showed that almost all changes in RIFs, including the naphthaquinone ring modifications, which change the conformation of the ansa bridge, lead to loss of activity (58, 61–63). Although RIF O retains its inhibitory activity against RNAP despite conformational changes in the ansa chain it is suggested that the in vitro activity of RIF O could be at least partly due to the hydrolyzed product, RIF S (64, 65). RMP was the result of many hundreds of imine, hydrazide, and oxime derivatives of 3-formyl RIF SV (Fig. 4). Due to its potency against M. tuberculosis and improved pharmacokinetic properties, RMP has remained the mainstay in TB therapy since its introduction in 1968 (13). After successful synthesis of RMP, many structure-activity relationship studies moved toward the synthesis of RMP analogues with enhanced half-life to permit intermittent dosing. These studies resulted in the synthesis of many C-3 substituted RMPs (66). RPT, which has a cyclopentyl group instead of a terminal methyl group at C-3, was the best derivative (Fig. 4). Not only is RPT as active as RMP against M. tuberculosis, but its longer half-life enables twice-weekly dosing for TB patients (67). However, RPT is completely cross-resistant with RMP (68, 69). RPT is currently used in combination with isoniazid to treat latent M. tuberculosis infections (70).
Figure 4.
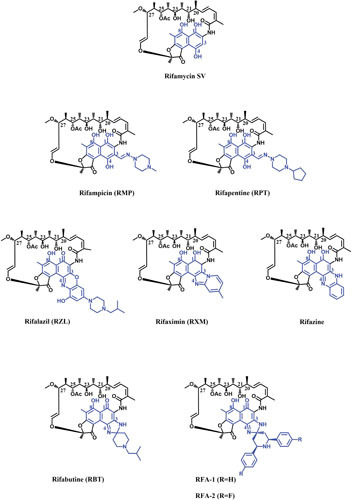
Chemical structures of different RIFs. The ansa chain and naphthalene moiety of the molecules are shown in black and blue, respectively.
Modifications of C-3/C-4 in RIFs
A variety of RIF analogues with an additional ring linking C-3 and C-4 have been synthesized. The antibacterial activity of different C-3/C-4 analogues of RIFs can differ by several orders of magnitude, and therefore, most of the recent attempts have been focused on making such derivatives of RIFs (71). Rifazine was one of the first rigidified benzannulated analogues of RIF which retained its activity against RNAP (Fig. 4). Rifazine has considerably higher potency against Staphylococcus aureus compared with RIF SV (49, 72). Rifaximin (RXM) was another RIF analogue with an additional ring (Fig. 4) which had similar activity as RMP in cell culture, though its activity against Staphylococcus epidermidis RNAP was about half of RMP activity (73). RXM has very low oral absorption due to the two oppositely charged nitrogens in addition to phenolic hydroxyls, which is beneficial for treatment of gastrointestinal infections where a high fecal concentration of drug is required (74, 75). RXM is approved in many countries to treat uncomplicated traveler’s diarrhea (E. coli-born irritable bowel syndrome with diarrhea) (75). Although resistant mutations for RXM are selected at a lower rate compared with RMP, cross-resistance between the two drugs is inevitable (76, 77).
A milestone in the optimization of RIF derivatives was the synthesis of spiro-piperidyl RIF analogues, out of which RBT was the most promising (Fig. 4) (78). The crystal structure of T. thermophilus RNAP holoenzyme in complex with RBT showed that, similar to RMP, the ansa moiety of RBT makes contact with β residues of RNAP, suggesting the same MOA as RMP. It is proposed that the C-3/C-4 tail of RBT makes contact with the σ-subunit of RNAP, which enables RBT to inhibit the elongation of RNA at earlier stages of transcription compared to RMP. However, there is no strong biochemical evidence to support that binding to the σ-subunit is responsible for this variation (19, 35). RBT is more potent against RMP-susceptible M. tuberculosis than RMP (20), but most of the RMPr M. tuberculosis strains are also resistant to RBT, albeit with lower MICs than those for RMP (79). The in vitro activity of RBT against RMPr RNAPs also confirmed its partial cross-resistance with RMP (20). Further studies of large numbers of clinically isolated RMPr strains indicated the potential of RBT for treatment of patients diagnosed with specific RMPr-TB (80, 81). One of the main drawbacks of RIFs is the interaction with human pregnane X receptor (hPXR), which induces the hepatic cytochrome P450 (Cyp450) and other proteins involved in xenobiotic metabolism. This is of great importance in patients coinfected with HIV and M. tuberculosis, as certain antiviral drugs are metabolized by Cyp450. Because of the low drug-drug interactions of RBT, especially with antiviral drugs, and the high level of in vivo toxicity, it is used as an alternative therapy against several mycobacterial infections, including M. avium-intracellulare complex (MAC) in patients with AIDS, and for treatment of MDR-TB (82, 83). More recently, synthesis of RBT analogues resulted in two promising candidates, RFA-1 and RFA-2 (Fig. 4), which display MIC values up to 100 times lower than that of RMP and 20 times lower than that of RBT against MDR-TB (84, 85). Preliminary molecular modelling calculations on an RMPr M. tuberculosis RNAP showed increased interaction energy between the RFA-1 compared to RBT and RMP. Therefore, the enhanced antimicrobial activity of the two derivatives against RMPr strains derives from their tighter binding to RNAP as a result of additional enzyme-ligand contacts (85).
Another breakthrough in RIF analogues was the synthesis of 3′-hydroxy-5′-aminobenzoxazinorifamycin derivatives. The benzoxazinorifamycins (bxRIFs) showed enhanced in vivo activity against slowly growing mycobacteria, including M. tuberculosis and MAC compared to RMP, and provided better absorption from the gastrointestinal tract. Among such derivatives, KRM-1648 (rifalazil [RLZ]) was the most promising analogue due to its excellent potency against M. tuberculosis in addition to the improved pharmacokinetic characteristics (Fig. 4) (86). RLZ strongly inhibits a spectrum of Gram-positive bacteria with MICs lower than or similar to those for RMP. Although the frequency of spontaneous mutation for RLZ is almost same as for RMP (87), it has somewhat enhanced activity against some of the isolates which are resistant to RMP and RBT (36, 88–90). Fujii et al. showed that RLZ similarly inhibits RNAPs of different bacterial origins in vitro and concluded that its improved antibacterial activity depends on cell wall permeability of the target bacteria (45). Despite many exceptional characteristics of RLZ, including high potency (up to 250-fold more potent than RMP) (91), its activity against some RMPr strains (88), high volume of distribution and tissue level (92), and lack of hepatic Cyp450 induction (93), RLZ development was suspended due to high toxicity observed in phases I and II of clinical trials (94).
In a study aimed at the synthesis of novel bxRIFs, Gill et al. showed three novel RLZ derivatives which have improved binding affinity to wild-type and rifamycin-resistant (RIFr) RNAPs of M. tuberculosis (94). It is suggested that bxRIFs are capable of making additional contacts with RNAP (possibly with σ region 3.2 and other regions of the RNAP complex) (36). However, all analogues exhibited high human pregnane X receptor (hPXR) activation and cytotoxicity and had low antitubercular activity in cell culture (94).
MECHANISM OF RESISTANCE TO RIFs
Shortly after the discovery of RIFs, the occurrence of resistant mutations creating a high level of resistance was observed in the laboratory and in infected patients (95, 96). Bacteria develop resistance to RIFs at frequencies of 10–10 to 10–7 depending on the organism, methodology, and type of RIF molecule (16, 97). Because of the rapid emergence of resistant isolates, RMP is used in drug combinations almost exclusively for treatment of TB (4).
Mutations Affecting the β Subunit
Almost all mutations conferring resistance to RIFs map to the rpoB gene (which encodes the β subunit of RNAP) in M. tuberculosis (29, 98), E. coli (22, 99), and other microorganisms examined (100–102) (Fig. 5). Although E. coli is not the main therapeutic target for most RIFs, well-studied RNAP of this organism in addition to the homology of its RNAP to the M. tuberculosis RNAP makes it a good model for genetic and physiological studies. RIFr spontaneous point mutations, mostly as single amino acid changes, occur within four regions of rpoB (known as the RIF resistance-determining region, RRDR): N-terminal cluster (cluster N, amino acids 146 to 148), cluster I (amino acids 504 to 538), cluster II (amino acids 562 to 575), and cluster III (amino acids 684 to 691) (Fig. 5) (103, 104). The vast majority of such mutations (∼95%) are within the 81-base pair region of cluster I, which is highly conserved among bacterial RNAPs but different from archaeal or eukaryotic RNAPs (98, 105). According to crystal structure of the T. aquaticus RNAP bound to RMP, almost all residues that directly interact with RMP are encoded by cluster I. Almost all residues involved in direct interaction with RMP are susceptible to mutations resulting in RMP resistance (Fig. 5) (3).
Figure 5.
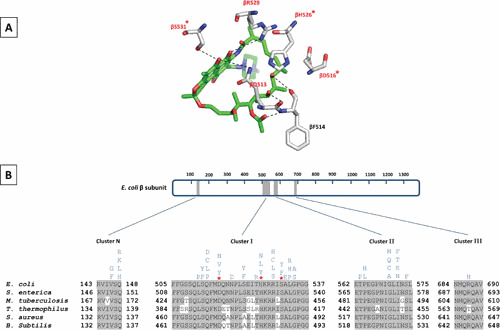
Rifamycin resistance-determining regions of different bacterial RNAPs. (A) Schematic representation of RMP in the stick model (green) bound to residues of E. coli RIF-binding pocket (gray stick model; PDBID: 5UAC) (48). The hydrogen bonds between RMP and residues are shown as dashed lines. Amino acid residues that are mutated in clinical RIFr isolates are highlighted in red. The three residues which are most frequently mutated to confer RIFr clinical isolates of M. tuberculosis are marked by an asterisk. (B) The schematic on top represents the primary sequence of the E. coli β subunit. The amino acid numbering is depicted. Gray boxes represent the four clusters (RMP resistance-determining regions; RRDRs) where RIFr mutations occur. A sequence alignment showing these clusters in E. coli, S. enterica, M. tuberculosis, T. thermophilus, S. aureus, and B. subtilis is depicted below the schematic bar. Amino acids that are identical to E. coli are highlighted in gray. Mutations that confer RIFr in E. coli are indicated above the sequence.
While point mutations have been found in 33 codons of RRDR, only three mutations, S531L, H526Y, and D516V, account for around 41%, 36%, and 9%, respectively, of all clinically isolated RIFr-TB strains (98). The three most frequent RMPr mutations have direct effect on interactions with oxygens at C-8 and C-21 of RMP (Fig. 5) (3). Depending on the point mutations, the mechanism of resistance to RIFs varies. For example, the βS531L does not impose a significant structural or functional impact on RNAP in the absence of RMP. The βS531L disorders the RIF-binding pocket upon RMP binding and therefore reduces the binding affinity for RMP. The collision between the leucine at β531 and the β subunit fork loop 2 (a conserved loop of the active center which plays a role in DNA strand separation) is suggested to be responsible for disordering the RIF-binding pocket. The H526Y mutation significantly changes the RIF-binding pocket and therefore causes a significant steric conflict for binding of RMP to RNAP. As a result, mutations at βH526 lead to very high levels of resistance to RMP. The less frequent mutation, βD516V, reduces the affinity of RIF binding only by changing the electrostatic surface of the RIF-binding pocket (48, 106). Other RIF-resistant rpoB mutations which do not make direct interactions with RIF are thought to act by changing the conformation of the RNAP.
Many studies focused on binding surfaces away from the RIF-binding pocket to target the RIFr bacterial RNAPs (40, 107). However, the RIF-binding pocket is still a promising target for the development of new RIFs with activity against RIFr pathogens. For example, the additional contacts of KglA with the RIF-binding pocket induces conformational changes of KglA which, compared with RMP, positions the drug slightly away from the RIF-binding pocket (Fig. 3). The increased distance between KglA and RNAP enables it to maintain its inhibitory activity against the most frequent RIF-resistant bacterial RNAP (βS531L). Due to additional contacts and slightly different modes of binding to RNAP, it was suggested that only an unlikely event of two concurrent mutations within the RIF-binding pocket can produce resistance to KglA (41). However, such mutations impose a great fitness cost and may compromise the transcriptional integrity and therefore make the pathogen nonviable (108).
Other Mechanisms of Resistance to RIFs
Some bacteria, such as certain soil-dwelling actinomycetes, are intrinsically resistant to RIFs. An example of such intrinsic resistance is the presence of asparagine at consensus codon 531 in comparison to serine in susceptible bacteria (109). Various RNAP-independent mechanisms have been described for RMP resistance, though mostly in clinically insignificant organisms. For example, the removal of RIFs by efflux pumps has been shown in different strains, including M. tuberculosis (110, 111). In Nocardia farcinica, an RMP monooxygenase (ROX) is recognized as a secondary mechanism of RMPr which, by adding an oxygen to the C3 side chain of RMP, converts it to a compound with lower antimicrobial activity (112). Furthermore, in certain species of mycobacteria, including the emerging pathogen Mycobacterium abscessus, the high level of resistance to RMP is partly associated with the ADP-ribosyltransferase (Arr) enzyme that can modify RMP with ADP-ribosyl, a group transferred from NAD+, which results in RIF inactivation (113). Since the ribosylation of RMP by Arr occurs at position C23, the presence of bulky side chains in the vicinity of the ribosylation site is suggested to create a steric hindrance for RMP inactivation. For instance, different 3-morpholino RIFs which possess a bulky carbamate group at C25 are resistant to RIF inactivation by Arr (114). KglA and its congeners which carry a relatively large sugar side group at C-27 (41, 42) are potential candidates for developing RIFs with the ability to overcome Arr-mediated RIF inactivation.
Fitness Cost of RIFr Mutations and Fitness-Compensatory Mechanisms
Most mutations conferring antibiotic resistance come with a cost, meaning the bacterial strains carrying resistant mutations have compromised fitness compared with the ancestral strains (115–118). In the absence of antibiotics, resistant bacteria bearing reduced fitness should be outcompeted by the drug-sensitive bacteria and be eliminated from the population. However, the fitness cost of antibiotic resistance is counterbalanced by compensatory evolution. Rapidly evolving secondary mutations provide the resistant bacteria the ability to become as fit as susceptible strains and hence stabilize the resistant bacterial population in the absence of antibiotic (116).
Almost all Rif-resistant mutations impose reduced fitness. The fitness cost of RIFr mutations is generally inevitable, as the process targeted by RIFs, the transcription, is vital to all aspects of bacterial life, including the growth and virulence (117–122). The fitness cost of RIFr mutations and their corresponding prevalence in nature have a negative correlation. Therefore, only the RIFr mutations with low fitness costs are found frequently in resistant clinical isolates (48, 123). Fitness in bacteria critically depends on their surrounding environment. For example, RIFr mutants, which reduce the bacterial fitness in exponential growth, can be advantageous in the later stages of growth (e.g., aging colonies), at high temperatures, or in low-glucose environments (124, 125). The fitness costs of RIFr mutations could be ameliorated by secondary mutations within RNAP β, β′, or α subunits and occur at higher frequencies than the mutations resulting in reversion of primary RIFr mutation (Fig. 6 and Table 1) (108, 122, 126, 127). The secondary mutations are concentrated in particular structural regions of RNAP and may act by different mechanisms, including restoring the properties of the RIF-binding pocket, changing the interaction of enzyme with substrate or RNA, or affecting the interactions between different RNAP subunits (e.g., mutations in the α-β′ interface) (108, 122, 127, 128). The secondary mutations do not considerably change the susceptibility of bacteria to RIFs, with the exception of those that are known RIFr mutations (e.g., βD516G) (122).
Figure 6.
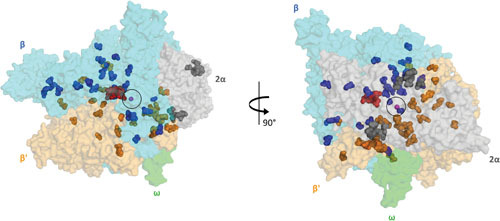
Two views of the RIF compensatory mutations from different RIFr bacteria (108, 121, 122, 128, 131) mapped onto the crystal structure of the E. coli RNAP-RMP complex (PDB:5UAC). The RNAP core enzyme is illustrated as a transparent surface (α subunits, gray; β subunit, cyan; β′ subunit, bright orange; ω subunit, gray). The active center of RNAP, marked by the presence of catalytic Mg2+ (magenta sphere) is circled. The RMP molecule is depicted as red spheres. Compensatory mutations found on the α, β, and β′ subunits of RNAP are shown as gray, blue, and orange spheres, respectively.
Table 1.
List of secondary mutations in different RIFr bacteria
| Strain | RMPr mutation | Secondary mutations in α | Secondary mutations in β | Secondary mutations in β′ | Reference |
|---|---|---|---|---|---|
| Salmonella enterica | βS531 | αA189EαT196S | βN339HβD340YβE546kβG809AβQ1264R | β′H419Pβ′A446Vβ′D622Eβ′R634Pβ′T674Iβ′I937Tβ′S942Lβ′R943Hβ′R1075Cβ′R1075Lβ′G1136A | 128 |
| Salmonella enterica | βR529 | αR191C | βD516GβP560LβP564SβE565AβR637CβH673Y | β′P64Lβ′L770Pβ′R1075Pβ′R1075Hβ′G1136β′R1140Hβ′V1198E | 122 |
| E. coli a | βD516 | βS574YβH554YβS574Y | 121 | ||
| E. coli a | βP564 | βR211PβS574FβL194R | 121 | ||
| E. coli a | βL511 | βD516G | 121 | ||
| E. coli a | βI572 | βG556G | 121 | ||
| M. tuberculosis c | βS531 | αA189VαR191WαV192GαT196A | βT1286IβQ490RβP25S | β′G257Rβ′G257Sβ′N341Sβ′P359Rβ′P359Vβ′F377Lβ′K370Rβ′V408Gβ′P420_V421insAβ′L432Vβ′L441Pβ′L452Vβ′D622Eβ′E658Gβ′E658Dβ′E658Aβ′P998Rβ′P998Sβ′A1312Vβ′N1350Sβ′A1312V | 108 |
| M. tuberculosis b | βS531 | β′D410Yβ′F377Cβ′G257Rβ′H450Qβ′V408Gβ′V408Aβ′I416Tβ′Q448E | 131 | ||
| M. tuberculosis b | βD516 | β′V408G | 131 | ||
| M. tuberculosis c | βH526 | αE29K | βE472Gβv650A | β′A919G | 108 |
| M. tuberculosis c | βQ513 | βN856SβR841H | 108 |
The rpoAC genes were not sequenced for compensatory mutations.
The rpoAB genes were not sequenced for compensatory mutations.
Only the mutated residues which are identical between E. coli and M. tuberculosis were included.
A combination of different factors determine the recurrence of RIFr mutations. In addition to low fitness cost, a high level of resistance to RIF, and acquiring secondary mutations to further reduce the fitness cost, all contribute to the success of specific RIFr mutations in clinical isolates of M. tuberculosis (129). Although the compensatory mutations play key roles in developing clinical RIFr bacteria with high fitness (118, 120, 127, 130), our knowledge of the MOA of such mutations is very limited. Investigating the MOA of compensatory mutations in different organisms could possibly shed new light on the activities of RNAP and allosteric switches within the enzyme and open new doors to potential novel approaches for tackling antibiotic resistance.
Although the importance of RIFr fitness compensatory mutations has been studied in different organisms, E. coli (121) and Salmonella are broadly used as genetically amenable model organisms to study the fitness cost in resistance to RIFs and the genetics of the corresponding compensatory evolution (122, 128, 129). The compensatory mutations found in the model organisms overlap with the mutations found in clinical isolates of M. tuberculosis, suggesting the conserved role of fitness compensatory mutations in bacteria (Table 1) (122, 127–129).
ACKNOWLEDGMENTS
This work was supported by Wellcome Trust Investigator Awards (102851/Z/13/Z and 217189/Z/19/Z), a MICA MRC grant (MR/T000740/1), and an EPSRC Programme grant (EP/T002778/1) to N.Z.
Contributor Information
Hamed Mosaei, Centre for Bacterial Cell Biology, Biosciences Institute, Newcastle University, Newcastle Upon Tyne, NE2 4AX, UK.
Nikolay Zenkin, Centre for Bacterial Cell Biology, Biosciences Institute, Newcastle University, Newcastle Upon Tyne, NE2 4AX, UK.
Susan T. Lovett, Brandeis University, Waltham, MA
Deborah Hinton, Laboratory of Cell and Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MA.
REFERENCES
- 1.Mosaei H, Harbottle J. 2019. Mechanisms of antibiotics inhibiting bacterial RNA polymerase. Biochem Soc Trans 47:339–350 10.1042/BST20180499. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2018. Global Tuberculosis Report. https://apps.who.int/iris/bitstream/handle/10665/274453/9789241565646-eng.pdf?ua=1&ua=1.
- 3.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 4.Floss HG, Yu T-W. 2005. Rifamycin-mode of action, resistance, and biosynthesis. Chem Rev 105:621–632 10.1021/cr030112j. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Sensi P, Margalith P, Timbal MT. 1959. Rifomycin, a new antibiotic; preliminary report. Farmaco Sci 14:146–147. [PubMed] [Google Scholar]
- 6.Thiemann JE, Zucco G, Pelizza G. 1969. A proposal for the transfer of Streptomyces mediterranei Margalith and Beretta 1960 to the genus Nocardia as Nocardia mediterranea (Margalith and Beretta) comb. nov. Arch Mikrobiol 67:147–155 10.1007/BF00409680. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Lechevalier MP, Prauser H, Labeda DP, Ruan J-S. 1986. Two new genera of nocardioform actinomycetes: Amycolata gen. nov. and Amycolatopsis gen. nov. Int J Syst Bacteriol 36:29–37 10.1099/00207713-36-1-29. [DOI] [Google Scholar]
- 8.Margalith P, Pagani H. 1961. Rifomycin. XIV. Production of rifomycin B. Appl Microbiol 9:325–334 10.1128/AEM.9.4.325-334.1961. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sensi P, Timbal MT, Maffii G. 1960. Rifomycin IX. Two new antibiotics of rifomycin family: rifomycin S and rifomycin SV. Preliminary report. Experientia 16:412 10.1007/BF02178838. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Greco AM, Ballotta R, Sensi P. 1961. Activation of rifomycin B and rifomycin 0. Production and properties of rifomycin S and rifomycin S. Farmaco 16:165–180. [Google Scholar]
- 11.Fueresz S, Timbal MT. 1963. Antibacterial activity of rifamycins. Chemotherapia (Basel) 257:200–208 10.1159/000220123. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Bergamini N, Fowst G. 1965. Rifamycin SV. A review. Arzneimittelforschung 15(Suppl):951–1002. [PubMed] [Google Scholar]
- 13.Sensi P. 1983. History of the development of rifampin. Rev Infect Dis 5(Suppl 3):S402–S406 10.1093/clinids/5.Supplement_3.S402. [PubMed] [DOI] [PubMed] [Google Scholar]
- 14.World Health Organization. 2019. Global tuberculosis report. https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1.
- 15.Hartmann G, Behr W, Beissner K-A, Honikel K, Sippel A. 1968. Antibiotics as inhibitors of nucleic acid and protein synthesis. Angew Chem Int Ed Engl 7:693–701 10.1002/anie.196806931. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Goldstein BP. 2014. Resistance to rifampicin: a review. J Antibiot (Tokyo) 67:625–630 10.1038/ja.2014.107. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Calvori C, Frontali L, Leoni L, Tecce G. 1965. Effect of rifamycin on protein synthesis. Nature 207:417–418 10.1038/207417a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Hartmann G, Honikel KO, Knüsel F, Nüesch J. 1967. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim Biophys Acta 145:843–844 10.1016/0005-2787(67)90147-5. [DOI] [PubMed] [Google Scholar]
- 19.Artsimovitch I, Vassylyeva MN, Svetlov D, Svetlov V, Perederina A, Igarashi N, Matsugaki N, Wakatsuki S, Tahirov TH, Vassylyev DG. 2005. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell 122:351–363 10.1016/j.cell.2005.07.014. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Gill SK, Garcia GA. 2011. Rifamycin inhibition of WT and Rif-resistant Mycobacterium tuberculosis and Escherichia coli RNA polymerases in vitro. Tuberculosis (Edinb) 91:361–369 10.1016/j.tube.2011.05.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 21.Wehrli W, Knüsel F, Schmid K, Staehelin M. 1968. Interaction of rifamycin with bacterial RNA polymerase. Proc Natl Acad Sci USA 61:667–673 10.1073/pnas.61.2.667. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezekiel DH, Hutchins JE. 1968. Mutations affecting RNA polymerase associated with rifampicin resistance in Escherichia coli. Nature 220:276–277 10.1038/220276a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Wehrli W, Knüsel F, Staehelin M. 1968. Action of rifamycin on RNA-polymerase from sensitive and resistant bacteria. Biochem Biophys Res Commun 32:284–288 10.1016/0006-291X(68)90382-3. [DOI] [PubMed] [Google Scholar]
- 24.Sippel AE, Hartmann GR. 1970. Rifampicin resistance of RNA polymerase in the binary complex with DNA. Eur J Biochem 16:152–157 10.1111/j.1432-1033.1970.tb01066.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Wehrli W, Handschin J, Wunderli W. 1976. Interaction between rifampicin and DNA-dependent RNA polymerase of E. coli, p 397–412. In Losick R, Chamberlin M (ed), RNA Polymerase. Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.McClure WR, Cech CL. 1978. On the mechanism of rifampicin inhibition of RNA synthesis. J Biol Chem 253:8949–8956. [PubMed] [Google Scholar]
- 27.Nolte O. 1997. Rifampicin resistance in Neisseria meningitidis: evidence from a study of sibling strains, description of new mutations and notes on population genetics. J Antimicrob Chemother 39:747–755 10.1093/jac/39.6.747. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Ramaswamy S, Musser JM. 1998. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis 79:3–29 10.1054/tuld.1998.0002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Yew W-W. 2015. Mechanisms of drug resistance in Mycobacterium tuberculosis: update 2015. Int J Tuberc Lung Dis 19:1276–1289 10.5588/ijtld.15.0389. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Morse R, O’Hanlon K, Virji M, Collins MD. 1999. Isolation of rifampin-resistant mutants of Listeria monocytogenes and their characterization by rpoB gene sequencing, temperature sensitivity for growth, and interaction with an epithelial cell line. J Clin Microbiol 37:2913–2919 10.1128/JCM.37.9.2913-2919.1999. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustaev A, Zaychikov E, Severinov K, Kashlev M, Polyakov A, Nikiforov V, Goldfarb A. 1994. Topology of the RNA polymerase active center probed by chimeric rifampicin-nucleotide compounds. Proc Natl Acad Sci USA 91:12036–12040 10.1073/pnas.91.25.12036. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Severinov K, Mustaev A, Severinova E, Kozlov M, Darst SA, Goldfarb A. 1995. The beta subunit Rif-cluster I is only angstroms away from the active center of Escherichia coli RNA polymerase. J Biol Chem 270:29428–29432 10.1074/jbc.270.49.29428. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Williams DL, Spring L, Collins L, Miller LP, Heifets LB, Gangadharam PR, Gillis TP. 1998. Contribution of rpoB mutations to development of rifamycin cross-resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 42:1853–1857 10.1128/AAC.42.7.1853. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Artsimovitch I, Vassylyev DG. 2006. Is it easy to stop RNA polymerase? Cell Cycle 5:399–404 10.4161/cc.5.4.2466. [PubMed] [DOI] [PubMed] [Google Scholar]
- 35.Feklistov A, Mekler V, Jiang Q, Westblade LF, Irschik H, Jansen R, Mustaev A, Darst SA, Ebright RH. 2008. Rifamycins do not function by allosteric modulation of binding of Mg2+ to the RNA polymerase active center. Proc Natl Acad Sci USA 105:14820–14825 10.1073/pnas.0802822105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molodtsov V, Nawarathne IN, Scharf NT, Kirchhoff PD, Showalter HD, Garcia GA, Murakami KS. 2013. X-ray crystal structures of the Escherichia coli RNA polymerase in complex with benzoxazinorifamycins. J Med Chem 56:4758–4763 10.1021/jm4004889. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zenkin N, Severinov K. 2004. The role of RNA polymerase σ subunit in promoter-independent initiation of transcription. Proc Natl Acad Sci USA 101:4396–4400 10.1073/pnas.0400886101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bochkareva A, Zenkin N. 2013. The σ70 region 1.2 regulates promoter escape by unwinding DNA downstream of the transcription start site. Nucleic Acids Res 41:4565–4572 10.1093/nar/gkt116. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pupov D, Kuzin I, Bass I, Kulbachinskiy A. 2014. Distinct functions of the RNA polymerase σ subunit region 3.2 in RNA priming and promoter escape. Nucleic Acids Res 42:4494–4504 10.1093/nar/gkt1384. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin W, Mandal S, Degen D, Liu Y, Ebright YW, Li S, Feng Y, Zhang Y, Mandal S, Jiang Y, Liu S, Gigliotti M, Talaue M, Connell N, Das K, Arnold E, Ebright RH. 2017. Structural basis of Mycobacterium tuberculosis transcription and transcription inhibition. Mol Cell 66:169–179.e8 10.1016/j.molcel.2017.03.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosaei H, Molodtsov V, Kepplinger B, Harbottle J, Moon CWCW, Jeeves RERE, Ceccaroni L, Shin Y, Morton-Laing S, Marrs ECL, Wills C, Clegg W, Yuzenkova Y, Perry JDJD, Bacon J, Errington J, Allenby NEE, Hall MJMJ, Murakami KSKS, Zenkin N. 2018. Mode of Action of kanglemycin A, an ansamycin natural product that is active against rifampicin-resistant Mycobacterium tuberculosis. Mol Cell 72:263–274.e5 10.1016/j.molcel.2018.08.028. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peek J, Lilic M, Montiel D, Milshteyn A, Woodworth I, Biggins JB, Ternei MA, Calle PY, Danziger M, Warrier T, Saito K, Braffman N, Fay A, Glickman MS, Darst SA, Campbell EA, Brady SF. 2018. Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat Commun 9:4147 10.1038/s41467-018-06587-2. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zenkin N, Kulbachinskiy A, Bass I, Nikiforov V. 2005. Different rifampin sensitivities of Escherichia coli and Mycobacterium tuberculosis RNA polymerases are not explained by the difference in the β-subunit rifampin regions I and II. Antimicrob Agents Chemother 49:1587–1590 10.1128/AAC.49.4.1587-1590.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harshey RM, Ramakrishnan T. 1976. Purification and properties of DNA-dependent RNA polymerase from Mycobacterium tuberculosis H37RV. Biochim Biophys Acta 432:49–59 10.1016/0005-2787(76)90040-X. [DOI] [PubMed] [Google Scholar]
- 45.Fujii K, Saito H, Tomioka H, Mae T, Hosoe K. 1995. Mechanism of action of antimycobacterial activity of the new benzoxazinorifamycin KRM-1648. Antimicrob Agents Chemother 39:1489–1492 10.1128/AAC.39.7.1489. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fábry M, Sümegi J, Venetianer P. 1976. Purification and properties of the RNA polymerase of an extremely thermophilic bacterium: Thermus aquaticus T2. Biochim Biophys Acta 435:228–235 10.1016/0005-2787(76)90104-0. [DOI] [PubMed] [Google Scholar]
- 47.Minakhin L, Nechaev S, Campbell EA, Severinov K. 2001. Recombinant Thermus aquaticus RNA polymerase, a new tool for structure-based analysis of transcription. J Bacteriol 183:71–76 10.1128/JB.183.1.71-76.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molodtsov V, Scharf NT, Stefan MA, Garcia GA, Murakami KS. 2017. Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol Microbiol 103:1034–1045 10.1111/mmi.13606. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wehrli W, Staehelin M. 1969. The rifamycins: relation of chemical structure and action on RNA polymerase. Biochim Biophys Acta 182:24–29 10.1016/0005-2787(69)90516-4. [DOI] [PubMed] [Google Scholar]
- 50.Wehrli W. 1977. Ansamycins. Chemistry, biosynthesis and biological activity. Top Curr Chem 72:21–49 10.1007/BFb0048448. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Rastogi N, Goh KS, Berchel M, Bryskier A. 2000. Activity of rifapentine and its metabolite 25-O-desacetylrifapentine compared with rifampicin and rifabutin against Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis and M. bovis BCG. J Antimicrob Chemother 46:565–570 10.1093/jac/46.4.565. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Nigam A, Almabruk KH, Saxena A, Yang J, Mukherjee U, Kaur H, Kohli P, Kumari R, Singh P, Zakharov LN, Singh Y, Mahmud T, Lal R. 2014. Modification of rifamycin polyketide backbone leads to improved drug activity against rifampicin-resistant Mycobacterium tuberculosis. J Biol Chem 289:21142–21152 10.1074/jbc.M114.572636. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilhelm JM, Oleinick NL, Corcoran JW. 1968. The inhibition of bacterial RNA synthesis by the rifamycin antibiotics. Biochim Biophys Acta 166:268–271 10.1016/0005-2787(68)90515-7. [DOI] [PubMed] [Google Scholar]
- 54.Maggi N, Furesz S, Sensi P. 1968. Rifomycins. XLIX. Influence of the carboxyl group on the antibacterial activity of rifomycins. J Med Chem 11:368–369 10.1021/jm00308a042. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Sensi P, Maggi N, Ballotta R, Fueresz S, Pallanza R, Arioli V. 1964. Rifamycins. XXXV. Amides and hydrazides of rifamycin B. J Med Chem 7:596–602 10.1021/jm00335a005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Pallanza R, Fürész S, Timbal MT, Carniti G. 1965. In vitro bacteriological studies on rifamycin B diethylamide (rifamide). Arzneimittelforschung 15:800–802. [PubMed] [Google Scholar]
- 57.Stanzani L, Venturini AP, Mantovani V. 1978. A new tolypomycin-Y derivative: in vitro and in vivo antimicrobial activity. J Antibiot (Tokyo) 31:1195–1200 10.7164/antibiotics.31.1195. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Brufani M, Cellai L, Cerrini S, Fedeli W, Segre A, Vaciago A. 1982. Structure-activity relationships in the ansamycins. Molecular structure and activity of 3-carbomethoxy rifamycin S. Mol Pharmacol 21:394–399. [PubMed] [Google Scholar]
- 59.Bellomo P, Marchi E, Mascellani G, Brufani M. 1981. Synthesis and antibacterial activity of some ester, amides, and hydrazides of 3-carboxyrifamycin S. Relationship between structure and activity of ansamycins. J Med Chem 24:1310–1314 10.1021/jm00143a010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 60.Dampier MF, Whitlock HW Jr. 1975. Letter: electronegative groups at C-3 of rifamycin S enhance its activity toward DNA-dependent RNA polymerase. J Am Chem Soc 97:6254–6256 10.1021/ja00854a057. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Arora SK. 1983. Correlation of structure and activity in ansamycins. Molecular structure of sodium rifamycin SV. Mol Pharmacol 23:133–140. [PubMed] [Google Scholar]
- 62.Bartolucci C, Cellai L, Di Filippo P, Lamba D, Segre AL, Bianco AD, Guiso M, Pasquali V, Brufani M. 2018. Hydrogenation of the ansa-chain of rifamycins. X-ray crystal structure of (16S)-16,17,18,19-tetrahydrorifamycin S. Helv Chim Acta 76:1459–1468 10.1002/hlca.19930760406. [DOI] [Google Scholar]
- 63.Brufani M, Cerrini S, Fedeli W, Vaciago A. 1974. Rifamycins: an insight into biological activity based on structural investigations. J Mol Biol 87:409–435 10.1016/0022-2836(74)90094-1. [DOI] [PubMed] [Google Scholar]
- 64.Bacchi A, Pelizzi G. 1999. Conformational variety for the ansa chain of rifamycins: comparison of observed crystal structures and molecular dynamics simulations. J Comput Aided Mol Des 13:385–396 10.1023/A:1008070316079. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Bacchi A, Pelizzi G, Nebuloni M, Ferrari P. 1998. Comprehensive study on structure-activity relationships of rifamycins: discussion of molecular and crystal structure and spectroscopic and thermochemical properties of rifamycin O. J Med Chem 41:2319–2332 10.1021/jm970791o. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Aristoff PA, Garcia GA, Kirchhoff PD, Showalter HD. 2010. Rifamycins: obstacles and opportunities. Tuberculosis (Edinb) 90:94–118 10.1016/j.tube.2010.02.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Temple ME, Nahata MC. 1999. Rifapentine: its role in the treatment of tuberculosis. Ann Pharmacother 33:1203–1210 10.1345/aph.18450. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Arioli V, Berti M, Carniti G, Randisi E, Rossi E, Scotti R. 1981. Antibacterial activity of DL 473, a new semisynthetic rifamycin derivative. J Antibiot (Tokyo) 34:1026–1032 10.7164/antibiotics.34.1026. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Bemer-Melchior P, Bryskier A, Drugeon HB. 2000. Comparison of the in vitro activities of rifapentine and rifampicin against Mycobacterium tuberculosis complex. J Antimicrob Chemother 46:571–576 10.1093/jac/46.4.571. [PubMed] [DOI] [PubMed] [Google Scholar]
- 70.Knoll BM, Nog R, Wu Y, Dhand A. 2017. Three months of weekly rifapentine plus isoniazid for latent tuberculosis treatment in solid organ transplant candidates. Infection 45:335–339 10.1007/s15010-017-1004-5. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Maggi N, Pallanza R, Sensi P. 1965. New derivatives of rifamycin SV. Antimicrob Agents Chemother 5:765–769. [PubMed] [Google Scholar]
- 72.Kradolfer F, Neipp L, Sackmann W. 1966. Chemotherapeutic activity of new derivatives of rifamycin. Antimicrob Agents Chemother 6:359–364. [PubMed] [Google Scholar]
- 73.Villain-Guillot P, Gualtieri M, Bastide L, Leonetti J-P. 2007. In vitro activities of different inhibitors of bacterial transcription against Staphylococcus epidermidis biofilm. Antimicrob Agents Chemother 51:3117–3121 10.1128/AAC.00343-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cellai L, Cerrini S, Segre A, Battistoni C, Cossu G, Mattogno G, Brufani M, Marchi E. 1985. A study of structure-activity relationships in 4-deoxypyrido[1′,2′-1,2]imidazo[5,4-c]rifamycin SV derivatives by electron spectroscopy for chemical analysis and 1H NMR. Mol Pharmacol 27:103–108. [PubMed] [Google Scholar]
- 75.Adachi JA, DuPont HL. 2006. Rifaximin: a novel nonabsorbed rifamycin for gastrointestinal disorders. Clin Infect Dis 42:541–547 10.1086/499950. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Ruiz J, Mensa L, Pons MJ, Vila J, Gascon J. 2008. Development of Escherichia coli rifaximin-resistant mutants: frequency of selection and stability. J Antimicrob Chemother 61:1016–1019 10.1093/jac/dkn078. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Kothary V, Scherl EJ, Bosworth B, Jiang Z-D, Dupont HL, Harel J, Simpson KW, Dogan B. 2013. Rifaximin resistance in Escherichia coli associated with inflammatory bowel disease correlates with prior rifaximin use, mutations in rpoB, and activity of Phe-Arg-β-naphthylamide-inhibitable efflux pumps. Antimicrob Agents Chemother 57:811–817 10.1128/AAC.02163-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanfilippo A, Della Bruna C, Marsili L, Morvillo E, Pasqualucci CR, Schioppacassi G, Ungheri D. 1980. Biological activity of a new class of rifamycins. Spiro-piperidyl-rifamycins. J Antibiot (Tokyo) 33:1193–1198 10.7164/antibiotics.33.1193. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Yang B, Koga H, Ohno H, Ogawa K, Fukuda M, Hirakata Y, Maesaki S, Tomono K, Tashiro T, Kohno S. 1998. Relationship between antimycobacterial activities of rifampicin, rifabutin and KRM-1648 and rpoB mutations of Mycobacterium tuberculosis. J Antimicrob Chemother 42:621–628 10.1093/jac/42.5.621. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Berrada ZL, Lin S-YG, Rodwell TC, Nguyen D, Schecter GF, Pham L, Janda JM, Elmaraachli W, Catanzaro A, Desmond E. 2016. Rifabutin and rifampin resistance levels and associated rpoB mutations in clinical isolates of Mycobacterium tuberculosis complex. Diagn Microbiol Infect Dis 85:177–181 10.1016/j.diagmicrobio.2016.01.019. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whitfield MG, Warren RM, Mathys V, Scott L, De Vos E, Stevens W, Streicher EM, Groenen G, Sirgel FA, Van Rie A. 2018. The potential use of rifabutin for treatment of patients diagnosed with rifampicin-resistant tuberculosis. J Antimicrob Chemother 73:2667–2674 10.1093/jac/dky248. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ungheri D, Della Bruna C, Sanfilippo A. 1984. Studies on the mechanism of action of the spiropiperidylrifamycin LM 427 on rifampicin-resistant M. tuberculosis. Drugs Exp Clin Res 10:681–689. [Google Scholar]
- 83.Baciewicz AM, Chrisman CR, Finch CK, Self TH. 2008. Update on rifampin and rifabutin drug interactions. Am J Med Sci 335:126–136 10.1097/MAJ.0b013e31814a586a. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Barluenga J, Aznar F, García A-B, Cabal M-P, Palacios JJ, Menéndez M-A. 2006. New rifabutin analogs: synthesis and biological activity against Mycobacterium tuberculosis. Bioorg Med Chem Lett 16:5717–5722 10.1016/j.bmcl.2006.08.090. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.García A-B, Palacios JJ, Ruiz M-J, Barluenga J, Aznar F, Cabal M-P, García JM, Díaz N. 2010. Strong in vitro activities of two new rifabutin analogs against multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:5363–5365 10.1128/AAC.00149-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamane T, Hashizume T, Yamashita K, Konishi E, Hosoe K, Hidaka T, Watanabe K, Kawaharada H, Yamamoto T, Kuze F. 1993. Synthesis and biological activity of 3′-hydroxy-5′-aminobenzoxazinorifamycin derivatives. Chem Pharm Bull (Tokyo) 41:148–155 10.1248/cpb.41.148. [PubMed] [DOI] [PubMed] [Google Scholar]
- 87.Fujii K, Tsuji A, Miyazaki S, Yamaguchi K, Goto S. 1994. In vitro and in vivo antibacterial activities of KRM-1648 and KRM-1657, new rifamycin derivatives. Antimicrob Agents Chemother 38:1118–1122 10.1128/AAC.38.5.1118. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luna-Herrera J, Reddy MV, Gangadharam PR. 1995. In vitro activity of the benzoxazinorifamycin KRM-1648 against drug-susceptible and multidrug-resistant tubercle bacilli. Antimicrob Agents Chemother 39:440–444 10.1128/AAC.39.2.440. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moghazeh SL, Pan X, Arain T, Stover CK, Musser JM, Kreiswirth BN. 1996. Comparative antimycobacterial activities of rifampin, rifapentine, and KRM-1648 against a collection of rifampin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Antimicrob Agents Chemother 40:2655–2657 10.1128/AAC.40.11.2655. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rothstein DM, Suchland RJ, Xia M, Murphy CK, Stamm WE, Murphy CK, Stamm WE. 2008. Rifalazil retains activity against rifampin-resistant mutants of Chlamydia pneumoniae. J Antibiot (Tokyo) 61:489–495 10.1038/ja.2008.65. [PubMed] [DOI] [PubMed] [Google Scholar]
- 91.Lounis N, Roscigno G. 2004. In vitro and in vivo activities of new rifamycin derivatives against mycobacterial infections. Curr Pharm Des 10:3229–3238 10.2174/1381612043383287. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Global Alliance for TB Drug Development (TB Alliance). 2008. Rifalazil. Tuberculosis (Edinb) 88:148–150 10.1016/S1472-9792(08)70023-4. [DOI] [Google Scholar]
- 93.Mae T, Hosoe K, Yamamoto T, Hidaka T, Ohashi T, Kleeman JM, Adams PE. 1998. Effect of a new rifamycin derivative, rifalazil, on liver microsomal enzyme induction in rat and dog. Xenobiotica 28:759–766 10.1080/004982598239173. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Gill SK, Xu H, Kirchhoff PD, Cierpicki T, Turbiak AJ, Wan B, Zhang N, Peng K-W, Franzblau SG, Garcia GA, Showalter HDH. 2012. Structure-based design of novel benzoxazinorifamycins with potent binding affinity to wild-type and rifampin-resistant mutant Mycobacterium tuberculosis RNA polymerases. J Med Chem 55:3814–3826 10.1021/jm201716n. [PubMed] [DOI] [PubMed] [Google Scholar]
- 95.Sensi P, Ballotta R, Greco AM, Gallo GG. 1961. Rifomycin. XV. Activation of rifomycin B and Rifomycin O. Production and properties of rifomycin S and rifomycin SV. Farm Ed Sci 16:165–180. [Google Scholar]
- 96.Malmborg A-S, Molin L, Nyström B. 1971. Rifampicin compared with penicillin in the treatment of gonorrhea. Chemotherapy 16:319–325 10.1159/000220742. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. 1970. Geldanamycin, a new antibiotic. J Antibiot (Tokyo) 23:442–447 10.7164/antibiotics.23.442. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Telenti A, Imboden P, Marchesi F, Lowrie D, Cole S, Colston MJ, Matter L, Schopfer K, Bodmer T. 1993. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 341:647–650 10.1016/0140-6736(93)90417-F. [DOI] [PubMed] [Google Scholar]
- 99.Heil A, Zillig W. 1970. Reconstitution of bacterial DNA-dependent RNA-polymerase from isolated subunits as a tool for the elucidation of the role of the subunits in transcription. FEBS Lett 11:165–168 10.1016/0014-5793(70)80519-1. [DOI] [PubMed] [Google Scholar]
- 100.Heep M, Rieger U, Beck D, Lehn N. 2000. Mutations in the beginning of the rpoB gene can induce resistance to rifamycins in both Helicobacter pylori and Mycobacterium tuberculosis. Antimicrob Agents Chemother 44:1075–1077 10.1128/AAC.44.4.1075-1077.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heep M, Beck D, Bayerdörffer E, Lehn N. 1999. Rifampin and rifabutin resistance mechanism in Helicobacter pylori. Antimicrob Agents Chemother 43:1497–1499 10.1128/AAC.43.6.1497. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cai X-C, Xi H, Liang L, Liu J-D, Liu C-H, Xue Y-R, Yu X-Y. 2017. Rifampicin-resistance mutations in the rpoB gene in Bacillus velezensis CC09 have pleiotropic effects. Front Microbiol 8:178 10.3389/fmicb.2017.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Severinov K, Soushko M, Goldfarb A, Nikiforov V. 1994. RifR mutations in the beginning of the Escherichia coli rpoB gene. Mol Gen Genet 244:120–126 10.1007/BF00283512. [PubMed] [DOI] [PubMed] [Google Scholar]
- 104.Severinov K, Soushko M, Goldfarb A, Nikiforov V. 1993. Rifampicin region revisited. New rifampicin-resistant and streptolydigin-resistant mutants in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem 268:14820–14825. [PubMed] [Google Scholar]
- 105.Sandgren A, Strong M, Muthukrishnan P, Weiner BK, Church GM, Murray MB. 2009. Tuberculosis drug resistance mutation database. PLoS Med 6:e2 10.1371/journal.pmed.1000002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sutherland C, Murakami KS. 2018. An introduction to the structure and function of the catalytic core enzyme of Escherichia coli RNA polymerase. Ecosal Plus 2018 10.1128/ecosalplus.ESP-0004-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl H-G, Dehò G, Donadio S, Ebright RH. 2017. Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase. Cell 169:1240–1248.e23 10.1016/j.cell.2017.05.042. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon H-S, Choi H, Lee M, Glynne RJ, Barnes SW, Walker JR, Batalov S, Yusim K, Feng S, Tung C-S, Theiler J, Via LE, Boshoff HIM, Murakami KS, Korber B, Barry CE III, Cho S-N. 2014. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol Microbiol 91:1106–1119 10.1111/mmi.12520. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim H, Kim S-H, Ying Y-H, Kim H-JH, Koh Y-H, Kim C-J, Lee S-H, Cha C-Y, Kook Y-H, Kim B-J. 2005. Mechanism of natural rifampin resistance of Streptomyces spp. Syst Appl Microbiol 28:398–404 10.1016/j.syapm.2005.02.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 110.Piddock LJV, Williams KJ, Ricci V. 2000. Accumulation of rifampicin by Mycobacterium aurum, Mycobacterium smegmatis and Mycobacterium tuberculosis. J Antimicrob Chemother 45:159–165 10.1093/jac/45.2.159. [PubMed] [DOI] [PubMed] [Google Scholar]
- 111.Li G, Zhang J, Guo Q, Wei J, Jiang Y, Zhao X, Zhao LL, Liu Z, Lu J, Wan K. 2015. Study of efflux pump gene expression in rifampicin-monoresistant Mycobacterium tuberculosis clinical isolates. J Antibiot (Tokyo) 68:431–435 10.1038/ja.2015.9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 112.Hoshino Y, Fujii S, Shinonaga H, Arai K, Saito F, Fukai T, Satoh H, Miyazaki Y, Ishikawa J. 2010. Monooxygenation of rifampicin catalyzed by the rox gene product of Nocardia farcinica: structure elucidation, gene identification and role in drug resistance. J Antibiot (Tokyo) 63:23–28 10.1038/ja.2009.116. [PubMed] [DOI] [PubMed] [Google Scholar]
- 113.Rominski A, Roditscheff A, Selchow P, Böttger EC, Sander P. 2017. Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J Antimicrob Chemother 72:376–384 10.1093/jac/dkw466. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Combrink KD, Denton DA, Harran S, Ma Z, Chapo K, Yan D, Bonventre E, Roche ED, Doyle TB, Robertson GT, Lynch AS. 2007. New C25 carbamate rifamycin derivatives are resistant to inactivation by ADP-ribosyl transferases. Bioorg Med Chem Lett 17:522–526 10.1016/j.bmcl.2006.10.016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr Opin Microbiol 2:489–493 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 116.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271 10.1038/nrmicro2319. [PubMed] [DOI] [PubMed] [Google Scholar]
- 117.Mariam DH, Mengistu Y, Hoffner SE, Andersson DI. 2004. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother 48:1289–1294 10.1128/AAC.48.4.1289-1294.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJM. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946 10.1126/science.1124410. [PubMed] [DOI] [PubMed] [Google Scholar]
- 119.Björkman J, Hughes D, Andersson DI. 1998. Virulence of antibiotic-resistant Salmonella typhimurium. Proc Natl Acad Sci USA 95:3949–3953 10.1073/pnas.95.7.3949. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Billington OJ, McHugh TD, Gillespie SH. 1999. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:1866–1869 10.1128/AAC.43.8.1866. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reynolds MG. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brandis G, Wrande M, Liljas L, Hughes D. 2012. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol Microbiol 85:142–151 10.1111/j.1365-2958.2012.08099.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 123.Heep M, Brandstätter B, Rieger U, Lehn N, Richter E, Rüsch-Gerdes S, Niemann S. 2001. Frequency of rpoB mutations inside and outside the cluster I region in rifampin-resistant clinical Mycobacterium tuberculosis isolates. J Clin Microbiol 39:107–110 10.1128/JCM.39.1.107-110.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Taddei F, Matic I, Radman M. 1995. cAMP-dependent SOS induction and mutagenesis in resting bacterial populations. Proc Natl Acad Sci USA 92:11736–11740 10.1073/pnas.92.25.11736. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rodríguez-Verdugo A, Gaut BS, Tenaillon O. 2013. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol Biol 13:50 10.1186/1471-2148-13-50. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hughes D, Brandis G. 2013. Rifampicin resistance: fitness costs and the significance of compensatory evolution. Antibiotics (Basel) 2:206–216 10.3390/antibiotics2020206. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2011. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44:106–110 10.1038/ng.1038. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brandis G, Hughes D. 2013. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J Antimicrob Chemother 68:2493–2497 10.1093/jac/dkt224. [PubMed] [DOI] [PubMed] [Google Scholar]
- 129.Brandis G, Pietsch F, Alemayehu R, Hughes D. 2015. Comprehensive phenotypic characterization of rifampicin resistance mutations in Salmonella provides insight into the evolution of resistance in Mycobacterium tuberculosis. J Antimicrob Chemother 70:680–685 10.1093/jac/dku434. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.O’Neill AJ, Huovinen T, Fishwick CWG, Chopra I. 2006. Molecular genetic and structural modeling studies of Staphylococcus aureus RNA polymerase and the fitness of rifampin resistance genotypes in relation to clinical prevalence. Antimicrob Agents Chemother 50:298–309 10.1128/AAC.50.1.298-309.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.de Vos M, Müller B, Borrell S, Black PA, van Helden PD, Warren RM, Gagneux S, Victor TC. 2013. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother 57:827–832 10.1128/AAC.01541-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]