

Abstract
Chemical etching of nano-sized metal clusters at the atomic level has a high potential for creating metal number-specific structures and functions that are difficult to achieve with bottom-up synthesis methods. In particular, precisely etching metal atoms one by one from nonmetallic element-centred metal clusters and elucidating the relationship between their well-defined structures, and chemical and physical properties will facilitate future materials design for metal clusters. Here we report the single-gold etching at a hypercarbon centre in gold(I) clusters. Specifically, C-centred hexagold(I) clusters protected by chiral N-heterocyclic carbenes are etched with bisphosphine to yield C-centred pentagold(I) (CAuI5) clusters. The CAuI5 clusters exhibit an unusually large bathochromic shift in luminescence, which is reproduced theoretically. The etching mechanism is experimentally and theoretically suggested to be a tandem dissociation-association-elimination pathway. Furthermore, the vacant site of the central carbon of the CAuI5 cluster can accommodate AuCl, allowing for post-functionalisation of the C-centred gold(I) clusters.
Subject terms: Organometallic chemistry, Chemical synthesis, Nanoscale materials
The control of atomically precise etching of nano-sized metal clusters is important for understanding their structure-specific properties. Here, the authors report the etching of a single gold atom on a hypercarbon centre of gold(I) clusters.
Introduction
Etching is a top-down method to downsize the structures at the atomic level and modify the chemical and physical properties of a wide range of nanomaterials such as nanocrystals and colloidal nanoparticles1–5, and nanoclusters6–11 for a variety of applications. For example, chemical etching methods that involve ligand engineering have made great advances, such as thiolate etching of phosphine- or thiolate-protected nanogold clusters6–8, reverse etching of thiolated-Au25 with phosphine (exchange of strong donor ligands for weaker donor ligands)9, and phosphine exchanged by N-heterocyclic carbene (NHC) to give NHC-containing Au1110,11. Rapid developments in X-ray crystallography have revealed that chemical etching alters the nanocluster structures of the metal core12 and ligand surface13–17. However, the control and understanding of chemical etching at the atomic level have only just begun18,19. Recently, Cao et al. used real-time electrospray ionisation mass spectrometry to reveal degradation and anomalous recombination processes in the chemical etching of Au25 nanoclusters19. It also remains controversial whether the ligand-exchange mechanisms in nanogold regions containing AuI and Au0 atoms is SN2-like bimolecular nucleophilic substitution or SN1-like type unimolecular nucleophilic substitution20–22. Despite the promise of chemical etching as a general technique to downsize metal clusters at the atomic level, little attention has been paid to ligand-protected AuI clusters.
Among the AuI cluster family, clusters radially coordinated to main-group elements23–29 such as O-centre23, N-centre24,25, C-centre26, and S-centre27–29 are attractive due to the polyhedral structures similar to nanogold clusters, AuI⋅⋅⋅AuI interactions30–34 and structure-dependent photophysical properties. In particular, the hypercoordinated carbon (hypercarbon)35-centred hexagold(I) (CAuI6) cluster [C(AuI-L)6]X2 (L = ligand; X = counterion)26 that bridges nano-sized metal clusters and organic molecules36,37 has attracted attention and significant advances have been made in this area. Many of these CAuI6 clusters exhibit structure-specific luminescence38–44 and can be used as bio-labels39,44 by using highly bottom-up designable shell ligands based on phosphines38–40 and NHCs41–45. However, there is only one isolated example of a C-centred pentagold(I) cluster [C(AuI-L)5]X (L = triphenylphosphine, TPP) produced by the bottom-up synthesis of aurating CH2[B(OCH3)2]2 with a gold(I) complex, and if the reaction time is extended, the TPP-protected CAuI6 cluster is the main product46. Thus, it is still difficult to control the number of AuI atoms bound to the hypercarbon centre. In particular, from the viewpoint of the effects of reducing the number of AuI atoms on photophysical properties and reactivity, the development of a highly generalised single-gold etching method for gold(I) clusters is an important research topic.
Here, we discovered that a chiral NHC-protected C-centred hexagold(I) cluster can be etched with a bisphosphine ligand to generate a chiral NHC-protected CAuI5 cluster. This was achieved by controlling the number of gold atoms centred at the hypercarbon at the atomic level (Fig. 1). Furthermore, this etching method is also useful for the synthesis of TPP-protected CAuI5 analogues. In general, smaller gold clusters show more blue-shifted emission than larger clusters, but the ligand-protected CAuI5 clusters show unusually red-shifted signals in both absorption and emission spectra compared to the CAuI6 counterparts, which was rationalised by theoretical calculations. Further experimental and theoretical studies suggest that a tandem dissociation-association-elimination pathway is involved in the etching mechanism. The NHC-protected CAuI6 clusters are generally chemically stable, and the active site had to be placed on the hypercarbon to confer reactivity. In fact, CAuI5 synthesised by this method was highly reactive with AuCl, producing a CAuI6 cluster with a different ligand. Thus, the chemical etching method is expected to be a way not only to reduce the size of metal ion clusters and significantly change their electronic structure, but also to asymmetrise the metal ion cluster structure and provide active sites.
Fig. 1. The schematic illustration of etching ligand-protected C-centred gold(I) cluster.
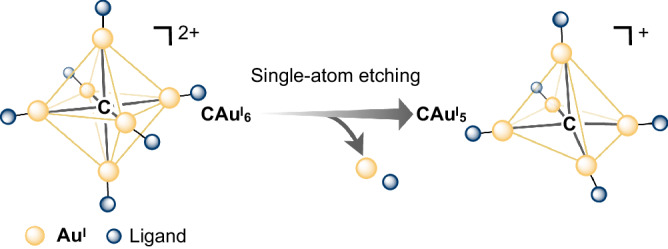
Etching-induced elimination of [LAuI] at the hypercarbon centre of the ligand-protected C-centred hexagold(I) (CAuI6) cluster results in the formation of the ligand-protected C-centred pentagold(I) (CAuI5) cluster, wherein the ligand can be an N-heterocyclic carbene or phosphine.
Results
Synthesis of CAuI5 by etching CAuI6 with bisphosphine
We previously reported enantiopure NHC-protected asymmetrically twisted CAuI6 clusters: [(C)(AuI-SS-NHC)6](BF4)2 and [(C)(AuI-RR-NHC)6](BF4)2 ( = SS- and RR-1NHC, SS-NHC = N,N’-bis[(S)-α-methylbenzyl]-benzimidazol-2-ylidene, RR-NHC = N,N’-bis[(R)-α-methylbenzyl]-benzimidazol-2-ylidene)43. Although NHCs are generally thought to bind strongly to coinage metal47,48, we investigated whether etching occurs when bisphosphine is added to NHC-protected CAuI6 clusters. For example, when 2.5 equiv. of cis-1,2-bis(diphenylphosphino)ethene (cis-depe) were added to a dichloromethane solution of SS-1NHC, the original pale-yellow solution immediately turned orange. Subsequent crystallisation from diethyl ether/dichloromethane at 4 °C gave orange-red blocky crystals of [(C)(AuI-SS-NHC)5](BF4) (SS-2NHC) in 80% yield on a hypercarbon basis. Its enantiomer [(C)(AuI-RR-NHC)5](BF4) (RR-2NHC) was also synthesised. Another product [(cis-depe)2AuI](BF4) (3) was obtained as colourless blocky crystals by prolonged recrystallisation. They were characterised by ESI-MS spectrometry, NMR spectroscopy and elemental analysis (Suppl. Figs. 2–12). The ESI-MS spectrum found the CAuI5 cluster SS-2NHC at m/z 2628.85 corresponding to [(C)(AuI-SS-NHC)5]+ (calcd. 2628.73). In the 1H NMR spectrum of the CAuI5 cluster SS-2NHC in d6-acetone, the signals attributed to the NHC ligand showed a significant downshift compared to those of the CAuI6 cluster SS-1NHC (Suppl. Fig. 6). This is attributed to the magnetic environment, which is deshielded from the shell ligands with less steric hindrance. Similarly in the 13C NMR spectra, the signal at 198.8 ppm attributed to the NHC carbon-donors of SS-2NHC was downshifted from those of SS-1NHC at 190.0 ppm (Suppl. Fig. 7), suggesting a marked influence on resonances from different gold nuclearities.
Moreover, a phosphine analogue [(C)(AuI-TPP)5](BF4) (2TPP) was also obtained in 90% yield by etching [(C)(AuI-TPP)6](BF4)2 (1TPP)26 instead. The 31P NMR spectrum of 2TPP in d6-acetone showed a singlet signal at 32.6 ppm, which was downshifted from that of 1TPP (Suppl. Fig. 8)46. Furthermore, etching SS-1NHC and 1TPP with excess cis-depe (50 equiv.) yielded corresponding pentagold(I) clusters in both cases, with no detectable smaller gold species such as tetragold(I) or trigold(I) clusters. Etching SS-1NHC and 1TPP with 1,2-bis(diphenylphosphino)benzene also yielded the corresponding CAuI5 clusters. These data indicate that etching the CAuI6 cluster with cis-depe provides high selectivity for the CAuI5 cluster.
Single-crystal structures and computational bonding analysis
The single-crystal X-ray diffraction (SCXRD) determined structures in Fig. 2 show the overall structure of SS- and RR-2NHC including a hypercarbon, five gold(I) ions, five ligands and a BF4– counterion. SS- and RR-2NHC are crystallised in the I4 space group with low flack parameters of 0.010(9) and –0.027(11), respectively. Their flack parameters are very low (Suppl. Table 1), suggesting that optically pure molecules are packed. Take the example of SS-2NHC as shown in Table 1, the AuI⋅⋅⋅AuI distances (2.8667(10)–3.3141(15) Å) and the CNHC–AuI bonds (2.03(2)–2.08(4) Å) are similar to those in SS-1NHC, but the Ccentre–AuI bonds (2.03(2)–2.075(8) Å) of SS-2NHC are slightly shorter than those of SS-1NHC (2.100(14)–2.126(12) Å)43, suggesting that the endohedral five Ccentre–AuI bonds in the CAuI5 cluster are more favourable. The [(C)(AuI-L)5]+ cation in SS-2NHC can be regarded as eliminating one [LAuI] moiety from the [(C)(AuI-L)6]2+ cation in SS-1NHC. As a result, the NHC ligands of SS-2NHC rearrange themselves to minimise mutual steric hindrance (in Fig. 2b, three grey-coloured benzimidazolylidene moieties on the same plane and two orange-coloured benzimidazolylidene moieties on two planes with a 63° crossing angle), forming a monocationic CAuI5 cluster with C2-symmetry (Suppl. Fig. 15). It should be noted that the hypercarbon of SS-2NHC is close to the centroid of the four gold atoms at the bottom of the square pyramid (0.46(3) Å), which could be an important coordinating site for post-functionalisation (vide infra). Meanwhile, the surface vacancy found in this SS-2NHC molecule is well shielded in its packing structure by intermolecular interactions with a ligand on the gold(I) at the apex of another cluster molecule (Suppl. Fig. 16), thus maintaining high chemical stability in the solid state. In contrast, the phosphine-protected analogue 2TPP crystallised in the P21/n space group (Fig. 2c)42 and exhibited AuI⋅⋅⋅AuI interactions (2.85528(18)–3.21332(19) Å) and P–AuI bonds (2.2546(8)–2.2735(8) Å). The Ccentre–AuI bonds (2.064(3)–2.082(3) Å) of 2TPP are slightly shorter than those of CAuI6 counterpart (average 2.12 Å)26, similar to the shorter Ccentre–AuI bonds of SS-2NHC compared to SS-1NHC.
Fig. 2. Synthesis and characterisation of C-centred pentagold(I) clusters.

a Etching syntheses of CAuI6 clusters26,43 to CAuI5 clusters using cis-1,2-bis(diphenylphosphino)ethene (cis-depe) (Suppl. Fig. 1). b Single-crystal X-ray diffraction (SCXRD) structures of the cations [(C)(AuI-L)5]+ (L = SS- and RR-NHC) and the CAuI5 cores of SS- and RR-2NHC with optically active NHC ligands. c SCXRD structures of the cation [(C)(AuI-TPP)5]+ 46 and the core of 2TPP with TPP. d Microscopy images of crystals of SS-2NHC (orange crystals) and 2TPP (yellow crystals) under ambient light. e Explanation and examples of geometry index τ values51, which is used for showing the geometric differences of CAuI5 cores with NHCs and TPP (Table 1). The τ value represents the geometric difference between the regular square pyramidal (τ = 0) and trigonal bipyramidal (τ = 1), τ = (β – α)/60°, where α and β are the two largest basal angles. Colour code: Au, yellow; C, grey; N, blue; P, orange; H, white. BF4– counterions and solvent molecules are omitted for clarity.
Table 1.
Selected structural parameters of SS2NHC and 2TPP
| SS-2NHC | 2TPP | |
|---|---|---|
| AuI⋅⋅⋅AuI (Å) | 2.8667(10)–3.3141(15) | 2.85528(18)–3.21332(19) |
| Ccentre–AuI (Å) | 2.03(2)–2.075(8) | 2.064(3)–2.082(3) |
| CNHC–AuI (Å) | 2.03(2)–2.08(4) | / |
| P–AuI (Å) | / | 2.2546(8)–2.2735(8) |
| τ | 0.32 | 0.68 |
Bond distances (Å) of AuI⋅⋅⋅AuI, Ccentre–AuI, CNHC–AuI and P–AuI, and geometry index τ values. The statistical significance of the errors for the bond distances is derived from the precision of the SCXRD data (Suppl. Table 1).
Moreover, the bonding characters of CAuIn (n = 5, 6) clusters were computationally studied based on the above crystallography data, and the bond distances of the crystal structures were well reproduced by the density functional theory (DFT) calculations. The calculated Ccentre–AuI and AuI⋅⋅⋅AuI distances of CAuIn (n = 5, 6) cores as well as Wiberg bond orders (WBO) suggested very interesting structural dependencies (Suppl. Table 3). The Ccentre–AuI and AuI⋅⋅⋅AuI distances of the N,N’-diisopropylimidazolidene (IiPr)-protected CAuI6 cluster41 are 2.19 and 3.10 Å, respectively. Their bond orders are 0.41 and 0.16, respectively, indicating that the Ccentre–AuI bond is stronger than each AuI⋅⋅⋅AuI interaction. In the CAuI6 cores of SS-1NHC and 1TPP, the binding characteristics obtained are largely unchanged. Of note, the Ccentre–AuI bonding in the CAuI5 cores of SS-2NHC and 2TPP is slightly stronger compared to the corresponding CAuI6 clusters. This is demonstrated by the shorter bond lengths (2.09–2.16 Å) and larger WBO values (0.50–0.57). Regarding the aurophilic interactions in the CAuI5 cores, the AuI⋅⋅⋅AuI distances and bond orders are nearly the same as those of CAuI6 cores. Therefore, missing one Au atom in the CAuI5 cores may result in stronger Ccentre–AuI bonds, which may be important for stabilising the CAuI5 clusters. Moreover, the orbital interactions of the CAuI6 cluster were previously discussed in detail49,50: the SS-1NHC and SS-2NHC clusters have [CAu6]2+ and [CAu5]+ cores, respectively, and the C–AuI bond orders exhibit an unusual C–AuI bond hypervalence.
In addition, to better understand the geometric differences of CAuI5 cores with NHCs and TPP, we introduced the index parameter τ (Fig. 2e, Table 1), wherein τ is 0 for perfect square pyramidal and 1 for perfect trigonal bipyramidal51. This evaluation method suggests that the CAuI5 cores of SS- and RR-2NHC have a distorted square pyramidal geometry (τ = 0.32), while the CAuI5 core of 2TPP is much closer to a trigonal bipyramidal geometry (τ = 0.68), indicating that NHCs and TPP exert different ligand effects on the CAuI5 core.
Absorption, emission profiles, and theoretical calculations
The CAuI6 clusters are known to be efficient emitters with intriguing structure-dependent properties38–45, while the photophysical properties of the CAuI5 clusters remain unknown. In general, reducing the metal core size is known to induce a blue shift in absorption and emission52. However, the UV-vis spectra of SS-2NHC and 2TPP in dichloromethane, in contrast, showed their maximum absorption wavelengths at 420 nm and 382 nm, respectively, and were significantly more red-shifted than those of CAuI6 clusters, SS-1NHC (λmax = 373 nm) and 1TPP (λmax = 365 nm) (Suppl. Fig. 17). Similarly, photoluminescence of the CAuI5 clusters showed a bathochromic shift signal in contrast to the CAuI6 clusters (Fig. 3a, Suppl. Fig. 18). The solid-state SS-2NHC exhibited orange-red emission (λemmax = 676 nm), which is 151 nm more red-shift than SS-1NHC. The acetone solution of SS-2NHC was also red-emissive, with no apparent solvation effects (Suppl. Fig. 19). On the other hand, the emission of 2TPP in the solid state is 59 nm more red-shifted than 1TPP and emits yellow at 365 nm excitation (λemmax = 584 nm). Neither 1TPP nor 2TPP emits light in solution. This is because the terminal coordination of phosphine to the gold(I) atom may facilitate nonradiative relaxation pathways38. Comparing the absolute quantum yields (Φ), SS-2NHC in the solid state showed the strongest emission. Here, the Φ values of SS-2NHC, SS-1NHC, 2TPP, and 1TPP were 0.61, 0.02, 0.29, and 0.19, respectively (Suppl. Fig. 20). They exhibit microsecond-level lifetimes (Suppl. Fig. 21), suggesting phosphorescence properties in the solid state.
Fig. 3. Photoluminescence of C-centred gold(I) clusters in the solid state and the theoretical study.

a Emission spectra: SS-1NHC (blue line, excited by 266 nm), SS-2NHC (orange line, excited by 510 nm), 1TPP (blue line, excited by 356 nm), 2TPP (yellow line, excited by 365 nm), insets: photographs (size: 12 mm ×12 mm) of crystals under 365 nm UV-light irradiation. b The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) distributions of SS-2NHC. c HOMO–LUMO gap values based on theoretical calculations: SS-1NHC (3.99 eV), SS-2NHC (3.30 eV), 1TPP (4.50 eV), and 2TPP (3.98 eV). Source data are provided as a Source Data file.
To gain insight into the optical properties specific to their electronic structures, we performed DFT and time-dependent (TD)-DFT calculations based on the SCXRD structures (Suppl. Figs. 22–27). The UV-vis spectrum of SS-2NHC calculated by TD-DFT (Suppl. Fig. 22) reproduces well the maximum band in the range from 393 to 415 nm, which is mainly due to the transitions of HOMO-1 → LUMO, HOMO-2 → LUMO and HOMO → LUMO + 1 (Suppl. Table 4). Orbital composition analysis by Mulliken partition (Suppl. Table 6) reveals that the occupied orbitals of HOMO-n (n = 0–2) in SS-2NHC are mainly derived from gold(I) ions (52.6–55.4%) with an increased contribution from the hypercarbon (26.9–32.5%) compared with that in SS-1NHC 43. The unoccupied orbitals of LUMO+n (n = 0, 1) in SS-2NHC are localised mainly at NHCs (64.7–74.8%), with small fractions of gold(I) ions (25.0–35.2%). Thus, the dominant metal-to-ligand charge-transfer (MLCT) mixed with a slight metal-centred (MC) charge-transfer was responsible for the low-energy absorption bands42. On the other hand, the compositions of the frontier orbitals of the TPP-protected CAuIn (n = 5, 6) clusters are also comparable (Suppl. Tables 7, 8, Suppl. Figs. 26, 27). These theoretical results suggest that the C-centred gold(I) core and ligands are essentially enrolled in their electronic structures, and thus different optical features can be explained by altering the gold nuclearity and ligands primarily via the MLCT transition42.
Importantly, the HOMO–LUMO gaps calculated for these CAuIn (n = 5, 6) clusters (Fig. 3c) show that the gaps of CAuI5 clusters are clearly smaller than those of the CAuI6 clusters: 3.30 eV (SS-2NHC) < 3.99 eV (SS-1NHC), 3.98 eV (2TPP) < 4.50 eV (1TPP). This is in good agreement with the fact that the absorptions of CAuI5 clusters are more red-shifted than those of the CAuI6 clusters. The calculated phosphorescence energies well reproduce the smaller phosphorescence energy of SS-2NHC (1.83 eV) than that of SS-1NHC (2.36 eV) (Suppl. Table 9) observed in experiments, which confirms that the emission of SS-2NHC is red-shifted and reveals that the C-centred gold(I) clusters have a pronounced size-dependent effect on the photoluminescence properties.
Probing the atomic-level etching process
To understand the mechanism of this efficient etching method to reshape C-centred gold(I) clusters, we investigated this process using UV-vis absorption spectroscopy, NMR spectroscopy and ESI-MS spectrometry. First, the UV-vis spectra of SS-1NHC etched with cis-depe showed a rapid change (Fig. 4a,b). When cis-depe was added to a dichloromethane solution of SS-1NHC (c = 5 × 10–5 M, 293 K), its characteristic peak (λmax = 345 nm) gradually decreased and new peaks appeared at 385 nm and 421 nm derived from SS-2NHC. Accordingly, the original pale-yellow solution turned bright yellow. In contrast, the time-course UV-vis spectra of 1TPP etched with cis-depe (Suppl. Fig. 28) showed that the peak at 382 nm appeared even more instantaneously for 2TPP. The colourless solution turned yellow within 5 s, a much faster change than etching SS-1NHC. This can be reasonably explained by the weaker binding of triphenylphosphine to gold than NHC48, consistent with the shorter CNHC–AuI bonds than P–AuI bonds in the single-crystal structures.
Fig. 4. Monitoring the etching process.

a Time-course UV-vis absorption spectra of the etching reaction of SS-1NHC (c = 5 × 10–5 M, 293 K) using cis-depe in dichloromethane: stage A, 1 s intervals; stage B, 5 min intervals; insets: photographs taken at the beginning of the reaction and after 70 min under ambient light, and a colour scale corresponding to the absorbance changes of the reaction. b The changes in absorbance at 345 nm and 421 nm as a function of time (corresponding to the spectra in a), suggesting that SS-2NHC was rapidly formed. c 1H NMR spectrum (d6-acetone, 300 K), showing signals of SS-1NHC (purple) and internal standard (IS, 1,3,5-trimethoxybenzene, blue-labelled). d Signals from SS-2NHC (pink-labelled, 53% yield) measured immediately after adding cis-depe. e After 0.5 h, SS-2NHC was formed in 98% yield. f A proposed etching mechanism with two intermediates Int1L and Int2L (L = NHC, TPP), with Int1TPP and Int2TPP detected by ESI-MS spectrometry (Suppl. Fig. 32). Colour code: Au, yellow; C, grey; L, cyan; P, orange. Source data are provided as a Source Data file.
Next, the 1H NMR spectra of SS-1NHC etched with cis-depe in d6-acetone were measured over time (Fig. 4c–e). The results showed that SS-2NHC was formed in 98 % yield 0.5 h after the addition of cis-depe (1,3,5-trimethoxybenzene as the internal standard), with the detection of another product [(cis-depe)2AuI](BF4) (3) (Suppl. Fig. 29). On the other hand, when 1TPP was etched with cis-depe, the time-course of the 1H NMR spectra in d6-acetone showed that 2TPP was formed in 88% yield after 18 min (Suppl. Fig. 30). The 31P NMR spectrum in d6-acetone after the reaction showed signals for complex 3 (δ 21.3 ppm) and free triphenylphosphine (δ –4.2 ppm). Furthermore, we turned to ESI-MS spectrometry to obtain more molecular information. As a result, a signal corresponding to SS-2NHC was observed at m/z 2628.71 (calcd. 2628.73 for [(C)(AuI-SS-NHC)5]+) immediately after adding cis-depe, and the very weak signals of two intermediates Int1NHC and Int2NHC were found (Suppl. Fig. 31). Interestingly, in the process of etching 1TPP, two di-cationic mass peaks, Int1TPP [(C)(AuI-TPP)6]2+ (m/z found 1252.08, calcd. 1252.12) and Int2TPP ([(C)(AuI-TPP)5(AuI-cis-depe)]2+ (m/z found 1450.62, calcd. 1450.86) were detected (Suppl. Fig. 32). However, no information was available for the association adduct [(C)(AuI-TPP)6(cis-depe)]2+ by binding cis-depe to 1TPP. This would suggest that the initial stage of etching phosphine-protected CAuI6 cluster with cis-depe is a dissociation process. Similarly, the formation of the association adduct [(C)(AuI-SS-NHC)6(cis-depe)]2+ would be difficult due to the steric hindrance from NHC ligands in SS-1NHC. Overall, as shown in Fig. 4f, the initial dissociation process similar to the ligand-exchange SN1-like mechanism22 generating the first intermediates Int1L (L = TPP or NHC) would occur when etching the ligand-protected CAuI6 clusters. Subsequent association with cis-depe would form the second intermediates Int2L (L = TPP or NHC), and then the elimination of AuI with cis-depe finally produce the corresponding CAuI5 clusters.
To illustrate the proposed etching mechanism as shown in Fig. 4f, we computed the energy profiles of the etching process in dichloromethane (Suppl. Fig. 33). In the first dissociation stage, to break one of the six AuI–L bonds from the original ligand-protected CAuI6 cluster, for example when L = TPP, Int1TPP formed in relatively high energy (30.2 kcal mol–1), suggesting the high stability of 1TPP. In the second association stage, when cis-depe coordinated to Int1TPP, the resulting Int2TPP was largely stabilised with a dramatically decreased energy of 10.9 kcal mol–1. Third, followed by the elimination of [cis-depeAu]+ to form a highly stable complex 3, 2TPP was finally formed with an energy of 16.4 kcal mol–1 via breaking the Ccentre–AuI bond and four AuI⋅⋅⋅AuI contacts of Int2TPP. The energy profiles were similarly illustrated when L = NHC (Suppl. Fig. 33b). Therefore, the theoretical data supported the tandem dissociation-association-elimination pathway in this etching process.
Stability study of CAuI5 clusters
SS-2NHC and 2TPP in the solid state are stable for more than a year under ambient conditions, but they are reactive in solution, in contrast to the more stable CAuI6 clusters43. The changes over time of 1H NMR spectra indicate that the solution of SS-2NHC in d6-acetone is stable for at least one week (Suppl. Fig. 34). On the other hand, once dissolved in CDCl3, both SS-2NHC and 2TPP partially reverted to the CAuI6 cluster (Suppl. Figs. 35, 36), and the original yellowish solutions gradually faded. These results suggest that the CAuI5 clusters are more reactive in solution than the corresponding CAuI6 clusters, which is consistent with the higher energies of the CAuI5 clusters in the calculated energy profiles described above.
Reactivity of the CAuI5 cluster
The metal clusters with exposed surfaces are of growing interest53. The more open-spaced coordination site at the bottom of the distorted square pyramidal structures of SS- and RR-2NHC were assumed to be the site where the sixth AuI species is most accessible to the hypercarbon. Therefore, we added an acetone solution of (tht)AuICl (1 equiv.) to an acetone solution of SS-2NHC at room temperature. The reaction was conducted in an ultrasonication bath for 20 min, and the crystallisation yielded a heteroleptic Cl-coordinated CAuI6 cluster [(C)(AuI-SS-NHC)5(AuICl)](BF4) (SS-4NHC) (Fig. 5, see characterisation data in Suppl. Figs. 37–41, Suppl. Table 10). Its enantiomer RR-4NHC was similarly obtained. In the overall SCXRD structures of SS- and RR-4NHC, in particular, the structures corresponding to the five NHC ligand parts of the CAuI5 clusters, SS- and RR-2NHC, were found to be largely intact (Suppl. Fig. 37). The sixth gold(I) atom was coordinated to the Cl-anion with bond distances of 2.308(6) Å and 2.310(7) Å in SS- and RR-4NHC, respectively. However, the introduction of AuCl into 2TPP was not successful, probably due to its low stability, and only the original CAuI6 cluster 1TPP was finally isolated. Given the NHC-protected monogold chloride complexes are extensively used as active catalysts54–56, this cluster-based analogue Cl-coordinated CAuI6 cluster protected by the NHC ligands will be a milestone in the development of highly reactive hexagold(I) clusters.
Fig. 5. Reactivity of the hypercarbon in CAuI5 clusters.
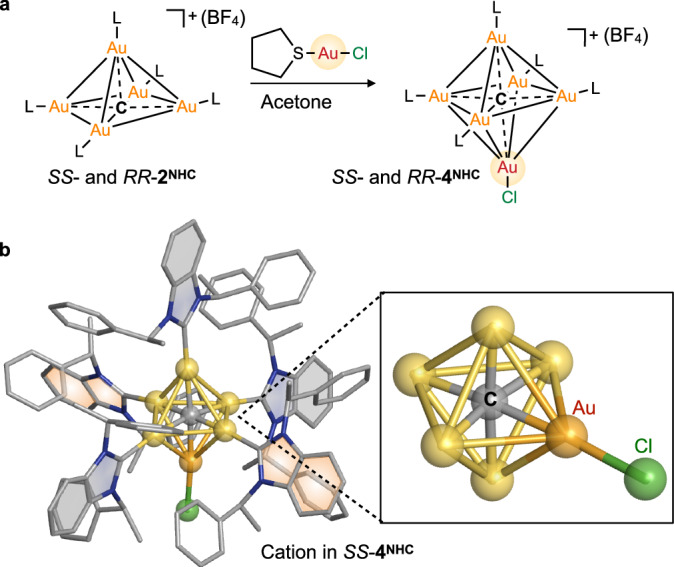
a Reaction of SS-(or RR-)2NHC with (tht)AuICl (tht = tetrahydrothiophene) yielded Cl-coordinated CAuI6 clusters: SS- and RR-4NHC [(C)(AuI-L)5(AuICl)](BF4) (L = SS- and RR-NHC). b The SCXRD structure of the [(C)(AuI-SS-NHC)5(AuICl)]+ cation and the Cl-coordinated CAuI6 core in SS-4NHC. The arrangement of three grey-coloured benzimidazolylidene moieties on the same plane and two orange-coloured benzimidazolylidene moieties on two planes in SS-4NHC is similar to that in SS-2NHC (Suppl. Fig. 37). Colour code: Au, yellow and orange; C, grey; N, blue; Cl, green. Hydrogen atoms, BF4– counterion, and solvent molecules are omitted for clarity.
In addition, the Cl-coordinated CAuI6 clusters SS- and RR-4NHC restored very weak green emission in the solid state and no emission was observed in the solution at room temperature. In particular, the circular dichroism spectra of SS- and RR-2NHC and SS- and RR-4NHC in dichloromethane (Suppl. Fig. 42) showed similar chiroptical signals with the strongest signal at 250 nm contributed mainly by the chiral ligands, which can be explained by the similar arrangement of the ligand shell and symmetric metal cores. In a word, the etch-produced CAuI5 clusters (SS- and RR-2NHC) and the post-functionalised Cl-coordinated CAuI6 clusters (SS- and RR-4NHC) exhibit different properties from the original CAuI6 clusters (SS- and RR-1NHC), thus revealing intriguing structure-property relationships by using etching as atomically precise “surgery” at the hypercarbon atom.
Discussion
In summary, we have shown that etching of the NHC-protected CAuI6 clusters allows size-selective synthesis of the corresponding CAuI5 clusters. The peculiar red-shift signals in the absorption and emission of CAuI5 clusters can be explained by theoretical calculations. A tandem dissociation-association-elimination pathway for the atomical-level etching was proposed based on experimental and theoretical studies. The envisaged coordination ability of the hypercarbon atom in CAuI5 clusters was confirmed by adding AuICl, leading to the novel heteroleptic Cl-coordinated CAuI6 clusters. These results of the single-gold etching of the CAuI6 clusters at the atomic level indicate a unique and highly generalised method using phosphine ligands for etching of NHC-protected gold clusters. This study not only elucidated the unusual photophysical properties of metal clusters containing fewer metal nuclei, but also provided opportunities to explore post-functionalisation and reactivities in surface-exposed metal ion clusters. This result shows that establishing a synthesis method using precision etching of CAuIn (n < 6) clusters is important for elucidating the chemical and physical properties and reactivity of unsymmetric clusters. Therefore, the chemical etching method is a way to reduce the size of metal ion clusters and will be developed to control the electronic structure, asymmetrisation of the metal ion cluster structure, catalytic reactions, and metal ion exchange.
Methods
NMR spectra
1H, 13C and 2D NMR spectra were measured on a Bruker AVANCE III-500 (500 MHz) spectrometer. The residual solvent signal was used to calibrate the 1H (7.26 ppm), 13C NMR (77.16 ppm) measurements when CDCl3 was used. The residual solvent signal was used to calibrate the 1H (2.05 ppm), 13C NMR (29.84 ppm) measurements when d6-acetone was used.
ESI-MS analysis
ESI-TOF-MS data were measured on a Micromass LCT Premier XE mass spectrometer. Unless otherwise noted, the experimental conditions were as follows (ion mode, positive; capillary voltage, 2400 V; sample cone voltage, 30 V; desolvation temperature, 150 °C; source temperature, 80 °C).
Elemental analysis
Elemental analyses (C, N, H) were conducted in the microanalytical laboratory, Department of Chemistry, School of Science, the University of Tokyo, using a Vario MICRO Cube elemental analyser with MgO added.
Single-crystal X-ray diffraction analysis
X-ray crystallographic analysis was performed using a Rigaku XtaLAB PRO MM007DW PILATUS diffractometer with MoKα and CuKα radiation (93 K), and the obtained data were calculated using the Crystal Structure crystallographic software package. The refinement was performed using an OLEX2 software57 with SHELXT58. All hydrogen atoms were geometrically placed and refined using the riding model.
Photophysical analysis
The UV-vis absorption spectra were measured on a JASCO V-770 UV-vis spectrophotometer, wherein the temperature is set at 293 K unless otherwise mentioned. The emission and excitation spectra were measured using a Jasco FP-8300 fluorometer. The absolute quantum yields and lifetime measurements in the solid state were measured using Quantaurus-QY (Hamamatsu C9920-02G) and Hamamatsu C11367-02, respectively. CD spectra were measured on a JASCO J-820 circular dichroism spectrometer. The experimental conditions of CD analysis were as follows: bandwidth, 1 nm; response, 0.5 s; data acquisition interval, 0.5 nm; scanning rate, 100 nm min–1.
DFT and TD-DFT calculations
We applied the B3LYP functional59 for geometry optimizations and TD-DFT calculations. The relativistic effective core potential LANL2DZ60 was used for the Au atoms, and the basis set for the other atoms was 6-31G*61. Since MLCTs in the Au clusters etched in this study do not show long-distance charge transfer and essentially correspond to charge reorganisation, global hybrid functionals such as B3LYP adequately describe these electronic transitions. For simulating absorption spectra, 200 excited states were solved to cover the spectrum in the energy range up to about 220 nm was calculated in the velocity form. TD-DFT calculation was conducted with the polarizable continuum model (PCM) and the non-equilibrium linear response scheme62, including the solvent effect of CH2Cl2. All calculations were conducted using the Gaussian 16 suite of programs63. The orbital composition was analysed using the Multiwfn program64.
Handling
All the syntheses were conducted under air unless otherwise mentioned.
Chemical reagents
Unless otherwise noted, all the solvents were purchased from WAKO Pure Chemical Industries Ltd. and used without further purification. The >96% (NMR) pure cis-1,2-bis(diethylphosphino)ethene (cis-depe) was purchased from WAKO Pure Chemical Industries Ltd. and the >98% (GC) pure1,2-bis(diethylphosphino)benzene was purchased from TCI Co., Ltd., and used without further purification. The starting materials of carbon-centred hexagold(I) clusters [(C)(AuI-SS-NHC)6](BF4)2 (SS-1NHC) [(C)(AuI-RR-NHC)6](BF4)2 (RR-1NHC)43 and [(C)(AuI-TPP)6](BF4)2 (1TPP)26 were synthesised according to the reported procedures.
Synthesis of [(C)(AuI-SS-NHC)5](BF4) (SS-2NHC) and [(C)(AuI-RR-NHC)5](BF4) (RR-2NHC)
To the solution of SS-1NHC [(C)(AuI-SS-NHC)6](BF4)2 (6.7 mg, 2 μmol) in CH2Cl2 (1 mL), a solution of 2.5 equiv. of cis-depe (5 μmol, 2.0 mg) in CH2Cl2 (1 mL) was added dropwise at room temperature. The original pale-yellow solution turned orange immediately. Next, the resulting reaction mixture was concentrated to 0.3 mL using an evaporator and then filtered into a tube through the cotton, finally layered with 3 mL Et2O for slow diffusion and stored in a refrigerator at 4 °C. After one day, orange-red blocky crystals of SS-2NHC [(C)(AuI-SS-NHC)5](BF4) were formed and isolated (4.3 mg, 80% yield, based on the hypercarbon). The RR-2NHC [(C)(AuI-RR-NHC)5](BF4) (4.6 mg, 85% yield, based on the hypercarbon) is obtained similarly by using RR-1NHC [(C)(AuI-RR-NHC)6](BF4)2 (6.7 mg, 2 μmol) as starting material. Anal. calcd. for [C116H110Au5BF4N10](CH2Cl2)2: C, 49.11; H, 3.98; N, 4.85. Found: C, 49.20; H, 4.23; N, 4.96. ESI-MS (CH2Cl2, positive): [SS-2NHC]+ [C116H110N10Au5]+, m/z 2628.85 (calcd. 2628.73). ESI-MS (CH2Cl2, positive): [RR-2NHC]+ [C116H110N10Au5]+, m/z 2628.92 (calcd. 2628.73). 1H NMR (500 MHz, 300 K, CDCl3): δ 7.66–7.60 (m, 4H), 7.38 (q, J = 7.4 Hz, 2H), 7.16–7.12 (m, 2H), 7.03–7.01 (m, 2H), 7.00 (d, J = 1.3 Hz, 1H), 6.95 (t, J = 7.4 Hz, 3H), 1.73 (d, J = 7.3 Hz, 5H). 13C NMR (126 MHz, 300 K, CDCl3): δ 206.1 (CNHC), 198.8, 140.1, 133.2, 129.3, 128.6, 128.1, 123.8, 114.1, 59.0 (–CH–), 17.5 (–CH3). In the 13C NMR spectrum, the signal of the hypercarbon atom was not detected even after a long-time accumulation.
Synthesis of [(C)(AuI-TPP)5](BF4) (2TPP)
The synthesis of 2TPP is similar to that of SS-2NHC by using 1TPP [(C)(AuI-TPP)6](BF4)2 as the starting material. After adding a solution of cis-depe (25 μmol, 9.9 mg, 2.5 equiv.) in CH2Cl2 (1 mL) to a solution of 1TPP (29.4 mg, 10 μmol) in CH2Cl2 (1 mL), the resulting mixture turned from colourless to yellow. Next, the resulting reaction mixture was concentrated to 0.3 mL using an evaporator, and then filtered into a tube through the cotton, finally layered with 3 mL Et2O for slow diffusion and stored in a refrigerator at 4 °C. After several days, the yellow blocky crystals of 2TPP were isolated (22.3 mg, yield 93%, based on the hypercarbon). Anal. calcd. for [C91H75Au5BF4P5]: C, 45.63; H, 3.16; N, 0. Found: C, 45.61; H, 3.24; N, 0.22. ESI-MS (CH2Cl2, positive): [2TPP]+ [C91H75P5Au6]+, m/z 2307.21 (calcd. 2307.29). 1H NMR (500 MHz, 300 K, CDCl3): δ 7.61–7.53 (m, 2H), 7.46–7.37 (m, 2H), 7.19 (td, J = 7.9, 1.8 Hz, 3H). 31P NMR (202 MHz, 300 K, CDCl3): δ 32.59 (s); 13C NMR (126 MHz, 300 K, CDCl3): δ 134.9 (d, J = 14.9 Hz), 133.0 (d, J = 50.3 Hz), 131.9, 129.9 (d, J = 11.4 Hz). In the 13C NMR spectrum, the signal of the hypercarbon atom was not detected even after a long-time accumulation.
Synthesis of complex [(cis-depe)2AuI](BF4) (3)
In the above-mentioned synthesis of SS-2NHC, after isolating the desired crystals of SS-2NHC. The residue was used for recrystallisation, and several colourless crystals of [(cis-depe)2AuI](BF4) (3) (yield 50% by 1H NMR) were formed after one week. Similarly, after isolating crystals of 2TPP from its crystallisation tube, wherein the residue was recrystallised to give 3 (yield 99%, by 1H NMR). Its single crystal structure was determined by SCXRD (Suppl. Fig. 14). Its 1H NMR spectrum is consistent with literature65.
Synthesis of [(C)(AuI-SS-NHC)5(AuCl)](BF4) (SS-4NHC) and [(C)(AuI-RR-NHC)5(AuCl)](BF4) (RR-4NHC)
To a solution of SS-2NHC (16 mg, 6 µmol) in 10 mL acetone, 1.2 equiv. of (tht)AuCl (2.3 mg, 7 µmol) in acetone (1 mL) was added dropwise (note: fast mixing caused decomposition and the formation of black precipitates) with continuous ultrasonic oscillation for 20 min. On completion, the resulting mixture was concentrated to 0.5 mL, and then filtered into a tube through the cotton, finally layered with 3 mL Et2O for slow diffusion and stored in a refrigerator at 4 °C. After several days, dark blocky crystals of [(C)(AuI-SS-NHC)5(AuCl)](BF4) SS-4NHC was isolated (10.0 mg, 57% yield, based on the hypercarbon). Anal. calcd. for [C116H110Au6BClF4N10](CH3COCH3)(H2O): C, 47.26; H, 3.93; N, 4.63. Found: C, 46.90; H, 4.34; N, 4.86. ESI-MS (CH2Cl2, positive): [SS-4NHC]+ [C116H110N10ClAu6]+, m/z 2860.71 (calcd. 2860.66). ESI-MS (CH2Cl2, positive): [RR-4NHC]+ [C116H110N10ClAu6]+, m/z 2860.70 (calcd. 2860.66). 1H NMR (500 MHz, 300 K, CDCl3): δ 7.64 (d, J = 7.6 Hz, 4H), 7.38 (q, J = 7.3 Hz, 2H), 7.23–7.22 (m, 2H), 7.11–7.02 (m, 4H), 6.96 (t, J = 7.6 Hz, 4H), 1.62 (d, J = 7.3 Hz, 6H). 13C NMR (126 MHz, 300 K, CDCl3): δ 189.93 (CNHC), 139.8, 132.8, 129.4, 128.8, 128.0, 124.4, 114.6, 59.8 (–CH–), 17.3 (–CH3). In the 13C NMR spectrum, the signal of the central carbon atom was not detected even after a long-time accumulation.
Kinetic studies of the etching process
Time-course experiments monitored by UV-vis spectroscopy
Etching [(C)(AuI-SS-NHC)6](BF4)2 (SS-1NHC) with cis-depe. A solution of SS-1NHC (5.0 ×10–5 M) in CH2Cl2 was prepared by dissolving SS-1NHC (1.7 mg, 0.5 µmol) in 10 mL CH2Cl2. We first measured this dichloromethane solution of SS-1NHC (5.0 ×10–5 M, 3 mL) by UV-vis spectroscopy at 293 K. Then, once 30 µL of cis-depe (2 equiv., 0.01 M) in CH2Cl2 was added (meanwhile, the solution was charged with a small magnetic stir and stirring at a rate of 60 rpm), the UV-vis spectra (Fig. 4a in the main text) of the resulting reaction were immediately measured at intervals of 1 s (stage A), and then at intervals of 5 min (stage B).
Etching [(C)(AuI-TPP)6](BF4)2 (1TPP) with cis-depe. A solution of 1TPP (5.0 ×10–5 M) in CH2Cl2 was prepared by dissolving 1TPP (1.5 mg, 0.5 µmol) in 10 mL CH2Cl2. We first measured this dichloromethane solution of 1TPP (5.0 ×10–5 M, 3 mL) by UV-vis spectroscopy at 293 K. Then, once 30 µL of cis-depe (2 equiv., 0.01 M) in CH2Cl2 was added (meanwhile, the solution was charged with a small magnetic stir and stirring at a rate of 60 rpm), the UV-vis spectra of the resulting reaction were immediately measured at intervals of 1 s (Suppl. Fig. 28).
Time-course experiments monitored by 1H NMR spectroscopy
Etching [(C)(AuI-SS-NHC)6](BF4)2 (SS-1NHC) with cis-depe. A solution of SS-1NHC (2.0 ×10–3 M) in d6-acetone was prepared by dissolving SS-1NHC (3.3 mg, 1 µmol) in d6-acetone (0.5 mL) in the presence of an internal standard (IS, 1,3,5-trimethoxybenzene, 6 equiv., 6 µmol, 1.0 mg). We firstly measured the solution of SS-1NHC in d6-acetone by 1H NMR spectroscopy at 300 K. Then, once the solution of cis-depe (2.5 equiv., 3.3 ×10–2 M, 75 µL) in d6-acetone was added, the 1H NMR spectra of the resulting mixed sample were immediately measured and then measured continuously at intervals of approx. 2 min. The 1H NMR spectra of this reaction were measured for 30 min (Suppl. Fig. 29).
Etching [(C)(AuI-TPP)6](BF4)2 (1TPP) with cis-depe. A solution of 1TPP (2.0 ×10–3 M) in d6-acetone was prepared by dissolving 1TPP (2.9 mg, 1 µmol) in 0.5 mL d6-acetone in the presence of an internal standard (IS, 1,3,5-trimethoxybenzene, 6 equiv., 6 µmol, 1.0 mg). We firstly measured the solution of 1TPP in d6-acetone 1H NMR spectroscopy at 300 K. Then, once the solution of cis-depe (2.5 equiv., 3.3 ×10–2 M, 75 µL) in d6-acetone was added, the 1H NMR spectra of the resulting mixed sample were immediately measured and then measured continuously at intervals of approx. 2 min. The 1H NMR spectra of this reaction were measured for 18 min (Suppl. Fig. 30).
Time-course experiments monitored by ESI-MS spectrometry
Etching [(C)(AuI-SS-NHC)6](BF4)2 (SS-1NHC) with cis-depe. A solution of SS-1NHC (5.0 × 10–5 M) in acetone was prepared by dissolving SS-1NHC (1.7 mg, 0.5 µmol) in 10 mL acetone. We first measured the original solution of SS-1NHC (5.0 × 10–5 M, 1 mL) in acetone by ESI-MS spectrometry. Then, once 10 µL of cis-depe (2 equiv., 0.01 M) in acetone was added (meanwhile the solution was charged with a small magnetic stir and stirring), the ESI-MS spectra of the resulting reaction were measured as shown in Suppl. Fig. 31.
Etching [(C)(AuI-TPP)6](BF4)2 (1TPP) with cis-depe. A solution of 1TPP (5.0 × 10–5 M) in acetone was prepared by dissolving 1TPP (1.5 mg, 5.0 × 10–4 mmol) in acetone. We first measured the original solution of 1TPP (5.0 × 10–5 M, 1 mL) in acetone by ESI-MS spectrometry. Then, once 10 µL of cis-depe (2 equiv., 0.01 M) in acetone was added (meanwhile the solution was charged with a small magnetic stir and stirring), the ESI-MS spectra of the resulting reaction were measured. The intermediate species was observed as shown in Suppl. Fig. 32.
Theoretical calculation details for etching mechanism
The proposed intermediates including Int1NHC, Int2NHC, Int1TPP, and Int2TPP were simulated and optimised using the Gaussian 16 suite of programs63. Optimisation was performed using the B3LYP functional59 combined with basis sets of 6-31G* (for C, N, P, H)61 and LANL2DZ (for Au)60. The solvent effects were evaluated by single-point calculations in the optimised structure using the polarisable continuum model (PCM). All chemical species involved were optimised in the singlet state. The calculated energy profiles were illustrated in Suppl. Fig. 33.
Supplementary information
Source data
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP21H05022 (M.S.), JP22K14691 (X.P.), JP22H05133 (M.E.), MEXT KAKENHI Grant Number JP16H06509 (M.S.), and JST, CREST Grant Number JPMJCR22B2 (M.S.), Japan.
Author contributions
M.S. and X.-L.P. designed the project, analysed the results, and prepared the manuscript. H.U. and Z.L. assisted in the synthetic experiments. P.Z. and M.E. performed the theoretical calculations and analyses. All authors were involved in revising the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. The X-ray crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2280948 to CCDC 2280952. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Masahiro Ehara, Email: ehara@ims.ac.jp.
Mitsuhiko Shionoya, Email: shionoya@chem.s.u-tokyo.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-49295-w.
References
- 1.Jin R, Zeng C, Zhou M, Chen Y. Atomically precise colloidal metal nanoclusters and nanoparticles: fundamentals and opportunities. Chem. Rev. 2016;116:10346–10413. doi: 10.1021/acs.chemrev.5b00703. [DOI] [PubMed] [Google Scholar]
- 2.Ye X, et al. Single-particle mapping of nonequilibrium nanocrystal transformations. Science. 2016;354:874–877. doi: 10.1126/science.aah4434. [DOI] [PubMed] [Google Scholar]
- 3.Zhou S, et al. Enabling complete ligand exchange on the surface of gold nanocrystals through the deposition and then etching of silver. J. Am. Chem. Soc. 2018;140:11898–11901. doi: 10.1021/jacs.8b06464. [DOI] [PubMed] [Google Scholar]
- 4.Lin L, Chen M, Qin H, Peng X. Ag nanocrystals with nearly ideal optical quality: synthesis, growth mechanism, and characterizations. J. Am. Chem. Soc. 2018;140:17734–17742. doi: 10.1021/jacs.8b10793. [DOI] [PubMed] [Google Scholar]
- 5.Godbold P, et al. Surfactant removal for colloidal nanocrystal catalysts mediated by N-heterocyclic carbenes. J. Am. Chem. Soc. 2021;143:2644–2648. doi: 10.1021/jacs.0c12278. [DOI] [PubMed] [Google Scholar]
- 6.Shichibu Y, Negishi Y, Tsukuda T, Teranishi T. Large-scale synthesis of thiolated Au25 clusters via ligand exchange reactions of phosphine-stabilized Au11 Clusters. J. Am. Chem. Soc. 2005;127:13464–13465. doi: 10.1021/ja053915s. [DOI] [PubMed] [Google Scholar]
- 7.Zhang B, Chen C, Chuang W, Chen S, Yang P. Size transformation of the Au22(SG)18 nanocluster and its surface-sensitive kinetics. J. Am. Chem. Soc. 2020;142:11514–11520. doi: 10.1021/jacs.0c03919. [DOI] [PubMed] [Google Scholar]
- 8.Cao Y, et al. Control of single-ligand chemistry on thiolated Au25 nanoclusters. Nat. Commun. 2020;11:5498. doi: 10.1038/s41467-020-19327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li M-B, Tian S-K, Wu Z, Jin R. Peeling the core–shell Au25 nanocluster by reverse ligand-exchange. Chem. Mater. 2016;28:1022–1025. doi: 10.1021/acs.chemmater.5b04907. [DOI] [Google Scholar]
- 10.Narouz MR, et al. N-heterocyclic carbene-functionalized magic-number gold nanoclusters. Nat. Chem. 2019;11:419–425. doi: 10.1038/s41557-019-0246-5. [DOI] [PubMed] [Google Scholar]
- 11.Kurashige W, Niihori Y, Sharma S, Negishi Y. Precise synthesis, functionalization and application of thiolate-protected gold clusters. Coord. Chem. Rev. 2016;320:238–250. doi: 10.1016/j.ccr.2016.02.013. [DOI] [Google Scholar]
- 12.Bootharaju MS, Joshi CP, Alhilaly MJ, Bakr OM. Switching a nanocluster core from hollow to nonhollow. Chem. Mater. 2016;28:3292–3297. doi: 10.1021/acs.chemmater.5b05008. [DOI] [Google Scholar]
- 13.Ni TW, Tofanelli MA, Phillips BD, Ackerson CJ. Structural basis for ligand exchange on Au25(SR)18. Inorg. Chem. 2014;53:6500–6502. doi: 10.1021/ic5010819. [DOI] [PubMed] [Google Scholar]
- 14.Zeng C, Chen Y, Das A, Jin R. Transformation chemistry of gold nanoclusters: from one stable size to another. J. Phys. Chem. Lett. 2015;6:2976–2986. doi: 10.1021/acs.jpclett.5b01150. [DOI] [PubMed] [Google Scholar]
- 15.Yao Q, et al. Precise control of alloying sites of bimetallic nanoclusters via surface motif exchange reaction. Nat. Commun. 2017;8:1555. doi: 10.1038/s41467-017-01736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Q, Yuan X, Chen T, Leong DT, Xie J. Engineering functional metal materials at the atomic level. Adv. Mater. 2018;30:1802751. doi: 10.1002/adma.201802751. [DOI] [PubMed] [Google Scholar]
- 17.Kang X, Zhu M. Transformation of atomically precise nanoclusters by ligand-exchange. Chem. Mater. 2019;31:9939–9969. doi: 10.1021/acs.chemmater.9b03674. [DOI] [Google Scholar]
- 18.Pettibone JM, Hudgens JW. Gold cluster formation with phosphine ligands: etching as a size-selective synthetic pathway for small clusters? ACS nano. 2011;5:2989–3002. doi: 10.1021/nn200053b. [DOI] [PubMed] [Google Scholar]
- 19.Cao Y, et al. Revealing the etching process of water-soluble Au25 nanoclusters at the molecular level. Nat. Commun. 2021;12:3212. doi: 10.1038/s41467-021-23568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song Y, Murray RW. Dynamics and extent of ligand exchange depend on electronic charge of metal nanoparticles. J. Am. Chem. Soc. 2002;124:7096–7102. doi: 10.1021/ja0174985. [DOI] [PubMed] [Google Scholar]
- 21.Montalti M, et al. Kinetics of place-exchange reactions of thiols on gold nanoparticles. Langmuir. 2003;19:5172–5174. doi: 10.1021/la034581s. [DOI] [Google Scholar]
- 22.Nan Y, Xia N, Wu Z. Metal nanoparticles confronted with foreign ligands: mere ligand exchange or further structural transformation? Small. 2021;17:2000609. doi: 10.1002/smll.202000609. [DOI] [PubMed] [Google Scholar]
- 23.Schmidbaur H, Hofreiter S, Paul M. Synthesis of the gold analogue of the elusive doubly protonated water molecule. Nature. 1995;377:503–504. doi: 10.1038/377503a0. [DOI] [Google Scholar]
- 24.Grohmann A, Riede J, Schmidbaur H. Electron-deficient bonding at pentacoordinate nitrogen. Nature. 1990;345:140–142. doi: 10.1038/345140a0. [DOI] [Google Scholar]
- 25.Shan H, Yang Y, James AJ, Sharp PR. Dinitrogen bridged gold clusters. Science. 1997;275:1460–1462. doi: 10.1126/science.275.5305.1460. [DOI] [Google Scholar]
- 26.Scherbaum F, Grohmann A, Huber B, Krüger C, Schmidbaur H. “Aurophilicity” as a consequence of relativistic effects: the hexakis(triphenylphosphaneaurio)methane dication [(Ph3PAu)6C]2⊕. Angew. Chem. Int. Ed. Engl. 1988;27:1544–1546. doi: 10.1002/anie.198815441. [DOI] [Google Scholar]
- 27.Gimeno MC, Laguna A. Chalcogenide centred gold complexes. Chem. Soc. Rev. 2008;37:1952–1966. doi: 10.1039/b708618k. [DOI] [PubMed] [Google Scholar]
- 28.Yan L-L, et al. Stimuli-induced reversible transformation between decanuclear and pentanuclear gold(I) sulfido complexes. J. Am. Chem. Soc. 2022;144:19748–19757. doi: 10.1021/jacs.2c05946. [DOI] [PubMed] [Google Scholar]
- 29.Yao L-Y, Hau FK-W, Yam VW-W. Addition reaction-induced cluster-to-cluster transformation: controlled self-assembly of luminescent polynuclear gold(I) μ3-sulfido clusters. J. Am. Chem. Soc. 2014;136:10801–10806. doi: 10.1021/ja505599v. [DOI] [PubMed] [Google Scholar]
- 30.Schmidbaur H, Schier A. A briefing on aurophilicity. Chem. Soc. Rev. 2008;37:1931–1951. doi: 10.1039/b708845k. [DOI] [PubMed] [Google Scholar]
- 31.Schmidbaur H, Schier A. Aurophilic interactions as a subject of current research: an up-date. Chem. Soc. Rev. 2012;41:370–412. doi: 10.1039/C1CS15182G. [DOI] [PubMed] [Google Scholar]
- 32.Pyykkö P. Strong closed-shell interactions in inorganic chemistry. Chem. Rev. 1997;97:597–636. doi: 10.1021/cr940396v. [DOI] [PubMed] [Google Scholar]
- 33.Andris E, et al. Aurophilic interactions in [(L)AuCl]…[(L′)AuCl] dimers: calibration by experiment and theory. J. Am. Chem. Soc. 2018;140:2316–2325. doi: 10.1021/jacs.7b12509. [DOI] [PubMed] [Google Scholar]
- 34.Mirzadeh N, Priver SH, Blake AJ, Schmidbaur H, Bhargava SK. Innovative molecular design strategies in materials science following the aurophilicity concept. Chem. Rev. 2020;120:7551–7591. doi: 10.1021/acs.chemrev.9b00816. [DOI] [PubMed] [Google Scholar]
- 35.Olah, G. A., Prakash, G. K. S., Wade, K., Molnár, Á. & Williams, R. E. Hypercarbon Chemistry 2nd. Edition Ch. 1 (Wiley, New York, 2011).
- 36.Hoffmann R. Building bridges between inorganic and organic chemistry (Nobel Lecture) Angew. Chem. Int. Ed. Engl. 1982;21:711–724. doi: 10.1002/anie.198207113. [DOI] [Google Scholar]
- 37.Raubenheimer HG, Schmidbaur H. Gold chemistry guided by the isolobality concept. Organometallics. 2012;31:2507–2522. doi: 10.1021/om2010113. [DOI] [Google Scholar]
- 38.Jia J-H, Wang Q-M. Intensely luminescent gold(I)−silver(I) cluster with hypercoordinated carbon. J. Am. Chem. Soc. 2009;131:16634–16635. doi: 10.1021/ja906695h. [DOI] [PubMed] [Google Scholar]
- 39.Chen M, et al. A phosphorescent silver(I)–gold(I) cluster complex that specifically lights up the nucleolus of living cells with FLIM imaging. Biomaterials. 2013;34:4284–4295. doi: 10.1016/j.biomaterials.2013.02.032. [DOI] [PubMed] [Google Scholar]
- 40.Lei Z, Wang Q-M. Homo and heterometallic gold(I) clusters with hypercoordinated carbon. Coord. Chem. Rev. 2019;378:382–394. doi: 10.1016/j.ccr.2017.11.001. [DOI] [Google Scholar]
- 41.Ube H, Zhang Q, Shionoya M. A carbon-centered hexagold(I) cluster supported by N-heterocyclic carbene ligands. Organometallics. 2018;37:2007–2009. doi: 10.1021/acs.organomet.8b00291. [DOI] [Google Scholar]
- 42.Lei Z, Pei X-L, Ube H, Shionoya M. Reconstituting the C-centered hexagold(I) clusters with N-heterocyclic carbene ligands. Bull. Chem. Soc. Jpn. 2021;94:1324–1330. doi: 10.1246/bcsj.20210060. [DOI] [Google Scholar]
- 43.Pei X-L, et al. Asymmetric twisting of C-centered octahedral gold(I) clusters by chiral N-heterocyclic carbene ligation. J. Am. Chem. Soc. 2022;144:2156–2163. doi: 10.1021/jacs.1c10450. [DOI] [PubMed] [Google Scholar]
- 44.Lei Z, et al. N-Heterocyclic carbene-based C-centered Au(I)-Ag(I) clusters with intense phosphorescence and organelle-selective translocation in cells. Nat. Commun. 2022;13:4288. doi: 10.1038/s41467-022-31891-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei Z, et al. Photoluminescence control by atomically precise surface metallization of C-centered hexagold(I) clusters using N-heterocyclic carbenes. Chem. Sci. 2023;14:6207–6215. doi: 10.1039/D3SC01976D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scherbaum F, Grohmann A, Müller G, Schmidbaur H. Synthesis, structure, and bonding of the cation [{(C6H5)3PAu}5C]⊕. Angew. Chem. Int. Ed. Engl. 1989;28:463–465. doi: 10.1002/anie.198904631. [DOI] [Google Scholar]
- 47.Smock SR, Alimento R, Mallikarjun Sharada S, Brutchey RL. Probing the ligand exchange of N-heterocyclic carbene-capped Ag2S nanocrystals with amines and carboxylic acids. Inorg. Chem. 2021;60:13699–13706. doi: 10.1021/acs.inorgchem.1c02018. [DOI] [PubMed] [Google Scholar]
- 48.Carey DM, Muñoz-Castro A. Evaluation of N-heterocyclic carbene counterparts of classical gold clusters; bonding properties of octahedral CAu6, icosahedral Au13Cl2, and bi-icosahedral Au25Cl2 cores from relativistic DFT calculations. J. Phys. Chem. C. 2019;123:12466–12473. doi: 10.1021/acs.jpcc.9b01254. [DOI] [Google Scholar]
- 49.Görling A, Rösch N, Ellis DE, Schmidbaur H. Electronic structure of main-group-element-centered octahedral gold clusters. Inorg. Chem. 1991;30:3986–3994. doi: 10.1021/ic00021a005. [DOI] [Google Scholar]
- 50.Häberien OD, Schmidbaur H, Rösch N. Stability of main-group element-centered gold cluster cations. J. Am. Chem. Soc. 1994;116:8241–8248. doi: 10.1021/ja00097a034. [DOI] [Google Scholar]
- 51.Addison, A. W., Rao, T. N., Reedijk, J., van Rijn, J. & Verschoor, G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua [1,7-bis(N-methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper (II) perchlorate. J. Chem. Soc., Dalton Trans. 1349–1356 (1984).
- 52.Ni J, et al. Deep-blue electroluminescence from phosphine-stabilized Au3 triangles and Au3Ag pyramids. Angew. Chem. Int. Ed. 2022;61:e202213826. doi: 10.1002/anie.202213826. [DOI] [PubMed] [Google Scholar]
- 53.Yonesato K, et al. Surface-exposed silver nanoclusters inside molecular metal oxide cavities. Nat. Chem. 2023;15:940–947. doi: 10.1038/s41557-023-01234-w. [DOI] [PubMed] [Google Scholar]
- 54.Waldvogel, S. R., Spurg, A. & Hahn, F. E. Activating Unreactive Substrates Ch. 7 (Wiley-VCH Verlag GmbH & Co. KGaA, Germany, 2009).
- 55.Nolan SP. The development and catalytic uses of N-heterocyclic carbene gold complexes. Acc. Chem. Res. 2011;44:91–100. doi: 10.1021/ar1000764. [DOI] [PubMed] [Google Scholar]
- 56.Ye R, et al. Supported Au nanoparticles with N-heterocyclic carbene ligands as active and stable heterogeneous catalysts for lactonization. J. Am. Chem. Soc. 2018;40:4144–4149. doi: 10.1021/jacs.8b01017. [DOI] [PubMed] [Google Scholar]
- 57.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JA, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [Google Scholar]
- 58.Sheldrick GM. Crystal structure refinement with SHELXL. Acta Cryst. C. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 60.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985;82:270–283. doi: 10.1063/1.448799. [DOI] [Google Scholar]
- 61.Binkley JS, Pople JA, Hehre WJ. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980;102:939–947. doi: 10.1021/ja00523a008. [DOI] [Google Scholar]
- 62.Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- 63.Frisch, M. J. et al. Gaussian, Inc., Gaussian 16 rev. A03, Wallingford, CT, (2016).
- 64.Lu T, Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 65.Healy PC, Bradley T, Michael LW, Peter GP. Synthesis, structure and cytotoxicity studies of four-coordinate bis(cis–bis(diphenylphosphino)ethene) gold(I) complexes. [Au(dppey)2]X. J. Inorg. Biochem. 2010;104:625–631. doi: 10.1016/j.jinorgbio.2010.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request. The X-ray crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2280948 to CCDC 2280952. Source data are provided with this paper.