

Abstract
Objective: Aggregating evidence highlights the strong genetic basis underpinning congenital heart disease (CHD). Here BMP4 was chosen as a prime candidate gene causative of human CHD predominantly because BMP4 was amply expressed in the embryonic hearts and knockout of Bmp4 in mice led to embryonic demise mainly from multiple cardiovascular developmental malformations. The aim of this retrospective investigation was to discover a novel BMP4 mutation underlying human CHD and explore its functional impact. Methods: A sequencing examination of BMP4 was implemented in 212 index patients suffering from CHD and 236 unrelated non-CHD individuals as well as the family members available from the proband carrying a discovered BMP4 mutation. The impacts of the discovered CHD-causing mutation on the expression of NKX2-5 and TBX20 induced by BMP4 were measured by employing a dual-luciferase analysis system. Results: A new heterozygous BMP4 mutation, NM_001202.6:c.318T>G;p.(Tyr106*), was found in a female proband affected with familial CHD. Genetic research of the mutation carrier’s relatives unveiled that the truncating mutation was in co-segregation with CHD in the pedigree. The nonsense mutation was absent from 236 unrelated non-CHD control persons. Quantitative biologic measurement revealed that Tyr106*-mutant BMP4 failed to induce the expression of NKX2-5 and TBX20, two genes whose expression is lost in CHD. Conclusion: The current findings indicate BMP4 as a new gene predisposing to human CHD, allowing for improved prenatal genetic counseling along with personalized treatment of CHD patients.
Keywords: Congenital heart disease, molecular genetics, signal transduction, BMP4, reporter gene assay
Introduction
Congenital heart disease (CHD) is the most frequent type of human birth malformation globally, occurring in approximately 0.8%-1.2% of all live births and roughly 10% of miscarriages globally, accounting for nearly 33% of all congenital deformations [1-3]. Notably, if minor cardiac developmental aberrations are encompassed, such as patent foramen ovale and aortic bicuspid valve, the prevalence of CHD rises to around 5% of all live births [4-6]. As an array of cardiovascular developmental deformations, CHD is clinically categorized into > 30 distinct isoforms, encompassing double-outlet right ventricle (DORV) and ventricular septal defect (VSD) [2,7-14]. Though certain mild/minor forms of CHD may resolve spontaneously [2], severe/complex forms of CHD usually lead to worse quality of life [15-17], reduced exercise performance [18-21], neurodevelopmental delay and structural brain anomaly [22-26], ischemic/thromboembolic stroke [27,28], acute renal injury/chronic kidney disease [29-32], hepatic fibrosis [33,34], pulmonary dysplasia/pulmonary arterial hypertension [35-37], bacterial endocarditis [38-42], chronic heart failure [43-45], supraventricular/ventricular arrhythmias [46-48], or premature cardiac death [49-54]. During recent decades, striking advances have been made in pediatric cardiac surgical procedures and catheter-based interventional techniques for CHD along with perioperative intensive care of cases with CHD, which substantially decrease the mortality of CHD cases, allowing almost 95% of live births with various forms of CHD to reach adulthood, and now adults outnumber children among the survivors living with CHD [55-60]. Despite the marked improvement, the mortality among adult survivors with CHD remains at high risk for miscellaneous cardiovascular comorbidities (encompassing cardiac dysrhythmias, heart failure, and infective endocarditis), neurodevelopmental disabilities, cerebrovascular thromboembolism, pulmonary arterial hypertension, chronic kidney disease, cancer, or demise in later life [61-65]. Consequently, CHD causes morbidity and socioeconomic encumbrances, highlighting a need to further decipher its molecular pathogenesis [2].
During vertebrate embryogenesis, cardiac organogenesis undergoes an exceedingly complex biologic process that predominantly relies on precise spatiotemporal interactions between different multipotent heart progenitors [66]. A finely-coordinated sophisticated signaling network, which principally consists of transforming growth factor-β (TGFβ), bone morphogenetic protein (BMP), Wnt family member (WNT), nodal growth differentiation factor (NODAL), and fibroblast growth factor (FGF), induces expression of a key cluster of cardiac transcription factors encompassing NKX2.5, TBX20/5/1, and GATA5/6/4 that work in a mutually-reinforcing cascade to drive cellular lineage restriction and proliferation, differentiation, and migration of different progenitor cell populations to the proper chambers for specific types of cardiac cells [66]. Both environmental pathogenic factors and inheritable defects can disturb the heart-development process, giving rise to a diverse array of CHD [1,3,66-71]. It is believed that non-inherited risk factors account for about 30% of CHD, although the molecular mechanisms of CHD caused by detrimental environmental exposure are largely elusive [71]. Well-recognized non-genetic factors that enhance vulnerability to CHD include maternal disease, maternal ingestion of medication, maternal consumption of toxic chemical, and maternal exposure of gaseous pollutants, atmospheric particulate substances, and heavy metals during early pregnancy [71]. However, increasing evidence demonstrates that genetically compromised components are responsible for most CHD [1,3,66,67]. In addition to chromosomal aneuploidies and copy number variations, a growing body of deleterious mutations in > 100 genes, including NKX2.5 and TBX20, have been found to contribute to CHD [1,3,66,67,72-96]. Nevertheless, known genetic causes of CHD can explain < 40% of CHD patients, and in most (> 60%) cases, the genetic determinants for CHD remain uncertain [66,96].
Recent investigations have established the essential role of BMP4 as an important member of the TGFβ superfamily of polypeptide signaling molecules in regulating the cardiovascular morphogenetic process, particularly at the early stages of cardiac development [97-101]. In mice, knockout of Bmp4 results in embryonic lethality, mainly because of abnormal cardiac development [98]; while conditional deletion of Bmp4 in cardiomyocytes leads to DORV, VSD, and atrioventricular canal defect [99]. Moreover, murine embryos with compound heterozygous deletion of Bmp4 and Bmp2 also manifest VSD [100,101]. Furthermore, in humans, BMP4 was found to be highly expressed in the heart with a similar expression level in the healthy and CHD-affected hearts (with no BMP4 mutation), and sustained mRNA and protein expressions of BMP4 were observed in CHD patients [97]. These findings highlight the crucial role of BMP4 in normal cardiac organogenesis. This suggests that genetically defective BMP4 predisposes to CHD in humans.
Materials and methods
Recruitment of research participants
The current retrospective case-control investigation was accomplished in line with the Declaration of Helsinki. The ethical review committee of Tongji Hospital in Shanghai approved the protocols involved in human research (with an approved protocol code of LL(H)-09-07 and an ethical approval date of July 27, 2009). After ethical approval, the research subjects or their parents signed an informed consent form, at the time of enrollment before the commencement of the current study. For the present human research, 212 probands suffering from CHD and 236 unrelated non-CHD volunteers were recruited from the Chinese Han-ethnicity population. The pedigree members available from the CHD-affected probands were also enlisted. Each research subject underwent a comprehensive clinical evaluation at study entry, including a systemic review of personal and familial histories along with medical records, meticulous physical examination, echocardiographic/electrocardiographic images, and routine laboratory tests. For all research subjects, CHD was diagnosed based on the echocardiographic images and/or operative reports. All the patients had an echocardiogram-documented CHD, of whom most had medical records indicating surgical or catheter-based treatment for CHD. The affected individuals’ CHD was categorized based on the international nomenclature [102]. The inclusion and exclusion criteria for the CHD patient group and non-CHD control group are described elsewhere [103]. The patients with syndromic CHD were excluded from the current study just because the genetic causes for most syndromic CHD were known and no evidence indicated the association of a BMP4 defect with syndromic CHD. Patients with known causes (including chromosomal aneuploidy and a copy number variation) associated with CHD were also excluded. Clinical data and demographic information together with 1-3 mL of venous blood were acquired from every research participant.
Sequencing assay of BMP4
Isolation of genomic DNA from the research participants’ circulating leucocytes was completed by utilizing a genomic DNA purification kit (Thermo Fisher, USA) according to the manual. The oligonucleotide primers applied to the amplification of the coding exons along with the splicing donors/acceptors of human BMP4 (GenBank accession number: NC_000014.9) are given in Table 1. Polymerase chain reaction (PCR)-amplification of BMP4 from a research participant’s genomic DNA was fulfilled on a thermal cycler apparatus (Bio-Rad, USA) with a Taq DNA polymerase kit (New England Biolabs, USA) as well as the BMP4-specific primers mentioned above. The amplicons were fragmented through 1.6% agarose gel electrophoresis and purified utilizing a gel extraction kit (Invitrogen, USA) following the manufacturer’s procedures. Direct PCR-DNA sequencing assay of the purified amplicons was performed as described elsewhere [103-105]. Additionally, for a detected human BMP4 mutation, the gnomAD database along with the SNP database was accessed to confirm its novelty as described previously [103].
Table 1.
Oligonucleotide primers for amplifying the coding sequences along with flanking introns of the human BMP4 gene
| Coding exon | Forward (from 5’ to 3’) | Reverse (from 5’ to 3’) | Amplicon size (bp) |
|---|---|---|---|
| 1 | CAGTTTGGGCAGCAGTTACAC | GGCTCGAGATAGCTTGGACG | 546 |
| 2 | GGGTGAGACTTTCCCGACCT | TAAAGGAGGTCCGACGGAAGG | 670 |
| 3 | TGCTTTCCATCTTGCCCCTC | CTGGACTGGGGCTTTGATGT | 606 |
| 4-a | GGTTTGTTAGCTGCCCCACT | CCCTTGAGGTAACGATCGGC | 602 |
| 4-b | GGCTAGCCATTGAGGTGACTC | ATAAAAGTCCAGCTATAAGGAAGC | 658 |
Production of gene-expressing constructs
As described previously [106], cDNA was produced by reverse transcription of the mRNA isolated from the excised myocardial issue, which originated from a case experiencing surgical therapy for Fallot’s tetralogy. A 1368-bp fragment including the entire coding region of wild-type human BMP4 (Nucleotide accession number: NM_001202.6) was PCR-amplified from human heart cDNA using the AccuPrime™ Taq DNA Polymerase Kit (Invitrogen, USA) and a human BMP4-specific pair of primers of 5’-GTCGAATTCAACGCACTGCTGCAGCTTC-3’ (forward) and 5’-GTCTCTAGAGTGTATATCTGTCTATCCTC-3’ (reverse). The amplified fragment containing whole human BMP4 cDNA and the pcDNATM3.1(+) vector (Invitrogen, USA) were doubly cut by EcoRI and XbaI, gel-purified, and recombined by T4 DNA ligase to construct the wild-type human BMP4-pcDNATM3.1(+) vector. Using the wild-type human BMP4-pcDNATM3.1(+) vector as a template, the Tyr106*-mutant human BMP4-pcDNATM3.1(+) vector was yielded employing a site-targeted mutagenesis kit (Thermo Scientific, USA) along with a complimentary pair of oligonucleotide primers (forward: 5’-CACTGGTCTTGAGTAGCCTGAGCGCCCGGCC-3’; backward: 5’-GGCCGGGCGCTCAGGCTACTCAAGACCAGTG-3’). The NKX2-5-luc and TBX20-luc constructs were generated as previously described [106]. All constructed vectors were verified by DNA sequencing analysis.
Cellular transfection with gene-expressing vectors and dual-luciferase analysis
HeLa cells (a cervical cancer cell line derived from Henrietta Lacks) were routinely maintained as described elsewhere [106]. Cells were counted using a hemocytometer (Invitrogen, USA) and seeded in a 24-well plate (Greiner Bio-One, USA), cultivated for 36 h, then transient cellular transfection with gene-expressing vectors was conducted using the Lipofectamine® LTX & PLUSTM Reagent (Invitrogen, USA). Specifically, cells were transfected with 15 ng of pGL4.75, 1.5 μg of NKX2-5-luc or TBX20-luc, and 0.6 μg of each gene-expressing vector (empty pcDNATM3.1(+), wild-type human BMP4-pcDNATM3.1(+), or Tyr106*-mutant human BMP10-pcDNATM3.1(+), alone or together). Here, the renilla luciferase-expressing plasmid of pGL4.75 (Promega, USA) was employed as an internal control to balance transfection efficiency. The empty pcDNATM3.1(+) plasmid was utilized as an external negative control. HeLa cells transfected with gene-expressing plasmids were harvested 36 h after cellular transfection and lysed in a lysis buffer (Promega, USA). Cell lysates were applied to the measurement of firefly/renilla luciferase activities, respectively, as described in detail elsewhere [106]. For each gene-expressing vector, a cellular transfection experiment was accomplished in three independent replicates.
Statistics
Quantitative variables (age and promoter activity) are given as means ± standard deviations (x̅ ± SD) throughout. Qualitative values (sex, race and positive family history of CHD) are expressed as frequency numbers (n) and percentages (%). An independently measured Student’s t-test was applied to compare continuous variables between two groups. To compare continuous variables among over three groups, a one-way analysis of variance followed by the Tukey-Kramer post-hoc test was used. Categorical variables were compared between two groups using Fisher’s exact test or Pearson’s chi-square (χ2) test when indicated. Statistical assay was done with SPSS v17 (SPSS, USA). In all cases, a 2-sided P < 0.05 denoted a statistical difference.
Results
Basic features of the CHD-affected proband cohort
In the current human investigation, 212 index patients with miscellaneous forms of CHD (including 95 female index patients and 117 male index patients, with an average age of 5.8 years) was evaluated clinically in comparison with 236 unrelated non-CHD volunteers without family history of CHD (including 106 female volunteers and 130 male volunteers, with an average age of 5.7 years). All the study individuals were enlisted from the Chinese Han-race population. The included index patients had echocardiographic/surgical documentations suggesting the presence of CHD, while the included volunteers showed normal echocardiograms, with no proof suggesting cardiovascular structural deformities. Of the 212 CHD-affected probands, 61 probands reported a positive familial history of CHD, whereas none of the 236 volunteers employed as control subjects had it. No research subjects had known non-heritable factors susceptible to CHD, including maternal obesity, diabetes mellitus, hypothyroidism, phenylketonuria, pre-eclampsia, primary hypertension, nutritional deficiency, epilepsy, connective tissue disease, acute febrile illness, along with exposure to therapeutic medications, toxicants, and ionizing radiation during gestation, and the vast majority of CHD-affected probands experienced catheter-based interventional/surgical treatment for CHD. The basic demographic and phenotypic features of the 212 probands with a wide spectrum of CHD are summarized in Table 2.
Table 2.
Basic characteristics of the research cohort comprising 212 index patients affected with a wide spectrum of congenital heart disease
| Variable | Number or mean ± SD | Percentage or range |
|---|---|---|
| Demographics | ||
| Female index patients | 95 | 44.8 |
| Male index patients | 117 | 55.2 |
| Age at initial recruitment (years) | 5.8 ± 3.27 | 0.6-11.3 |
| Having a family history of CHD | 61 | 28.8 |
| Distribution of distinct forms of CHD | ||
| VSD | 53 | 25.0 |
| ASD | 48 | 22.6 |
| PDA | 16 | 7.5 |
| TOF | 12 | 5.7 |
| DORV | 10 | 4.7 |
| TGA | 4 | 1.9 |
| HLV | 4 | 1.9 |
| TAPVC | 2 | 0.9 |
| PS | 2 | 0.9 |
| CoA | 1 | 0.5 |
| PTA | 1 | 0.5 |
| AS | 1 | 0.5 |
| VSD + PDA | 18 | 8.5 |
| DORV + VSD | 16 | 7.5 |
| VSD + ASD | 9 | 4.2 |
| ASD + PDA | 5 | 2.4 |
| TOF + ASD | 5 | 2.4 |
| TGA + VSD | 4 | 1.9 |
| PTA + VSD | 1 | 0.5 |
| Dysrhythmias | ||
| AVB | 15 | 7.1 |
| AF | 11 | 5.2 |
| Medical management | ||
| Catheter-based treatment for CHD | 117 | 55.2 |
| Surgical procedures for CHD | 72 | 34.0 |
| Follow-up observation | 23 | 10.8 |
AF: atrial fibrillation; AS: aortic stenosis; ASD: atrial septal defect; AVB: atrioventricular block; CHD: congenital heart disease; CoA: coarctation of the aorta; DORV: double-outlet right ventricle; HLV: hypoplastic left ventricle; PDA: patent ductus arteriosus; PS: pulmonary stenosis; PTA: persistent truncus arteriosus; TAPVC: total anomalous pulmonary venous connection; TGA: transposition of the great arteries; TOF: tetralogy of Fallot; VSD: ventricular septal defect.
Identification of a CHD-causative mutation in BMP4
By DNA sequencing examination of human BMP4 in 212 probands with a diverse array of CHD, a heterozygous BMP4 mutation, NM_001202.6:c.318T>G;p.(Tyr106*), was found in one female proband affected with congenital DORV and VSD. Sequencing assay of BMP4 in the mutation carrier’s available relatives demonstrated that the truncating mutation co-segregated with CHD in the family (arbitrarily designated as Family C002). Genetic assay of Family C002 revealed that CHD was inherited in an autosomal-dominant mode. Notably, all the six CHD-affected members (I-1, II-3, II-8, III-3, III-7, and IV-2 from Family C002) of the mutation-harboring proband underwent surgical treatment for CHD. The proband’s father’s grandfather (I-1) died of sudden cardiac death when he was 63 years old. The BMP4 mutation was absent from the 236 unrelated non-CHD controls or from the gnomAD and SNP databases, verifying the novelty of the discovered BMP4 nutation responsible for CHD. The DNA sequencing chromatograms displaying the heterozygous c.318T>G mutation in BMP4 in contrast to its wild type are illustrated in Figure 1A. The structural motifs of the wild-type and Tyr106*-mutant human BMP4 proteins are shown in Figure 1B. The pedigree of the proband carrying the discovered human BMP4 mutation is shown in Figure 1C. In Family C002, there were 21 family members available, including 10 male and 11 female members, with ages varying from 1 to 78 years. All the six affected members suffered DORV and VSD and underwent surgery for CHD. No recognized environmental factors vulnerable to CHD were unmasked in each pedigree member. The clinical characteristic profile as well as BMP4 mutation status of the living relatives with CHD from Family C002 are summed in Table 3.
Figure 1.

A new BMP4 mutation causing familial congenital cardiovascular malformations. A. Sequence chromatogram traces revealing the heterozygous BMP4 mutation in the CHD-affected proband (Mutant) along with corresponding homozygous control in an unaffected family member of the proband (Wild type). A vertical arrow points to the nucleotide position where the heterozygous BMP4 mutation (c.318T>G) occurs. B. Schematic diagrams delineating the structural domains of human BMP4 proteins. The Tyr106*-mutant BMP4 protein (Mutant) was predicted to lose 303 amino acids at the carboxyl terminus (COOH). TGFβ: transforming growth factor-beta; NH2: amino terminus. C. Pedigree manifesting autosomal-dominant inheritance of ventricular septal defect and double-outlet right ventricle. An oblique arrow points to the index patient. A family member’s genotype is marked with “+” or “-”, of which “+” signifies a member harboring the heterozygous BMP4 mutation, while “-” signifies a member with no BMP4 mutation.
Table 3.
Clinical characteristic profile and BMP4 mutation status of the living relatives from Family C002 suffering congenital heart defects
| Individual (Family C002) | Sex | Age (years) | Cardiovascular structural deformities | BMP4 mutation (Tyr106*) |
|---|---|---|---|---|
| II-3 | Male | 54 | DORV, VSD | +/- |
| II-8 | Female | 50 | DORV, VSD | +/- |
| III-3 | Male | 27 | DORV, VSD | +/- |
| III-7 | Male | 26 | DORV, VSD | +/- |
| IV2 | Female | 2 | DORV, VSD | +/- |
VSD: ventricular septal defect; DORV: double-outlet right ventricle; +/-: heterozygote for the human BMP4 mutation.
Functional failure of Tyr106*-mutant BMP4 in inducing expression of NKX2-5
As presented in Figure 2, in grown HeLa cells expressing multiple constructs, encompassing empty pcDNATM3.1(+) as an external negative control (-), wild-type human BMP4-pcDNATM3.1(+) (BMP4), and Tyr106*-mutant human BMP4-pcDNATM3.1(+) (Tyr106*), alone or together, BMP4 and Tyr106* induced the transcriptional activity of the NKX2-5 promoter by ~19-fold and ~2-fold, respectively (BMP4 versus Tyr106*: t = 12.7416; P = 0.0002). When BMP4 and Tyr106* were co-expressed, the induced transactivation on the NKX2-5 promoter was ~10-fold (BMP4 versus Tyr106* + BMP4: t = 5.9808; P = 0.0039). Equivalent statistical results were obtained when multiple comparisons were carried out (F = 94.983, P = 6.514 × 10-8). Specifically, for (-) versus BMP4, t = 17.1833; P < 0.0001; for (-) versus Tyr106*, t = 0.3167; P = 0.9978; for (-) versus BMP4 + (-), t = 8.9933; P < 0.0001; for (-) versus Tyr106* + BMP4, t = 8.6367; P < 0.0001; for BMP4 versus Tyr106*, t = 16.8667; P < 0.0001; for BMP4 versus BMP4 + (-), t = 8.1900; P = 0.0001; for BMP4 versus Tyr106* + BMP4, t = 8.5467; P < 0.0001; for Tyr106* versus BMP4 + (-), t = 8.6767; P < 0.0001; for Tyr106* versus Tyr106* + BMP4, t = 8.3200; P < 0.0001; for BMP4 + (-) versus Tyr106* + BMP4, t = 0.3567; P = 0.9965.
Figure 2.
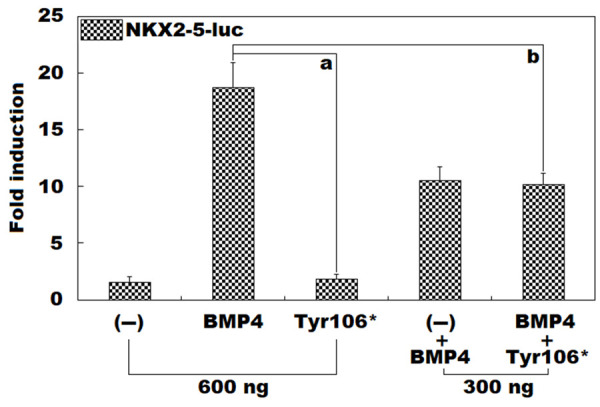
No induction of NKX2-5 expression by Tyr106*-mutant BMP4. In maintained HeLa cells, dual-luciferase measurement of the expression of the NKX2-5 promoter-driven firefly luciferase (NKX2-5-luc) in the presence of wild-type human BMP4-pcDNATM3.1(+) vector (BMP4) or Tyr106*-mutant human BMP10-pcDNATM3.1(+) vector (Tyr106*), separately or in combination, showed that Tyr106* failed to induce the expression of NKX2-5. For every expression vector, functional experiments were performed three times in triplicate. Here, “a” means P < 0.001, and “b” means P < 0.005, in comparison to BMP4 (600 ng).
Inability of Tyr106*-mutant BMP4 to induce transcription of TBX20
As illustrated in Figure 3, in maintained HeLa cells expressing multiple plasmids, encompassing empty pcDNATM3.1(+) plasmid (-), wild-type human BMP4-pcDNATM3.1(+) (BMP4), and Tyr106*-mutant human BMP4-pcDNATM3.1(+) (Tyr106*), separately or together, BMP4 and Tyr106* induced the transactivation on the TBX20 promoter by ~14-fold and ~2-fold, respectively (BMP4 versus Tyr106*: t = 13.1716; P = 0.0002). When BMP4 and Tyr106* were expressed in combination, the elicited transcriptional activity of the TBX20 promoter was ~6-fold (BMP4 versus Tyr106* + BMP4: t = 7.4809; P = 0.0017). Parallel statistical results were achieved when multiple comparisons were made (F = 76.560, P = 1.847 × 10-7). Specifically, for (-) versus BMP4, t = 12.1267; P < 0.0001; for (-) versus Tyr106*, t = 0.0400; P = 1.0000; for (-) versus BMP4 + (-), t = 5.8167; P = 0.0002; for (-) versus Tyr106* + BMP4, t = 4.7833; P = 0.0011; for BMP4 versus Tyr106*, t = 12.0867; P < 0.0001; for BMP4 versus BMP4 + (-), t = 6.3100; P = 0.0001; for BMP4 versus Tyr106* + BMP4, t = 7.3433; P < 0.0001; for Tyr106* versus BMP4 + (-), t = 5.7767; P = 0.0002; for Tyr106* versus Tyr106* + BMP4, t = 4.7433; P = 0.0012; for BMP4 + (-) versus Tyr106* + BMP4, t = 1.0333; P = 0.7116.
Figure 3.
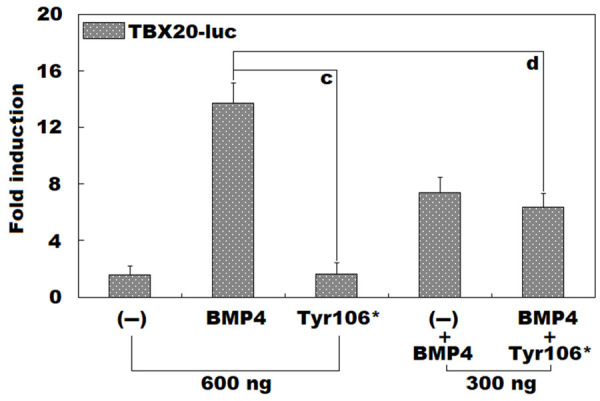
Inability of Tyr106*-mutant BMP4 to induce TBX20 expression. Dual-reporter analysis of the expression of the TBX20 promoter-driven firefly luciferase (TBX20-luc) in grown HeLa cells in the presence of wild-type human BMP4-pcDNATM3.1(+) vector (BMP4) or Tyr106*-mutant human BMP10-pcDNATM3.1(+) vector (Tyr106*), singly or in both, revealed that Tyr106* failed to induce the expression of TBX20. For each expression vector, three independent biological measurements were conducted in triplicate. Here, “c” signifies P < 0.001, and “d” indicates P < 0.005, when compared with BMP4 (600 ng).
Discussion
In humans, the BMP4 gene is mapped at chromosome 14q22.2, which codes for a growth factor comprising 408 amino acids, a secreted ligand with multiple functions belonging to the TGFβ superfamily. It consists of over 30 ligands recognized so far [97]. The TGFβ superfamily is classified into several subcategories, including BMPs, TGFβs, growth and differentiation factors (GDFs), as well as activins/inhibins. TGFβ superfamily signaling occurs through a heterodimeric complex comprising two types of receptors (types I and II) [97]. BMPs were initially demonstrated to account for the development of bone as well as cartilage. Further research has substantiated their pivotal roles in embryonic development with a diverse array of effects, encompassing growth and differentiation along with apoptosis of multiple cell types, such as chondroblasts, osteoblasts, epithelial cells, and neuronic cells [97]. In the heart, BMP signaling first activates the endocardium by the establishment of a proper environment, followed by the promotion of epithelial-mesenchymal transition as well as invasion of the mesenchymal cells into the endocardial cushion with the aid of TGFβ as well as Notch signaling [97]. The BMP ligands bind receptors (type II), resulting in activation of receptors (type I), which phosphorylates the SMAD signal transducers, playing a crucial role in inducing transcription of target genes [97]. Additionally, non-SMAD signaling pathways are also involved in governing BMP signaling [97,107]. Although all BMP proteins possess a similar protein structure, each BMP protein has a unique tissue expression spectrum along with a distinct physiologic function [108,109]. To date, six BMP members have been validated to be amply expressed in the embryonic heart, encompassing BMP4, BMP6, BMP2, BMP5, BMP7, and BMP10, of which BMP4 was shown to be highly expressed in the mesoderm as well as the outflow tract myocardium [110]. It exerts multiple functions during cardiac organogenesis, particularly during the early period of cardiac organogenesis [97,103,111]. Specifically, BMP4 functions to induce the formation of endocardial cushion and outflow tract cushion mainly by increasing epithelial-mesenchymal transition of endocardial cells at the outflow tract and inducing the invasion of cardiac neural crest into the outflow tract and aortopulmonary septum cushions during embryogenesis [97,99]. BMP4 was shown to play an essential role in regulating cardiovascular morphogenesis by interaction with transcription factors [97]. Furthermore, recent investigations have shown that BMP4 can induce the expression of multiple important downstream genes, encompassing NKX2-5 and TBX20 [97,112-114], which encodes transcription factors crucial for normal cardiovascular development, and disease-causing mutations in both NKX2-5 and TBX20 have been involved in the occurrence of CHD [85,115-117]. In the current study, the discovered Tyr106* mutation was anticipated to yield a truncated BMP4 protein lacking pivotal structural motifs. Results from functional experiments showed that Tyr106*-mutant BMP4 failed to induce the expression of NKX2-5 as well as TBX20. Collectively, these findings suggest that BMP4 haploinsufficiency is a molecular determinant of CHD in humans.
In experimental mice, functionally defective BMP4 results in CHD. Deletion of Bmp4 caused embryonic lethality, mainly due to cardiovascular developmental abnormalities [98]; while conditional ablation of Bmp4 in cardiomyocytes caused early embryonic death, along with DORV, VSD, and atrioventricular canal defect [99]. Moreover, loss of Bmp4 from the anterior heart field in mice led to a remarkably reduced number of cells in the developing endocardial cushions within the outflow tract, abnormal cushion remodeling, persistent truncus arteriosus, VSD, and semilunar valve deformity [118]. Additionally, murine embryos with the compound heterozygous knockout of both Bmp4 and Bmp2 also manifested VSD [100,101]. Conditional deletion of both Bmp4 and Bmp7 in the murine second heart field led to defective epithelial to mesenchymal transition, decreased cardiac neural crest ingress, and persistent truncus arteriosus [119]. Conditional inactivation of Bmp4 from TBX1-expressing cells in mouse embryos resulted in a spectrum of malformations resembling the cardiovascular anomalies of patients with DiGeorge syndrome, mainly affecting the remodeling of outflow tract and pharyngeal arch arteries [120]. Taken together, these experimental animal studies underscore the essential role of BMP4 in cardiovascular development, so that BMP4 insufficiency may be a molecular basis of CHD.
In humans, BMP4 was validated to be amply expressed in the heart, with a similar expression level in normal and CHD-affected hearts (with no BMP4 mutation). Sustained mRNA and protein expressions of BMP4 were observed in CHD patients [97]. Furthermore, a common BMP4 intronic SNP (rs762642) was reported to confer an enhanced vulnerability to sporadic CHD in a Chinese population [119], though the functional effect of the rs762642 polymorphism was not explored [121]. These results support that genetically compromised BMP4 contributes to CHD.
Limitations of the current study are as follows. First, we could not rule out that other genetic defects might also contribute to CHD in the patients harboring a BMP4 mutation. Whole-genome or whole-exome sequencing analysis would be needed to assess this. Second, more cellular functional experiments should be performed, especially for the further cell experiments performed in cardiomyocytes. Third, establishment of a mouse model with the BMP4 mutation knocked in would help to validate the causative effects of the BMP4 mutation. Finally, BMP4 should be screened in populations of different ethnicities to evaluate the mutational spectrum and prevalence of BMP4.
Conclusion
This research indicates BMP4 as a causative gene responsible for human CHD and unveils a new molecular mechanism underpinning human CHD. This has hypothetical clinical implications for prenatal diagnosis and personalized prophylaxis of CHD in a subgroup of patients.
Acknowledgements
This human research was funded by the Natural Science Foundation of Ningbo City, Zhejiang Province, China (202003N4230), the Zhejiang Province-Ningbo City Joint-Construction Medical Key Discipline Construction Plan Project (2022-B17), and the First-Batch Major Tackling Project for Senior Medical Health Teams in Ningbo City, Zhejiang Province, China (2022020405).
Disclosure of conflict of interest
None.
References
- 1.Albesher N, Massadeh S, Hassan SM, Alaamery M. Consanguinity and congenital heart disease susceptibility: insights into rare genetic variations in Saudi Arabia. Genes (Basel) 2022;13:354. doi: 10.3390/genes13020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Fugar S, Generoso G, Heard DG, Hiremath S, Ho JE, Kalani R, Kazi DS, Ko D, Levine DA, Liu J, Ma J, Magnani JW, Michos ED, Mussolino ME, Navaneethan SD, Parikh NI, Poudel R, Rezk-Hanna M, Roth GA, Shah NS, St-Onge MP, Thacker EL, Virani SS, Voeks JH, Wang NY, Wong ND, Wong SS, Yaffe K, Martin SS American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2023 update: a report from the American Heart Association. Circulation. 2023;147:e93–e621. doi: 10.1161/CIR.0000000000001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Y, Deng W, Wang Z, Wang Y, Zheng H, Zhou K, Xu Q, Bai L, Liu H, Ren Z, Jiang Z. Genetics of congenital heart disease. Clin Chim Acta. 2024;552:117683. doi: 10.1016/j.cca.2023.117683. [DOI] [PubMed] [Google Scholar]
- 4.Mahmoud H, Cinteză E, Voicu C, Mărgărint I, Rotaru I, Aria A, Youssef T, Nicolescu A. Challenging diagnosis of anomalous origin of the right coronary artery from the pulmonary artery. Diagnostics (Basel) 2022;12:2671. doi: 10.3390/diagnostics12112671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spaziani G, Girolami F, Arcieri L, Calabri GB, Porcedda G, Di Filippo C, Surace FC, Pozzi M, Favilli S. Bicuspid aortic valve in children and adolescents: a comprehensive review. Diagnostics (Basel) 2022;12:1751. doi: 10.3390/diagnostics12071751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bray JJH, Freer R, Pitcher A, Kharbanda R. Family screening for bicuspid aortic valve and aortic dilatation: a meta-analysis. Eur Heart J. 2023;44:3152–3164. doi: 10.1093/eurheartj/ehad320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasielska-Trojan A, Święchowicz B, Antoszewski B. Coexistence of thumb aplasia and cleft lip and alveolus with aortopulmonary Window-A tip for prenatal diagnostics for rare heart anomalies. Diagnostics (Basel) 2022;12:569. doi: 10.3390/diagnostics12030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szabo TM, Heidenhoffer E, Kirchmaier Á, Pelok B, Frigy A. Cor triatriatum sinister presenting as cardioembolic stroke in a young woman. Diagnostics (Basel) 2022;13:97. doi: 10.3390/diagnostics13010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giuffrida MG, Goldoni M, Genovesi ML, Carpentieri G, Torres B, Deac AD, Cecchetti S, Martinelli A, Vaisfeld A, Flex E, Bernardini L. 3’UTR deletion of NONO leads to corpus callosum anomaly, left ventricular non-compaction and ebstein’s anomaly in a male fetus. Diagnostics (Basel) 2022;12:2354. doi: 10.3390/diagnostics12102354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li ZZ, Zhang WL, Zhu M, Liang H. A case of congenital left ventricular diverticulum in an adult patient. Ann Noninvasive Electrocardiol. 2023;28:e13025. doi: 10.1111/anec.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bekheet S, Al-Radi OO, Abou Zahr R. Congenital giant left ventricular aneurysm. Cardiol Young. 2023;33:1203–1205. doi: 10.1017/S1047951122003651. [DOI] [PubMed] [Google Scholar]
- 12.Mery CM, Well A, Taylor K, Carberry K, Colucci J, Ulack C, Zeiner A, Mizrahi M, Stewart E, Dillingham C, Cook T, Hartounian A, McCullum E, Affolter JT, Van Diest H, Lamari-Fisher A, Chang S, Wallace S, Teisberg E, Fraser CD Jr. Examining the real-life journey of individuals and families affected by single-ventricle congenital heart disease. J Am Heart Assoc. 2023;12:e027556. doi: 10.1161/JAHA.122.027556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda T, Ikeda T, Yoshizawa K. Tricuspid valve repair with papillary muscle approximation for congenital tricuspid valve regurgitation due to tricuspid valve dysplasia. Cardiol Young. 2023;33:660–662. doi: 10.1017/S1047951122002499. [DOI] [PubMed] [Google Scholar]
- 14.Bourcier T, Willoteaux S, Furber A, Biere L. Anomalous origin of the left circumflex artery from the pulmonary artery associated with non-compaction of the left ventricle: usefulness of multimodality imaging-a case report. Eur Heart J Case Rep. 2023;7:ytad250. doi: 10.1093/ehjcr/ytad250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Connor AM, Cassedy A, Wray J, Brown KL, Cohen M, Franklin RCG, Gaynor JW, MacGloin H, Mahony L, Mussatto K, Newburger JW, Rosenthal DN, Teitel D, Ernst MM, Wernovsky G, Marino BS. Differences in quality of life in children across the spectrum of congenital heart disease. J Pediatr. 2023;263:113701. doi: 10.1016/j.jpeds.2023.113701. [DOI] [PubMed] [Google Scholar]
- 16.Ly R, Karsenty C, Amedro P, Cohen S, Domanski O, Godart F, Radojevic J, Vaksmann G, Naccache N, Boubrit A, Bataille V, Hascoet S, Ladouceur M. Health-related quality of life and its association with outcomes in adults with congenital heart disease and heart failure: insight from FRESH-ACHD registry. J Am Heart Assoc. 2023;12:e027819. doi: 10.1161/JAHA.122.027819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu HC, Chaou CH, Lo CW, Chung HT, Hwang MS. Factors affecting psychological and health-related quality-of-life status in children and adolescents with congenital heart diseases. Children (Basel) 2022;9:578. doi: 10.3390/children9040578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Genuchten WJ, Helbing WA, Ten Harkel ADJ, Fejzic Z, Md IMK, Slieker MG, van der Ven JPG, Boersma E, Takken T, Bartelds B. Exercise capacity in a cohort of children with congenital heart disease. Eur J Pediatr. 2023;182:295–306. doi: 10.1007/s00431-022-04648-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villaseca-Rojas Y, Varela-Melo J, Torres-Castro R, Vasconcello-Castillo L, Mazzucco G, Vilaró J, Blanco I. Exercise capacity in children and adolescents with congenital heart disease: a systematic review and meta-analysis. Front Cardiovasc Med. 2022;9:874700. doi: 10.3389/fcvm.2022.874700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masood IR, Detterich J, Cerrone D, Lewinter K, Shah P, Kato R, Sabati A. Reduced forced vital capacity and the number of chest wall surgeries are associated with decreased exercise capacity in children with congenital heart disease. Pediatr Cardiol. 2022;43:54–61. doi: 10.1007/s00246-021-02692-0. [DOI] [PubMed] [Google Scholar]
- 21.Sheng SP, Feinberg JL, Bostrom JA, Tang Y, Sweeney G, Pierre A, Katz ES, Whiteson JH, Haas F, Dodson JA, Halpern DG. Adherence and exercise capacity improvements of patients with adult congenital heart disease participating in cardiac rehabilitation. J Am Heart Assoc. 2022;11:e023896. doi: 10.1161/JAHA.121.023896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peyvandi S, Rollins C. Fetal brain development in congenital heart disease. Can J Cardiol. 2023;39:115–122. doi: 10.1016/j.cjca.2022.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patt E, Singhania A, Roberts AE, Morton SU. The genetics of neurodevelopment in congenital heart disease. Can J Cardiol. 2023;39:97–114. doi: 10.1016/j.cjca.2022.09.026. [DOI] [PubMed] [Google Scholar]
- 24.Sadhwani A, Wypij D, Rofeberg V, Gholipour A, Mittleman M, Rohde J, Velasco-Annis C, Calderon J, Friedman KG, Tworetzky W, Grant PE, Soul JS, Warfield SK, Newburger JW, Ortinau CM, Rollins CK. Fetal brain volume predicts neurodevelopment in congenital heart disease. Circulation. 2022;145:1108–1119. doi: 10.1161/CIRCULATIONAHA.121.056305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parekh SA, Cox SM, Barkovich AJ, Chau V, Steurer MA, Xu D, Miller SP, McQuillen PS, Peyvandi S. The effect of size and asymmetry at birth on brain injury and neurodevelopmental outcomes in congenital heart disease. Pediatr Cardiol. 2022;43:868–877. doi: 10.1007/s00246-021-02798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peyvandi S, Xu D, Barkovich AJ, Gano D, Chau V, Reddy VM, Selvanathan T, Guo T, Gaynor JW, Seed M, Miller SP, McQuillen P. Declining incidence of postoperative neonatal brain injury in congenital heart disease. J Am Coll Cardiol. 2023;81:253–266. doi: 10.1016/j.jacc.2022.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yelton SEG, Flores S, Sun LR, Nelson-McMillan K, Loomba RS. Association between congenital heart disease and stroke: insights from a national database. Pediatr Cardiol. 2024;45:1–7. doi: 10.1007/s00246-023-03315-6. [DOI] [PubMed] [Google Scholar]
- 28.Yeh HR, Kim EH, Yu JJ, Yun TJ, Ko TS, Yum MS. Arterial ischemic stroke in children with congenital heart diseases. Pediatr Int. 2022;64:e15200. doi: 10.1111/ped.15200. [DOI] [PubMed] [Google Scholar]
- 29.Patel SB, Webber Z, Strah DD, Hellinger RD, Yrun-Duffy M, Kowalek KA, Seckeler MD. Acute hospital outcomes for renal transplantation in patients with moderate or severe congenital heart disease. Am J Cardiol. 2023;186:87–90. doi: 10.1016/j.amjcard.2022.10.034. [DOI] [PubMed] [Google Scholar]
- 30.Gillesén M, Fedchenko M, Giang KW, Dimopoulos K, Eriksson P, Dellborg M, Mandalenakis Z. Chronic kidney disease in patients with congenital heart disease: a nationwide, register-based cohort study. Eur Heart J Open. 2022;2:oeac055. doi: 10.1093/ehjopen/oeac055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Sayegh S, Ephrem G, Wish JB, Moe S, Lim K. Kidney disease and congenital heart disease: partnership for life. Front Physiol. 2022;13:970389. doi: 10.3389/fphys.2022.970389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kourelis G, Kanakis M, Samanidis G, Tzannis K, Bobos D, Kousi T, Apostolopoulou S, Kakava F, Kyriakoulis K, Bounta S, Rammos S, Papagiannis J, Giannopoulos N, Orfanos SE, Dimopoulos G. Acute kidney injury predictors and outcomes after cardiac surgery in children with congenital heart disease: an observational cohort study. Diagnostics (Basel) 2022;12:2397. doi: 10.3390/diagnostics12102397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reiter FP, Hadjamu NJ, Nagdyman N, Zachoval R, Mayerle J, De Toni EN, Kaemmerer H, Denk G. Congenital heart disease-associated liver disease: a narrative review. Cardiovasc Diagn Ther. 2021;11:577–590. doi: 10.21037/cdt-20-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Egbe AC, Miranda WR, Jain CC, Kamath PS, Katta RR, Andi K, Goda AY, Connolly HM. Improvement in hepatic and renal function following isolated heart transplant in adults with congenital heart disease. Int J Cardiol. 2022;364:44–49. doi: 10.1016/j.ijcard.2022.06.024. [DOI] [PubMed] [Google Scholar]
- 35.Li DB, Xu XX, Hu YQ, Cui Q, Xiao YY, Sun SJ, Chen LJ, Ye LC, Sun Q. Congenital heart disease-associated pulmonary dysplasia and its underlying mechanisms. Am J Physiol Lung Cell Mol Physiol. 2023;324:L89–L101. doi: 10.1152/ajplung.00195.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiu SN, Lu CW, Lin MT, Chen CA, Wu MH, Wang JK. Pulmonary hypertension in adult congenital heart disease in Asia: a distinctive feature of complex congenital heart disease. J Am Heart Assoc. 2022;11:e022596. doi: 10.1161/JAHA.121.022596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jone PN, Ivy DD, Hauck A, Karamlou T, Truong U, Coleman RD, Sandoval JP, Del Cerro Marín MJ, Eghtesady P, Tillman K, Krishnan US. Pulmonary hypertension in congenital heart disease: a scientific statement from the American Heart Association. Circ Heart Fail. 2023;16:e00080. doi: 10.1161/HHF.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 38.Havers-Borgersen E, Butt JH, Østergaard L, Petersen JK, Torp-Pedersen C, Køber L, Fosbøl EL. Long-term incidence of infective endocarditis among patients with congenital heart disease. Am Heart J. 2023;259:9–20. doi: 10.1016/j.ahj.2023.01.012. [DOI] [PubMed] [Google Scholar]
- 39.van Melle JP, Roos-Hesselink JW, Bansal M, Kamp O, Meshaal M, Pudich J, Luksic VR, Rodriguez-Alvarez R, Sadeghpour A, Hanzevacki JS, Sow R, Timóteo AT, Morgado MT, De Bonis M, Laroche C, Boersma E, Lancellotti P, Habib G EURO-ENDO Investigators Group. Infective endocarditis in adult patients with congenital heart disease. Int J Cardiol. 2023;370:178–185. doi: 10.1016/j.ijcard.2022.10.136. [DOI] [PubMed] [Google Scholar]
- 40.Arvanitaki A, Ibrahim W, Shore D, Diller GP, Li W, Rafiq I, Gatzoulis M, Montanaro C. Epidemiology and management of Staphylococcus Aureus infective endocarditis in adult patients with congenital heart disease: a single tertiary center experience. Int J Cardiol. 2022;360:23–28. doi: 10.1016/j.ijcard.2022.04.078. [DOI] [PubMed] [Google Scholar]
- 41.Moscatelli S, Leo I, Bianco F, Surkova E, Pezel T, Donald NA, Triumbari EKA, Bassareo PP, Pradhan A, Cimini A, Perrone MA. The role of multimodality imaging in patients with congenital heart disease and infective endocarditis. Diagnostics (Basel) 2023;13:3638. doi: 10.3390/diagnostics13243638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brida M, Balint HO, Bence A, Panfile E, Prokšelj K, Kačar P, Lebid IH, Šimkova I, Bobocka K, Meidrops K, Strenge A, Perčin L, Kapleriene L, Gumbiene L, Tomkiewicz-Pająk L, Komar M, Roos-Hesselink JW, Gatzoulis MA, Diller GP Study Group on Adult Congenital Heart Disease in Central and South-Eastern Europe. Infective endocarditis in adults with congenital heart disease: contemporary management and related outcomes in Central and South-Eastern European region. Int J Cardiol. 2023;377:45–50. doi: 10.1016/j.ijcard.2023.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Brida M, Lovrić D, Griselli M, Riesgo Gil F, Gatzoulis MA. Heart failure in adults with congenital heart disease. Int J Cardiol. 2022;357:39–45. doi: 10.1016/j.ijcard.2022.03.018. [DOI] [PubMed] [Google Scholar]
- 44.Bergh N, Skoglund K, Fedchenko M, Bollano E, Eriksson P, Dellborg M, Wai Giang K, Mandalenakis Z. Risk of heart failure in congenital heart disease: a nationwide register-based cohort study. Circulation. 2023;147:982–984. doi: 10.1161/CIRCULATIONAHA.122.061546. [DOI] [PubMed] [Google Scholar]
- 45.Dhingra NK, Mazer CD, Connelly KA, Verma S. Chronic heart failure management in adult patients with congenital heart disease. Curr Opin Cardiol. 2023;38:82–87. doi: 10.1097/HCO.0000000000001011. [DOI] [PubMed] [Google Scholar]
- 46.Wu MH, Chiu SN, Tseng WC, Lu CW, Kao FY, Huang SK. Atrial fibrillation in adult congenital heart disease and the general population. Heart Rhythm. 2023;20:1248–1254. doi: 10.1016/j.hrthm.2023.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Bessière F, Waldmann V, Combes N, Metton O, Dib N, Mondésert B, O’Leary E, De Witt E, Carreon CK, Sanders SP, Moore JP, Triedman J, Khairy P. Ventricular arrhythmias in adults with congenital heart disease, part I: JACC State-of-the-Art review. J Am Coll Cardiol. 2023;82:1108–1120. doi: 10.1016/j.jacc.2023.06.034. [DOI] [PubMed] [Google Scholar]
- 48.Bessière F, Waldmann V, Combes N, Metton O, Dib N, Mondésert B, O’Leary E, De Witt E, Carreon CK, Sanders SP, Moore JP, Triedman J, Khairy P. Ventricular arrhythmias in adults with congenital heart disease, part II: JACC State-of-the-Art review. J Am Coll Cardiol. 2023;82:1121–1130. doi: 10.1016/j.jacc.2023.06.036. [DOI] [PubMed] [Google Scholar]
- 49.Khairy P, Silka MJ, Moore JP, DiNardo JA, Vehmeijer JT, Sheppard MN, van de Bruaene A, Chaix MA, Brida M, Moore BM, Shah MJ, Mondésert B, Balaji S, Gatzoulis MA, Ladouceur M. Sudden cardiac death in congenital heart disease. Eur Heart J. 2022;43:2103–2115. doi: 10.1093/eurheartj/ehac104. [DOI] [PubMed] [Google Scholar]
- 50.Van Bulck L, Goossens E, Morin L, Luyckx K, Ombelet F, Willems R, Budts W, De Groote K, De Backer J, Annemans L, Moniotte S, de Hosson M, Marelli A, Moons P BELCODAC consortium. Last year of life of adults with congenital heart diseases: causes of death and patterns of care. Eur Heart J. 2022;43:4483–4492. doi: 10.1093/eurheartj/ehac484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucron H, Brard M, d’Orazio J, Long L, Lambert V, Zedong-Assountsa S, Le Harivel de Gonneville A, Ahounkeng P, Tuttle S, Stamatelatou M, Grierson R, Inamo J, Cuttone F, Elenga N, Bonnet D, Banydeen R. Infant congenital heart disease prevalence and mortality in French Guiana: a population-based study. Lancet Reg Health Am. 2023;29:100649. doi: 10.1016/j.lana.2023.100649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agasthi P, Van Houten HK, Yao X, Jain CC, Egbe A, Warnes CA, Miranda WR, Dunlay SM, Stephens EH, Johnson JN, Connolly HM, Burchill LJ. Mortality and morbidity of heart failure hospitalization in adult patients with congenital heart disease. J Am Heart Assoc. 2023;12:e030649. doi: 10.1161/JAHA.123.030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miles KG, Liu J, Tseng SY, DeFranco EA, Divanovic AA, Jones HN, Ollberding NJ, Cnota JF. Neonatal depression is associated with 1-year mortality in critical congenital heart disease. J Am Heart Assoc. 2023;12:e028774. doi: 10.1161/JAHA.122.028774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Damkjær M, Garne E, Loane M, Urhoj SK, Ballardini E, Cavero-Carbonell C, Coi A, García-Villodre L, Given J, Gissler M, Heino A, Jordan S, Limb E, Neville AJ, Pierini A, Rissmann A, Tan J, Scanlon I, Morris JK. Timing of cardiac surgical interventions and postoperative mortality in children with severe congenital heart defects across Europe: data from the EUROlinkCAT Study. J Am Heart Assoc. 2023;12:e029871. doi: 10.1161/JAHA.122.029871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao PS. Advances in the diagnosis and management of congenital heart disease in children. Children (Basel) 2023;10:753. doi: 10.3390/children10040753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fesslova V, Cavoretto PI. Recent advances in the diagnosis and management of congenital heart defects. J Clin Med. 2022;11:5534. doi: 10.3390/jcm11195534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moisa SM, Burlacu A, Brinza C, Țarcă E, Butnariu LI, Trandafir LM. An up-to-date narrative review on congenital heart disease percutaneous treatment in children using contemporary devices. Diagnostics (Basel) 2022;12:1189. doi: 10.3390/diagnostics12051189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Divekar AA, Arar YM, Clark S, Tandon A, Zellers TM, Veeram Reddy SR. Transcatheter device therapy and the integration of advanced imaging in congenital heart disease. Children (Basel) 2022;9:497. doi: 10.3390/children9040497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouma BJ, Mulder BJ. Changing landscape of congenital heart disease. Circ Res. 2017;120:908–922. doi: 10.1161/CIRCRESAHA.116.309302. [DOI] [PubMed] [Google Scholar]
- 60.Triedman JK, Newburger JW. Trends in congenital heart disease: the next decade. Circulation. 2016;133:2716–2733. doi: 10.1161/CIRCULATIONAHA.116.023544. [DOI] [PubMed] [Google Scholar]
- 61.Niwa K, Kaemmerer H, von Kodolitsch Y. Current diagnosis and management of late complications in adult congenital heart disease. Cardiovasc Diagn Ther. 2021;11:478–480. doi: 10.21037/cdt-21-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joye R, Beghetti M, Wacker J, Malaspinas I, Bouhabib M, Polito A, Bordessoule A, Shah DC. Early and late postoperative tachyarrhythmias in children and young adults undergoing congenital heart disease surgery. Pediatr Cardiol. 2023;44:312–324. doi: 10.1007/s00246-022-03074-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karazisi C, Dellborg M, Mellgren K, Giang KW, Skoglund K, Eriksson P, Mandalenakis Z. Risk of cancer in young and older patients with congenital heart disease and the excess risk of cancer by syndromes, organ transplantation and cardiac surgery: Swedish health registry study (1930-2017) Lancet Reg Health Eur. 2022;18:100407. doi: 10.1016/j.lanepe.2022.100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campolo J, Annoni G, Giaccardi M, Andreassi MG. Congenital heart disease and the risk of cancer: an update on the genetic etiology, radiation exposure damage, and future research strategies. J Cardiovasc Dev Dis. 2022;9:245. doi: 10.3390/jcdd9080245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams RG. Late causes of death after congenital heart defects: a population-based study from Finland. J Am Coll Cardiol. 2016;68:499–501. doi: 10.1016/j.jacc.2016.05.037. [DOI] [PubMed] [Google Scholar]
- 66.Choudhury TZ, Garg V. Molecular genetic mechanisms of congenital heart disease. Curr Opin Genet Dev. 2022;75:101949. doi: 10.1016/j.gde.2022.101949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill MC, Kadow ZA, Long H, Morikawa Y, Martin TJ, Birks EJ, Campbell KS, Nerbonne J, Lavine K, Wadhwa L, Wang J, Turaga D, Adachi I, Martin JF. Integrated multi-omic characterization of congenital heart disease. Nature. 2022;608:181–191. doi: 10.1038/s41586-022-04989-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lv Y, Gao RF, Yang CX, Xu YJ, Yang YQ. Increased gestational palmitic acid predisposes offspring to congenital heart disease. Cell Rep Med. 2023;4:100984. doi: 10.1016/j.xcrm.2023.100984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.García-Flores E, Rodríguez-Pérez JM, Borgonio-Cuadra VM, Vargas-Alarcón G, Calderón-Colmenero J, Sandoval JP, García-Montes JA, Espinoza-Gutiérrez VM, Reyes-García JG, Cazarín-Santos BG, Miranda-Duarte A, Gamboa-Domínguez A, Pérez-Hernández N. DNA methylation levels of the TBX5 gene promoter are associated with congenital septal defects in mexican paediatric patients. Biology (Basel) 2022;11:96. doi: 10.3390/biology11010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang G, Wang B, Yang P. Epigenetics in congenital heart disease. J Am Heart Assoc. 2022;11:e025163. doi: 10.1161/JAHA.121.025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boyd R, McMullen H, Beqaj H, Kalfa D. Environmental exposures and congenital heart disease. Pediatrics. 2022;149:e2021052151. doi: 10.1542/peds.2021-052151. [DOI] [PubMed] [Google Scholar]
- 72.Digilio MC, Dentici ML, Loddo S, Laino L, Calcagni G, Genovese S, Capolino R, Bottillo I, Calvieri G, Dallapiccola B, Marino B, Novelli A, Versacci P. Congenital heart defects in the recurrent 2q13 deletion syndrome. Eur J Med Genet. 2022;65:104381. doi: 10.1016/j.ejmg.2021.104381. [DOI] [PubMed] [Google Scholar]
- 73.Debiec RM, Hamby SE, Jones PD, Safwan K, Sosin M, Hetherington SL, Sprigings D, Sharman D, Lee K, Salahshouri P, Wheeldon N, Chukwuemeka A, Boutziouka V, Elamin M, Coolman S, Asiani M, Kharodia S, Skinner GJ, Samani NJ, Webb TR, Bolger AP. Contribution of NOTCH1 genetic variants to bicuspid aortic valve and other congenital lesions. Heart. 2022;108:1114–1120. doi: 10.1136/heartjnl-2021-320428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang RT, Guo YH, Yang CX, Gu JN, Qiu XB, Shi HY, Xu YJ, Xue S, Yang YQ. SOX7 loss-of-function variation as a cause of familial congenital heart disease. Am J Transl Res. 2022;14:1672–1684. [PMC free article] [PubMed] [Google Scholar]
- 75.Shi HY, Xie MS, Yang CX, Huang RT, Xue S, Liu XY, Xu YJ, Yang YQ. Identification of SOX18 as a new gene predisposing to congenital heart disease. Diagnostics (Basel) 2022;12:1917. doi: 10.3390/diagnostics12081917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma Q, Yang Y, Liu Y. Associations between NKX2-5 gene polymorphisms and congenital heart disease in the Chinese Tibetan population. Am J Transl Res. 2022;14:8407–8415. [PMC free article] [PubMed] [Google Scholar]
- 77.Meerschaut I, Steyaert W, Bové T, François K, Martens T, De Groote K, De Wilde H, Muiño Mosquera L, Panzer J, Vandekerckhove K, Moons L, Vermassen P, Symoens S, Coucke PJ, De Wolf D, Callewaert B. Exploring the mutational landscape of isolated congenital heart defects: an exome sequencing study using cardiac DNA. Genes (Basel) 2022;13:1214. doi: 10.3390/genes13071214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abhinav P, Zhang GF, Zhao CM, Xu YJ, Wang J, Yang YQ. A novel KLF13 mutation underlying congenital patent ductus arteriosus and ventricular septal defect, as well as bicuspid aortic valve. Exp Ther Med. 2022;23:311. doi: 10.3892/etm.2022.11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okashah S, Vasudeva D, El Jerbi A, Khodjet-El-Khil H, Al-Shafai M, Syed N, Kambouris M, Udassi S, Saraiva LR, Al-Saloos H, Udassi J, Al-Shafai KN. Investigation of genetic causes in patients with congenital heart disease in Qatar: findings from the sidra cardiac registry. Genes (Basel) 2022;13:1369. doi: 10.3390/genes13081369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ke ZP, Zhang GF, Guo YH, Sun YM, Wang J, Li N, Qiu XB, Xu YJ, Yang YQ. A novel PRRX1 loss-of-function variation contributing to familial atrial fibrillation and congenital patent ductus arteriosus. Genet Mol Biol. 2022;45:e20210378. doi: 10.1590/1678-4685-GMB-2021-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abdelrahman HA, Akawi N, Al-Shamsi AM, Ali A, Al-Jasmi F, John A, Hertecant J, Al-Gazali L, Ali BR. Bi-allelic null variant in matrix metalloproteinase-15, causes congenital cardiac defect, cholestasis jaundice, and failure to thrive. Clin Genet. 2022;101:403–410. doi: 10.1111/cge.14107. [DOI] [PubMed] [Google Scholar]
- 82.De Ita M, Gaytán-Cervantes J, Cisneros B, Araujo MA, Huicochea-Montiel JC, Cárdenas-Conejo A, Lazo-Cárdenas CC, Ramírez-Portillo CI, Feria-Kaiser C, Peregrino-Bejarano L, Yáñez-Gutiérrez L, González-Torres C, Rosas-Vargas H. Clustering of genetic anomalies of cilia outer dynein arm and central apparatus in patients with transposition of the great arteries. Genes (Basel) 2022;13:1662. doi: 10.3390/genes13091662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Z, Qiao XH, Xu YJ, Liu XY, Huang RT, Xue S, Qiu HY, Yang YQ. SMAD1 loss-of-function variant responsible for congenital heart disease. Biomed Res Int. 2022;2022:9916325. doi: 10.1155/2022/9916325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walton NA, Nguyen HH, Procknow SS, Johnson D, Anzelmi A, Jay PY. Repurposing normal chromosomal microarray data to harbor genetic insights into congenital heart disease. Biology (Basel) 2023;12:1290. doi: 10.3390/biology12101290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bolunduț AC, Lazea C, Mihu CM. Genetic alterations of transcription factors and signaling molecules involved in the development of congenital heart defects-a narrative review. Children (Basel) 2023;10:812. doi: 10.3390/children10050812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang H, Xiao F, Qian Y, Wu B, Dong X, Lu Y, Cheng G, Wang L, Yan K, Yang L, Chen L, Kang W, Li L, Pan X, Wei Q, Zhuang D, Chen D, Yin Z, Yang L, Ni Q, Liu R, Li G, Zhang P, Li X, Peng X, Wang Y, Chen H, Ma X, Liu F, Cao Y, Huang G, Zhou W. Genetic architecture in neonatal intensive care unit patients with congenital heart defects: a retrospective study from the China neonatal genomes project. J Med Genet. 2023;60:247–253. doi: 10.1136/jmedgenet-2021-108354. [DOI] [PubMed] [Google Scholar]
- 87.Padua MB, Helm BM, Wells JR, Smith AM, Bellchambers HM, Sridhar A, Ware SM. Congenital heart defects caused by FOXJ1. Hum Mol Genet. 2023;32:2335–2346. doi: 10.1093/hmg/ddad065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin XY, Chen HX, Chen Z, Yang Q, Han J, He GW. Genetic variants of ISL1 gene promoter identified from congenital tetralogy of fallot patients alter cellular function forming disease basis. Biomolecules. 2023;13:358. doi: 10.3390/biom13020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li YJ, Wang J, Ye WG, Liu XY, Li L, Qiu XB, Chen H, Xu YJ, Yang YQ, Bai D, Huang RT. Discovery of GJC1 (Cx45) as a new gene underlying congenital heart disease and arrhythmias. Biology (Basel) 2023;12:346. doi: 10.3390/biology12030346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li N, Li YJ, Guo XJ, Wu SH, Jiang WF, Zhang DL, Wang KW, Li L, Sun YM, Xu YJ, Yang YQ, Qiu XB. Discovery of TBX20 as a novel gene underlying atrial fibrillation. Biology (Basel) 2023;12:1186. doi: 10.3390/biology12091186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suo MJ, Chen WC, Xu ZQ, Tian GX, Li T, Li P, Sheng W, Huang GY, Ma XJ. X-linked BCOR variants identified in Chinese Han patients with congenital heart disease. J Gene Med. 2023;25:e3461. doi: 10.1002/jgm.3461. [DOI] [PubMed] [Google Scholar]
- 92.Wu H, Wu H, He Y, Sun W, Meng Y, Wen B, Chu M. Functional characterization of GATA6 genetic variants associated with mild congenital heart defects. Biochem Biophys Res Commun. 2023;641:77–83. doi: 10.1016/j.bbrc.2022.12.004. [DOI] [PubMed] [Google Scholar]
- 93.Deng Q, Wang X, Gao J, Xia X, Wang Y, Zhang Y, Chen Y. Growth restriction and congenital heart disease caused by a novel TAB2 mutation: a case report. Exp Ther Med. 2023;25:258. doi: 10.3892/etm.2023.11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Y, Dai X, Liu H, Peng J, Chen J. A novel ZIC3 mutation in a Chinese family with heterotaxy and multiple types of congenital heart defect. Prenat Diagn. 2023;43:275–279. doi: 10.1002/pd.6294. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Xu YJ, Yang CX, Huang RT, Xue S, Yuan F, Yang YQ. SMAD4 loss-of-function mutation predisposes to congenital heart disease. Eur J Med Genet. 2023;66:104677. doi: 10.1016/j.ejmg.2022.104677. [DOI] [PubMed] [Google Scholar]
- 96.Jang MY, Patel PN, Pereira AC, Willcox JAL, Haghighi A, Tai AC, Ito K, Morton SU, Gorham JM, McKean DM, DePalma SR, Bernstein D, Brueckner M, Chung WK, Giardini A, Goldmuntz E, Kaltman JR, Kim R, Newburger JW, Shen Y, Srivastava D, Tristani-Firouzi M, Gelb BD, Porter GA Jr, Seidman CE, Seidman JG. Contribution of previously unrecognized RNA splice-altering variants to congenital heart disease. Circ Genom Precis Med. 2023;16:224–231. doi: 10.1161/CIRCGEN.122.003924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bobos D, Soufla G, Angouras DC, Lekakis I, Georgopoulos S, Melissari E. Investigation of the role of BMP2 and -4 in ASD, VSD and complex congenital heart disease. Diagnostics (Basel) 2023;13:2717. doi: 10.3390/diagnostics13162717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- 99.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–2367. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goldman DC, Donley N, Christian JL. Genetic interaction between Bmp2 and Bmp4 reveals shared functions during multiple aspects of mouse organogenesis. Mech Dev. 2009;126:117–127. doi: 10.1016/j.mod.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Uchimura T, Komatsu Y, Tanaka M, McCann KL, Mishina Y. Bmp2 and Bmp4 genetically interact to support multiple aspects of mouse development including functional heart development. Genesis. 2009;47:374–384. doi: 10.1002/dvg.20511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jacobs JP, Franklin RCG, Béland MJ, Spicer DE, Colan SD, Walters HL 3rd, Bailliard F, Houyel L, St Louis JD, Lopez L, Aiello VD, Gaynor JW, Krogmann ON, Kurosawa H, Maruszewski BJ, Stellin G, Weinberg PM, Jacobs ML, Boris JR, Cohen MS, Everett AD, Giroud JM, Guleserian KJ, Hughes ML, Juraszek AL, Seslar SP, Shepard CW, Srivastava S, Cook AC, Crucean A, Hernandez LE, Loomba RS, Rogers LS, Sanders SP, Savla JJ, Tierney ESS, Tretter JT, Wang L, Elliott MJ, Mavroudis C, Tchervenkov CI. Nomenclature for pediatric and congenital cardiac care: unification of clinical and administrative nomenclature - The 2021 International Paediatric and Congenital Cardiac Code (IPCCC) and the Eleventh Revision of the International Classification of Diseases (ICD-11) Cardiol Young. 2021;31:1057–1188. doi: 10.1017/S104795112100281X. [DOI] [PubMed] [Google Scholar]
- 103.Dong BB, Li YJ, Liu XY, Huang RT, Yang CX, Xu YJ, Lv HT, Yang YQ. Discovery of BMP10 as a new gene underpinning congenital heart defects. Am J Transl Res. 2024;16:109–125. doi: 10.62347/IVRF4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang J, Xie X, Zhong Z, Yuan H, Xu P, Gao H, Lai Y. Prevalence of antibiotic resistance of Helicobacter pylori isolates in Shanghai, China. Am J Transl Res. 2022;14:7831–7841. [PMC free article] [PubMed] [Google Scholar]
- 105.Wang J, Wang XC, Gu ZH, Ren GW, Zhao XH, Qu XK, Xu YJ, Yang YQ. A novel GJA5 variant associated with increased risk of essential hypertension. Am J Transl Res. 2023;15:1259–1270. [PMC free article] [PubMed] [Google Scholar]
- 106.Gu JN, Yang CX, Ding YY, Qiao Q, Di RM, Sun YM, Wang J, Yang L, Xu YJ, Yang YQ. Identification of BMP10 as a novel gene contributing to dilated cardiomyopathy. Diagnostics (Basel) 2023;13:242. doi: 10.3390/diagnostics13020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang J, Greene S, Martin JF. Bmp signalling in congenital heart disease: new developments and future directions. Birth Defects Res A Clin Mol Teratol. 2011;91:441–448. doi: 10.1002/bdra.20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 109.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 110.Liu W. BMP4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc Natl Acad Sci U S A. 2004;101:4489–4494. doi: 10.1073/pnas.0308466101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsaytler P, Liu J, Blaess G, Schifferl D, Veenvliet JV, Wittler L, Timmermann B, Herrmann BG, Koch F. BMP4 triggers regulatory circuits specifying the cardiac mesoderm lineage. Development. 2023;150:dev201450. doi: 10.1242/dev.201450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jamali M, Karamboulas C, Rogerson PJ, Skerjanc IS. BMP signaling regulates Nkx2-5 activity during cardiomyogenesis. FEBS Lett. 2001;509:126–130. doi: 10.1016/s0014-5793(01)03151-9. [DOI] [PubMed] [Google Scholar]
- 113.Takei S, Ichikawa H, Johkura K, Mogi A, No H, Yoshie S, Tomotsune D, Sasaki K. Bone morphogenetic protein-4 promotes induction of cardiomyocytes from human embryonic stem cells in serum-based embryoid body development. Am J Physiol Heart Circ Physiol. 2009;296:H1793–H1803. doi: 10.1152/ajpheart.01288.2008. [DOI] [PubMed] [Google Scholar]
- 114.Cagavi E, Bartulos O, Suh CY, Sun B, Yue Z, Jiang Z, Yue L, Qyang Y. Functional cardiomyocytes derived from Isl1 cardiac progenitors via Bmp4 stimulation. PLoS One. 2014;9:e110752. doi: 10.1371/journal.pone.0110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chung IM, Rajakumar G. Genetics of congenital heart defects: the NKX2-5 gene, a key player. Genes (Basel) 2016;7:6. doi: 10.3390/genes7020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reamon-Buettner SM, Borlak J. NKX2-5: an update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD) Hum Mutat. 2010;31:1185–1194. doi: 10.1002/humu.21345. [DOI] [PubMed] [Google Scholar]
- 117.Chen Y, Xiao D, Zhang L, Cai CL, Li BY, Liu Y. The role of Tbx20 in cardiovascular development and function. Front Cell Dev Biol. 2021;9:638542. doi: 10.3389/fcell.2021.638542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McCulley DJ, Kang JO, Martin JF, Black BL. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev Dyn. 2008;237:3200–3209. doi: 10.1002/dvdy.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bai Y, Wang J, Morikawa Y, Bonilla-Claudio M, Klysik E, Martin JF. Bmp signaling represses Vegfa to promote outflow tract cushion development. Development. 2013;140:3395–3402. doi: 10.1242/dev.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nie X, Brown CB, Wang Q, Jiao K. Inactivation of Bmp4 from the Tbx1 expression domain causes abnormal pharyngeal arch artery and cardiac outflow tract remodeling. Cells Tissues Organs. 2011;193:393–403. doi: 10.1159/000321170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qian B, Mo R, Da M, Peng W, Hu Y, Mo X. Common variations in BMP4 confer genetic susceptibility to sporadic Congenital Heart Disease in a Han Chinese Population. Pediatr Cardiol. 2014;35:1442–1447. doi: 10.1007/s00246-014-0951-1. [DOI] [PMC free article] [PubMed] [Google Scholar]