

Abstract
Rice tungro bacilliform virus (RTBV) is a plant pararetrovirus whose DNA genome contains four genes encoding three proteins and a large polyprotein. The function of most of the viral proteins is still unknown. To investigate the role of the gene II product (P2), we searched for interactions between this protein and other RTBV proteins. P2 was shown to interact with the coat protein (CP) domain of the viral gene III polyprotein (P3) both in the yeast two-hybrid system and in vitro. Domains involved in the P2-CP association have been identified and mapped on both proteins. To determine the importance of this interaction for viral multiplication, the infectivity of RTBV gene II mutants was investigated by agroinoculation of rice plants. The results showed that virus viability correlates with the ability of P2 to interact with the CP domain of P3. This study suggests that P2 could participate in RTBV capsid assembly.
Rice tungro bacilliform virus (RTBV) is a reverse-transcribing DNA virus which, in association with an RNA virus, Rice tungro spherical virus (RTSV), is responsible for rice tungro disease (22), the most important viral disease of rice in South and Southeast Asia. In rice tungro, RTBV induces most of the symptoms (yellowing and reddening of the leaves, stunting of rice plants) and RTSV is mainly involved in the transmission of both viruses via the green leafhopper Nephotettix virescens (5).
RTBV is the type and only known member of the “RTBV-like viruses” genus, which has been classified in the Caulimoviridae family comprising caulimoviruses, badnaviruses, and two other genera (29, 31). The plant viruses which belong to this family have many features in common with retroviruses and are also often referred to, together with the human and animal hepadnaviruses, as pararetroviruses (23, 40, 42).
The bacilliform RTBV particles are elongated icosahedrons with a diameter of 30 nm and a length of approximately 130 nm, which varies with the virus isolate (22). The RTBV genome is a circular double-stranded DNA molecule of about 8 kbp, containing two site-specific discontinuities resulting from the replication process by reverse transcription and four large open reading frames (ORFs) (Fig. 1A) (1, 17, 39). The corresponding proteins, P1, P2, P3, and P4, are synthesized by specialized translation mechanisms (10–12) from a pregenomic RNA which is used as the template for viral replication and also serves as a polycistronic mRNA (22).
FIG. 1.

Schematic representations of the RTBV genome and P3 polyprotein. (A) Genome organization. Viral DNA is represented by a thin double line with the sites of the two discontinuities (Δ1 and Δ2) indicated. The thick arrows outside the DNA represent the four viral genes (I, II, III, and IV). The pregenomic RNA is shown as a thin arrow inside the DNA. (B) P3 polyprotein. The locations of the domains corresponding to MP, CP, PR, RT, RH within P3 are shown. Domains with unknown functions are indicated by question marks. Positions of the cleavage sites characterized by Laco et al. (27) and Marmey et al. (30) are indicated by vertical lines and arrows. Other presumed cleavage sites are symbolized by zigzag lines and question marks. Positions of the amino and carboxy termini of CP (p37) and RT (p55 and p62) are indicated. The black circle indicates the position of the zinc finger motif in CP.
The roles of P1 (24 kDa) and P4 (46 kDa) are still unknown. P3 is a large polyprotein of 196 kDa (Fig. 1B). Sequence comparisons with retroviral and other pararetroviral proteins suggest that P3 contains domains corresponding to the movement protein (MP), coat protein (CP), aspartic protease (PR), reverse transcriptase (RT), and RNase H (RH), ordered from the N terminus to the C terminus (17, 26, 39, 45). The viral protease is at least partly responsible for the processing of P3. The cleavage sites at the N- and C-terminal extremities of the RT-RH domain have been characterized. It has been demonstrated that the PR-RT-RH polyprotein can be processed to yield two proteins of 55 and 62 kDa (p55 and p62) when expressed in insect cells from the 3′ part of gene III (27).
First reports indicated that RTBV particles contain two major CP species of 33 and 37 kDa (p33 and p37) (39). The N terminus of p33 was determined to be at amino acid 502. Considering its size and the position of its N-terminal residue within P3, p33 should contain, in its C-terminal region, the basic domain and the Cys-His motif which are conserved in plant pararetrovirus CPs. This motif is the equivalent of the zinc finger motif of retroviral Gag proteins and consequently is thought to be involved in specific RNA binding during packaging of the pregenomic RNA into virions (40). Recently, Marmey et al. (30) showed that RTBV virions contain only a single coat protein species of 37 kDa, with the second peptide (of 34 kDa) most probably being a degradation product of the 37-kDa protein generated during virus purification. Amino acids 477 and 791 of P3 were deduced, from mass spectral analysis, to correspond to the N- and C-terminal residues, respectively, of the 37-kDa coat protein (p37).
ORF II encodes a 12-kDa protein (P2) for which no definite function has been assigned. P2 of RTBV and of the badnavirus commelina yellow mottle virus (CoYMV) were shown to be associated with purified virions (3, 22; A. Druka and R. Hull, personal communication). P2 of RTBV and of the badnavirus cacao swollen shoot virus (CSSV) were also described as sequence-nonspecific nucleic acid binding proteins (24, 25). The C termini of RTBV and CSSV P2, which possess basic, hydrophobic, and proline residues, support the nucleic acid binding activity. Such residues are also present at the C termini of caulimovirus gene III products and of bacterial histone-like proteins (34). Moreover, the C-terminal extremity of cauliflower mosaic virus (CaMV) P3 possesses a nonspecific nucleic acid binding activity (33, 34), suggesting a common role for this protein and the P2 of RTBV or badnaviruses in their respective life cycles.
To investigate the role of RTBV P2, we searched for possible interactions between this protein and other RTBV proteins. P2 was shown to interact with the CP domain of P3 both in the yeast two-hybrid system and in vitro. We have characterized this interaction and identified peptide motifs involved in the binding on both proteins. To evaluate the importance of this interaction in the context of viral infection, we introduced point mutations within gene II of the RTBV genome and investigated the infectivity of these mutants by agroinoculation of rice plants. Our results showed that virus viability correlates with the ability of P2 to interact with the CP domain of P3.
MATERIALS AND METHODS
Virus and plants.
All RTBV sequences were derived from the sequenced infectious clone pJIIS2 (6, 17). Rice (Oryza sativa) cv. Taichung Native 1 (TN1) was used for agroinoculation experiments. Plants were grown in an air-conditioned room with 12 h of illumination by 400-W lamps (HPI-T; Philips), at temperatures of 26 and 22°C during the light and dark periods, respectively.
Bacteria, yeast, and transformations.
For cloning and mutagenesis experiments, all plasmids were propagated in Escherichia coli DH5α. Glutathione S-transferase (GST) and GST-P2 fusion protein were expressed in E. coli BL21 (Pharmacia Biotech). The Agrobacterium tumefaciens strain LBA4301(pTiC58) was used for agroinoculation experiments (6). Plasmids were introduced into agrobacteria by electroporation. For the two-hybrid experiments, yeast strains HF7c and SFY526 (Clontech) were used. Yeast transformation was performed as specified by the Clontech Matchmaker Two-Hybrid System protocol (PT1265-1) for small-scale transformations.
Construction of recombinant plasmids.
The recombinant plasmids used in the two-hybrid system experiments were created by insertion of viral sequences into vectors pGAD424 and pGBT9 (Clontech). PCR was used to generate DNA fragments flanked by appropriate restriction sites for cloning into the two-hybrid plasmids, either directly or after subcloning into pET-3a (41) or pBIIKS(+) (Stratagene). Oligonucleotides used for PCR amplifications are listed in Table 1.
TABLE 1.
Oligonucleotides used for the PCR amplification of complete or partial RTBV ORFs
| ORF | Oligonucleotidesa |
|---|---|
| P1 |
AAGTCTTACCCGGGT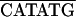 GTTCCAAAGAGGGATCTT-86 (+) GTTCCAAAGAGGGATCTT-86 (+) |
| _______ | |
| AACATGGATCCGCGCTCATGTAGCTTGATG-654 (−) | |
| _______ | |
| P2 | TAAGCATCAAGCCATATGATGCGCTGATTAC-679 (+) |
| _______ | |
| TAGAAGTACCAGCATATGGTCTAAGGCTCA-995 (−) | |
| _______ | |
| MP | GTAAACCCGGGCAAACATATGAGCCTTAGACCATTT-1011 (+) |
GACA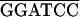 TTATTCTATAGATTTGGC-2371 (−) TTATTCTATAGATTTGGC-2371 (−) |
|
| _______ | |
| 40-CP | CGCGGATCCAGCCATGGCCAAATCTATAGAAGAAAT-2390 (+) |
| _______ | |
| GGCGAATTCCAAGATCTTATATTTCAAGGAAATTTTCAA-3443 (−) | |
| _______ | |
| PR | ACGCGAATTCACAACCTCATTAGCTATGAGAGCAAAT-3849 (+) |
| _______ | |
| ACGCGGATCCTTAATATCCTAATATTGGTTGAGGCTG-4384 (−) | |
| _______ | |
| RT-RH p55 | ACCGGAATTCAAGAATGAAATAGGAAATCAAAGT-4431 (+) |
| _______ | |
| ACGCGGATCCTTATTTTAAAATAAAATCTCCCTCACG-5827 (−) | |
| RT-RH p62 |
ACCG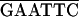 AAGAATGAAATAGGAAATCAAAGT-4431 (+) AAGAATGAAATAGGAAATCAAAGT-4431 (+) |
ACGC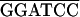 TTAGGCACTCTTTTCCTTTAGCCTTTC-5995 (−) TTAGGCACTCTTTTCCTTTAGCCTTTC-5995 (−) |
|
| P4 |
GATC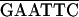 ATCCATATGAATATAGAGTACCCG-6059 (+) ATCCATATGAATATAGAGTACCCG-6059 (+) |
GCTTT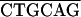 ACCCGGGTTAAGCATTGTCCATACG-7194 (−) ACCCGGGTTAAGCATTGTCCATACG-7194 (−) |
The sequence of the oligonucleotides is given 5′ to 3′. RTBV-derived sequences are underlined, and the position of the 3′-end nucleotide of each oligonucleotide in the plus- or minus-strand RTBV sequence is indicated on the right. The restriction sites used for cloning are overlined. In some cases, additional restriction sites are present but not indicated.
(i) pGAD-P1 and pGBT-P1.
The DNA fragment resulting from PCR amplification of ORF I was digested with NdeI and BamHI and cloned in pET-3a digested with the same enzymes. The resulting recombinant plasmid was linearized with NdeI, treated with the Klenow fragment of E. coli DNA polymerase I, and then digested with BamHI. The ORF I-derived fragment was finally inserted into the SmaI and BamHI sites of pGAD424 and pGBT9 to create pGAD-P1 and pGBT-P1, respectively.
(ii) pGAD-P2 and pGBT-P2.
The ORF II DNA fragment was digested with NdeI and cloned into the NdeI site of pET-3a to create pET-P2. pET-P2 was digested with NdeI, and the ORF II fragment obtained was treated with the Klenow enzyme and inserted into the SmaI site of pGAD424 and of pGBT9 as an in-frame fusion with the Gal4 activation domain and Gal4 binding domains (Gal4AD and Gal4BD, respectively) to generate pGAD-P2 and pGBT-P2, respectively.
(iii) pGAD-MP and pGBT-MP.
The ORF III DNA fragment corresponding to the movement protein domain was digested with NdeI and BamHI and cloned into pET-3a digested with the same enzymes. The resulting recombinant plasmid was linearized with NdeI, treated with Klenow enzyme, and then cut with BamHI. pGAD-MP and pGBT-MP were prepared by inserting the ORF III fragment obtained into the SmaI and BamHI sites of pGAD424 and pGBT9, respectively.
(iv) pGAD-CP and pGBT-CP.
The fragment of ORF III encoding the CP domain of the P3 polyprotein was digested with BamHI and EcoRI and cloned into pBIIKS(+) digested with the same enzymes to create pBKS-CP. The ORF III BamHI-SalI fragment from pBKS-CP was cloned into BamHI-SalI-restricted pGBT9. The recombinant plasmid obtained was digested with BamHI, treated with the Klenow enzyme, and religated to restore the frame between the Gal4BD and the CP domain, to finally give pGBT-CP. pGAD-CP was obtained by cloning the 1,132-bp EcoRI-EcoRI fragment from pGBT-CP, as an in-frame fusion with the Gal4AD, into EcoRI-linearized pGAD424.
(v) pGAD-PR, pGBT-PR, pGAD-p55, pGBT-p55, pGAD-p62, and pGBT-p62.
ORF III fragments encoding the protease, p55, and p62 proteins were digested by EcoRI and BamHI and inserted into pGAD242 and pGBT9 digested with the same enzymes.
(vi) pGAD-P4 and pGBT-P4.
The ORF IV fragment was digested with EcoRI and PstI and inserted into EcoRI-PstI-restricted pGAD424 and pGBT9.
(vii) pGEX-P2.
The GST-P2 expression plasmid (pGEX-P2) was constructed by ligating the NdeI-NdeI (Klenow enzyme-treated) ORF II fragment from pET-P2, as an in-frame fusion with the GST domain, into pGEX-2TK (Pharmacia Biotech) linearized with SmaI.
(viii) pUC19-RTBV.
To facilitate the preparation of the mutagenized constructs used for agroinoculation experiments, the SalI-SalI fragment from pRTRB1162 (6), corresponding to the complete RTBV genome, was subcloned into SalI-linearized pUC19 to create pUC19-RTBV.
All constructs were verified by appropriate restriction enzyme digestions and partial or complete sequencing of the cloned inserts.
Mutagenesis.
Mutant pGBT-P2Δ1 was prepared by digesting pGBT-P2 with PstI and religating the biggest fragment obtained. pGBT-P2Δ2 was created by an in-frame insertion, into PstI-linearized pGBT9, of the smallest fragment produced by the PstI digestion of pGBT-P2. Mutants pGBT-P2Δ3 and pGBT-Δ4 were produced by cloning the DNA fragments obtained by PCR with oligonucleotides 5′-CCGGAATTCACAACTAGTGGAAGTAGTTCAG-3′, 5′-CCGGAATTCAAGTTTAATTGGGTATTTACTC-3′, and 5′-ACGCGGATCCGGCTCATGCTGGATATTTTC-3′ and digested with EcoRI and BamHI into pGBT9 digested with the same enzymes.
Mutants pGBT-P2M1 to pGBT-P2M19 were created by digesting pGBT-P2 with SpeI (which cuts within the ORF II sequence) and BamHI (which cuts in the cloning site) and exchanging the fragment corresponding to the C-terminal region of ORF II for a mutagenized DNA fragment digested with the same enzymes. The different mutated DNA fragments were produced by PCR with oligonucleotide 5′-AGATTAATAATCTTACAACCCAAG-3′, which corresponds to nucleotides 849 to 872 (located in front of the SpeI site) of the RTBV positive-strand sequence, and oligonucleotides containing the desired mutation and a BamHI site. Sequences of mutagenic oligonucleotides used are available on request.
pGBT-CPΔ1 was produced by insertion of the 790-bp SmaI-NsiI fragment from pGBT-CP into the SmaI and PstI sites of pGBT9. pGBT-CPΔ2 was created by ligating the 386-bp EcoRV-NsiI fragment from pBKS-CP into SmaI- and PstI-digested pGBT9. Small deletions and point mutations were introduced by replacing the 550-bp AflII-PstI fragment from pGAD-CP with a mutagenized fragment digested with the same enzymes. Mutated DNA fragments were obtained by overlap extension PCR (20) with oligonucleotide 5′-AAGATAGGTTCTTAATAGA-3′, which corresponds to nucleotides 2831 to 2849 of the RTBV positive-strand sequence (located in front of the AflII site), and oligonucleotide 5′-TTAATAATAAAAATCATAAATCATAAG-3′, which corresponds to a region of the minus-strand sequence of pGBT-CP located just downstream of the PstI site, and the different pairs of mutagenic primers. Sequences of mutagenic oligonucleotides used are available on request.
Plasmid constructs used for agroinoculations were prepared in two steps. Mutations were first introduced into pUC19-RTBV using overlap extension PCR (20). The mutated fragments were synthesized by using oligonucleotides 5′-GTTTAATTGGGTATTTACTCTAG-3′ (nucleotides 745 to 767 of the RTBV plus-strand sequence) and 5′-CTTCCTATAGTTTTGATTGCTAC-3′ (nucleotides 1613 to 1591 of the RTBV minus-strand sequence) and four different pairs of mutagenic oligonucleotides: 5′-GGAATTAAAAGAAAATAGCCAGCATGAGCCTTAGAC-3′ plus 5′-GGCTCATGCTGGCTATTTTCTTTTAATTCCTTTC-3′; 5′-GAAAGGAATTAAAGCAAAATATCCAGCATGAGC-3′ plus 5′-TGCTGGATATTTTGCTTTAATTCCTTTCTTAGG-3′; 5′-CCTAAGAAAGGAGCTAAAAGAAAATATCCAGC-3′ plus 5′-GCTGGATATTTTCTTTTAGCTCCTTTCTTAGGTGGTGC-3′; and 5′-CAAAGCACCACCTGCGGCAGGAATTAAAAGAAAATATCCAGC-3′ plus 5′-CTTTTAATTCCTGCCGCAGGTGGTGCTTTGTACTG-3′ (the first and second oligonucleotides of each pair correspond to plus- and minus-strand RTBV sequences, respectively; the mutations are underlined). PCR products were digested with SacII and AvrII and inserted, in place of the wild-type sequence, into pUC19-RTBV digested with the same enzymes. SalI-SalI RTBV mutated sequences from the mutagenized pUC19-RTBV plasmids were cloned in place of the wild-type sequence of pRTRB1162 (6) to create pRT-P2M5, pRT-P2M8, pRT-P2M11, and pRT-P2M13.
All constructs were verified by digestions with appropriate restriction enzymes and partial or complete sequencing of the cloned inserts. For pRT-P2M5, pRT-P2M8, pRT-P2M11, and pRT-P2M13, the complete region generated by PCR and cloned was sequenced.
Detection of protein-protein interactions in yeast.
Protein-protein interactions were detected in yeast strains by two approaches: (i) growth on selective media lacking histidine, due to the activation of HIS3 reporter gene, and (ii) detection of β-galactosidase activity resulting from activation of the lacZ reporter gene.
HF7c cells transformed with each of the recombinant pGAD424 and pGBT9 plasmid pairs were plated on medium deficient in leucine, tryptophan, and histidine and incubated at 30°C for 3 to 12 days. Stimulation of yeast growth compared to a control HF7c strain transformed with empty pGAD424 and pGBT9 vectors indicated an interaction between the proteins tested.
β-Galactosidase activity was measured by colony lift filter and liquid assays as described in the Clontech yeast protocols handbook PT3024-1. For the filter assay, transformed HF7c or SFY526 cells were plated on synthetic dropout (SD) medium deficient in leucine and tryptophan but containing histidine and incubated at 30°C for 3 to 4 days. Colonies obtained were transferred to nitrocellulose filters, frozen in liquid nitrogen, thawed at room temperature, and finally assayed for β-galactosidase activity by incubating the filters at 30°C in the presence of the substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). The times taken for the appearance of blue color and the intensity of the blue color after 6 h were both assessed. Alternatively, the colonies (at least 10 per plasmid pair tested) were restreaked on fresh plates, allowed to grow for a further 2 days, and then tested. For the β-galactosidase liquid assay, transformants were first grown for 18 h at 30°C in SD medium deficient in leucine and tryptophan and then used to inoculate cultures with a starting optical density OD of approximately 0.25. When the cultures reached an OD of between 0.5 and 0.8, the cells were harvested by centrifugation, washed, and subjected to four cycles of freezing (in liquid nitrogen) and thawing (at 42°C) for lysis. The yeast lysates were assayed for β-galactosidase enzymatic activity with o-nitrophenyl-β-d-galactopyranoside (ONPG) as a substrate for color development. The enzymatic activity was determined in five independent transformants. β-Galactosidase units are calculated as: (optical density at 420 nm [OD420] × 1,000)/(OD600 × time [in minutes] × volume [in milliliters]).
In vitro transcription.
EcoRI-linearized pBKS-CP was transcribed by incubation with T7 polymerase (Biofinex) by the method of Gurevich (14). Transcripts were purified by precipitation with 3 M lithium chloride followed by precipitation with ethanol. The integrity of the synthesized transcripts was evaluated on a 6% denaturing polyacrylamide gel. RNA was quantified by measuring the absorbance at 260 nm.
In vitro translation.
Transcripts were translated in wheat germ extract (Promega) (40 ng of transcript/μl of translation medium) or in reticulocyte lysate (Promega) (80 ng of transcript/μl of translation medium) as described by the supplier. Translation products were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized by autoradiography.
In vitro binding experiments.
A fresh 10-ml overnight culture of E. coli BL21 transformed with pGEX-2TK or pGEX-P2 was added to 90 ml of Luria broth containing ampicillin (100 μg/ml). The cultures were then incubated for 1 h at 37°C with shaking. Expression of GST or GST-P2 was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. After a further 3 h of incubation, bacteria were harvested by centrifugation at 5,000 × g for 5 min at 4°C and resuspended in 30 ml of binding buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, 1 mM dithiothreitol, 1× protease inhibitors [Complete; Roche Molecular Biochemicals]). The bacteria were then lysed on ice by mild sonication, and the lysates were centrifuged for 10 min at 15,000 × g. Supernatants were mixed with 100 μl of glutathione-Sepharose 4B beads, previously washed and resuspended in NETN buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40), and incubated for 1 h at 4°C with gentle shaking. The beads were then washed three times by short centrifugation and resuspension in NETN buffer. Approximately equal amounts (25 μg), as judged by Coomassie blue staining, of GST and GST-P2 bound to the beads and resuspended in 500 μl of binding buffer were incubated with 10 μl of in vitro translation reaction mixture for 1 h at 4°C with gentle shaking. The beads were then washed three times as described above, resuspended in 100 μl of dissociation buffer (125 mM Tris-HCl [pH 6.8], 10% SDS, 25% β-mercaptoethanol), boiled for 5 min to dissociate the protein-bead complexes, and finally pelleted. Aliquots (10 μl) of the supernatants were subjected to SDS-PAGE, and the [35S]methionine-labelled proteins were detected by autoradiography.
The induction and purification steps were monitored by visualizing the bacterial proteins by SDS-PAGE separation and Coomassie blue staining.
Agroinoculations.
Agroinoculations were performed essentially as described by Dasgupta et al. (6). Agrobacteria transformed with one of the constructs (pRTRB1162, pRT-P2M5, pRT-P2M8, pRT-P2M11, and pRT-P2M13) were plated on YEB medium (0.5% Bacto Beef Extract [Difco], 0.1% Bacto Yeast Extract [Difco], 0.5% Bacto Peptone [Difco], 0.5% sucrose, 2 mM MgSO4, pH 7.2) containing kanamycin (50 μg/ml) and rifampin (100 μg/ml) and supplemented with 4 mM MgSO4. For plant injections, 48-h precultures were used to inoculate cultures in 100 ml of YEB containing kanamycin (50 μg/ml), rifampin (100 μg/ml), and 4 mM MgSO4. After 24 h of incubation at 28°C with vigorous shaking, the cells were harvested by centrifugation at 5,000 × g for 20 min, washed once with deionized water, and then resuspended in 100 μl of water. The viscous suspension was immediately used for agroinoculations of 3- to 4-week-old rice plants. Three injections of 10 μl were performed with Hamilton syringes at the base of the stem of each plant. At 7 and 10 days after the inoculations, the plants were thoroughly sprayed and watered with a solution of cefotaxime (500 μg/ml) and vancomycin (500 μg/ml) to kill the agrobacteria.
Detection of viral antigens in yeast and in plants.
Yeast protein extracts were prepared as described in the Clontech yeast protocols handbook PT3024-1, with the following modifications. A 5-ml volume of SD selective medium was inoculated with a single colony and incubated at 30°C overnight with shaking. A 500-μl volume of the overnight culture was added to 4.5 ml of fresh medium and incubated until the OD reached 0.4 to 0.6. The cultures were then centrifuged, and the pellets were washed once with ice-cold water and then frozen immediately at −70°C. The pellets were then resuspended in prewarmed complete cracking buffer and placed on ice. Cell suspensions were added to tubes containing glass beads and vortexed for 1 min. Dissociation buffer (125 mM Tris-HCl [pH 6.8], 10% SDS, 25% β-mercaptoethanol) containing 3 mM phenylmethylsulfonyl fluoride and 1× protease inhibitors (Complete; Boehringer Mannheim) was added to the suspensions (1:1, vol/vol), which were then boiled for 5 min and vortexed again for 1 min. The tubes were then stored at −70°C before being subjected to further analyses.
Plant protein extracts were prepared by grinding pieces of leaf directly in the dissociation buffer and boiling them for 5 min. The presence of P2 and P2 derivatives in yeast and plant extracts was analysed by Western blotting. Total proteins of the extracts were separated by SDS-PAGE and transferred to nitrocellulose. P2 and P2 fusion proteins were detected using antibodies raised against P2 (kindly provided by A. Druka and R. Hull, (John Innes Centre, Norwich, United Kingdom) and the ECL detection kit (Amersham).
Detection of viral DNA in plants and analysis of viral progeny.
The viral genome was detected by PCR analysis. Small pieces of infected leaves were added directly to 100-μl PCR mixtures containing 1× PCR buffer, 0.2 mM each deoxynucleoside triphosphate, 1.5 mM MgCl2, 2.5 U of Taq DNA polymerase (Gibco BRL), and oligonucleotides 5′-ACCGGAATTCTACATGAGCGCTGATTACCCAACTTTCAAGGAAGCCC698-3′ and 5′-CGCGGATCCTAGATTCAGAGGTTGAATCTTGGG1034-3′ (2.5 ng/μl each; the restriction sites are in italics, and the position of the 3′-end nucleotide in the RTBV plus- or minus-strand sequence is indicated), which hybridyze to the 5′ and 3′ extremities of the RTBV gene II, respectively. PCR was performed under to the following conditions: 94°C for 5 min and 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s. The PCR products were digested with EcoRI and BamHI and purified from agarose gels by using the QIAquick gel extraction kit (Qiagen). PCR fragments obtained from the leaves of five different plants agroinoculated with the same mutant were combined and cloned into the BamHI and EcoRI sites of pBII-KS(+). After transformation of E. coli, plasmid DNA from 10 independent clones was analyzed by sequencing.
RESULTS
RTBV P2 interacts with the CP domain of the P3 polyprotein in the yeast two-hybrid system.
We used the yeast two-hybrid system (9) to search for interactions between P2 and other RTBV proteins. The genes corresponding to P1, P2, and P4 and the regions of gene III corresponding to the MP, CP, PR and RT-RH (p55 and p62) (Fig. 1) were fused in frame to the Gal4 DNA binding domain in the yeast vector pGBT9 and to the Gal4 transcription activation domain of pGAD424 (Fig. 2). At the time this work was initiated, the exact localization of the CP domain within P3 was not known. The region of P3 chosen for our experiments corresponds to p33, described by Qu et al. (39), extended at its N- and C-terminal extremities by 35 and about 21 amino acids, respectively. This domain (40-CP) encodes a protein of 40 kDa which encompasses the p37 amino acid sequence now characterized by Marmey et al. (30) as the bona fide CP.
FIG. 2.

Interaction between P2 and other RTBV proteins in the yeast two-hybrid system. Yeast strain HF7c was cotransformed with a plasmid encoding the Gal4AD-P2 fusion protein and a plasmid expressing one of the other RTBV proteins fused to the Gal4BD (A) or with plasmid pairs encoding the various Gal4 domain-RTBV protein fusions in the opposite combination (B). The β-galactosidase activity produced by the resulting transformants was assessed by filter assay, with the appearance of a blue color indicating interaction between the proteins tested. The strength of the interactions was estimated by the intensity of the blue coloration after 6 h: +++, dark blue; +, light blue; −, no color. The time taken for the blue color to appear is indicated in parentheses.
Yeast strain HF7c was cotransformed with all possible pairwise combinations of the different recombinant plasmids. Colonies obtained were assayed for β-galactosidase activity resulting from the activation of the lacZ reporter gene, indicating a physical interaction between RTBV-derived proteins. Colonies turned blue when pGAD-P2/pGBT-CP or pGAD-CP/pGBT-P2 plasmid pairs were used for cotransformations (Fig. 2). No blue color was observed in any other case. Moreover, no β-galactosidase activity could be detected when the plasmids were used individually or in combination with the pGAD424 or pGBT9 empty vectors. The yeast clones harboring the pGAD-P2/pGBT-CP or pGAD-CP/pGBT-P2 plasmid pairs were able to grow three to four times faster on medium lacking histidine than was a control clone transformed with pGAD424 and pGBT9, indicating a positive induction of HIS3 reporter gene expression. Similar β-galactosidase activities were also observed when these two plasmid pairs were used to transform the yeast strain SFY526 (data not shown). All of these results clearly show that the P2 and CP domains of the fusion proteins are able to interact with each other in yeast.
Depending on the nature of the fusion protein pairs tested, differences in intensity and in the time required for appearance of the blue color (possibly due to changes in protein conformation, stability, or efficiency of nuclear targeting) were observed during the filter assays (Fig. 2). Since the AD-CP/BD-P2 combination consistently gave rise to a higher β-galactosidase activity than did the AD-P2/BD-CP pair, we generally used plasmids expressing the former fusion proteins for further experiments.
P2 interacts with the 40-CP in vitro.
To validate the results obtained with the two-hybrid system, we examined the interaction of P2 with 40-CP in vitro. We cloned the RTBV gene II into the bacterial pGEX-2TK vector to express P2 as a GST fusion protein. GST and the GST-P2 fusion protein were overexpressed in E. coli and purified from soluble fractions of bacterial lysates by using glutathione-Sepharose 4B beads (Fig. 3A). The purity of the proteins was assessed after dissociation from the beads and separation by SDS-PAGE (Fig. 3A, lanes 5 and 9). GST-P2 had an apparent molecular mass of 38 kDa, which is consistent with the size of P2 (12 kDa) plus 26 kDa (corresponding to GST). GST or GST-P2 bound to beads was then incubated in the presence of [35S]methionine-labelled 40-CP, produced by in vitro translation in either wheat germ extract (Fig. 3B, lane 1) or reticulocyte lysate (lane 2). After the mixtures were washed, the proteins present in the bead-bound complexes were separated by SDS-PAGE and visualized by autoradiography (Fig. 3C). The results show that 40-CP translated in vitro in either system associates with the GST-P2 fusion protein (Fig. 3C, lanes 3 and 4) but not with GST alone (lanes 1 and 2). Two additional smaller products (Fig. 3C), which probably correspond to degradation products or to polypeptides produced in vitro by internal initiation events, were also specifically detected in the GST-P2 fractions (lanes 3 and 4). On the other hand, neither the 46-kDa protein (Fig. 3B), which is nonspecifically synthesized in the reticulocyte lysate (lanes 2 and 4), nor the majority of putative CP truncated products, produced in both in vitro translation systems, could bind to GST-P2. These observations argue in favor of a specific binding of 40-CP to P2.
FIG. 3.

In vitro binding of P2 to 40-CP. (A) Expression and purification of the GST-P2 fusion protein. P2 fused to GST was expressed in E. coli and isolated by binding to glutathione-Sepharose 4B beads. GST alone was also expressed and purified. The proteins present in the total bacterial lysates before (lanes 2 and 6) and after (lanes 3 and 7) induction, in the soluble fractions (lanes 4 and 8), and in the fractions bound to the beads (lanes 5 and 9) were subjected to SDS-PAGE and visualized by Coomassie blue staining. The positions of GST (26 kDa) and GST-P2 (38 kDa) are indicated on the right; the sizes of the protein markers (lanes 1 and 10) are indicated on the left. (B) Synthesis of 40-CP by in vitro translation. 40-CP was translated from a transcript corresponding to nucleotides 2370 to 3462 of the RTBV plus-strand sequence in the presence of [35S]methionine in either wheat germ extract (lane 1) or reticulocyte lysate (lane 2). Labelled translation products were analyzed by SDS-PAGE and autoradiography. Lanes 3 and 4 correspond to the translation controls (without addition of RNA) for the wheat germ extract and reticulocyte lysate, respectively. The positions of 40-CP and the size markers are indicated on the right and left, respectively. ⧫, position of the 46-kDa protein (lanes 2 and 4) mentioned in the text. (C) Interaction of 40-CP with GST-P2. 40-CP synthesized in vitro was incubated with GST-P2 or GST bound to the glutathione-Sepharose 4B beads. The labelled proteins produced in either wheat germ extract (lanes 1 and 3) or in reticulocyte lysate (lanes 2 and 4) and coprecipitated with the GST-P2- or GST-bead complexes were separated by SDS-PAGE and visualized by autoradiography. The positions of 40-CP and the size markers are indicated on the right and left, respectively. Asterisks, positions of the truncated forms of CP (lanes 3 and 4), which are discussed in the text.
Thus, our in vitro results are consistent with those obtained in the yeast two-hybrid system and confirm that P2 can interact specifically with 40-CP.
Mapping of the CP-interacting domain of P2.
To localize the domain(s) of P2 mediating the binding to 40-CP, we examined the effects of mutations introduced into the P2 coding sequence of pGBT-P2 by using the yeast two-hybrid system. In this system, β-galactosidase activity can be correlated, to a certain extent, with the affinity between the interacting proteins (7). For this study, the expression level of the different modified proteins was controlled by Western blotting (see Materials and Methods) and no significant variations in the amount of protein expressed were observed compared to the wild-type fusion protein (data not shown).
Two truncated forms of the BD-P2 protein (BD-P2Δ1 and BD-P2Δ2), containing the first three-quarters and the last quarter of P2, respectively, were tested for their interaction with the AD-CP protein expressed from pGAD-CP (Fig. 4A). The results of the β-galactosidase assays showed that the N-terminal three-quarters (amino acids 1 to 88) of P2 is not sufficient to establish the interaction with the CP. BD-P2Δ2 retains a binding activity which corresponds, according to our liquid-assay quantifications, to about 50% of the activity of the complete P2 fusion protein. This result indicates that the C-terminal region of P2 plays an important role in establishing the interaction with 40-CP. However, an additional domain(s) located upstream of amino acid 87 could also contribute to the efficiency of the association. Therefore, two larger fusions (BD-P2Δ3 and BD-P2Δ4) were tested (Fig. 4A). BD-P2Δ3 possesses a CP binding activity similar to that of BD-P2Δ2 (around 50%), and BD-P2Δ4 shows a slight increase (about 10%) in its affinity for the CP. These results indicate that the addition of residues 27 to 86 to the C-terminal part of P2 does not restore the full CP binding activity, suggesting either that the first 26 amino acids have the potential to substantially improve the affinity of P2 for the CP or, most probably, that the folding of the partial fusion proteins differs from that of the complete BD-P2 fusion.
FIG. 4.

Mapping of the CP-interacting domain of P2 with the yeast two-hybrid system. (A and B) Yeast strain HF7c was cotransformed with the plasmids encoding CP fused to Gal4AD (pGAD-CP) and P2 or modified P2 fused to the Gal4BD (pGBT-P2, pGBT-P2Δ1 to pGBT-P2Δ4, pGBT-P2M1 to pGBT-P2M19), and the transformants obtained were tested for β-galactosidase (β-gal.) activity. Mutations introduced in the P2 coding sequence of pGBT-P2 correspond to large deletions (A) and small deletions or amino acid substitutions (B). (A) P2 and truncated P2 forms are depicted schematically by open boxes, and Gal4BD is depicted by the interrupted black boxes. (B) Amino acid substitutions and small deletions in the C-terminal region of P2 fusion proteins are indicated by bold letters and bold hyphens, respectively. Numbers above the open boxes and the amino acid sequences refer to positions in P2. β-Galactosidase activity was determined by filter and liquid assay procedures. For the filter assay quantifications, β-galactosidase activity was assessed by the intensity of the blue color of yeast clones after 6 h: +++, dark blue; ++, intermediate color; +, light blue; +/− pale blue; −, no color. For the liquid-assay quantifications, the values indicate percent β-galactosidase activities relative to those obtained with the wild-type P2 construct, measured in extracts from yeast cells expressing AD-CP and one of the BD-P2 versions. Values given are the mean of the relative β-galactosidase activities (and their standard deviations) of five independent clones. The β-galactosidase activity of yeast clones harboring the P2 wild-type construct was, on average, 2.7 β-galactosidase units, as defined in Materials and Methods. The control corresponds to a yeast clone transformed with pGBT9 and pGAD424; n.d., not determined. (C) Role of the C-terminal residues of P2 in establishing the interaction with CP. Residues which are involved are shown in bold type. Boxed residues are crucial and underlined residues are important for the association with CP. The importance of residues QYK97 was not assessed.
To determine more precisely the peptide motifs and individual residues of the C-terminal part of P2 which are involved in its CP-binding activity, small deletions and point mutations (in most cases substitutions by alanines) were introduced into the complete P2 coding sequence of pGBT-P2 (Fig. 4B). Analysis of the CP binding capacity of the modified proteins allowed us to assign the C-terminal residues of P2 to one of three classes: (i) those which are not essential for binding, (ii) those which are important for the strength of the interaction, and (iii) those which play a crucial role in binding. The replacement of the C-terminal alanine by a glutamine (BD-P2M1) or the deletion of this residue (BD-P2M2) did not affect the affinity of P2 for the CP. Replacement of Y108 or P109 by an alanine (BD-P2M4 and BD-P2M3) reduced the strength of the interaction, and deletion of the three last amino acids (BD-P2M5) resulted in a very low β-galactosidase activity. A series of mutants with double or triple amino acid substitutions (BD-P2M6, BD-P2M10, BD-P2M13, BD-P2M14, BD-P2M18, and BD-P2M19) and the replacement of A98 with a glycine (BD-P2M17) were prepared. Repeated attempts to prepare a construct expressing a P2 fusion protein with substitution of QYK97 by alanines were unsuccessful for unknown reasons. Analysis of the binding capacity of these modified proteins allowed a stretch of critical residues, extending from P99 to K107, to be defined. A series of mutants, each with a single point mutation, were then generated. Individual substitution of P99 or I104 by an alanine (BD-P2M16 and BD-P2M11) totally abolished the interaction, and substitution of K105 or R106 (BD-P2M9 and BD-P2M8) resulted in a strong decrease in the binding efficiency. Substitution of the other residues had less impact, but significant variations could nevertheless be observed. We can conclude from these mapping experiments that the essential domain of P2 required for CP binding resides within the last 12 residues (Fig. 4C). Three types of residues (prolines, hydrophobic, and basic amino acids) are particularly important for the CP binding capacity of P2, and P99 and I104 play key roles in establishing the interaction.
Identification of CP residues important for the interaction with P2.
To identify the domain(s) of 40-CP involved in the interaction with P2, we prepared two deletion mutants with mutations in pGBT-CP (Fig. 5A). pGBT-CPΔ1 and pGBT-CPΔ2 express fusion proteins containing the N-terminal two-thirds (BD-CPΔ1) and the middle part (BD-CPΔ2), respectively, of 40-CP. Neither of these truncated forms was able to interact with AD-P2, encoded by pGAD-P2, suggesting a role for the C-terminal portion of the 40-CP in binding. This region contains the conserved basic domain and zinc finger motif present in all pararetrovirus CPs and is also rich in hydrophobic amino acids. Such residues are often involved in protein-protein interactions. Moreover, we have shown that two hydrophobic residues in P2 (I104 and Y108) are crucial for or contribute to the efficiency of CP binding (see above). Therefore, we evaluated the importance of some of the hydrophobic residues present within the CP C-terminal region by generating small deletions within the 3′ part of the CP coding sequence of pGAD-CP (pGAD-CPΔ730–732, pGAD-CPΔ736–739, and pGAD-CPΔ799–803) (Fig. 5B). We also evaluated the importance of the basic domain by deleting residues 749 to 769 (pGAD-CPΔ749–769) and of the zinc finger motif by replacing the conserved cysteines (C772, C774, and C777) with alanines (pGAD-CP[C772A;C774A;C777A]). The affinity of modified AD-CP proteins for BD-P2 encoded by pGBT-P2 was then analyzed. The results of β-galactosidase assays indicate that deletion of residues 799 to 803 or deletion of the basic domain does not modify the affinity of the CP for P2. Modification of residues in the zinc finger motif did not have a significant effect, indicating that formation of this structure is not required for interaction with P2. On the other hand, deletions of amino acids LAF732 (AD-CPΔ730–732) and IYTI739 (AD-CPΔ736–739) resulted in undetectable β-galactosidase activities. AD-CP proteins in which the residues of these two small regions were individually replaced by alanines (AD-CPM1 to AD-CPM6; A731 was not modified) were also tested (Fig. 5C). Apart from L730, modification of any of these amino acids abolished the interaction. The possibility of detecting the 40-CP/40-CP homotypic interaction in the yeast two-hybrid system (E. Herzog, unpublished data) allowed us to control the stability of the different AD-CP variants expressed. These results point out the essential role of F732 and of the IYTI739 residues for the association of the CP with P2.
FIG. 5.

Modifications of the CP affecting its interaction with P2 in the yeast two-hybrid system. Yeast strain HF7c was cotransformed with plasmids encoding P2 in fusion with the Gal4AD (pGAD-P2) and the CP or CP derivatives in fusion with the Gal4BD (pGBT-CP, pGBT-CPΔ1, and pGBT-CPΔ2) (A) or with plasmids encoding P2 in fusion with the Gal4BD (pGBT-P2) and the CP versions in fusion with Gal4AD (pGAD-CP, pGAD-CPΔ730–732, pGAD-CPΔ736–739, pGAD-CPΔ749–769, pGAD-CP[C772A;C774A;C777A], and pGAD-CPΔ799–803; pGAD-CPM1 to pGAD-CPM6) (B and C). The β-galactosidase (β-gal.) activity was estimated by filter assay. Open boxes represent CP or CP derivatives in the CP fusion proteins, and interrupted black and gray boxes represent Gal4BD and Gal4AD, respectively. The basic domain of CP is indicated by a shaded box, and the zinc finger motif is indicated by a black circle. (B) Small deletions and replacements by alanines are indicated by V and arrows, respectively. (C) Alanine substitutions introduced in Gal4AD-CP fusion protein are shown in bold. Numbers indicated above open boxes and amino acid sequences refer to positions in the P3 polyprotein.
Infectivity of RTBV gene II mutants in rice.
The importance of P2 residues involved in the CP interaction for the activity of the protein in vivo and consequently for virus multiplication was assessed by agroinoculation (6) of rice plants with wild-type RTBV or one of four different RTBV gene II mutants (Table 2). The three first mutants express modified P2 proteins that either retain a weak CP interaction capacity in the two-hybrid system (pRT-P2M5 and pRT-P2M8) or have lost the ability to interact with the CP (pRT-P2M11). The fourth mutant (pRT-P2M13) was prepared to specifically assess the importance of the nonspecific nucleic acid binding activity of P2 for virus viability; Jacquot et al. (25) have shown that replacement of KK102 by alanines, which has only a limited effect on the capacity of P2 to interact with the CP, totally abolished the affinity of the protein for nucleic acids in vitro. Plant infection was determined by Western blot detection of gene II products and by PCR analysis to detect the presence of the viral genome. The results of two independent experiments showed that no viral components (P2 or viral genome) could be detected in extracts from plants agroinoculated with mutant pRT-P2M11 (Table 2). Hence, the presence of the isoleucine at position 104 in P2 is critical for both interaction with the CP and virus infectivity. For the mutants pRT-P2M5, pRT-P2M8, and pRT-P2M13, the infection efficiency was similar to that of wild-type virus (Table 2), and gene II products and the viral genome could be detected in similar amounts in infected plants (data not shown). The progeny of the viable mutants were analyzed by PCR amplification and sequencing of gene II (see Materials and Methods). In all cases, the mutations were conserved in the genomes of the mutant virus progeny and no reversions were observed. Considering the quantification experiments in the two-hybrid system, these results indicate that a low level of P2-CP interaction is sufficient for RTBV infectivity and that, surprisingly, the modification of an important element (KK102) of the nucleic acid binding domain of P2 can be tolerated.
TABLE 2.
Agroinoculations of rice with RTBV gene II mutants
| Plasmida | P2 versionb | No. of plants infected/no. inoculatedc in:
|
Infection (%)d | |
|---|---|---|---|---|
| Expt 1 | Expt 2 | |||
| pRTRB1162 | P2 | 11/15 | 20/25 | 77 |
| pRT-P2M5 | P2M5 (Δ108–110) | 8/15 | 21/25 | 69 |
| pRT-P2M8 | P2M8 (R106A) | 10/15 | 19/25 | 72 |
| pRT-P2M11 | P2M11 (I104A) | 0/15 | 0/25 | 0 |
| pRT-P2M13 | P2M13 (K101A; K102A) | 9/15 | 19/25 | 68 |
Plasmid constructs used to transform A. tumefaciens.
P2 variants encoded by the RTBV mutants. Modifications are indicated in parentheses.
Infection was detected by testing the plants for the presence of the RTBV gene II product and the viral genome.
The values correspond to the mean percentages of infected plants in the two independent experiments.
DISCUSSION
The results presented here demonstrate that RTBV P2 can interact specifically with the CP domain of the P3 polyprotein. The region of P3 that we have chosen for our experiments completely encompasses the amino acid sequence of the p37 coat protein, the basic subunit of RTBV particles (30). An essential CP-interacting region of P2, extending from amino acid 99 to residue 109, has been characterized. In the yeast two-hybrid system, P99 and I104 were shown to be crucial and K105, R106, Y108, and P109 were shown to contribute significantly to the association with the CP. The 12 last amino acids of P2 also support nucleic acid binding activity in vitro (25). Thus, the C-terminal extremity of P2 seems to play a central role in its function. Residues of the CP (F732, I736 to I739) important for its interaction with P2 were also identified. They correspond mostly to hydrophobic amino acids and are located just in front of the basic region and the zinc finger motif, which are thought to be involved in the recognition of the RTBV pregenomic RNA (or genomic DNA) during the encapsidation process (40). Secondary-structure predictions (not shown) have revealed that CP residues 732 to 739 could potentially form a β-sheet. Considering that every modification that we introduced into this potential β-sheet had a severe impact (the contribution of CRS735 residues was not assessed), and considering that alanine substitutions often have the propensity to destabilize this type of secondary structure (8), it is likely that the complete structural motif formed by residues 732 to 739 participates in P2 association.
The results of the agroinoculation experiments with gene II mutants indicate that I104 also plays a crucial role in P2 function in vivo. The fact that the P2 I104 substitution variant which cannot interact with the CP is not viable suggests an important role for this interaction in the RTBV life cycle. Surprisingly, replacement of R106 by an alanine and truncation of P2 by removal of YPA110, which in the two-hybrid experiments strongly affected the interaction between P2 and the CP, had no significant effect on virus multiplication. These results indicate that in the context of viral infection the situation is certainly more complex compared to the two-hybrid system or in vitro experimental conditions. The molecular mechanisms involved in the different steps of a virus multiplication cycle often require the formation of multicomponent macromolecular complexes, containing several viral and/or cellular proteins. It is possible that the interaction of P2 with the CP is involved in processes (see below) which require additional partners in vivo, i.e., other RTBV and/or cellular proteins. Moreover, replacement of KK102 by alanines, which has only a slight effect on the P2-CP interaction but totally abolished the interaction of P2 with nucleic acids in vitro (25), did not prevent viral infection. Whether P2 still has some remaining nucleic acid binding activity functioning in vivo, perhaps also involving other viral or cellular proteins, and what the relevance of this interaction is to the infection cycle are consequently still open questions.
In terms of structure, P2 can be divided into two subdomains. The N-terminal two-thirds has the propensity to form α-helices, whereas the C-terminal region seems not to be particularly structured (not shown). This dual organization certainly has to be taken into consideration during analyses of the structure/function relationship of P2. Sequence comparisons have revealed that residues 56 to 73 exhibit the heptad periodicity characteristic of coiled-coil structures (28), and Leclerc et al. have found that P2, similar to the corresponding P3 of CaMV, can form a tetramer in vitro (28). We were not able to show self-association of P2 in the yeast two-hybrid system. Either P2 does not self-assemble in vivo or a masking effect resulting from the fusion to Gal4 domains, which renders the putative coiled-coil motifs of P2 monomers inaccessible to each other, might explain the absence of the P2 homomeric interaction in this system.
Leclerc et al. (28) have proposed that the tetramerization of CaMV P3 in a parallel orientation could improve the affinity of its C-terminal domain for nucleic acids by a cooperative binding effect, and Tsuge et al. (44) have shown recently that CaMV P3 can form a tetramer in planta. Therefore, it is likely that the tetramerization of P2 also occurs in vivo and that, particularly in the context of the viral infection, this could reinforce the affinity and perhaps also the specificity of the interaction either with nucleic acids or with the CP (or both) by a spatial condensation of interacting domains. Such a property has been described for the ICP35 assembly protein of herpes simplex virus type 1 (HSV-1) (38) and has been used to create new high-affinity binding molecules (43). This could explain why mutants expressing P2 forms which do not retain strong affinity for the CP are still infectious.
Parallel studies in our laboratory have demonstrated that CaMV P3 also possesses the capacity to interact with the CaMV CP through its C-terminal extremity (D. Leclerc, unpublished data). When using the yeast two-hybrid system, we were not able to show cross-interactions between RTBV and CaMV proteins despite the similarities between these proteins (39; E. Herzog and D. Leclerc, unpublished data). This observation argues in favor of highly specific molecular interactions between P2 or P3 and their corresponding CPs.
The fact that P2 is associated with virions (22) and that it interacts with nucleic acids (25) and with the CP, strongly suggests a role for this protein in RTBV particle formation. To date, we have limited information on the general structure of RTBV particles, and the molecular interactions leading to virus assembly are unknown. From optical diffraction experiments, Hull (22) proposed that RTBV particles have a structure which is based on an icosahedral symmetry. More information is available for CaMV. Assembly of the icosahedral CaMV particles (4) takes place in inclusion bodies (40). The main component of these inclusions is the multifunctional viral protein P6, which interacts with the CP and was proposed to play an accessory role in virus assembly (19). Such inclusion bodies are not found in RTBV-infected tissues, and the precise location of virion assembly is still unknown. In addition to the CP, participation of other RTBV proteins may be required for virus assembly. Immunolabelling experiments have revealed the presence of P1 in disrupted RTBV virions (16). Similar results have shown that CoYMV P1 is also associated with immature virions (3). P2 was found closely associated with immature and mature virions in both viruses (3, 22; Druka and Hull, unpublished).
We propose that P2 could play a role similar to that of the scaffolding proteins of double-stranded DNA bacteriophages and to that of certain animal DNA viruses (18). P2 shares two main properties with scaffolding proteins of bacteriophage P22, HSV-1, and cytomegaloviruses (36, 38, 46). It can interact with the CP through its C-terminal extremity (our work) and can oligomerize (28). Consequently, P2 might participate in capsid assembly in two successive steps. First, P2 monomers could interact with the CP. P2 tetramerization may then promote interaction between CP subunits. P2 could thus recruit the CP subunits to the nascent capsid structure and facilitate the assembly of the capsid shell.
It has been shown that scaffolding proteins associate with their respective CP through either electrostatic (bacteriophage P22) (37) or hydrophobic (HSV-1, cytomegalovirus) (2, 21) interactions. For P2, we have shown in the yeast two-hybrid system that hydrophobic residues (I104, Y108) are important for CP binding and that two basic residues (K105, R106) contribute to it. This indicates that P2-CP association could combine both hydrophobic and electrostatic interactions.
Scaffolding proteins are normally only transiently associated with maturing virus particles but are not generally present in the mature virions (18). However, as shown by electron microscopy experiments (22; Druka and Hull, unpublished), some P2 remains associated with the capsid. Its role in the mature particles could be (i) inert, (ii) to stabilize the virions, or (iii) possibly to act in an additional step of the RTBV infection cycle.
It is interesting that for both P2 and the CP, the domains involved in nucleic acid interactions and in protein-protein interactions are overlapping or are very close to each other. The proximity of the functional domains could facilitate sequential molecular interactions during virus assembly. It has been shown recently in our laboratory that the CaMV CP can bind specifically to a region of the 35S pregenomic RNA leader (13). The zinc finger motif of the CP and the preceding basic domain were essential for this interaction, which is thought to play a role in either encapsidation, replication, or both; the two processes are probably coupled. Based on the fact that P3 of CaMV and the P2 proteins of badnaviruses and RTBV can interact nonspecifically with nucleic acids in vitro, Mesnard et al. (32, 33) and Jacquot et al. (24) have suggested that these proteins could be involved in condensing the newly synthesized double-stranded DNA genome, as proposed for the hepadnavirus core protein (15, 35). In the light of our results, it is likely that RTBV P2 is also involved in another, albeit closely related process during RTBV morphogenesis: assembly of the capsid shell.
ACKNOWLEDGMENTS
We are grateful to Helen Rothnie, Livia Stavolone, and Johannes Fütterer for critical reading of the manuscript and to David Kirk for technical assistance. We also thank Denis Leclerc, Lyubov Ryabova, Mikhail Pooggin, the other members of our laboratory, Vitaly Boyko, Arnis Druka, and Roger Hull for helpful discussions and unpublished data. We thank Arnis Druka, Margaret Boulton, and Roger Hull (John Innes Centre, Norwich, United Kingdom) for providing RTBV clones, antibodies, Agrobacterium tumefaciens strains, and rice seeds.
Orlene Guerra-Peraza was funded by the “Eidgenössische stipendienkomission für Ausländische Studierende”. Etienne Herzog is the recipient of an EMBO long-term fellowship and is also partially supported by the Roche Research Foundation.
REFERENCES
- 1.Bao Y, Hull R. Characterization of the discontinuities in rice tungro bacilliform virus DNA. J Gen Virol. 1992;73:1297–1301. doi: 10.1099/0022-1317-73-5-1297. [DOI] [PubMed] [Google Scholar]
- 2.Beaudet-Miller M, Zhang R, Durkin J, Gibson W, Kwong A D, Hong Z. Virus-specific interaction between the human cytomegalovirus major capsid protein and the C terminus of the assembly protein precursor. J Virol. 1996;70:8081–8088. doi: 10.1128/jvi.70.11.8081-8088.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng C P, Lockhart B E, Olszewski N E. The ORF I and II proteins of Commelina yellow mottle virus are virion-associated. Virology. 1996;223:263–271. doi: 10.1006/viro.1996.0478. [DOI] [PubMed] [Google Scholar]
- 4.Cheng R H, Olson N H, Baker T S. Cauliflower mosaic virus: a 420 subunit (T=7), multilayer structure. Virology. 1992;186:655–668. doi: 10.1016/0042-6822(92)90032-k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahal G, Hibino H, Saxena R C. Association of leafhopper feeding behavior with transmission of rice tungro to susceptible and resistant rice cultivars. Phytopathology. 1990;80:371–377. [Google Scholar]
- 6.Dasgupta I, Hull R, Eastop S, Poggi-Pollini C, Blackebrough M L, Boulton M I, Davies J W. Rice tungro bacilliform virus DNA independently infects rice after Agrobacterium-mediated transfer. J Gen Virol. 1991;72:1215–1221. doi: 10.1099/0022-1317-72-6-1215. [DOI] [PubMed] [Google Scholar]
- 7.Estojak J, Brent R, Golemis E A. Correlation of two-hybrid affinity data with in vitro measurements. Mol Cell Biol. 1995;15:5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fersht A. Structure and mechanism in protein science. W. H. New York, N.Y: Freeman & Co.; 1999. [Google Scholar]
- 9.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 10.Fütterer J, Rothnie H M, Hohn T, Potrykus I. Rice tungro bacilliform virus open reading frames II and III are translated from polycistronic pregenomic RNA by leaky scanning. J Virol. 1997;71:7984–7989. doi: 10.1128/jvi.71.10.7984-7989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fütterer J, Potrykus I, Bao Y, Li L, Burns T M, Hull R, Hohn T. Position-dependent ATT initiation during plant pararetrovirus Rice tungro bacilliform virus translation. J Virol. 1996;70:2999–3010. doi: 10.1128/jvi.70.5.2999-3010.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fütterer J, Potrykus I, Valles-Brau M P, Dasgupta I, Hull R, Hohn T. Splicing in a plant pararetrovirus. Virology. 1994;198:663–670. doi: 10.1006/viro.1994.1078. [DOI] [PubMed] [Google Scholar]
- 13.Guerra-Peraza O, de Tapia M, Hohn T, Hemmings-Mieszczak M. Interaction of the cauliflower mosaic virus coat protein with the pregenomic RNA leader. J Virol. 2000;74:2067–2072. doi: 10.1128/jvi.74.5.2067-2072.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurevich V V. Use of bacteriophage RNA polymerase in RNA synthesis. Methods Enzymol. 1996;275:382–397. doi: 10.1016/s0076-6879(96)75023-1. [DOI] [PubMed] [Google Scholar]
- 15.Hatton T, Zhou S, Standring D N. RNA- and DNA-binding activities in hepatitis B virus capsid protein: a model for their roles in viral replication. J Virol. 1992;66:5232–5241. doi: 10.1128/jvi.66.9.5232-5241.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay J M, Grieco F, Druka A, Pinner M, Lee S-C, Hull R. Detection of rice tungro bacilliform virus gene products in vivo. Virology. 1994;205:430–437. doi: 10.1006/viro.1994.1663. [DOI] [PubMed] [Google Scholar]
- 17.Hay J M, Jones M C, Blackebrough M L, Dasgupta I, Davies J W, Hull R. An analysis of the sequence of an infectious clone of rice tungro bacilliform virus, a plant pararetrovirus. Nucleic Acids Res. 1991;19:2615–2621. doi: 10.1093/nar/19.10.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendrix R W, Garcea R L. Capsid assembly of dsDNA viruses. Semin Virol. 1994;5:15–26. [Google Scholar]
- 19.Himmelbach A, Chapdelaine Y, Hohn T. Interaction between cauliflower mosaic virus inclusion body protein and capsid protein: implications for viral assembly. Virology. 1996;217:147–157. doi: 10.1006/viro.1996.0102. [DOI] [PubMed] [Google Scholar]
- 20.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 21.Hong Z, Beaudet-Miller M, Durkin J, Zhang R, Kwong A D. Identification of a minimal hydrophobic domain in the herpes simplex virus type 1 scaffolding protein which is required for interaction with the major capsid protein. J Virol. 1996;70:533–540. doi: 10.1128/jvi.70.1.533-540.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hull R. Molecular biology of rice tungro viruses. Annu Rev Phytopathol. 1996;34:275–297. doi: 10.1146/annurev.phyto.34.1.275. [DOI] [PubMed] [Google Scholar]
- 23.Hull R, Will H. Molecular biology of viral and nonviral retroelements. Trends Genet. 1989;5:357–359. doi: 10.1016/0168-9525(89)90151-0. [DOI] [PubMed] [Google Scholar]
- 24.Jacquot E, Hagen L S, Jacquemond M, Yot P. The open reading frame 2 product of cacao swollen shoot badnavirus is a nucleic acid-binding protein. Virology. 1996;225:191–195. doi: 10.1006/viro.1996.0587. [DOI] [PubMed] [Google Scholar]
- 25.Jacquot E, Keller M, Yot P. A short basic domain supports a nucleic acid-binding activity in the rice tungro bacilliform virus open reading frame 2 product. Virology. 1997;239:352–359. doi: 10.1006/viro.1997.8859. [DOI] [PubMed] [Google Scholar]
- 26.Laco G S, Beachy R N. Rice tungro bacilliform virus encodes reverse transcriptase, DNA polymerase, and ribonuclease H activities. Proc Natl Acad Sci USA. 1994;91:2654–2658. doi: 10.1073/pnas.91.7.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laco G S, Kent S B H, Beachy R N. Analysis of the proteolytic processing and activation of the rice tungro bacilliform reverse transcriptase. Virology. 1995;208:207–214. doi: 10.1006/viro.1995.1144. [DOI] [PubMed] [Google Scholar]
- 28.Leclerc D, Burri L, Kajava A V, Mougeot J L, Hess D, Lustig A, Kleemann G, Hohn T. The open reading frame III product of cauliflower mosaic virus forms a tetramer through a N-terminal coiled-coil. J Biol Chem. 1998;273:29015–29021. doi: 10.1074/jbc.273.44.29015. [DOI] [PubMed] [Google Scholar]
- 29.Lockhart B E L. Evidence for a double-stranded circular DNA genome in a second group of plant viruses. Phytopathology. 1990;80:127–131. [Google Scholar]
- 30.Marmey P, Bothner B, Jacquot E, de Kochko A, Ong C A, Yot P, Siuzdak G, Beachy R N, Fauquet C M. Rice tungro bacilliform virus open reading frame 3 encodes a single 37-kDa coat protein. Virology. 1999;253:319–326. doi: 10.1006/viro.1998.9519. [DOI] [PubMed] [Google Scholar]
- 31.Mayo M A, Pringle C R. Virus taxonomy—1997. J Gen Virol. 1998;79:649–657. doi: 10.1099/0022-1317-79-4-649. [DOI] [PubMed] [Google Scholar]
- 32.Mesnard J M, Carriere C. Comparison of packaging strategy in retroviruses and pararetroviruses. Virology. 1995;213:1–6. doi: 10.1006/viro.1995.1540. [DOI] [PubMed] [Google Scholar]
- 33.Mesnard J M, Kirchherr D, Wurch T, Lebeurier G. The cauliflower mosaic virus gene III product is a non-sequence-specific DNA binding protein. Virology. 1990;174:622–624. doi: 10.1016/0042-6822(90)90118-b. [DOI] [PubMed] [Google Scholar]
- 34.Mougeot J L, Guidasci T, Wurch T, Lebeurier G, Mesnard J M. Identification of C-terminal amino acid residues of cauliflower mosaic virus open reading frame III protein responsible for its DNA binding activity. Proc Natl Acad Sci USA. 1993;90:1470–1473. doi: 10.1073/pnas.90.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive- strand DNA synthesis but not for virus assembly. J Virol. 1992;66:4107–4116. doi: 10.1128/jvi.66.7.4107-4116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker M H, Casjens S, Prevelige P E J. Functional domains of bacteriophage P22 scaffolding protein. J Mol Biol. 1998;281:69–79. doi: 10.1006/jmbi.1998.1917. [DOI] [PubMed] [Google Scholar]
- 37.Parker M H, Prevelige P E J. Electrostatic interactions drive scaffolding/coat protein binding and procapsid maturation in bacteriophage P22. Virology. 1998;250:337–349. doi: 10.1006/viro.1998.9386. [DOI] [PubMed] [Google Scholar]
- 38.Pelletier A, Do F, Brisebois J J, Lagace L, Cordingley M G. Self-association of herpes simplex virus type 1 ICP35 is via coiled-coil interactions and promotes stable interaction with the major capsid protein. J Virol. 1997;71:5197–5208. doi: 10.1128/jvi.71.7.5197-5208.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu R D, Bhattacharyya M, Laco G S, de Kochko A, Rao B L, Kaniewska M B, Elmer J S, Rochester D E, Smith C E, Beachy R N. Characterization of the genome of rice tungro bacilliform virus: comparison with Commelina yellow mottle virus and caulimoviruses. Virology. 1991;185:354–364. doi: 10.1016/0042-6822(91)90783-8. [DOI] [PubMed] [Google Scholar]
- 40.Rothnie H M, Chapdelaine Y, Hohn T. Pararetroviruses and retroviruses: a comparative review of viral structure and gene expression strategies. Adv Virus Res. 1994;44:1–67. doi: 10.1016/s0065-3527(08)60327-9. [DOI] [PubMed] [Google Scholar]
- 41.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 42.Temin H M. Reverse transcription in the eukaryotic genome: retroviruses, pararetroviruses, retrotransposons and retrotranscripts. Mol Biol Evol. 1985;2:455–468. doi: 10.1093/oxfordjournals.molbev.a040365. [DOI] [PubMed] [Google Scholar]
- 43.Terskikh A V, Le Doussal J M, Crameri R, Fisch I, Mach J P, Kajava A V. “Peptabody”: a new type of high avidity binding protein. Proc Natl Acad Sci USA. 1997;94:1663–1668. doi: 10.1073/pnas.94.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuge S, Kobayashi K, Nakayashiki H, Mise K, Furusawa I. Cauliflower mosaic virus ORF III product forms a tetramer in planta: its implication in viral DNA folding during encapsidation. Microbiol Immunol. 1999;43:773–780. doi: 10.1111/j.1348-0421.1999.tb02469.x. [DOI] [PubMed] [Google Scholar]
- 45.Tzafrir I, Ayala-Navarrete L, Lockhart B E, Olszewski N E. The N-terminal portion of the 216-kDa polyprotein of Commelina yellow mottle badnavirus is required for virus movement but not for replication. Virology. 1997;232:359–368. doi: 10.1006/viro.1997.8569. [DOI] [PubMed] [Google Scholar]
- 46.Wood L J, Baxter M K, Plafker S M, Gibson W. Human cytomegalovirus capsid assembly protein precursor (pUL80.5) interacts with itself and with the major capsid protein (pUL86) through two different domains. J Virol. 1997;71:179–190. doi: 10.1128/jvi.71.1.179-190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]