

Abstract
Purpose:
Curcumin is an ideal chemopreventive and antitumor agent characterized by poor bioavailability and low stability. The development of synthetic structural analogues like dimethoxycurcumin (DMC) could overcome these drawbacks. In this study we compared the cytotoxicity, metabolism and the epigenetic changes induced by both drugs in leukemia cells.
Methods:
Apoptosis and cell cycle analysis were analyzed by flow cytometry. Real-time PCR was used for gene expression analysis. DNA methylation was analyzed by DNA pyrosequencing. The metabolic stability was determined using human pooled liver microsomes. Chromatin Immunoprecipitation was used to quantify histone methylation.
Results:
Clinically relevant concentration of curcumin and DMC were not cytotoxic to leukemia cells and induced G2/M cell cycle arrest. DMC was more metabolically stable than curcumin. Curcumin and DMC were devoid of DNA hypomethylating activity. DMC induced the expression of promoter methylated genes without reversing DNA methylation and increased H3K36me3 mark near the promoter region of hypermethylated genes.
Conclusion:
DMC is a more stable analogue of curcumin that can induce epigenetic changes not induced by curcumin. DMC induced the expression of promoter methylated genes. The combination of DMC with DNA methyltransferase inhibitors could harness their combined induced epigenetic changes for optimal re-expression of epigenetically silenced genes.
Keywords: Curcumin, dimethoxycurcumin, DNA methylation, DNA pyrosequencing, histone methylation
Introduction
Curcumin, a hydrophobic polyphenol derived from the rhizome of the herb Curcuma longa (turmeric) has a wide spectrum of pharmacological activities (1). Curcumin is the active ingredient of the Indian spice turmeric in addition to the other two ingredients, demethoxycurcumin and bisdemethoxycurcumin. Curcumin demonstrated anti-inflammatory (2), antimicrobial (3), antiviral (4), antioxidant (5) and anti-tumorigenic activity (6–8) in several studies. Curcumin is considered an ideal chemopreventive and antitumor agent because of its multiple targets. Curcumin is safe when administered at high doses; however, its low bioavailability due to poor absorption and rapid metabolism is a major drawback. Different formulation based approaches were adopted to overcome its low bioavailability like liposomal curcumin and curcumin nanoparticles. Additionally, several structural analogues like pyrimidine-substituted curcumin analogues (9), carbonyl moiety modified analogues (10), dimethoxycurcumin (DMC) (11), T63 (12), EF31 (13), UBS109 (13) and C086 (14) were also synthesized to improve the solubility and bioavailability of curcumin.
Curcumin and its analogues were shown to induce epigenetic changes in tumor cells. Curcumin modulated histone acetylation by inhibiting histone deacetylase (HDAC) and histone acetyltransferase (HAT) enzymes in tumor cells (15). Curcumin modulated microRNAs (miRNAs) expression in tumor cells (16). Moreover, curcumin demonstrated a DNA hypomethylating effect and induced the expression of silenced promoter-methylated genes in several studies (13, 17–22). However, other reports demonstrated that curcumin lacks a DNA hypomethylating effect or only modify DNA methylation in partially methylated loci (23, 24). The controversy regarding the DNA hypomethylating effect of curcumin and its analogues remains to be further elucidated.
Curcumin was also shown to induce apoptosis and cell cycle arrest in tumor cells (1, 8). Unfortunately, most of the previous reports that studied the epigenetic changes or the cytotoxic effect of curcumin used very high concentrations of curcumin (5–30 μM) that are considered clinically irrelevant. Curcumin achievable concentration in plasma was reported as 1.77 ± 1.87 μmol/L.
In this study, we are reporting the DNA and histone methylation changes induced by clinically relevant concentrations of pure curcumin and its synthetic analogue DMC in leukemia cells. Clinically relevant concentrations of curcumin and DMC were not cytotoxic to leukemia cells but induced G2/M cell cycle arrest. DMC but not curcumin induced the expression of promoter-methylated genes like p15 and CDH-1, indicating a possible DNA hypomethylating effect of DMC. Surprisingly, both drugs lacked any significant gene-specific and global DNA hypomethylating activity. Analysis of histone methylation in the CpG island near the promoter region of the p15 and CDH-1 genes showed an increase in H3K36me3 mark after DMC treatment, a mark associated with actively transcribed genes. Our results demonstrate that although both compounds lack a DNA hypomethylating effect, DMC induced the expression of promoter-methylated genes by a mechanism that does not involve DNA methylation reversal.
Materials and methods
Cell culture and chemicals
CEM, BV-173 and Kasumi-1 leukemia cells were grown in RPMI medium supplemented with 10% fetal bovine serum (life Technologies, CA) in a humidified atmosphere containing 5% CO2 at 37°C. Curcumin analytical standard grade (Sigma, WI) was dissolved in DMSO as 10 mM stock. DMC (Cayman, MI) was dissolved in DMSO as a 10mM stock. 5-azacytidine (5AC, Sigma, WI) was dissolved in PBS as 10 mM stock. PCR primers were purchased from Integrated DNA Technologies (Coralville, IA).
Apoptosis quantitation
Apoptosis quantitation was performed by double staining and fluorescence detection using flow cytometry as described previously (25). Briefly, 1× 106 cells were stained by Guava Nexin Reagent (EMD Millipore, MA) and incubated at room temperature in the dark for 20 minutes. Guava Nexin Reagent is a mixture of Annexin V-phosphatidylethanolamine (PE) and the cell impermeant dye, 7-aminoactinomycin D (7-AAD). Samples were acquired on a Guava easyCyte 5 system.
Cell cycle analysis
Analysis of cell cycle populations was performed by propidium iodide (PI) staining as described previously (26). Sample acquisition was performed on a Guava easyCyte 5 system.
Metabolism of curcumin and DMC by human liver microsomal enzymes
The time course metabolism of curcumin and DMC was evaluated using human pooled liver microsomes followed by HPLC analysis. A validated HPLC method with fluorescence detector was used to quantify both curcumin and DMC. The HPLC system consisted of a quaternary pump and a Waters H – class autosampler (Milford, MA). A Waters Symmetry C – 18 analytical column (4.6 X 150 mm, 5 μm) was used. The mobile phase was run on an isocratic condition and consisted of acetonitrile and 10 mM potassium phosphate containing 0.1 % TEA (pH = 4.5) (70:30 (v/v)) at a flow rate of 1.0 ml/min. Fluorescence detection wavelengths were 420 nm for excitation and 549 nm for emission. The lower limit of quantification was 0.0025 μg/ml and 0.005 μg/ml, with a linearity range of 0.0025 to 15 μg/ml and 0.005 to 10 μg/ml for curcumin and DMC, respectively. Accuracy and precision were determined by replicate injection of quality control samples. Both precision and accuracy were of satisfactory results below 17 % of CV.
The metabolism studies for both curcumin (2 μM) and DMC (2 μM) were conducted in triplicates using human pooled liver microsomes (0.5 mg/ml). The reaction mixture consisted of NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride) and 10 mM potassium phosphate buffer (pH 7.4). The metabolism studies for both compounds were conducted in the absence and presence of 3 mM of uridine 5’-diphosphoglucuronic acid (UDPGA) to account for phase II metabolism process. The reactions were initiated by adding the drug to the pre-warmed reaction mixture. After 0, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55 and 60 min of incubation at 37°C, 100μL of the reaction was sampled, immediately mixed with 100μL of acetonitrile to terminate the reaction and centrifuged at 14,000 rpm for 20 min. Aliquots of the supernatant were collected for HPLC analysis. The intrinsic clearance values were calculated using the formula:
Where indicates intrinsic clearance, indicates the maximum rate achieved at maximum substrate concentration, indicates the substrate concentration when the reaction rate is half of indicates the initial metabolic rate and is the substrate concentration at time 0. The intrinsic clearance values were calculated separately from each of the replicates. values are presented as mean ± SD from three replicates performed for each reaction.
Quantitative Real-time PCR analysis of gene expression
RNA was extracted using RNeasy kit according to the manufacturer’s instructions (Qiagen, CA). RNA was treated with DNase enzyme to remove any DNA contamination associated with the process of RNA extraction using the Turbo DNA-free kit (Ambion-Life Technologies, NY) according to the manufacturer’s instructions. cDNA was generated using the Verso cDNA synthesis kit (ThermoScientific, MA). Real-time PCR was performed on a RealPlex II thermal cycler (Eppendorf, NY) using Kapa SYBR Fast qPCR kit (KapaBiosystems, MA) and a two-step cycling protocol with annealing/extension temperature of 60 °C. Supplementary table I shows the primers sequences used for p15, CDH-1, DNA methyltransferase (DNMT) and Ten Eleven Translocation (TET ) isotypes.
DNA pyrosequencing
Pyrosequencing is a sequencing by synthesis method used for quantitation of DNA methylation (27). Biotin-labelled, single-stranded PCR products generated from bisulfite-treated DNA are used as a template with an internal primer to perform the pyrosequencing reaction. DNA was extracted using the Quick-gDNA microprep kit (Zymo Research, CA). Bisulfite treatment of DNA (500 ng) was performed using the EZ DNA Methylation-Gold kit (Zymo Research). Amplification of template DNA for pyrosequencing was performed using a PyroMark PCR kit (Qiagen). Primers for CDH-1 and p15 genes were designed using the PyroMark Assay Design Software (Qiagen) and the reverse primer was biotin labelled. The amplicon length was 170 and 207 bp for the p15 and the CDH-1, respectively. The amplification and the size of the amplicon were verified by agarose gel electrophoresis (supplementary figure 1). The sequence to analyze for the p15 gene was CGGGCCGCTGCGCGTCTGGGGGCTGCGGAATGCGCGA and included seven CpG sites (underlined). The sequence to analyze for the CDH-1 gene was CGGCAGCGCGCCCTCACCTCTGCCCAGGACGCGGC and included five CpG sites (underlined). Pyrosequencing was performed on a PyroMark Q24 instrument (Qiagen).
Analysis of global DNA methylation was performed using the Long Interspersed Nuclear Element-1 (LINE-1) assay (28). The CpG sites in LINE-1 sequences are normally heavily methylated and can be used as a surrogate marker for global DNA methylation. PyroMark Q24 CpG LINE-1 kit (Qiagen) was used to quantify the methylation level of three CpG sites in positions 318 to 331 of LINE-1 sequence (GenBank accession number X58075). Briefly, bisulfite treated DNA was used as a template to amplify a 146 bp fragment by PCR using a biotin-labelled reverse primer. The sequence to analyze after bisulfite conversion was TTYGTGGTGYGTYGTTT (Y indicates C or T) and included three CpG sites (underlined).
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the EZ-Magna ChIP G kit (Millipore). 1 × 107 cells were fixed using non-methanol formaldehyde ampules (ThermoScientific). The cells were incubated in a final concentration of 1% formaldehyde for 5 minutes at room temperature with shaking. The cross-linked DNA was sheared using a Covaris S2 Focused-ultrasonicator (Covaris, MA). The shearing conditions were duty cycle: 2%, intensity: 3 and cycles per burst: 200 for 12 minutes using 130 μL tubes. The sheared DNA was analyzed by Agilent 2100 Bioanalyzer to determine the average size of the sheared DNA. 1% of the sheared cross-linked DNA was saved as input. Immunoprecipitation of the cross-linked DNA was performed using overnight incubation with a ChIP grade rabbit monoclonal anti-trimethyl-histone H3-lysine 36 (H3K36me3) (Millipore, Cat#17–10032) and rabbit monoclonal anti-trimethyl-histone H3-lysine 4 (H3K4me3) (Millipore, Cat#17–614) in presence of protein G magnetic beads. Normal rabbit IgG was used as a negative control (Millipore). The immuno-precipitated cross-linked DNA was pulled down using a magnetic separator followed by reversal of the cross linking to free DNA. Real-time PCR analysis of the pulled down DNA was performed. The ChIP primers for the p15 gene were designed to amplify a 155 bp amplicon within the exon 1-associated CpG island. The forward primer was 5’ AGGGTAATGAAGCTGAG-CCC3’ and the reverse primer 5’ CTTGTTCTCCTC-GCGCATTC3’. The ChIP primers for the CDH-1 gene amplified a 207 bp amplicon within the exon 1-associated CpG island and the forward primer was 5’ GGGTGTGGAGAA-GGGGTG3’ and the reverse primer was 5’ GGAATGCACCACTCCTCAGA3’. The Input DNA was amplified using GAPDH primers; forward primer 5’TACTAGCGGTTTTACGGGCG3’ and reverse primer 5’TCGAACAGGAGGAGCAGAGAGCGA3’. The cycle threshold (Ct) for the ChIP antibodies and the negative control (IgG) were normalized to the input Ct using the equation
Where IP indicates the Ct for the ChIP antibodies or the IgG and DF indicates the input dilution factor. The ChIP yield was calculated as a percentage of the input using the equation
Statistical analyses
Data are represented as the average of the number of replicates ± the standard deviation (SD). Statistical difference between the control and drug-treated samples was calculated using Student’s t-test or ANOVA followed by Bonferroni post-hoc test where appropriate. p<0.05 was considered statistically different.
Results
Clinically relevant concentrations of curcumin does not induce apoptosis in tumor cells
Curcumin is known to induce apoptosis in different types of tumors like prostate, lungs, colon cancer and leukemia cell. However, the IC50 of curcumin in these tumor cells is many times higher than the detected peak plasma concentration of curcumin in human (29). Since our aim is to study the epigenetic changes induced by clinically relevant concentration of curcumin and DMC, it is important to use drug concentrations that are not cytotoxic. Leukemia cells (BV-173, Kasumi-1 and CEM) were incubated with different concentrations of curcumin and DMC for different time points (figure 1). Clinically relevant concentrations of curcumin (1 and 2 μM) did not induce significant apoptosis after 72 h of treatment in leukemia cells (figure 1a). Longer exposure time of leukemia cells to curcumin (168 h) was also non-toxic to leukemia cells except for BV-173, which showed significant apoptosis induction after curcumin treatment. Equimolar concentrations of the synthetic curcumin analogue DMC were more toxic to leukemia cells (figures 1a and 1b). DMC (1 μM) induced minor apoptosis in Kasumi-1 and CEM cells even after 168 h treatment but was highly toxic to BV-173 cells. DMC (2 μM) was highly toxic to all tested leukemia cells and consequently the 1 μM concentration was chosen to be used in all the experiments thereafter. These results demonstrate that curcumin (2 μM) and DMC (1 μM) concentrations are non-toxic to CEM and Kasumi-1 cells and can be used to study the epigenetic changes associated with their treatment of leukemia cells.
Figure 1. Clinically relevant concentrations of curcumin do not induce apoptosis in leukemia cells.
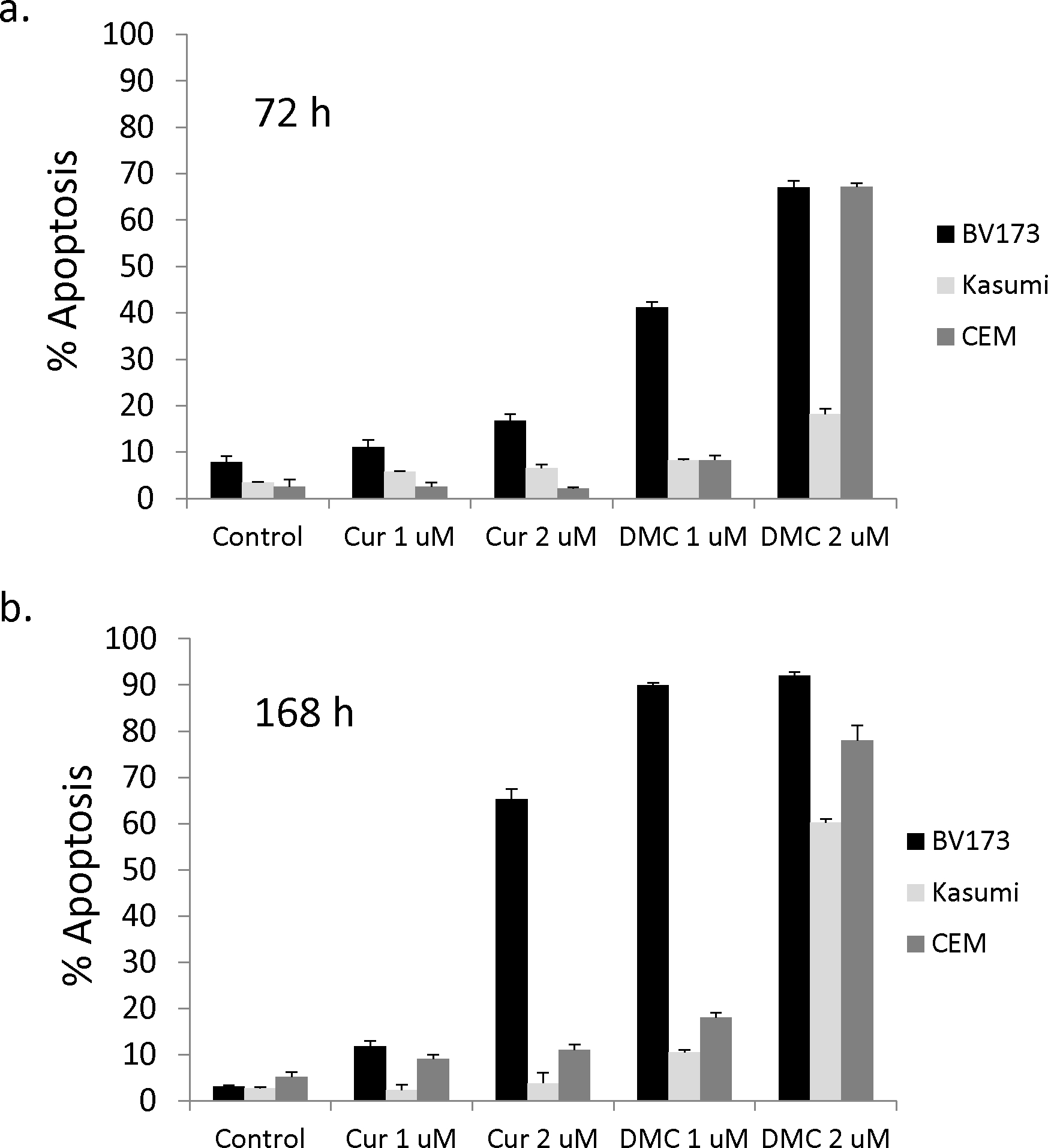
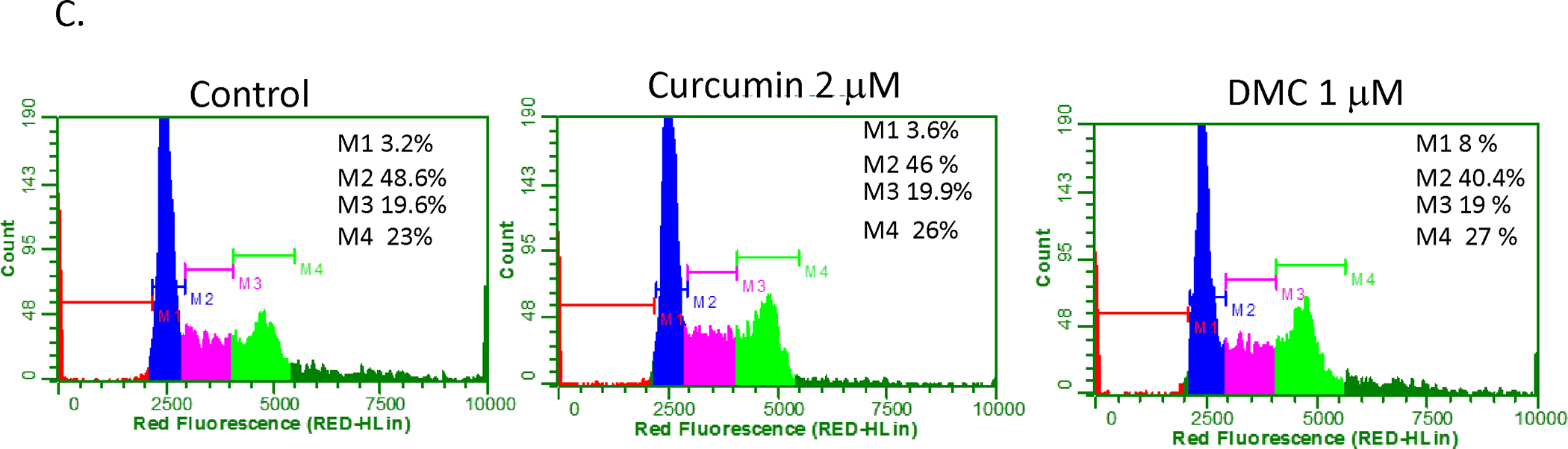
Kasumi-1, BV-173 and CEM leukemia cells were treated with different concentrations of curcumin (Cur) and DMC for 72 h (1a) and 168 h (1b). Fresh aliquots of Cur and DMC were added daily to the cells and apoptosis induction was measured as described under methods. Data represent the average of 3 replicates ± SD. C. CEM cells treated with Cur (2 uM) and DMC (1 uM) for 72 h followed by PI staining and cell cycle analysis as described under methods. Apoptotic cells are defined by the M1 marker, G1 phase population defined by the M2 marker, S phase population defined by the M3 marker and the G2/M population defined by the M4 marker. The percentage of each population is listed on the upper right corner of each figure. The data is a representative of 3 replicates. The average of 3 replicates for each marker was used to determine statistical difference by Student’s t-test.
Curcumin was reported to induce cell cycle arrest in different tumors (30). Although curcumin (2 μM) and DMC (1 μM) did not induce significant apoptosis in CEM and Kasumi-1 cells, their effect on cell cycle arrest is unknown. Figure 1c shows that both drugs increased the M4 marker population significantly (p<0.05) after 72 h from 22.8 ± 0.3 % to 26.1 ± 0.2 % for curcumin and to 26.9 ± 0.2 % for DMC, indicating G2/M phase cell cycle arrest. The same effect was also observed with Kasumi-1 cells (data not shown). These results show that although low concentrations of curcumin and DMC are non-toxic to leukemia cells, they induce G2/M arrest which may contribute to their antitumor effect.
Curcumin is more rapidly cleared than DMC by the human liver microsomal enzymes
Lower concentrations of DMC demonstrated more potency than curcumin in inducing apoptosis and cell cycle arrest. This may be attributable to the difference in metabolic stability of curcumin and its synthetic analogue, DMC. In order to verify that, the metabolism of both drugs was compared using human pooled liver microsomal enzymes at different time points followed by HPLC quantitative analysis of the intact drug as described under methods. The chromatographic analysis indicated that the retention times for curcumin and DMC were 2.3 and 4.8 min, respectively (supplementary figure 2). The intrinsic clearance of curcumin and DMC followed first order reaction kinetics with the concentration of the substrate declining mono-exponentially with time (supplementary figures 3). The calculated intrinsic clearance values for curcumin and DMC in the absence and presence of UDPGA are presented in Table I. These values demonstrate that in the absence of UDPGA (which accounts for phase II metabolism), DMC is less stable than curcumin; while in presence of UDPGA, DMC is more stable than curcumin.
Table I.
Intrinsic clearance values for curcumin and DMC in human pooled liver microsomes.
| Intrinsic clearance (microliters per minute per milligram of protein) | |||
|---|---|---|---|
| Curcumin | DMC | ||
| Absence of UDPGA | Presence of UDPGA | Absence of UDPGA | Presence of UDPGA |
| 140.18±2.32 | 196.37±19.13a | 177.48±6.52b | 173.80±4.88c |
P< 0.05 based on one - way ANOVA followed by Bonferroni post – hoc test. Comparison of intrinsic clearance values of curcumin in presence and absence of UDPGA.
P< 0.05 based on one - way ANOVA followed by Bonferroni post – hoc test. Comparison of intrinsic clearance values of curcumin and DMC in absence of UDPGA.
P< 0.05 based on one - way ANOVA followed by Bonferroni post – hoc test. Comparison of intrinsic clearance values of curcumin and DMC in presence of UDPGA.
DMC induces the expression of promoter-methylated genes in leukemia cells
Promoter DNA methylation is a well-established mechanism of epigenetic gene silencing. Reversal of promoter methylation to re-express silenced tumor suppressor genes using DNA hypomethylating agents is a rational approach in cancer therapy (31). The activity of curcumin and its analogues as DNA hypomethylating agent is controversial and unclear. In order to understand the impact of curcumin treatment on the expression of promoter-methylated genes, we monitored the expression of p15 and CDH-1 genes that are known to be frequently methylated in leukemia cells (32) after treatment with curcumin, DMC and 5AC. 5 AC is a potent DNA hypomethylating agent and used as a positive control. p15 and CDH-1 promoter methylation in CEM and kasumi-1 cells was confirmed by DNA pyrosequencing as described below. DMC induced the expression of both p15 and CDH-1 after 72 h in CEM cells (figure 2). The induction was comparable to that induced by the potent DNA hypomethylating agent 5AC and was also detected in Kasumi-1 cells (data not shown). On the other hand, curcumin induction of both genes was not statistically significant (figure 2). Higher concentrations of curcumin (5 and 10 μM) did not also induce the expression of both genes significantly (data not shown). These data show that DMC but not curcumin, induces the gene expression of promoter methylated genes, suggesting a possible DNA hypomethylating effect by DMC, similar to 5AC.
Figure 2. DMC induces the expression of promoter-methylated genes.
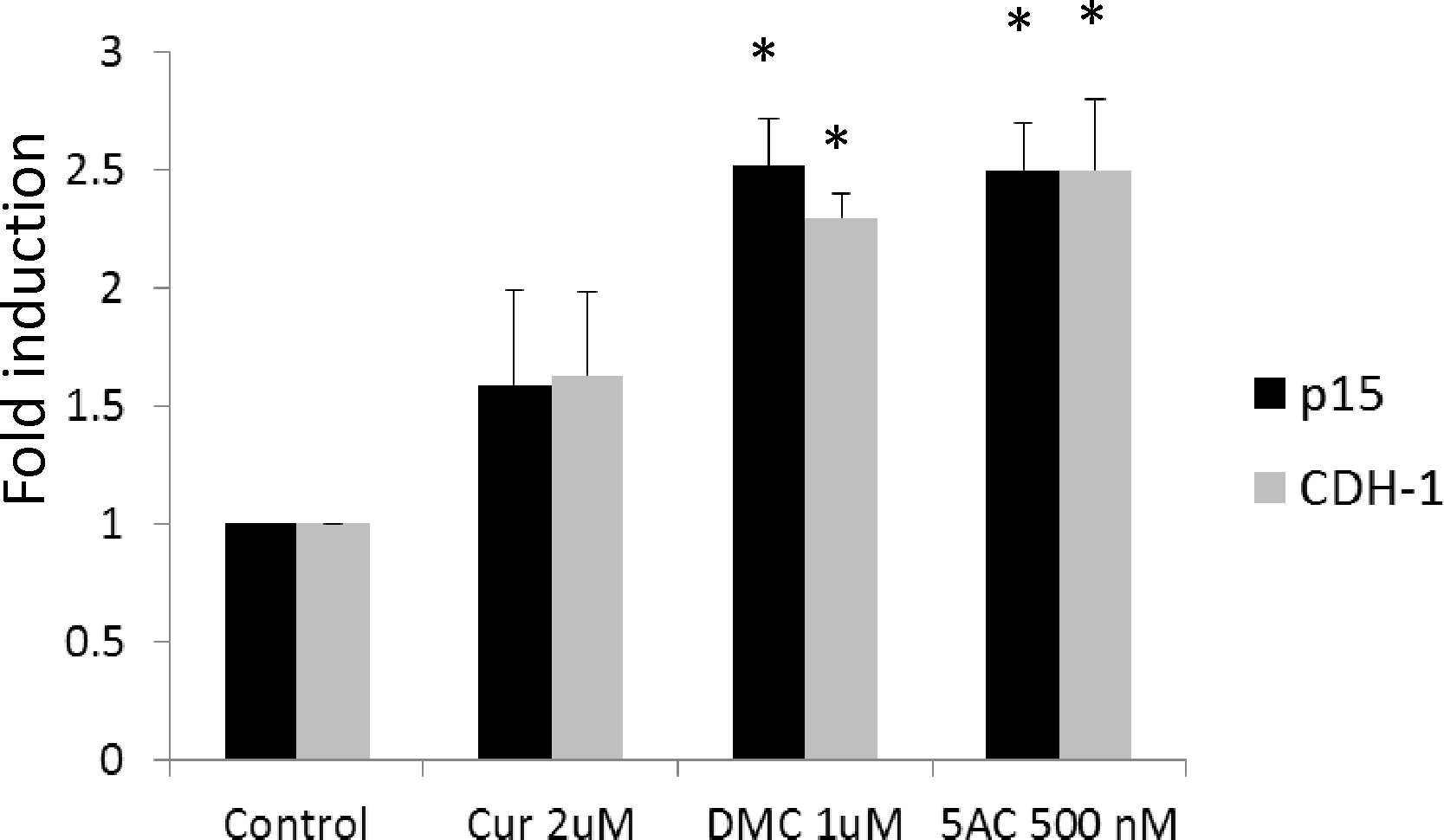
Real-time gene expression analysis of the p15 and CDH-1 genes in CEM cells after treatment with curcumin (Cur) 2 uM, DMC 1 uM and 5-azacytidine (5AC) 500 nM for 72 h. Data represent the average of 3 replicates ± SD and * indicates a significant difference from the corresponding control cells at p<0.05.
Curcumin and DMC lack DNA hypomethylating activity
In order to directly study the effect of curcumin and DMC on DNA methylation reversal of p15 and CDH-1 promoters, DNA pyrosequencing was used to quantitate DNA methylation reversal after curcumin and DMC treatment. DNA pyrosequencing is considered the gold standard in the quantitative analysis of DNA methylation (33). CEM and Kasumi-1 cells were incubated with curcumin or DMC for 72, 120 and 168 h (fresh drug was added daily to the cells) and the methylation status of seven CpG sites within the exon 1-associated CpG island of the p15 gene was monitored by DNA pyrosequencing. All the CpG sites in the CEM control cells were almost completely methylated (above 90%) except for CpG 4 and CpG 7 sites which showed an average methylation of 86 ± 2.6 and 75 ± 1.5 (figures 3a). Curcumin (1 and 2 μM) and DMC (0.5 and 1 μM) did not significantly reverse DNA methylation at any of the seven CpG sites after 72 h in CEM cells (figure 3a). On the other hand, 5AC (positive control) reversed DNA methylation at all the CpG sites. Longer duration of treatment for 120 h (supplementary figure 4a) and 168 h (data not shown) did not also show any significant methylation reversal by curcumin or DMC, while 5AC reversed methylation in all CpG sites. The same pattern of results was also observed in Kasumi-1 cells after 72 h (supplementary figure 4b), 120 and 168 h (data not shown).
Figure 3. Curcumin and DMC lack gene-specific DNA hypomethylating activity.
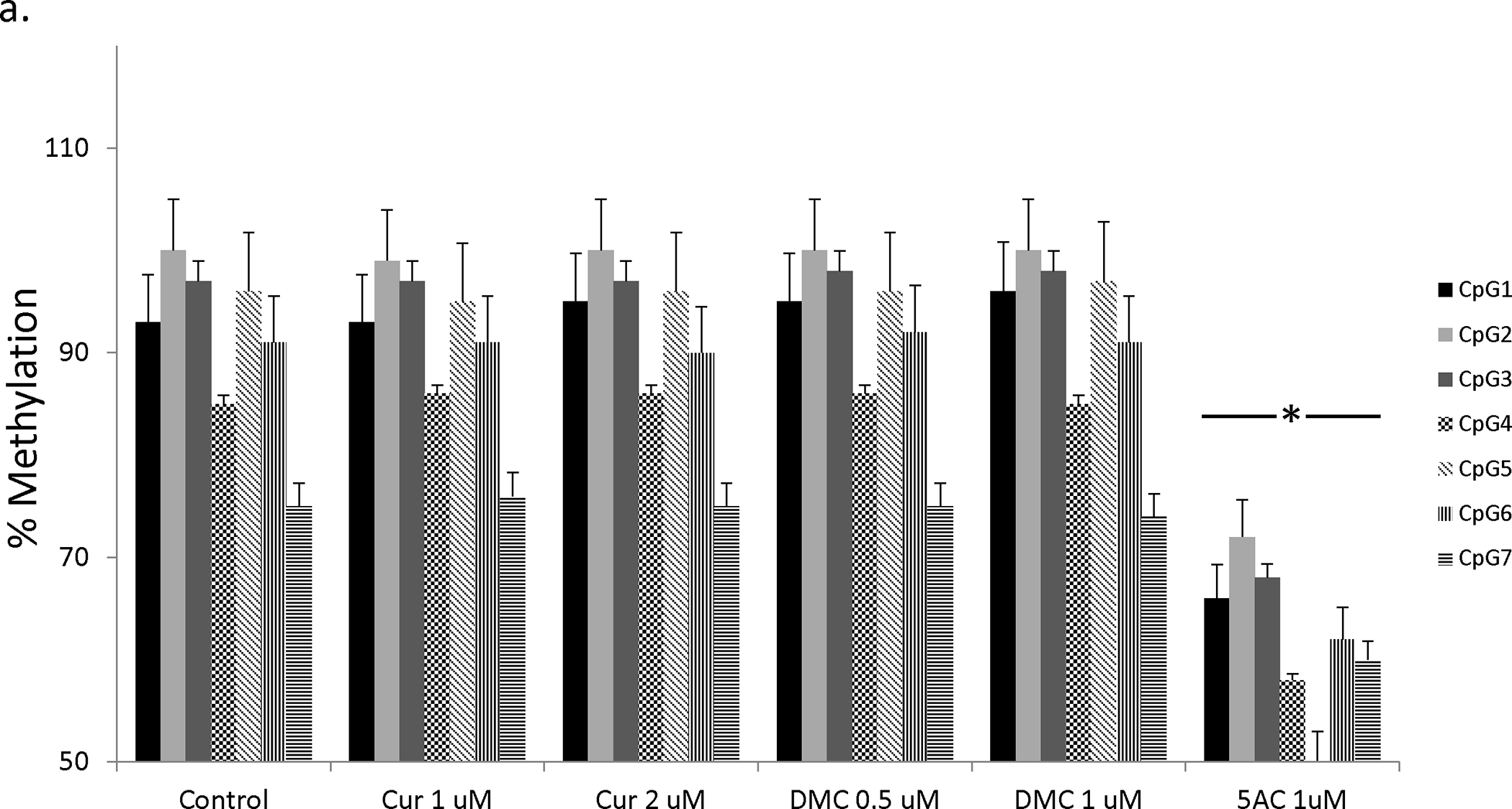
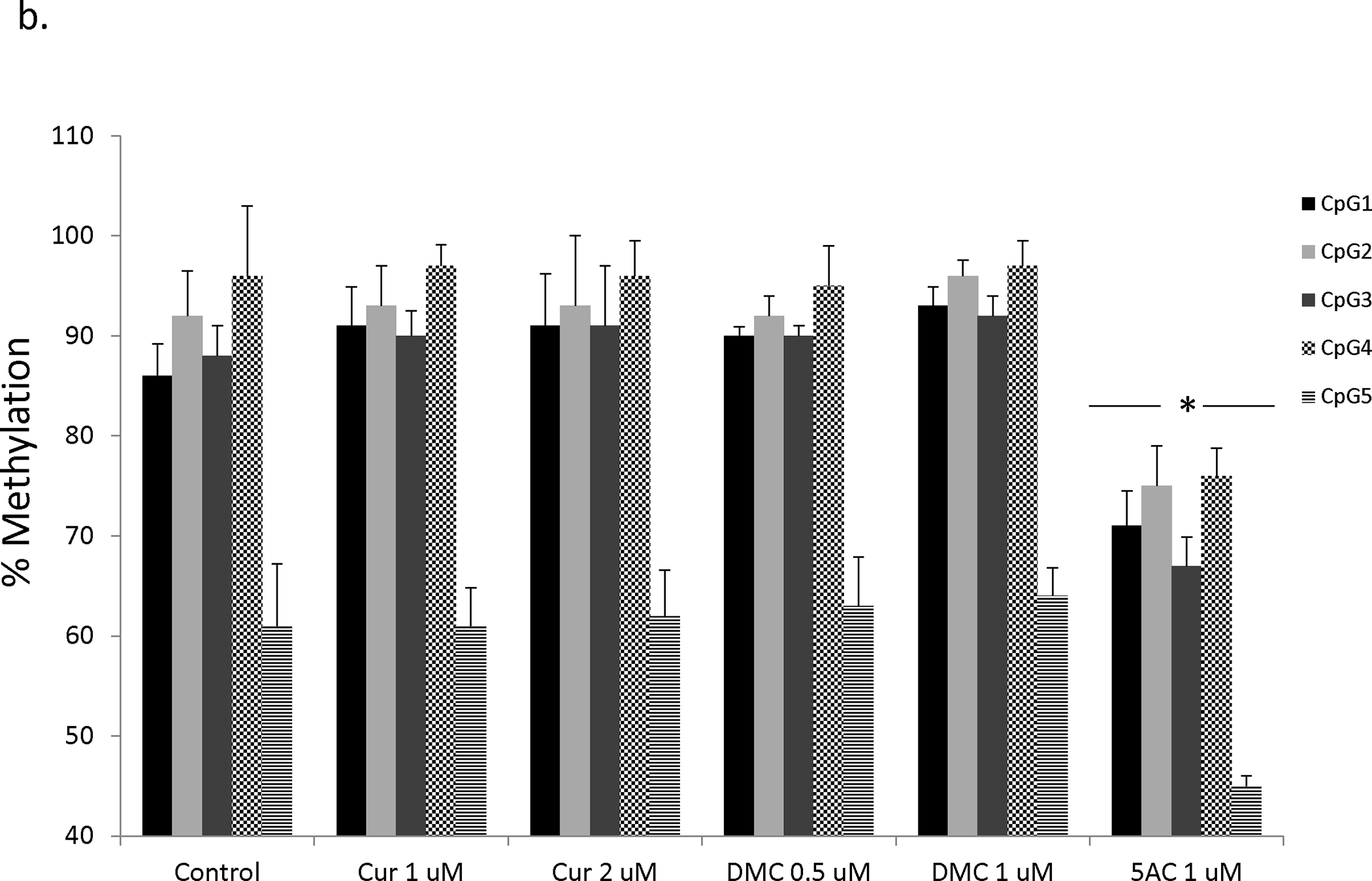
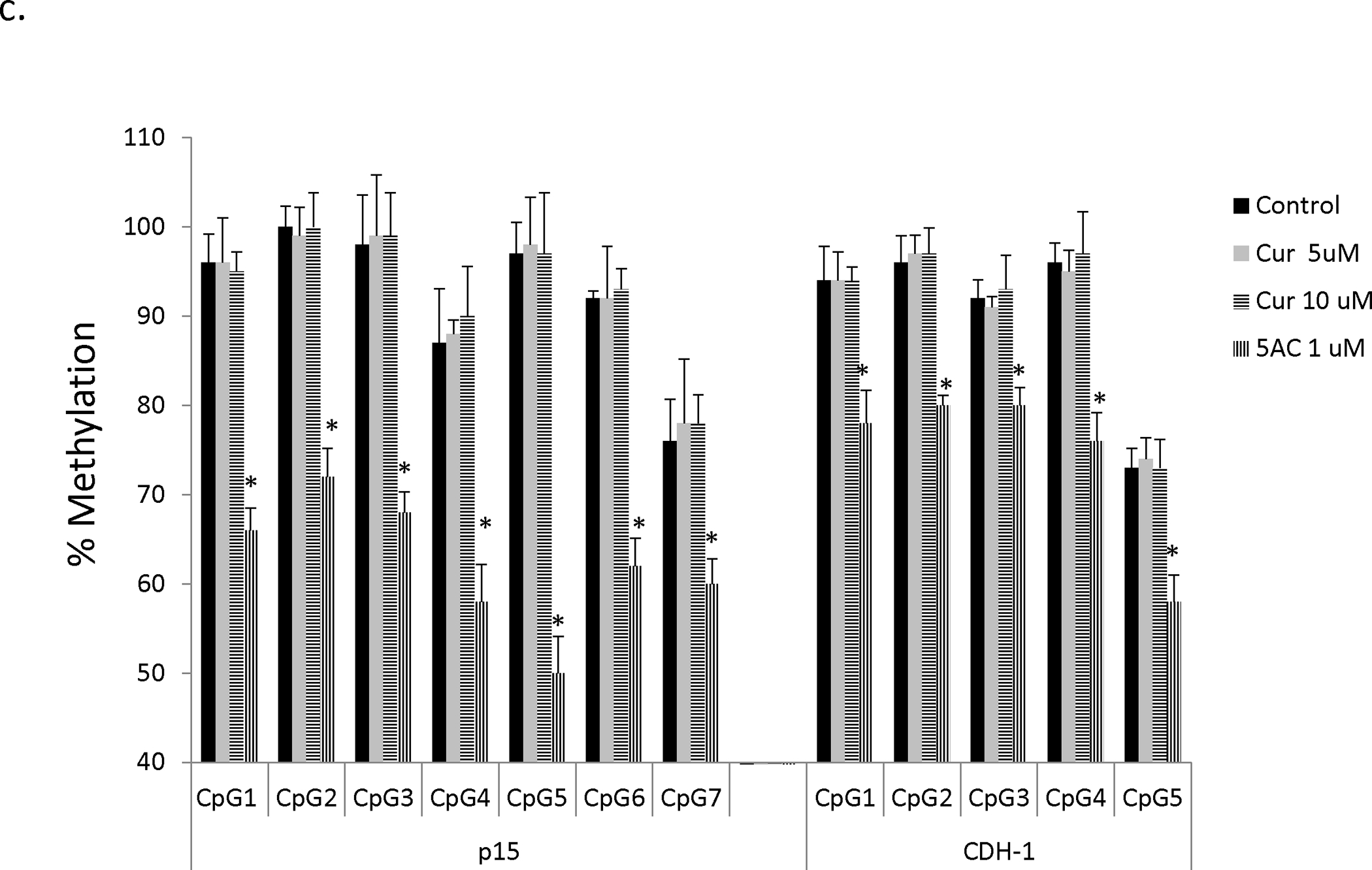
a. CEM cells were treated with different concentrations of curcumin (Cur), DMC and 5AC for 72 h followed by DNA extraction, DNA bisulfite treatment and DNA pyrosequencing of seven CpG sites in the CpG island of the p15 gene as described under methods. Data represent the average of duplicates ± SD and * indicates statistically significant difference from the control at p<0.05. b. CEM cells were treated with different concentrations of curcumin (Cur), DMC and 5AC for 72 h followed by DNA extraction, DNA bisulfite treatment and DNA pyrosequencing of five CpG sites in the CpG island of the CDH-1 gene as described under methods. Data represent the average of duplicates ± SD and * indicates statistically significant difference from the control at p<0.05. c. CEM cells were treated with high concentrations (5 and 10 uM) of curcumin (Cur) and 5AC (1 uM) for 72 h followed by DNA extraction, DNA bisulfite treatment and DNA pyrosequencing of seven CpG sites in the CpG island of the p15 gene and five CpG sites in the CpG island of the CDH-1 gene as described under methods. Data represent the average of duplicates ± SD and * indicates statistically significant difference from the control at p<0.05.
To further confirm the lack of hypomethylating effect of curcumin and DMC in other genes, we investigated their effect on CDH-1 gene methylation reversal in both CEM and Kasumi-1 cells. CEM cells were treated with curcumin (1 and 2 μM) and DMC (0.5 and 1 μM) for 72, 120 and 168 h and the methylation changes in five CpG sites within exon 1-associated CpG island of CDH-1 gene were monitored. The first four CpG sites were highly methylated (above 85 %) and the average methylation of the fifth CpG site was 61 ± 5.2 % in the CEM control cells (figure 3b). Similar to the p15 gene, no significant DNA methylation reversal was observed in any of the CpG sites after 72 h (figure 3b), 120 h and 168 h (data not shown). On the other hand, 5 AC reversed methylation in all the five CpG sites. Similar results were also observed in Kasumi-1 cells after 72 h (supplementary figure 5), 120 h and 168 h (data not shown). Collectively, these data confirm that clinically relevant concentration of curcumin and DMC lack significant gene-specific DNA hypomethylating activity.
It is possible that we did not observe any hypomethylating activity of curcumin because of the low concentrations used. To address this concern, CEM cells were treated with high concentrations of curcumin (5 and 10 μM) for 72 and 120 h and methylation reversal in both p15 and CDH-1 genes was monitored. It is worth mentioning here that similar high concentrations of DMC are highly toxic to leukemia cells and cannot be used. Figure 3c shows that even high concentrations of curcumin did not reverse p15 or CDH-1DNA methylation in CEM cells after 72 h and after 120 h (data not shown). Kasumi-1 cells showed similar results as CEM cells after 72 h (Supplementary figure 6).
Curcumin and DMC do not reverse global DNA methylation
Gene promoter methylation and global DNA methylation (heterochromatic DNA repeats and dispersed retrotransposons) are controlled by different pools of DNMT enzymes (34). Preferential demethylation of euchromatic regions over heterochromatic centromeric repeats was observed with other DNA hypomethylating agents like RG-108 (35). Consequently, it is possible that curcumin and DMC may exhibit preferential demethylation of heterochromatic DNA repeats. In order to test this, CEM cells were treated with different concentration of curcumin (1, 2, 5 and 10 μM) and DMC (0.5 and 1 μM) for 72, 120 and 168 h and reversal of global DNA methylation was monitored by LINE-1 DNA pyrosequencing (three CpG sites) as described under methods. Curcumin and DMC did not significantly reverse any of the CpG sites after 72 h treatment, while 5AC reversed DNA methylation in all the CpG sites (figure 4). Treatment for longer duration 120 h and 168 h did not also reverse DNA methylation (data not shown). Similar effect was observed with Kasumi-1 cells, further confirming that curcumin and DMC do not reverse global DNA methylation (supplementary figure 7). Collectively, both curcumin and DMC lack global DNA hypomethylating activity.
Figure 4. Curcumin and DMC do not reverse LINE-1 methylation.
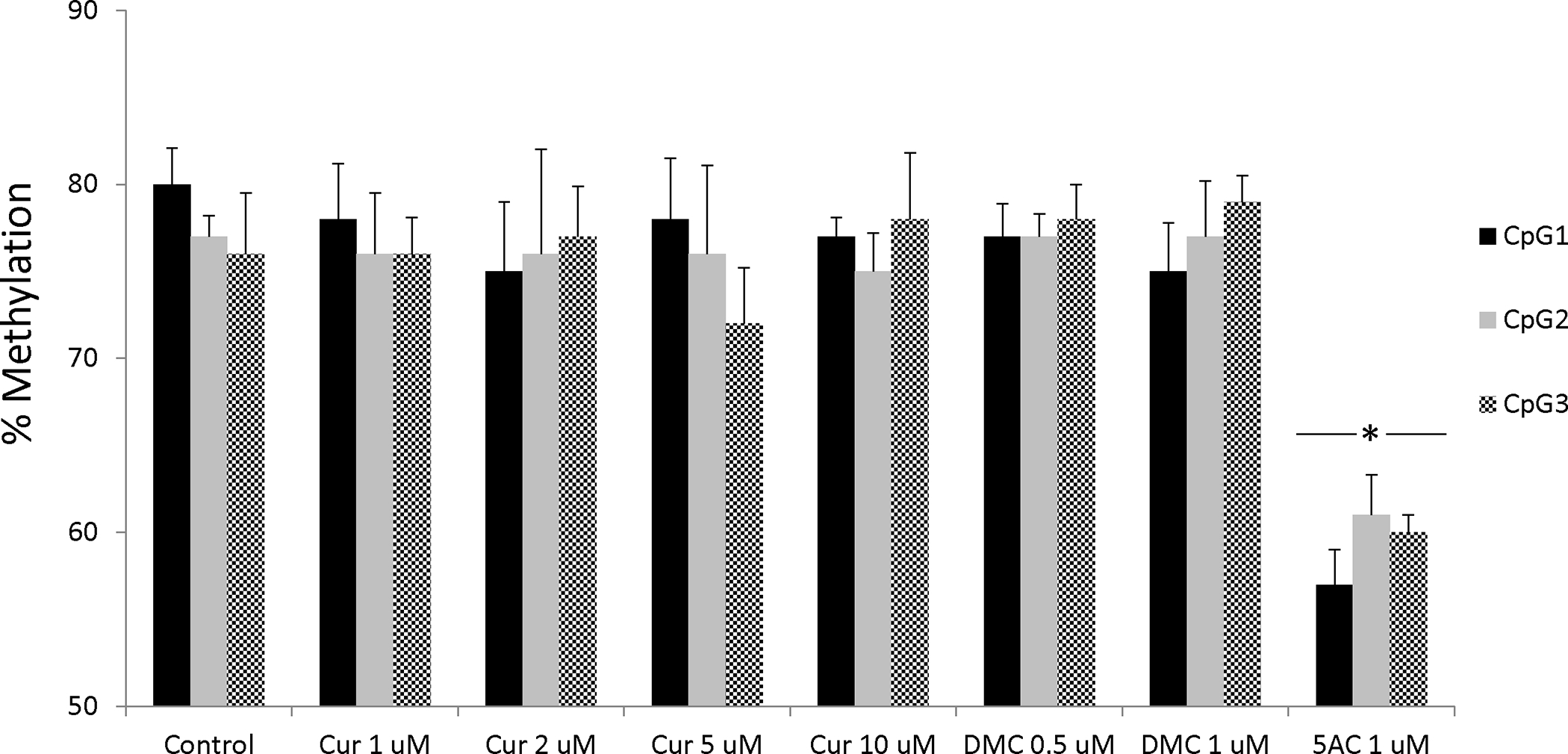
CEM cells were treated with different concentrations of curcumin (Cur), DMC and 5AC (1 uM) for 72 h followed by DNA extraction, DNA bisulfite treatment and DNA pyrosequencing of three CpG sites in the LINE-1 sequence (GenBank accession number X58075) as described under methods. Data represent the average of duplicates ± SD and * indicates statistically significant difference from the control at p<0.05.
Curcumin and DMC upregulate the expression of TET enzymes
Although both curcumin and DMC lack DNA hypomethylating activity, it is still important to study their effect on the gene expression of the enzymes that control the DNA methylation machinery; viz DNMT and TET enzymes, before considering their combination with other epigenetic modifiers like DNMT inhibitors. Curcumin and DMC did not significantly alter the expression of any of the three isoforms of DNMT in CEM cells (Table II). On the other hand, both drugs induced the expression of TET1 and TET2 enzymes (Table II). TET3 was slightly but significantly induced by curcumin and DMC. Additionally, 5AC induced the expression of the three isoforms of the TET enzymes. Induction of the TET isoforms was also observed in kasumi-1 cells after curcumin and DMC treatment (data not shown). These data demonstrate that both curcumin and DMC induce TET enzymes expression, which could be of value when combining them with DNMT inhibitors.
Table II.
Effect of curcumin, DMC and 5AC on the expression of the enzymes controlling DNA methylation.
| Fold Induction | |||
|---|---|---|---|
| Gene | Cur | DMC | 5AC |
| DNMT1 | 1.21 ± 0.2 | 1.39 ± 0.3 | 1.1 ± 0.1 |
| DNMT3a | 1.28 ± 0.2 | 1.47 ± 0.3 | 1.51 ± 0.3 |
| DNMT3b | 1.15 ± 0.1 | 1.29 ± 0.1 | 1.49 ± 0.3 |
| TET1 | 2.2 ± 0.1* | 2.3 ± 0.4* | 2.4 ± 0.2* |
| TET2 | 1.9 ± 0.1* | 1.8 ± 0.2* | 2 ± 0.2* |
| TET3 | 1.4 ± 0.1* | 1.64 ± 0.3* | 1.65 ± 0.2* |
CEM cells were treated with curcumin (Cur) 2 μM, DMC 1 μM and 5AC 500 nM for 72 hours. Real-time PCR analysis of gene expression was performed as described under methods. Data represent the mean ± standard deviation of three replicates.
indicates significant difference from the control at p<0.05.
DMC increases H3K36me3 mark in the putative CpG island of p15 and CDH-1 genes
The induction of the promoter-methylated genes p15 and CDH-1 by DMC (figure 2c) cannot be explained by DNA methylation reversal as DMC lacks any significant DNA hypomethylating activity. In order to understand the mechanism of induction, we hypothesized that other DMC-induced histone modifications like histone methylation may contribute to the observed induction in expression. H3K4 trimethylation (H3K4me3) and H3K36 trimethylation (H3K36me3) are known to correlate with open states of transcriptionally active chromatin (36). ChIP analyses were conducted using control IgG, anti-H3K4me3, and anti-H3K36me3 antibodies prior to and after treatment with curcumin, DMC and 5AC in CEM cells as described under methods. ChIP primers for both genes were designed to amplify a product within the exon-1 associated CpG island and overlapping with the pyrosequenced strand (figure 5a). Optimal chromatin shearing was confirmed as described under methods and the average size of sheared DNA was between 300 – 600 bp (figure 5b). Immunoprecipitated DNA fragments located within the promoter CpG island of the p15 and CDH-1 genes were amplified via Real-time PCR. No significant change in the H3K4me3 mark was detected in both genes after treatment with curcumin, DMC or 5AC (figure 5c). On the other hand, there was a significant increase in the H3K36me3 mark in the CpG island of both the p15 and CDH-1 genes after treatment with DMC and 5AC, but not with curcumin (figure 5d). Moreover, kasumi-1 cells demonstrated similar increase in H3K36me3 mark after treatment with DMC and 5AC (supplementary figure 8).Taken together, these data demonstrate that the effect on DMC and curcumin on histone methylation is different, which may consequently result in different gene expression changes induced by both drugs.
Figure 5. DMC and 5AC increase H3K36me3 mark within the putative CpG islands of p15 and CDH-1.
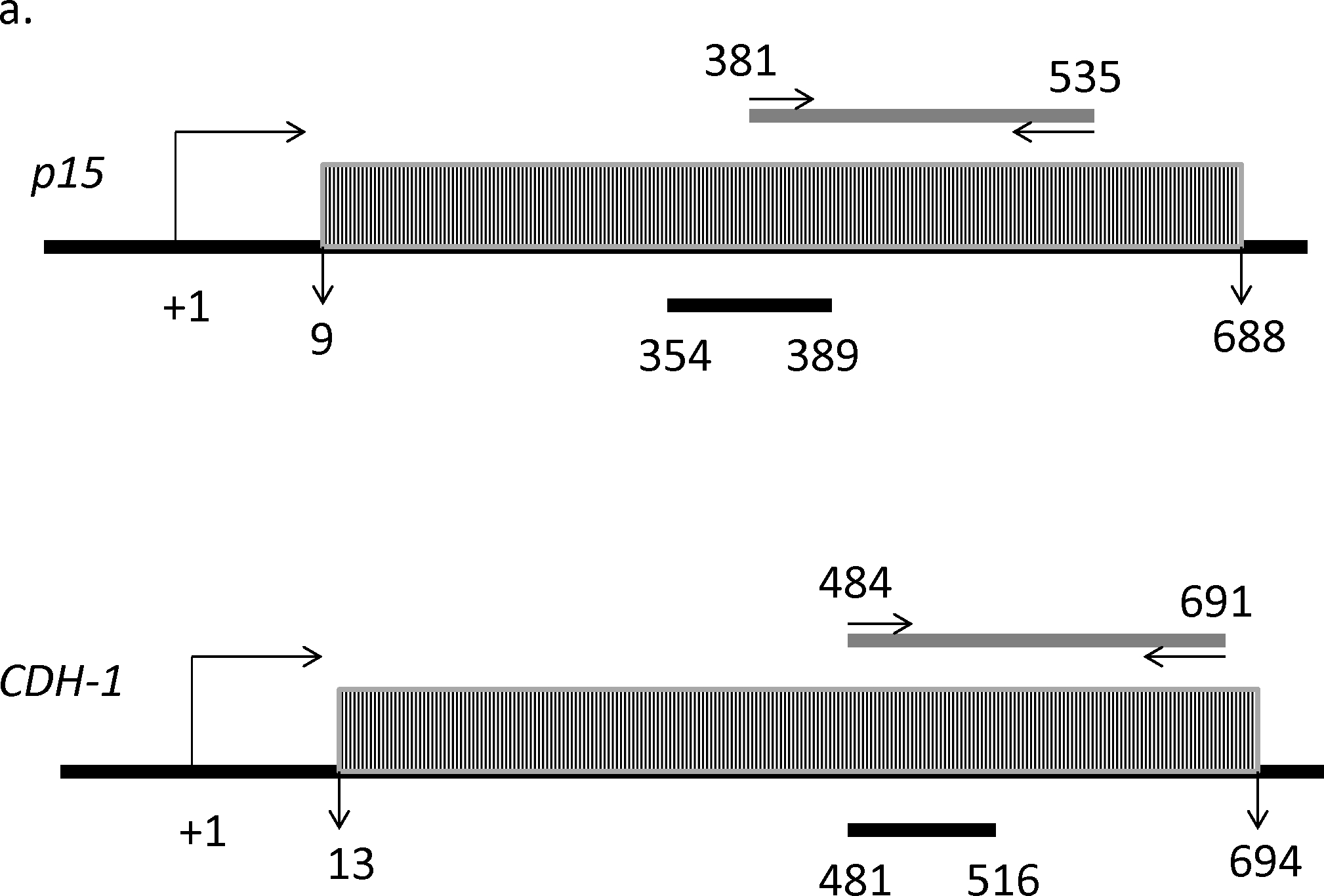
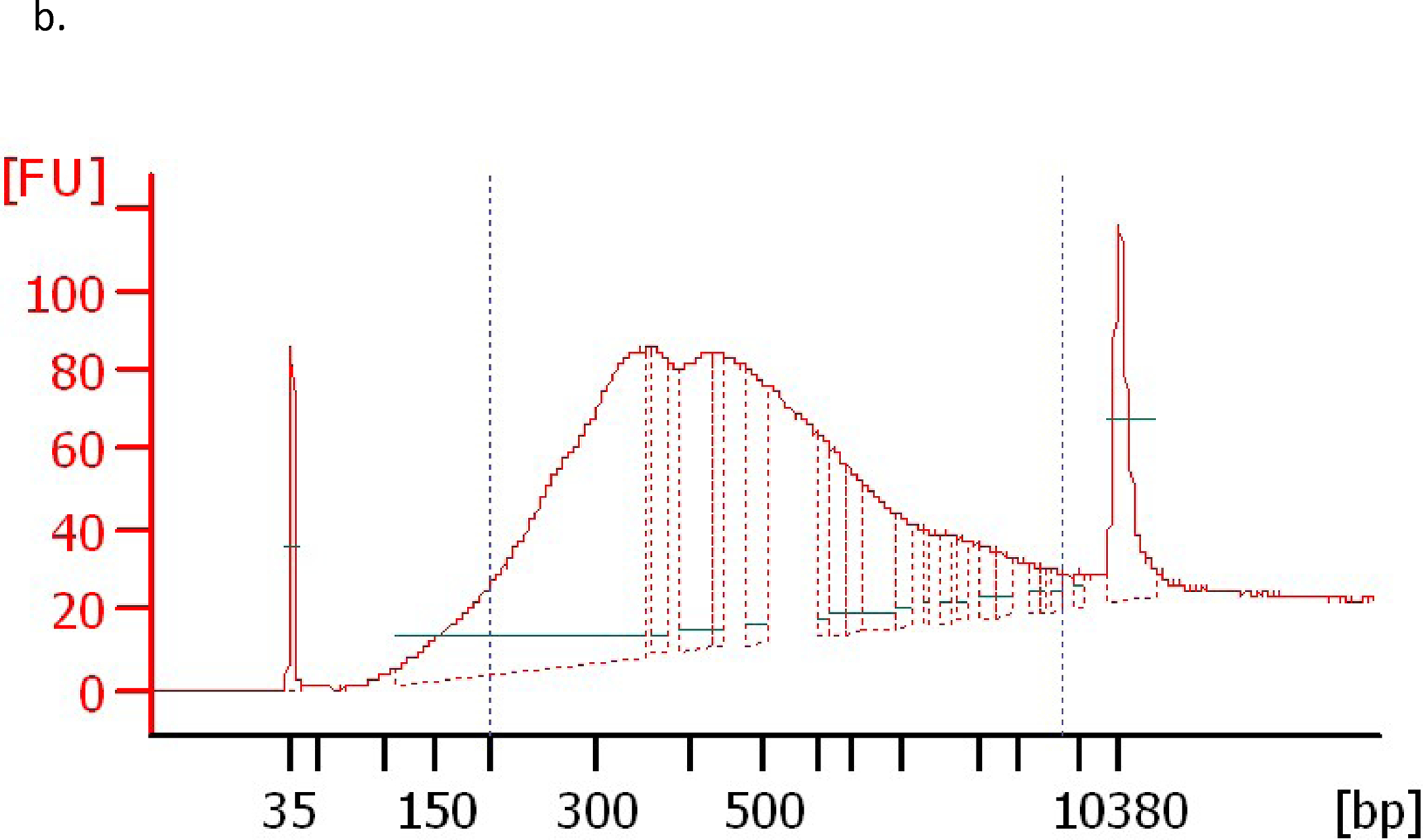
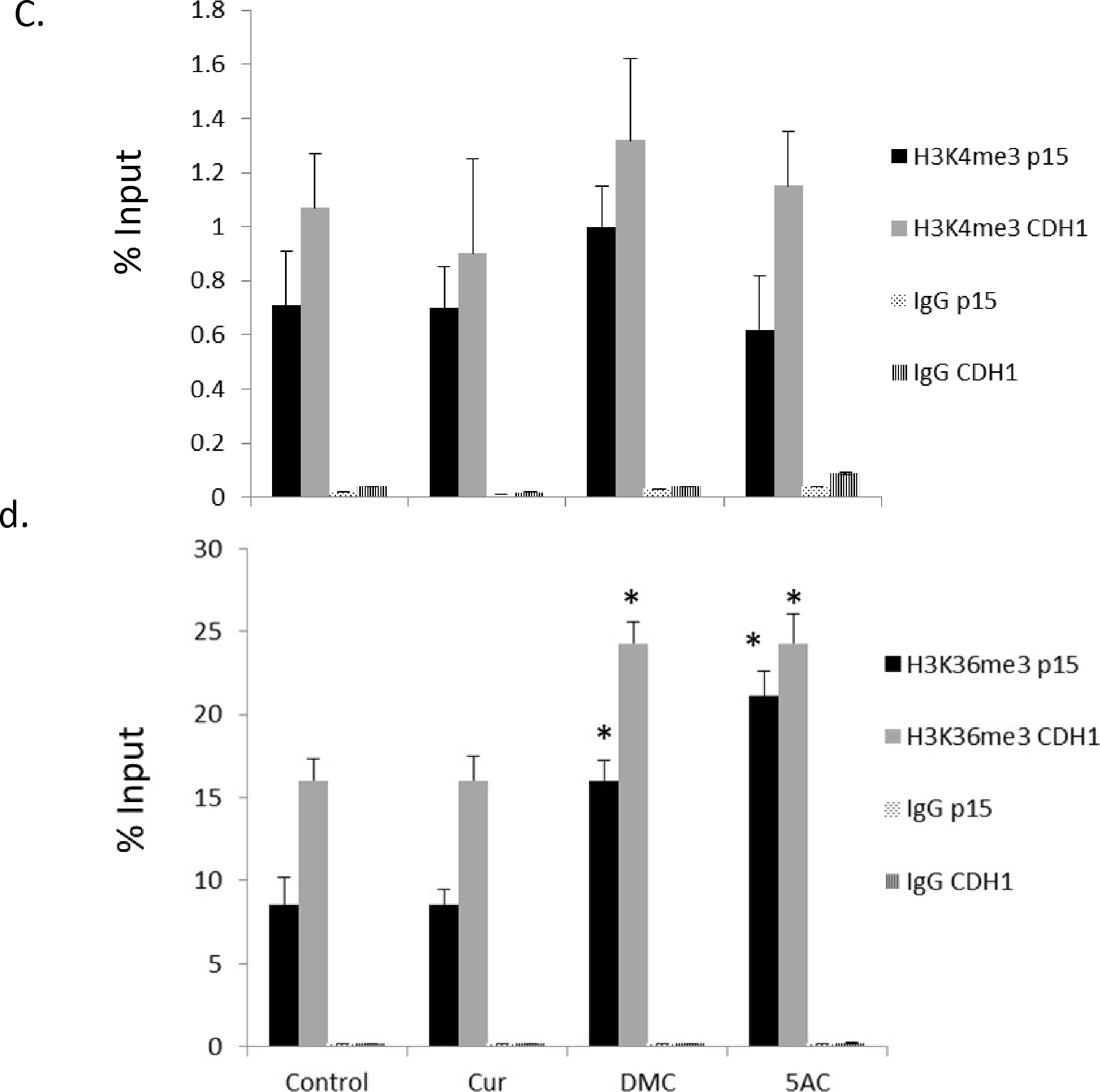
a. Schematic representation of the position of the putative CpG islands (grey rectangles filled with black narrow vertical lines) for the p15 and CDH-1 genes relative to the transcription start site (TSS) represented by the bending arrow (+1). The grey horizontal bar shows the position of the ChIP primers amplicon (155 bp) and the arrows represent start position of the forward and reverse primers relative to the TSS. The black horizontal bar shows the position of the DNA sequence analyzed by DNA pyrosequencing relative to the TSS. b. Optimal chromatin shearing by Covaris ultrasonicator. A representative electropherogram showing the size distribution of the sheared chromatin under the conditions specified in the methods section with major fragments size between 300–600 bp. FU indicates fluorescence units and bp indicates the size in base pairs. The two peaks at 35 bp and 10380 bp represent the lower and upper markers (ladders). c. CEM cells were treated with curcumin (Cur) 2 uM, DMC 1 uM and 5AC 500 nM for 72 h. ChIP using H3K4me3 antibody followed by qPCR using primers for p15 and CDH-1 genes was performed as described under methods and the % of input was calculated. H3K4me3 p15 indicates immunoprecipitation with H3K4me3 antibody followed by PCR amplification using primers for the p15 gene. H3K4me3 CDH1 indicates immunoprecipitation with H3K4me3 antibody followed by PCR amplification using primers for the CDH-1 gene. Data represent the mean of 3 replicates ± SD. Rabbit IgG antibody was used as a negative control. d. CEM cells were treated with curcumin (Cur) 2 uM, DMC 1 uM and 5AC 500 nM for 72 h. ChIP using H3K36me3 antibody followed by qPCR using primers for p15 and CDH-1 genes was performed as described under methods and the % of input was calculated. Data represent the mean of 3 replicates ± SD. Rabbit IgG was used as a negative control. * indicates significant difference from the control at p<0.05.
Discussion
An ideal chemopreventive and chemotherapeutic agent should affect multiple molecular targets in cancer cells with minimal toxicity in normal healthy cells. Chemotherapeutic agents lack the multiple target effect and are highly non selective. On the other hand, the polyphenolic phytochemical curcumin meets both criteria. Curcumin was shown to inhibit multiple vital pathways in cancer cells and was found to be cytoprotective for normal cells because of its antioxidant effect. However, the poor bioavailability of curcumin is a major hurdle in its successful use as an antitumor agent and the development of modified synthetic analogues provides a feasible strategy to improve its pharmacokinetic properties. In this study, we are comparing the cytotoxicity, metabolism and the epigenetic changes induced by clinically relevant concentrations of curcumin and its synthetic analogue, DMC. DMC was more toxic to leukemia cells than curcumin; concordant with being more metabolically stable. Unlike curcumin, DMC induced the expression of promoter-methylated genes like p15 and CDH-1; similar to the potent DNA hypomethylating agent 5AC. Both curcumin and DMC lacked any significant gene-specific or global DNA hypomethylating activity in leukemia cells. DMC increased the H3K36me3 mark near the promoter region of the p15 and CDH-1 genes, while curcumin did not show any significant changes in H3K36me3 or H3k436me3 level. These data elucidate significant differences between curcumin and DMC and demonstrate that although both compounds lack DNA hypomethylating activity, DMC can induce the expression of promoter methylated genes.
Previous reports investigated the epigenetic changes and cytotoxicity induced by curcumin using high concentrations (5–50 μM) that are considered clinically irrelevant (15, 18, 21). In this study, we used a lower concentration of curcumin (2 μM) that is close to the achievable plasma concentration of curcumin (29). A serious concern for using such low concentration would be the lack of any pharmacological action associated with curcumin treatment. However, our data provide four evidences that refute this concern. First, low concentrations of curcumin induced G2/M cell cycle arrest that was comparable to the more stable DMC (figure 1c). Second, treatment of BV-173 cells with 2 μM curcumin for 168 h induced massive apoptosis comparable to DMC. This also demonstrates the differential sensitivity of leukemia cells to curcumin and that resistance to curcumin-induced apoptosis in kasumi-1 and CEM leukemia cells could be attributed to factors other than curcumin poor bioavailability. Third, low concentration of curcumin induced the expression of the three TET isoforms similar to DMC and the potent DNA hypomethylating agent 5AC. Finally, when low curcumin concentrations failed to reverse DNA methylation and induce the expression of promoter-methylated genes, we used higher concentrations of curcumin (5 and 10 μM) and could not detect any methylation reversal or induction of gene expression, indicating that the lack of DNA hypomethylating activity is not attributed to the low concentration of curcumin used. Collectively, low concentration of curcumin was generally non-toxic to leukemia cells, induced cell cycle arrest and gene expression changes, which are all considered desirable effects for an epigenetic modifier.
DMC chemical structure differs from curcumin in that the two phenolic hydroxyl groups in curcumin are replaced by methoxy groups in DMC (supplemental figure 2). Since compounds with hydroxyl groups can be readily subjected to phase II metabolism (e.g., glucuronidation); it was expected that under physiological conditions and at equimolar concentrations, that DMC would be more resistant to phase II metabolism as opposed to curcumin. Indeed, our metabolism studies demonstrated that DMC has significantly lower intrinsic clearance values (P<0.05), and hence is more metabolically stable than curcumin in presence of UDPGA, which accounts for phase II metabolism (p<0.05) (Table I). These results are in line with our data reported herein which shows DMC as a more potent compound than curcumin in inducing apoptosis in leukemia cells. Nevertheless, other factors like the impact of both drugs on the expression of pro-apoptotic and anti-apoptotic genes could also contribute to that. Also, It is worth mentioning that the intrinsic clearance values of DMC in presence and absence of UDPGA were comparable (P>0.05), demonstrating the minimal effect of UDPGA on the metabolic stability of DMC; which was not the case with curcumin (Table I). Consistently, previous in vivo reports that employed higher concentrations (10–20 μM) of DMC and curcumin indicated that DMC is more metabolically stable and has higher bioavailability than curcumin (37, 38).
DNA hypomethylating agents induce methylation reversal and re-expression of epigenetically silenced genes. Molecular docking studies suggested a possible covalent interaction between curcumin and the catalytic pocket of DNMT1 (22). Global DNA methylation analysis demonstrated a comparable methylation reversal to the potent DNA hypomethylating agent decitabine after treatment of leukemia cells with 3 and 30 μM of a commercial curcumin mixture (consists of curcumin 80.3%, demethoxycurcumin 20.2% and bisdemethoxycurcumin 10.8%). Another study also used the curcumin mixture in prostate cancer cells but in different proportions (curcumin 70% and 30% for dimethoxycurcumin and bisdemethoxycurcumin) and demonstrated significant reversal of Nrf2 gene promoter methylation after treatment with the curcumin mixture (21). In our study, we used pure analytical grade curcumin and not a commercial mixture of curcumin and this may contribute to the contradicting results. It is possible that demethoxycurcumin and/or bisdemethoxycurcumin are the active DNA hypomethylating agents in the mixture. Another possibility is that the presence of the three compounds together may have a synergistic effect on DNA hypomethylation.
A different study used pure analytical grade curcumin to monitor DNA methylation reversal at the promoter region of the Neurog1 gene in LNCaP prostate cancer cells (20). Curcumin induced CpG demethylation and induced the expression of Neurog1 in LNCaP cells contrary to our findings in leukemia cells. We believe that the contribution of using cells from different tissues to the conflicting results is minimal if any, based on the documented activity of other DNA hypomethylating agents (decitabine and 5-azacytidine) in both leukemia cells and solid tumors. A possible explanation could be provided by the findings of another group that showed that curcumin selectively demethylate partially-methylated loci and not fully-methylated CpG sites (23). Indeed, the CpG sites studied in this report were almost fully methylated compared to 26% average CpG methylation in 47 CpG sites at the Neurog1 promoter region (20). On the other hand and in support of this study, curcumin was shown to have no significant global DNA demethylating activity in both leukemia and colorectal cancer cells after 6 days of treatment (23, 24). Taken together, factors like using curcumin mixture versus pure curcumin and the methylation density of the CpG sites to be analyzed could contribute to the controversy of the activity of curcumin as a DNA hypomethylating agent. Nonetheless, we are presenting solid evidence that DMC and pure curcumin, used at either low or high concentrations, did not reverse DNA methylation of highly methylated (70–95%) CpG sites, which is consistent with previous findings (23).
The analysis method used to quantitate DNA methylation reversal could also contribute to the conflicting results. In this study, we are using DNA pyrosequencing, which is considered the gold standard in quantitative analysis of both gene-specific and global DNA methylation (33). Other studies used techniques like bisulfite genomic sequencing (21), which is considered semi-quantitative and LC-MS/MS (39), which measures only global methylation changes without any information about gene-specific methylation. It is worth mentioning that DNA pyrosequencing was also used by Link et al (23) and their results are concordant with our findings.
The combination of curcumin with other epigenetic modifiers like DNMT inhibitors (decitabine and 5AC) could harness the effect of curcumin on histone acetylation and the effect of DNMT inhibitors on DNA methylation to induce optimum re-expression of epigenetically silenced tumor suppressor genes; similar to the sequential administration of DNMT inhibitors and HDAC inhibitors (32). Consequently, it is important to study the effect of curcumin on the expression of genes that control the DNA methylation machinery as this could augment or antagonize the action of DNMT inhibitors. DNMT isotypes (DNMT1, DNMT3a and DNMT3b) catalyze the transfer of methyl group from the universal methylation donor S-adenosyl-L-methionine (SAM) to cytosine. On the other hand, the TET isotypes (TET1, TET2 and TET3) catalyze the conversion of methylcytosine to 5-hydroxymethylcytosine with consequent active DNA demethylation (40). Curcumin and DMC did not induce the expression of any of the DNMT isotypes in leukemia cells suggesting the absence of an antagonistic effect between curcumin and DNMT inhibitors. On the other hand, the expression of the TET enzymes was induced by both curcumin and DMC suggesting a possible potentiation of the action of DNMT inhibitors on DNA methylation reversal when combined with curcumin, which needs to be validated.
The impact of curcumin and DMC on histone methylation is largely unknown. The position of the histone amino acid residue and its degree of methylation (mono, di or trimethylation) affects chromatin configuration. H3K4me3 and H3k36me3 are associated with transcriptionally active chromatin; while H3K9me3 and H3K27me3 mark closed heterochromatin domains (36). Curcumin induced a decrease of the H3K27me3 mark near the promoter region of Neurog1 and globally in prostate cancer cells (20). The decrease in H3K27me3 was associated with induction of Neurog1 expression. In this study, both DMC and 5AC increased the H3K36me3 but not the H3K4me3 mark near the methylated promoter region of p15 and CDH-1 genes in leukemia cells and that was associated with induction of their expression. In contrast to 5AC, DMC did not reverse promoter DNA methylation in both genes, indicating that induction of expression by DMC is independent on DNA methylation reversal. Previous reports demonstrated induction of gene expression in presence of promoter methylation. The expression of the estrogen receptor alpha (ER-alpha) gene is silenced by DNA methylation in breast cancer. ER-alpha expression was induced by treatment with the HDAC inhibitor trichostatin A (TSA) without reversing DNA methylation (41). Moreover, the class III HDAC SIRT1 was shown to localize to promoter methylated silenced tumor suppressor genes. Inhibition of SIRT1 re-expressed the silenced genes despite retention of promoter methylation (42). Furthermore, treatment of colorectal cancer cells with different HDAC inhibitors was able to induce the expression of promoter methylated genes without reversing DNA methylation, indicating that DNA methylation could not prevent gene reactivation by drug-induced resetting of the chromatin state (43). Other DMC-induced epigenetic changes like histone acetylation cannot be ruled out and may also contribute to the observed induction of gene expression. The mechanism of H3K36me3 increase by DMC and 5AC is not clear. A direct effect on the expression or activity of histone methyltransferases or histone demethylases is possible and needs to be investigated.
In conclusion, we highlighted major differences between curcumin and its synthetic analogue, DMC. The fact that DMC can induce the expression of promoter methylated genes without reversing DNA methylation suggests a possible synergistic induction mechanism of gene expression upon combining DMC with DNMT inhibitors; similar to the previously reported combination of HDAC inhibitors and DNMT inhibitors.
Supplementary Material
Acknowledgements & Disclosures
This work was supported by the Scholarship of Discovery Intramural Research Grant Program (SDIRGP) from Albany College of Pharmacy to TEF and University of Maryland intramural research grant to HH. The work was also supported by NIH Grant Numbers NIGMS R15GM104865 to KCG, 5 P30 RR032135 from the COBRE Program of the National Center for Research Resources and 8 P30 GM 103498 from the National Institute of General Medical Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Abbreviations
- 5AC
5-azacytidine
- 7-AAD
7-aminoactinomycin D
- Annexin V-PE
Annexin V-phosphatidylethanolamine
- ChIP
chromatin Immunoprecipitation
- DMC
Dimethoxycurcumin
- DNMT
DNA methyltransferase
- HAT
Histone acetyltransferase
- HDAC
Histone deacetylase
- TET
Ten Eleven Translocation
- UDPGA
Uridine 5’-diphosphoglucuronic acid
Footnotes
The authors have no financial disclosures or conflicts of interest to declare.
References
- 1.Shankar S, Srivastava RK. Bax and Bak genes are essential for maximum apoptotic response by curcumin, a polyphenolic compound and cancer chemopreventive agent derived from turmeric, Curcuma longa. Carcinogenesis. 2007;28(6):1277–1286. [DOI] [PubMed] [Google Scholar]
- 2.Shehzad A, Rehman G, Lee YS. Curcumin in inflammatory diseases. Biofactors. 2013;39(1):69–77. [DOI] [PubMed] [Google Scholar]
- 3.Bukhari SN, Franzblau SG, Jantan I, Jasamai M. Current prospects of synthetic curcumin analogs and chalcone derivatives against mycobacterium tuberculosis. Med Chem. 2013;9(7):897–903. [DOI] [PubMed] [Google Scholar]
- 4.Ou JL, Mizushina Y, Wang SY, Chuang DY, Nadar M, Hsu WL. Structure-activity relationship analysis of curcumin analogues on anti-influenza virus activity. Febs J. 2013;280(22):5829–5840. [DOI] [PubMed] [Google Scholar]
- 5.Gazal M, Valente MR, Acosta BA, Kaufmann FN, Braganhol E, Lencina CL, Stefanello FM, Ghisleni G, Kaster MP. Neuroprotective and antioxidant effects of curcumin in a ketamine-induced model of mania in rats. Eur J Pharmacol. 2014;724:132–139. [DOI] [PubMed] [Google Scholar]
- 6.Shankar S, Ganapathy S, Chen Q, Srivastava RK. Curcumin sensitizes TRAIL-resistant xenografts: molecular mechanisms of apoptosis, metastasis and angiogenesis. Mol Cancer. 2008;7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srivastava RK, Chen Q, Siddiqui I, Sarva K, Shankar S. Linkage of curcumin-induced cell cycle arrest and apoptosis by cyclin-dependent kinase inhibitor p21(/WAF1/CIP1). Cell Cycle. 2007;6(23):2953–2961. [DOI] [PubMed] [Google Scholar]
- 8.Shankar S, Chen Q, Sarva K, Siddiqui I, Srivastava RK. Curcumin enhances the apoptosis-inducing potential of TRAIL in prostate cancer cells: molecular mechanisms of apoptosis, migration and angiogenesis. J Mol Signal. 2007;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu P, Xu L, Gao L, Zhang M, Wang S, Tong S, Sun Y, Zhang L, Jiang T. Exploring pyrimidine-substituted curcumin analogues: design, synthesis and effects on EGFR signaling. Bioorg Med Chem. 2013;21(17):5012–5020. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborti S, Dhar G, Dwivedi V, Das A, Poddar A, Chakraborti G, Basu G, Chakrabarti P, Surolia A, Bhattacharyya B. Stable and potent analogues derived from the modification of the dicarbonyl moiety of curcumin. Biochemistry. 2013;52(42):7449–7460. [DOI] [PubMed] [Google Scholar]
- 11.Chen WF, Deng SL, Zhou B, Yang L, Liu ZL. Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenyl groups. Free Radic Biol Med. 2006;40(3):526–535. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Liu YZ, Zhang F, Wang HS, Zhang G, Zhou BH, Zuo YL, Cai SH, Bu XZ, Du J. Identification of potential pathways involved in the induction of cell cycle arrest and apoptosis by a new 4-arylidene curcumin analogue T63 in lung cancer cells: a comparative proteomic analysis. Mol Biosyst. 2014. [DOI] [PubMed] [Google Scholar]
- 13.Nagaraju GP, Zhu S, Wen J, Farris AB, Adsay VN, Diaz R, Snyder JP, Mamoru S, El-Rayes BF. Novel synthetic curcumin analogues EF31 and UBS109 are potent DNA hypomethylating agents in pancreatic cancer. Cancer Lett. 2013;341(2):195–203. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Liu Y, Chen Y, Xu J. C086, a novel analog of curcumin, induces growth inhibition and down-regulation of NFkappaB in colon cancer cells and xenograft tumors. Cancer Biol Ther. 2011;12(9):797–807. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007;101(6):427–433. [DOI] [PubMed] [Google Scholar]
- 16.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7(3):464–473. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Peng Y, Wu LC, Xie Z, Deng Y, Hughes T, He S, Mo X, Chiu M, Wang QE, He X, Liu S, Grever MR, Chan KK, Liu Z. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS One. 2013;8(2):e55934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du L, Xie Z, Wu LC, Chiu M, Lin J, Chan KK, Liu S, Liu Z. Reactivation of RASSF1A in breast cancer cells by curcumin. Nutr Cancer. 2012;64(8):1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parashar G, Parashar NC, Capalash N. Curcumin causes promoter hypomethylation and increased expression of FANCF gene in SiHa cell line. Mol Cell Biochem. 2012;365(1–2):29–35. [DOI] [PubMed] [Google Scholar]
- 20.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY, Saw CL, Cheung KL, Kong AN. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. Aaps J. 2011;13(4):606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem Pharmacol. 2011;82(9):1073–1078. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, Li PK, Lin J, Fuchs JR, Marcucci G, Li C, Chan KK. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett. 2009;19(3):706–709. [DOI] [PubMed] [Google Scholar]
- 23.Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, Goel A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS One. 2013;8(2):e57709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medina-Franco JL, Lopez-Vallejo F, Kuck D, Lyko F. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Mol Divers. 2011;15(2):293–304. [DOI] [PubMed] [Google Scholar]
- 25.Fandy TE, Jiemjit A, Thakar M, Rhoden P, Suarez L, Gore SD. Decitabine Induces Delayed Reactive Oxygen Species (ROS) Accumulation in Leukemia Cells and Induces the Expression of ROS Generating Enzymes. Clin Cancer Res. 2014;20(5):1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiemjit A, Fandy TE, Carraway H, Bailey KA, Baylin S, Herman JG, Gore SD. p21(WAF1/CIP1) induction by 5-azacytosine nucleosides requires DNA damage. Oncogene. 2008;27(25):3615–3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007;2(9):2265–2275. [DOI] [PubMed] [Google Scholar]
- 28.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moiseeva EP, Almeida GM, Jones GD, Manson MM. Extended treatment with physiologic concentrations of dietary phytochemicals results in altered gene expression, reduced growth, and apoptosis of cancer cells. Mol Cancer Ther. 2007;6(11):3071–3079. [DOI] [PubMed] [Google Scholar]
- 30.Liu HS, Ke CS, Cheng HC, Huang CY, Su CL. Curcumin-induced mitotic spindle defect and cell cycle arrest in human bladder cancer cells occurs partly through inhibition of aurora A. Mol Pharmacol. 2011;80(4):638–646. [DOI] [PubMed] [Google Scholar]
- 31.Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem. 2009;16(17):2075–2085. [DOI] [PubMed] [Google Scholar]
- 32.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, Licht J, McConnell MJ, Nasrallah C, Kim MK, Zhang W, Sun Y, Murgo A, Espinoza-Delgado I, Oteiza K, Owoeye I, Silverman LR, Gore SD, Carraway HE. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Migheli F, Stoccoro A, Coppede F, Wan Omar WA, Failli A, Consolini R, Seccia M, Spisni R, Miccoli P, Mathers JC, Migliore L. Comparison study of MS-HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS One. 2013;8(1):e52501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 2004;5(12):1181–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brueckner B, Garcia Boy R, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M, Lyko F. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 2005;65(14):6305–6311. [DOI] [PubMed] [Google Scholar]
- 36.Sarris M, Nikolaou K, Talianidis I. Context-specific regulation of cancer epigenomes by histone and transcription factor methylation. Oncogene. 2014;33(10):1207–1217. [DOI] [PubMed] [Google Scholar]
- 37.Tamvakopoulos C, Dimas K, Sofianos ZD, Hatziantoniou S, Han Z, Liu ZL, Wyche JH, Pantazis P. Metabolism and anticancer activity of the curcumin analogue, dimethoxycurcumin. Clin Cancer Res. 2007;13(4):1269–1277. [DOI] [PubMed] [Google Scholar]
- 38.Mach CM, Chen JH, Mosley SA, Kurzrock R, Smith JA. Evaluation of liposomal curcumin cytochrome p450 metabolism. Anticancer Res. 2010;30(3):811–814. [PubMed] [Google Scholar]
- 39.Liu Z, Liu S, Xie Z, Blum W, Perrotti D, Paschka P, Klisovic R, Byrd J, Chan KK, Marcucci G. Characterization of in vitro and in vivo hypomethylating effects of decitabine in acute myeloid leukemia by a rapid, specific and sensitive LC-MS/MS method. Nucleic Acids Res. 2007;35(5):e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohr F, Dohner K, Buske C, Rawat VP. TET genes: new players in DNA demethylation and important determinants for stemness. Exp Hematol. 2011;39(3):272–281. [DOI] [PubMed] [Google Scholar]
- 41.Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, Herman JG, Davidson NE. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer Res. 2000;60(24):6890–6894. [PubMed] [Google Scholar]
- 42.Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006;2(3):e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raynal NJ, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, Estecio MR, Issa JP. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012;72(5):1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.