

SUMMARY
Proper preimplantation development is essential to assemble a blastocyst capable of implantation. Live-imaging has uncovered major events driving early development in mouse embryos, yet studies in human have been limited by restrictions on genetic manipulation and lack of imaging approaches. We have overcome this barrier by combining fluorescent dyes with live-imaging to reveal the dynamics of chromosome segregation, compaction, polarization, blastocyst formation, and hatching in the human embryo. We also show that blastocyst expansion mechanically constrains trophectoderm cells causing nuclear budding and DNA shedding into the cytoplasm. Furthermore, cells with lower peri-nuclear keratin levels are more prone to undergo DNA loss. Moreover, applying trophectoderm biopsy, a mechanical procedure performed clinically for genetic testing, increases DNA shedding. Thus, our work reveals distinct processes underlying human development, compared to mouse, and suggests that aneuploidies in human embryos may not only originate from chromosome segregation errors during mitosis, but also nuclear DNA shedding.
INTRODUCTION
The global decline in fertility constitutes a major public health concern and is reflected in the exponential rise in babies born from in vitro fertilization (IVF)1. Therefore, it is imperative to understand the mechanisms patterning human preimplantation development and the determinants of embryo quality. Following fertilization, the embryo develops as a self-contained structure and undergoes a series of cleavage divisions culminating in the formation of the blastocyst2. This preimplantation stage of development has thus far been studied primarily in the mouse embryo. Classically analyzed using histology in fixed specimens, advances in genetic tools for fluorescently labeling cells and live-imaging have made it possible to visualize the dynamic course of events driving early mouse development3–9.
Similar approaches would prove powerful to discover the cellular and morphogenetic events underlying early human development. Revealing this is important as studies have indicated differences between mouse and human preimplantation development. For example, it has been observed that the timing of compaction, a process involving extensive changes in cell shape, likely differs in mouse and human embryos10. Thus, it remains unknown how human embryo compaction may be coordinated with other key events occurring during these stages, such as the onset of apical-basal polarization or the first spatial segregation of cells into inner and outer positions, which establishes the inner cells mass (ICM) and trophectoderm. Furthermore, preimplantation genetic testing (PGT) for aneuploidy, which is widely used in assisted reproduction to assess embryo quality has shown that preimplantation human embryos often contain a larger number of aneuploid cells compared to somatic tissues11–13. Whereas entirely aneuploid embryos derive from chromosome segregation errors during meiosis11,14, mosaic aneuploid patterns have been proposed to result from chromosome segregation errors during mitosis15. However, we have lacked the tools to track how mitotic errors occur in real time in the human embryo.
Currently, there are numerous restrictions on the use of genetic manipulation and microinjection of DNA or mRNA into human embryos, which would allow expression of fluorescent proteins for imaging studies. Moreover, while microinjections are typically performed in the 1-cell embryo, the majority of human embryos donated for research are obtained at the blastocyst stage, consisting of 100–200 cells2, which is incompatible with microinjection techniques. Thus, establishing approaches that combine non-invasive fluorescent labeling of cells with live-imaging remains an open challenge to uncover the processes patterning preimplantation human development.
RESULTS
Bypassing the need for genetic manipulation to image preimplantation development with cellular resolution
To establish the application of non-invasive imaging with subcellular resolution in live preimplantation embryos, we first test fluorescent dyes which are membrane permeable and label specific cellular structures in the mouse embryo, from the 2-cell to blastocyst stage (Figure 1A; Figure S1A; Video S1). Incubation with the dye SPY650-DNA, which labels genomic DNA, and SPY555-actin, which labels F-actin, produces high signal to noise ratios (Figure S1B). Furthermore, cell cycle durations, developmental timing, and rates of blastocyst progression are similar between dyed and non-dyed embryos (Figure 1B and 1C). Following transfer into pseudopregnant mice, dyed embryos produce offspring at a similar rate to non-dyed control embryos (Figure S1C and S1D). Moreover, labeling patterns using the dyes are equivalent to those produced using microinjection of mRNAs coding for fluorescently-tagged histone 2B (H2B-GFP) and utrophin-RFP (Utr-RFP) (Figure 1D–1F), which have been extensively used for visualizing the dynamics of chromatin and F-actin in live embryos, respectively3–8. The fluorescent dyes also produce images similar to fixed specimens stained with 4′,6-diamidino-2-phenylindole (DAPI) and Phalloidin-Rhodamine, well established markers of chromatin and F-actin, respectively (Figure S1B).
Figure 1. Imaging live mouse embryos with fluorescent dyes can bypass the need for genetic manipulation or mRNA injection.
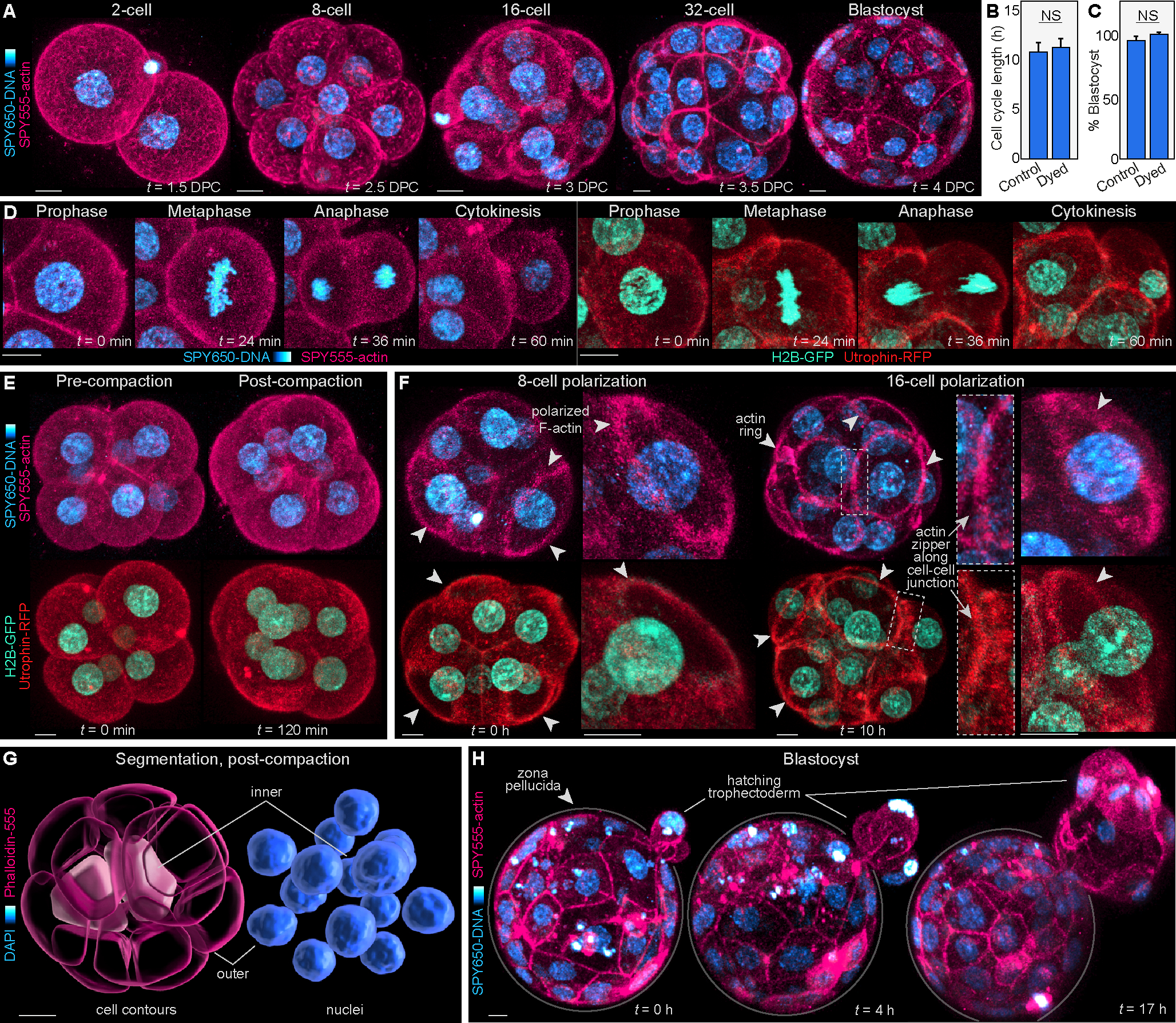
(A) Live-imaging of mouse embryos labeled with SPY650-DNA and SPY555-actin at various developmental stages.
(B) Embryos stained with the SPY dyes display similar cell cycle lengths at the 4- to 8-cell stage. The control group was measured using brightfield in non-dyed embryos (N = 18 embryos per group; NS = not significant by student’s t-test).
(C) Blastocyst progression rates were similar between dyed and non-dyed embryos (N = 3 independent experiments; NS = not significant by student’s t-test).
(D–H) The combination of SPY650-DNA and SPY555-actin allows visualization of central events characterizing preimplantation development, similar to embryos microinjected with H2B-GFP and Utrophin-RFP mRNAs. These include the main phases of mitosis (D), embryo compaction (E), formation of F-actin-rich apical domains and of F-actin rings that undergo zippering along cell-cell junctions (F), visualization of the first inner cells of the embryo that will form the ICM, which can be computationally segmented within the 16-cell embryo (G), and blastocyst hatching (H). In (D-F), embryos microinjected with mRNA for H2B-GFP and Utr-RFP are shown for comparison with the SPY650-DNA and SPY555-actin approach.
Scale bars, 10 μm.
See also Figures S1 and S2, and Videos S1 and S2.
Analysis of the live-imaging data confirms that SPY650-DNA and SPY555-actin enables confocal three-dimensional (3D) scans of the embryo (at 5–10 minute intervals) producing accurate visualization of central events during preimplantation development. These include: capturing the main phases of mitosis (Figure 1D; Video S1); visualizing the major changes in cell shape that characterize embryo compaction at the 8-cell stage16 (Figure 1E; Video S1); detecting cell polarization at the 8-cell stage identified by the formation of a disk-shaped apical domain17, and establishing apical F-actin rings at the 16-cell stage17 (Figure 1F; Figure S2; Videos S1 and S2); tracking the expansion and zippering of F-actin rings, which seal the embryo prior to blastocyst formation5 (Figure 1F; Figure S2; Videos S1 and S2); detecting the first internalized cells within the 16-cell embryo, which will form the inner cell mass (ICM)4,18 (Figure 1G; Figure S1E–I); and visualizing blastocyst expansion and hatching from the zona pellucida19 (Figure 1H; Video S1). Thus, the combination of fluorescent dyes with live-imaging permits the study of key events during preimplantation development, and can bypass cell-labeling approaches relying on mRNA or DNA microinjection.
Cellular and morphogenetic processes underlying human preimplantation development
We next use this approach to uncover the cellular and morphogenetic processes underlying human development using cleavage-stage embryos derived from IVF. First, we study cell cycle dynamics and capture the main phases of mitosis (Figure 2A), which reveals that the duration of human mitosis is similar to mouse (Figure 2B). By contrast, interphase is 27 ± 4% longer in human (16.1 ± 0.9 h versus 12.7 ± 0.4 h, P < 0.01) (Figure 2B). This is consistent with previous reports showing conservation of the timing of mitosis across species20, and with studies proposing that the duration of morphogenetic processes in different species is determined predominantly by interphase21,22.
Figure 2. Non-invasive imaging reveals cell dynamics underlying early human preimplantation development.
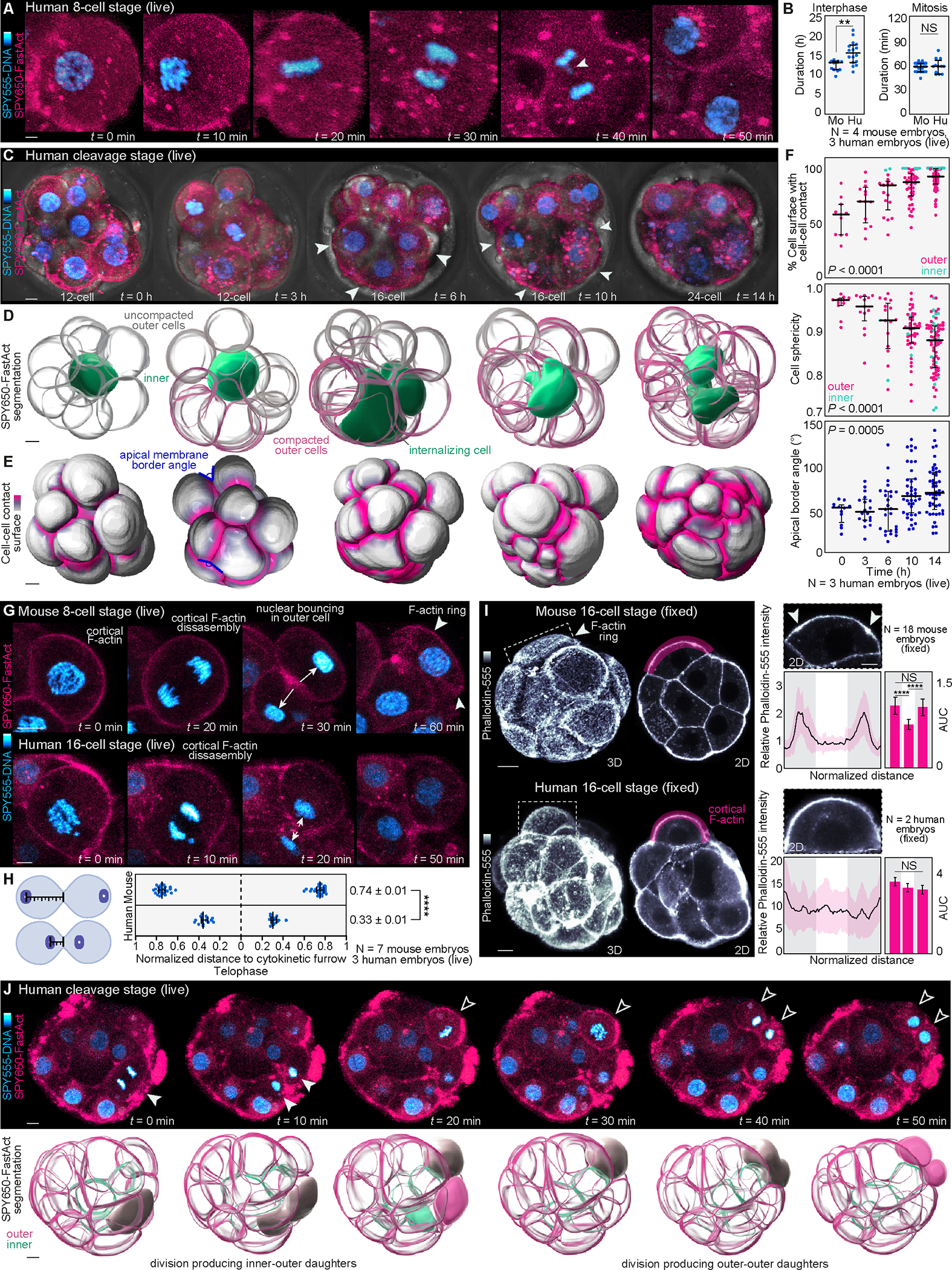
(A) Live-imaging of cleavage stage human embryos labeled with SPY555-DNA and SPY650-FastAct allows visualization of the main phases of mitosis.
(B) Comparison of interphase and mitosis duration between mouse (Mo) and human (Hu) cleavage stage (16- to 32-cell) embryos (N = 4 mouse and 3 human embryos, n = 12 and 16 cells (interphase) and n = 31 and 12 cells (mitosis) for mouse and human, respectively; **P< 0.01, NS = not significant by student’s t-test).
(C) Example of a cleavage-stage human embryo undergoing compaction. Arrowheads show compacting cells.
(D) Computational segmentation of embryos shown in (C) enables visualization of changes in cell morphology during compaction. Note that compaction occurs in an asynchronous manner and that an inner cell becomes completely enclosed by its neighbors during the compaction process.
(E) and (F) Analysis of changes in cell-cell contact, cell sphericity and angle between apical membranes as proxies for compaction (N = 3 cleavage stage human embryos, and n = 11, 13, 17, 44, 77 cells at 0, 3, 6, 10, 14h, respectively; ****P< 0.0001 by one-way ANOVA, Kruskal-Wallis test).
(G) Analysis of nuclear position and apical polarization following division in live embryos. In mouse embryos, the cell nucleus bounces against the apical cell cortex at the end of cytokinesis triggering formation of an F-actin ring7. In the human embryo cell nuclei remain closer to the cytokinetic furrow (arrows) without bouncing against the cortex. Arrowheads highlight an actin ring in mouse.
(H) Distance of the nucleus to the cytokinetic furrow is measured for each mitotic pair at telophase (N = 7 mouse and 3 human embryos, n = 50 and 32 cells for mouse and human, respectively; ****P< 0.0001 by two-tailed unpaired student’s t-test).
(I) Most outer cells display F-actin rings in mouse embryos shown by Phalloidin-555 staining. The human apical domain enriches F-actin but does not form ring-like structures. Quantification of F-actin intensity along the apical cortex (magenta region) to highlight the presence (arrows) or absence of F-actin rings. For comparisons, the apical cortex was divided into thirds and the area under the curve (AUC) was calculated (N = 18 mouse embryos and 2 human embryos; n = 29 mouse cells and 11 human cells; ****P< 0.0001, NS = not significant by one-way ANOVA test).
(J) Live-imaging also exposes the first lineage segregation events generating outer-outer and inner-outer progeny in human. Upper images show 2D planes with dividing cells. Lower panels show segmented 3D reconstructions. The examples demonstrate a division producing outer-inner progeny and a second division producing outer-outer progeny.
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
The dye and live-imaging approach also enables cell contour tracking, including inner cells, which we apply to quantitatively dissect compaction dynamics in the human embryo (Figure 2C and 2D). The onset of compaction is known to differ between species, starting at the 8-cell stage in mouse23, and 16- to 32-cell stage in monkey and bovine24. Consistent with compaction in the mouse embryo25,26, dyed human embryos demonstrate an increase in cell-cell contact and angle between apical membranes with a decrease in cell sphericity, which all serve as proxies for compaction (Figure 2E and 2F). Clinical studies of 2D time-lapse images (Figure S3A) and of fixed embryos indicate that human embryo compaction is heterogeneous10,27, and most frequently begins at the 12-cell stage28. In line with this, we tracked compaction beginning at the 12- to 14-cell stage over 6 to 10 hours (Figure 2C–2F). Indeed, the timing of compaction initiation determines inner-outer cell segregation due to laws of spherical packing. The Newton number, or kissing number29,30, can be used to infer the minimum number of cells (12) that can be arranged within a sphere such that all cells maintain contact with each other and an inner cell (Figure S3B). Thus, at the 12-cell stage human compaction begins with at least one cell occupying an inner position, unlike the 8-cell stage mouse embryo which does not yet contain an inner cell (Figure S3C). This suggests that the mechanisms underlying inner-outer segregation, and plausibly cell polarization, differ between species.
In the mouse, we and others have shown that compaction is synchronized with asymmetric cell divisions7, polarization, and apical constriction4. Key cytoskeletal dynamics underlie these processes, including actin rings and actin zippers5, which can be readily resolved in dyed mouse embryos (Figure S2; Video S2). Furthermore, the formation of F-actin rings in mouse is mediated by bouncing of the cell nucleus against the apical cell pole at the end of cytokinesis7. By contrast, we found that cell nuclei do not bounce against the cortex in the human embryo and instead, remain closer to the cytokinetic furrow (Figure 2G and 2H; Figure S3D; Video S3). Moreover, analysis of live dyed (N = 3) and stained fixed (N = 2) human morulae shows F-actin enrichment at the apical pole, but is homogenously distributed across the cortex and does not form clear ring-like structures (Figure 2I). Although access to human morulae is significantly limited (see Limitations of the study), these results are consistent with recent studies of human embryo polarization27 and models proposing that apical domains may not be asymmetrically inherited during cell division to determine ICM versus trophectoderm fate5,6, unlike previous suggestions31,32.
Our approach also enables tracking of cell divisions and internalization, which was previously unresolvable by simple light microscopy. Importantly, dyed human embryos demonstrate different types of divisions producing outer-outer and inner-outer daughter cells that occur concomitant to compaction (Figure 2J; Figure S3E–S3G). Furthermore, inner cells retain their inner position and do not return to an outer location, as proposed in some earlier studies33 (Figure S3F). Together, these results highlight differences in the sequence of compaction, inner-outer cell segregation, and cell polarization during mouse and human development. In the mouse embryo, compaction starts at the 8-cell stage and is linked with cell polarization and inner-outer segregation culminating in a synchronous and relatively rapid process. By contrast, in the human embryo, compaction starts later and is more asynchronous, without clear links to apical polarization or inner-outer segregation.
We also analyze cell dynamics during formation of the human blastocyst (Figure 3A). While SPY650-DNA provides immediate nuclear visualization similar to mouse, SPY555-actin requires 1–2 hours to localize to cell membranes in human (Figure 3A). This is likely due to species differences in zona pellucida content and structure34–36 leading to binding and prolonged diffusion through the human zona pellucida (Figure S4A and S4B). In addition to visualizing the outer trophectoderm layer, our approach allows us to track cells undergoing divisions deeper in the embryo located within the ICM (Figure 3B). Moreover, we identify long F-actin tethers traversing the entire blastocoel (Figure 3B; Figure S4C). Our movies show that these projections form during cavitation and serve as tethers linking the mural trophectoderm to the ICM region, in line with previous observations in mouse37. However, while mouse blastocysts also present these tethers, they are more frequent and abundant in human (Figure S4D and S4E). Finally, the use of SPY555-actin does not interfere with blastocyst hatching, a process indicative of embryo quality38,39 and ultimately required for implantation (Figure 3C).
Figure 3. Dynamics of human blastocyst formation.
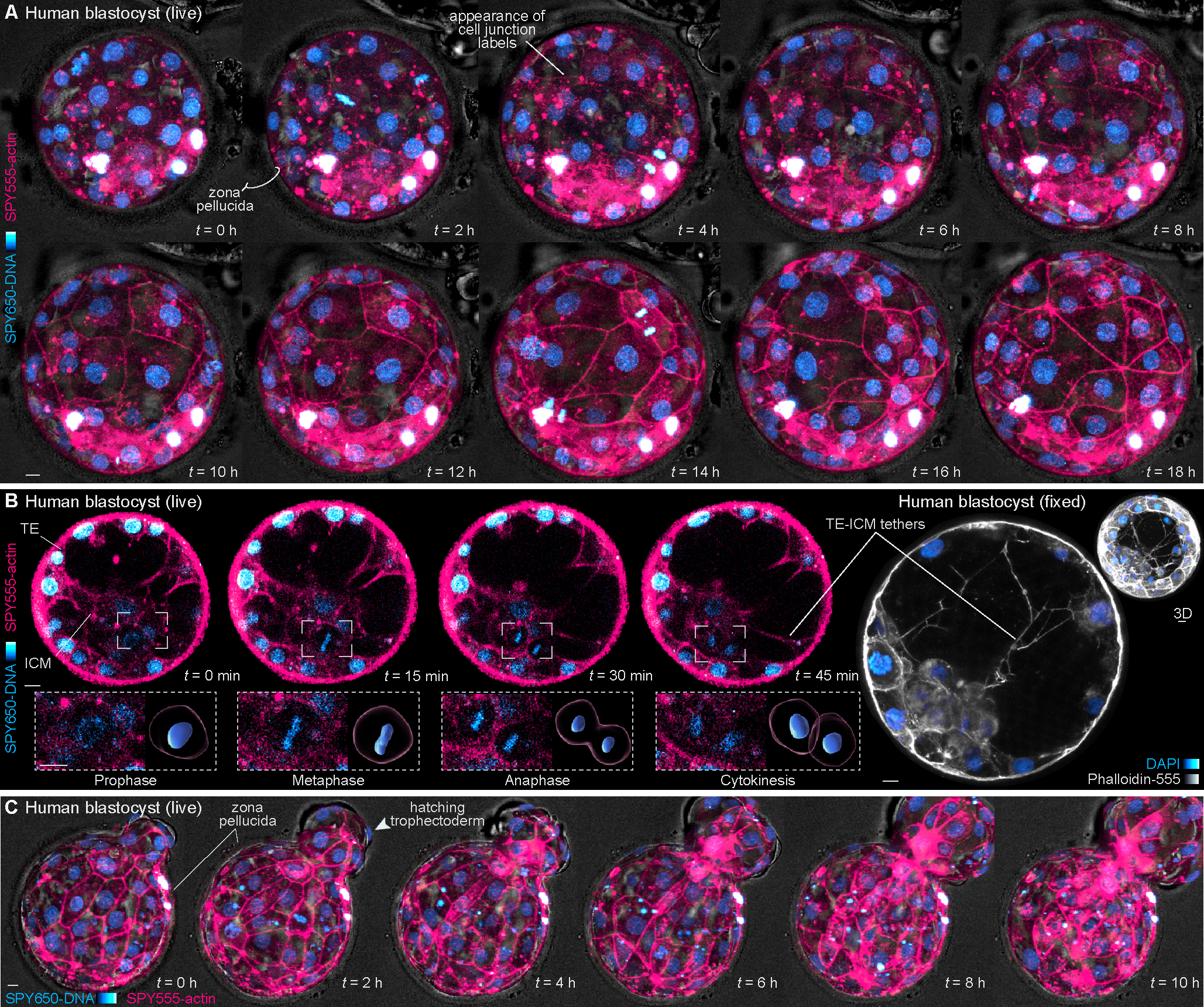
(A) Live-imaging of a human blastocyst stained with SPY650-DNA and SPY555-actin. After 1–2 hours cell-cell junctions become clearly labeled by SPY555-actin.
(B) 2D planes of a blastocyst during cavitation. A cell division within the ICM can be tracked over time and computationally segmented. Imaging through the blastocyst also reveals tether-like structures projecting between the trophectoderm and ICM validated by Phalloidin-555 staining in fixed embryos.
(C) Selected frames of a live human embryo undergoing hatching.
The zona pellucida was masked out in live human embryos to improve embryo visualization. Scale bars, 10 μm.
See also Figure S4.
Similar to cell cycle analysis at the cleavage stage, we investigated the timing of cell divisions at the blastocyst stage (Figure 4A; Video S3). The duration of mitosis is similar in both trophectoderm and ICM and between mouse and human (Figure 4B and 4C). However, the duration of interphase in human is longer than in mouse for both trophectoderm and ICM (15.4 ± 0.5 h versus 13.0 ± 0.5 h in the trophectoderm P < 0.01; 15.8 ± 0.7 h versus 12.0 ± 0.5 h in the ICM, P < 0.05) (Figure 4B and 4C). Trophectoderm cell divisions in the mouse embryo have been proposed to trigger recurrent collapses observed in the blastocoel40. By contrast, analysis of division patterns and blastocyst volume in the human shows that trophectoderm mitoses do not display high temporal synchronicity or correlate with blastocoel collapse (Figure 4D–4F; Figure S4F and S4G). Thus, trophectoderm cells undergo divisions without overtly disrupting trophectoderm layer cohesiveness. This is consistent with studies showing that outer cells retain their zippered junctions with neighboring cells during division5 and with maintenance of permeability barriers during cell divisions in other epithelial tissues41.
Figure 4. Cell divisions and chromosome segregation errors in the human blastocyst.
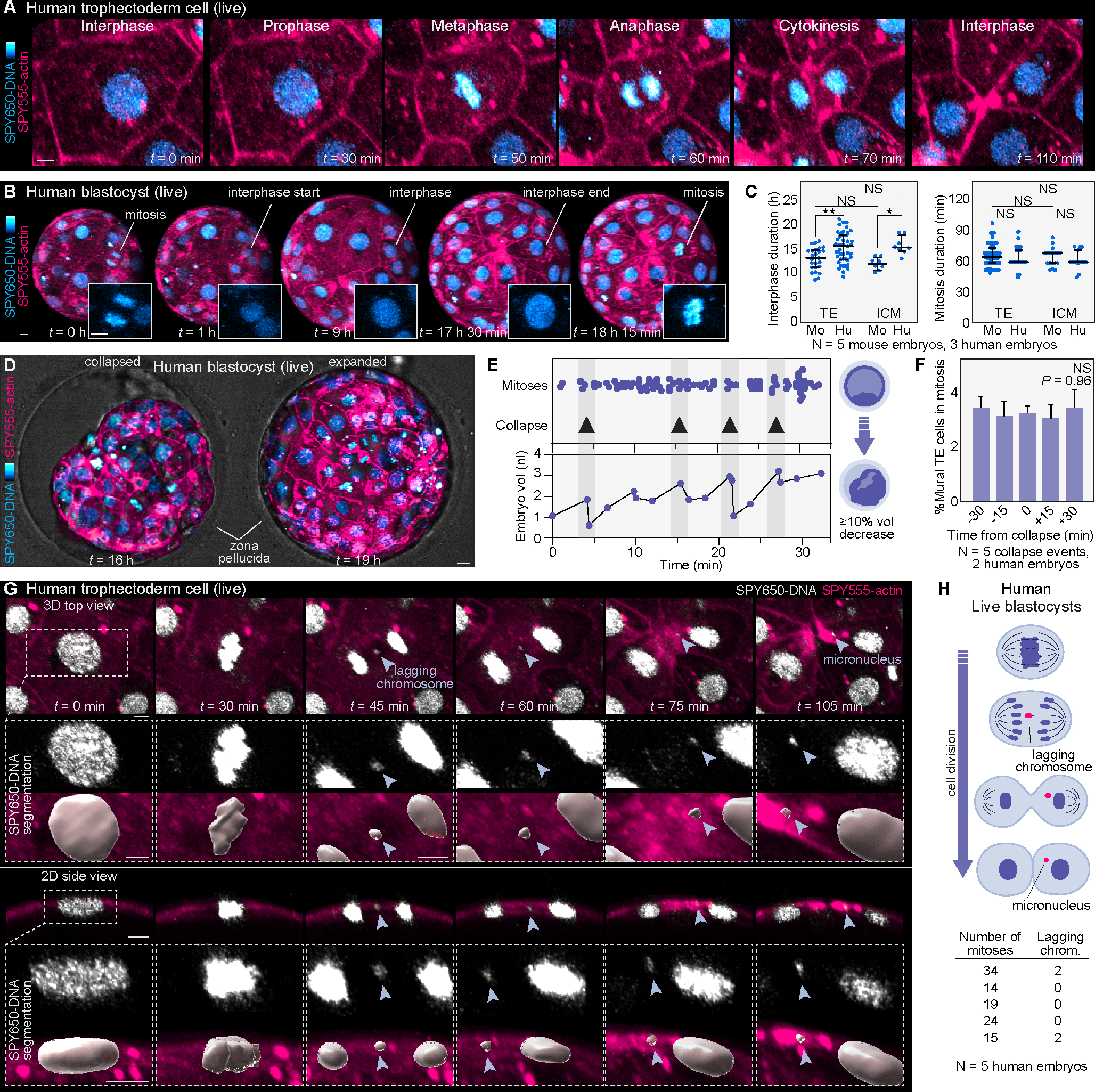
(A) Example of mitosis in the human trophectoderm.
(B) Tracking interphase duration in the human trophectoderm by identifying nuclei with interphase morphology and measuring the time between mitoses. Insets show chromatin morphology.
(C) Comparison of interphase and mitosis duration between mouse (Mo) and human (Hu) trophectoderm and ICM (N = 5 mouse and 3 human embryos, n = 25, 45, 8, 8 cells (interphase) and n = 96, 187, 18, 15 cells (mitosis); **P< 0.01, *P< 0.05, NS = not significant by one-way ANOVA test).
(D) Examples of blastocyst collapse and expansion.
(E) Analysis of cell divisions relative to embryo collapse events (probed by measuring embryo volume). Note the lack of correlation between divisions and embryo volume.
(F) Quantification and comparison of mural trophectoderm mitoses at different time points before and after embryo collapse (N = 5 collapse events in 2 human embryos; NS = not significant by Kruskal-Wallis test).
(G) Live-imaging with SPY650-DNA and SPY555-actin showing the formation of a micronucleus from a lagging chromosome during the mitosis of a trophectoderm cell in a human blastocyst in 3D top and 2D sectional views. Insets show magnified images of the process and surface segmentations of the DNA signal.
(H) Scheme representing micronucleus formation from a lagging chromosome during mitosis. Quantification of the number of mitoses with lagging chromosomes relative to the total number of mitotic events analyzed in human blastocysts (N = 5 human embryos).
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Compared to somatic cells, aneuploidy is more prevalent in cells of the embryo, with 5–15% of all embryos identified as mosaic aneuploid on PGT results42–45. We therefore explore whether our approach allows detection of chromosome segregation errors in the human embryo. Tracking cells permits direct visualization of lagging chromosomes during mitosis. These errors occur in 3.8 ± 2.6% of tracked cell divisions and subsequently form micronuclei (Figure 4G and 4H; Figure S4H). The lagging chromosomes detected in human embryos by SPY-DNA appear morphologically similar to those found in mouse embryos (Figure S4I–S4K). Furthermore, their segregation dynamics are spatiotemporally consistent with those found in mouse, using both dye (Figure S4I–S4K) and microinjection46. Together, these results unveil cellular and morphogenetic processes underlying human preimplantation development and highlight differences between mouse and human as the blastocyst expands and hatches.
Trophectoderm cell nuclei undergo budding and DNA shedding during blastocyst expansion and biopsy
Formation of the fluid-filled blastocoel results in the physical segregation of putative ICM and trophectoderm cells. Furthermore, the rising intracavitary pressure causes shape changes in trophectoderm cell nuclei from spherical to more ellipsoid (Figure 5A). Live-imaging using SPY650-DNA allows us to visualize nuclear morphology during blastocyst expansion, and reveals bud-like structures protruding from the cell nucleus (Figure 5B; Figure S5A). Notably, tracking these nuclear buds over time shows that they are shed into the cytoplasm, producing cytoplasmic DNA structures (Figure 5B and 5C; Figure S5A–S5C; Videos S4 and S5). We refer to these structures as cytDNA to distinguish them from mitochondrial and nuclear DNA. Over 15 hours, 2–5 cytDNA structures are generated per blastocyst, representing 2.9% ± 0.3% of all trophectoderm cells. Per time point, this equates to 1.3% ± 0.2% of trophectoderm cell nuclei per embryo (Figure 5D). We found a similar frequency of nuclear budding and DNA loss in cultured mouse embryos that did and did not undergo live-imaging, indicating no overt effects from the dyes or live-imaging (Figure 5E). Furthermore, nuclear buds are also detected in blastocysts flushed from the uterus 4 days post coitum (d.p.c.) and immediately fixed, suggesting no effects from culture itself (Figure 5E, Figure S5D). Therefore, these data suggest an additional process of DNA loss different from chromosome segregation errors during mitosis.
Figure 5. Identification of nuclear budding and DNA shedding.
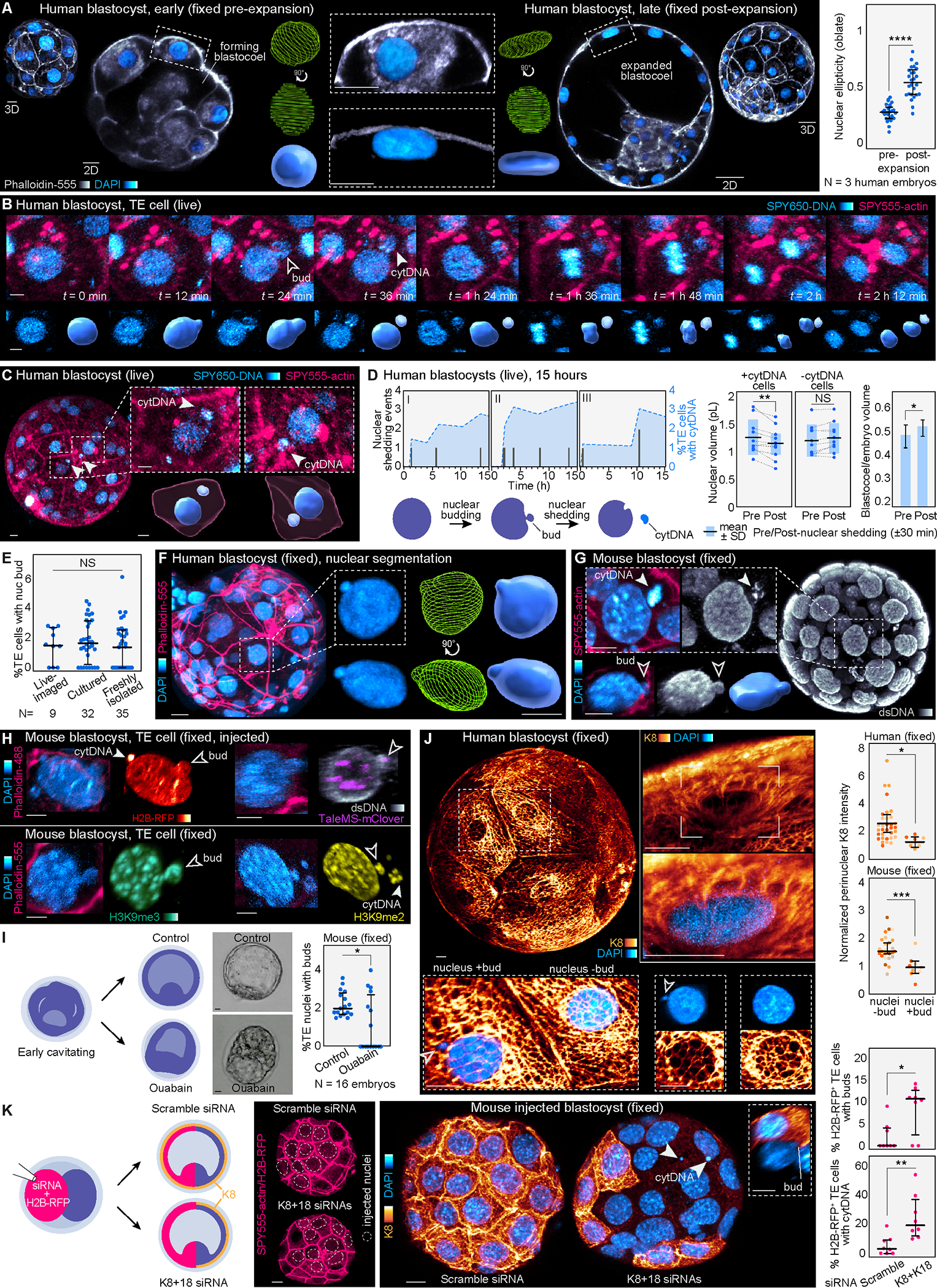
(A) Images of early and late human blastocysts stained for DAPI and Phalloidin-555. Nuclear segmentation highlights the change in nuclear morphology. Analysis of nuclear morphology in live embryos labeled with SPY650-DNA reveals flattening of trophectoderm nuclei in the expanded human blastocyst (N = 3 human embryos, n = 27 and 26 cells for pre- and post-expansion, respectively; ****P< 0.0001 by two-tailed unpaired t-test).
(B) Live-imaging in human blastocysts demonstrates the appearance of nuclear buds and cytDNA followed by cell division. Lower panels show the SPY650-DNA signal segmented.
(C) Images of live human blastocysts uncover SPY650-DNA–labeled structures within the cytoplasm in expanded blastocysts. Insets show the segmented SPY650-DNA and SPY555-actin signals.
(D) Quantification of nuclear DNA shedding events producing cytDNA in three human embryos over time. Analysis of nuclear and blastocoel volumes pre- and post-DNA loss. Note the reduction in nuclear volume following DNA loss. Neighboring control nuclei (without DNA loss) maintain the same volume (N = 11 DNA loss events in 3 human embryos; **P< 0.01, *P< 0.05, NS = not significant by two-tailed paired student’s t-test). Scheme depicts nuclear budding and cytDNA formation.
(E) Quantification of the percentage of trophectoderm cells with nuclear buds in mouse embryos dyed and live-imaged, cultured without dyes, and freshly isolated and immediately fixed at 4 d.p.c. (N = 9 live-imaged, 32 cultured and 35 freshly isolated embryos, NS = not significant by Kruskal-Wallis test).
(F) Computational segmentation of human trophectoderm nuclei showing nuclear buds.
(G) Detection of cytDNA structures and nuclear buds in an expanded mouse blastocyst fixed and stained with DAPI, and with antibodies against double-stranded DNA.
(H) Immunostaining showing the presence of histone H2B-RFP, the pericentromeric marker TaleMS-mClover, and histone modifications H3K9me2 and H3K9me3 in nuclear buds and cytDNA in trophectoderm cells of expanded mouse blastocysts.
(I) Treatment with ouabain prevents cavity expansion and reduces the percentage of trophectoderm cells with buds (N = 16 mouse embryos per group; *P< 0.05 by Mann-Whitney U test).
(J) Human blastocyst showing keratin network around most trophectoderm cell nuclei. Higher magnification images highlight the cage-like organization of keratin filaments surrounding the nucleus. Analysis of K8 fluorescence intensity shows lower perinuclear K8 levels in cells with nuclear buds (N = 3 human and 7 mouse embryos labeled by color, n = 33 and 7 cells (human) and n = 26 and 9 cells (mouse); ***P< 0.001, *P< 0.05, by Mann-Whitney U test).
(K) siRNAs for K8+K18 injected in half of the embryo disrupt the keratin network and cause an increased number of cells with nuclear buds and cytDNA compared to scramble siRNA-injected embryos (N = 8 embryos per group, **P< 0.01, *P< 0.05, by Mann-Whitney U test). Dashed circles show injected nuclei confirmed by H2B-RFP signal. Inset shows a nuclear bud in a knockdown cell.
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm
See also Figures S5–S7, and Videos S4–S6.
Following DNA shedding, cells containing cytDNA can undergo division (Figure 5B; Video S5). Similar to previous work tracking micronuclei in mouse embryos46, cytDNA structures are not recaptured by the mitotic spindle or reincorporated into the newly formed nuclei, but become inherited in the cytoplasm of daughter cells following division (Figure 5B). Furthermore, computational segmentation of cell nuclei (Figure 5F; Figure S5E) demonstrates a decrease in nuclear volume following DNA shedding, consistent with loss of nuclear material into the cytoplasm (Figure 5D). Human and mouse embryos stained with DAPI, Hoechst-33342 or antibodies recognizing double stranded DNA or histone 3 lysine 9 di- and tri-methylation (H3K9me2 and H3K9me3) confirms the presence of similar nuclear buds and cytDNA structures (Figure 5G and 5H; Figure S5F; Video S6). Moreover, they are also detectable in embryos injected with mRNA for H2B-RFP (Figure 5H; Figure S5F; Video S6) and TaleMS-mClover, which marks major satellite repeats enriched in pericentromeric chromosome regions47 (Figure 5H; Figure S5F). Whereas it is currently unfeasible to predict if nuclear budding and DNA loss involves whole or partial chromosomes, these results indicate that cytDNA structures can contain DNA, heterochromatin, and pericentromeric regions.
We next explore whether cytDNA structures are associated with programmed cell death (PCD), typically characterized by pyknotic DNA fragments48. These fragments are marked by cleaved caspase-3 and gradually cleared from apoptotic cells49,50. Immunofluorescence in mouse embryos shows cleaved caspase-3 staining surrounding cytDNA (Figure S6A and S6B). However, within cells containing cytDNA, 80 ± 13% of the nuclei remain intact and do not display pyknotic morphology or cleaved Caspase-3 staining (Figure S6A and S6B). To track these cells in real time, we use an active-caspase fluorescent reporter (CellEvent Caspase-3/7 Green), which demonstrates that a subset of cells generating cytDNA can activate caspase-associated signaling (Figure S6C). Analysis of the DNA damage marker γH2AX, typically associated with cell death in other systems, yields similar results to those obtained with caspase detection. A subset of cells in the preimplantation embryo display γH2AX signal in their nucleus, consistent with previous work51. However, cells with cytDNA do not display increased γH2AX staining in their nuclei, compared to cells devoid of cytDNA (6.0 ± 0.6% vs 6.2 ± 1.0%, P = 0.815) (Figure S6D and S6E). Together, these results show that cytDNA structures are not generated from chromosome segregation errors during mitosis, or produced by cells undergoing stereotypical PCD or extensive DNA damage. Instead, they bud from intact-appearing nuclei, can persist within the cytoplasm and be inherited following cell division.
Interestingly, DNA shedding is only observed in expanded blastocysts and not in earlier cavitating stages in both mouse and human (Figure 5C; Figure S5B). Recent work showed that when cultured cells migrate through mechanically confined microenvironments their nuclei deform causing transient ruptures52,53. Cells in the embryo also become mechanically constrained when the blastocoel expands and the trophectoderm layer flattens along its apical-basal axis (Figure 5A). Similar to nuclear rupture in cultured cells52,53, nuclear budding in the embryo occurs along the axis of mechanical stress (Figure S5G). Diminishing cavity expansion with ouabain, a Na/K-ATPase inhibitor40, decreases nuclear budding (Figure 5I) and points further to a link between mechanical stress and DNA shedding. At the stage of maximal blastocoel expansion, trophectoderm cells breach the zona pellucida during hatching. Once hatched, cells adopt a more spherical morphology, which is indicative of reduced mechanical stress4,54,55 (Figure S7A). Consistent with this, cells in the hatched region do not display increased nuclear budding compared to trophectoderm cells remaining within the zona pellucida (Figure S7B–S7E). Moreover, cell nuclei within the ICM do not undergo such pronounced morphological deformation and nuclear budding during blastocoel expansion is not observed (Figure S5H). We note however, that the number of ICM cells within the embryo is markedly lower than trophectoderm cells, thus precluding extensive imaging of ICM cells.
To further understand why a subset of trophectoderm cell nuclei are susceptible to nuclear budding during blastocyst expansion, we analyze the organization of the keratin filament network. Keratins confer cellular stability in various tissues experiencing mechanical stress4,56,57 and can mechanically support cell nuclei in various systems58,59. Furthermore, we previously showed that keratins stabilize the apical cortex of the trophectoderm6. Analysis of human embryos reveals that, in addition to the cortical keratin network, a peri-nuclear keratin network develops starting at the early cavitation stage (Figure S5I), which forms a prominent cage-like framework surrounding trophectoderm nuclei in expanded blastocysts (Figure 5J; Figure S5I). However, perinuclear keratin expression is heterogeneous throughout the trophectoderm and cells with cytDNA structures have lower peri-nuclear keratin levels (Figure 5J). Moreover, using siRNAs for K8 and K18 microinjected into one cell of 2-cell stage embryos disrupts the keratin network6 and causes an increase in nuclear budding and cytDNA formation, without affecting the number of caspase 3-positive cells (Figure 5K; Figure S6F–S6H). Thus, this indicates that trophectoderm cells with lower perinuclear keratin expression are more prone to DNA shedding in response to the mechanical stressors of blastocyst expansion. The peri-nuclear keratin cage develops in parallel to rising Lamin-A levels (Figure S5J), a filamentous protein important for nuclear structure60. In embryos overexpressing K8-Emerald + K18-Emerald, Lamin-A is significantly upregulated (Figure S5K). Furthermore, cells with cytDNA have lower Lamin-A levels compared to cells without cytDNA, consistent with results correlating lower peri-nuclear keratin levels with increased nuclear budding (Figure S5L; Figure 5J). Together these results indicate a role for these cytoskeletal components in protecting cell nuclei against budding, and further support the proposal that mechanical stress induces DNA shedding into the cytoplasm.
Finally, we investigate the relevance of mechanically-induced DNA shedding to assisted reproductive technologies (ART). Prior to transfer, PGT is widely used to screen for aneuploidy in blastocysts61. Testing requires trophectoderm biopsy, whereby a small number of cells are mechanically removed for sequencing. We thus employed the biopsy procedure as an additional mechanical stressor in both mouse and human embryos. Suction and traction were applied to the mural trophectoderm and prior to complete transection, biopsied cells were retained to track embryo-biopsy pairs (Figure 6A; Figure S7F; Video S7). Notably, trophectoderm cells in biopsied mouse embryos display an increased number of nuclear buds compared to cells in equivalently staged, non-biopsied hatching blastocysts (3.95% vs 1.65% TE cells, P < 0.0001, Figure 6B–6D). Furthermore, nuclear buds generated after biopsy could be tracked to shed cytDNA by live-imaging (Figure 6E). Nuclear buds proximal (<20 μm) to the point of biopsy were more likely to be oriented towards the biopsy pipette (<45°) compared to distal buds (>20 μm) (Figure S7G–S7I). This is compared to hatching control blastocysts (not biopsied), where there is no difference in bud angle comparing nuclei proximal and distal to the hatching site (Figure S7C–S7E), which suggests the mechanical stress of biopsy induces DNA shedding. Similar to mouse experiments, biopsy of human blastocysts results in a significant increase in nuclear budding (6.0% vs 0.70%, P = 0.0022, Figure 6F and 6G) with an equal distribution of nuclear buds between mural and polar trophectoderm (Figure S7J–S7K). Together, these results further demonstrate a link between mechanical stress and DNA shedding and suggest that in tested blastocysts, cytDNA could be found in both biopsied cells and those retained in the trophectoderm.
Figure 6. Trophectoderm biopsy causes nuclear budding.
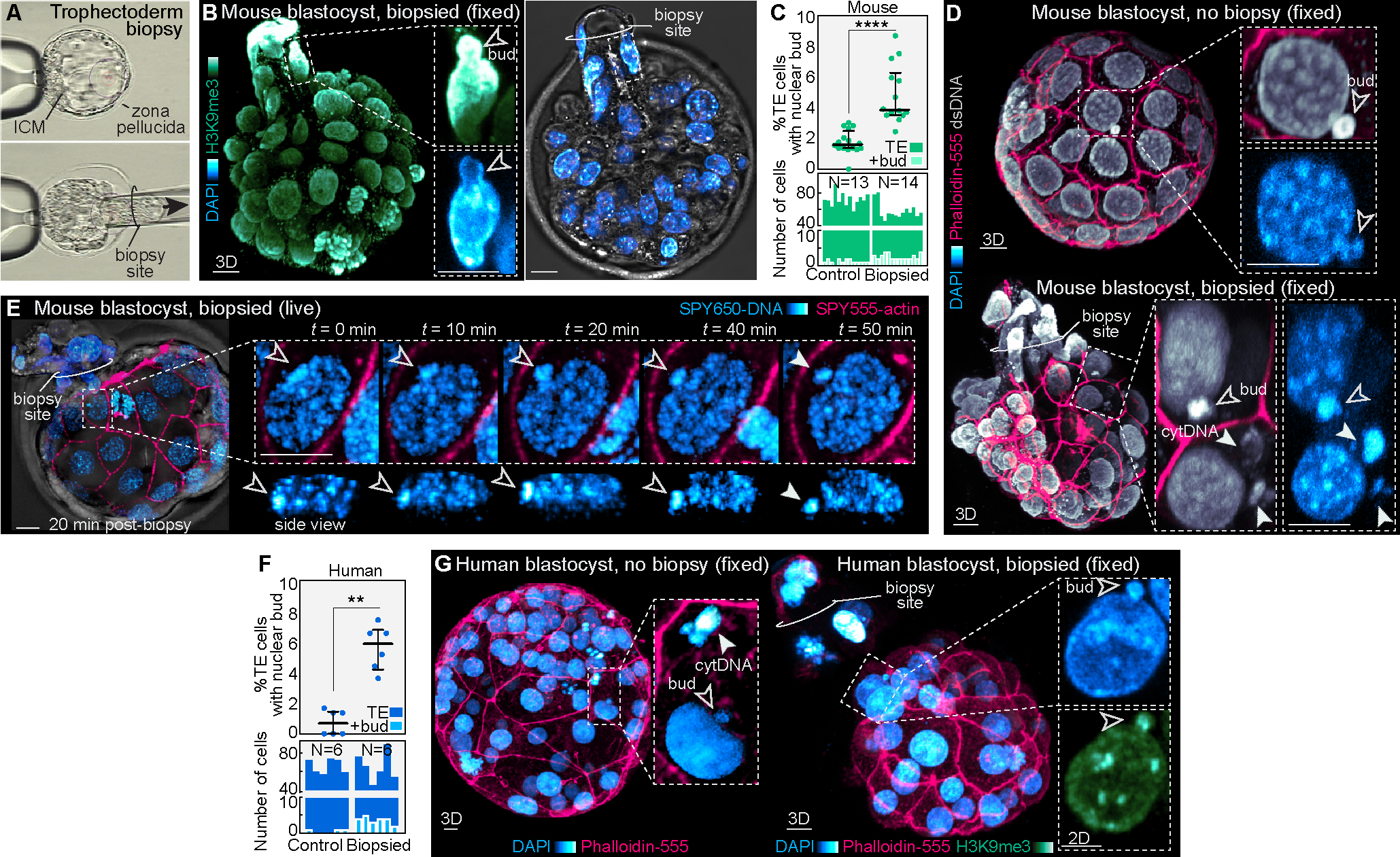
(A) Example of biopsy procedure performed on a mouse blastocyst.
(B) Representative image of a fixed biopsied embryo labeled for H3K9me3 and DAPI reveals nuclear buds.
(C) and (D) Analysis shows an increased percentage of trophectoderm cells with nuclear buds in biopsied versus non-biopsied mouse blastocysts, visualized by DAPI staining and immunofluorescence for double-stranded DNA. Dot plot of percent trophectoderm cells with nuclear buds above and bar graph depicting absolute number of trophectoderm cells with nuclear buds per embryo below. Each bar represents one embryo (N = 13 control and 14 biopsied mouse embryos; ****P< 0.0001 by Mann-Whitney U test).
(E) Live-imaging of a mouse blastocyst labeled with SPY650-DNA and SPY555-actin 20 min post-biopsy reveals the appearance of nuclear buds and generation of cytDNA structures.
(F) and (G) Analysis of biopsied human embryos reveal a similar increase in nuclear budding (N = 6 human embryos per group; **P< 0.01 by Mann-Whitney U test).
Graphs show median with interquartile range. Scale bars, 10 μm.
DISCUSSION
The main events driving preimplantation development have thus far been inferred form work in the mouse embryo62–64. The use of fluorescent dyes and live-imaging allows us to reveal some of the key cellular and morphogenetic processes patterning the preimplantation human embryo with high spatiotemporal resolution. Our data expose differences between human and mouse development, including features of the cell cycle, compaction, polarization, and inner-outer segregation. While the duration of mitosis is relatively conserved, interphase is longer in human, consistent with the longer course of human preimplantation development. Additionally, the timing of compaction differs with it preceding inner-outer lineage segregation18 in mouse, and occurring after inner-outer segregation in human. During this period cell nuclei in human embryos remain spatially closer during cytokinesis, without bouncing against the cell poles, as is the case in mouse7. This may explain why human cleavage-stage embryos display a more homogeneous enrichment of F-actin at the apical cortex following polarization, but lack the prominent F-actin rings found in mouse5. Furthermore, human blastocysts exhibit a greater number of F-actin projections between the trophectoderm and ICM compared to mouse. An outstanding question is whether these tethers mediate cell-cell signaling similar to cytonemes in Drosophila and stem cells65–67, or mechanical interactions similar to protrusions in the early embryo3,68. Thus, while the mouse remains an important model of preimplantation development, these findings reveal how key events in human development differ.
Our data also identify a process of nuclear DNA shedding. Recent studies have demonstrated transient ruptures of cell nuclei when cells are mechanically constrained by microfabricated chips, yet it remains to be determined if cell nuclei respond to similar stressors in vivo52,53. Our finding of nuclear budding and cytDNA formation show that some nuclei shed DNA into the cytoplasm in both mouse and human blastocysts, as trophectoderm cells become mechanically constrained during blastocoel expansion. Most trophectoderm nuclei are surrounded by a peri-nuclear keratin filament network, and cells with a lower peri-nuclear keratin density or microinjected with K8+K18 siRNAs display more nuclear buds and cytDNA structures. This indicates that the keratin peri-nuclear network mechanically protects the nucleus against DNA budding and shedding, consistent with functions found for other intermediate filaments in cultured cells69,70. The location of nuclear budding may not only be determined by the point of maximal mechanical stress, but also nuclear and peri-nuclear structure. Future studies will test whether specific nuclear regions, such as those with high curvature71, dense chromatin packing, or lower levels of keratins or lamins are more prone to undergo budding.
It remains technically unfeasible to track and isolate single trophectoderm cells producing cytDNA for sequencing. However, we could detect nuclear buds and cytDNA structures using markers for DNA, chromatin and pericentromeric chromosome regions. In addition, we measured a reduction in nuclear volume following DNA loss and show that cytDNA can be inherited by future daughter cells resulting in presumptive abnormal genomic complements (Figure 7). Our study also highlights that the best approach to follow this nuclear DNA shedding process and distinguish it from other events like chromosome segregation errors is via direct embryo live-imaging. Thus, we propose that DNA shedding from trophectoderm nuclei could represent an additional process accounting for mosaic aneuploidy, thus far linked to chromosome segregation errors in mitosis or cell death13,44,72–82. Currently, mosaic aneuploidy in human embryos is reported if 20–80% of tested cells are aneuploid61. Our results show DNA shedding in <5% of cells. Nevertheless, these live-imaging experiments only capture a sample of chromosome segregation errors and nuclear DNA shedding events, whereas mosaic PGT results reflect the cumulative effect of these events. Our data reveal DNA shedding similarly occurs in the mouse, yet it remains a common assumption that mouse embryos have a lower incidence of aneuploidy compared to human. However, spectral karyotyping reveals a 25% aneuploidy rate in mouse blastocysts, with 20% of them mosaic83, suggesting that the segregation errors and DNA shedding visualized here may contribute to mosaicism in both species.
Figure 7. Scheme of processes producing DNA loss in the embryo.
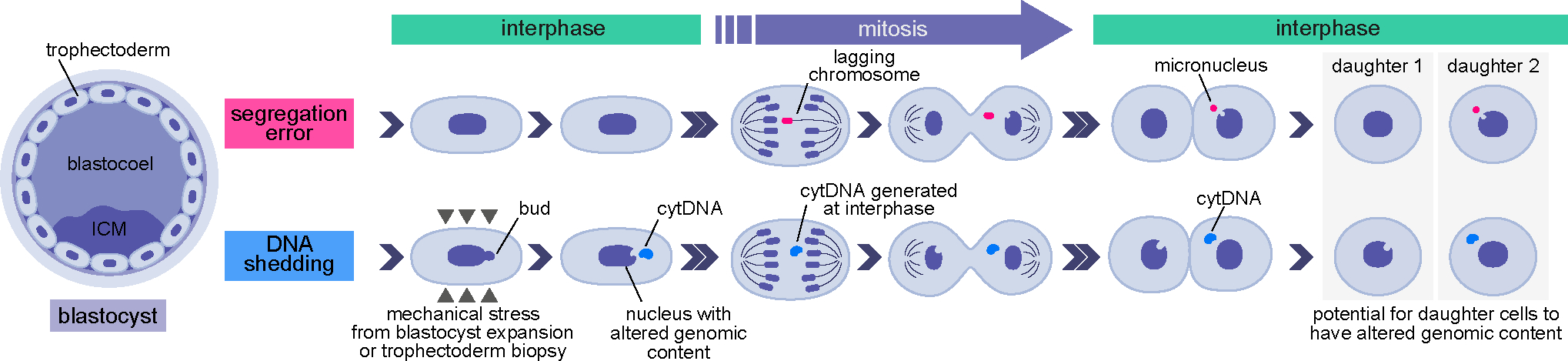
Chromosome segregation errors occurring during cell mitosis can frequently account for aneuploidy in the preimplantation embryo. Our data show that cell nuclei can also shed DNA into the cytoplasm during interphase, as a consequence of mechanical stress experienced during blastocyst cavity expansion or biopsy. Following subsequent cell divisions, cells with chromosome segregation errors or DNA shedding may produce progeny with abnormal genomic contents.
Our results open the question of whether cytDNA influences cell function or has detrimental effects on embryo quality. cytDNA is detected by caspases, yet these cells do not undergo stereotypical PCD or display higher levels of DNA damage. Instead, cytDNA can persist within the cytoplasm and become inherited by daughter cells following cell division. Thus, instead of eliminating cells with cytDNA, the blastocyst maintains trophectoderm integrity. Furthermore, cytDNA could have functions within the cytoplasm, such as recruitment of caspases to trigger non-apoptotic signaling, as found during caspase-dependent cell shape changes in Drosophila84. We also do not exclude the possibility of downstream mechanisms eventually depleting these cells from the trophectoderm, such as relative dilution via increasing cell cycle duration, as proposed for aneuploid cells in the trophectoderm85,86.
PGT for aneuploidy is widely used as a proxy for embryo quality prior to transfer61. Consistent with our proposal that trophectoderm nuclei can release DNA as a result of mechanical stress, we demonstrate that trophectoderm biopsy itself increases DNA budding and shedding from cell nuclei. Although the fate of these cells remains to be determined, these results show that mechanical stress, both physiologic (blastocoel expansion) and induced (trophectoderm biopsy) alters nuclear structure. Moreover, the exponential adoption of IVF worldwide1 calls for a comprehensive understanding of how embryo manipulations may alter subcellular processes. Finally, as the imaging approach established here can bypass the requirement for microinjection or genetic manipulation to fluorescently label cells, it may be applied in the future to determine how the cellular and morphogenetic processes that pattern the human embryo confer developmental potential and thus enable non-invasive prediction of embryo quality.
Limitations of the study
Here, we provide an analysis of preimplantation human embryos at cellular resolution. Mouse embryos can be readily isolated at any stage of development and the availability of hundreds of well-defined genetic strains significantly reduces embryo-to-embryo variability. By contrast, a primary challenge in studying human embryos is the scarcity of early cleavage-stage samples and their substantial embryo-to-embryo heterogeneity. While freezing embryos at the cleavage stage was previously common protocol in IVF clinics, in 2013 it became recommended practice to freeze and transfer at the blastocyst stage87. Hence, human cleavage stage embryos have become especially difficult to obtain. This limits our ability to perform extensive analyses of the processes regulating patterning of the human embryo prior to the blastocyst stage.
We also show that the use of fluorescent dyes can bypass the need for genetic-based manipulations to fluorescently mark specific cellular structures in live embryos. Yet, only a handful of fluorescent dyes may be available and suitable for live-embryo imaging, limiting the number of structures that can be investigated.
Finally, we demonstrate that trophectoderm nuclei undergo budding leading to DNA shedding into the cytoplasm. It remains experimentally challenging to determine whether the shed DNA constitutes random sequences, specific chromosomes, or genomic regions more prone to undergo shedding. This could be explored in future studies by combining live-embryo imaging to identify cells undergoing DNA shedding, followed by laser capture of these cells or their progeny to analyze their genetic and epigenetic contents. Along the same line, our study is limited to defined imaging windows ranging in the order of 12 to 24 hours to minimize photodamage. Advances in imaging detection requiring lower intensities may enable longer cell tracking in vivo. Combined with light-inducible marking of cells undergoing chromosome segregation errors or DNA nuclear shedding, this would allow tracking the fate of these cells, their progeny, and their genetic contents following implantation.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nicolas Plachta (Nicolas.Plachta@pennmedicine.upenn.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data supporting the findings of this study are available from the corresponding author on reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mouse embryo work
Mouse embryo experimentation was approved by the Biological Resource Center Institutional Animal Care and Use Committee (IACUC Protocol 806983). Superovulated wild-type female mice (Hsd:NSA(CF-1)) were used following animal ethics guidelines of the University of Pennsylvania (PA, USA). Embryos were flushed from oviducts with M2 medium (Merck) and cultured in KSOM+AA (Merck) at 37°C and 5% CO2 covered by mineral oil (Sigma). Five-week-old female mice were superovulated using 5 IU of Pregnant Mare Serum Gonadotropin (PMSG, Prospec) followed by 5 IU of recombinant human Chorionic Gonadotropin (CG, Sigma) given intraperitoneally 46 h after and immediately before mating. Two-cell stage embryos were flushed from oviducts with M2 medium (Merck) and cultured in KSOM+AA (Merck) covered by mineral oil (Sigma), at 37 °C and 5% CO2. Rates of blastocyst formation were analyzed by staining embryos at the 8-cell stage with the two dyes and quantifying the number of embryos progressing to the blastocyst stage following 48 h, compared to non-dyed controls. For ouabain treatment, early cavitating blastocysts were treated with 300 μM ouabain or vehicle for 18 hours followed by fixation. For some experiments, blastocysts were isolated from the uterus at 4 d.p.c. and fixed for analysis.
Human embryo work
Discarded human blastocysts were donated for research under determinations by the New England institutional review boards (WO 1–6450-1). All samples were de-identified prior to the thawing process. Discarded embryos were determined as “Not Human Subjects Research.” All human embryo experiments were performed at Boston IVF, Waltham MA. Previously vitrified embryos were thawed according to the manufacturer’s protocol (90137-SO—Vit Kit-Thaw, FUJIFILM Irvine Scientific, USA) and cultured for 1 h in individual drops of 75 μl of Continuous Single Culture Complete (CSC) media with human serum albumin (HSA) (FUJIFILM Irvine Scientific, USA), covered with mineral oil in an incubator at 37°C, 7% CO2 and 6% O2 until further staining. All human embryo experiments were performed under private funding by Boston IVF and iGenomix.
METHOD DETAILS
Non-Surgical Embryo Transfer (NSET)
Mouse blastocysts were transferred to CF1 pseudopregnant females via NSET using the NSET Non-Surgical Embryo Transfer Device (ParaTechs Corporation) according to the manufacturer’s description. All embryos used in the NSET experiments were obtained from B6SJLF1/J × Hsd:NSA(CF-1) mating and therefore had a mixed genetic background as shown by a different color coat of born pups. The embryos were isolated at the 2-cell stage and cultured in vitro for 72 hours before the transfer. Twenty-four hours prior to transfer the embryos from the experimental group were stained with 1:2,000 SPY650-DNA and SPY555-actin for 1 hour, imaged for 1 hour to assess signal and washed. Each recipient female received 18–22 blastocysts from either the stained (experimental) or unstained (control) group. Carrier embryos were not used. After the embryo transfer, the foster mothers were allowed to deliver full-term pups.
Trophectoderm biopsy
Embryos were collected at the 2-cell stage and cultured for 3 days. Those reaching the blastocyst stage without significant fragmentation or degeneration were biopsied four days after fertilization. Briefly, a blastocyst was positioned in the holding pipette and a laser pulse (Zilos-tk, UK) was used to generate an opening in the zona pellucida (ZP) away from the inner cell mass. The extraembryonic cells were allowed to herniate through the ZP and then mechanically partially removed with a biopsy needle and laser pulses between cell junctions. Biopsied segments remained connected to the trophectoderm proper for imaging purposes. Embryos were either cultured for an additional 30 minutes to allow re-expansion and then fixed or dyed for live imaging. All procedures were done using Multipurpose Handling Medium Complete (MHM-C, Irvine Scientific) with Gentamicin, under gas mineral oil. For image analysis, only blastocysts with >50 trophectoderm nuclei were included. Discarded human blastocysts were similarly biopsied using the procedure and materials above.
DNA and mRNA
DNA constructs were cloned into a pCS2+ vector for mRNA production. The mMESSAGE mMACHINE® SP6 kit (Ambion) was used to synthesize RNA using linearized plasmids as templates then purified using the RNAeasy kit (Qiagen) following the manufacturer’s instructions. Embryos were microinjected with 0.1 to 0.3 pL RNA diluted in injection buffer (5 mM Tris, 5 mM NaCl, 0.1mM EDTA) with a FemtoJet (Eppendorf) as follows: 10 ng μl−1 H2B-RFP5; 5 ng μl−1 H2B-GFP5; 75 ng μl−1 Utrophin-RFP6; 300 ng μl−1 K8-Emerald and K18-Emerald6; 100 ng μl−1 TaleMS-mClover47.
siRNAs (Qiagen)
Mm_Krt2–8_1 (AACCATGTACCAGATTAAGTA, 200 nM)
Mm_Krt2–8_2 (ATGGATGGCATCATCGCTGAA, 200 nM)
Mm_Krt1–18_1 (CAGAGTGGTGTCCGAGACTAA, 200 nM)
Mm_Krt1–18_3 (CCGGGAACATCTGGAGAAGAA, 200 nM)
AllStars negative control siRNA (Sequence undisclosed by Qiagen, 800 nM)
Live embryo imaging
Live mouse and human embryos were stained with the live cell stain probes SPY555-Actin (1:2,000, Spirochrome), SPY650-DNA (1:1,000, Spirochrome), SPY555-DNA (1:1,000, Spirochrome), SPY650-FastAct (1:2000, Spirochrome), CellEvent Caspase-3/7 Green Detection Reagent (1:1,000, Invitrogen) for 1–2 h depending on the embryo stage, then imaged in a 1:5,000–10,000 solution in KSOM. Human embryos were left to recover and re-expand for 1–2 h after thawing. Staining was performed for 1 hour at the 8-to-16 cell for cleavage stage studies and for 2 hours at morula or early blastocyst stage for blastocyst formation and hatching analyses. For imaging, embryos were cultured in μ-Slide 8/18 Well Glass Bottom (Ibidi) at 37 °C and 5% CO2 in an incubator adapted for the microscope system (Leica SP8 or Nikon A1RHD25). Mouse embryos were imaged using a laser scanning confocal (Leica SP8) with water Apochromat 40X 1.1 NA objectives and highly sensitive HyD detectors (Leica). Human embryos were imaged on a Nikon A1RHD25 point scanning confocal microscope in the Nikon BioImaging Lab (NBIL, Cambridge MA). Confocal three-dimensional (3D) scans of the embryos were performed at 5–15 minute intervals for 20–40 hours.
Staining and immunofluorescence
For immunostaining, embryos were fixed with 4% paraformaldehyde in DPBS-0.1% Triton X-100 for 30 min at room temperature, permeabilized in DPBS-0.5% Triton X-100 for 30 min, incubated in blocking solution (2% bovine serum albumin in DPBS-0.1% Triton X-100) for 1 h and incubated with primary antibodies in blocking solution overnight at 4°C: dsDNA (1:1,000, Abcam), Keratin-8 (1:100, DSHB), Lamin A (1:200, Santa Cruz Biotechnology), cleaved Caspase-3 (1:200, Cell Signaling), γH2A.X (Ser139) (1:200, Cell Signaling), H3K9me2 (1:500, Abcam), H3K9me3 (1:700, Abcam). Embryos were rinsed in DPBS and incubated with secondary Alexa Fluor 488 or 647 conjugated antibodies (Invitrogen) in blocking solution (1:1,000) for 2 h. To label F-actin, fixed embryos were incubated with Phalloidin-Rhodamine (1:500, Molecular Probes), Phalloidin-Alexa Fluor 555 (1:500, Invitrogen), Phalloidin-Alexa Fluor 488 (1:500, Invitrogen) or SPY555-Actin (1:1,000, Spirochrome), and for nuclear staining with DAPI (Sigma) or Hoechst-33342 at 1:1,000.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis and measurements of cell and nuclear volume and position
3D visualizations of embryos were performed using Imaris 9.7 software (Bitplane AG). The Gaussian filter was applied prior to analysis on human embryos included in Figure 5G, Figure S4E and S4F. The manual surface rendering module was used for cell segmentation. Cell and nuclear volumes and shape were derived from the segmented data using the Imaris statistics module. Nuclear ellipticity (oblate) was used to evaluate nuclear flattening during blastocyst expansion. Nuclear sphericity was used to measure nuclear rounding during hatching; nuclei from the hatched area were compared to those of unhatched trophectoderm cells withing the same embryo. To assess nuclear volume after DNA loss, the same nucleus was tracked 30 min before and after a nuclear bud was visualized. Nuclear budding was defined as a prominent deformation of the cell nucleus leading to the formation of a protrusion. For each nuclear budding event, a neighboring nucleus that did not undergo DNA loss was analyzed at the same time points to serve as a control. The ratio of blastocoel-to-embryo volume was used as a proxy for cavity expansion. Analysis of nuclear position relative to the cytokinetic furrow was performed at the stage of compaction and polarization for mouse (8-cell stage) and human (16-cell stage) embryos using Imaris. A 5 μm thick plane in the direction of cell division was used to visualize both nuclei of the mitotic pair, their cell poles and the cytokinetic furrow. The distance from the nucleus center to the cytokinetic furrow was measured at telophase in the division plane. An additional measurement to the cleavage point was performed 1 hour after telophase. In order to compare mouse and human nuclear positioning, distances were normalized to the maximum distance from the cell pole to the cytokinetic furrow. This division plane was also used to determine cell division orientation by measuring the angle between the line connecting the center of mass of the embryo and the midpoint between the two daughter cells. For the analysis of inner-outer cell position, cells were computationally segmented and tracked over time. Outer cells were defined following previous criteria4 as those having a portion of their surface exposed to the outer zona pelucida. Inner cells were defined as those having their entire surface covered by other cells. As cells internalized, they gradually decrease their apical surface area until they are defined as an inner cell4. Tethers connecting the ICM to the trophectoderm at the blastocyst stage were visualized and measured in 3D in Imaris. Only blastocysts displaying SPY555-actin positive signal and a good signal-to-noise ratio were analyzed. In order to compare the length of TE-ICM tethers between mouse and human, lengths were normalized to the maximum diameter of the embryos.
Quantification of fluorescence intensities
Following segmentation, the Imaris statistics module was used to obtain values for total and mean fluorescence intensities, cell volumes, and nuclear volumes. The fluorescence intensity profiles for Phalloidin-555 were measured in Fiji using a line width of 25 pixels along the apical cortex. The distance between cell-cell junctions was normalized and divided into thirds for comparison. For keratin and lamin A fluorescence, intensity is reported as a normalized ratio to nuclear DAPI/Hoechst fluorescence to correct for weaker intensity with increasing depth through the embryo. Normalized perinuclear keratin fluorescence was then compared between cells that displayed nuclear buds to cells with intact nuclei.
Cell cycle measurements
Total cell cycle duration in Figure 1B was obtained by measuring the time between a cell division to the next division of one of the resulting daughter cells in dyed and control (non-dyed) 4-to-8 cell embryos. Brightfield was used to identify cytokinesis in both dyed and non-dyed embryos. The duration of interphase was measured by tracking a cell dividing in a live embryo and then following one or both daughter cells throughout their entire interphase until the next mitosis. Nuclear, chromatin and cell morphologies were used to identify interphase start after the previous telophase and interphase end just before the next prophase. Cells already in interphase at the start of the imaging session were excluded for these analyses as this would have yielded falsely shorter interphase times. Mitotic phases were measured in regions of embryos imaged at higher magnification to clearly visualize the dynamic changes in chromatin organization that distinguish interphase, prophase, metaphase, anaphase, telophase and cytokinesis. Mitosis duration was measured from the first time point displaying a prophase morphology until telophase and cytokinesis were completed. Cell cycle measurements factor in a ± 10 min inbuilt error due to temporal resolution during image acquisition.
Quantification of cell-cell contact, sphericity and angle between apical membranes
The cytoplasm of all cells in an embryo were segmented and converted into 3D meshes of approximately evenly spaced vertices. For each vertex in each cell the distance between it and the vertices of neighboring cells was calculated. If the distance between any two vertices on the surface of neighboring cells was below a threshold of 2 μm these points were considered to be in cell-cell contact. The percentage of points in cell-cell contact was then calculated for each cell. The volume and surface area of these meshes were also used to calculate the sphericity of each cell. The angle between bordering apical membranes was measured as previously described4. Intercellular and adjacent apical membranes were traced using two straight lines and the angle between these lines calculated in Imaris. Compaction was defined using previously established criteria3,4.
Analysis of nuclear bud formation
cytDNA structures were segmented and the value of all pixels outside of these segmented regions set to 0. The apical-basal axis of each cell was approximated as a line that passes through the center of mass of the embryo and the center of mass of the cell’s nucleus. A 2D slice along this axis was then taken through the embryo and rotated around the apical-basal axis by 180° in 1° increments to produce a stack of images. This stack of images was then collapsed into a single image producing a ‘rotational maximum projection’ of cytDNA structures in the perinuclear space. Images from multiple nuclei were than combined creating a heatmap of where cytDNA structures form with respect to the cells apical-basal axis. Nuclear bud angles were measured in Imaris using measurement points. A line was drawn from the nuclear center of mass to the center of biopsy or hatching point and defined as 0°. A second line was then drawn from the nuclear center of mass to the nuclear bud. From this, the angle between lines and distances were measured.
Statistical analyses
Statistical analyses were performed in Excel and GraphPad Prism. Data were analyzed for normality using a D’Agostino-Pearson omnibus normality test. Variables displaying a normal distribution were analyzed using an unpaired, two-tailed Student’s t-test for two groups, and ANOVA with Dunnett’s multiple comparisons test for more than two groups. Variables that did not follow a normal distribution were analyzed using an unpaired, two-tailed Mann-Whitney U test for two groups, and Kruskal-Wallis test with Dunn’s multiple comparisons test for more than two groups. Correlation was analyzed with a two-tailed Pearson’s Correlation test. Reproducibility was confirmed by independent experiments. Graphs show median ± interquartile range.
Supplementary Material
Video S1. Live imaging of mouse embryos, related to Figure 1.
Combining the use of SPY650-DNA and SPY555-actin enables live imaging with high spatiotemporal resolution in live mouse embryos. These examples show: (1) Cell divisions during the 2-to-4 cell stage. The imaging spatiotemporal resolution allows us to capture the main phases of mitosis and formation of the cytokinetic furrow. (2) Compaction at the 8-cell stage. SPY555-actin allows visualization of the main changes in cell shape; note the flattening of the cells against each other during the compaction process. (3) Formation of F-actin rich apical domains at the 8-to-16 cell stage. Note the disassembly of the apical domains prior to cell division. (4) Formation of F-actin rings during the 16-cell stage. Imaging over time shows the gradual expansion and subsequent zippering of these rings along cell-cell junctions across most cells of the embryo. (5) Formation of the blastocoel at the 32-cell and expansion of the blastocyst. Note the periodic collapses in blastocoel volume. Cell divisions across the trophectoderm layer can be observed during the expansion process.
Video S2 Live imaging of F-actin rings formation the 16-cell stage mouse embryo, related to Figure 1 and Figure S2.
Live mouse embryo shows formation, expansion, and zippering of F-actin rings during the 16-cell stage labeled by SPY650-FastAct, until the next round of mitosis.
Video S3. Live imaging of cell divisions at the cleavage and blastocyst stage, related to Figures 2 and 4.
Live imaging of a cell division in a mouse and human cleavage stage embryo stained with SPY555-DNA and SPY650-FastAct. In mouse, the cell nucleus bounces against the apical cell cortex of the prospective outer cell at the end of cytokinesis. By contrast, in human, cell nuclei remain closer to the cytokinetic furrow without bouncing against the cortex. The use of SPY650-DNA and SPY555-actin also allows live imaging of cell divisions within the human blastocyst. The main phases of mitosis can be captured in this trophectoderm cell.
Video S4. Live imaging of the human blastocyst, related to Figure 4 and Figure 5.
Example of live imaging in a human blastocyst shows cell divisions across the trophectoderm. Arrowheads identify the appearance of cytoplasmic DNA structures in a subset of trophectoderm cells.
Video S5, Live imaging of nuclear budding and DNA shedding in the human trophectoderm, related to Figure 5.
Live imaging demonstrates the formation of a nuclear bud with subsequent loss into the cytoplasm of a trophectoderm cell in a live human blastocyst. The second part of the movie shows computational segmentation of the DNA and actin signals to highlight the loss of DNA. In this example the cell undergoes mitosis, and one daughter cell inherits cytDNA.
Video S6, Chromatin markers in nuclear buds, related to Figure 5.
Three-dimensional rotations show a structure budding from cell nuclei labeled with H3K9me3 and H2B-RFP in mouse trophectoderm cells.
Video S7. Trophectoderm biopsy in mouse blastocysts, related to Figure 6.
Example of the trophectoderm biopsy process in a mouse blastocyst.
Figure S1. Additional characterization of fluorescent dyes, related to Figure 1.
(A) Brightfield images corresponding to the embryos shown in Figure 1A.
(B) Incubation of live mouse embryos with SPY650-DNA and SPY555-actin followed by washing of the dyes produces similar labeling patterns to those found in live embryos microinjected with mRNA for H2B-GFP and Utr-RFP, or fixed embryos stained with DAPI and Phalloidin-Rhodamine.
(C) and (D), Liveborn pups derived from control (unstained) and embryos stained with SPY650-DNA and SPY555-actin following in utero transfer (NS = not significant by two-tailed unpaired student’s t-test).
(E) 2D planes showing SPY555-actin and SPY650-FastAct signal in 16-cell stage live mouse embryos. This allows for the identification of inner cells.
(F) Computational segmentation of the 16-cell stage embryo shown in Figure 1G. Cell contours were defined by the Phalloidin-555 signal. The embryo is shown as a 3D projection and brightfield image. A representative z-plane (0.4 μm slice) is shown with the cell contours of an outer cell overlaying the Phalloidin-555 signal.
(G-I) Representative image of a single slice through the center of the embryo stained with Phalloidin-555 shown in Figure 1G and S1F. Segmentation allows identification of inner versus outer cells. The series of images along the z-plane are projected to create three-dimensional segmentation of cell contours.
Scale bars, 10 μm.
Figure S2. Embryos dyed with SPY555-DNA and SPY650-FastAct display normal actin ring expansion and zippering, related to Figure 1.
(A) Live-imaging of a 16-cell stage mouse embryo with SPY555-DNA and SPY650-FastAct displaying formation and expansion of actin rings, and zippering along the cell-cell junctions. Note that the dyes allow for normal morphological changes including actin ring expansion and zippering along cell-cell junctions, indicating no overt effects on actin dynamics.
(B-D) High magnification images of three pairs of cells at selected time points. 2D sections show actin rings and zippering as bright fluorescence regions along the cell cortex and at the cell-cell junctions. </p/> Scale bars, 10 μm.
Figure S3. Compaction and early inner cell allocation in human embryos, related to Figure 2.
(A) Brightfield images of a human embryo at different stages of preimplantation development (2-cell, 8-cell, 16-cell, compacted morula and expanded blastocyst). An inner cell can be seen at the 16-cell stage, before the initiation of compaction (arrowhead).
(B) Schematic diagram of Newton’s kissing number problem. At the 12-cell stage, at least one blastomere must occupy an inner position to maintain the spherical organization and cell-cell contacts of the embryo. In three-dimensions, this dictates that outer cells align along the vertices of a regular icosahedron.
(C) Brightfield images and schematic displaying morphological differences between mouse and human embryos at the 8- and 16-cell stages. Note that at the 16-cell stage, the mouse embryo is completely compacted. Human embryos have not yet completed compaction and contain spherical blastomeres. Embryos must contain 2–3 inner cells at the 16-cell stage.
(D) Analysis of nuclear position following division in live cleavage stage embryos. Distance of the nucleus to the cytokinetic furrow/cleavage point is measured for each mitotic pair 1 hour after telophase and normalized to the maximum distance from the cell pole to the cytokinetic furrow (N = 7 mouse and 3 human embryos, n = 46 and 30 cells for mouse and human, respectively; ****P< 0.0001 by two-tailed unpaired student’s t-test). SPY555-DNA and SPY650-FastAct live staining show nuclear positioning 1h after telophase. Note that an additional time point after telophase is shown from embryos in Figure 2G.
(E) Radial histogram of human morulae showing the number of divisions (x-axis) versus division angle relative to the center of mass of the embryo (y-axis) (n = 16 divisions, N = 3 human embryos). Division planes with angles ≤45° to the center of mass are classified as asymmetric (inner-outer progeny), while angles >45° are classified as symmetric (outer-outer progeny).
(F) Live-imaging of human cleavage stage embryos stained with SPY555-DNA and SPY650-FastAct allows for tracking of inner-outer and outer-outer progeny over time. 2D sections show the trophectoderm fate of outer cells and the ICM fate of inner cells in the early blastocyst.
(G) Live-imaging with SPY555-DNA and SPY650-FastAct reveals apical constriction in the human embryo. 2D sections of a cleavage stage embryo show how a newly generated outer cell reduces its apical external contact, gets internalized and becomes an inner cell. Computational segmentation of the SPY650-FastAct signal showing the outer undergoing apical constriction and becoming an inner cell. The top views highlight the reduction in apical surface.
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S4. Analysis of blastocyst structures and chromosome segregation errors during mitosis in the living embryo, related to Figure 3 and Figure 4.
(A) Live-imaging of a human blastocyst stained with SPY650-DNA and SPY555-actin. 2D sections of the embryo displayed in Figure 3A show that some of the SPY555-actin dye is retained in the zona pellucida. Note that as the zona pellucida thins down with blastocyst development, trophectoderm cells and cell-cell junctions improve their staining.
(B) Live-imaging of a mouse early blastocyst stained with SPY650-DNA and SPY555-actin. Note that the SPY555-actin signal is barely detectable in the murine zona pellucida, and cell-cell junctions can be detectable early on.
(C) 2D plane through a live mouse blastocyst stained with SPY650-DNA and SPY555-actin reveals tether-like structures projecting between the trophectoderm and ICM similar to those observed in human.
(D) and (E) Quantification of the number and length of TE-ICM tethers shows that they are more frequent and longer in human blastocysts (N = 7 mouse and 3 human embryos; ****P< 0.0001, *P< 0.05 by two-tailed unpaired student’s t-test). Tether length is normalized to embryo diameter.
(F) Examples of blastocyst collapse and expansion. Quantification of trophectoderm mitoses at different time points before and after embryo collapse (N = 10 collapse events in 6 mouse embryos; NS = not significant by Kruskal-Wallis test).
(G) Analysis of cell divisions relative to embryo collapse events (probed by measuring embryo volume) in different mouse embryos.
(H) Live-imaging with SPY650-DNA and SPY555-actin showing that the micronucleus formed in the human trophectoderm cell shown in Figure 4G can still be detected hours later. The cell is shown in 3D top and 2D sectional views with magnified images of the cell and surface segmentations of the DNA signal.
(I) Live-imaging in mouse 8-cell embryos microinjected with mRNA for the centromere marker TaleMS-mClover and stained with SPY650-DNA allow the identification of lagging chromosomes during mitosis.
(J) Live-imaging in mouse blastocysts with SPY650-DNA and SPY555-actin showing the formation of micronuclei in trophectoderm cells.
(K) Quantification of the number of mitoses with lagging chromosomes relative to the total number of mitotic events analyzed in mouse blastocysts (N = 8 mouse embryos).
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S5. Additional characterization of nuclear buds and cytDNA structures, related to Figure 5.
(A) Live-imaging of mouse blastocysts show the appearance of nuclear buds and cytDNA. Lower panels show SPY650-DNA signal segmented.
(B) Images of live mouse blastocysts reveal SPY650-DNA–labeled structures within the cytoplasm. Smaller insets show segmented embryos highlighting the increase in cavity volume at the advanced stage.
(C) 1 μm-thick planes in both xy and xz show nuclear shedding events in live mouse and human embryos (shown in 3D in Figure 5B and Figure S5A).
(D) Bar graphs of number of trophectoderm cells and cells with nuclear buds in cultured and in 4 d.p.c. freshly isolated, fixed mouse embryos (shown in Figure 5E, N = 32 cultured and 35 freshly isolated embryos). Each bar represents an individual embryo.
(E) Confocal image stack showing each 2D at 0.5 μm interval. Note the presence of the bud in the middle sections originating from the nucleus.
(F) Immunostaining for dsDNA in combination with histone modifications H3K9me2 and H3K9me3, histone H2B-RFP and the centromere marker TaleMS-mClover in advanced mouse blastocysts. 400 nm-thick planes in both xy and xz show the presence of these chromatin markers in nuclear buds and cytDNA structures of trophectoderm cells.
(G) Segmentation and analysis of nuclear bud position relative to the embryo’s center of mass shows that buds are produced along the xy axis (i.e., orthogonal to the apical-basal axis of the cell).
(H) Analysis of nuclear morphology in live embryos labeled with SPY650-DNA shows that ICM cell nuclei do not undergo significant morphological change during blastocyst expansion (N = 3 human embryos, n = 16 and 19 cells for pre- and post-expansion, respectively; NS = not significant by two-tailed unpaired t-test). Quantification of nuclear ellipticity (oblate) serves as a proxy for flattening.
(I) Staining for keratin-8 in the early and late human blastocyst. Note the prominent keratin tethers connecting the nucleus to the cortex at the early stage. As the blastocyst expands and the trophectoderm cell flattens, the keratin network forms a cage around the nucleus.
(J) Some trophectoderm cells in the mouse blastocyst display lower keratin levels. Perinuclear K8 levels strongly correlate with lamin-A levels (N = 8 mouse embryos, n = 73 cells; r = 0.69, ****P< 0.0001 by two-tailed Pearson’s correlation). 2D sections reveal higher perinuclear K8 in nuclei with higher lamin-A levels.
(K) Representative image of an embryo after injection with K8-Emerald and K18Emerald at the 2-cell stage. Embryos were then fixed at the 8-cell stage and stained for lamin-A, with fluorescence intensity normalized to DAPI. Cells with K8/K18 overexpression show higher lamin-A levels compared to uninjected cells (N = 7 embryos, n = 29 uninjected cells, 26 injected cells. **P < 0.01 by Mann-Whitney U test).
(L) Trophectoderm cells with cytDNA display lower levels of lamin-A (N = 7 mouse embryos, n = 60 control and 9 +cytDNA cells; **P < 0. 01 by two-tailed unpaired student’s t-test).
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S6. Analysis of caspase activity and γH2AX, related to Figure 5.
(A) Immunostaining for cleaved Caspase-3 with SPY555-actin and DAPI in fixed mouse blastocysts. Example of trophectoderm cell showing cleaved Caspase-3 surrounding a cytDNA structure. Note that the corresponding cell nucleus remains intact and does not display cleaved Caspase-3 staining (top panel). By contrast, the bottom panel shows an example of a pyknotic nucleus recognized by cleaved Caspase-3 undergoing PCD in the ICM.
(B) Quantification of cytDNA structures positive for cleaved Caspase-3 in cells displaying either an intact or pyknotic nucleus (N = 5 mouse embryos; **P< 0.01, *P< 0.05 by Mann-Whitney U test).
(C) Live-imaging of mouse blastocysts with SPY650-DNA, SPY555-actin and CellEvent Caspase-3/7 Green reveals caspase activity following the formation of cytDNA.
(D) Immunostaining for γH2AX and dsDNA in advanced mouse blastocysts highlighting the presence or absence of γH2AX in cells displaying nuclear buds and cytDNA.
(E) Analysis of nuclear γH2AX intensity in cells displaying or not cytDNA structures (N = 5 mouse embryos per group; NS = not significant by Mann-Whitney U test).
(F) Immunostaining for cleaved Caspase-3 and Hoechst in scramble and K8+K18 siRNAs injected embryos.
(G) and (H) Analysis of cleaved Caspase-3 positive cells and pyknotic nuclei after K8+K18 downregulation in mouse blastocysts (N = 11 control and 21 K8+K18 siRNAs embryos; NS = not significant by two-tailed unpaired student’s t-test).
Scale bars, 10 μm.
Figure S7. Nuclear budding during blastocyst hatching and trophectoderm biopsy, related to Figure 5 and Figure 6.
(A) Analysis of nuclear shape during hatching. Staining for DAPI and Phalloidin-555 reveals nuclei round as trophectoderm cells hatch from the zona pellucida and are subject to less tension. Sphericity is used to assess nuclear shape change (N = 5 mouse embryos, n = 50 non-hatched and 50 hatched nuclei; ****P< 0.0001 by two-tailed unpaired student’s t-test).
(B) Quantification of the percentage of trophectoderm cells with nuclear buds in mouse expanded and hatching blastocysts (N = 17 expanded and 15 hatched mouse embryos; NS = not significant by two-tailed unpaired student’s t-test).
(C) Representative image of a hatching blastocyst (brightfield) with overlayed masked nucleus stained with DAPI. Example measurements of the distance between nucleus center and hatching site (dotted line) as well as the angle between bud, nuclear center and hatching site (yellow spheres).
(D) Scatter plot of distance to hatching site versus bud angle. Dots represent individual nuclei with buds. Dot plot of bud angle comparing nuclear buds proximal (<20 μm) and distal (>20 μm) to the hatching site. Dots represent individual nuclear buds (NS = not significant by two-tailed Mann-Whitney U test).
(E) Radial histogram of hatching mouse blastocysts showing the number of nuclear buds (x-axis) versus bud angle (y-axis); n = 44 nuclear buds, N = 12 mouse embryos for (C-F).
(F) Brightfield images showing trophectoderm biopsy in a mouse blastocyst. Arrows indicate forces exerted by the holding and biopsy pipets as well as movement of the biopsy pipet along the y axis to facilitate cell-cell separation. Images correspond to Video S7.
(G) Representative image of a fixed biopsied mouse blastocyst with an example nuclear bud masked and stained for H3K9me3. Sample measurements of the distance between nucleus center and point of biopsy (dotted line) as well as the angle between bud, nuclear center and biopsy point (yellow spheres).
(H) Plot of distance to biopsy point versus bud angle. Dot plot of bud angle comparing nuclear buds proximal (<20 μm) and distal (>20 μm) to the biopsy point. Dots represent individual nuclear buds (*P = 0.02 by two-tailed Mann-Whitney U test).
(I) Radial histogram of biopsied mouse blastocysts showing the number of nuclear buds (x-axis) versus bud angle (y-axis); n = 43 nuclear buds, N = 15 mouse embryos for (H) and (I).
(J) Representative fixed human blastocyst after biopsy stained with Phalloidin-555. Location of nuclei with buds are marked by yellow spheres. Mural and polar trophectoderm cells are outlined, as determined relative to ICM and biopsy site orientation (dotted lines).
(K) Quantification of nuclear bud localization in biopsied human (n = 22 nuclear buds, 7 embryos; P = 0.75 by two-tailed Wilcoxon test) and mouse (n = 38 nuclear buds, 15 embryos; P = 0.51 by two-tailed Wilcoxon test) embryos represented in 10 × 10 dot plots.
Graphs show median with interquartile range. Scale bars, 10 μm.
ACKNOWLEDGEMENTS
The mouse work was carried out in the Plachta lab supported by grants from the NIGMS (GM139970–01) and NICHD (HD102013–01A1) to N.P., P50 NICHD (5P50HD068157–10) to N.P., R.M.S., and M.S.B., F32HD107914 to E.A.R.C., and NICHD RSDP (K12HD849–36) and ASRM to R.M.S.; Igenomix Foundation postdoctoral fellowship to A.D.-M.; and Anatomical Society (UK) doctoral scholarship to A.A.M. The human embryo work was performed in the embryology lab at Boston IVF. We thank the Nikon BioImaging Lab (NBIL, Cambridge MA) for help with embryo imaging experiments.
Footnotes
DECLARATION OF INTERESTS
D.S. is on the Scientific Advisory Board of Igenomix, consults to ALIFE and Legacy and is Deputy Editor of Human Reproduction. C.S. is Scientific Advisor of Igenomix.
REFERENCES
- 1.Adamson GD, de Mouzon J, Chambers GM, Zegers-Hochschild F, Mansour R, Ishihara O, Banker M, and Dyer S (2018). International Committee for Monitoring Assisted Reproductive Technology: world report on assisted reproductive technology, 2011. Fertil. Steril. 110, 1067–1080. 10.1016/j.fertnstert.2018.06.039. [DOI] [PubMed] [Google Scholar]
- 2.Niakan KK, Han J, Pedersen RA, Simon C, and Pera RA (2012). Human pre-implantation embryo development. Development 139, 829–841. 10.1242/dev.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fierro-Gonzalez JC, White MR, Silva J, and Plachta N (2013). Cadherin-dependent filopodia control preimplantation embryo compaction Nat. Cell Biol. 15, 1424–1433. [DOI] [PubMed] [Google Scholar]
- 4.Samarage CR, White MD, Alvarez YD, Fierro-Gonzalez JC, Henon Y, Jesudason EC, Bissiere S, Fouras A, and Plachta N (2015). Cortical Tension Allocates the First Inner Cells of the Mammalian Embryo. Dev. Cell 34, 435–447. [DOI] [PubMed] [Google Scholar]
- 5.Zenker J, White MD, Gasnier M, Alvarez YD, Lim HYG, Bissiere S, Biro M, and Plachta N (2018). Expanding Actin Rings Zipper the Mouse Embryo for Blastocyst Formation. Cell 173, 776–791 e717. 10.1016/j.cell.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 6.Lim HYG, Alvarez YD, Gasnier M, Wang Y, Tetlak P, Bissiere S, Wang H, Biro M, and Plachta N (2020). Keratins are asymmetrically inherited fate determinants in the mammalian embryo. Nature 585, 404–409. 10.1038/s41586-020-2647-4. [DOI] [PubMed] [Google Scholar]
- 7.Pomp O, Lim HYG, Skory RM, Moverley AA, Tetlak P, Bissiere S, and Plachta N (2022). A monoastral mitotic spindle determines lineage fate and position in the mouse embryo. Nat. Cell Biol. 24, 155–167. 10.1038/s41556-021-00826-3. [DOI] [PubMed] [Google Scholar]
- 8.Gu B, Bradshaw B, Zhu M, Sun Y, Hopyan S, and Rossant J (2022). Live imaging YAP signalling in mouse embryo development. Open Biol. 12, 210335. 10.1098/rsob.210335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumortier JG, Le Verge-Serandour M, Tortorelli AF, Mielke A, de Plater L, Turlier H, and Maitre JL (2019). Hydraulic fracturing and active coarsening position the lumen of the mouse blastocyst. Science 365, 465–468. 10.1126/science.aaw7709. [DOI] [PubMed] [Google Scholar]
- 10.Iwata K, Yumoto K, Sugishima M, Mizoguchi C, Kai Y, Iba Y, and Mio Y (2014). Analysis of compaction initiation in human embryos by using time-lapse cinematography. J. Assist. Reprod. Genet. 31, 421–426. 10.1007/s10815-014-0195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hassold T, and Hunt P (2001). To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2, 280–291. 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 12.Weaver BA, and Cleveland DW (2006). Does aneuploidy cause cancer? Curr. Opin. Cell. Biol. 18, 658–667. 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Franasiak JM, Forman EJ, Hong KH, Werner MD, Upham KM, Treff NR, and Scott RT Jr. (2014). The nature of aneuploidy with increasing age of the female partner: a review of 15,169 consecutive trophectoderm biopsies evaluated with comprehensive chromosomal screening. Fertil. Steril. 101, 656–663 e651. 10.1016/j.fertnstert.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Thomas C, Cavazza T, and Schuh M (2021). Aneuploidy in human eggs: contributions of the meiotic spindle. Biochem. Soc. Trans. 49, 107–118. 10.1042/BST20200043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavez SL, Loewke KE, Han J, Moussavi F, Colls P, Munne S, Behr B, and Reijo Pera RA (2012). Dynamic blastomere behaviour reflects human embryo ploidy by the four-cell stage. Nat. Commun. 3, 1251. 10.1038/ncomms2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducibella T, Albertini DF, Anderson E, and Biggers JD (1975). The preimplantation mammalian embryo: characterization of intercellular junctions and their appearance during development. Dev. Biol. 45, 231–250. [DOI] [PubMed] [Google Scholar]
- 17.Johnson MH, and Maro B (1985). A dissection of the mechanisms generating and stabilizing polarity in mouse 8- and 16-cell blastomeres: the role of cytoskeletal elements. J. Embryol. Exp. Morphol. 90, 311–334. [PubMed] [Google Scholar]
- 18.Johnson MH, and Ziomek CA (1981). The foundation of two distinct cell lineages within the mouse morula. Cell 24, 71–80. 0092–8674(81)90502-X [pii]. [DOI] [PubMed] [Google Scholar]
- 19.Leonavicius K, Royer C, Preece C, Davies B, Biggins JS, and Srinivas S (2018). Mechanics of mouse blastocyst hatching revealed by a hydrogel-based microdeformation assay. Proc. Natl. Acad. Sci. U.S.A. 115, 10375–10380. 10.1073/pnas.1719930115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wieser S, and Pines J (2015). The biochemistry of mitosis. Cold Spring Harb. Perspect. Biol. 7, a015776. 10.1101/cshperspect.a015776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuda M, Hayashi H, Garcia-Ojalvo J, Yoshioka-Kobayashi K, Kageyama R, Yamanaka Y, Ikeya M, Toguchida J, Alev C, and Ebisuya M (2020). Species-specific segmentation clock periods are due to differential biochemical reaction speeds. Science 369, 1450–1455. 10.1126/science.aba7668. [DOI] [PubMed] [Google Scholar]
- 22.Rayon T, Stamataki D, Perez-Carrasco R, Garcia-Perez L, Barrington C, Melchionda M, Exelby K, Lazaro J, Tybulewicz VLJ, Fisher EMC, and Briscoe J (2020). Species-specific pace of development is associated with differences in protein stability. Science 369. 10.1126/science.aba7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White MD, Bissiere S, Alvarez YD, and Plachta N (2016). Mouse Embryo Compaction. Curr. Top. Dev. Biol. 120, 235–258. 10.1016/bs.ctdb.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Koyama H, Suzuki H, Yang X, Jiang S, and Foote RH (1994). Analysis of polarity of bovine and rabbit embryos by scanning electron microscopy. Biol. Reprod. 50, 163–170. 10.1095/biolreprod50.1.163. [DOI] [PubMed] [Google Scholar]
- 25.Ducibella T, Ukena T, Karnovsky M, and Anderson E (1977). Changes in cell surface and cortical cytoplasmic organization during early embryogenesis in the preimplantation mouse embryo. J. Cell Biol. 74, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson MH, Maro B, and Takeichi M (1986). The role of cell adhesion in the synchronization and orientation of polarization in 8-cell mouse blastomeres. J. Embryol. Exp. Morphol. 93, 239–255. [PubMed] [Google Scholar]
- 27.Zhu M, Shahbazi M, Martin A, Zhang C, Sozen B, Borsos M, Mandelbaum RS, Paulson RJ, Mole MA, Esbert M, et al. (2021). Human embryo polarization requires PLC signaling to mediate trophectoderm specification. eLife 10. 10.7554/eLife.65068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coticchio G, Ezoe K, Lagalla C, Shimazaki K, Ohata K, Ninomiya M, Wakabayashi N, Okimura T, Uchiyama K, Kato K, and Borini A (2021). Perturbations of morphogenesis at the compaction stage affect blastocyst implantation and live birth rates. Hum. Reprod. 36, 918–928. 10.1093/humrep/deab011. [DOI] [PubMed] [Google Scholar]
- 29.Newton I (1846). Mathematical Principles of Natural Philosophy. Daniel Adee; New York [Google Scholar]
- 30.Bender C (1874). Bestimmung der grössten Anzahl gleich Kugeln, welche sich auf eine Kugel von demselben Radius, wie die übrigen, auflegen lassen. Archiv. Math. Physik. (Grunert) 56, 302–306. [Google Scholar]
- 31.Maitre JL, Turlier H, Illukkumbura R, Eismann B, Niwayama R, Nedelec F, and Hiiragi T (2016). Asymmetric division of contractile domains couples cell positioning and fate specification. Nature 536, 344–348. 10.1038/nature18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korotkevich E, Niwayama R, Courtois A, Friese S, Berger N, Buchholz F, and Hiiragi T (2017). The Apical Domain Is Required and Sufficient for the First Lineage Segregation in the Mouse Embryo. Dev. Cell 40, 235–247 e237. 10.1016/j.devcel.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe T, Biggins J, Tannan N, and Srinivas S (2014). Limited predictive value of blastomere angle of division in trophectoderm and inner cell mass specification. Development 141, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Familiari G, Relucenti M, Heyn R, Micara G, and Correr S (2006). Three-dimensional structure of the zona pellucida at ovulation. Microsc. Res. Tech. 69, 415–426. 10.1002/jemt.20301. [DOI] [PubMed] [Google Scholar]
- 35.Gupta SK (2018). The Human Egg’s Zona Pellucida. Curr. Top. Dev. Biol. 130, 379–411. 10.1016/bs.ctdb.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Wassarman PM, and Litscher ES (2018). The Mouse Egg’s Zona Pellucida. Curr. Top. Dev. Biol. 130, 331–356. 10.1016/bs.ctdb.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Salas-Vidal E, and Lomeli H (2004). Imaging filopodia dynamics in the mouse blastocyst. Dev. Biol. 265, 75–89. 10.1016/j.ydbio.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 38.Balaban B, Urman B, Sertac A, Alatas C, Aksoy S, and Mercan R (2000). Blastocyst quality affects the success of blastocyst-stage embryo transfer. Fertil. Steril. 74, 282–287. 10.1016/s0015-0282(00)00645-2. [DOI] [PubMed] [Google Scholar]
- 39.Dokras A, Sargent IL, Ross C, Gardner RL, and Barlow DH (1991). The human blastocyst: morphology and human chorionic gonadotrophin secretion in vitro. Hum. Reprod. 6, 1143–1151. 10.1093/oxfordjournals.humrep.a137500. [DOI] [PubMed] [Google Scholar]
- 40.Chan CJ, Costanzo M, Ruiz-Herrero T, Monke G, Petrie RJ, Bergert M, Diz-Munoz A, Mahadevan L, and Hiiragi T (2019). Hydraulic control of mammalian embryo size and cell fate. Nature 571, 112–116. 10.1038/s41586-019-1309-x. [DOI] [PubMed] [Google Scholar]
- 41.Ragkousi K, and Gibson MC (2014). Cell division and the maintenance of epithelial order. J. Cell Biol. 207, 181–188. 10.1083/jcb.201408044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coll L, Parriego M, Mateo S, Garcia-Monclus S, Rodriguez I, Boada M, Coroleu B, Polyzos NP, Vidal F, and Veiga A (2021). Prevalence, types and possible factors influencing mosaicism in IVF blastocysts: results from a single setting. Reprod. Biomed. Online 42, 55–65. 10.1016/j.rbmo.2020.09.025. [DOI] [PubMed] [Google Scholar]
- 43.Leigh D, Cram DS, Rechitsky S, Handyside A, Wells D, Munne S, Kahraman S, Grifo J, Katz-Jaffe M, Rubio C, et al. (2022). PGDIS position statement on the transfer of mosaic embryos 2021. Reprod. Biomed. Online 45, 19–25. 10.1016/j.rbmo.2022.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Rodrigo L, Clemente-Ciscar M, Campos-Galindo I, Peinado V, Simon C, and Rubio C (2020). Characteristics of the IVF Cycle that Contribute to the Incidence of Mosaicism. Genes 11. 10.3390/genes11101151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fragouli E, Munne S, and Wells D (2019). The cytogenetic constitution of human blastocysts: insights from comprehensive chromosome screening strategies. Hum. Reprod. Update 25, 15–33. 10.1093/humupd/dmy036. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez-Diez C, Yamagata K, Trivedi S, Haverfield J, and FitzHarris G (2016). Micronucleus formation causes perpetual unilateral chromosome inheritance in mouse embryos. Proc. Natl. Acad. Sci. U.S.A. 113, 626–631. 10.1073/pnas.1517628112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyanari Y, Ziegler-Birling C, and Torres-Padilla ME (2013). Live visualization of chromatin dynamics with fluorescent TALEs. Nat. Struct. Mol. Biol. 20, 1321–1324. 10.1038/nsmb.2680. [DOI] [PubMed] [Google Scholar]
- 48.Ellis RE, Yuan JY, and Horvitz HR (1991). Mechanisms and functions of cell death. Annu. Rev. Cell Biol. 7, 663–698. 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 49.Kerr JF, Wyllie AH, and Currie AR (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raff M (1998). Cell suicide for beginners. Nature 396, 119–122. 10.1038/24055. [DOI] [PubMed] [Google Scholar]
- 51.Ziegler-Birling C, Helmrich A, Tora L, and Torres-Padilla ME (2009). Distribution of p53 binding protein 1 (53BP1) and phosphorylated H2A.X during mouse preimplantation development in the absence of DNA damage. Int. J. Dev. Biol. 53, 1003–1011. 10.1387/ijdb.082707cz. [DOI] [PubMed] [Google Scholar]
- 52.Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, and Lammerding J (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon-Dumenil AM, Manel N, and Piel M (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362. 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- 54.Krieg M, Arboleda-Estudillo Y, Puech PH, Kafer J, Graner F, Muller DJ, and Heisenberg CP (2008). Tensile forces govern germ-layer organization in zebrafish. Nat. Cell Biol. 10, 429–436. 10.1038/ncb1705. [DOI] [PubMed] [Google Scholar]
- 55.Manning ML, Foty RA, Steinberg MS, and Schoetz EM (2010). Coaction of intercellular adhesion and cortical tension specifies tissue surface tension. Proc. Natl. Acad. Sci. U.S.A. 107, 12517–12522. 10.1073/pnas.1003743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang L, and Goldman RD (2004). Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 5, 601–613. 10.1038/nrm1438. [DOI] [PubMed] [Google Scholar]
- 57.Broussard JA, Jaiganesh A, Zarkoob H, Conway DE, Dunn AR, Espinosa HD, Janmey PA, and Green KJ (2020). Scaling up single-cell mechanics to multicellular tissues - the role of the intermediate filament-desmosome network. J. Cell Sci. 133. 10.1242/jcs.228031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacob JT, Nair RR, Poll BG, Pineda CM, Hobbs RP, Matunis MJ, and Coulombe PA (2020). Keratin 17 regulates nuclear morphology and chromatin organization. J. Cell Sci. 133. 10.1242/jcs.254094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laly AC, Sliogeryte K, Pundel OJ, Ross R, Keeling MC, Avisetti D, Waseem A, Gavara N, and Connelly JT (2021). The keratin network of intermediate filaments regulates keratinocyte rigidity sensing and nuclear mechanotransduction. Sci. Adv. 7. 10.1126/sciadv.abd6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, and Spann TP (2002). Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 16, 533–547. 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- 61.van Montfoort A, Carvalho F, Coonen E, Kokkali G, Moutou C, Rubio C, Goossens V, and De Rycke M (2021). ESHRE PGT Consortium data collection XIX-XX: PGT analyses from 2016 to 2017(dagger). Hum. Reprod. Open 2021, hoab024. 10.1093/hropen/hoab024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamanaka Y, Ralston A, Stephenson RO, and Rossant J (2006). Cell and molecular regulation of the mouse blastocyst. Dev. Dyn. 235, 2301–2314. 10.1002/dvdy.20844. [DOI] [PubMed] [Google Scholar]
- 63.White MD, Zenker J, Bissiere S, and Plachta N (2018). Instructions for Assembling the Early Mammalian Embryo. Dev. Cell 45, 667–679. 10.1016/j.devcel.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 64.Zernicka-Goetz M, Morris SA, and Bruce AW (2009). Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat. Rev. Genet. 10, 467–477. nrg2564 [pii] 10.1038/nrg2564. [DOI] [PubMed] [Google Scholar]
- 65.Hsiung F, Ramirez-Weber FA, Iwaki DD, and Kornberg TB (2005). Dependence of Drosophila wing imaginal disc cytonemes on Decapentaplegic. Nature 437, 560–563. 10.1038/nature03951. [DOI] [PubMed] [Google Scholar]
- 66.Roy S, Hsiung F, and Kornberg TB (2011). Specificity of Drosophila cytonemes for distinct signaling pathways. Science 332, 354–358. 10.1126/science.1198949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Junyent S, Garcin CL, Szczerkowski JLA, Trieu TJ, Reeves J, and Habib SJ (2020). Specialized cytonemes induce self-organization of stem cells. Proc. Natl. Acad. Sci. U.S.A. 117, 7236–7244. 10.1073/pnas.1920837117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma BX, Yang L, Tian Y, Jin L, and Huang B (2022). Cytoplasmic strings between ICM and mTE are a positive predictor of clinical pregnancy and live birth outcomes: A time-lapse study. Front. Med. 9, 934327. 10.3389/fmed.2022.934327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patteson AE, Vahabikashi A, Pogoda K, Adam SA, Mandal K, Kittisopikul M, Sivagurunathan S, Goldman A, Goldman RD, and Janmey PA (2019). Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 218, 4079–4092. 10.1083/jcb.201902046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pogoda K, Byfield F, Deptula P, Ciesluk M, Suprewicz L, Sklodowski K, Shivers JL, van Oosten A, Cruz K, Tarasovetc E, et al. (2022). Unique Role of Vimentin Networks in Compression Stiffening of Cells and Protection of Nuclei from Compressive Stress. Nano Lett. 22, 4725–4732. 10.1021/acs.nanolett.2c00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pfeifer CR, Tobin MP, Cho S, Vashisth M, Dooling LJ, Vazquez LL, Ricci-De Lucca EG, Simon KT, and Discher DE (2022). Gaussian curvature dilutes the nuclear lamina, favoring nuclear rupture, especially at high strain rate. Nucleus 13, 129–143. 10.1080/19491034.2022.2045726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Munne S, Lee A, Rosenwaks Z, Grifo J, and Cohen J (1993). Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum. Reprod. 8, 2185–2191. 10.1093/oxfordjournals.humrep.a138001. [DOI] [PubMed] [Google Scholar]
- 73.Fragouli E, and Wells D (2011). Aneuploidy in the human blastocyst. Cytogenet Genome Res. 133, 149–159. 10.1159/000323500. [DOI] [PubMed] [Google Scholar]
- 74.Taylor TH, Gitlin SA, Patrick JL, Crain JL, Wilson JM, and Griffin DK (2014). The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 20, 571–581. 10.1093/humupd/dmu016. [DOI] [PubMed] [Google Scholar]
- 75.Viotti M, Victor AR, Barnes FL, Zouves CG, Besser AG, Grifo JA, Cheng EH, Lee MS, Horcajadas JA, Corti L, et al. (2021). Using outcome data from one thousand mosaic embryo transfers to formulate an embryo ranking system for clinical use. Fertil. Steril. 115, 1212–1224. 10.1016/j.fertnstert.2020.11.041. [DOI] [PubMed] [Google Scholar]
- 76.Practice C, and Genetic Counseling Professional Group of the American Society for Reproductive Medicine. Electronic address a.a.o. (2020). Clinical management of mosaic results from preimplantation genetic testing for aneuploidy (PGT-A) of blastocysts: a committee opinion. Fertil. Steril. 114, 246–254. 10.1016/j.fertnstert.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 77.Handyside AH, McCollin A, Summers MC, and Ottolini CS (2021). Copy number analysis of meiotic and postzygotic mitotic aneuploidies in trophectoderm cells biopsied at the blastocyst stage and arrested embryos. Prenat. Diagn. 41, 525–535. 10.1002/pd.5816. [DOI] [PubMed] [Google Scholar]
- 78.Popovic M, Dhaenens L, Boel A, Menten B, and Heindryckx B (2020). Chromosomal mosaicism in human blastocysts: the ultimate diagnostic dilemma. Hum. Reprod. Update 26, 313–334. 10.1093/humupd/dmz050. [DOI] [PubMed] [Google Scholar]
- 79.Capalbo A, Poli M, Rienzi L, Girardi L, Patassini C, Fabiani M, Cimadomo D, Benini F, Farcomeni A, Cuzzi J, et al. (2021). Mosaic human preimplantation embryos and their developmental potential in a prospective, non-selection clinical trial. Am. J. Hum. Genet. 108, 2238–2247. 10.1016/j.ajhg.2021.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Munne S, and Wells D (2017). Detection of mosaicism at blastocyst stage with the use of high-resolution next-generation sequencing. Fertil. Steril. 107, 1085–1091. 10.1016/j.fertnstert.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 81.Greco E, Minasi MG, and Fiorentino F (2015). Healthy Babies after Intrauterine Transfer of Mosaic Aneuploid Blastocysts. N. Engl. J. Med. 373, 2089–2090. 10.1056/NEJMc1500421. [DOI] [PubMed] [Google Scholar]
- 82.Vazquez-Diez C, and FitzHarris G (2018). Causes and consequences of chromosome segregation error in preimplantation embryos. Reproduction 155, R63–R76. 10.1530/REP-17-0569. [DOI] [PubMed] [Google Scholar]
- 83.Lightfoot DA, Kouznetsova A, Mahdy E, Wilbertz J, and Hoog C (2006). The fate of mosaic aneuploid embryos during mouse development. Dev. Biol. 289, 384–394. 10.1016/j.ydbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 84.Saias L, Swoger J, D’Angelo A, Hayes P, Colombelli J, Sharpe J, Salbreux G, and Solon J (2015). Decrease in Cell Volume Generates Contractile Forces Driving Dorsal Closure. Dev. Cell 33, 611–621. 10.1016/j.devcel.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 85.Bolton H, Graham SJL, Van der Aa N, Kumar P, Theunis K, Fernandez Gallardo E, Voet T, and Zernicka-Goetz M (2016). Mouse model of chromosome mosaicism reveals lineage-specific depletion of aneuploid cells and normal developmental potential. Nat. Commun. 7, 11165. 10.1038/ncomms11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang M, Rito T, Metzger J, Naftaly J, Soman R, Hu J, Albertini DF, Barad DH, Brivanlou AH, and Gleicher N (2021). Depletion of aneuploid cells in human embryos and gastruloids. Nat. Cell Biol. 23, 314–321. 10.1038/s41556-021-00660-7. [DOI] [PubMed] [Google Scholar]
- 87.Practice Committees of the American Society for Reproductive M, and the Society for Assisted Reproductive T (2013). Blastocyst culture and transfer in clinical-assisted reproduction: a committee opinion. Fertil. Steril. 99, 667–672. 10.1016/j.fertnstert.2013.01.087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Live imaging of mouse embryos, related to Figure 1.
Combining the use of SPY650-DNA and SPY555-actin enables live imaging with high spatiotemporal resolution in live mouse embryos. These examples show: (1) Cell divisions during the 2-to-4 cell stage. The imaging spatiotemporal resolution allows us to capture the main phases of mitosis and formation of the cytokinetic furrow. (2) Compaction at the 8-cell stage. SPY555-actin allows visualization of the main changes in cell shape; note the flattening of the cells against each other during the compaction process. (3) Formation of F-actin rich apical domains at the 8-to-16 cell stage. Note the disassembly of the apical domains prior to cell division. (4) Formation of F-actin rings during the 16-cell stage. Imaging over time shows the gradual expansion and subsequent zippering of these rings along cell-cell junctions across most cells of the embryo. (5) Formation of the blastocoel at the 32-cell and expansion of the blastocyst. Note the periodic collapses in blastocoel volume. Cell divisions across the trophectoderm layer can be observed during the expansion process.
Video S2 Live imaging of F-actin rings formation the 16-cell stage mouse embryo, related to Figure 1 and Figure S2.
Live mouse embryo shows formation, expansion, and zippering of F-actin rings during the 16-cell stage labeled by SPY650-FastAct, until the next round of mitosis.
Video S3. Live imaging of cell divisions at the cleavage and blastocyst stage, related to Figures 2 and 4.
Live imaging of a cell division in a mouse and human cleavage stage embryo stained with SPY555-DNA and SPY650-FastAct. In mouse, the cell nucleus bounces against the apical cell cortex of the prospective outer cell at the end of cytokinesis. By contrast, in human, cell nuclei remain closer to the cytokinetic furrow without bouncing against the cortex. The use of SPY650-DNA and SPY555-actin also allows live imaging of cell divisions within the human blastocyst. The main phases of mitosis can be captured in this trophectoderm cell.
Video S4. Live imaging of the human blastocyst, related to Figure 4 and Figure 5.
Example of live imaging in a human blastocyst shows cell divisions across the trophectoderm. Arrowheads identify the appearance of cytoplasmic DNA structures in a subset of trophectoderm cells.
Video S5, Live imaging of nuclear budding and DNA shedding in the human trophectoderm, related to Figure 5.
Live imaging demonstrates the formation of a nuclear bud with subsequent loss into the cytoplasm of a trophectoderm cell in a live human blastocyst. The second part of the movie shows computational segmentation of the DNA and actin signals to highlight the loss of DNA. In this example the cell undergoes mitosis, and one daughter cell inherits cytDNA.
Video S6, Chromatin markers in nuclear buds, related to Figure 5.
Three-dimensional rotations show a structure budding from cell nuclei labeled with H3K9me3 and H2B-RFP in mouse trophectoderm cells.
Video S7. Trophectoderm biopsy in mouse blastocysts, related to Figure 6.
Example of the trophectoderm biopsy process in a mouse blastocyst.
Figure S1. Additional characterization of fluorescent dyes, related to Figure 1.
(A) Brightfield images corresponding to the embryos shown in Figure 1A.
(B) Incubation of live mouse embryos with SPY650-DNA and SPY555-actin followed by washing of the dyes produces similar labeling patterns to those found in live embryos microinjected with mRNA for H2B-GFP and Utr-RFP, or fixed embryos stained with DAPI and Phalloidin-Rhodamine.
(C) and (D), Liveborn pups derived from control (unstained) and embryos stained with SPY650-DNA and SPY555-actin following in utero transfer (NS = not significant by two-tailed unpaired student’s t-test).
(E) 2D planes showing SPY555-actin and SPY650-FastAct signal in 16-cell stage live mouse embryos. This allows for the identification of inner cells.
(F) Computational segmentation of the 16-cell stage embryo shown in Figure 1G. Cell contours were defined by the Phalloidin-555 signal. The embryo is shown as a 3D projection and brightfield image. A representative z-plane (0.4 μm slice) is shown with the cell contours of an outer cell overlaying the Phalloidin-555 signal.
(G-I) Representative image of a single slice through the center of the embryo stained with Phalloidin-555 shown in Figure 1G and S1F. Segmentation allows identification of inner versus outer cells. The series of images along the z-plane are projected to create three-dimensional segmentation of cell contours.
Scale bars, 10 μm.
Figure S2. Embryos dyed with SPY555-DNA and SPY650-FastAct display normal actin ring expansion and zippering, related to Figure 1.
(A) Live-imaging of a 16-cell stage mouse embryo with SPY555-DNA and SPY650-FastAct displaying formation and expansion of actin rings, and zippering along the cell-cell junctions. Note that the dyes allow for normal morphological changes including actin ring expansion and zippering along cell-cell junctions, indicating no overt effects on actin dynamics.
(B-D) High magnification images of three pairs of cells at selected time points. 2D sections show actin rings and zippering as bright fluorescence regions along the cell cortex and at the cell-cell junctions. </p/> Scale bars, 10 μm.
Figure S3. Compaction and early inner cell allocation in human embryos, related to Figure 2.
(A) Brightfield images of a human embryo at different stages of preimplantation development (2-cell, 8-cell, 16-cell, compacted morula and expanded blastocyst). An inner cell can be seen at the 16-cell stage, before the initiation of compaction (arrowhead).
(B) Schematic diagram of Newton’s kissing number problem. At the 12-cell stage, at least one blastomere must occupy an inner position to maintain the spherical organization and cell-cell contacts of the embryo. In three-dimensions, this dictates that outer cells align along the vertices of a regular icosahedron.
(C) Brightfield images and schematic displaying morphological differences between mouse and human embryos at the 8- and 16-cell stages. Note that at the 16-cell stage, the mouse embryo is completely compacted. Human embryos have not yet completed compaction and contain spherical blastomeres. Embryos must contain 2–3 inner cells at the 16-cell stage.
(D) Analysis of nuclear position following division in live cleavage stage embryos. Distance of the nucleus to the cytokinetic furrow/cleavage point is measured for each mitotic pair 1 hour after telophase and normalized to the maximum distance from the cell pole to the cytokinetic furrow (N = 7 mouse and 3 human embryos, n = 46 and 30 cells for mouse and human, respectively; ****P< 0.0001 by two-tailed unpaired student’s t-test). SPY555-DNA and SPY650-FastAct live staining show nuclear positioning 1h after telophase. Note that an additional time point after telophase is shown from embryos in Figure 2G.
(E) Radial histogram of human morulae showing the number of divisions (x-axis) versus division angle relative to the center of mass of the embryo (y-axis) (n = 16 divisions, N = 3 human embryos). Division planes with angles ≤45° to the center of mass are classified as asymmetric (inner-outer progeny), while angles >45° are classified as symmetric (outer-outer progeny).
(F) Live-imaging of human cleavage stage embryos stained with SPY555-DNA and SPY650-FastAct allows for tracking of inner-outer and outer-outer progeny over time. 2D sections show the trophectoderm fate of outer cells and the ICM fate of inner cells in the early blastocyst.
(G) Live-imaging with SPY555-DNA and SPY650-FastAct reveals apical constriction in the human embryo. 2D sections of a cleavage stage embryo show how a newly generated outer cell reduces its apical external contact, gets internalized and becomes an inner cell. Computational segmentation of the SPY650-FastAct signal showing the outer undergoing apical constriction and becoming an inner cell. The top views highlight the reduction in apical surface.
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S4. Analysis of blastocyst structures and chromosome segregation errors during mitosis in the living embryo, related to Figure 3 and Figure 4.
(A) Live-imaging of a human blastocyst stained with SPY650-DNA and SPY555-actin. 2D sections of the embryo displayed in Figure 3A show that some of the SPY555-actin dye is retained in the zona pellucida. Note that as the zona pellucida thins down with blastocyst development, trophectoderm cells and cell-cell junctions improve their staining.
(B) Live-imaging of a mouse early blastocyst stained with SPY650-DNA and SPY555-actin. Note that the SPY555-actin signal is barely detectable in the murine zona pellucida, and cell-cell junctions can be detectable early on.
(C) 2D plane through a live mouse blastocyst stained with SPY650-DNA and SPY555-actin reveals tether-like structures projecting between the trophectoderm and ICM similar to those observed in human.
(D) and (E) Quantification of the number and length of TE-ICM tethers shows that they are more frequent and longer in human blastocysts (N = 7 mouse and 3 human embryos; ****P< 0.0001, *P< 0.05 by two-tailed unpaired student’s t-test). Tether length is normalized to embryo diameter.
(F) Examples of blastocyst collapse and expansion. Quantification of trophectoderm mitoses at different time points before and after embryo collapse (N = 10 collapse events in 6 mouse embryos; NS = not significant by Kruskal-Wallis test).
(G) Analysis of cell divisions relative to embryo collapse events (probed by measuring embryo volume) in different mouse embryos.
(H) Live-imaging with SPY650-DNA and SPY555-actin showing that the micronucleus formed in the human trophectoderm cell shown in Figure 4G can still be detected hours later. The cell is shown in 3D top and 2D sectional views with magnified images of the cell and surface segmentations of the DNA signal.
(I) Live-imaging in mouse 8-cell embryos microinjected with mRNA for the centromere marker TaleMS-mClover and stained with SPY650-DNA allow the identification of lagging chromosomes during mitosis.
(J) Live-imaging in mouse blastocysts with SPY650-DNA and SPY555-actin showing the formation of micronuclei in trophectoderm cells.
(K) Quantification of the number of mitoses with lagging chromosomes relative to the total number of mitotic events analyzed in mouse blastocysts (N = 8 mouse embryos).
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S5. Additional characterization of nuclear buds and cytDNA structures, related to Figure 5.
(A) Live-imaging of mouse blastocysts show the appearance of nuclear buds and cytDNA. Lower panels show SPY650-DNA signal segmented.
(B) Images of live mouse blastocysts reveal SPY650-DNA–labeled structures within the cytoplasm. Smaller insets show segmented embryos highlighting the increase in cavity volume at the advanced stage.
(C) 1 μm-thick planes in both xy and xz show nuclear shedding events in live mouse and human embryos (shown in 3D in Figure 5B and Figure S5A).
(D) Bar graphs of number of trophectoderm cells and cells with nuclear buds in cultured and in 4 d.p.c. freshly isolated, fixed mouse embryos (shown in Figure 5E, N = 32 cultured and 35 freshly isolated embryos). Each bar represents an individual embryo.
(E) Confocal image stack showing each 2D at 0.5 μm interval. Note the presence of the bud in the middle sections originating from the nucleus.
(F) Immunostaining for dsDNA in combination with histone modifications H3K9me2 and H3K9me3, histone H2B-RFP and the centromere marker TaleMS-mClover in advanced mouse blastocysts. 400 nm-thick planes in both xy and xz show the presence of these chromatin markers in nuclear buds and cytDNA structures of trophectoderm cells.
(G) Segmentation and analysis of nuclear bud position relative to the embryo’s center of mass shows that buds are produced along the xy axis (i.e., orthogonal to the apical-basal axis of the cell).
(H) Analysis of nuclear morphology in live embryos labeled with SPY650-DNA shows that ICM cell nuclei do not undergo significant morphological change during blastocyst expansion (N = 3 human embryos, n = 16 and 19 cells for pre- and post-expansion, respectively; NS = not significant by two-tailed unpaired t-test). Quantification of nuclear ellipticity (oblate) serves as a proxy for flattening.
(I) Staining for keratin-8 in the early and late human blastocyst. Note the prominent keratin tethers connecting the nucleus to the cortex at the early stage. As the blastocyst expands and the trophectoderm cell flattens, the keratin network forms a cage around the nucleus.
(J) Some trophectoderm cells in the mouse blastocyst display lower keratin levels. Perinuclear K8 levels strongly correlate with lamin-A levels (N = 8 mouse embryos, n = 73 cells; r = 0.69, ****P< 0.0001 by two-tailed Pearson’s correlation). 2D sections reveal higher perinuclear K8 in nuclei with higher lamin-A levels.
(K) Representative image of an embryo after injection with K8-Emerald and K18Emerald at the 2-cell stage. Embryos were then fixed at the 8-cell stage and stained for lamin-A, with fluorescence intensity normalized to DAPI. Cells with K8/K18 overexpression show higher lamin-A levels compared to uninjected cells (N = 7 embryos, n = 29 uninjected cells, 26 injected cells. **P < 0.01 by Mann-Whitney U test).
(L) Trophectoderm cells with cytDNA display lower levels of lamin-A (N = 7 mouse embryos, n = 60 control and 9 +cytDNA cells; **P < 0. 01 by two-tailed unpaired student’s t-test).
Graphs show median with interquartile range. The zona pellucida was masked out in human live embryos to improve visualization. Scale bars, 10 μm.
Figure S6. Analysis of caspase activity and γH2AX, related to Figure 5.
(A) Immunostaining for cleaved Caspase-3 with SPY555-actin and DAPI in fixed mouse blastocysts. Example of trophectoderm cell showing cleaved Caspase-3 surrounding a cytDNA structure. Note that the corresponding cell nucleus remains intact and does not display cleaved Caspase-3 staining (top panel). By contrast, the bottom panel shows an example of a pyknotic nucleus recognized by cleaved Caspase-3 undergoing PCD in the ICM.
(B) Quantification of cytDNA structures positive for cleaved Caspase-3 in cells displaying either an intact or pyknotic nucleus (N = 5 mouse embryos; **P< 0.01, *P< 0.05 by Mann-Whitney U test).
(C) Live-imaging of mouse blastocysts with SPY650-DNA, SPY555-actin and CellEvent Caspase-3/7 Green reveals caspase activity following the formation of cytDNA.
(D) Immunostaining for γH2AX and dsDNA in advanced mouse blastocysts highlighting the presence or absence of γH2AX in cells displaying nuclear buds and cytDNA.
(E) Analysis of nuclear γH2AX intensity in cells displaying or not cytDNA structures (N = 5 mouse embryos per group; NS = not significant by Mann-Whitney U test).
(F) Immunostaining for cleaved Caspase-3 and Hoechst in scramble and K8+K18 siRNAs injected embryos.
(G) and (H) Analysis of cleaved Caspase-3 positive cells and pyknotic nuclei after K8+K18 downregulation in mouse blastocysts (N = 11 control and 21 K8+K18 siRNAs embryos; NS = not significant by two-tailed unpaired student’s t-test).
Scale bars, 10 μm.
Figure S7. Nuclear budding during blastocyst hatching and trophectoderm biopsy, related to Figure 5 and Figure 6.
(A) Analysis of nuclear shape during hatching. Staining for DAPI and Phalloidin-555 reveals nuclei round as trophectoderm cells hatch from the zona pellucida and are subject to less tension. Sphericity is used to assess nuclear shape change (N = 5 mouse embryos, n = 50 non-hatched and 50 hatched nuclei; ****P< 0.0001 by two-tailed unpaired student’s t-test).
(B) Quantification of the percentage of trophectoderm cells with nuclear buds in mouse expanded and hatching blastocysts (N = 17 expanded and 15 hatched mouse embryos; NS = not significant by two-tailed unpaired student’s t-test).
(C) Representative image of a hatching blastocyst (brightfield) with overlayed masked nucleus stained with DAPI. Example measurements of the distance between nucleus center and hatching site (dotted line) as well as the angle between bud, nuclear center and hatching site (yellow spheres).
(D) Scatter plot of distance to hatching site versus bud angle. Dots represent individual nuclei with buds. Dot plot of bud angle comparing nuclear buds proximal (<20 μm) and distal (>20 μm) to the hatching site. Dots represent individual nuclear buds (NS = not significant by two-tailed Mann-Whitney U test).
(E) Radial histogram of hatching mouse blastocysts showing the number of nuclear buds (x-axis) versus bud angle (y-axis); n = 44 nuclear buds, N = 12 mouse embryos for (C-F).
(F) Brightfield images showing trophectoderm biopsy in a mouse blastocyst. Arrows indicate forces exerted by the holding and biopsy pipets as well as movement of the biopsy pipet along the y axis to facilitate cell-cell separation. Images correspond to Video S7.
(G) Representative image of a fixed biopsied mouse blastocyst with an example nuclear bud masked and stained for H3K9me3. Sample measurements of the distance between nucleus center and point of biopsy (dotted line) as well as the angle between bud, nuclear center and biopsy point (yellow spheres).
(H) Plot of distance to biopsy point versus bud angle. Dot plot of bud angle comparing nuclear buds proximal (<20 μm) and distal (>20 μm) to the biopsy point. Dots represent individual nuclear buds (*P = 0.02 by two-tailed Mann-Whitney U test).
(I) Radial histogram of biopsied mouse blastocysts showing the number of nuclear buds (x-axis) versus bud angle (y-axis); n = 43 nuclear buds, N = 15 mouse embryos for (H) and (I).
(J) Representative fixed human blastocyst after biopsy stained with Phalloidin-555. Location of nuclei with buds are marked by yellow spheres. Mural and polar trophectoderm cells are outlined, as determined relative to ICM and biopsy site orientation (dotted lines).
(K) Quantification of nuclear bud localization in biopsied human (n = 22 nuclear buds, 7 embryos; P = 0.75 by two-tailed Wilcoxon test) and mouse (n = 38 nuclear buds, 15 embryos; P = 0.51 by two-tailed Wilcoxon test) embryos represented in 10 × 10 dot plots.
Graphs show median with interquartile range. Scale bars, 10 μm.
Data Availability Statement
Data supporting the findings of this study are available from the corresponding author on reasonable request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request