

Abstract
Mitochondria must maintain adequate amounts of metabolites for protective and biosynthetic functions. However, how mitochondria sense the abundance of metabolites and regulate metabolic homeostasis is not well understood. In this work, we focused on glutathione (GSH), a critical redox metabolite in mitochondria, and identified a feedback mechanism that controls its abundance through the mitochondrial GSH transporter, SLC25A39. Under physiological conditions, SLC25A39 is rapidly degraded by mitochondrial protease AFG3L2. Depletion of GSH dissociates AFG3L2 from SLC25A39, causing a compensatory increase in mitochondrial GSH uptake. Genetic and proteomic analysis identified a putative iron-sulfur cluster in the matrix-facing loop of SLC25A39 as essential for this regulation, coupling mitochondrial iron homeostasis to GSH import. Altogether, our work revealed a paradigm for the autoregulatory control of metabolic homeostasis in organelles.
Cells require the ability to sense changes in the abundance of nutrients to ensure their efficient use for survival and growth under environmental perturbations (1). Although several nutrient sensing mechanisms have been described for cytosolic metabolites such as amino acids and cholesterol (2), whether organelles sense and regulate metabolite availability is not fully understood. Mitochondria are semiautonomous compartments with endosymbiotic origins. As a source of oxidative reactions and with an independent genetic system, mitochondria must maintain optimal concentrations of nucleotides, amino acids, and antioxidant molecules to perform critical protective and biosynthetic functions. Indeed, previous work provided evidence for such homeostatic mechanisms for a subset of redox-active molecules (3, 4). Among these, glutathione (GSH) is a small-molecule thiol that is highly abundant in mitochondria and is required for antioxidant defense and iron homeostasis (5). Mitochondrial GSH availability is largely controlled by its import through the SLC25A39 transporter on the mitochondrial inner membrane (6, 7). SLC25A39 protein accumulates upon GSH depletion, which strongly indicates a potential feedback mechanism to control the availability of mitochondrial GSH (6). In line with this observation, cells treated with buthionine sulfoximine (BSO; a GSH synthesis inhibitor) alone or in combination with erastin (an antagonist of uptake of the GSH precursor cystine) increased the abundance of SLC25A39 commensurate with the intensity of GSH depletion (fig. S1, A and B). However, how GSH levels are sensed and maintained through SLC25A39 in mitochondria is unknown.
SLC25A39 is a short–half-life protein regulated by mitochondrial GSH availability
To understand how SLC25A39 is regulated in mitochondria, we compared global mRNA and protein abundance using the OpenCell database (8). This revealed a small subset of posttranscriptionally regulated proteins with low protein-to-mRNA ratios, such as Hypoxia Inducible Factor 1 Subunit Alpha (HIF1A), DAP3 binding cell death enhancer 1 (DELE1), Activating Transcription Factor 4 (ATF4), ornithine decarboxylase antizyme (OAZ1), and F-box and leucine-rich repeat protein 5 (FBXL5). These proteins are rapidly degraded or translationally repressed under basal conditions but accumulate upon diverse environmental stimuli, which enables their dynamic response to cellular stress. Among these proteins, SLC25A39 was the only mitochondrial transporter, indicating that a posttranscriptional mechanism may underlie its regulation (Fig. 1A). To determine the mode of SLC25A39 regulation in response to GSH depletion, we generated a reporter construct in which a 3xFLAG-tagged SLC25A39 cDNA was cotranslated with an internal control (RFP), separated by a 2A self-cleaving peptide from porcine teschovirus-1 (P2A). GSH depletion in human embryonic kidney HEK293T cells expressing this reporter strongly induced the accumulation of the 3xFLAG-SLC25A39 protein relative to the abundance of RFP (Fig. 1B). This effect was independent of oxidative stress, as only supplementation of GSH, but not other antioxidants, suppressed SLC25A39 accumulation (Fig. 1B and fig. S1C). Because these observations point to a protein-level control, we measured the half-life of SLC25A39 in cycloheximide chase assays. SLC25A39 protein was unstable, with an estimated half-life of 15 min (Fig. 1C). GSH depletion significantly extended the half-life of SLC25A39 (>300 min), and supplementing cells with GSH restored this rapid degradation (Fig. 1C). These data indicate that SLC25A39 has a short half-life and becomes stabilized when GSH is depleted.
Fig. 1. SLC25A39 is a short–half life protein regulated by mitochondrial GSH availability.
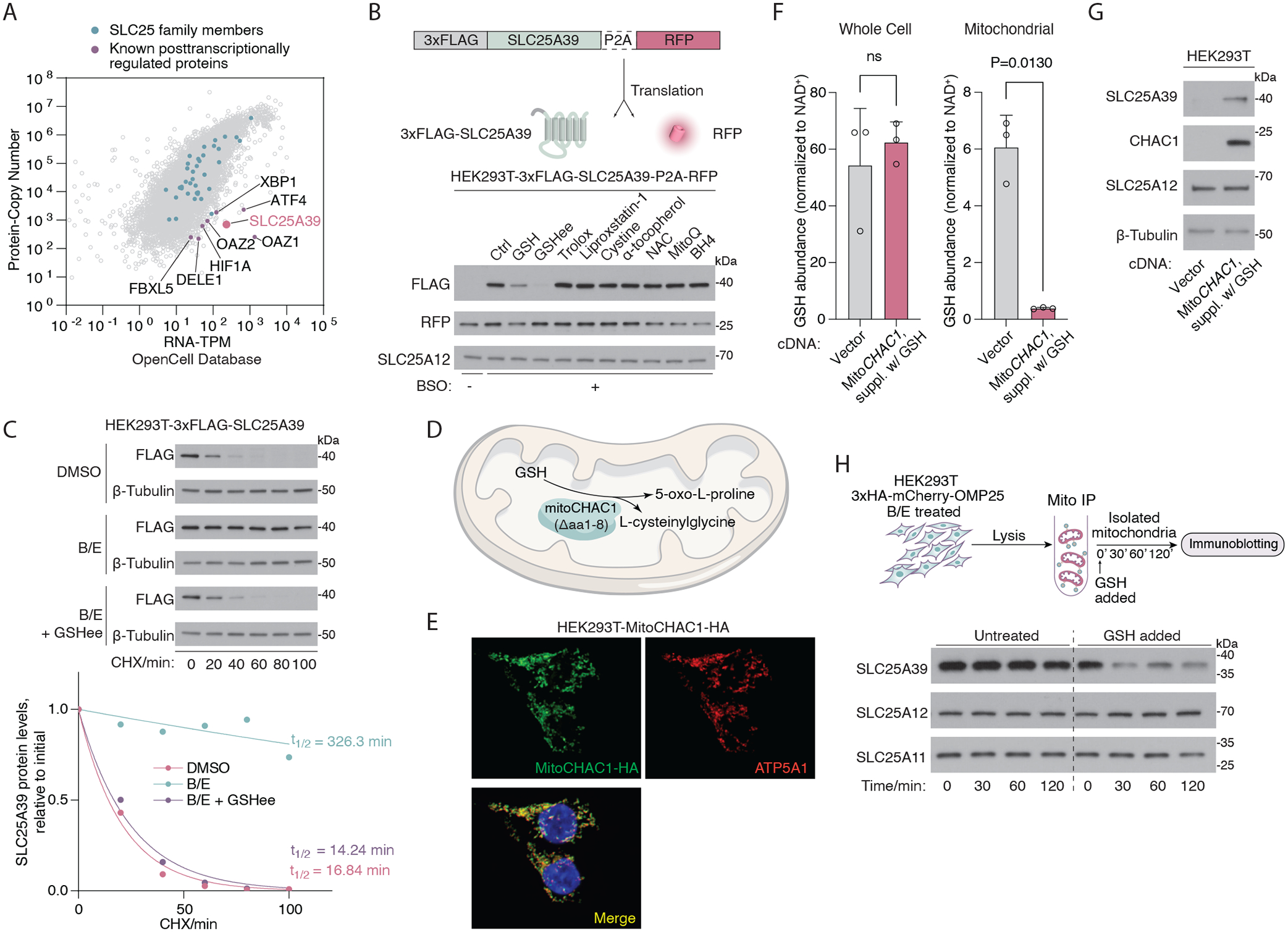
(A) Scatter plot showing the protein copy number versus mRNA abundance (TPM) for all genes in HEK293T cells detectable across the proteome. Original data were retrieved from the OpenCell database. Green dots denote SLC25 family proteins, and purple dots denote representative proteins known to be regulated post transcriptionally. (B) (Top) Schematic showing the construct for cotranslational expression of 3xFLAG-tagged SLC25A39 and RFP, separated by a self-cleaving P2A peptide. (Bottom) Immunoblots of the indicated proteins in HEK293T cells expressing the aforementioned construct. Cells were treated with BSO (1 mM) for 48 hours and were then treated with GSH (10 mM), GSH ethyl ester (GSHee, 10 mM), Trolox (50 μM), Liproxstatin-1 (1 μM), cystine (200 μM), N-acetylcysteine (NAC, 1 mM), α-tocopherol (5 μM), MitoQ (30 nM), or BH4 (4 μM) for 8 hours. RFP was used as an internal control for the translational levels of the construct, and SLC25A12 was used as a loading control. (C) (Top) Immunoblots of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39 cDNA treated with cycloheximide (CHX, 50 μg/ml) for the indicated times. Prior to CHX treatment, cells were treated with BSO (1 mM) and erastin (5 μM) for 24 hours and GSH ethyl ester (GSHee, 10 mM) for 8 hours. DMSO was used as the control. β-tubulin was used as a loading control. (Bottom) Quantification of FLAG band signal intensity from the immunoblots above. Half-life (t1/2) was calculated by the nonlinear fitting of FLAG band signal intensity versus time to one phase decay exponential model. (D) Schematic showing the localization and the catalytic reaction of engineered MitoCHAC1 protein. (E) Immunofluorescence images of MitoCHAC1 (HA, green), ATP5A1 (red), and DAPI (blue) in HEK293T cells. (F) Whole-cell and mitochondrial abundance of GSH (normalized to NAD+ abundance) in HEK293T cells expressing empty vector or MitoCHAC1 (in the presence of exogenous GSH). Data are mean ± SD representing three biologically independent samples. P values were calculated from Welch’s t test. (G) Immunoblots showing the amounts of the indicated proteins in HEK293T cells that express an empty vector or MitoCHAC1 (in the presence of exogenous GSH). SLC25A12 and β-tubulin were used as loading controls. (H) (Top) The schematic of the cell-free assay that uses immunopurified mitochondria (mito IP) from HEK293T cells to analyze SLC25A39 stability. (Bottom) Immunoblots of the indicated proteins from purified mitochondria after treating them with GSH (20 mM) for the indicated times. SLC25A12 and SLC25A11 were used as loading controls.
Given that the mitochondrial matrix harbors a distinct GSH pool (9), we sought to determine whether SLC25A39 stability is regulated specifically by changes in the availability of local mitochondrial GSH, thus forming a feedback loop in which the substrate controls its own uptake. To test this possibility, we engineered the human GSH-specific gamma-glutamylcyclotransferase (CHAC1), a cytosolic enzyme that catalyzes the conversion of GSH into 5-oxoproline and L-cysteinylglycine (10, 11), to be expressed in mitochondria (hereby referred to as MitoCHAC1) (Fig. 1, D and E, and fig. S1D). This engineered enzyme allowed us to specifically deplete mitochondrial GSH without significantly altering whole-cell GSH levels (Fig. 1F and fig. S1, E to G). Confirming the robust enzymatic activity of MitoCHAC1, we observed 5-oxoproline accumulation in the mitochondria (fig. S1H). Depletion of mitochondrial GSH with MitoCHAC1 expression was sufficient to induce SLC25A39 accumulation (Fig. 1G and fig. S1I). Expression of MitoCHAC1 did not affect the abundance of SLC25A39 mRNA and only slightly decreased cell proliferation (fig S1, J and K). Moreover, expression of MitoCHAC1 did not impact the abundance of the master regulator of the antioxidant response, nuclear factor erythroid 2–related factor 2 (NRF2), or that of its downstream transcriptional targets (12), indicating that NRF2 activity is not controlled by mitochondrial GSH availability (fig. S1L). To formally test whether SLC25A39 stability can be regulated independently of any signaling input from the cytosol, we developed a cell-free system with immunopurified mitochondria (Fig. 1H). Although isolated mitochondria from GSH-depleted cells displayed highly stable SLC25A39, direct supplementation with exogenous glutathione led to rapid degradation of SLC25A39, indicating that signaling input from cytosol or nucleus is dispensable for such regulation. Thus, a compartmentalized feedback mechanism appears to enable mitochondria to rapidly respond to the changes in the concentration of GSH in the matrix by tuning SLC25A39 stability.
AFG3L2 degrades SLC25A39 in a GSH-dependent manner
SLC25A39, unlike other SLC25 family members, has an exceptionally low protein copy number to mRNA-TPM ratio and is distinctly sensitive to GSH availability. We therefore considered that a particular structural feature on SLC25A39 might allow this regulation. We aligned AlphaFold (13)–predicted structures of SLC25A39 with those of highly expressed SLC25 family members as well as that of SLC25A40, a paralog of SLC25A39 that is not sensitive to GSH availability (Fig. 2A and fig. S2, A and B). This analysis revealed a protruding loop on the matrix side of the SLC25A39 protein that corresponded to amino acids 42 to 106 (aa42–106) (Fig. 2A and fig. S2A). Although this domain is not critical for GSH transport activity (fig. S2C), its deletion completely abrogated GSH sensitivity and extended protein half-life (>250 min), thereby uncoupling the GSH-mediated regulation of SLC25A39 from its transport function (Fig. 2B and fig. S2D, lanes 1 to 4). Splicing this fragment into a distant SLC25 family member (SLC25A11) that does not respond to perturbations in GSH levels rendered it GSH responsive (Fig. 2C). The differential responses of SLC25A39 and its close paralog, SLC25A40, to GSH availability raised the possibility that the matrix loop may be the critical attribute for the diversification of these two genes. Indeed, a significant portion of the SLC25A39 loop domain responsible for GSH sensitivity is evolutionarily new, with no homologs outside vertebrates (data S1). Using a maximum likelihood method, we reconstructed the ancestral sequence of SLC25A39 (fig. S2, E to G). When spliced into SLC25A11, the loop domain in the ancestral SLC25A39 sequence conferred GSH responsiveness, indicating that this feature arose early in the evolution of SLC25A39 and might be the key driving force behind the diversification of mitochondrial GSH transporters (fig. S2H). Thus, the conserved matrix-facing loop domain is necessary and sufficient for the GSH-mediated regulation of SLC25A39.
Fig. 2. AFG3L2 binds and degrades SLC25A39 through a matrix loop domain in a GSH-dependent manner.
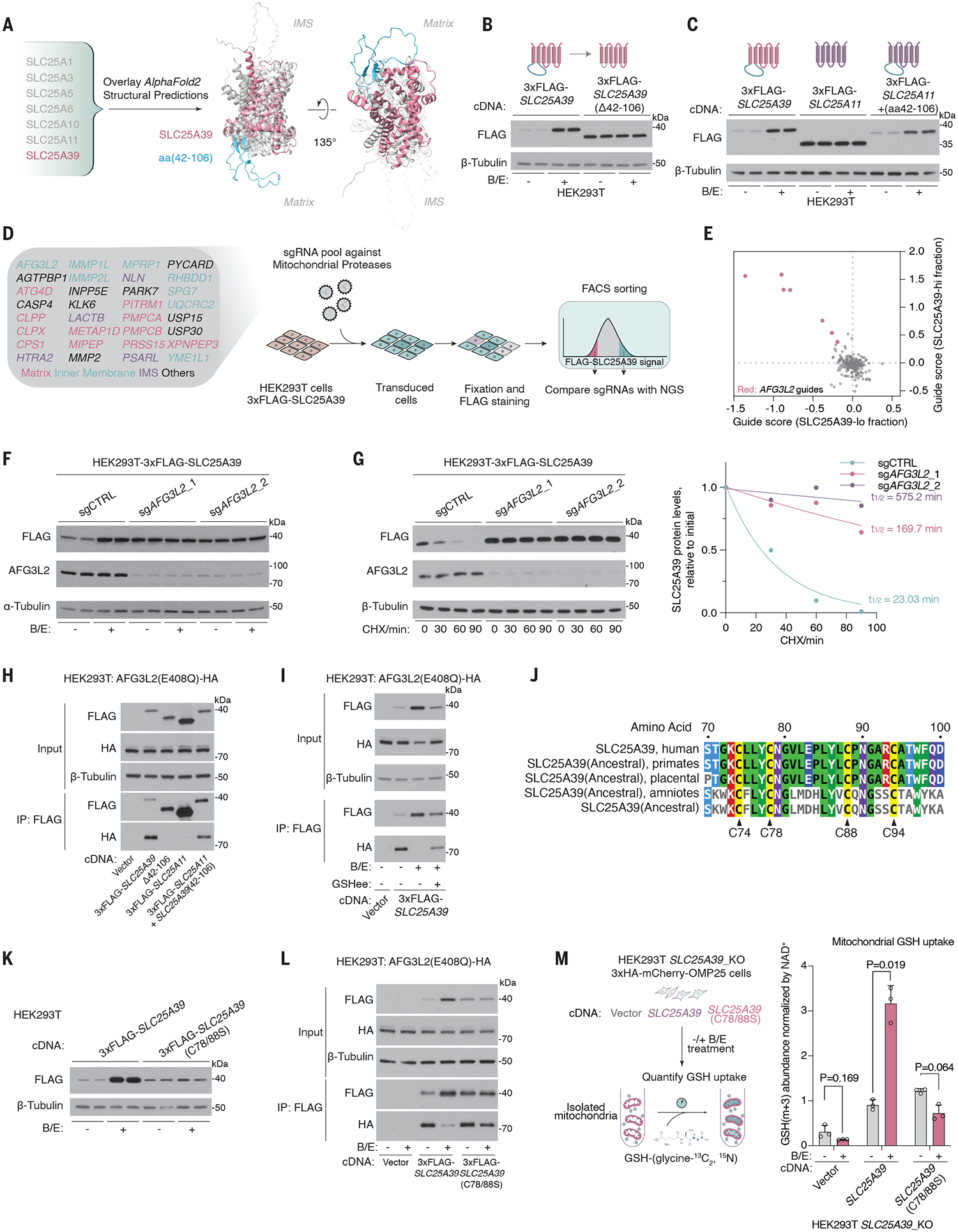
(A) Schematic showing the alignment of AlphaFold2-predicted structural models of the indicated proteins. SLC25A39 is highlighted in pink, with aa42–106 highlighted in green. (B) Immunoblots of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39 or 3xFLAG-SLC25A39 without the matrix-facing loop (Δaa42–106) after 24-hour treatment with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (C) Immunoblots of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39, 3xFLAG-SLC25A11, or a chimeric protein in which aa42–106 of SLC25A39 is spliced into SLC25A11 after 24-hour treatment with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (D) Schematic showing the library design of the mitochondrial peptidase sgRNA library and the workflow of the FACS-based CRISPR screen for 3xFLAG-SLC25A39 stability. (E) Scatter plot showing the enrichment of sgRNAs targeting mitochondrial proteases in the SLC25A39-lo cell fraction (x axis) and SLC25A39-hi cell fraction (y axis). Red dots represent sgRNAs targeting AFG3L2. (F) Immunoblots of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39 and sgRNAs targeting control or AFG3L2 after 24-hour treatment with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (G) (Left) Immunoblots of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39 and sgRNAs targeting AFG3L2 or control upon treatment with cycloheximide (CHX, 50 μg/ml) for the indicated times. β-tubulin was used as a loading control. (Right) Quantification of FLAG bands signal intensity from the immunoblots. Half-life (t1/2) was calculated by the nonlinear fitting of FLAG band signal intensity versus time to one phase decay exponential model. (H) Immunoblots of the indicated proteins from whole-cell lysates or FLAG-immunoprecipitation from HEK293T cells stably expressing cDNAs for vector, 3xFLAG-SLC25A39, 3xFLAG-SLC25A39 lacking matrix-facing loop (Δaa42–106), 3xFLAG-SLC25A11, or a chimeric protein in which aa42–106 of SLC25A39 is spliced into SLC25A11 and transiently transfected with AFG3L2 (E408Q)-HA cDNA. (I) Immunoblots of the indicated proteins from whole-cell lysates or FLAG immunoprecipitation from HEK293T cells stably expressing cDNA for empty vector or 3xFLAG-SLC25A39 and are transiently transfected with AFG3L2(E408Q)-HA cDNA. Cells were treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control. Indicated cells were then treated for 8 hours with GSHee (10 mM). (J) Multiple sequence alignment between SLC25A39 and the inferred ancestral sequence of SLC25A39 reconstructed from amino acid sequences of SLC25A39 homologs in the indicated taxa. Four conserved cysteines in the matrix-facing loop are highlighted. (K) Immunoblots for the indicated proteins in HEK293T cells that express 3xFLAG-SLC25A39 or 3xFLAG-SLC25A39(C78/88S). Cells were treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (L) Immunoblot of the indicated proteins from whole-cell lysates or FLAG immunoprecipitation from HEK293T cells stably expressing cDNAs for vector, 3xFLAG-SLC25A39, or 3xFLAG-SLC25A39(C78/88S) and are transiently transfected with AFG3L2(E408Q)-HA cDNA. Cells were treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (M) (Left) Schematic showing the GSH uptake assay that uses immunopurified mitochondria from HEK293T-SLC25A39_KO cells expressing cDNAs for empty vector, SLC25A39, or SLC25A39(C78/88S). (Right) Abundance of GSH-(glycine-13C2 ,15N) taken up by isolated mitochondria. Data are mean ± SD representing three biologically independent samples. P values were calculated from Welch’s multiple t test with the Holm-Šídák method.
We next sought to determine the proteolytic machinery involved in SLC25A39 degradation. Inhibition of proteasomal or lysosomal function did not impact SLC25A39 stability (fig. S3A). Given these results and the mitochondria-localized regulation of SLC25A39 (Fig. 1H), we focused on proteolysis systems in the mitochondria. We designed a fluorescence-activated cell sorting (FACS)–based CRISPR screen for 3xFLAG-SLC25A39 stability with a single guide RNA (sgRNA) library targeting all annotated mitochondrial proteins with peptidase activity (34 genes; 7 sgRNAs per gene) (Fig. 2D and fig. S3, B and C). Transduced cells were stained with a FLAG antibody, and sgRNA sequences from cells with the highest (SLC25A39-hi) and the lowest (SLC25A39-lo) FLAG signal were quantified by next-generation sequencing. Among all candidates, AFG3L2, a mitochondrially localized protease, was the only target whose loss significantly altered the abundance of SLC25A39 (q value = 0.009) (Fig. 2E, fig. S3D, and data S2). Consistent with the screen results, the loss of AFG3L2 completely abolished its GSH-mediated regulation (Fig. 2F and fig. S3E) and stabilized SLC25A39 even at normal GSH concentrations (Fig. 2G). SPG7, the paralog of AFG3L2, was dispensable for this process (fig. S3, F to H). Furthermore, immunoprecipitation experiments with an ATP triphosphatase–mutant AFG3L2(E408Q), which allows the detection of transient protease-target interactions (14), revealed that SLC25A39 associated with AFG3L2 through its loop domain (aa42–106) (Fig. 2H). When this loop was removed from SLC25A39, the loss of AFG3L2 did not further stabilize the protein (fig S2D). This interaction was highly sensitive to changes in GSH availability and was abrogated by GSH depletion (Fig. 2I). Unbiased proteomic profiling further confirmed that SLC25A39, but not other mitochondrial membrane proteins, was specifically targeted for degradation by AFG3L2 in a GSH-dependent manner (fig S4, A to F, and data S3). GSH availability therefore determines the turnover of SLC25A39 protein by enabling the recruitment of AFG3L2 through the matrix-facing loop domain of SLC25A39.
CRISPR screen identifies [2Fe-2S] cluster assembly as essential for SLC25A39 stability
To further dissect the function of the loop domain in the GSH-mediated regulation of SLC25A39, we used two orthogonal approaches. We used a reporter assay for mitochondrial protein stability (fig. S5A) to identify a short fragment on the GSH-responsive loop of SLC25A39 (aa72–86) necessary for its recruitment to AFG3L2 and proteolysis (fig. S5, B to E). Additionally, a detailed conservation analysis revealed four highly conserved cysteines—Cys74, Cys78, Cys88, and Cys94—within the vicinity of this fragment (Fig. 2J and fig. S5F). Introducing individual cysteine-to-serine mutations, particularly on Cys88 and Cys94, partially abrogated SLC25A39 stabilization upon GSH depletion (Fig. 2J and fig. S6A). More notably, mutating two cysteines in the loop domain was sufficient to completely abolish the regulation (Fig. 2K and fig. S6, B and C). The absence of these conserved cysteines led to the constitutive association of SLC25A39 and AFG3L2, independent of GSH abundance (Fig. 2L). To investigate the functional role of these cysteines in maintaining GSH homeostasis in the mitochondria, we performed uptake assays with isotope-labeled GSH [GSH-(glycine-13C2,15N)] in mitochondria isolated from control cells or cells in which GSH was depleted. Consistent with the strong feedback mechanism, in the presence of wildtype SLC25A39, GSH depletion led to a 3.5-fold compensatory increase in mitochondrial GSH uptake (Fig. 2M). By contrast, mutating cysteine residues (C78/88S) completely abolished this homeostatic mechanism (Fig. 2M and fig. S6D), confirming the essential role of conserved cysteines in the feedback regulation of SLC25A39-mediated GSH uptake.
Cysteine residues on proteins have critical roles in redox sensing, posttranslational modifications, and cofactor binding (15). However, our initial interrogation showed that disulfide bond formation and relay or hypoxic response did not impact SLC25A39 regulation (fig. S7, A to E). To identify the precise mechanism by which cysteines in the loop domain enable GSH-mediated regulation of SLC25A39 stability, we performed a FACS-based CRISPR screen for SLC25A39 stability under GSH depletion in HEK293T cells expressing a 3xFLAG-tagged SLC25A39 cDNA (Fig. 3A). Given that regulation of SLC25A39 can occur independently of any cytosolic machinery (Fig. 1H), for these screens, we generated an sgRNA library that contained all annotated mitochondrial proteins (MITO-sgRNA) (16, 17). After immunostaining with a FLAG antibody, transduced cells with the highest (SLC25A39-hi) and lowest (SLC25A39-lo) FLAG signal intensity were isolated and their sgRNA abundances were quantified. Among the genes whose disruption led to a lower amount of SLC25A39 were many assembly factors for [2Fe-2S] clusters such as NFS1, ISCU, HSCB, and GLRX5 (Fig. 3, B and C, fig. S8A, and data S2). Additionally, one of the genes whose loss most strongly enhanced SLC25A39 stability was ABCB7, a putative transporter involved in the export of iron-sulfur clusters from mitochondria (18–20). A gene ontology analysis further confirmed [2Fe-2S] cluster assembly as the most enriched pathway for the scoring genes in the SLC25A39-lo fraction (FDR = 1.8 × 10−5) (Fig. 3B and fig. S8A). Consistent with the screen results, acute loss of Fe-S cluster assembly factors (ISCU, GLRX5, or HSCB) abolished SLC25A39 stabilization under GSH depletion (Fig. 3D and fig. S8, B to F), whereas ABCB7 loss constitutively stabilized SLC25A39, likely owing to the mitochondrial accumulation of [2Fe-2S] clusters (Fig. 3D and fig. S8G) (21). Unbiased proteomics experiments under GSH-depleted conditions also revealed SLC25A39 as the only mitochondrial transporter whose abundance decreased upon blocking synthesis of [2Fe-2S] clusters (Fig. 3E, fig. S8H, and data S4). The loss of [2Fe-2S] assembly factor HSCB (22) led to the constitutive association of SLC25A39 with AFG3L2 (Fig. 3F). Altogether, these results suggest that [2Fe-2S] cluster synthesis is necessary for stabilizing SLC25A39 when cells are depleted of GSH.
Fig. 3. CRISPR screen identifies [2Fe-2S] cluster assembly as essential for the regulation of SLC25A39 stability.
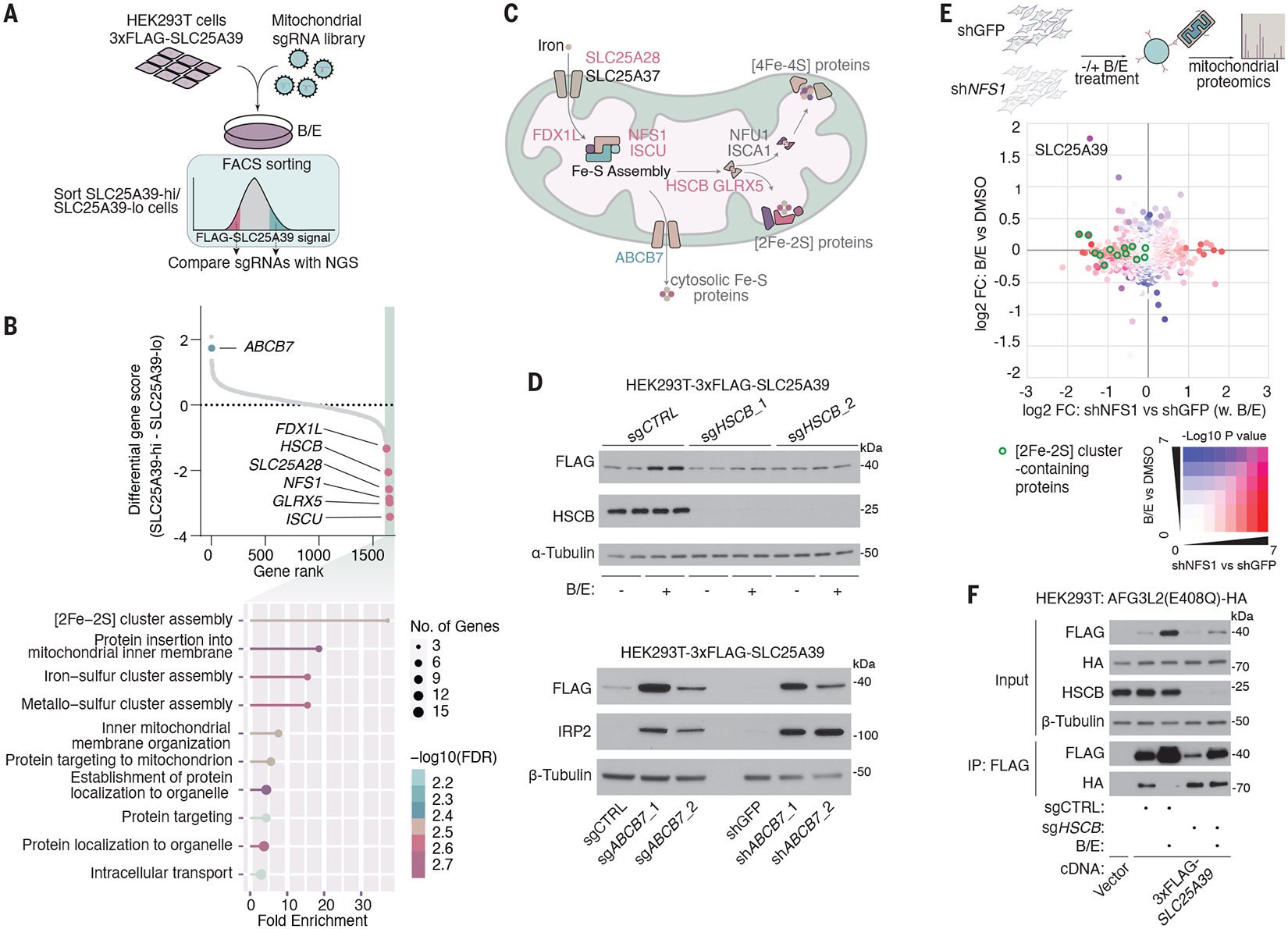
(A) Schematic of the CRISPR screen workflow with mitochondrial sgRNA library for SLC25A39 stability under GSH depletion. (B) (Top) Dot plot showing the distribution of differential CRISPR gene score calculated as (median guide enrichment in the SLC25A39-hi fraction) – (median guide enrichment in the SLC25A39-lo fraction). Pink dots indicate genes in the [2Fe-2S] cluster assembly pathway. The green dot indicates the putative mitochondrial [2Fe-2S] cluster exporter ABCB7. (Bottom) Gene Ontology enrichment analysis of genes with a differential CRISPR gene score lower than −1. (C) Schematic of the mitochondrial Fe-S cluster assembly pathways that highlights the genes that scored (pink and green) in the CRISPR screen. (D) Immunoblots of the indicated proteins in HEK293T cells that express 3xFLAG-SLC25A39. (Top) Cells were transduced with lentivirus expressing Cas9 and control sgRNA or sgRNAs that target iron-sulfur cluster assembly factor HSCB. Cells were treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (Bottom) Cells were transduced with lentivirus expressing control sgRNA or sgRNAs targeting ABCB7 (left), or shRNAs targeting GFP or ABCB7 (right). (E) Scatter plot showing log2 fold change and −log10(P value) of proteomics analysis from immunopurified mitochondria of the indicated samples. The x axis represents log2 protein fold change (FC) in isolated mitochondria from HEK293T cells that express shRNA targeting GFP or cysteine desulfurase NFS1 after 24-hour treatment with BSO (1 mM) and erastin (5 μM). The y axis represents log2 fold change (FC) in mitochondrial protein abundance after treating cells with BSO (1 mM) and Erastin (5 μM) versus DMSO as the control. The color grid indicates −log10(P values) and green circles represent [2Fe-2S] cluster–containing proteins. (F) Immunoblots of the indicated proteins from the whole-cell lysates or FLAG-immunoprecipitation from HEK293T cells stably expressing cDNAs for vector or 3xFLAG-SLC25A39, are infected with lentivirus-expressing control sgRNA or sgRNAs for HSCB, and are transiently transfected with AFG3L2(E408Q)-HA cDNA. Cells were treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control.
Iron-sulfur clusters are typically coordinated by cysteines and are essential for the stability of the holoprotein. Because iron-sulfur cluster assembly and conserved cysteines are indispensable for SLC25A39 stability, we considered the possibility that SLC25A39 might be associated with a [2Fe-2S] prosthetic group. To test the presence of potential Fe-containing cofactors associated with SLC25A39, we labeled HEK293T cells expressing 3xFLAG-tagged SLC25A39 cDNA F4 with radioactive 55FeCl3 (Fig. 4A). Although we did not observe evidence of increased iron-sulfur cluster synthesis or accumulated mitochondrial iron (fig. S9, A to C), the amount of 55Fe immunoprecipitated with SLC25A39 was increased upon GSH depletion (Fig. 4B). This increase of bound iron appeared to precede the stabilization of SLC25A39 because similar results were observed in AFG3L2-knockout cells with a comparable amount of immunoprecipitated SLC25A39 protein (Fig. 4B). This factor is unlikely to exist as heme iron or [4Fe-4S] clusters as depleting cells of ferrochelatase (FECH), a critical enzyme of heme synthesis or [4Fe-4S] cluster-assembly factor NFU1 had no effect on SLC25A39 regulation (fig. S9, D and E). By contrast, the loss of [2Fe-2S] assembly factor HSCB almost completely blocked iron binding to SLC25A39 (fig. S9F). The iron-binding activity of SLC25A39 lies within its glutathione-sensing matrix loop. Deleting this loop abolished the iron-binding of SLC25A39, and splicing the loop domain of SLC25A39 to SLC25A11, which normally does not bind iron, enabled it to associate with iron (fig. S9G). Mutating the critical cysteines in the loop domain (Cys74/78/88/94) prevented the association of 55Fe to SLC25A39 upon glutathione depletion (Fig. 4C). Furthermore, with a synthetic peptide encompassing the GSH-responsive cysteines of SLC25A39 (aa73–95), we were able to reconstitute the peptide-[2Fe-2S] cluster complex in vitro that displayed characteristic spectral features of a [2Fe-2S] cluster (Fig. 4D) in which all four cysteines could engage in metal coordination (fig. S9H). We conclude that SLC25A39 associates with a GSH-sensitive iron-sulfur cluster through its cysteine residues.
Fig. 4. A GSH-sensitive iron-sulfur cluster associates with SLC25A39 and mediates its regulation.

(A) Schematic showing the method of tracking iron-sulfur cluster bound to SLC25A39 by means of 55Fe tracing. (B) (Left) Immunoblots of the indicated proteins from whole-cell lysates or FLAG immunoprecipitation from HEK293T cells stably expressing cDNAs for empty vector or 3xFLAG-SLC25A39 and are infected with lentivirus-expressing control sgRNA or sgRNA targeting AFG3L2. Cells were labeled with 55FeCl3 in the culture media and treated for 24 hours with BSO (1 mM) and erastin (5 μM) or DMSO as the control. (Right) The amount of 55Fe bound to FLAG immunoprecipitant, from the identical cells as those in the immunoblot, quantified by liquid scintillation assay. (C) (Left) Immunoblots of the indicated proteins from whole-cell lysates or FLAG immunoprecipitation from HEK293FS cells stably expressing cDNAs for empty vector, 3xFLAG-SLC25A39, 3xFLAG-SLC25A39(C74/78/88/94S), or 3xFLAG-SLC25A11. Cells were labeled with 55FeCl3 in the culture media and treated for 24 hours with BSO (1 mM) and erastin (5 μM). (Right) The amount of 55Fe bound to FLAG immunoprecipitant, from the identical cells as those used for the immunoblot, quantified by liquid scintillation assay. (D) (Left) Schematic showing the location and peptide sequence of SLC25A39(aa73–95) used to reconstitute peptide-[2Fe-2S] cluster complex in vitro. (Right) UV-visible spectrum (with the calculated contribution of the indicated species) and Mössbauer spectrum of the reconstituted peptide-[2Fe-2S] complex. The Mössbauer spectrum was least-squares fit to extract hyperfine parameters: isomer shift (δ), quadrupole splitting (Δ), full width at half-maximum (FWHM), and intensities (I). The red and green curves of the Mössbauer spectrum plot represent two Fe3+ doublets with slightly different shifts and quadrupole splitting fitted to the spectral data. Red: δ = 0.23 mm/s, Δ = 0.51 mm/s, and I = 70%. Green: δ = 0.30 mm/s, Δ = 0.90 mm/s, and I = 30%. (E) Schematic for cysteine reactivity profiling for SLC25A39 with an iodoacetamide-desthiobiotin (IA-DTB) probe. (F) Reactivity of SLC25A39-Cys94, Cys88, and Cys202 after a 24-hour treatment with indicated reagents, quantified by mass spectrometry as the intensity ratio between IA-DTB–labeled versus unlabeled peptide containing SLC25A39-Cys94 and normalized to DMSO-treated samples. [(B), (C), and (F)] Data are mean ± SD and represent three biologically independent samples. P values were calculated from one-way analysis of variance.
As a small-molecule thiol, GSH could exchange with the cysteine ligands of a [2Fe-2S] cluster, resulting in partial or complete dissociation of the cluster from the holoprotein (21, 23, 24) (fig. S9I). In line with these previous observations, GSH supplementation induced a notable change in the spectral feature of the SLC25A39 (aa73–95)–[2Fe-2S] cluster complex (fig S9, J and K), indicating the displacement of one or more cysteine residues in the loop domain. Additionally, supplementation of cell-permeable GSH to GSH-depleted cells significantly reduced the amount of iron bound to SLC25A39 (fig. S9L). To further support these findings, we used chemical proteomics to assess the reactivity of cysteines with an iodoacetamide-desthiobiotin probe, given that coordination with iron-sulfur clusters could limit cysteine reactivity (Fig. 4E) (25). Cys94, a conserved cysteine in the SLC25A39 matrix loop that was most reliably detected by mass spectrometry, displayed a strong decrease in reactivity upon GSH depletion, which was restored by iron chelation (Fig. 4F and data S5). We observed a similar trend for Cys88; by contrast, Cys202, a cysteine residue that lies outside of the matrix-facing loop, showed no significant change in reactivity. These observations support a model in which GSH depletion leads to the tight association of a [2Fe-2S] cluster to SLC25A39 through cysteines (C74/78/88/94) in the loop domain, which in turn prevents the recruitment of protease AFG3L2, thus stabilizing SLC25A39.
SLC25A39-mediated GSH import maintains mitochondrial Fe-GSH balance
We considered why such a feedback mechanism might have evolved and how it might contribute to mitochondrial function. In addition to its antioxidant role, GSH is a major endogenous iron ligand in cells (26). In yeast, the absence or an excess of GSH can both lead to abnormal iron metabolism and the activation of cellular stress responses (27). Given the essential role of iron-sulfur clusters in SLC25A39 stability, this feedback mechanism might help maintain a proper balance between the availability of iron and GSH in mitochondria. Consistent with this idea, when we increased the mitochondrial iron/GSH ratio by overexpressing Mitoferrin 1 (SLC25A37) or Mitoferrin 2 (SLC25A28), the two major mitochondrial iron importers, SLC25A39 protein levels increased (Fig. 5A and fig. S10A). This regulation occurs through the GSH-responsive cysteines on SLC25A39 because mutating these cysteines in situ dampened the response of SLC25A39 to iron overload (fig. S10B). The increase in mitochondrial iron accompanied a compensatory increase in the abundance of GSH in the mitochondria (fig. S10C). By contrast, iron chelation, which decreases the mitochondrial iron/GSH ratio, abolished the stabilization of SLC25A39 upon GSH depletion (Fig. 5B and fig. S10D). We used multiple orthogonal approaches to perturb mitochondrial iron and GSH pools, and the abundance of SLC25A39 responded in accordance with the change in iron/GSH ratio, indicating that a feedback mechanism maintains iron/GSH balance (Fig. 5, C and D). We tested whether GSH limitation and iron overload imposed similar stresses on mitochondrial function by comparing mitochondrial proteomes from cells depleted of GSH depletion (6) or exposed to excess mitochondrial iron after the overexpression of Mitoferrin 2 (fig. S11A). These conditions led to the depletion of similar proteins, particularly those that function in the mitochondrial translation machinery, ETC components, and iron-sulfur proteins (fig. S11, B and C, and data S6). Decreases in the abundance of mitochondrial proteins induced by mitochondrial iron overload could be largely restored by boosting mitochondrial GSH uptake, supporting a critical buffering role of GSH in iron overload (Fig. 5, E and F; fig. S11, D and E; and data S7). SLC25A39 appeared to protect mitochondrial function at least partially through its adaptive response to iron/GSH balance because defects in the GSH-sensing function of SLC25A39 led to suboptimal mitochondrial function and impaired viability under GSH limitation (fig. S12, A to D). Thus, a feedback mechanism for SLC25A39 stability enables cells to maintain optimal mitochondrial functions by restoring iron/GSH balance.
Fig. 5. SLC25A39-mediated GSH import maintains iron/GSH balance in mitochondria.
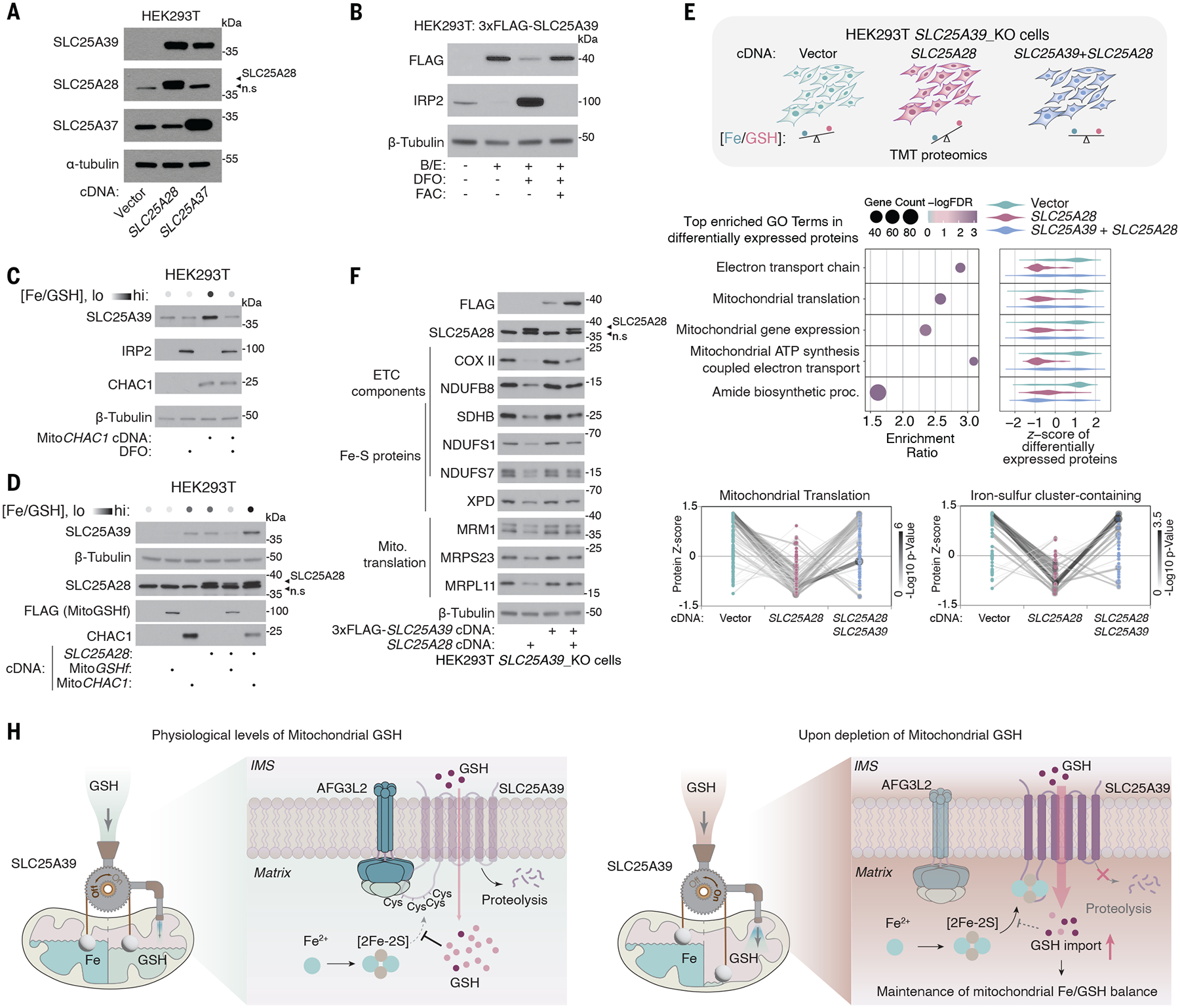
(A) Immunoblots of the indicated proteins from HEK293T cells overexpressing cDNAs of Mitoferrin 1 (SLC25A37), Mitoferrin 2 (SLC25A28), or an empty vector. (B) Immunoblot of the indicated proteins in HEK293T cells expressing 3xFLAG-SLC25A39 cDNA after 24-hours treatment with BSO (1 mM) and Erastin (5 μM); BSO, Erastin, and deferoxamine (50 μM); or BSO, Erastin, deferoxamine (50 μM), and Ferric Ammonium Citrate (FAC, 10 μg/ml). (C) Immunoblots of the indicated proteins from HEK293T cells overexpressing cDNAs of MitoCHAC1 or empty vector after 4 hours of treatment with 50-μM iron chelator deferoxamine (DFO) or control. (D) Immunoblots of the indicated proteins from HEK293T cells overexpressing cDNAs of SLC25A28, MitoCHAC1, or FLAG-tagged MitoGSHf, an engineered bacterial GSH synthase localized to the mitochondria. (E) (Top) Schematic of the experiment setup of TMT proteomics for cells with different iron/GSH ratios. Three biologically independent samples per condition from HEK293T-SLC25A39_KO cells expressing the indicated cDNAs were used. (Middle) Gene ontology enrichment analysis of the most differentially expressed proteins across the three conditions that shows the top five most significantly enriched biological processes. The violin plots indicate the relative protein abundance (z-scores) of the differentially expressed proteins in the indicated biological processes. (Bottom) Dot plot representing protein levels (z-scores) of the mitochondrial translation machinery and iron-sulfur cluster–containing proteins. The darkness of the lines represents the statistical significance of the changes in protein abundance. (F) Immunoblots of the indicated proteins in HEK293T-SLC25A39_KO cells overexpressing empty vector, SLC25A39 cDNA, Mitoferrin 2 (SLC25A28) cDNA, or both. (G) Schematic for the model describing the autoregulatory control of mitochondrial iron/GSH balance by SLC25A39.
Discussion
Metabolic homeostasis is maintained by negative feedback regulation at both the cellular and organismal levels. We provide evidence that organelles use similar principles to control their internal metabolite pools. In mitochondria, autoregulatory control of GSH availability occurs by coupling GSH sensing in the matrix to the degradation of its transporter, SLC25A39. This regulation pattern mirrors the homeostatic mechanisms for redox potential (28) or pH (29) in other organelles. Conceivably, other cellular compartments, the internal chemical environments of which differ from that of cytosol, may harbor similar mechanisms for sensing and regulating metabolites abundance (30).
Concentrations of metabolites are often maintained within strict limits. Deviations from the optimal range, either through deficiency or excess, can damage cellular functions. We propose that feedback regulation of mitochondrial GSH may primarily serve to maintain an appropriate amount of GSH to accompany free iron. This is consistent with the chemical properties of GSH, which is predicted to be the major iron ligand in cells (26). Labile iron in mitochondria is essential for enzymatic activities and macromolecular complex assembly in many pathways. By contrast, excess iron can cause oxidative damage to proteins, DNA, and membrane lipids (31). This dual effect of free iron implies that its concentration in the mitochondria must be properly buffered. Feedback regulation of the availability of mitochondrial GSH is absent in lower organisms and appears to arise more recently in vertebrates. Given the essential role of SLC25A39 in erythropoiesis (6, 32), this feature may have evolved to cope with more complex iron metabolism in vertebrates. Understanding how such closed-loop regulation functions in other physiological or pathological processes may provide important insights into the systemic role of metabolic compartmentalization.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Birsoy laboratory for helpful suggestions. Data were generated by the Proteomics Resource Center (RRID:SCR_017797), Flow Cytometry Resource Center, Drug Discovery Resource Center, and Genomics Resource Center at The Rockefeller University. We thank T. Carroll and all staff of the Bioinformatics Resource Center for their help in data analysis. We thank Y. Shen for the illustration of the summary figure.
Funding:
National Cancer Institute F99CA284249 (Y.L.); Medical Scientist Training Program, National Institute of General Medical Sciences award T32GM007739 (F.S.Y. and R.A.W.); Damon Runyon Cancer Research Foundation DRG-2431–21 (G.U.); NIH/NIDDK F32 fellowship DK127836 (T.C.K.); Merck Postdoctoral Fellowship at The Rockefeller University (T.C.K.); The Shapiro-Silverberg Fund for the Advancement of Translational Research (T.C.K.); Pershing Square Sohn Foundation (R.K.H.); National Cancer Institute Cancer Center Support Grant P30-CA008748 (R.K.H.); Rockefeller University start-up funds (E.V.V. and N.R.); Robertson Foundation (E.V.V.); Simons Foundation 290358FY18 and 290358FY19 (S.S.M.); Natural Sciences and Engineering Research Council of Canada RGPIN-2020–04375 (S.S.M.); European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska–Curie grant agreement no. 813873 (S.S.M.); Mark Foundation Emerging Leader Award (K.B.); Searle Scholar (K.B.); Pew-Stewart Scholar (K.B.); National Cancer Institute, R01CA273233 (K.B.); Richard Hite Pershing Square Sohn Foundation (R.K.H.); NIH-National Cancer Institute Cancer Center Support grant P30-CA008748; Rockefeller University start-up funds (E.V.V. and N.R.); Robertson Foundation (E.V.V.); Boehringer Ingelheim Fonds PhD fellowship (A.K.).
Footnotes
Competing interests: K.B. is a scientific advisor to Nanocare Pharmaceuticals and Atavistik Bio. The other authors declare no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Efeyan A, Comb WC, Sabatini DM, Nature 517, 302–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown MS, Goldstein JL, Science 232, 34–47 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Mårtensson J, Lai JC, Meister A, Proc. Natl. Acad. Sci. U.S.A 87, 7185–7189 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu Z et al. , Mol. Microbiol 51, 149–158 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Beer SM et al. , J. Biol. Chem 279, 47939–47951 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Wang Y et al. , Nature 599, 136–140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi X et al. , Nat. Commun 13, 2483 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho NH et al. , Science 375, eabi6983 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffith OW, Meister A, Proc. Natl. Acad. Sci. U.S.A 82, 4668–4672 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crawford RR et al. , J. Biol. Chem 290, 15878–15891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsunoda S et al. , eLife 3, e03421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torrente L, DeNicola GM, Annu. Rev. Pharmacol. Toxicol 62,279–300 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Jumper J et al. , Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puchades C et al. , Mol. Cell 75, 1073–1085.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topf U et al. , Nat. Commun 9, 324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calvo SE, Clauser KR, Mootha VK, Nucleic Acids Res. 44 (D1), D1251–D1257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith AC, Robinson AJ, Nucleic Acids Res. 47 (D1), D1225–D1228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srinivasan V, Pierik AJ, Lill R, Science 343, 1137–1140 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Lee JY, Yang JG, Zhitnitsky D, Lewinson O, Rees DC, Science 343, 1133–1136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P et al. , Nat. Commun 13, 4339 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Cowan JA, Chem. Commun 51, 2253–2255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maio N et al. , Cell Metab. 19, 445–457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Que L Jr., Bobrik MA, Ibers JA, Holm RH, J. Am. Chem. Soc 96, 4168–4178 (1974). [DOI] [PubMed] [Google Scholar]
- 24.Que L Jr., Holm RH, Mortenson LE, J. Am. Chem. Soc 97, 463–464 (1975). [DOI] [PubMed] [Google Scholar]
- 25.Bak DW, Weerapana E, Nat. Chem. Biol 356–366 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hider RC, Kong XL, Biometals 24, 1179–1187 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Kumar C et al. , EMBO J. 30, 2044–2056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sevier CS et al. , Cell 129, 333–344 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Hu M et al. , Cell 185, 2292–2308.e20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bar-Peled L, Kory N, Nat. Metab 4, 1232–1244 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walter PB et al. , Proc. Natl. Acad. Sci. U.S.A 99, 2264–2269 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.