

SUMMARY
Phosphorus Fluoride Exchange (PFEx) represents a cutting-edge advancement in catalytic click-reaction technology. Drawing inspiration from Nature’s phosphate connectors, PFEx facilitates the reliable coupling of P(V)–F loaded hubs with aryl alcohols, alkyl alcohols, and amines to produce stable, multidimensional P(V)–O and P(V)–N linked products. The rate of P–F exchange is significantly enhanced by Lewis amine base catalysis, such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). PFEx substrates containing multiple P–F bonds are capable of selective, serial exchange reactions via judicious catalyst selection. In fewer than four synthetic steps, controlled projections can be deliberately incorporated along three of the four tetrahedral axes departing from the P(V) central hub, thus taking full advantage of the potential for generating three-dimensional diversity. Furthermore, late-stage functionalization of drugs and drug fragments can be achieved with the polyvalent PFEx hub, hexafluorocyclotriphosphazene (HFP), as has been demonstrated in prior research.
INTRODUCTION
Click chemistry is a versatile and powerful synthesis-based discovery method that relies on the formation of stable molecular connections. At its core, click chemistry encompasses a diverse and expanding set of robust and reliable reactions that enable the precise connection of discrete molecular modules. This approach mirrors the biogenesis of Nature’s essential biopolymers, such as DNA, RNA, proteins, and carbohydrates1,2. In fact, several of the processes scoring click status1 can be traced back to reversible chemistries commonly found in Nature, such as Michael additions, Diels–Alder cycloadditions, and condensation reactions. However, it was the advent of the CuAAC (copper-catalyzed azide-alkyne cycloaddition)2–5 reaction that solidified click chemistry as a leading paradigm for the rapid discovery of functional molecules. This unrivaled and irreversible process lacks a natural counterpart and has earned the reputation as the “cream of the crop” within the click chemistry toolbox.
The world of sulfur-based connective click chemistry was launched in 2014 with the development of Sulfur Fluoride Exchange (SuFEx) by Sharpless and co-workers8. SuFEx capitalizes on the latent reactivity of high oxidation state sulfur-fluoride bonds, which can be triggered by catalyst activation, to facilitate nearly perfect exchange8 with diverse nucleophiles including aryl and alkyl alcohols9, amines10–12, and carbanions13–15. This ground-breaking technique has opened new possibilities for chemical synthesis and holds tremendous potential for the development of novel functional materials and therapeutic agents.
SuFEx reactions classically occur between sulfur-centered hubs16 — sulfuryl fluoride (SO2F2)8, thionyl tetrafluoride (SOF4)17,18, ethenesulfonyl fluoride (ESF)8,19,20, and 2-substituted-alkynyl-1-sulfonyl fluorides (SASFs)21 — and aryl silyl ether nucleophiles. These reactions are typically activated by a suitable Lewis base amine (e.g., DBU)22,23, bifluoride ion14,24, or other catalysts12,23,25. While the direct S–F exchange between sulfur-containing hubs and aryl and alkyl alcohols is more challenging, modified SuFEx conditions reported by Moses and co-workers have made it possible by employing BTMG catalyst with HMDS additive, termed “Accelerated SuFEx Click Chemistry (ASCC)”9,26,27.
Among Nature’s most essential connectors are the phosphate esters and anhydrides. These unions are important in the makeup of nucleic acids, nucleotide coenzymes, nucleoside triphosphates (i.e., ATP), metabolic intermediates, and intermediates in many biochemical processes28. While phosphorus reagents are ubiquitous in synthetic organic chemistry, carbon5 and sulfur1,8,29 are more prevalent as synthetic connectors, a sentiment expressed in Westheimer’s thesis on Why Nature Chose Phosphates: “We can understand the choices made both by chemists and by the process of natural selection. They are both correct”28,30.
The first synthetic phosphate esters were prepared in France over 200 years ago31–34, laying the foundation for the rich body of chemistry that followed35–52. Today, organophosphates are indispensable molecules, with several notable examples including the lifesaving antiviral drug (e.g., (–)-remdesivir (1)), anticancer chemotherapy agents (e.g., (±)-cyclophosphamide (2)), and pesticides (e.g., terbufos (3)). The chemical, physical, and biological properties are modulated by the three other substituents projecting out along tetrahedral exit vectors from the phosphorus core.
The laboratory synthesis of phosphorus linkages typically hinges on the nucleophilic exchange of P(V) electrophiles. For example, the reaction between phosphoryl chloride (POCl3) with both primary and secondary amines to afford the P(V)–N linked products. However, this halide substitution event is not always optimal; preventing unwanted degradation or over-substitution can be difficult.
At this point, one can take direction from the genesis of SuFEx chemistry. In their seminal work, Sharpless and co-workers revisited early reports on the exceptional stability of sulfonyl fluorides to aqueous conditions by Steinkopf53,54, Davies and Dick55,56, and others. Analogous to the P–Cl substitution chemistry, S–Cl exchange often leads to poor outcomes. However, the staggering reactivity gap offered by switching from S–Cl bonds to S–F bonds opened the door to SuFEx – a second near-perfect click reaction alongside CuAAC.
This disparity in reactivity of sulfur-halide bond-containing species can be accounted for by considering the unique properties of the S(VI)–F bond. The shorter S–F bond (1.54 Å vs. S–Cl = 1.99 Å57,58) has a predicted bond dissociation energy (BDE) almost double that of the chloride6 (Figure 1A) and exclusively cleaves heterolytically due to the strongly electronegative fluorine8. This makes S(VI)–F groups stable toward nucleophilic addition (i.e., hydrolysis)59, thermolysis, oxidation55, and reduction53. Crucially, however, S(VI)–F bonds can be reliably activated for nucleophilic exchange when the correct catalyst-reagent combination is employed8.
Figure 1. Selected organophosphorus hubs.
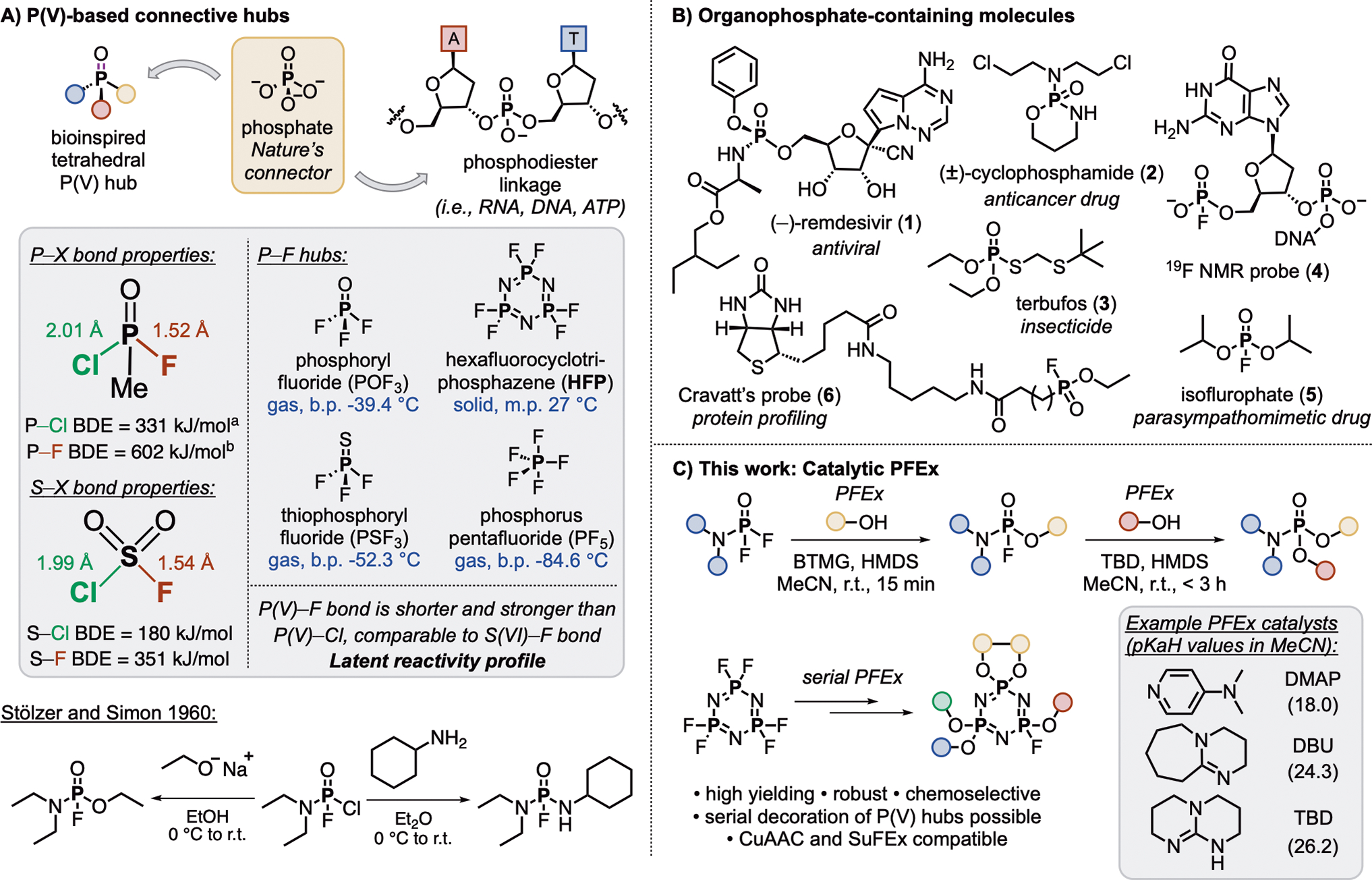
(A) P(V)-based connective hubs and associated physicochemical properties. A = adenine, T = thymine.
(B) Selected organophosphorus-containing molecules.
(C) This work: Catalytic phosphorus(V) fluoride exchange (PFEx).
[a]Calculated BDE of P–Cl bond in POCl36.
[b]Calculated BDE of P–F bond in POF37.
A similar pre-disposition exists when phosphorus is considered instead of sulfur. The shorter P–F bond (1.52 Å vs. P–Cl = 2.01 Å in CH3POFCl60, Figure 1A) has a higher predicted BDE of 602 kJ/mol7. Consequentially, in compounds bearing both P–Cl and P–F bonds, it is the P–Cl bond (BDE = 331 kJ/mol61) that preferentially reacts with incoming nucleophiles (i.e., amines and alkoxides) and hydrolyzes with KOH at 0 °C62, leaving the P–F bond untouched. Further, P–F bonds are found to be more thermally stable than P–Cl compounds63, survive refluxing in aniline64, and remain intact under reductive conditions64 (see Scheme 1C). However, activation of P–F bonds toward exchange with nucleophiles can be facilitated in a similar fashion to S–F compounds (i.e., trifluoromethylation with TMSCF3 mediated by KF)14,65,66. This pattern of reactivity is then, of course, sufficient to entice curiosity for the amenability of the P–F bond for click chemistry reaction development.
Scheme 1. Synthesis of PFEx substrates.

(A) Synthesis of phosphoramidic difluorides, cyclic phosphoramidofluoridates, and cyclic thiophosphoramidofluoridates.
(B) Synthesis of phosphoramidofluoridates and thiophosphoramidofluoridates. General reaction conditions: POCl3 or PSCl3 (1.0 equiv) and Et3N (1.0 equiv) were added to relevant phenol (1.0 equiv) in CH2Cl2 (0.25 M) at −78 °C then stirred overnight at room temperature. The required amine (1.0 equiv) was added, followed by Et3N (1.0 equiv) dropwise at −78 °C. The reaction was stirred at room temperature until complete (31P NMR). The reaction was filtered and concentrated. KF (8.0 equiv) and nBu4NCl (0.10 equiv) were added to the crude in acetone (0.25 M). After completion (31P NMR), the reaction was filtered, concentrated, and purified by silica column chromatography. Isolated yields are reported. Reactions were performed on 5.0 mmol following general procedures detailed in the supporting information unless stated otherwise. See supporting information for a complete list of products.
(C) Stability studies of representative P–X compounds.
[a]Product had limited stability.
[b]11a was completely consumed after 1 h; P–Cl exchange product was identified.
Organo(fluoro)phosphates are highly versatile molecules, but their historic association as toxic nerve agents67,68 has overshadowed their more favorable applications. For example, the resistance of P–F bonds to hydrolysis under biological conditions has been exploited to develop nucleoside phosphate prodrugs that selectively activate upon enzymatic cleavage69. P–F bonds have also found application in 19F NMR-based probes (4)70, therapeutics (e.g., isofluorophate (5)71), and probes used in protein profiling (e.g., Cravatt’s probe (6)72,73).
Exploiting the innate tunability of the P–F bond environment, we now bring phosphorus into the click chemistry fold and report catalyst-accelerated Phosphorus Fluoride Exchange (PFEx), a new click technology emulating Nature’s exemplary use of phosphate connectors (Figure 1C). PFEx is characterized by the Lewis base-catalyzed exchange of P(V)–F bonds with incoming nucleophiles to afford stable, tetrahedral P(V)–O and P(V)–N linked products with defined multidimensional projections departing from the tetrahedral phosphorus core. The reactivity profile of P–F hubs surpasses that of their P–Cl counterparts in terms of both reaction rate and performance, qualifying PFEx as a promising click reaction. Further, the controlled and sequential decoration of the central phosphorus atom achieved through PFEx, allows for the rapid construction of multidimensional connections under mild conditions, making PFEx an ideal biomimetic candidate for Diversity Oriented Clicking (DOC)21 and function-driven discovery projects.
RESULTS AND DISCUSSION
Synthesis of PFEx Substrates
Phosphoryl fluoride (POF3)74,75 and thiophosphoryl fluoride (PSF3) are conceptually ideal PFEx hubs with multiple P–F offerings76,77, but as highly toxic gases (b.p. −39.4 °C78 and, b.p. −52.3 °C79, respectively), are impractical for routine click chemistry80. We elected to use the widely available and bench-stable POCl3 (b.p. 103 °C)81 as a convenient starting point for PFEx substrate synthesis.
A selection of phosphoramidic difluorides82 (9a–9g) was prepared by the addition of secondary amines to POCl3 and Et3N, followed by fluoride-chloride halogen exchange using an optimized protocol [KF (8.0 equiv) in acetone at room temperature (see supplementary information Table S1)] (Scheme 1A)83. The cyclic fluoridates 10a–10g were prepared following an identical sequence using the corresponding 2-(aminomethyl)phenol. The solid cyclic fluoridates were bench stable for at least 2 months, whereas the liquid phosphoramidic difluoride substrates (9a–9g) were found to decompose over several hours at room temperature84. However, 9a–9g were perfectly useable substrates if freshly prepared and delivered to the next step crude following simple Celite® filtration. The phosphoramidofluoridates 12a–12l were prepared from POCl3 or PSCl3 by the sequential treatment with an aryl alcohol followed by an amine to yield the corresponding phosphoramidochloridates or thiophosphoramidochloridates, respectively. These chloridates were readily converted to the corresponding fluoridates in the presence of KF (8.0 equiv) and tetrabutylammonium chloride (10 mol%) as a phase transfer catalyst (Scheme 1B).
The resistance of ‘FExable’ substrates to hydrolysis under biological conditions is necessary for application in covalent drugs and ‘Sleeping Beauty’-type probes. Hence, we evaluated the stability of representative P(V)–F and P(V)–Cl substrates in phosphate buffer solutions at room temperature (see Scheme 1C and supplementary information Table S2–S7 for full experimental details). The phosphoramidofluoridate 12a was stable for over 24 hours when exposed to buffers with pH values of 4.8, 7.4, and 8.8, whereas the chloridate 11a was not (refer to Scheme 3B for structures). A similar trend was observed for the cyclic phosphoramidofluoridates; 10c hydrolyzed only in the basic buffer after an extended reaction time, while the analogous chloridate 8c hydrolyzed across the range of buffer systems tested. Even the least stable PFEx substrates prepared (i.e., 12c) — phosphoramidofluoridates derived from primary amines — demonstrated superior stability to hydrolytic decomposition when contrasted to their chloride equivalent (Table S4). Each P–F substrate tested was stable when stirred in anhydrous ethanol, while the P–Cl analogs completely degraded after 24 hours85.
Scheme 3. Comparison of the reactivity of P(V)–F and P(V)–Cl substrates with 3,5-xylenol under catalyst-accelerated PFEx conditions.
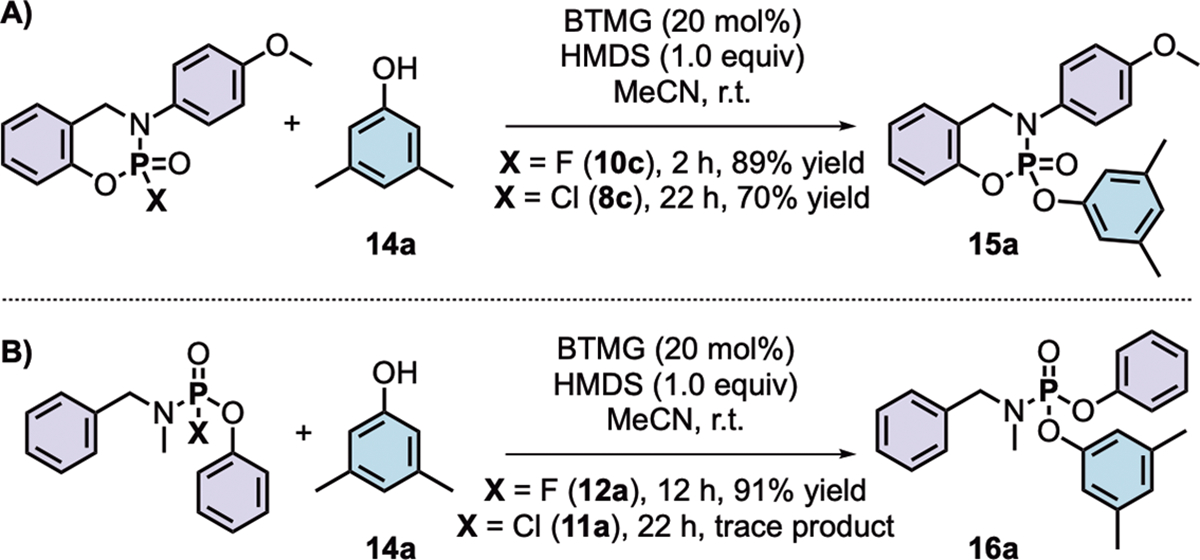
(A) Cyclic PFEx substrates.
(B) PFEx substrates.
Collectively, the stability of P(V)–F bonds over P(V)–Cl bonds to both hydrolysis86 and uncatalyzed nucleophilic displacement by alcohols, supports a window of reactivity akin to SuFEx, positioning PFEx as a standout candidate for a click reaction.
PFEx Reaction Development
To investigate catalyst-accelerated Phosphorus Fluoride Exchange, we drew inspiration from lessons learned in SuFEx click chemistry8. A test reaction was first performed with freshly prepared phosphoramidic difluoride 9b and the TBS-ether of 4-methoxyphenol in the presence of 20 mol% DBU catalyst at room temperature, which gave the P(V)–O linked PFEx product 12m in 81% isolated yield in just 1 hour (see Table S9 in the SI for full optimization). No reaction was observed in the absence of the DBU catalyst (entry 6, Table S9). The same catalyst-activated PFEx conditions worked well with a range of electron-rich and electron-deficient aromatic and heteroaromatic aryl silyl ethers, giving the products 12m–12u in good conversion (Scheme S1).
To streamline the new PFEx protocol and eliminate the need for prerequisite aryl silyl ether synthesis, we adopted the same accelerated conditions developed for SuFEx9,26. Phosphoramidates 12m–12u were prepared directly from the corresponding phenols using a synergistic combination of 20 mol% BTMG (2-tert-butyl-1,1,3,3-tetramethylguanidine, Barton’s base) and 1 equivalent of HMDS (Scheme 2); a 15-minute reaction time afforded the PFEx products in good to excellent yields. Of note is the preference for PFEx reaction with the difluoride substrates over the corresponding mono-fluoride products, mirroring the reactivity trend observed with the multidimensional iminosulfur oxydifluorides derived from thionyl tetrafluoride (SOF4)18. In each instance, the PFEx reaction stopped following the first substitution, leaving the remaining P(V)–F bond untouched. This impressive selectivity can be explained by considering the attenuation of the phosphorus’s electrophilicity after replacing an electron-withdrawing fluoride with an aryl alcohol. Control reactions with analogous dichlorides gave complex product mixtures.
Scheme 2. PFEx reaction of phosphoamidic difluorides with phenols.
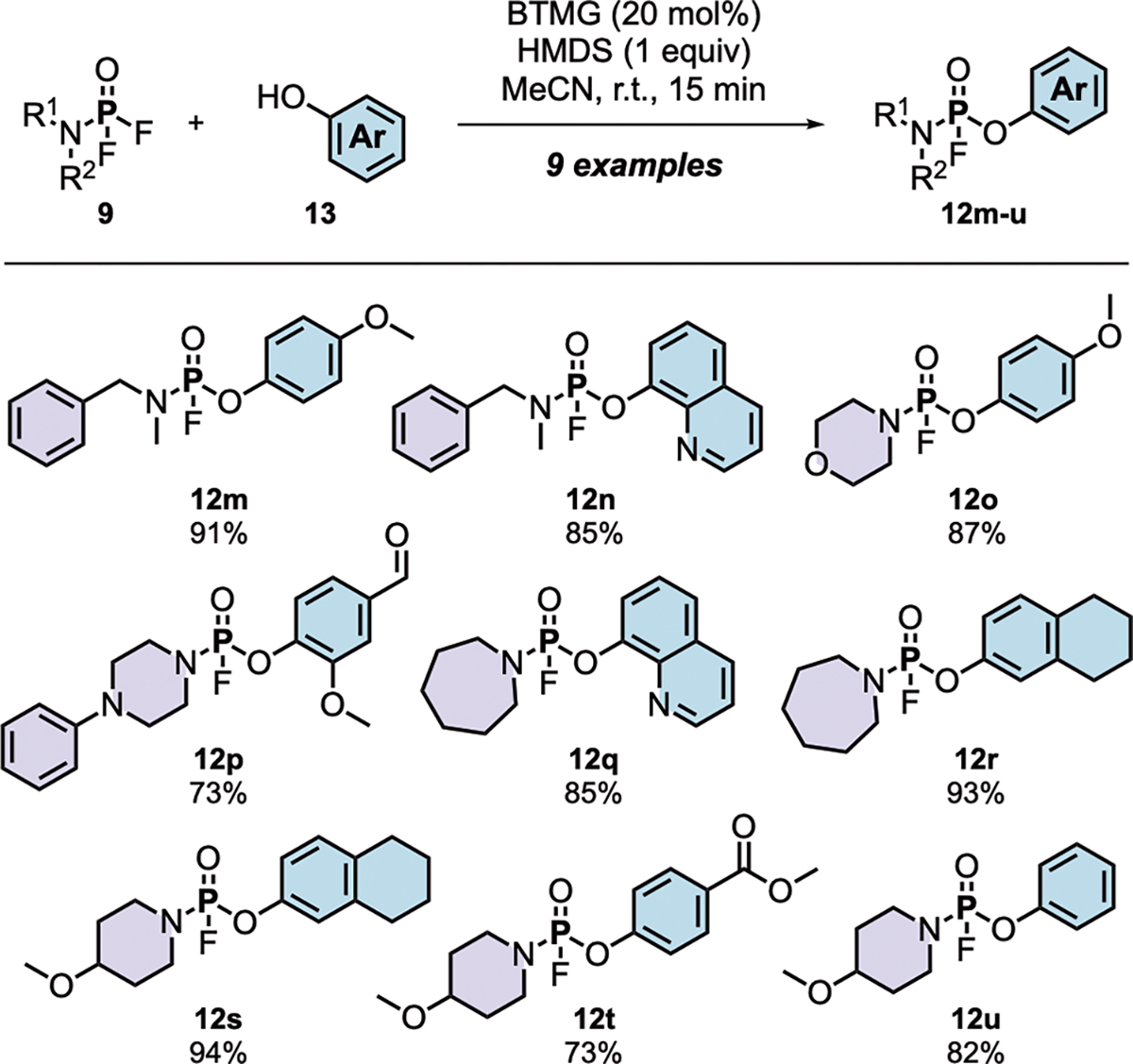
General reaction conditions: P(V)–F derivative (1.2 equiv) and phenol (1.0 equiv) were stirred in acetonitrile (0.4 M) in the presence of BTMG (20 mol%) and HMDS (1.0 equiv) for 15 min at room temperature. Isolated yields are reported. Reactions were conducted on a 1.20 mmol scale unless otherwise stated.
The cyclic phosphoramidofluoridate 10c was next reacted with 3,5-xylenol (14a) in the presence of BTMG and HMDS (Scheme 3A). In just 2 hours at room temperature, full consumption of the substrate 10c was observed, with the phosphoramidate product 15a isolated in 89% yield. The reaction with the analogous chloride 8c required 22 hours under similar conditions to reach a yield of just 70%. The PFEx substrate 12a was found to be less reactive under BTMG catalysis, requiring 12 hours at room temperature to achieve complete conversion to phosphoramidate 16a. In contrast, chloride 11a failed to yield any discernable product and degraded over the 22-hour reaction period (Scheme 3B). The enhanced reactivity of selected cyclic phosphates over their acyclic counterparts is well known87–92; with rates of hydrolysis and solvolysis up to a million times faster for cyclic species. The stability of phosphoramidic difluorides (9) relative to phosphoramidofluoridates (10 and 12) can be explained using an electronic rationale; having two highly electronegative fluorine atoms bonded to the phosphorus atom creates a substantially more positive P(V) core, leading to increased rates of hydrolysis and decomposition93. Replacing one fluorine atom with a less electronegative amino or phenoxy substituent stabilizes the phosphorus core toward hydrolysis/solvolysis.
PFEx Reaction Optimization and Scope
We next investigated a selection of catalysts to optimize the rate of the PFEx reaction. Due to their slow exchange reaction, the phosphoramidofluoridate 12a and 3,5-xylenol (14a) were chosen as model PFEx substrates. The catalyst screen revealed that 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and the phosphazene bases P4-tBu and P2-tBu were the superior catalysts, delivering quantitative yields of the PFEx product 16a in under 2 hours at room temperature (see Table 1, Tables S10–S12, and Figure S4 for full optimization, catalyst structures, and associated pKaH values in MeCN).
Table 1.
PFEx catalyst screen.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Catalyst | pKaH (MeCN) | Time (h) | Conversion (%)a |
|
| ||||
| 1 | P4-tBu | 42.7 | 1 | >99 |
| 2 | P2-tBu | 33.5 | 1.5 | >99 |
| 3 | TBD | 26.2 | 5 | >99 |
| 4 | BTMG | ~26 | 14 | 91 |
| 5 | DBU | 24.3 | 14 | 80 |
| 6 | P1-tBu | 26.9 | 14 | 10 |
| 7 | TMG | 23.7 | 14 | 9 |
| 8 | DPG | 18.8 | 14 | Trace |
| 9 | BEMP | 27.5 | 14 | 66 |
| 10 | MTBD | 25.0 | 14 | 64 |
| 11 | DMAP | 18.0 | 14 | 0 |
| 12 b | TBD | 26.2 | 2 | >99 |
| 13c | TBD | 26.2 | 7 | 16 |
| 14d | TBD | 26.2 | 7 | 20 |
Reactions were conducted on a 0.10 mmol scale in acetonitrile (0.25 M). Refer to supplementary information Figure S4 for a list of catalyst structures.
Conversions were determined by 31P NMR and 19F NMR.
1.20 equiv HMDS and 3, 5-dimethyphenol in MeCN (0.5 M) were employed.
HMDS was replaced with Et3N (1.0 equiv).
Without HMDS.
In the absence of the HMDS additive, none of the catalysts performed well, suggesting that synergism between the silicon reagent and catalyst is crucial for PFEx (cf. accelerated SuFEx)9. TBD was chosen as the preferred catalyst for further studies due to its relatively lower cost, tolerability of a wide selection of functional groups, and its position in a ‘sweet spot’ in terms of pKaH (TBD = 26.2 in MeCN) between the phosphazene superbases (pKaH 26.0 to 42.7) and guanidine/amidine bases (pKaH 18.8 to 25.0)99.
Monitoring the TBD-accelerated PFEx reaction between 12c and 3,5-xylenol (14a) by 1H NMR revealed a clean conversion to the phosphoramidate product 16c in just 60 minutes (Figure S6 and S7). No intermediates were identified on the NMR time scale9,27. Conversely, the reaction between 14a and chloridate 11c failed to deliver significant product over 24 hours, as determined by 1H NMR analysis (Figure S8–S10). Unreacted chloridate 11c instead decomposed to phosphorodiamidate S4, likely arising from competing P–Cl exchange with the stoichiometric HMDS reagent and subsequent N–Si bond cleavage (Figure S11). These results demonstrate the superior performance of catalyst-accelerated PFEx over P–Cl equivalents.
The optimized TBD-catalyzed PFEx conditions were successful with a range of P(V)–F substrates, affording P(V)–O linked products in excellent yield (Scheme 4). Reactions involving thiophosphoramidofluoridates required longer reaction times (i.e., 6 h required to form product 15b compared to 15 min for 15a)95. PFEx is tolerant to a range of functional groups, including aldehydes (15n), ketones (15p and 16g), esters (16h), and amides (16f). Aryl alcohols react chemoselectively as PFEx nucleophiles in the presence of secondary alcohols (15o) and anilines (15e). Noteworthy are the PFEx products incorporating natural products, including (+)-estrone (15j, dr = 1:1), (+)-estriol (15o, dr = 1:1), (+)-totarol (15f, dr = 1:1), and (–)-cholesterol (16e, dr = 2:1).
Scheme 4. Substrate scope for TBD-catalyzed PFEx reaction.
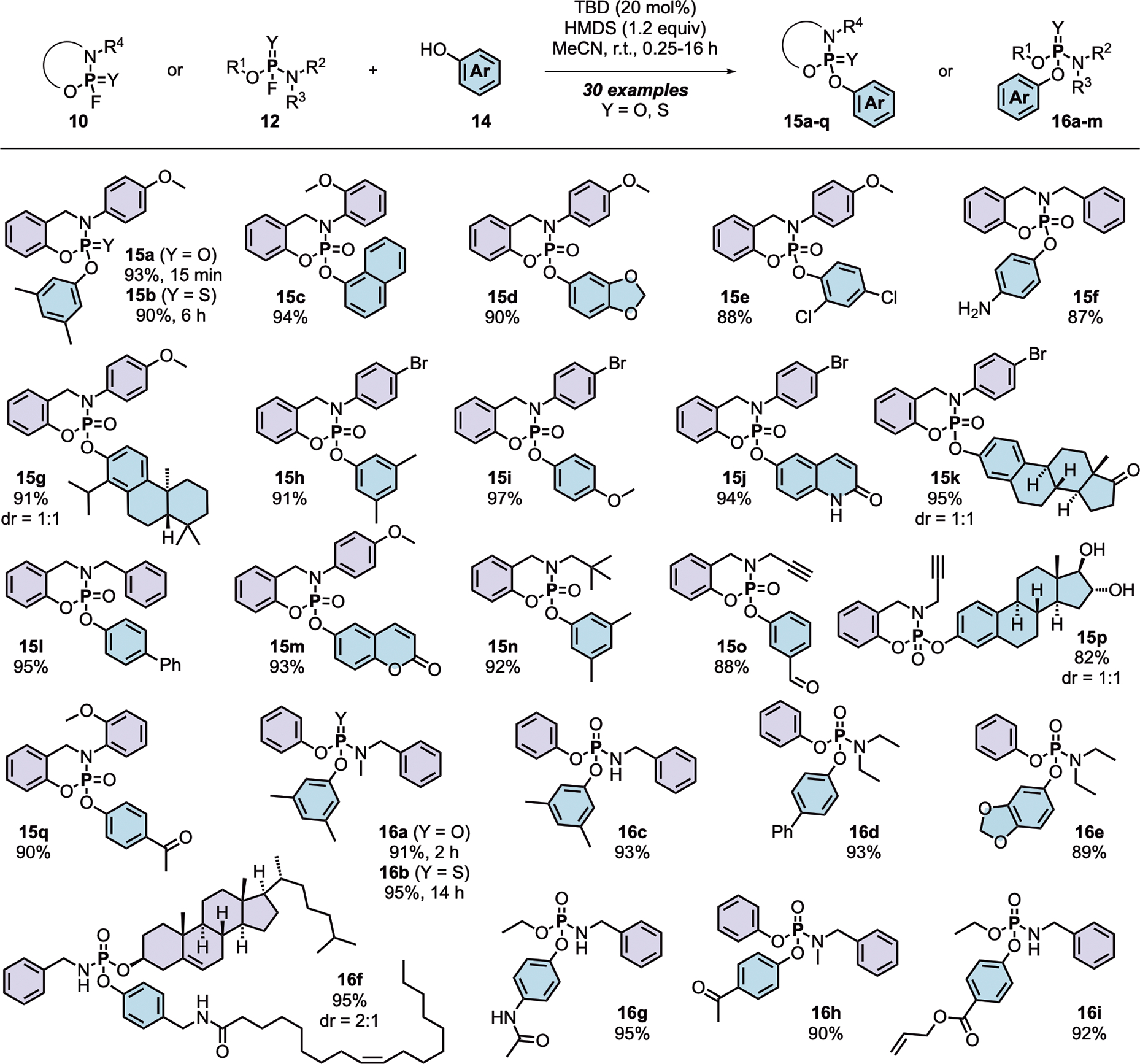
General reaction conditions: P(V)–F derivative (1.0 equiv) and phenol (1.2 equiv) were stirred in acetonitrile (0.4 M) in the presence of TBD (20 mol%) and HMDS (1.2 equiv) until completed as determined by TLC and both 19F and 31P NMR. Isolated yields are reported. See supporting information for a complete list of products.
Sequential PFEx Click Chemistry
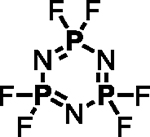
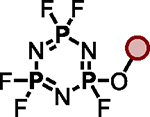
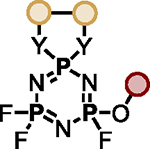
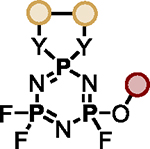
Polyfluorinated organophosphorus compounds offer significant potential in Diversity Oriented Clicking (DOC) strategies centered around PFEx21. Having demonstrated the robust mono-PFEx reaction of phosphoramidic difluorides (Scheme 2), we next explored the serial decoration of hexafluorocyclotriphosphazene (HFP) — a hub bearing six P–F bonds. Early studies by Shreeve and co-workers on the substitution of HFP by silyl-protected diols and dithiols found catalytic cesium fluoride facilitated this transformation96, while Chandrasekhar and Nagendran utilized phosphazenes to prepare a collection of multi-site coordinating ligands97. We discovered that diols could also be reacted with HFP in the presence of 2 equivalents of Et3N, affording the spirocyclophosphazene products (i.e., 17a) in good conversions without the need for silyl-protection (Scheme 5A). Alkyl alcohols and amines, such as 2-phenylethanol, benzylamine, azidothymidine (AZT), and cholesterol, behaved similarly with 1 equivalent of Et3N as a base, delivering compounds 17b–17e, respectively. Phenols underwent selective mono-PFEx with HFP when reacted in the presence of DMAP (10 mol%) and HMDS (1.0 equiv) (i.e., 17f) but generated intractable mixtures of PFEx products with HFP in the presence of stoichiometric Et3N or under TBD-catalyzed conditions.
Scheme 5. PFEx reactions of hexafluorocyclotriphosphazene (HFP).
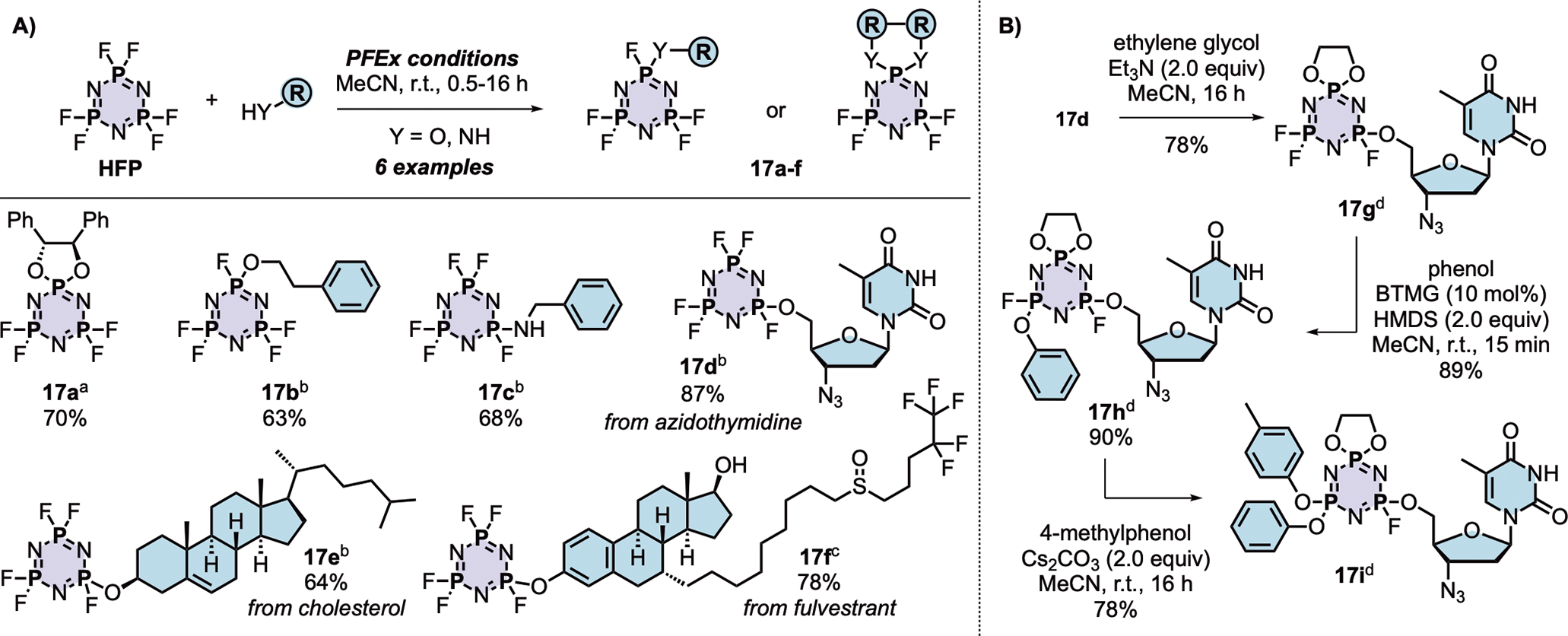
(A) Reaction with alcohols and phenols. PFEx conditions = [a]Et3N (2.0 equiv); [b]Et3N (1.0 equiv); [c]DMAP (10 mol%), HMDS (1.0 equiv).
(B) Sequential PFEx functionalization of HFP. [d]Inseparable, unquantifiable mixture of diastereoisomers obtained.
The multifunctional AZT-containing PFEx substrate 17d was further reacted with both ethylene glycol (17g, Scheme 5B) and ethylene diamine (17j, see SI). Substitution of the last remaining P–F bonds of 17g required more forcing conditions. Adding phenol to 17g required 10 mol% BTMG in the presence of HMDS. In contrast, 2 equivalents of cesium carbonate were required to react the geminal P–F bond of 17h with 4-methylphenol to give 17i98 — the product of 5 successive PFEx reactions.
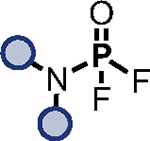
The serial functionalization of HFP highlights that judicious catalyst selection is crucial to obtain selective reactivity, especially when employing substrates with multiple P–F bonds. Tabulated below (Table 2) are the optimized reaction conditions for each substrate pair explored, which we believe will serve as a helpful resource when designing PFEx strategies.
Table 2.
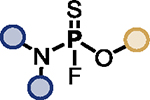
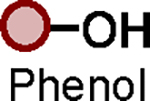
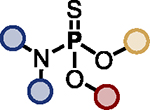
Optimized PFEx conditions.
| Entry | Phosphate | Substrate | Catalyst | Additive | Product |
|---|---|---|---|---|---|
|
| |||||
| 1 |
|
|
BTMG (20%) | HMDS (1.0 eq.) |
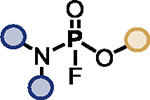
|
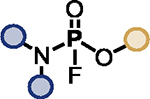
| 2 |
|
|
TBD (20%) | HMDS (1.2 eq.) |
|
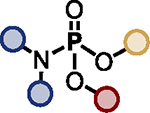
| 3 |
|
|
TBD (20%) | HMDS (1.2 eq.) |
|
| 4 |
|

|
DMAP (10%) | HMDS (1.0 eq.) |
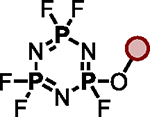
|
| 5 |
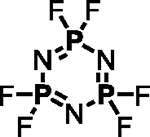
|
|
– | Et3N (1.0 eq.) |
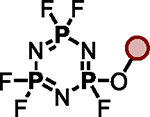
|
| 6 |
|
|
– | Et3N (2.0 eq.) |
|
| 7 |
|

|
BTMG (20%) | HMDS (1.0 eq.) |
|
| 8 |
|

|
– | Cs2CO3 (2.0 eq.) |
|
Orthogonal Click Chemistry
A key criterion of click reactions — and perhaps the most challenging to meet — is a requirement for chemoselective reactivity that allows connections to be made with control, ideally perfect control. To explore the resilience of PFEx as a click-compatible reaction, we prepared 18 as a model hub that is primed for three consecutive click reactions via i) a terminal alkyne for CuAAC, ii) a ‘SuFExable’ fluorosulfate, and iii) a ‘PFExable’ phosphoramidofluoridate (Scheme 6). First, under accelerated SuFEx conditions9 [BTMG (20 mol%), HMDS (1.0 equiv) in acetonitrile at room temperature, 30 min], the reaction of 18 with 4-phenylphenol afforded compound 19, exclusively; the incoming nucleophile reacting selectively with the fluorosulfate group. Next, 19 was subjected to TBD-accelerated PFEx conditions in the presence of allyl 4-hydroxybenzoate, giving the expected phosphoramidate 20 in an excellent 91% yield after 5 h. Finally, 20 was reacted with benzyl azide under standard CuAAC conditions [CuSO4•5H2O (10 mol%), sodium ascorbate (40 mol%) in DMF at room temperature, 3 h] yielding the ‘multi-clicked’ product 21. The sequential reaction of the click hub 18 under controlled conditions exemplifies the exquisite precision of chemoselective click transformations; the striking difference in reactivity between P–F and S–F clickable moieties creates a window of opportunity for orthogonal connective chemistry.
Scheme 6. The orthogonal reactivity between PFEx, SuFEx, and CuAAC catalysis.
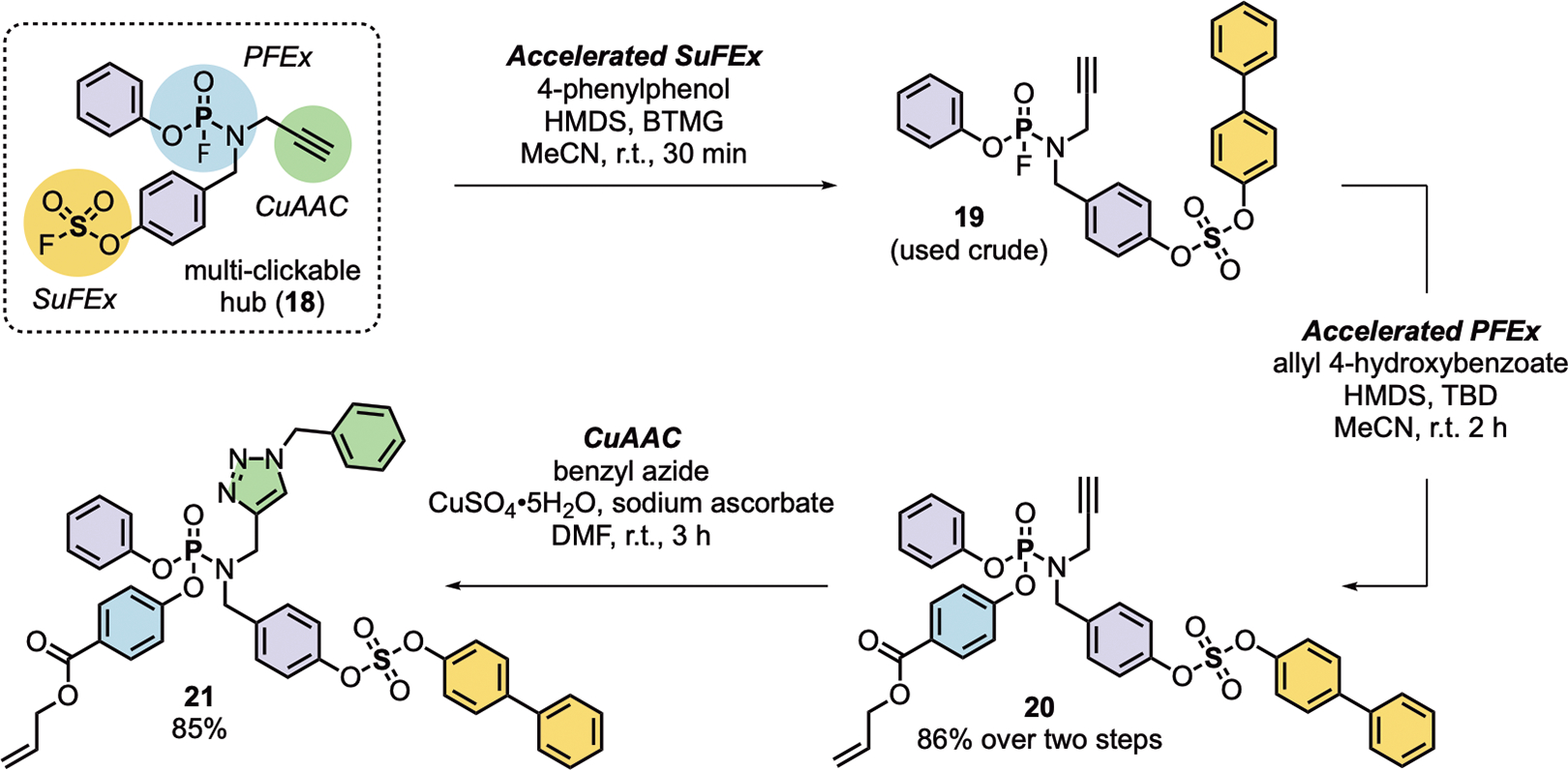
Accelerated SuFEx conditions: HMDS (1.0 equiv), BTMG (20 mol%), MeCN, r.t., 30 min. Accelerated PFEx conditions: HMDS (1.2 equiv), TBD (20 mol%), MeCN, r.t., 2 h. CuAAC conditions: CuSO4•5H2O (10 mol%), sodium ascorbate (40 mol%), DMF, r.t., 3 h.
Conclusion
In this work, we present PFEx (Phosphorus Fluoride Exchange) as a potent click reaction for discovering functional molecules. PFEx transformations proceed smoothly under Lewis nitrogen base catalysis, giving P–O and P–N linked products in high yield and in the absence of unwanted side-products. The superior reactivity of P(V)–F-containing compounds relative to their P(V)–Cl counterparts provides a unique compound class that can be selectively activated by appropriate catalysts, akin to SuFEx click reactions. Substrates with multiple P–F bonds offer an opportunity for Diversity Oriented Clicking, allowing for up to 5 successive steps of serial exchange reactions to create “multi-clickable” hubs, enabling selective PFEx reactions in the presence of SuFExable functional groups. This innovation in P(V)–F chemistry will undoubtedly spark new research and developments in the field.
EXPERIMENTAL PROCEDURES
Resource availability
Lead contact
Further information and requests for resources should be directed to, and will be fulfilled by, the lead contact, John E. Moses (moses@cshl.edu).
Materials availability
Full experimental details and characterization data can be found in the supplemental information.
Data and code availability
All data supporting this study are available in the manuscript or supplemental information.
Supplementary Material
ACKNOWLEDGMENTS
J.E.M. is thankful to the NCI Cancer Center Support Grant 5P30CA045508, Cold Spring Harbor Laboratory Northwell Health Affiliation, the F. M. Kirby Foundation, the Sunshine Foundation, S. J. Edwards, The Starr Foundation, The Wasily Family Foundation, the Australian Research Council (Future Fellowship; FT170100156), and La Trobe University for their generous support. K.B.S. is thankful for the financial support of the National Institutes of Health R01GM117145. This work was performed with assistance from the CSHL Mass Spectrometry and Metabolomics and CSHL Animal Imaging Shared Resources, which are supported in part by the Cancer Center Support Grant 5P30CA045508. We acknowledge Dr. Andrew S. Barrow, Dr. Grant A. L. Bare, and Dr. Nitin Upadhyay for their insights and contribution to PFEx click chemistry. The authors also thank Rebecca Koelln and Dr. Adam D. Moorhouse for assisting with the preparation of the manuscript and supporting information.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online.
A Note on Safety:
The work described in this manuscript involves the synthesis and handling of organophosphorus compounds, including organofluorophosphates. The authors strongly advise colleagues to acquaint themselves with the extensive literature36 on the toxicological properties of known representative compounds and to adhere to strict safety protocols. See supporting information for more details.
REFERENCES
- 1.Kolb HC, Finn MG, and Sharpless KB (2001). Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- 2.Moses JE, and Moorhouse AD (2007). The growing applications of click chemistry. Chem. Soc. Rev. 36, 1249–1262. 10.1039/B613014N. [DOI] [PubMed] [Google Scholar]
- 3.Rostovtsev VV, Green LG, Fokin VV, and Sharpless KB (2002). A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- 4.Moorhouse AD, and Moses JE (2008). Click Chemistry and Medicinal Chemistry: A Case of “Cyclo-Addiction.” ChemMedChem 3, 715–723. 10.1002/cmdc.200700334. [DOI] [PubMed] [Google Scholar]
- 5.Meng G, Guo T, Ma T, Zhang J, Shen Y, Sharpless KB, and Dong J (2019). Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89. 10.1038/s41586-019-1589-1. [DOI] [PubMed] [Google Scholar]
- 6.Takacs GA (1978). Heats of formation and bond dissociation energies of some simple sulfur- and halogen-containing molecules. J. Chem. Eng. Data 23, 174–175. 10.1021/je60077a020. [DOI] [Google Scholar]
- 7.Grant DJ, Matus MH, Switzer JR, Dixon DA, Francisco JS, and Christe KO (2008). Bond Dissociation Energies in Second-Row Compounds. J. Phys. Chem. A 112, 3145–3156. 10.1021/jp710373e. [DOI] [PubMed] [Google Scholar]
- 8.Dong J, Krasnova L, Finn MG, and Sharpless KB (2014). Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- 9.Smedley CJ, Homer JA, Gialelis TL, Barrow AS, Koelln RA, and Moses JE (2022). Accelerated SuFEx Click Chemistry for Modular Synthesis. Angew. Chem. Int. Ed. 61, e202112375. 10.1002/anie.202112375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei M, Liang D, Cao X, Luo W, Ma G, Liu Z, and Li L (2021). A Broad-Spectrum Catalytic Amidation of Sulfonyl Fluorides and Fluorosulfates. Angew. Chem. Int. Ed. 60, 7397–7404. 10.1002/anie.202013976. [DOI] [PubMed] [Google Scholar]
- 11.Luy J-N, and Tonner R (2020). Complementary Base Lowers the Barrier in SuFEx Click Chemistry for Primary Amine Nucleophiles. ACS Omega 5, 31432–31439. 10.1021/acsomega.0c05049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahapatra S, Woroch CP, Butler TW, Carneiro SN, Kwan SC, Khasnavis SR, Gu J, Dutra JK, Vetelino BC, Bellenger J, et al. (2020). SuFEx Activation with Ca(NTf2)2: A Unified Strategy to Access Sulfamides, Sulfamates, and Sulfonamides from S(VI) Fluorides. Org. Lett. 22, 4389–4394. 10.1021/acs.orglett.0c01397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao B, Li S, Wu P, Moses JE, and Sharpless KB (2018). SuFEx Chemistry of Thionyl Tetrafluoride (SOF4) with Organolithium Nucleophiles: Synthesis of Sulfonimidoyl Fluorides, Sulfoximines, Sulfonimidamides, and Sulfonimidates. Angew. Chem. Int. Ed. 57, 1957–1961. 10.1002/ange.201712145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smedley CJ, Zheng Q, Gao B, Li S, Molino A, Duivenvoorden HM, Parker BS, Wilson DJD, Sharpless KB, and Moses JE (2019). Bifluoride Ion Mediated SuFEx Trifluoromethylation of Sulfonyl Fluorides and Iminosulfur Oxydifluorides. Angew. Chem. Int. Ed. 58, 4552–4556. 10.1002/anie.201813761. [DOI] [PubMed] [Google Scholar]
- 15.Zeng D, Ma Y, Deng W-P, Wang M, and Jiang X (2022). Divergent sulfur(VI) fluoride exchange linkage of sulfonimidoyl fluorides and alkynes. Nat. Synth. 1, 455–463. 10.1038/s44160-022-00060-1. [DOI] [Google Scholar]
- 16.Barrow AS, Smedley CJ, Zheng Q, Li S, Dong J, and Moses JE (2019). The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 48, 4731–4758. 10.1039/C8CS00960K. [DOI] [PubMed] [Google Scholar]
- 17.Moissan H, and Lebeau P (1902). Invesigation of sulfur fluorides and sulfur oxyfluorides. Ann. Chim. Phys. 26, 145–178. [Google Scholar]
- 18.Li S, Wu P, Moses JE, and Sharpless KB (2017). Multidimensional SuFEx Click Chemistry: Sequential Sulfur(VI) Fluoride Exchange Connections of Diverse Modules Launched From An SOF4 Hub. Angew. Chem. Int. Ed. 56, 2903–2908. 10.1002/anie.201611048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krutak JJ, Burpitt RD, Moore WH, and Hyatt JA (1979). Chemistry of ethenesulfonyl fluoride. Fluorosulfonylethylation of organic compounds. J. Org. Chem. 44, 3847–3858. 10.1021/jo01336a022. [DOI] [Google Scholar]
- 20.Giel M-C, Smedley CJ, Mackie ERR, Guo T, Dong J, Costa T.P.S. da, and Moses JE. (2020). Metal-Free Synthesis of Functional 1-Substituted-1,2,3-Triazoles from Ethenesulfonyl Fluoride and Organic Azides. Angew. Chem. Int. Ed. 59, 1181–1186. 10.1002/ange.201912728. [DOI] [PubMed] [Google Scholar]
- 21.Smedley CJ, Li G, Barrow AS, Gialelis TL, Giel M-C, Ottonello A, Cheng Y, Kitamura S, Wolan DW, Sharpless KB, et al. (2020). Diversity Oriented Clicking (DOC): Divergent Synthesis of SuFExable Pharmacophores from 2-Substituted-Alkynyl-1-Sulfonyl Fluoride (SASF) Hubs. Angew. Chem. Int. Ed. 59, 12460–12469. 10.1002/anie.202003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gembus V, Marsais F, and Levacher V (2008). An Efficient Organocatalyzed Interconversion of Silyl Ethers to Tosylates Using DBU and p-Toluenesulfonyl Fluoride. Synlett 10, 1463–1466. 10.1055/s-2008-1078407. [DOI] [Google Scholar]
- 23.Lee C, Cook AJ, Elisabeth JE, Friede NC, Sammis GM, and Ball ND (2021). The Emerging Applications of Sulfur(VI) Fluorides in Catalysis. ACS Catal. 11, 6578–6589. 10.1021/acscatal.1c01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao B, Zhang L, Zheng Q, Zhou F, Klivansky LM, Lu J, Liu Y, Dong J, Wu P, and Sharpless KB (2017). Bifluoride-catalysed sulfur(VI) fluoride exchange reaction for the synthesis of polysulfates and polysulfonates. Nat. Chem. 9, 1083–1088. 10.1038/nchem.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Revathi L, Ravindar L, Leng J, Rakesh KP, and Qin H-L (2018). Synthesis and Chemical Transformations of Fluorosulfates. Asian J. Org. Chem. 7, 662–682. 10.1002/ajoc.201700591. [DOI] [Google Scholar]
- 26.Liu C, Yang C, Hwang S, Ferraro SL, Flynn JP, and Niu J (2020). A General Approach to O-Sulfation by a Sulfur(VI) Fluoride Exchange Reaction. Angew. Chem. Int. Ed. 59, 18435–18441. 10.1002/anie.202007211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang D-D, Streefkerk DE, Jordaan D, Wagemakers J, Baggerman J, and Zuilhof H (2020). Silicon-Free SuFEx Reactions of Sulfonimidoyl Fluorides: Scope, Enantioselectivity, and Mechanism. Angew. Chem. Int. Ed. 59, 7494–7500. 10.1002/ange.201915519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westheimer FH (1987). Why Nature Chose Phosphates. Science 235, 1173–1178. 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- 29.Knouse KW, Flood DT, Vantourout JC, Schmidt MA, Mcdonald IM, Eastgate MD, and Baran PS (2021). Nature Chose Phosphates and Chemists Should Too: How Emerging P(V) Methods Can Augment Existing Strategies. ACS Cent. Sci. 7, 1473–1485. 10.1021/acscentsci.1c00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamerlin SCL, Sharma PK, Prasad RB, and Warshel A (2013). Why nature really chose phosphate. Q. Rev. Biophys. 46, 1–132. 10.1017/S0033583512000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franz Anton Voegeli accessed triethyl phosphate (ca. 1848), while Clermont and Moschnin synthesized tetraethyl pyrophosphate in 1854. [Google Scholar]
- 32.Organophosphorus compounds appeared in the literature more frequently following the Second World War when the element’s importance was recognized. [Google Scholar]
- 33.Petroianu GA (2009). History of methyl phosphoric esters: Hall, Weger, and Lossen. Pharmazie 64, 840–845. [PubMed] [Google Scholar]
- 34.Petroianu GA (2009). The synthesis of phosphor ethers: who was Franz Anton Voegeli? Pharmazie, 269–275. 10.1691/ph.2009.8244. [DOI] [PubMed] [Google Scholar]
- 35.Cadogan JIG (1979). Organophosphorus Reagents in Organic Synthesis (Academic Press; ). [Google Scholar]
- 36.Timperley C (2014). Best Synthetic Methods: Organophosphorus (V) Chemistry (Newnes; ). [Google Scholar]
- 37.Murphy PJ (2004). Organophosphorus Reagents: A Practical Approach in Chemistry (Oxford University Press; ). [Google Scholar]
- 38.Corbridge DEC (2013). Phosphorus: Chemistry, Biochemistry and Technology, Sixth Edition (CRC Press; ). [Google Scholar]
- 39.Kurti L, and Czako B (2005). Strategic Applications of Named Reactions in Organic Synthesis (Elsevier; ). [Google Scholar]
- 40.Kolodiazhnyi OI (2008). Phosphorus Ylides: Chemistry and Applications in Organic Synthesis (John Wiley & Sons; ). [Google Scholar]
- 41.Juge S, and Genet JP (1989). Asymmetric synthesis of phosphinates, phosphine oxides and phosphines by Michaelis Arbuzov rearrangement of chiral oxazaphospholidine. Tetrahedron Lett. 30, 2783–2786. 10.1016/S0040-4039(00)99124-X. [DOI] [Google Scholar]
- 42.Juge S, Stephan M, Laffitte JA, and Genet JP (1990). Efficient asymmetric synthesis of optically pure tertiary mono and diphosphine ligands. Tetrahedron Lett. 31, 6357–6360. 10.1016/S0040-4039(00)97063-1. [DOI] [Google Scholar]
- 43.Han ZS, Goyal N, Herbage MA, Sieber JD, Qu B, Xu Y, Li Z, Reeves JT, Desrosiers J-N, Ma S, et al. (2013). Efficient Asymmetric Synthesis of P-Chiral Phosphine Oxides via Properly Designed and Activated Benzoxazaphosphinine-2-oxide Agents. J. Am. Chem. Soc. 135, 2474–2477. 10.1021/ja312352p. [DOI] [PubMed] [Google Scholar]
- 44.Corey EJ, Chen Z, and Tanoury GJ (1993). A new and highly enantioselective synthetic route to P-chiral phosphines and diphosphines. J. Am. Chem. Soc. 115, 11000–11001. 10.1021/ja00076a072. [DOI] [Google Scholar]
- 45.Knouse KW, deGruyter JN, Schmidt MA, Zheng B, Vantourout JC, Kingston C, Mercer SE, Mcdonald IM, Olson RE, Zhu Y, et al. (2018). Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 361, 1234–1238. 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu D, Rivas-Bascón N, Padial NM, Knouse KW, Zheng B, Vantourout JC, Schmidt MA, Eastgate MD, and Baran PS (2020). Enantiodivergent Formation of C–P Bonds: Synthesis of P-Chiral Phosphines and Methylphosphonate Oligonucleotides. J. Am. Chem. Soc. 142, 5785–5792. 10.1021/jacs.9b13898. [DOI] [PubMed] [Google Scholar]
- 47.Kuwabara K, Maekawa Y, Minoura M, Maruyama T, and Murai T (2020). Chemoselective and Stereoselective Alcoholysis of Binaphthyl Phosphonothioates: Straightforward Access to Both Stereoisomers of Biologically Relevant P-Stereogenic Phosphonothioates. J. Org. Chem. 85, 14446–14455. 10.1021/acs.joc.0c00687. [DOI] [PubMed] [Google Scholar]
- 48.Koizumi T, Yanada(nee Ishizaka) R, Takagi H, Hirai H, and Yoshii E. (1981). Grignard reaction of 2-phenyl-tetrahydropyrrolo-1,5,2-oxazaphospholes, observation of the stereospecific inversion of configuration. Tetrahedron Lett. 22, 571–572. 10.1016/S0040-4039(01)90157-1. [DOI] [Google Scholar]
- 49.Mondal A, Thiel NO, Dorel R, and Feringa BL (2022). P-chirogenic phosphorus compounds by stereoselective Pd-catalysed arylation of phosphoramidites. Nat. Catal. 5, 10–19. 10.1038/s41929-021-00697-9. [DOI] [Google Scholar]
- 50.DiRocco DA, Ji Y, Sherer EC, Klapars A, Reibarkh M, Dropinski J, Mathew R, Maligres P, Hyde AM, Limanto J, et al. (2017). A multifunctional catalyst that stereoselectively assembles prodrugs. Science 356, 426–430. 10.1126/science.aam7936. [DOI] [PubMed] [Google Scholar]
- 51.Featherston AL, Kwon Y, Pompeo MM, Engl OD, Leahy DK, and Miller SJ (2021). Catalytic asymmetric and stereodivergent oligonucleotide synthesis. Science 371, 702–707. 10.1126/science.abf4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forbes KC, and Jacobsen EN (2022). Enantioselective hydrogen-bond-donor catalysis to access diverse stereogenic-at-P(V) compounds. Science 376, 1230–1236. 10.1126/science.abp8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinkopf W (1927). Über Aromatische Sulfofluoride. J. Prakt. Chem. 117, 1–82. 10.1002/prac.19271170101. [DOI] [Google Scholar]
- 54.Steinkopf W, and Jaeger P (1930). Über Aromatische Sulfofluoride. II. Mitteilung. J. Prakt. Chem. 128, 63–88. 10.1002/prac.19301280104. [DOI] [Google Scholar]
- 55.Davies W, and Dick JH (1932). 285. Benzenesulphonyl fluoride derivatives. J. Chem. Soc, 2042–2046. 10.1039/JR9320002042. [DOI] [Google Scholar]
- 56.Davies W, and Dick JH (1932). 57. Aliphatic sulphonyl fluorides. J. Chem. Soc, 483–486. 10.1039/JR9320000483. [DOI] [Google Scholar]
- 57.Fernández LE, and Varetti EL (2005). A scaled quantum mechanical force field for the sulfuryl halides: II. The SO2XF (X=Cl, Br) halides. Spectrochim. Acta A Mol. Biomol. Spectrosc. 62, 221–225. 10.1016/j.saa.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 58.Müller HSP, and Gerry MCL (1994). Microwave spectroscopic investigation of thionyl chloride, SOCl2: hyperfine constants and harmonic force field. J. Chem. Soc., Faraday Trans. 90, 3473–3481. 10.1039/FT9949003473. [DOI] [Google Scholar]
- 59.Ciuffarin E, Senatore L, and Isola M (1972). Nucleophilic substitution at four-co-ordinate sulphur. Mobility of the leaving group. J. Chem. Soc., Perkin Trans. 2, 468–471. 10.1039/P29720000468. [DOI] [Google Scholar]
- 60.Durig JR, and Casper JM (1971). Vibrational spectra and structure of organophosphorus compounds. X. Methyl torsional frequencies and barriers to internal rotation of some CH3PXY2 compounds. J. Phys. Chem. 75, 1956–1963. 10.1021/j100682a009. [DOI] [PubMed] [Google Scholar]
- 61.Huang X, Zhao X, Zhang M, Xu Y, Zhi H, and Yang J (2017). Green Synthesis of Triaryl Phosphates with POCl3 in Water. ChemistrySelect 2, 11007–11011. 10.1002/slct.201702215. [DOI] [Google Scholar]
- 62.Stölzer C, and Simon A (1960). Über Fluorphosphorverbindungen, I. Chem. Ber. 93, 1323–1331. 10.1002/cber.19600930613. [DOI] [Google Scholar]
- 63.Dehnicke K, and Shihada A-F (1976). Structural and bonding aspects in phosphorus chemistry-inorganic derivatives of oxohaloqeno phosphoric acids. In Electrons in Oxygen- and Sulphur-Containing Ligands Structure and Bonding. (Springer; ), pp. 51–82. 10.1007/3-540-07753-7_2. [DOI] [Google Scholar]
- 64.Refer to Table S8 for more details. [Google Scholar]
- 65.worowska I, Dąbkowski W, and Michalski J (2001). Synthesis of Tri- and Tetracoordinate Phosphorus Compounds Containing a PCF3 Group by Nucleophilic Trifluoromethylation of the Corresponding PF Compounds. Angew. Chem. Int. Ed. 40, 2982–2984. . [DOI] [PubMed] [Google Scholar]
- 66.Abbott A, Sierakowski T, Kiddle JJ, Clark KK, and Mezyk SP (2010). Detailed Investigation of the Radical-Induced Destruction of Chemical Warfare Agent Simulants in Aqueous Solution. J. Phys. Chem. B 114, 7681–7685. 10.1021/jp101720j. [DOI] [PubMed] [Google Scholar]
- 67.Delfino RT, Ribeiro TS, and Figueroa-Villar JD (2009). Organophosphorus compounds as chemical warfare agents: a review. J. Braz. Chem. Soc. 20, 407–428. 10.1590/S0103-50532009000300003. [DOI] [Google Scholar]
- 68.Franca TCC, Kitagawa DAS, Cavalcante S.F. de A., da Silva JAV, Nepovimova E, and Kuca K. (2019). Novichoks: The Dangerous Fourth Generation of Chemical Weapons. Int. J. Mol. Sci. 20, 1222. 10.3390/ijms20051222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Egron D, Arzumanov AA, Dyatkina NB, Aubertin A-M, Imbach J-L, Gosselin G, Krayevsky A, and Périgaud C (2001). Synthesis, Anti-HIV Activity, and Stability Studies of 5′-Phosphorofluoridate Derivatives of AZT. Bioorg. Chem. 29, 333–344. 10.1006/bioo.2001.1220. [DOI] [PubMed] [Google Scholar]
- 70.Baranowski MR, Warminski M, Jemielity J, and Kowalska J (2020). 5′-fluoro(di)phosphate-labeled oligonucleotides are versatile molecular probes for studying nucleic acid secondary structure and interactions by 19F NMR. Nucleic Acids Res. 48, 8209–8224. 10.1093/nar/gkaa470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malatová Z, Gottlieb M, and Marsala J (1999). Depression of acetylcholinesterase synthesis following transient cerebral ischemia in rat: pharmacohistochemical and biochemical investigation. Gen. Physiol. Biophys. 18, 57–71. [PubMed] [Google Scholar]
- 72.Liu Y, Patricelli MP, and Cravatt BF (1999). Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. U.S.A. 96, 14694–14699. 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grigoryan H, Li B, Anderson EK, Xue W, Nachon F, Lockridge O, and Schopfer LM (2009). Covalent binding of the organophosphorus agent FP-biotin to tyrosine in eight proteins that have no active site serine. Chem. Biol. Interact. 180, 492–498. 10.1016/j.cbi.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schultze H (1880). Ueber die Oxydation von Haloidsalzen. J. Prakt. Chem. 21, 407–443. [Google Scholar]
- 75.Moissan H (1886). Action d’un courant électrique sur l’acide fluorhydrique anhydre. C. R. Acad. Sci. 102, 1543–1544. [Google Scholar]
- 76.Olah RG, Oswald AA, and Kuhn S (1959). Untersuchung organischer Phosphorverbindungen, III Darstellung von Difluorphosphorsäure- und von Difluorthiophosphorsäure-alkylamiden. Justus Liebigs Ann. Chem. 625, 88–91. 10.1002/jlac.19596250111. [DOI] [Google Scholar]
- 77.Cavell RG (1967). Chemistry of phosphorus fluorides. Part II. Secondary alkylamino derivatives of phosphoryl fluoride. Can. J. Chem. 45, 1309–1319. 10.1139/v67-217. [DOI] [Google Scholar]
- 78.Seel F, Ballreich K, and Peters W (1959). Darstellung anorganischer Fluorverbindungen mittels Benzoylfluorids (und Benzolsulfofluorids). Chem. Ber. 92, 2117–2122. [Google Scholar]
- 79.Booth HS, and Cassidy MC (1940). The Fluorination of Thiophosphoryl Trichloride: The Thiophosphoryl Chlorofluorides. J. Am. Chem. Soc. 62, 2369–2372. 10.1021/ja01866a031. [DOI] [Google Scholar]
- 80.The analogous SuFEx gases (i.e., SO2F2) are also toxic though are considerably less reactive than POF3.
- 81.Anderson HH (1953). Exchange Reactions in Volatile Isocyanates and Isothiocyanates of Silicon, Germanium and Phosphorus. J. Am. Chem. Soc. 75, 1576–1578. 10.1021/ja01103a016. [DOI] [Google Scholar]
- 82.Refer to Supplementary Information Figure S1 for naming conventions of P(V) compounds.
- 83.The existing literature methods to convert phosphoramidic dichlorides to the corresponding difluorides employ reagents like triethylamine hydrofluoride, sodium fluoride, or silver fluoride and are generally plagued by poor yields. [Google Scholar]
- 84.The limited stability of phosphoramidic difluorides is well reported. For example, these compounds readily decompose to give nitriles and orthophosphoric difluoride. As such, phospharmidic difluorides are often used without purification. See Smaliy RV, Chaikovskaya AA, and Pinchuk AM. (2006). Reactions of isocyanatophosphoryl difluoride with π-abundant nitrogen heterocycles and carbonyl compounds. Russ. Chem. Bull. 55, 585–587. 10.1007/s11172-006-0297-9. [DOI] [Google Scholar]
- 85.The phosphoramidates 15d and 16a were found to be comparably stable to the fluoridates 10c and 12a, respectively. The HFP-containing product 17d, was significantly less stable, likely due to decomposition of the HFP ring in aqueous conditions. Refer to Tables S5–S7. [Google Scholar]
- 86.Heap R, and Saunders BC (1948). 261. Esters containing phosphorus. Part VII. Substituted diaminofluorophosphine oxides. J. Chem. Soc, 1313–1316. 10.1039/JR9480001313. [DOI] [PubMed] [Google Scholar]
- 87.Verkade JG (1974). Phosphate basicity and nucleophilicity loss upon constraint: The role of the alkoxy oxygens. Bioinorg. Chem. 3, 165–182. 10.1016/S0006-3061(00)80040-X. [DOI] [PubMed] [Google Scholar]
- 88.Chang N, and Lim C (1998). Factors Governing the Enhanced Reactivity of Five-Membered Cyclic Phosphate Esters. J. Am. Chem. Soc. 120, 2156–2167. 10.1021/ja9729802. [DOI] [Google Scholar]
- 89.Dudev T, and Lim C (1998). Ring Strain Energies from ab Initio Calculations. J. Am. Chem. Soc. 120, 4450–4458. 10.1021/ja973895x. [DOI] [Google Scholar]
- 90.Núñez A, Berroterán D, and Núñez O (2003). Hydrolysis of cyclic phosphoramides. Evidence for syn lone pair catalysis. Org. Biomol. Chem. 1, 2283–2289. 10.1039/B300916E. [DOI] [PubMed] [Google Scholar]
- 91.Uchimaru T, Kawahara S, Tsuzuki S, Matsumura K, and Taira K (1999). Solution-phase energy profiles for trigonal bipyramidal species postulated as intermediates for the hydrolysis of methyl ethylene phosphate. J. Mol. Struct. THEOCHEM 469, 215–221. 10.1016/S0166-1280(99)00072-X. [DOI] [Google Scholar]
- 92.Lim C (1999). Ring Strain VS. Solvent Effects in Phosphate Base Hydrolysis. Phosphorus Sulfur Silicon Relat. Elem. 144, 769–773. 10.1080/10426509908546358. [DOI] [Google Scholar]
- 93.Crunden EW, and Hudson RF (1962). 702. The mechanism of hydrolysis of phosphorochloridates and related compounds. Part III. Phosphoramidochloridates. J. Chem. Soc, 3591–3599. 10.1039/JR9620003591. [DOI] [Google Scholar]
- 94.Ishikawa T (2009). Superbases for organic synthesis: guanidines, amidines and phosphazenes and related organocatalysts (Wiley; ). [Google Scholar]
- 95.Harger MJP (2005). A new mechanism for nucleophilic substitution at a thiophosphoryl centre revealed by the reaction of diisopropylamine with PSCl3. Chem. Commun, 2863–2865. 10.1039/B502615F. [DOI] [PubMed] [Google Scholar]
- 96.Vij A, Geib SJ, Kirchmeier RL, and Shreeve JM (1996). Fluoride Ion Induced Reactions of Silicon–Oxygen and Silicon–Sulfur Bonds with Hexafluorocyclotriphosphazenes: Synthesis, Reactivity, and X-ray Structural Analyses of Sulfur/Oxygen-Containing Monospirofluorophosphazenes. Inorg. Chem. 35, 2915–2929. 10.1021/ic951065j. [DOI] [Google Scholar]
- 97.Chandrasekhar V, and Nagendran S (2001). Phosphazenes as scaffolds for the construction of multi-site coordination ligands. Chem. Soc. Rev. 30, 193–203. 10.1039/B004872K. [DOI] [Google Scholar]
- 98.Products 17g–17i were obtained as diastereomeric mixtures that were inseparable by either flash column chromatography or LC-MS (refer to Figure S12 for representative 31P NMR spectra). [Google Scholar]
- 99.In multi-click sequences, SuFEx reactions must often be conducted prior to PFEx reactions. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting this study are available in the manuscript or supplemental information.