

Abstract
Transfer RNAs (tRNAs) are key adaptor molecules that decipher the genetic code during translation of messenger RNAs (mRNAs) in protein synthesis. In contrast to the traditional view of tRNAs as ubiquitously expressed housekeeping molecules, awareness is now growing that tRNA-encoding genes display tissue-specific and cell-type-specific patterns of expression, and that tRNA gene expression and function are both dynamically regulated by post-transcriptional RNA modifications. Moreover, dysregulation of tRNAs, mediated by alterations in either their abundance or function, can have deleterious consequences that contribute to several distinct human diseases, including neurological disorders and cancer. Accumulating evidence shows that reprogramming of mRNA translation through altered tRNA activity can drive pathological processes in a codon-dependent manner. This Review considers the emerging evidence in support of the precise control of functional tRNA levels as an important regulatory mechanism that coordinates mRNA translation and protein expression in physiological cell homeostasis, and highlight key examples of human diseases that are linked directly to tRNA dysregulation.
Introduction
Discovered more than 60 years ago1, transfer RNAs (tRNAs) are key molecules involved in deciphering the genetic code in messenger RNAs (mRNAs) and in the synthesis of proteins. Soon after the initial determination of tRNA structure2, tRNAs were shown to be highly structured, commonly consisting of ~76 nucleotides arranged in a cloverleaf secondary structure containing three stem loops, known as the D-loop (dihydrouridine-containing loop), anticodon loop and T-loop (thymidine, pseudouridine and cytidine-containing or TΨC loop). These stem loops fold onto themselves to form the classic L-shaped tRNA conformation, in which one arm is composed of the acceptor stem and T-loop, and a second arm is formed by the D-loop and anticodon stem loop (Figure 1a). Besides their compact shape, another defining characteristic of some tRNAs is the presence of inosine (a modified adenosine) in their anticodon triplet, which forms the basis for the wobble hypothesis3. Moreover, tRNAs contain numerous post-transcriptional chemical modifications that influence tRNA stability, structure, folding, translational fidelity and translational fine-tuning4-6. These tRNA modifications respond dynamically to changes in cellular metabolism or environmental cues and dysregulation of tRNA modifications can have major biological consequences (reviewed elsewhere7).
Figure 1. tRNA molecular structure and variance in isodecoder prevalence in eukaryotic species.

a ∣ Cloverleaf structure of a tRNA molecule. The colours denote different structural domains. b ∣ The number of tRNA isodecoders increases along the phylogenetic spectrum. Here, numbers of tRNA isodecoders are depicted as the fraction of all tRNA genes in different eukaryotes. Data for Part b were obtained from ref9.
Strikingly, the human genome contains ~500 genes encoding nuclear tRNAs, which are transcribed by RNA polymerase III (Pol III) and contain two internal promoter regions, Box-A (nucleotides 8–19) and Box-B (nucleotides 52–62), as well as 22 tRNAs encoded by the mitochondrial genome8. The nuclear-encoded tRNAs form 21 isoacceptor families, each consisting of tRNAs that encode the same amino acid but have a different anticodon; therefore, one isoacceptor family exists for every amino acid, including selenocysteine (an analogue of cysteine that contains selenium instead of sulfur, and is critical for formation of selenoproteins). Isodecoder families, by contrast, are tRNAs that share the same anticodon but possess nucleotide differences elsewhere in their structure. Interestingly, in eukaryotes the number of isodecoders (as a fraction of all tRNA genes) varies widely and tends to increase across the phylogenetic tree9; in humans, for instance, ~50% of tRNA genes encode isodecoders, whereas in budding yeast (Saccharomyces cerevisiae) only about 3% of tRNA genes encode isodecoders (Figure 1b). In light of the presumed functional redundancy between different isodecoders, the reasons why human genomes (and those of other higher eukaryotes) contain so many tRNA genes remain unclear. However, this situation hints at the possibility of tissue-specific or cell-type-specific expression of different subsets of tRNA genes and suggests that tRNA isodecoder function could be less redundant and/or more diverse than previously anticipated. Indeed, the exact number, particular repertoire and relative expression levels of tRNA genes in different cells remain largely unaddressed. Available data from newly developed bulk next-generation tRNA sequencing techniques suggest that a single human cell could have 300–400 tRNA genes that are actively transcribed8,10,11 but, as yet, no data focusing on tRNA abundance at single-cell resolution are available. Nevertheless, an emerging concept based on accumulating evidence from tRNA sequencing studies is that (similarly to mRNA expression or microRNA expression) tRNA expression can be influenced by context, including the cellular environment and tissue type9,11,12. Furthermore, although the extent to which each individual human tRNA contributes to protein synthesis remains undetermined, accumulating evidence indicates that not all tRNAs isoacceptors or isodecoders are functionally equivalent9,13,14. In consequence, changes in the abundance or function of specific tRNAs can result in codon-biased reprogramming of mRNA translation, whereby distinct sets of mRNAs that are enriched in a particular codon are particularly affected by alterations in specific tRNA genes and contribute to human disease15-20.
In this Review, we focus on the canonical role of tRNAs in mRNA translation, and provide a particular emphasis on the dysregulation of tRNA. The major sources of tRNA dysregulation can be categorized as causing changes in either tRNA abundance or tRNA function. The first group involves mutations in the molecular machinery that affect the transcription, processing or stability of tRNAs, as well as mutations in tRNA genes. The second group includes mutations in enzymes that affect tRNA function through the deposition and/or alteration of RNA modifications and defects in amino acid charging of tRNAs. We highlight the implications of tRNA dysregulation for human disease with a few illustrative examples, and discuss potential routes for therapeutic intervention. The non-canonical roles of tRNAs are not discussed as they have been extensively reviewed elsewhere21.
Changes in tRNA abundance
Mutations in RNA polymerase III complex genes
Pol III is responsible for the transcription of tRNAs as well as several other non-coding RNAs including 5S rRNA, U6 snRNA, 7SL RNA, 7SK RNA, and RNase P RNA22. Various inherited mutations, occurring in 9 of the 17 subunits of Pol III (also known as POLR3)23 have been primarily associated with a wide variety of neurodegenerative disorders (Figure 2a) (reviewed extensively elsewhere24,25). Mutations in the genes encoding six of these Pol III subunits (POLR3A26, POLR3B27, POLR1C28, POLR3K29, POLR3H30 and POLR3GL31,32) cause a spectrum of rare genetic disorders, including hypomyelinating leukodystrophy (HLD), which is the most common POLR3-related disorder. Mutations in BRF1, which encodes the 90 kDa subunit of transcription factor IIIB (TFIIIB, isoform 1 of which is involved in tRNA transcription as an activator of POLR3) are associated with a cerebellar-facial-dental syndrome that overlaps clinically with POLR3-related disorders33.
Figure 2. RNA polymerase III involvement in disease.

a ∣ Structure of RNA polymerase III (Pol III) (Protein Data Bank entry 7AST)23. The colours identify Pol III subunits that harbour pathological mutations in humans. b ∣ The regulatory effect of oncoproteins (red) and tumour suppressors (yellow) on Pol III-mediated transcription of tRNA genes. The three subunits of TFIIIB: TATA-box-binding protein (TBP), B double prime 1 (BDP1), and B-related factor-1 (BRF1), are shown interacting with TFIIIC. mTORC1 indirectly stimulates Pol III transcription via inhibition of MAF1, a negative regulator of Pol III. Ras small GTPases activate their downstream effector extracellular signal-regulated kinase (ERK), which induces BRF1 expression. Rb binds directly to BRF1, which inhibits Pol III recruitment. NOTCH1 stimulates tRNAVal transcription both directly and via targeting MYC. MYC directly interacts with BRF1 and activates the histone-modifying enzymes transformation and transcription domain-associated protein (TRRAP) and GCN5. P53 directly inhibits TBP and Pol III recruitment. Part a is adapted from ref23.
One proposed hypothesis to explain the pathophysiological consequences of hypofunctional Pol III proposes that mutations in Pol III lead to either globally reduced levels of tRNAs or reduced levels of specific tRNA species25. These deficits would have consequences for overall protein production, particularly during early development, where (for example) they cause defects in myelin formation that lead to the characteristic hypomyelination phenotype of HLD34. This hypothesis is supported by several reports that different disease-causing mutations result in decreased levels of some tRNAs; however, the identity of these tRNAs varies from study to study. For instance, a homozygous splice-site mutation (c.1771-6C>G) in POLR3A results in a global decrease of mature tRNA levels that reaches statistical significance for tRNAGly, tRNALeu, tRNAHis and the initiator tRNAMet, despite the reduction in tRNA levels being mild overall35. Similarly, fibroblasts from patients carrying mutations in POLR3K showed decreased levels of initiator tRNAMet (Ref.29). Moreover, HEK293 cells used to model a disease-causing mutation in POLR3A (c.25546A>G) using CRISPR–Cas9 uncovered a widespread decrease in precursor tRNA levels36. Although these studies provide a snapshot of the tRNA landscape in POLR3-related disorders, the reader should note that they were not carried out in neuronal cells; such mutations are most likely to have a profound impact on the Pol III transcriptome in the disease-relevant cell type(s).
Transcriptional regulation by oncogenes and tumour suppressors
Commitment to cell division is preceded by attainment of a critical cell size, a process that is directly coordinated with an increase in overall protein synthesis. A major contributor to this increase in translation rate is the transcription of tRNA genes, which is elevated in cells that possess altered tumour suppressor or oncogene pathways (reviewed elsewhere37). Increasing evidence indicates that Pol III can be directly regulated by well-known oncoproteins and tumour suppressors, which are encoded by some of the most commonly mutated genes in human cancers, such as those encoding mTORC1 components or involved in Ras signalling, MYC, TP53, NOTCH1 and RB1 (Figure 2b). For example, stimulation of Pol III transcription is mediated by direct or indirect phosphorylation of key components of the Pol III machinery by mTORC1 components (namely, the negative Pol III regulator MAF1) or its downstream effectors38-43. MYC is a transcription factor that drives increased cell division prior to protein synthesis and has been shown to control Pol III transcription by directly localizing at tRNA genes through association with BRF1, a key component of the TFIIIB complex44. This interaction can trigger the localization of histone-modifying enzymes such as histone acetyltransferase GCN5 (also known as KAT2A) and the adapter protein TRRAP, which helps to promote tRNA gene transcription45. Ras signalling can also increase Pol III transcription via increased expression of a component of the TFIIIB complex46,47. By contrast, the tumour suppressor proteins p53 and Rb both negatively regulate Pol III transcription by directly binding to members of the TFIIIB complex (TBP or BRF1), thereby inhibiting the recruitment of Pol III to tRNA genes48-53. In 2021, NOTCH1 (aberrant activation of which is a key event in T cell acute lymphoblastic leukemia (T-ALL)) was identified as a positive regulator of tRNA biogenesis (particularly tRNAVal)19. Taken together, this body of work supports the concept that modulation of tRNA levels via Pol III regulation is an important mechanism in the control of growth, malignant transformation and tumorigenesis. Whether the regulation of Pol III transcripts by oncogenes and tumour suppressor proteins is global or biased towards certain tRNA families, isoacceptors or isodecoders remains unclear.
Epigenetic regulation of tRNA gene expression
Gene expression can be positively or negatively regulated by epigenetic mechanisms such as DNA methylation and histone modifications (Figure 3a). Epigenetic regulation of tRNA transcription has not been investigated thoroughly; nonetheless, some evidence suggests that these processes might be partially responsible for regulating tRNA transcription in physiological states and in disease. For instance, hypermethylation of the TRX-CAT1-4 gene (also known as tRNA-iMet (anticodon CAT) 1–4), was identified as an age-related DNA methylation change within a disease-associated region identified in a genome-wide association study54. Further studies of a tRNA locus showed genomic enrichment for CG DNA hypermethylation that was age-related and tissue-specific55. In further support of this notion, early research performed in Xenopus laevis oocytes using hypermethylated tRNALys-encoding genes showed that Pol III-dependent transcription of tRNA genes could be affected by their methylation status56. More research is needed to clarify the role of gene methylation in transcriptional regulation of tRNA loci.
Figure 3. Epigenetic mechanisms and sequence variability might influence nuclear tRNA expression.

a ∣ Epigenetic mechanisms might regulate tRNA expression. For example, methylation of tRNA genes could block DNA polymerase III (Pol III) transcription. Similarly, histone marks (such as K3K27 acetylation) and overall nucleosome occupancy could influence the accessibility of chromatin at tRNA loci. b ∣ tRNA genes show high variability in their flanking regions and these hyper-variable regions69 overlap with Pol III occupancy and transcription start sites (TSSs)70. Top plot shows the frequency of single nucleotide polymorphisms (SNPs) for active and inactive tRNA loci. Bottom plot represents the distribution of Pol III occupation relative to each position within the tRNA gene and its flanking regions. The acceptor stem (blue), D-stem (red), anticodon stem (green), and T-stem (orange) are highlighted in the 2D tRNA structure for both plots. Flanking tRNA regions containing relatively high mutation rates can overlap with Pol III TSSs, which raises the important question of whether variable flanking regions influence tRNA expression. c ∣ Mutations in nuclear encoded tRNAs are rare, but one example is a mutation in the acceptor stem region (red) of the tRNASec gene that causes reduced U34 modifications and a deficit in stress-related translation of selenoproteins73. Patients with mutant tRNASec experience low plasma selenium levels, abdominal pain, fatigue, and muscle weakness. Part b is adapted from ref69 (top graph) and ref70 (bottom graph).
The influence of histone modifications on transcription of tRNA genes has been characterized in more detail (reviewed elsewhere57,58). For example, nucleosome occupancy or positioning is reportedly similar between Pol II transcribed genes and tRNA genes, and histone regulation might also play an important role in tRNA expression. The dynamics of this level of regulation are illustrated during macrophage differentiation; chromatin accessibility is positively correlated with the level of histone H3 acetylation at lysine 27 (H3K27ac), a histone mark positively associated with transcription levels, including tRNA transcription. Downregulation of many tRNA genes occurs during monocyte-to-macrophage differentiation, which suggests that their transcription is subject to dynamic control during cell differentiation12. Although the dynamic epigenetic regulation of tRNA expression is becoming increasingly appreciated, its relevance to disease states remains to be addressed.
Pol III-transcribed genes are found in intergenic regions that are relatively depleted of nucleosomes59, hinting that local chromatin accessibility might not play an important role in the transcription of tRNA genes, but studies performed in yeast suggest otherwise. For example, global nucleosome depletion in yeast cells leads to transcriptional activation of several loci containing tRNA genes60. Because most tRNA genes (along with intergenic factor binding sites) are about the size of a DNA molecule wrapped around a single nucleosome, the Pol III machinery has to compete with nucleosomes for access to tRNA genes61. Early research suggested that nucleosome depletion at loci containing tRNA genes was a consequence of active Pol III transcription; however, consistent evidence indicates that nucleosomes are actively excluded from tRNA genes by chromatin remodellers to facilitate their transcription62. Altogether, multiple lines of evidence support the role of dynamic chromatin remodelling at loci containing tRNA genes as a regulatory mechanism that modulates tRNA gene transcription. This regulatory mechanism has the capacity to respond quickly to changes in the cellular state and further research is warranted to evaluate its physiological role.
Mutations in tRNA-encoding genes
mt-tRNA genes are hotspots for mutations associated with multiple diseases (reviewed elsewhere63). Over the past 24 years, approximately 370 mutations in all 22 mt-tRNA genes have been reported64. These mutations have been linked with altered levels of tRNA modifications and with functional alterations in mitochondrial protein translation65.
The first observations showing that point mutations in mt-rRNA genes can lead to aberrant mt-tRNA function mediated by defects in the deposition of RNA modifications came from the study of tRNAs isolated from patients with two important mitochondrial disorders: myoclonus epilepsy associated with ragged red fibres (MERRF), and mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS)66. Mutations in mt-tRNALeu(UUR), including A3243G and U3271C (amongst other mutations associated with MELAS), hinder its recognition by tRNA modification GTPase GTPBP3–MTO1, the enzyme complex responsible for creating the modification 5-taurinomethyluridine (τm5U) at ‘wobble’ position 34. Mutation-dependent τm5U hypomodification at position 34 in mt-tRNALeu(UUR) results in impaired recognition of UUG codons, which are enriched in MT-ND6 mRNA (which encodes NADH-ubiquinone oxidoreductase chain 6, an essential component of mitochondrial complex I), thereby leading to defects in respiratory activity66,67.
Several other associations have been reported between mt-tRNA gene mutations and impaired RNA modifications. Some of these mutations do not overlap with the modified sites, suggesting that disease-causing point mutations can have an adverse effect on substrate recognition or alter the activity of tRNA-modifying enzymes. In 2020, an in-depth study of mt-tRNA modification in all 22 mt-tRNA species reported finding 18 types of modified nucleosides at 137 positions68. This study also identified 23 disease-associated point mutations in commonly modified bases that are likely to abolish the modification68. Further understanding of human mt-tRNA modifications is necessary to fully understand the basis of mitochondrial diseases.
Compared with mutations in mt-tRNA genes, relatively little is known about either the prevalence or the role in disease of mutations in cytosolic tRNA genes (Figure 3b). A 2018 study that focused on the mutation rates of cytosolic tRNA genes showed that these genes are subject to transcription-associated mutagenesis. Strikingly, this report showed that tRNA gene loci have a mutation rate up to tenfold higher than the genome-wide average69. Most of this genetic variability is accounted for by mutations in the flanking regions of tRNA genes, which are defined as the 20 nucleotides either upstream of the transcription start site or upstream of the mature tRNA sequence, plus the 10 nucleotides downstream of the 3ʹ end of the mature tRNA sequence. Interestingly, the flanking region with the highest variability is located at the 5ʹ end of the mature tRNA, a region that also harbours the transcription start site (which is only 5 nucleotides upstream of the mature tRNA sequence)69,70. This observation raises several interesting questions: can mutations in flanking regions affect tRNA expression? Do mutations in tRNA gene loci contribute to survival fitness and/or natural variation in humans? Although some evidence suggests that mutations in flanking regions can affect tRNA expression in human cells71, the effects of such mutations on transcription of tRNA genes has not yet been addressed.
By contrast, the mature tRNA sequence is highly conserved, an observation that is in line with the existence of strong selection pressure to preserve tRNA functionality69. Mutations in the mature tRNA sequence are, therefore, expected to be highly deleterious. One of the most interesting examples is the isodecoder tRNA-Arg (anticodon TCT) 4-1, which is specifically expressed in the central nervous system (CNS)13. A study in mice identified a point mutation (T>C; rs46447118) in the Mod205 locus that harbours the n-TRtct5 (nuclear encoded tRNA Arg5 (anticodon TCT) gene. This SNP is localized at position 50 of the tRNA T-loop and was shown to interfere with the maturation process, leading to accumulation of pre-tRNAs and decreased aminoacylation levels72. Interestingly, the mature sequence of tRNA-Arg (anticodon TCT) 4-1 is highly conserved in both vertebrates and invertebrates. One other mutation has been reported in a nuclear-encoded tRNA gene: a single nucleotide change (C65G) in the only functional selenocysteine tRNASec identified in a homozygous patient who had low plasma selenium levels, abdominal pain, fatigue, muscle weakness, and markedly decreased levels of stress-related selenoproteins (Figure 3c)73. Interestingly, this mutation, which is located in the acceptor stem, caused a reduction in U34 modification, which is essential for wobble pairing. These studies showcase the disease-causing potential of mutations in nuclear tRNA genes.
Mutations in non-coding sequences in the human genome are often overlooked and could contribute to the poor characterization of the variability of the nuclear tRNAome. Moreover, several studies suggest that tRNA gene copy number variation is observable in human genomes and might be a source of phenotypic variation between individuals. A cluster of five tRNA genes located on chromosome 1, including various Glu-CTC, Gly-TCC, Asp-GTC, Leu-CAG, and Gly-GCC genes, showed higher copy numbers compared to the hg19 reference genome74. Moreover, about 50% of the genotyped individuals had a deletion of at least one allele of the tRNALysCUU gene on chromosome 1174. However, owing to the identical amino acid sequences of mature tRNA isoacceptors arising from different genes, studying the contributions of individual tRNA loci remains very challenging. tRNA gene copy number variation might represent an underappreciated layer of genetic diversity and could potentially contribute to human disease and genetic fitness.
In summary, the cellular tRNA pool can be affected by alterations at multiple stages in the biogenesis pathway, including mutations in tRNA genes as well as defects in transcription or maturation. Dysregulation of post-transcriptional tRNA modifications, such as N7-methylguanosine (m7G), can also lead to changes in tRNA abundance by affecting the stability of tRNA (Box 1). Often tRNA levels are assumed to be produced in excess, meaning that their levels might not represent a limiting factor in protein homeostasis. However, it is now becoming increasingly clear that small alterations in the tRNA pool can have profound physiological consequences and can be linked directly to many human diseases, including neurological disorders and cancer.
Box 1. Altered tRNA stability and disease.
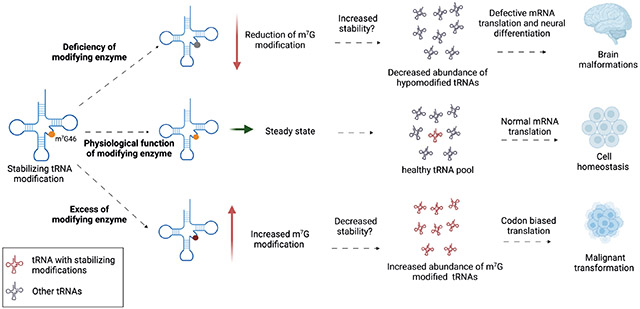
tRNAs are subject to extensive post-transcriptional modifications that can affect their function. Some of these modifications alter tRNA stability; for example, N-7-methylguanosine (m7G) at position 46 in a subset of tRNAs that harbour the RAGGU motif in their variable loop6,17,124. The crystal structure of tRNAPhe showed that m7G46 forms a base-triple interaction with C13 and G22 that helps stabilize the tertiary structure of the tRNA125. In mammals, the heterodimeric METTL1–WDR4 enzyme complex is responsible for depositing m7G in tRNAs. In humans, pathogenic mutations in WDR4 cause impaired tRNA m7G modification and are associated with microcephalic primordial dwarfism characterized by brain malformation, encephalopathy, and facial dimorphism126,127, and with Galloway–Mowat syndrome, a heterogeneous disorder characterized by neurodevelopmental defects combined with renal glomerular disease128. Deletion of either component of the MTase complex in yeast or in mouse stem cell models results in decreased abundance of m7G-modified tRNAs6,125,129. In yeast, the degradation of hypomodified tRNAs is carried out by a quality control pathway known as rapid tRNA decay pathway, whose counterpart in humans remains unknown130,131. In mice, deletion of the m7G methyltransferase (MTase) complex leads to defective stem cell renewal and neural differentiation, which is consistent with the severe brain malformations observed in patients with WDR4 mutations60. Moreover, expression of METTL1 and WDR4 is highly upregulated in human cancers, and METTL1 can act as an oncogene17,118. Mechanistically, overexpression of METTL1 leads to increased m7G levels and increased abundance of a subset of tRNAs. Surprisingly, one of these tRNAs with increased abundance is the CNS-specific tRNA-Arg (anticodon TCT) 4-1. Overexpression of this tRNA partially reproduced the malignant transformation observed with METTL1 overexpression, thus supporting the notion that changes in the tRNA pool and codon-biased mRNA translation are responsible for this malignant phenotype17.
Altered tRNA maturation and splicing
tRNA biogenesis begins with the transcription of premature tRNA (pre-tRNA), which then undergoes a multi-step maturation process involving trimming of the flanking region at the 5ʹ end (leader sequence) by nuclear ribonuclease (RNase) P75 and at the 3ʹ end (trailer sequence) by RNase Z1 and RNase Z2 (also known as ELAC1 and ELAC2)76. About 6% of pre-tRNAs also undergo the removal of a short intron by dedicated tRNA splicing machinery. This step is followed by the addition of a CCA (cytosine–cytosine–adenine) sequence to the 3ʹ end by the tRNA nucleotidyltransferase TRNT1 and deposition of a particular subset of the multitude of possible chemical modifications by specific RNA-modifying enzymes. Each mature tRNA contains an average of ~13 such modifications per molecule. Aminoacyl tRNA synthetase (aaRS) enzymes then charge each mature tRNA with an amino acid on their 3ʹ end (a process termed aminoacylation) that matches a particular trinucleotide anticodon sequence.
Maturation of pre-tRNA is critical for the regulation of normal cell homeostasis, division and growth, and pre-tRNA maturation is deregulated in various human diseases (Figure 4). Whereas no disease-associated mutations have yet been reported in genes encoding RNase P complex components, elevated expression levels of both the catalytically active RNase P RNA component H1 (also known as RPPH1) as well as the protein components of the complex have been found in cancer cells77. However, many mutations have been identified in mitochondrial RNase P (mtRNase P), the complex that carries out trimming of pre-tRNAs in mitochondria. Mutations in all three protein components (MRPP1, MRPP2 and MRPP3) of mtRNase P have been associated with a wide range of diseases. For instance, several mutations in MRPP2 have been linked with HSD10 disease (also known as 2-methyl-3-hydroxybutyric aciduria), which has complex features that resemble severe mitochondrial disorders78. The hallmarks of HSD10 include cardiomyopathy, loss of cognitive and motor functions, epilepsy and blindness. Functional studies performed in fibroblasts from patients with HSD10 showed that disease-causing mutations in MRPP2 can affect the processing of transcripts of mtRNA genes, tRNA modifications and the overall activity of mtRNase P79-81. Pathological mutations in MRPP1 (encoded by TRMT10C) have been associated with hypotonia, lactic acidosis and deafness. Similarly, studies conducted in patient-derived fibroblasts revealed normal activity of m1R9 methyltransferase (MTase), which catalyses N1-methylation of guanine or adenine in position 9 in mt-tRNAs, but impaired activity of mtRNase P due to decreased stability of MRPP1 protein, leading to accumulation of unprocessed precursor mt-tRNAs82. Furthermore, mutations in MRPP3 (encoded by PRORP) cause defects in mt-tRNA processing. Patients bearing these mutations present with variable phenotypes that include hearing loss, primary ovarian insufficiency, developmental delay and changes in brain white matter. Functionally, mutations in PRORP lead to decreased levels of MRPP3 in patient fibroblasts with concomitant accumulation of unprocessed mt-tRNAs. In vitro reconstitution assays also indicated that the pathogenic mutations impair mt-tRNA processing83. Strong evidence from animal models further supports the observations that pathogenic variants in all three subunits of mtRNAse P lead to mitochondrial dysfunction and a variety of clinical presentations84-86.
Figure 4. tRNA maturation and splicing defects in disease.

tRNA maturation involves the removal of 5' leader and 3' trailer sequences, CCA addition, and in some instances splicing of a short intron. Subsequently the mature tRNA is exported to the cytoplasm for aminoacylation (AA) by aminoacyl tRNA synthetases (aaRS). Some nuclear encoded tRNAs are imported into mitochondria. Mutations in proteins involved in the maturation of tRNAs (red) can lead to disease (shaded boxes). Levels of ribonuclease P (RNAse P) and ribonuclease P RNA component 1 (RPPH1) enzymes are increased in cancer but the physiological relevance of these alterations remains unknown.
Of the two nuclear RNase Z isoforms in humans, ELAC1 and ELAC2, ELAC2 is likely to be the enzyme primarily responsible for removal of the tRNA trailer sequence87. ELAC2 was first identified as a candidate gene associated with susceptibility to prostate cancer76,88, and a link between ELAC2 mutations and cardiac disease has also been established89. By contrast, ELAC1 has not yet been implicated in disease. ELAC1 functions to recycle tRNAs released from stalled ribosomes by removing the 2ʹ,3ʹ cyclic phosphate moiety (found on tRNAs that have been cleaved from the ribosome P-site by the tRNA endonuclease ANKZF1) that permits CCA reincorporation87.
Processed and recycled tRNAs are substrates for TRNT1-mediated addition or reincorporation of the CCA trinucleotide at the 3ʹ end. TRNT1 mutations have been linked with a spectrum of rare metabolic disorders that have a variety of clinical manifestations, including childhood-onset sideroblastic anaemia, B-lymphocyte immunodeficiency, retinitis pigmentosa, hepatomegaly, deafness, cerebellar atrophy, and deficiencies of pancreatic and renal function. Mechanistically, these mutations cause defective post-transcriptional modifications of both cytosolic and mitochondrial tRNAs, particularly in the addition of the 3' CCA trinucleotide to mitochondrial tRNACys, tRNALeuUUR and tRNAHis 90.
Lastly, about 6% of human tRNA genes contain a small intron that is removed by the tRNA splicing endonuclease (TSEN) complex to generate a functional mature tRNA91,92. Human TSEN consists of two catalytic subunits, TSEN2 and TSEN34, and two structural subunits, TSEN15 and TSEN5491. Mutations in the genes encoding each of these subunits have been found in patients with pontocerebellar hypoplasia (PCH), a group of developmental disorders that share clinical characteristics, including cerebellar hypoplasia and microcephaly93-97. Additionally, the mammalian TSEN complex associates with the RNA kinase CLP198, and CLP1 has been found to harbour mutations in patients with neuropathologies and tRNA splicing defects99-101. In the past year, biochemical reconstitution of the human TSEN–CLP1 complex along with analyses of fibroblasts from patients with PCH have provided insight into the molecular mechanism of the splicing defects observed in these patients. This work showed that although mutant TSEN retains its endonuclease activity, complex assembly and pre-tRNA cleavage are altered, leading to changes in the pre-tRNA pool101. Altogether, strong evidence indicates that that polymorphisms in the TSEN–CLP1 complex (and, therefore, defects in tRNA splicing) are the underlying cause of PCH102.
Changes in tRNA function
Besides changes in tRNA levels, changes that alter tRNA function can cause disease in humans. These include errors incorporated during amino acid charging as well as errors in post-transcriptional modifications that are essential for cognate codon recognition. In this section, we highlight key examples illustrating how these processes are directly linked to human disorders.
Altered amino acid charging
For a tRNA to be functional, it must be charged with an amino acid that matches its anticodon sequence. This well-characterized process is performed by aaRS enzymes74. Defects in amino acid charging can have devastating consequences in protein synthesis and are known to cause several disorders in humans (Figure 5). Pathological mutations in at least 17 different mitochondrial aaRSs have been described and predominantly result in CNS symptoms (reviewed elsewhere103).
Figure 5. Defects in tRNA modifications or aminoacylation influence translation.

Translation elongation factors (principally eIF1α) capture aminoacylated tRNAs (with the exceptions of initiator tRNAMet, which requires eIF218, and tRNASec, which requires eIFSec19) and deliver them to ribosomes to be utilized in the synthesis of proteins. Inside the A-site of the ribosome, the tRNA recognizes cognate codon sequences by forming hydrogen bonds with its anticodon triplet at positions 34, 35, and 36. Codon–anticodon interactions in positions 35 and 36 follow Watson–Crick base pairing, whereas those at position 34 sometimes do not (wobble pairing)3. Thus, the canonical function of tRNAs as adapter molecules during protein synthesis is influenced by changes in tRNA modifications. Absence of the mcm5U modification in the anticodon results in incorrect decoding, whereas I34 modifications in the anticodon expand the tRNA decoding capabilities. Furthermore, tRNAs lacking t6A37 modifications leads to reduced aminoacylation levels. Moreover, defects in aminoacyl tRNA synthetases (aaRS) can cause mischarging (red circle), resulting in either incorporation of an incorrect amino acid or the absence of amino acid charging followed by tRNA degradation, depletion of the corresponding tRNA, and ribosome stalling at the cognate codon. The best-understood example is Charcot–Marie–Tooth disease type 2D, in which mutant glycyl-tRNA synthetase (GARS) fails to release bound tRNAGly, thereby sequestering it and leading to depletion of functional tRNAGly 105.
The first reported disease-associated mutation in a cytosolic aaRS was described in patients with Charcot–Marie–Tooth disease (CMT) type 2D, a hereditary peripheral neuropathy characterized by progressive degeneration of motor and sensory neurons causing muscle weakness, and upper and lower limb atrophy. Mutations in any of the five cytoplasmic glycyl-tRNA synthetases (GARSs) have been implicated in CMT104 and a potential underlying molecular mechanism has been described. Although still a matter of debate, details of the molecular consequences of mutations in GARSs are emerging. New research supports the idea that aberrant GARS activity is related to a gain-of-function mechanism that results in impaired translation. Mutated GARS binds to tRNAGly and sequesters it, thereby depleting the tRNAGly pool and causing ribosome stalling on glycine codons, which results in chronic activation of the integrated stress response via the sensor kinase GCN2. The integrated stress response can be diminished by either pharmacological inhibition of GCN2 or ectopic expression of tRNAGly, which alleviate peripheral neuropathy in animal models105,106. Whether chronic activation of the integrated stress response is a mechanism shared by pathological mutations in other aaRS remains unclear, but aberrant aaRS activity is now accepted to be an important contributor to tRNA-related diseases.
Altered tRNA modification landscapes
tRNA molecules include numerous chemical modifications (involving 10–20% of their nucleotides) that play various roles in their stability, structure, folding, translational fidelity and fine-tuning of translation4-6. Human tRNA modifications are both tissue-specific and cell-type specific, and their levels and activity can change dynamically in response to cellular stressors or environmental cues107,108. Up to 39 types of RNA modification exist in cytoplasmic tRNAs and 18 types in mitochondrial tRNAs, and many of the enzymes responsible for these modifications are dysregulated in disease (reviewed elsewhere7). Study of the chemical modifications of RNA is known as epitranscriptomics and diseases caused by aberrant tRNA modifications are referred to as tRNA modopathies. The main pursuit of these two fields is to understand the molecular mechanisms of particular tRNA modifications.
Most tRNA modopathies involve a loss-of-function mechanism due to mutation or deletion of the responsible tRNA-modifying enzyme. Although understanding of tRNA modifications and their corresponding enzymes is still incomplete, aberrant modification of tRNAs can affect translation in three main ways: aberrant modifications in the anticodon that directly restrict or expand decoding functions; aberrant modifications in the tRNA body that alter its folding characteristics or structural stability (discussed above); and aberrant tRNA modifications that alter charging specificity (Figure 5). For example, aberrant modifications in the wobble position of the anticodon loop (position 34) can either restrict or expand the potential of a tRNA to decode a particular codon. The absence of 5-methoxycarbonylmethyluridine (mcm5U) or 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) results in translational infidelity leading to amino acid sequence errors and activation of the unfolded protein response and heat shock response109,110. By contrast, a change of adenosine to inosine at position 34 (that is, A43 to I34) enables the resulting anticodon to recognize codons that have U, C, or A in the third position3,111. Complete ablation of I34 is incompatible with life, which implies that I34 is essential for correct protein synthesis. In humans, some evidence indicates that the modification status of position 37 might also be important for correct aminoacylation, as abolishing N6-threonylcarbamoyl-adenosine (t6A) at position 37 (for instance) results in markedly reduced aminoacylation levels in mt-tRNALys 112.
Cancer is by far one of the most thoroughly studied pathologies in humans, and large multi-omic cancer datasets have been collected over the years. Global analysis of mRNA expression in patient-derived samples, using data included in The Cancer Genome Atlas (TCGA), showed that tRNA-modifying enzymes are highly dysregulated in cancer113. tRNA pseudouridine synthase A (PUS1, which converts uridine to pseudouridine (Ψ)), tRNA (guanine(26)-N(2))-dimethyltransferase (TRMT1, which converts guanosine to N2,N2-dimethylguanosine (m2,2G)) and tRNA (guanine-N(7)-)-methyltransferase (METTL1, which converts guanosine to 7-methylguanosine (m7G)) are the most strongly upregulated tRNA-modifying enzymes across different cancer types, whereas the most downregulated enzymes in TCGA dataset are the putative tRNA MTase TRMT9B (which catalyses an unknown modification, probably uridine modifications at the wobble position 34), tRNA (cytosine(38)-C(5))-methyltransferase (TRDMT1, which generates 5-methylcytosine (m5C) at position 38 in the anticodon loop of tRNAAsp, and tRNA methyltransferase 10 homolog A (TRMT10A), which catalyzes the formation of N1-methylguanine at position 9 (m9G)113. Further research is needed to fully understand the individual contributions of tRNA modifications to human diseases.
Altered tRNA pools
Repurposed microRNA sequencing data from TCGA has been used to estimate the changes in tRNA levels in diseased versus healthy tissues. The results indicate that an altered abundance of specific tRNAs is a widespread phenomenon in different human cancers113. This observation has implications for many human cancers as increasing evidence suggests that, rather than having global effects on protein synthesis, changes in the tRNA pool lead to codon-biased reprogramming of mRNA translation that can specifically drive oncogenic programmes. The examples presented below highlight the underlying role of disease mechanisms that exploit alterations in tRNA supply and demand to preferentially translate mRNAs involved in metastasis, drug resistance, cell growth and bioenergetics.
Translation initiation is the first rate-limiting step in protein synthesis and can be hijacked to support tumorigenesis114,115. For example, overexpression of tRNAiMet causes changes in metabolic and growth rates in immortalized human breast cells116; overexpression of tRNAiMet also promotes metastasis in melanoma tumours117 and furthermore increases tumour burden and vascularization in mice72.
Another regulatory layer in the translation of oncogenic mRNAs is the alteration of elongator tRNAs and the resulting changes in cognate codon usage (Figure 6a). Overexpression of tRNAGlu(UUC) and tRNAArg(CCG) promotes a pro-metastatic state in breast cancer by enhancing the translation of transcripts that are enriched in their cognate codons16. Moreover, reprogramming of mRNA translation through alteration of wobble modifications might play an important role in resistance to targeted therapy in melanoma driven by the BRAFV600E oncogene. Overexpression of U34 tRNA-modifying enzymes promote glycolysis through codon-dependent regulation of translation of HIF1A mRNA15. Another example of translational reprogramming is the accumulation of m7G-modified tRNAs upon overexpression of METTL1 and/or WDR4. Accumulation of tRNAArg(TCT) or tRNALys(CTT) leads to biased translation of growth-promoting proteins that are enriched in their respective cognate codons, thereby supporting malignant transformation17,118. A genome-wide loss-of-function CRISPR–Cas9 screen of components involved in tRNA biogenesis identified tRNAVal as a critical adaptation in the pathobiology of T-ALL19. Mechanistically, increased NOTCH1 signalling is responsible for the upregulation of tRNAVal 19. This change affects the translation of mRNAs involved in the electron transport chain, which are enriched with valine codons, thereby enhancing mitochondrial bioenergetics19. Interestingly, dietary restriction of valine consumption reduced the tumour burden in a mouse model of T-ALL, suggesting a potential avenue for treatment19. The relevance to disease of the emerging concept of codon-biased translation (first described in yeast in response to cellular stresses109,110) is becoming increasingly apparent and might help to explain the varied phenotypes associated with various diseases caused by altered tRNA expression or function.
Figure 6. Alterations in the tRNA pool can drive disease and offer avenues for therapeutic intervention.

a ∣ Dysregulation of the levels of specific tRNAs leads to codon-biased translation. During steady state, the abundance of certain tRNAs could be a rate-limiting factor for the translation of oncogenic mRNAs enriched in their cognate codon (AGA highlighted in red), which restricts the translation rate and results in normal protein synthesis. Upregulation of the corresponding tRNA (red modified tRNAArg(TCT) versus other tRNAs (blue) removes this restriction, raising translation rates and increasing the production of oncogenic protein. This mechanism can result from increased ribosome occupancy or faster (enhanced) translation of mRNAs containing its cognate codon (red AGA). b ∣ Potential avenues for therapeutic intervention include pharmacological inhibition of tRNA modifying enzymes or aminoacyl tRNA synthetases (aaRSs) using small molecules or biological agents. These approaches could correct dysfunctional tRNA levels in cancer and various neurological diseases. Similarly, modulation of the expression of downstream tRNA effectors, either by ectopic restoration (green tRNA molecules) or inhibition of overexpressed tRNAs (red) might offer new strategies to treat diseases caused by altered levels of functional tRNAs. Potential approaches include pharmacological inhibition of pathological tRNAs by small molecules, RNA interference (RNAi), or antisense oligonucleotides (green). Another approach is the expression of anticodon engineered tRNAs to suppress premature termination codons and achieve normal protein synthesis.
Avenues for therapeutic intervention
Proteins involved in the biogenesis and function of tRNAs are attractive therapeutic targets to treat human diseases (Figure 6b). For example, aaRSs have received considerable attention as potential therapeutic targets in drug development, not only those targeting human aaRSs but also those targeting aaRSs produced by human pathogens (reviewed elsewhere119). Moreover, considerable effort is now being dedicated to the identification and development of small-molecule inhibitors of RNA-modifying enzymes, including tRNAs120. Another promising approach involves precise editing of the mitochondrial genome, which is opening the door to treatments for mitochondrial disorders121.
A currently overlooked area of potential therapeutic intervention is the direct modulation of tRNA molecules. This prospect could involve one of three approaches: restoration of functional tRNAs in patients with diseases caused by loss-of-function of a specific tRNA; tRNA modulation or inhibition in diseases caused by elevated levels of particular tRNAs (termed onco-tRNAs); or expression of anticodon-engineered tRNAs that can suppress premature termination codons, (Figure 6b). The clearest preclinical example of the first approach is the rescue of animal models of CMT due to aberrant GARSs by overexpression of a transgenic tRNAGly molecule105. This strategy remains to be tested in a clinical setting, and more research into its feasibility is needed. The second approach relies on the inhibition of pathogenic tRNAs such as tRNA-Arg (anticodon-TCT)-4-1, which our group has identified as a key tRNA that is largely responsible for the oncogenic activity of METTL117. Potential strategies to modulate tRNA-Arg (anticodon-TCT)-4-1 levels in tumours include the use of small molecules that target this tRNA, RNA interference (RNAi) or the use of antisense oligonucleotides to block tRNA function.122,123
Conclusions and future perspectives
Owing to their high cellular abundance and stability, tRNAs have been commonly considered to be housekeeping molecules that are intimately linked to cell proliferation and cell-cycle control. However, it is becoming increasingly clear that tRNAs are highly regulated, and that even small changes in their abundance or their nucleotide modification levels can have profound effects, leading to aberrant translation, changes in protein expression and disease states. Accumulating evidence suggests that aberrant tRNAs can drive various different human pathologies, including pro-tumorigenic programmes and neurodegenerative diseases. Nonetheless, our understanding of the role of many tRNA modifications and the corresponding modifying enzymes is currently limited. Available data highlight the tRNA epitranscriptome and the functional tRNA pool as important regulatory layers in the translation of the human genome. With the development of new high-throughput technologies (including RNA sequencing methods and RNA mass spectrometry) that are capable of detecting and quantifying tRNA modifications and tRNA abundance, we anticipate that the field will gain increased understanding of the epitranscriptome and tRNAome in both physiological and disease states. We envision that tRNA-focused analyses will become part of routine multi-omics studies, similarly to mRNAs and microRNAs, that aim to identify potential treatment targets as well as biomarkers for early diagnosis.
Key points.
tRNA-encoding genes display tissue-specific and cell-type-specific patterns of expression.
tRNA gene expression and function are both dynamically regulated by post-transcriptional RNA modifications.
Dysregulation of tRNAs, mediated by alterations in either their abundance or function, can have deleterious consequences that contribute to several distinct human diseases.
Reprogramming of mRNA translation through altered tRNA activity can drive pathological processes in a codon-dependent manner.
Transfer RNA (tRNA) function and gene expression profiles are dynamically regulated by post-transcriptional tRNA modifications. Here, Orellana and colleagues discuss the canonical role of tRNAs in mRNA translation, focusing on alterations in tRNA abundance or function implicated in human diseases.
Acknowledgements
E.A.O. is supported by the Pew Latin American Fellows Program in the Biomedical Sciences from Pew Charitable Trusts and by a fellowship from the Damon Runyon Cancer Research Foundation (DRG-2378–19). R.I.G. is supported by an Outstanding Investigator Award (R35CA232115) from the National Cancer Institute (NCI) of the NIH.
Footnotes
Competing interests
R.I.G. is a co-founder and scientific advisory board member of 28/7 Therapeutics and Theonys Therapeutics.
References
- 1. Hoagland MB, Stephenson ML, Scott JF, Hecht LI & Zamecick PC A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem 231, 241–257 (1958). This paper described the discovery of tRNAs.
- 2. Holley RW et al. Structure of a ribonucleic acid. Science 147, 1462–1465 (1965). This paper described the structure of tRNAs and the finding of a modified base (inosine).
- 3. Crick FHC Codon-anticodon pairing: the wobble hypothesis. J. Mol. Biol 19, 548–555 (1966). This paper described the possibility of non-canonical (wobble) pairing of the anticodon loop with its cognate codon sequence on mRNAs.
- 4.Chou H-J, Donnard E, Gustafsson HT, Garber M & Rando OJ Transcriptome-wide analysis of roles for tRNA modifications in translational regulation. Mol. Cell 68, 978–992.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Crécy-Lagard V. et al. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 47, 2143–2159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrov A, Martzen MR & Phizicky EM Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 8, S1355838202024019 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol 22, 375–392 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Chan PP & Lowe TM GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 44, D184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodenbour JM & Pan T Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 34, 6137–6146 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canella D, Praz V, Reina JH, Cousin P & Hernandez N Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 20, 710–721 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gogakos T. et al. Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 20, 1463–1475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Bortle K, Phanstiel DH & Snyder MP Topological organization and dynamic regulation of human tRNA genes during macrophage differentiation. Genome Biol. 18, 180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ishimura R. et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459 (2014). This paper showed for the first time the existence of tissue-specific tRNA isodecoder expression and hinted at the differential functions of isodecoders.
- 14.Geslain R & Pan T Functional analysis of human tRNA isodecoders. J. Mol. Biol 396, 821–831 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rapino F. et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Goodarzi H. et al. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165, 1416–1427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orellana EA et al. METTL1-mediated m7G modification of Arg-TCT tRNA drives oncogenic transformation. Mol. Cell 81, 3323–3338.e14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loayza-Puch F. et al. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530, 490–494 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Thandapani P. et al. Valine tRNA levels and availability regulate complex I assembly in leukaemia. Nature 601, 428–433 (2021). doi: 10.1038/s41586-021-04244-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng W. et al. Trm9-catalyzed tRNA modifications regulate global protein expression by codon-biased translation. PLoS Genet. 11, e1005706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Z, Wilson B, Kumar P & Dutta A Noncanonical roles of tRNAs: tRNA fragments and beyond. Annu Rev Genet. 54, 47–69 10.1146/annurev-genet-022620-101840 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dieci G, Fiorino G, Castelnuovo M, Teichmann M & Pagano A The expanding RNA polymerase III transcriptome. Trends Genet. 23, 614–622 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Ramsay EP et al. Structure of human RNA polymerase III. Nat. Commun 11, 6409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeganeh M & Hernandez N RNA polymerase III transcription as a disease factor. Genes Dev. 34, 865–882 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lata E. et al. RNA polymerase III subunit mutations in genetic diseases. Front. Mol. Biosci 8, 696438 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernard G. et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet 89, 415–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saitsu H. et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am. J. Hum. Genet 89, 644–651 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thiffault I. et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun 6, 7623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorboz I. et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet 4, e289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franca MM et al. Exome sequencing reveals the POLR3H gene as a novel cause of primary ovarian insufficiency. J. Clin. Endocrinol. Metab 104, 2827–2841 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terhal PA et al. Biallelic variants in POLR3GL cause endosteal hyperostosis and oligodontia. Eur. J. Hum. Genet 28, 31–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beauregard-Lacroix E. et al. A variant of neonatal progeroid syndrome, or Wiedemann-Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. Eur. J. Hum. Genet 28, 461–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borck G. et al. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 25, 155–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin W & Popko B Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 12, 379–385 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azmanov DN et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum. Mol. Genet 25, 4302–4314 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Choquet K. et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem 294, 7445–7459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White RJ RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 24, 622–629 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN & White RJ mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl Acad. Sci. USA 107, 11823–11828 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shor B. et al. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem 285, 15380–15392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michels AA et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol. Cell. Biol 30, 3749–3757 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Y, Tsang CK & Zheng XFS Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 28, 2220–2230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huber A. et al. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 23, 1929–1943 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J, Moir RD, McIntosh KB & Willis IMW TOR signaling regulates ribosome and tRNA synthesis via LAMMER/Clk and GSK-3 family kinases. Mol. Cell 45, 836–843 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomez-Roman N, Grandori C, Eisenman RN & White RJ Direct activation of RNA polymerase III transcription by c-Myc. Nature 421, 290–294 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Kenneth NS et al. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc. Natl Acad. Sci. USA 104, 14917–14922 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goodfellow SJ et al. Regulation of RNA polymerase III transcription during hypertrophic growth. EMBO J. 25, 1522–1533 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong S, Zhang C & Johnson DL Epidermal growth factor enhances cellular TATA binding protein levels and induces RNA polymerase I- and III-dependent gene activity. Mol. Cell. Biol 24, 5119–5129 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cairns CA & White RJ p53 is a general repressor of RNA polymerase III transcription. EMBO J. 17, 3112–3123 (1998). This work showed that the tumour suppressor p53 can regulate Pol III transcription.
- 49.Crighton D. et al. p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J. 22, 2810–2820 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutcliffe JE, Brown TRP, Allison SJ, Scott PH & White RJ Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol. Cell. Biol 20, 9192–9202 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larminie CGC et al. Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J. 16, 2061–2071 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirsch HA, Gu L & Henry RW The retinoblastoma tumor suppressor protein targets distinct general transcription factors to regulate RNA polymerase III gene expression. Mol. Cell. Biol 20, 9182–9191 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chu WM, Wang Z, Roeder RG & Schmid CW RNA polymerase III transcription repressed by Rb through its interactions with TFIIIB and TFIIIC2. J. Biol. Chem 272, 14755–14761 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Bell CG et al. Novel regional age-associated DNA methylation changes within human common disease-associated loci. Genome Biol. 17, 193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acton RJ et al. The genomic loci of specific human tRNA genes exhibit ageing-related DNA hypermethylation. Nat. Commun 12, 2655 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waddington CH The epigenotype. 1942. Int. J. Epidemiol 41, 10–13 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Shukla A & Bhargava P Regulation of tRNA gene transcription by the chromatin structure and nucleosome dynamics. Biophys Acta Gene Regul Mech 1861, 295–309 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Park JL et al. Epigenetic regulation of noncoding RNA transcription by mammalian RNA polymerase III. Epigenomics 9, 171–187 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Lee CK, Shibata Y, Rao B, Strahl BD & Lieb JD Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet 36, 900–905 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Guffanti E. et al. Nucleosome depletion activates poised RNA polymerase III at unconventional transcription sites in Saccharomyces cerevisiae. J. Biol. Chem 281, 29155–29164 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Kaplan N. et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar Y & Bhargava P A unique nucleosome arrangement, maintained actively by chromatin remodelers facilitates transcription of yeast tRNA genes. BMC Genomics 14, 402 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor RW & Turnbull DM Mitochondrial DNA mutations in human disease. Nat. Rev. Genet 6, 389–402 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ruiz-Pesini E. et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–D828 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suzuki T, Nagao A & Suzuki T Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet 45, 299–329 (2011). [DOI] [PubMed] [Google Scholar]
- 66. Tsutomu S, Asuteka N & Takeo S Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. Wiley Interdiscip. Rev. RNA 2, 376–386 (2011). This review discusses modopathies linked to mitochondrial tRNAs, which were the first modopathies to be described in humans.
- 67.Kirino Y. et al. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl Acad. Sci. USA 101, 15070–15075 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki T. et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun 11, 4269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thornlow BP et al. Transfer RNA genes experience exceptionally elevated mutation rates. Proc. Natl Acad. Sci. USA 115, 8996–9001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan B, Tzertzinis G, Schildkraut I & Ettwiller L Comprehensive determination of transcription start sites derived from all RNA polymerases using ReCappable-seq. Genome Res. 32, 162–174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doran JL, Bingle WH & Roy KL Two human genes encoding tRNA(GCCGly). Gene 65, 329–336 (1988). [DOI] [PubMed] [Google Scholar]
- 72.Clarke CJ et al. The initiator methionine tRNA drives secretion of type II collagen from stromal fibroblasts to promote tumor growth and angiogenesis. Curr. Biol 26, 755–765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schoenmakers E. et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Invest 126, 992–996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iben JR & Maraia RJ tRNA gene copy number variation in humans. Gene 536, 376–384 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frank DN & Pace NR Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Annu Rev Biochem 67, 153–180 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Takaku H, Minagawa A, Takagi M & Nashimoto M A candidate prostate cancer susceptibility gene encodes tRNA 3ʹ processing endoribonuclease. Nucleic Acids Res. 31, 2272–2278 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Y, Cheng K, Liang W & Wang X lncRNA RPPH1 promotes non-small cell lung cancer progression through the miR-326/WNT2B axis. Oncol. Lett 20, 105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zschocke J HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J. Inherit. Metab. Dis 35, 81–89 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Chatfield KC et al. Mitochondrial energy failure in HSD10 disease is due to defective mtDNA transcript processing. Mitochondrion 21, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oerum S. et al. Novel patient missense mutations in the HSD17B10 gene affect dehydrogenase and mitochondrial tRNA modification functions of the encoded protein. Biochim. Biophys. Acta. Mol. Basis Dis 1863, 3294–3302 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Vilardo E & Rossmanith W Molecular insights into HSD10 disease: impact of SDR5C1 mutations on the human mitochondrial RNase P complex. Nucleic Acids Res. 43, 5112–5119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Metodiev MD et al. Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am. J. Hum. Genet 98, 993–1000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hochberg I. et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am. J. Hum. Genet 108, 2195–2204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rackham O. et al. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 16, 1874–1890 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Rauschenberger K. et al. A non-enzymatic function of 17β-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol. Med 2, 51–62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saoji M, Sen A & Cox RT Loss of individual mitochondrial ribonuclease P complex proteins differentially affects mitochondrial tRNA processing in vivo. Int. J. Mol. Sci 22, 6066 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yip MCJ, Savickas S, Gygi SP & Shao S ELAC1 repairs tRNAs cleaved during ribosome-associated quality control. Cell Rep. 30, 2106–2114.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rebbeck TR et al. Association of HPC2/ELAC2 genotypes and prostate cancer. Am. J. Hum. Genet 67, 1014–1019 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haack TB et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet 93, 211–223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wedatilake Y. et al. TRNT1 deficiency: clinical, biochemical and molecular genetic features. Orphanet J. Rare Dis 11, 90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paushkin SV, Patel M, Furia BS, Peltz SW & Trotta CR Identification of a human endonuclease complex reveals a link between tRNA splicing and pre-mRNA 3ʹ end formation. Cell 117, 311–321 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Lowe TM & Eddy SR tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Budde BS et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 40, 1113–1118 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Namavar Y. et al. Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134, 143–156 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bierhals T, Korenke GC, Uyanik G & Kutsche K Pontocerebellar hypoplasia type 2 and TSEN2: review of the literature and two novel mutations. Eur. J. Med. Genet 56, 325–330 (2013). [DOI] [PubMed] [Google Scholar]
- 96.Alazami AM et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10, 148–161 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Breuss MW et al. Autosomal-recessive mutations in the tRNA splicing endonuclease subunit TSEN15 cause pontocerebellar hypoplasia and progressive microcephaly. Am. J. Hum. Genet 99, 228–235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weitzer S & Martinez J The human RNA kinase hClp1 is active on 3ʹ transfer RNA exons and short interfering RNAs. Nature 447, 222–226 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Hanada T. et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 495, 474–480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karaca E. et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157, 636–650 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sekulovski S. et al. Assembly defects of human tRNA splicing endonuclease contribute to impaired pre-tRNA processing in pontocerebellar hypoplasia. Nat. Commun 12, 5610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van Dijk T, Baas F, Barth PG & Poll-The BT What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J. Rare Dis 13, 92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sissler M, González-Serrano LE & Westhof E Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med 23, 693–708 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Antonellis A. et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet 72, 1293–1299 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zuko A. et al. tRNA overexpression rescues peripheral neuropathy caused by mutations in tRNA synthetase. Science 373, 1161–1166 (2021). This work showcases a potential approach to treat Charcot-Marie-Tooth disease by inducing overexpression of a tRNA.
- 106.Spaulding EL et al. The integrated stress response contributes to tRNA synthetase-associated peripheral neuropathy. Science 373, 1156–1161 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoffmann A. et al. Accurate mapping of tRNA reads. Bioinformatics 34, 1116–1124 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Pinkard O, McFarland S, Sweet T & Coller J Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun 11, 4104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patil A. et al. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol. 9, 990–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Begley U. et al. Trm9 catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell 28, 860–870 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Murphy FV & Ramakrishnan V Structure of a purine–purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol 11, 1251–1252 (2004). [DOI] [PubMed] [Google Scholar]
- 112.Lin H. et al. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun 9, 1875 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z. et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun. Biol 1, 234 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wolfe AL et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513, 65–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Truitt ML et al. Differential requirements for eIF4E dose in normal development and cancer. Cell 162, 59–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pavon-Eternod M. et al. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 37, 7268–7280 (2009). This paper demonstrated that overexpression of tRNAs can result in malignant phenotypes.
- 117.Birch J. et al. The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biol. Open 5, 1371–1379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dai Z. et al. N7-methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol. Cell 81, 3339–3355.e8 (2021). [DOI] [PubMed] [Google Scholar]
- 119.Kwon NH, Fox PL & Kim S Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discov 18, 629–650 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Yankova E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mok BY et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lueck JD et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun 10, 822 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Porter JJ, Heil CS & Lueck JD Therapeutic promise of engineered nonsense suppressor tRNAs. Wiley Interdiscip. Rev. RNA 12, e1641 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin S. et al. Mettl1/Wdr4-Mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 71, 244–255.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jovine L, Djordjevic S & Rhodes D The crystal structure of yeast phenylalanine tRNA at 2.0 Å resolution: cleavage by Mg2+ in 15-year old crystals. J. Mol. Biol 301, 401–414 (2000). [DOI] [PubMed] [Google Scholar]
- 126.Shaheen R. et al. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 16, 210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Trimouille A. et al. Further delineation of the phenotype caused by biallelic variants in the WDR4 gene. Clin. Genet 93, 374–377 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Braun DA et al. Mutations in WDR4 as a new cause of Galloway–Mowat syndrome. Am. J. Med. Genet. A 176, 2460–2465 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Alexandrov A, Grayhack EJ & Phizicky EM tRNA m7G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA 11, 821–830 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Alexandrov A. et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96 (2006). This report describes the existence of a quality control mechanism that degrades hypomodified tRNAs in yeast (namely, the rapid tRNA decay pathway).
- 131.de Zoysa T & Phizicky EM Hypomodified tRNA in evolutionarily distant yeasts can trigger rapid tRNA decay to activate the general amino acid control response, but with different consequences. PLOS Genet. 16, e1008893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]