

ABSTRACT
The intestinal microbiota is an important environmental factor implicated in CRC development. Intriguingly, modulation of DNA methylation by gut microbiota has been reported in preclinical models, although the relationship between tumor-infiltrating bacteria and CIMP status is currently unexplored. In this study, we investigated tumor-associated bacteria in 203 CRC tumor cases and validated the findings using The Cancer Genome Atlas datasets. We assessed the abundance of Bacteroides fragilis, Escherichia coli, Fusobacterium nucleatum, and Klebsiella pneumoniae through qPCR analysis and observed enrichment of all four bacterial species in CRC samples. Notably, except for E. coli, all exhibited significant enrichment in cases of CIMP. This enrichment was primarily driven by a subset of cases distinguished by high levels of these bacteria, which we labeled as “Superhigh”. The bacterial Superhigh status showed a significant association with CIMP (odds ratio 3.1, p-value = 0.013) and with MLH1 methylation (odds ratio 4.2, p-value = 0.0025). In TCGA CRC cases (393 tumor and 45 adj. normal), bacterial taxa information was extracted from non-human whole exome sequencing reads, and the bacterial Superhigh status was similarly associated with CIMP (odds ratio 2.9, p < 0.001) and MLH1 methylation (odds ratio 3.5, p < 0.001). Finally, 16S ribosomal RNA gene sequencing revealed high enrichment of Bergeyella spp. C. concisus, and F. canifelinum in CIMP-Positive tumor cases. Our findings highlight that specific bacterial taxa may influence DNA methylation, particularly in CpG islands, and contribute to the development and progression of CIMP in colorectal cancer.
KEYWORDS: Gut microbiota, Colorectal cancer, CpG island methylator phenotype, DNA methylation, Epigenetics
Introduction
Colorectal cancer (CRC) poses a significant global health burden due to its high prevalence and mortality rates.1 While hereditary factors, such as germline mutations, contribute to a small proportion of CRC cases,2 the majority are sporadic, indicating a multifaceted interaction between genetic and environmental factors in its development.3 Numerous studies have aimed to understand the epigenetic landscape of CRC, revealing a complex relationship between DNA methylation patterns and the pathogenesis of CRC.4 CpG island methylator phenotype (CIMP) is characterized by a distinct pattern of hypermethylation in multiple CpG loci throughout the cancer genome. Initially identified in CRC patients,5 CIMP has been observed in various cancers. In gliomas, CIMP is predominantly caused by IDH1/2 mutations, which lead to the accumulation of the oncometabolite 2-hydroxyglutarate, a competitive inhibitor of the TET family of DNA dioxygenases.6 Mutations in succinate dehydrogenase are also associated with CIMP in stromal tumors.7 While mutations in the TCA cycle genes have been linked to CIMP in other cancers, CIMP in CRC lacks well-defined genetic alterations. This suggests that external factors may potentially contribute to CIMP in CRC, as genetic aberrations alone cannot be solely account for its cause.
The human gut microbiota comprises a densely populated and metabolically active microbial community that plays a pivotal role in protecting the intestinal mucosal barrier, supplying essential nutrients, and modulating the host immune system for proper immune function.8 Dysbiosis, characterized by an imbalance in composition and disturbance function of the gut microbiota, has been associated with chronic inflammation and tumorigenesis in the gut.9 Studies have revealed hypermethylation of DNA in the gastric mucosa of patients infected with Helicobacter pylori, as well as in patients with Epstein-Barr virus (EBV). Fusobacterium nucleatum, frequently associated with colorectal cancer pathogenesis, has been found to be disproportionately enriched in colorectal cancer patients with CIMP compared to those without CIMP. Previous investigations identified a significant association between F. nucleatum and Bacteroides fragilis and CIMP in colorectal cancer,10,11 suggesting a distinct gut microbiota as a key player that associates with CIMP in colorectal cancer (without inferring cause vs. effect). To test this hypothesis, we selected four bacterial species-Bacteroides fragilis, Escherichia coli, Fusobacterium nucleatum, and Klebsiella pneumoniae-with significant enrichment in CRC patients12–15 to examine the association between CIMP and bacterial enrichment in CRC tumor tissue. We evaluated the relationship between CIMP status and bacterial enrichment in colorectal cancer tumor specimens, as well as adjacent normal samples. Our findings were verified using data from The Cancer Genome Atlas. Furthermore, we performed 16S rRNA gene sequencing to identify additional bacterial taxa displaying enrichment in colorectal cancer tumor tissue, particularly those characterized by CIMP.
Materials and Methods
Human colorectal cancer tissue samples
A total of 169 fresh frozen colorectal adenocarcinoma tumors and 165 adjacent normal tissues (cohort 1 and 2) were obtained from patients undergoing surgery at the Fox Chase Cancer Center. Tissue samples were selected to include approximately equal numbers from the proximal and distal colons. Additionally, 34 colorectal adenocarcinoma tumor genomic DNA samples were obtained from a previous study.10
DNA extraction
Fresh frozen human CRC tumor or adjacent normal tissue samples were lysed in 2% SDS and 25 mM EDTA tissue lysate solution with proteinase K (20 ug/mL). The lysates were incubated at 56°C for an hour and then transferred to 37°C until tissues were completely lysed. Proteins were precipitated with 10 M ammonium acetate and removed from homogenized tissue lysates by centrifugation at 12000RPM. Subsequently, DNA was precipitated using isopropanol, washed with 70% ethanol, and dissolved in LTET buffer (10 mM Tris pH 8.0, 0.1 mM EDTA, 0.1% Tween20). The DNA concentration was measured with the Qubit dsDNA Broad Range assay kit (Thermo Fisher, #Q32853).
Digital Restriction Enzyme Analysis of Methylation (DREAM)
Digital Restriction Enzyme Analysis of Methylation (DREAM) was performed on 115 CRC tumor and 15 adjacent normal genomic DNA samples (Table S1), following a previously described method.16 Briefly, the CRC genomic DNA samples were purified with 1X AMPure XP beads to remove fragments shorter than 200 base pairs to obtain high molecular weight DNA.17 The purified genomic DNA underwent sequential digestion with SmaI and XmaI, both recognize CCCGGG sites in DNA. This sequential digestion yielded distinct sequence patterns for methylated and unmethylated sites: SmaI only cuts unmethylated sites with blunt ends, while XmaI targets any remaining methylated sites with a 5’ CCGG overhang. These digested DNA fragments were utilized to construct Illumina libraries using NEBNext adapters and indexed primers (NEB #E7335S/E7500S). Methylation percentage was calculated as the fraction of methylated signatures determined by XmaI digestion out of the total of enzyme-digested sites as previously described.16
Bacterial strains
Bacterial genomic DNA samples were obtained from the American Type Culture Collection (Bacteroides fragilis ATCC 25285D–5, Fusobacterium nucleatum ATCC 25586D–5, and Klebsiella pneumoniae ATCC 700721D–5) (Table S2). Escherichia coli genomic DNA was isolated from E. coli K12 lab strain and E. coli NC101 adherent-invasive strain cultured in LB medium.
Quantitative PCR
Genomic DNA was extracted from the human CRC tumor and adjacent normal tissues and quantified with the Qubit dsDNA BR Assay (Thermo Fisher #Q32850). TaqMan primers and probes were used to quantify the abundance of B. fragilis, F. nucleatum, and K. pneumoniae in the genomic DNA of human CRC tumor and adjacent normal tissue as previously described.18–20 The primer set for E. coli was designed based on previously described primers21 (Table S3). Each bacterial primer/probes set was tested for the primer efficiency and specificity with bacterial genomic DNA (Table S2). The E. coli primers were evaluated against both the E. coli K12 lab strain and the E. coli NC101 adherent-invasive strain. The cycle at threshold (Ct) values of each bacterial species were normalized to the Ct values of human DNA detected by SLC24A3 or PGT genes.10,22 All assays were performed twice in triplicates, and the results were averaged.
16S rRNA gene sequencing
A total of 114 pairs of tumor and adjacent normal samples were evaluated for the sequencing from the cohort 1 (Table S1). The V3-V4 hypervariable region of 16S rRNA gene was amplified using the selected universal primers for Illumina barcode indexes, following previously described methods23 (Table S4). To ensure proper microbial genomic DNA extraction and library generation, ZymoBIOMICS Microbial Community Standard (Zymo, #D6300), ZymoBIOMICS Microbial Community DNA Standard (Zymo, #D6305), B. fragilis gDNA, E. coli gDNA, F. nucleatum gDNA, and K. pneumoniae gDNA were included in the library generation. The generated library samples were sequenced using the Illumina MiSeq (300bp paired-end).
Illumina EPIC array analysis
EPIC array data files of 76 CRC tumor tissue and 77 CRC adjacent normal tissue samples were processed and normalized using the ChAMP R package.24 We filtered out low-quality probes and imputed missing values with champ.filter()24 and normalized with champ.norm()24 using the beta-mixture quantile normalization (BMIQ) method.25
The cancer genome atlas data analysis
TCGA Level 2 Illumina HumanMethylation 450K array data for 295 colon adenocarcinoma (COAD) and 98 rectum adenocarcinoma (READ) patients, and Level 1 whole exome sequencing (WXS) data were downloaded using TCGAbiolinks.26 From the methylation array datasets, 393 primary solid tumor samples and 45 solid normal tissue samples were selected based on the availability, and technical replicates were averaged. To identify cancer-specific sites, 370,964 sites were filtered based on low methylation variability (STDEV <0.2), low average β-value (β-value <0.1) in primary solid normal samples, and high methylation variability in tumor samples (STDEV >0.2). These 18,289 cancer-specific CpG sites were converted to binary methylation status at the threshold of 0.2 β-value to compensate for the tumor purity.
The unaligned reads from TCGA-COAD and TCGA-READ WXS data were downloaded and re-aligned to the human CHM13 reference genome27 with bwa-mem2 to ensure the removal of human read contamination in the initial extraction. Subsequently, the unaligned reads were re-extracted and classified using Kraken2 with the full KrakenUniq database containing human, archaea, bacteria, viral, plasmid, and UniVec_Core.28 The classified counts were summarized at the genus level and estimated by Bayesian Re-estimation of Abundance with Bracken.29 Genus-level abundance values for each sample were normalized using the number of reads assigned to human (counts per million) for downstream analysis.
16S rRNA sequencing data analysis
The 16S rRNA sequencing data obtained from the paired 114 CRC tumor and adjacent normal samples were trimmed using cutadapt30 (Table S1). DADA2 was used to filter, trim, estimate error profiles, merge paired end reads, remove chimeras, and assign taxonomy by fitting amplicon sequencing variant (ASV) sequences to a pre-trained naïve Bayes classifier from the Silva v138 database. Species assignments were made by DADA2-formatted Silva v138 reference sequences.31 Community standards described above were used to assess the accuracy of the species-level assignment and determine possible contaminants. ASV-level counts, taxonomic information, and sample metadata were imported into R using the phyloseq package.32 ASVs not assigned to at least the Phylum level and ASVs present in fewer than 1% of samples were filtered out. ASV-level counts were agglomerated to the species level and analysis of compositions of microbiota with bias correction 2 (ANCOM-BC2)33 was used for differential abundance testing and to produce adjusted log-counts for ordination by PCA. Taxa were defined as significantly differentially abundant in each condition following testing with ANCOM-BC2 if their p-value was smaller than 0.05. The adjusted log abundances were calculated by adding a pseudocount (log(count +1)) to avoid taking log(0) and then adjust the log counts by subtracting the estimated samples fraction. Previously reported common laboratory contaminants in 16S rRNA sequencing data were removed before the further downstream analysis.34
Statistical analyses
All statistical analyses were performed using R.35 Unsupervised hierarchical clustering of DREAM methylation and TCGA methylation array data was performed using ward.D2 clustering method in the pheatmap R package.36 Group-level comparisons were performed using the Wilcoxon signed rank test or Kruskal-Wallis test where appropriate. Principal component analysis (PCA) was used to show variance between the Superhigh and the Non-Superhigh bacterial cases and to compare bacterial enrichment across different CIMP classes. Permutational multivariate analysis of variance (PERMANOVA) was used to confirm differences in the microbiota between tissue types or CIMP statuses with the vegan R package.37 The ANCOM-BC2 R package33 was used to address zero-inflation and over-dispersion in count data.38 Odds ratios and p-values for clinical and molecular associations with the Superhigh group were calculated by creating a contingency table and the Fisher’s Exact Test. Correlations between tumor and adjacent normal, as well as between 16S rRNA gene sequencing and bacterial qPCR, were tested by Pearson correlation. Volcano plots comparing abundances between tissues and CIMP statuses were constructed using the adjusted log-counts and the negative log10 of p-values determined by the ANCOM-BC2 R package. Shannon’s diversity index for 16S rRNA gene sequencing abundance counts of CRC tumor and CIMP tumors was calculated using the vegan R package. All plots were generated using the ggplot2 R package.39 No significant influence by technical artifacts was observed in any of the datasets studied.
Data availability
Sequence data from this article are available in the NCBI GEO (accession number: GSE237525, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE237525). Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jean-Pierre J. Issa, MD (jpissa@coriell.org).
Results
CIMP classification of CRC tumor samples
Based on previous work, we hypothesized the role for a distinct gut microbiota in CIMP in colorectal cancer. Kelly et al. (2017) have previously developed an enhanced CIMP classifier in AML,40 which we subsequently adapted for CRC methylation analysis. Utilizing the DREAM platform, we detected the methylation status of 26,370 CpG sites following the initial quality filtering, ensuring a minimum of 30 reads/site in at least 85% of the 115 tumor samples (Table S1), including individual technical replicates to ensure high confidence and high coverage sites. Given that much of the methylation variation is attributed to aging,5 we used the adjacent normal samples to eliminate aging-specific methylation sites. CpG sites were selected based on high variability (STDEV >20%) of methylation across tumor samples, coupled with low variability (STDEV <10%) and low average (average <2%) methylation in the adjacent normal samples, resulting in the identification of 1,317 cancer-specific CpG sites. To mitigate variability due to normal cell infiltration, we transformed these cancer-specific sites using binary values denoting methylated (1) and unmethylated (0) states, with methylated defined as greater than or equal to 10%, and unmethylated defined as less than 10%. The samples were assigned to CIMP categories based on unsupervised hierarchical clustering. The cluster with the highest number of methylated sites was labeled as CIMP-High (left cluster), the cluster with an intermediate number as CIMP-Low (right cluster), and the cluster with sparsely methylated sites as CIMP-Negative (middle cluster) (Figure 1a).
Figure 1.
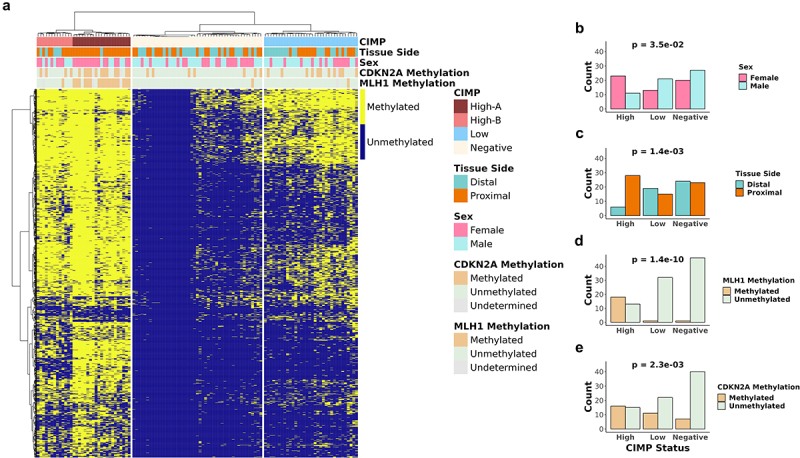
Categorical analysis of hierarchical clustering of methylation data CIMP status of CRC patient tumor samples.
(a) Unsupervised hierarchical clustering of 115 CRC tumor samples based on 1317 CpG sites from DREAM methylation data. b-e) Bar plots showing the patient statistics (b) sex, (c) tissue side, (d) MLH1 methylation status, and (e) CDKN2A methylation status in each CIMP status. Fisher’s exact test was used to test for statistical significance for each comparison in b-e.
As previously reported,41,42 we observed that CIMP-High cluster exhibited enrichment for female patients, proximal colon tumors, and methylation of MLH1 (Table S5, Figure 1b-e). The CIMP-High cluster showed a higher frequency of hypermethylated MLH1 (High 58.1%, Low 3.0%, Negative 2.1% p-value <0.001) and CDKN2A (High 51.6%, Low 33.3%, Negative 14.9% p-value <0.001) compared to the CIMP-Low or the CIMP-Negative cluster. Furthermore, proximal tumors were more prevalent in the CIMP-High cluster (High 84.2%, Low 44.1%, Negative 48.9, p-value <0.01) compared to the distal tumors, along with an older average age (High 72.2 years old, Low 70.0 years old, Negative 63.4 years old, p-value <0.01).
CIMP-High were further categorized into two clusters: CIMP-High-A and CIMP-High-B. The CIMP-High-A cluster showed a higher frequency of hypermethylated MLH1 (High-A 89.5%, High-B 8.3%, p-value <0.001) and CDKN2A (High-A 63.2%, High-B 33.3%, p-value >0.05) compared to the CIMP-High-B cluster (Table S6). The CIMP-High-A cluster was enriched for proximal tumors (High-A 95.2%, High-B 61.5%, p-value <0.05) and an older average age (High-A 77.6 years old, High-B 63.5 years old, p-value <0.001).
To increase the sample size, we analyzed the Illumina EPIC array data from previously reported seventy-six cases43 (Table S1). 27474 cancer-specific CpG sites were selected and assigned to binary methylation status at a 0.2 Beta-value threshold. Unsupervised hierarchical clustering of the binary cancer-specific sites revealed three distinct clusters (CIMP-High, CIMP-Low, and CIMP-Negative) with patterns consistent with those observed in the DREAM data (Figure S1). Furthermore, analysis of the EPIC array data also showed distinct CIMP-High-A and CIMP-High-B classes. Similar to the DREAM methylation data, the CIMP-High-A cluster displayed a higher prevalence in the proximal colon (High-A 100%, High-B 40%), a higher proportion of female patients (High-A 75%, High-B 60%), and a greater frequency of MLH1 methylated samples (High-A 50%, High-B 10%) in the EPIC array data. We merged the two cohorts for bacterial associations analysis.
Enrichment of multiple bacterial species in CIMP-Positive CRC
Previously, our laboratory reported an association between CIMP-High CRC and Fusobacterium.10 Here, we aimed to confirm this finding and establish an association between additional bacterial species associated with CRC and CIMP-Positive tumors. Using quantitative real-time PCR (qPCR), we detected Bacteroides fragilis, Escherichia coli, Fusobacterium nucleatum, and Klebsiella pneumoniae in 203 CRC tumor and 165 paired adjacent normal samples (Material and Methods) (Table S1 and S7). Consistent with previous findings,10,12,15,44 we observed a significant enrichment of all individual bacterial species in tumor tissue compared to adjacent normal tissue (Figure 2a). Next, we compared the bacterial enrichment in CIMP-Positive (CIMP-High and CIMP-Low) and CIMP-Negative groups. We observed a significantly higher enrichment of B. fragilis, F. nucleatum, and K. pneumoniae in CIMP-Positive compared to CIMP-Negative tumor samples (Wilcoxon signed-rank test, p < 0.05, Figure 2b and Figure S2A). In contrast, E. coli did not show a significant association with CIMP (Figure 2b).
Figure 2.
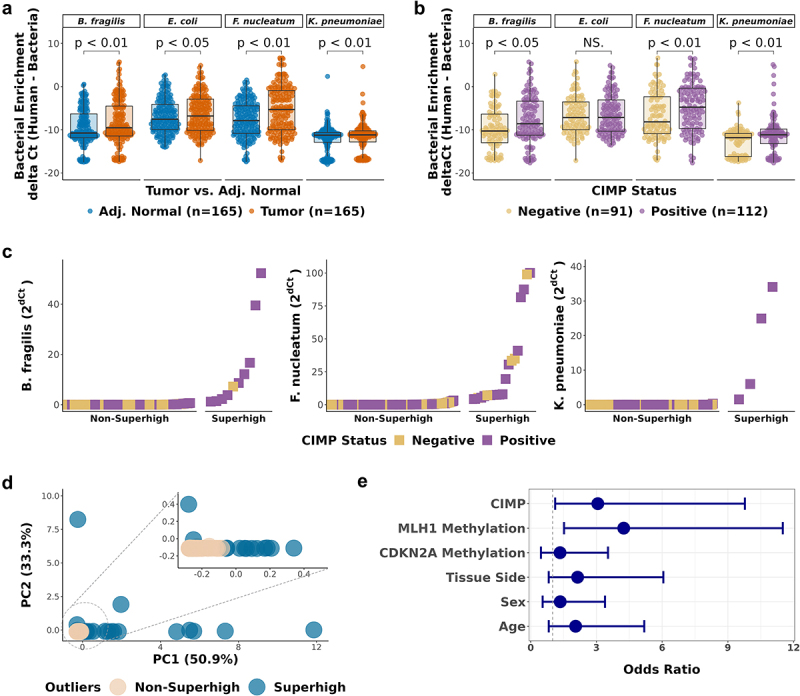
Enrichment of CRC-associated bacteria in CIMP-Positive CRC tumor samples.
a-b. Comparison of bacterial enrichment by qPCR between (a) paired tumor and adjacent normal samples (n = 165), (b) CIMP-Positive and CIMP-Negative tumor samples (n = 203). Significance was calculated by Wilcoxon test. (c) Ranking of the CRC tumor samples by bacterial enrichment. Tumor samples with high bacterial enrichment are classified as Superhigh samples. (d) Principal component analysis (PCA) of the CRC-associated bacteria (B. fragilis, F. nucleatum, and K. pneumoniae) enrichment. The circled area is expanded in the inset PCA plot. (e) Odds ratios of the association of the bacterial Superhigh status, determined by the three bacterial species, with different parameters including the CIMP status, MLH1 methylation, CDKN2A methylation, colorectal tissue side, sex, and age. Blue dots represent the point estimates of the odds ratio, and the lines represent the 95% confidence intervals around the estimates. Fisher’s exact test was used to test for statistical significance for each comparison.
Bacterial superhigh enrichment in CIMP-Positive CRC
It has previously been suggested that bacterial biofilms underlie the development of some CRCs, and such biofilms would result in highly aberrant bacterial enrichment, as observed previously for F. nucleatum.10 To study this in the current dataset, we ranked the bacterial enrichment of 203 tumor samples for B. fragilis, F. nucleatum, and K. pneumoniae (Figure 2c). Following the approach used in our previous study,10 we determined thresholds at the inflection point of bacterial abundance and identified a subset of samples with high enrichment, which we termed as bacterial Superhigh cases. To confirm the effectiveness of the enrichment thresholds for the Superhigh cases using an unbiased statistical approach, we performed principal component analysis (PCA) on the bacterial enrichment qPCR data. PCA plots were color-coded by the binary Superhigh status, as shown in Figure 2c and by the CIMP status in Figure S2C (Figure 2d). Outlying points in the PCA plot predominantly belonged to the Superhigh group, while tightly clustered data points around the origin primarily represented the Non-Superhigh samples. This analysis highlights that the enrichment thresholds effectively identified the Superhigh cases and successfully segregated them into a distinct group of bacterial outliers in the PCA. For B. fragilis, F. nucleatum, and K. pneumoniae, a total of twenty-six bacterial Superhigh cases were identified, and they showed significant associations with CIMP and CIMP characteristics such as MLH1 methylation (Table 1). The Superhigh group had 3.1 times higher odds (95% CI, 1.19–9.78) of being CIMP-Positive and 4.2 times higher odds (95% CI, 1.52–11.5) of having MLH1 methylation (Figure 2e). Other CIMP characteristics, including CDKN2A methylation, sex, tissue side, and age all showed trends for higher odds in the Superhigh cases.
Table 1.
Association between characteristics of CIMP and the Superhigh bacterial group.
| Superhigh (n = 26) | Non-Superhigh (n = 177) | p-value | |
|---|---|---|---|
| CIMP status | 0.013 | ||
| Positive | 20 (76.9%) | 92 (52.0%) | |
| Negative | 6 (23.1%) | 85 (48.0%) | |
| MLH1 methylation status* | 0.0025 | ||
| Methylated | 10 (38.5%) | 22 (12.7%) | |
| Unmethylated | 16 (61.5%) | 151 (87.3%) | |
| CDKN2A methylation status* | 0.33 | ||
| Methylated | 8 (30.8%) | 43 (24.9%) | |
| Unmethylated | 18 (69.2%) | 130 (75.1%) | |
| Sex | 0.31 | ||
| Female | 14 (53.8%) | 82 (46.3%) | |
| Male | 12 (46.2%) | 95 (53.7%) | |
| Tissue Side† | 0.066 | ||
| Distal colon | 8 (32.0%) | 86 (50.3%) | |
| Proximal colon | 17 (68.0%) | 85 (49.7%) | |
| Pathologic Stage‡ | 0.098 | ||
| Stage 1 | 2 (7.7%) | 6 (3.8%) | |
| Stage 2 | 4 (15.4%) | 29 (18.2%) | |
| Stage 3 | 13 (50.0%) | 106 (66.7%) | |
| Stage 4 | 7 (26.9%) | 18 (11.3%) | |
| Age | 0.066 | ||
| Over 70 | 14 (53.8%) | 64 (36.2%) | |
| Under 70 | 12 (46.2%) | 113 (63.8%) |
*.missing 4 data points.
†.missing 7 data points.
‡.missing 18 data points.
Fisher’s exact test for all characteristics.
Bold denotes the significance
Bacterial enrichment in CRC adjacent normal tissue
A previous study reported that tumor type-specific bacteria have a higher bacterial load in tumor-adjacent normal breast cancer tissue than in healthy breast samples.45 Other previous studies have also shown the enrichment of Fusobacterium in adjacent normal tissue of colorectal cancer samples.10,15,22 Based on this, we investigated whether CIMP-specific bacteria are also enriched in adjacent normal tissue samples. Our analysis revealed statistically significant Pearson correlation coefficients between bacterial abundance of paired tumor and adjacent normal samples in B. fragilis (r = 0.76, p < 0.001), F. nucleatum (r = 0.71, p < 0.001), and K. pneumoniae (r = 0.71, p < 0.001) (Figure 3a). The significance of correlation testing and the adjusted p-values were both statistically significant (p < 0.001, adjusted.p < 0.001). We further investigated whether the Superhigh tumor cases had higher bacterial enrichment in their adjacent normal pairs. Most of the adjacent normal pairs of the Superhigh cases had higher bacterial enrichment than the median (Figure 3b). The adjacent normal pairs of the Superhigh tumor samples had significantly higher B. fragilis and F. nucleatum enrichment than the Non-Superhigh adjacent normal sample pairs (Figure 3c, p < 0.001). No significant difference was observed between the adjacent enrichments of the Superhigh and the Non-Superhigh in K. pneumoniae, possibly due to an insufficient number of samples.
Figure 3.
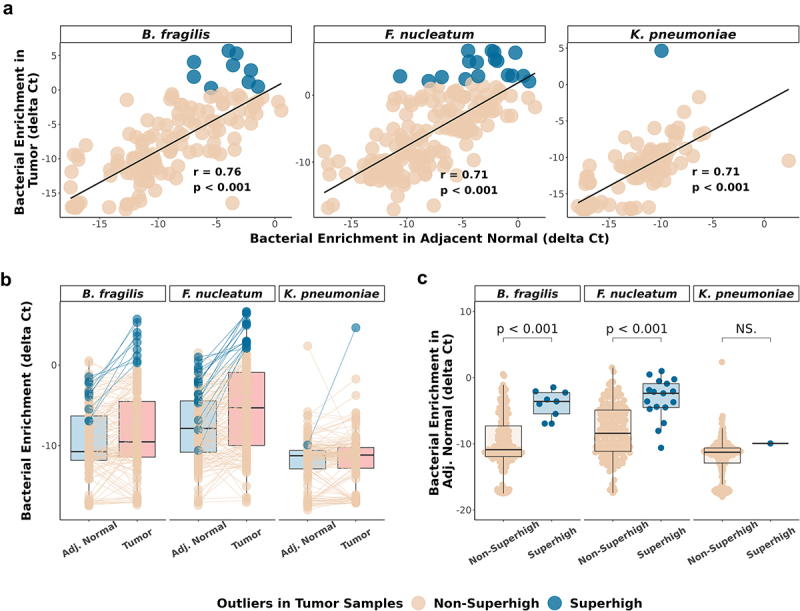
Bacterial enrichment in CRC adjacent normal is associated with CIMP.
(a) Scatterplot showing the enrichment of 115 paired tumor samples (y-axis) and adjacent normal samples (x-axis) by qPCR in B. fragilis, F. nucleatum, and K. pneumoniae. Linear regression lines (black) are built on each correlation. Pearson correlation coefficients and p-values are indicated in each plot. (b) Comparison of the delta Ct values between 115 paired adjacent normal and the tumor samples by bacterial qPCR. Blue and cream lines connect each paired adjacent normal and tumor samples. Blue dots represent bacterial Superhigh cases determined by high bacterial enrichment in tumor samples across the three bacterial species. (c) Comparison of the Superhigh and the Non-Superhigh cases determined by tumor samples in adjacent normal samples. Significance was calculated by Wilcoxon test.
Association between bacteria and CIMP in TCGA data
We analyzed the 450K methylation array data from 393 tumor and 45 normal tissue samples in TCGA-COAD and TCGA-READ datasets to verify the findings from the CRC human tissue samples (Table S1, see Materials and Methods). Unsupervised hierarchical clustering of the 18,289 selected sites (see Materials and Methods) resulted in three CIMP classes and clearly distinct classification of CIMP-High subsets, CIMP-High-A and CIMP-High-B (Figure 4a). CIMP-High group exhibited a higher frequency of female patients, was more localized in the proximal colon, and had a greater MLH1 methylation and CDKN2A methylation frequency than CIMP-Low or CIMP-Negative groups (Figure 4a, Table S8). Similarly, a significantly higher frequency of MLH1 methylated samples was observed in CIMP-High-A cluster than in CIMP-High-B cluster (88% (30/34), 4% (1/23), p-value <0.001, data not shown). These classifications were concordant with the observed clusters in DREAM data. Additionally, we compared our genome-wide approach with two previously reported CIMP gene panel classifications: the 8-gene panel from Nosho et al.46 and the 5-gene panel from Weisenberger et al..47 Using the Nosho panel, 38 out of 393 samples were classified as CIMP-High (348 CIMP-Low/Negative samples, 7 undetermined). Most of these were also classified as CIMP-High using our approach, but the panel misclassified 46% of the CIMP-High cases identified by the genome wide approach. Using the Weisenberger panel, 167 out of 383 samples were classified as CIMP-Positive but this panel misclassified 38.9% of the CIMP-Positive samples we identified.
Figure 4.
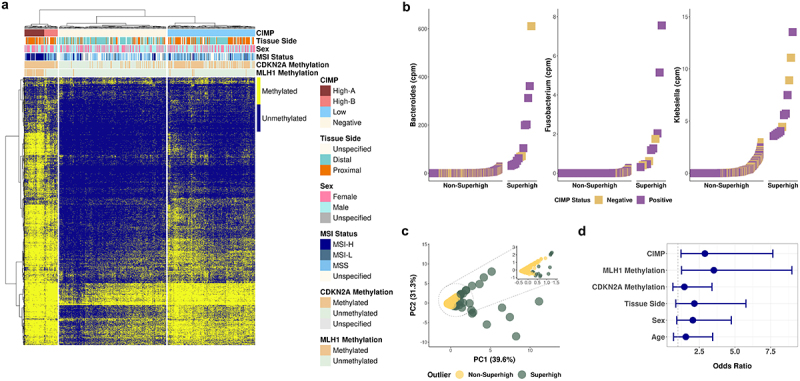
Validation of the CIMP classification method by DREAM using TCGA colon adenocarcinoma and rectum adenocarcinoma methylation array data set.
(a) Unsupervised hierarchical clustering of 393 primary solid tumor and 45 solid tissue normal samples based on 18,289 CpG sites from TCGA COAD and READ 450K methylation array data. (b) Superhigh ranking analysis of bacterial reads from 392 TCGA COAD and READ tumor whole exome sequencing data. (c) PCA of the enrichment of the three CRC-associated bacteria in TCGA datasets. The circled area is expanded in the inset PCA plot. (d) Odds ratios of the association of the bacterial Superhigh cases by TCGA cohorts with different parameters such as the CIMP status, MLH1 methylation, CDKN2A methylation, colorectal tissue side, sex, and age. Fisher’s exact test was used to test for statistical significance for each comparison.
We next analyzed the extracted bacterial reads from the whole exome sequencing data (see Materials and Methods) at the genus level. Ranking analysis of the 392 tumor samples revealed that the abundance of Bacteroides, Fusobacterium, and Klebsiella resembled the pattern of the Superhigh cases in the bacterial qPCR (Figure 4b). The abundance counts encompass all species within the three genera. The Superhigh group exhibited a higher dispersion than the aggregated grouping of the Non-Superhigh group in the PCA plot (Figure 4c). Among the bacterial Superhigh group, the odds ratio for association with CIMP was 2.9 (95% CI 1.2–7.7, p < 0.01) and MLH1 methylation was 3.5 (95% CI 1.3–9.0, p < 0.01) (Figure 4d, Table S9).
CIMP-specific microbiota
We performed 16S rRNA gene sequencing on the same set of human CRC tumor (n = 114) and adjacent normal samples (n = 114) (Table S1) to identify additional bacterial taxa in the colon that are associated with CIMP. We used ANCOM-BC233 to evaluate differential abundance at the species level. PCA of a total of 1,212 identified genera/species did not reveal discernible clustering between tumor and adjacent normal (p-value = 0.081, PERMANOVA) (Figure S4A), indicating that the observed associations are limited to specific bacteria. To identify these, we searched for cancer-specific significant taxa based on the level of enrichment and statistical significance (|log2FC| > 0.5, p-value <0.05) determined by ANCOM-BC2 (Figure 5b). Twenty-three significant genera/species were identified (Table S10). We confirmed the distinct clustering of tumor and adjacent normal samples by the selected taxa using PCA clustering and PERMANOVA test (p-value <0.001) (Figure 5a). Campylobacter, Fusobacterium nucleatum, Hungatella hathewayi, Enterobacter, and Leptotrichia were all significantly more enriched in tumor tissue than in adjacent normal tissue (Figure 5c, Figure S4), and all six bacterial taxa were previously reported to exhibit co-occurrence and association with CRC tumor tissue.48–50
Figure 5.
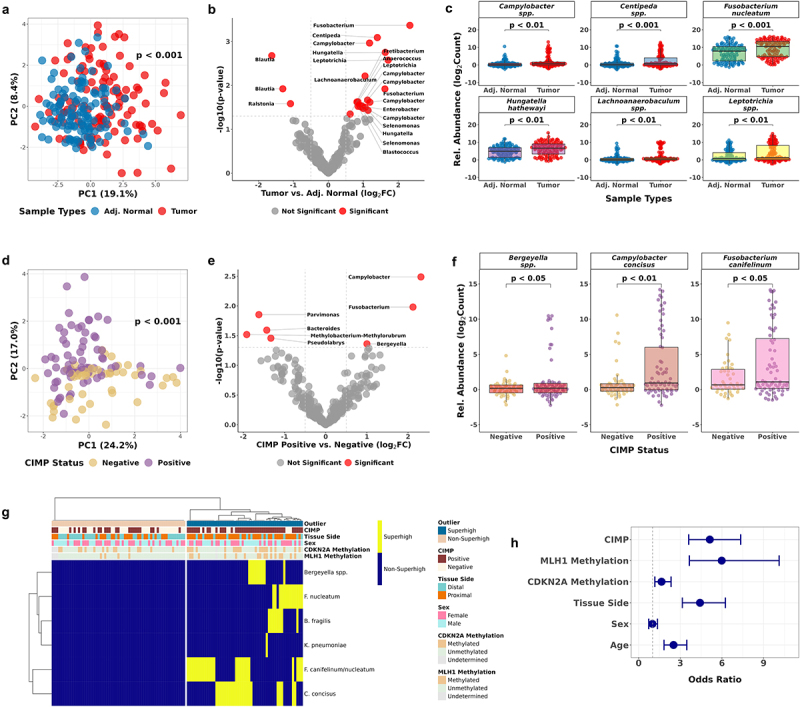
Discovery of CIMP-associated taxa in CRC tumor samples by 16S ribosomal RNA sequencing.
(a) PCA of 23 bacterial taxa selected by the tissue type comparison using ANCOM-BC2. P-values were calculated by PERMANOVA. (b) Volcano plot showing the 23 significant bacterial taxa enriched in tumor or adjacent normal CRC cases. (c) Bacterial enrichment comparison by 16S rRNA gene sequencing paired tumor and adjacent normal samples (n = 228). (d) PCA of 7 bacterial taxa selected by the comparison between CIMP statuses using ANCOM-BC2. (e) Volcano plot showing the 7 significant bacterial taxa enriched in CIMP-Positive or CIMP-Negative tumor samples. (f) Bacterial enrichment comparison between CIMP statuses by 16S rRNA gene sequencing tumor samples (n = 114). (g) Unsupervised hierarchical clustering of the binary Superhigh data of B. fragilis, F. nucleatum, K. pneumoniae, Bergeyella, C. concisus, and F. canifelinum. (h) Odds ratios of the bacterial Superhigh group to be associated with different parameters such as the CIMP status, MLH1 methylation, CDKN2A methylation, colorectal tissue side, sex, and age compared to the Non-Superhigh group. Fisher’s exact test was used to test for statistical significance for each comparison.
The enrichment of F. nucleatum in CRC tumor samples, as determined by 16S rRNA gene sequencing, was concordant with the qPCR analysis result (Figures 5c and 2a). In contrast, the results for B. fragilis and K. pneumoniae showed less concordance (Figure 2a, Table S10). We performed a Pearson correlation analysis to illustrate the relationship between the qPCR and the 16S rRNA gene sequencing results for the three bacterial species (Figure S6). The Pearson correlation coefficients revealed a moderate positive correlation between the 16S relative abundance and bacterial qPCR enrichment in B. fragilis (r = 0.56, p < 0.001) and F. nucleatum (r = 0.68, p < 0.001), and a weaker positive correlation in K. pneumoniae (r = 0.39, p < 0.001).
We then examined the taxa associated with CIMP-Positive tumors and identified seven significant genera/species (Table S11). A PCA plot revealed distinct clusters representing CIMP-Positive and CIMP-Negative clusters (p < 0.001, PERMANOVA, Figure 5d). Bergeyella, Campylobacter concisus, and Fusobacterium canifelinum were significantly enriched in CIMP-Positive tumor samples (p < 0.05, p < 0.01, p < 0.05, ANCOM-BC2 (Figure 5f)). To explore whether these additional bacterial taxa could strengthen the association between the gut microbiota and CIMP in CRC, we combined the binary bacterial Superhigh status defined by the bacterial qPCR and the 16S rRNA gene sequencing analyses.
We ranked each taxon based on the relative abundance of 114 tumor samples to determine the bacterial Superhigh cases from the CIMP-Positive enriched taxa (Figure S5A). We classified 45 Superhigh cases based on the enrichment of the three taxa (Figure S5B). The 16S Superhigh group had 5.6 times higher odds of being CIMP-Positive than the Non-Superhigh group (95% CI 2.2–16.0, p < 0.001, Figure S5C). Hierarchical clustering of the binary bacterial Superhigh status of 114 CRC tumor samples, determined by the combined six bacterial taxa, showed a distinct separation between the Superhigh and the Non-Superhigh cases (Figure 5g). The collective Superhigh group had 5.1 times higher odds of being CIMP-Positive (95% CI 3.6–7.3, p < 0.001) and 6 times higher odds of having MLH1 methylation (95% CI 3.7–10.1, p < 0.001) than the Non-Superhigh group, and primarily localized on the proximal colon and showed stronger association with older patients (Figure 5h).
We examined the association between CIMP-High subgroups and the bacterial Superhigh group as well. The odds of being CIMP-High-A were 2.7 times higher (95% CI 1.4–5.3, p < 0.01) in the Superhigh group than being CIMP-High-B. Thirteen taxa were identified in the comparison between CIMP-High vs. CIMP-Low&Negative classes (Figure S3B). A PCA plot showed clear distinct dispersion patterns between CIMP-High and CIMP-Low&Negative (Figure S3A) (p < 0.001, PERMANOVA). Including Campylobacter concisus, eight out of the thirteen were enriched in CIMP-High tumors (Figure S3C, Table S12). Parasutterella and Selenomonas have been previously associated with CRC and reported to have a significance co-occurrence with F. nucleatum, Campylobacter, and Leptotrichia.51,52
A reduction of alpha diversity has been commonly observed in tumor microbiota.53,54 We therefore determined alpha diversity in our dataset. Shannon’s diversity index was calculated (see the Materials and Methods) based on the 16S rRNA gene sequencing abundance counts (Figure S7). We observed a decrease in alpha diversity in the tumor samples compared to the adjacent normal samples (Figure S7A, p = 0.0014). Interestingly, the diversity index showed the most significant difference in CIMP-Low tumor samples (Figure S7B, p = 0.013). As expected, based on our hypothesis, the diversity index was lower in the Superhigh tumors compared to the Non-Superhigh tumors (Figure S7C, p = 0.012).
Discussion
CpG island methylator phenotype in colorectal cancer cannot solely be attributed to genetic aberrations. This highlights the significant impact of environmental factors on the aberrant changes in DNA methylation. Notably, the gut microbiota has been implicated in colorectal cancer,55,56 with the previous study on a significant association between CIMP and F. nucleatum.10 In this study, we identified CRC tumor samples with high bacterial abundance as bacterial Superhigh based on the enrichment of CRC-associated bacterial species, B. fragilis, F. nucleatum, and K. pneumoniae. We found a significant association between the bacterial Superhigh cases and CIMP characteristics. The classification of CIMP using DREAM was consistent with previously known CIMP characteristics, including its common localization in the proximal colon, higher prevalence in female patients, and MLH1 hypermethylation (Table S5). Our genome-wide analysis of CpG sites yielded a consistent and successful clustering of CIMP classification. This resolved the limited consensus on the CIMP definition previously observed due to the utilization of various CIMP gene sets in determining the CIMP status in tumor samples. Additionally, the genome-wide analysis identified two distinct subclasses of CIMP-High, High-A and High-B, where High-A is more significantly enriched with MLH1-methyated cases (Table S6). Interestingly, two MLH1-methylated tumor samples were in the CIMP-Low and CIMP-Negative groups (Figure 1a). The CIMP-Low MLH1-methylated sample had very low bacterial detection, and it may be an example of MLH1 methylation that develops independently of CIMP. The MLH1-methylated CIMP-Negative sample had a very high F. nucleatum enrichment. Although it could also be an example of MLH1 methylation that develops by non-CIMP mechanisms, this case could also be a missed CIMP-High case due to low tumor purity.
We examined the association between CIMP and CRC-associated bacterial species: B. fragilis, E. coli, F. nucleatum, and K. pneumoniae, and we observed a statistically significant association between B. fragilis, F. nucleatum, and K. pneumoniae, and CIMP and MLH1 methylation. This finding reinforces the previous report on F. nucleatum and B. fragilis in CIMP-Positive CRC tumors10,11,57 and introduces K. pneumoniae as a CIMP-associated bacteria for the first time to our knowledge. K. pneumoniae is both commensal and opportunistic microbial organism in the human body.58,59 The enrichment of K. pneumoniae in tumor tissue in our study might raise concerns about potential contamination in healthcare settings. While this argument cannot be entirely ruled out, numerous studies have demonstrated a specific association between K. pneumoniae and inflammation, as well as tumorigenesis in the colon.12,60
It is worth noting that the association of these species is based on the bacterial Superhigh status of CRC tumor samples, determined by high bacterial abundance of the three bacterial species collectively (Figure S2B). In contrast to the other three species, we did not observe a significant association between E. coli and CIMP (Figure 2b). E. coli in CRC has been associated with DNA damage by its colibactin-producing strains. A significant association between APC:c.835–8 A > G somatic mutation and an induction of a mutational signature in human intestinal organoids repeatedly exposed to genotoxic pks+ E. coli have been reported.11,61 Our finding supports a role of E. coli in mutagenesis through a CIMP-independent mechanism in CRC.
We performed a comparative analysis on bacterial enrichment between paired tumor and adjacent normal tissue samples to determine whether methylation changes associated with bacterial species are also present in the adjacent normal tissue. A high correlation between the bacterial enrichment in paired tumor and adjacent normal samples (Figure 3a) suggests that the bacterial enrichment found in the Superhigh tumor cases may have originated from the adjacent normal tissue and then amplified as the tumor developed. Notably, the adjacent normal pairs of the Superhigh bacterial tumor samples showed higher bacterial abundance than other adjacent normal samples (Figure 3b-c). Considering a crucial role of CIMP during the early stage of colorectal tumor formation,62,63 the bacterial enrichment in the tumor-adjacent tissue may significantly contribute to the development and progression of CIMP in colorectal cancer.
A recent study by Gihawi et al.64 showed that a significant methodological errors in a previous study, which had led to an overestimation of the amount of bacterial DNA present in TCGA tumor sequencing data. The authors demonstrated that contamination from mislabeled sequences and human reads introduced false positive bacterial reads in the analyzed samples. To address this issue, we re-aligned the WXS reads from TCGA-COAD and TCGA-READ datasets to the CHM13 human reference genome. The resulting unaligned reads were then classified using Kraken2 with the KrakenUniq database (see Methods and Materials). Genus-level counts were estimated using Bayesian re-estimation of abundances with Braken.29 Our TCGA data analysis revealed that the bacterial Superhigh cases had 2.9 times higher odds of being CIMP-Positive and 3.5 times higher odds of being MLH1 methylated than the Non-Superhigh cases (Figure 4d, Table S9). Although the overall data analysis revealed significant odds regarding the bacterial Superhigh cases being CIMP-Positive and frequency of MLH1 promoter methylation, it is important to acknowledge that TCGA data collection was not initially designed for bacterial enrichment study, which poses limitations including less accurate representation of the bacteria taxa presented in the samples.65
Furthermore, we identified enrichment of a distinct group of bacterial taxa that might play a role in CIMP development in CRC tumor by comparing the abundance counts from 16S rRNA gene sequencing. A high enrichment of Campylobacter, Fusobacterium nucleatum, Hungatella hathewayi, Enterobacter, Leptotrichia, and Selonomonas were detected in CRC tumor tissues, and the co-occurrence of these bacteria has been reported in CRC.50,52 Bergeyella, Campylobacter concisus, and Fusobacterium canifelinum were significantly enriched and associated with CIMP-Positive tumor tissues. A combined group of six CIMP-associated bacterial taxa from the qPCR and 16S rRNA gene sequencing showed a strong association, which supports the polymicrobial hypothesis14 regarding the gut microbiota’s association with CIMP in CRC tumorigenesis. Additionally, we observed a decrease in alpha diversity of colon tissue microbiota represented by Shannon’s diversity index in CRC tumor sample comparisons using the abundance counts from the 16S rRNA gene sequencing. Alpha diversity specifically decreased in the Superhigh tumor samples compared to the Non-Superhigh tumor samples (Figure S7C), as expected from aberrantly high enrichment of a limited group of bacterial taxa.
While the causal relationship between gut microbiota and CIMP is not fully understood, the strong association between the Superhigh enrichment of the bacterial taxa in CIMP-Positive CRC tumor samples supports the close interaction between the colonic epithelium and distinct polymicrobial groups in the gut. This interaction often involves bacterial invasion into the protective mucosal layer, a phenomenon commonly observed in tumorigenic biofilms. Biofilms are bacterial communities that adhere to the intestinal mucosa and form a protective matrix.66 Previous studies have indicated an increase in the tumorigenic potential of biofilms in the progression from normal mucosa to tumor tissue, and a high enrichment of distinct bacterial taxa within these tumor-associated biofilms.14,67 The strong correlation between the bacterial Superhigh tumor samples and their paired adjacent normal samples (Figure 3c) suggests a preexisting bacterial field defect that facilitates a formation of biofilm. Interestingly, the mucus layer from healthy individuals is devoid of bacteria and shows consistent biofilm formation across different colon locations.68 However, a notable difference in bacterial invasion into the mucosal layer was observed between tumor and adjacent normal tissues in CRC patients, with significantly higher levels in the proximal colon compared to the distal colon. The concurrent overlap between CIMP-Positive tumors and tumorigenic biofilms suggests that members of the invasive bacterial biofilms in tumor samples may be linked to CIMP in CRC. Further investigation into the CIMP status of biofilm-positive CRC tumor samples and the association between bacterial taxa in the biofilm and CIMP tumors would provide additional insights into the etiology of CIMP.
Recent work by Tricarico et al. demonstrated that inactivation of Tet1-Tdg induced hypermethylation in CpG islands using the Tet1–/– TdgN151/+ApcMin/+ mutant mouse model.69 TET family inactivation by the oncometabolite, D-2-hydroxyglutarate (D-2HG), resulting from IDH1/2 mutation, is a well-known cause of CIMP in glioma.6 The absence of TET or IDH mutations in CRC CIMP70 suggests that the gut microbiota may play a role in TET inactivation. The gut microbiota produces various metabolic products that influence host immunity and epigenetic regulation in the surrounding colonic epithelium.71 This implies that bacterial metabolites may interfere with epigenetic regulators such as TET, contributing to CIMP and cancer progression and initiation. Further investigation into bacterial metabolites produced by the CIMP-associated bacterial taxa observed in our study will be essential to identify potential oncometabolites and to provide insight into bacterial contributions to CIMP-positive cancer formation.
This study has several limitations. Antibiotics administration to patients prior to colonoscopy or surgical resection is common practice and the samples were not controlled for this, which could potentially skew the composition of gut microbiota. Also, while 16S rRNA gene sequencing is a valuable tool for gut microbiota classification, it lacks the ability to discern the ratio between human and bacterial DNA, which limits the ability to identify relatively high-level bacteria enrichment. Thus, validation of the sequencing results with targeted qPCR analysis is needed. Our data do not address the mechanism of the bacteria-DNA methylation link, which should be explored in future studies. Another limitation arises from adding small numbers to avoid taking the log of zero, which can affect data interpretation.72 To address this common and unresolved limitation with count data, we used ANCOM-BC2 to manage zeros and to address over-dispersion of count data through a linear model on log-transformed counts.33,38
In conclusion, our study revealed a significant enrichment of a distinct group of bacterial taxa, including B. fragilis, F. nucleatum, and K. pneumoniae, in CIMP-Positive CRC tumor samples, which we termed as the bacterial Superhigh. We identified a significant association between these bacterial Superhigh cases and CIMP in CRC. Given that CIMP in CRC is not characterized by frequent mutations in the DNA methylation regulatory pathways, our findings emphasize the gut microbiota as a prominent environment factor in the etiology of CIMP in CRC and highlight their potential as a valuable diagnostic and therapeutic target to mitigate CRC risk.
Supplementary Material
Funding Statement
This study was supported by the National Institutes of Health grants U54 CA221704(5) (to C. Sapienza, J.-P.J. Issa), R21 CA264213 (to C. Sapienza and J. Ghosh), P50-CA254897 (to J.-P.J. Issa and J. Jelinek), and R01-CA214005 (to J.-P.J. Issa and C. Jobin), and two PA Cure grant 914103047 (to C. Sapienza) from Pennsylvania Department of Health.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
PP, CJ, and J-P JI contributed to study design. PP generated all the data except the Illumina EPIC array data which were provided by JG and CS. PP, KK, GC, JM, and JJ have contributed to the data acquisition and bioinformatics analysis. PP and J-P JI analyzed and interpreted the data. PP and J-P JI prepared figures, tables, and the manuscript. PP, KK, GC, JJ, JM, RZG, JG, CS, CJ, and J-P JI contributed to editing the paper. All authors discussed the results, contributed to finalizing the manuscript, and declared no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2024.2363012
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F.. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–18. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Talseth-Palmer BA. The genetic basis of colonic adenomatous polyposis syndromes. Hered Cancer Clin Pract. 2017;15(1). doi: 10.1186/s13053-017-0065-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I. et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68(1):31–54. doi: 10.3322/caac.21440. [DOI] [PubMed] [Google Scholar]
- 4.Müller MF, Ibrahim AEK, Arends MJ. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016;469(2):125–134. doi: 10.1007/s00428-016-1956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer,” (in eng. Proc Natl Acad Sci U S A. 1999. Jul. 96(15):8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010. Dec. 18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jahromi MS, Xekouki P. et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013. June. 3(6):648–657. doi: 10.1158/2159-8290.cd-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thursby E, Juge N. Introduction to the human gut microbiota,” (in eng. Biochem J. [2017 May 16]. 474(11):1823–1836. doi: 10.1042/BCJ20160510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkins LJ, Monga M, Miller AW. Defining dysbiosis for a cluster of chronic diseases. Sci Rep. 2019;9(1):2019–09–09. doi: 10.1038/s41598-019-49452-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano H-O, Sugai T, An B. et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma,” (in eng. Cancer Res. 2014. Mar. 74(5):1311–1318. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joo JE,Chu YL, Georgeson P, Walker R, Mahmood K, Clendenning M, Meyers AL, Como J, Joseland S, Preston SG. et al. Intratumoral presence of the genotoxic gut bacteria pks+ E. coli, enterotoxigenic bacteroides fragilis, and fusobacterium nucleatum and their association with clinicopathological and molecular features of colorectal cancer. Br J Cancer. 2024.. doi: 10.1038/s41416-023-02554-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pope JL, Yang Y, Newsome RC, Sun W, Sun X, Ukhanova M, Neu J, Issa J-P, Mai V, Jobin C. Microbial colonization coordinates the pathogenesis of a Klebsiella pneumoniae infant isolate. Sci Rep. 2019;9(1). doi: 10.1038/s41598-019-39887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, Campbell BJ, Abujamel T, Dogan B, Rogers AB. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012. Oct. 338(6103):120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M. et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Nat Acad Of Sci. 2014;111(51):18321–18326. doi: 10.1073/pnas.1406199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012. Feb. 22(2):292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jelinek J, Madzo J. DREAM: a simple method for DNA methylation profiling by high-throughput sequencing. In: Tost J, editor. Methods in Molecular Biology. (NY): Springer; 2016. p. 111–127. [DOI] [PubMed] [Google Scholar]
- 17.Quail MA, Swerdlow H, Turner DJ. Improved protocols for the illumina genome analyzer sequencing system. Current Protocols In Human Genetics. 2009;62(1). doi: 10.1002/0471142905.hg1802s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee CS, Lee J. Evaluation of new gyrB-based real-time PCR system for the detection of B. fragilis as an indicator of human-specific fecal contamination, (in eng. J Microbiol Methods. 2010. Sep. 82(3):311–318. doi: 10.1016/j.mimet.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Martin FE, Nadkarni MA, Jacques NA, Hunter N. Quantitative microbiological study of human carious dentine by culture and real-time PCR: association of anaerobes with histopathological changes in chronic pulpitis,” (in eng. J Clin Microbiol. 2002. May. 40(5):1698–1704. doi: 10.1128/JCM.40.5.1698-1704.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartman LJ, Selby EB, Whitehouse CA, Coyne SR, Jaissle JG, Twenhafel NA, Burke RL, Kulesh DA. Rapid real-time PCR assays for detection of Klebsiella pneumoniae with the rmpa or maga genes associated with the hypermucoviscosity phenotype. J Mol Diagn. 2009;11(5):464–471. doi: 10.2353/jmoldx.2009.080136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 2014;5(1):4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. [2013 Jan 7]. 41(1):e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, Teschendorff AE. ChAMP: updated methylation analysis pipeline for Illumina BeadChips,” (in eng. Bioinformatics. [2017 Dec 15]. 33(24):3982–3984. doi: 10.1093/bioinformatics/btx513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data,” (in eng. Bioinformatics. [2013 Jan 15]. 29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. [2016 May 5]. 44(8):e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A. et al. The complete sequence of a human genome,” (in eng. Science. 2022. Apr. 376(6588):44–53. doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Rincon N, Wood DE, Breitwieser FP, Pockrandt C, Langmead B, Salzberg SL, Steinegger M. Metagenome analysis using the Kraken software suite. Nat Protoc. 2022;17(12):2815–2839. doi: 10.1038/s41596-022-00738-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Breitwieser FP, Thielen P, Salzberg SL. Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 2017;3:e104. doi: 10.7717/peerj-cs.104. [DOI] [Google Scholar]
- 30.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 31.Callahan BJ, Mcmurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mcmurdie PJ, Holmes S, Watson M. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1). doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chrisman B, He C, Jung JY, Stockham N, Paskov K, Washington P, Wall DP.. The human “contaminome”: bacterial, viral, and computational contamination in whole genome sequences from 1000 families. Sci Rep. 2022;12(1). doi: 10.1038/s41598-022-13269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.R core team. R: a language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. 2019). [Online]. Available: https://www.R-project.org.
- 36.Kolde R. Pheatmap: pretty heatmaps. R Package Version. 2019;1(2):726. [Google Scholar]
- 37.Vegan: community Ecology Package_. R Package. 2022). [Online]. Available: https://CRAN.R-project.org/package=vegan.
- 38.Lin H, Peddada SD. Multigroup analysis of compositions of microbiomes with covariate adjustments and repeated measures. Nat Methods. 2024;21(1):83–91. doi: 10.1038/s41592-023-02092-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ggplot2: elegant graphics for data analysis. (NY). ISBN 978-3-319-24277-4. [Online]. Available: Springer-Verlag; 2016. https://ggplot2.tidyverse.org. [Google Scholar]
- 40.Kelly AD, Kroeger H, Yamazaki J, Taby R, Neumann F, Yu S, Lee JT, Patel B, Li Y, He R. et al. A. D. Kelly et al. “A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia. 2017;31(10):2011–2019. doi: 10.1038/leu.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grady WM, Yu M, Markowitz SD. Epigenetic alterations in the gastrointestinal tract: Current and emerging use for biomarkers of cancer. Gastroenterology. 2021;160(3):690–709. doi: 10.1053/j.gastro.2020.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P. et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh J, Schultz B, Chan J, Wultsch C, Singh R, Shureiqi I, Chow S, Doymaz A, Varriano S, Driscoll M. et al. Epigenome-wide study identifies epigenetic outliers in normal mucosa of patients with colorectal cancer. (In Eng), Cancer Prev Res (Phila). [2022 Oct 10]. OF1–OF12. doi: 10.1158/1940-6207.CAPR-22-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL. et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018;359(6375):592–597. doi: 10.1126/science.aah3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E. et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science. [2020 May 29]. 368(6494):973–980. doi: 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES, Hazra A, Hunter DJ, Quackenbush J, Spiegelman D. et al. Comprehensive biostatistical analysis of CpG Island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One. 2008;3(11):e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D. et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006. Jul. 38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 48.Xia X, Wu WKK, Wong SH, Liu D, Kwong TNY, Nakatsu G, Yan PS, Chuang Y-M, Chan MWY, Coker OO. et al. Bacteria pathogens drive host colonic epithelial cell promoter hypermethylation of tumor suppressor genes in colorectal cancer. Microbiome. 2020;8(1):2020–12–01. doi: 10.1186/s40168-020-00847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, Pons B, Mirey G, Vignard J, Hendrixson DR. et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019. Feb. 68(2):289–300. doi: 10.1136/gutjnl-2018-317200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warren RL, Freeman DJ, Pleasance S, Watson P, Moore RA, Cochrane K, Allen-Vercoe E, Holt RA. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome. 2013;1(1):16. doi: 10.1186/2049-2618-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, De’angelis N, Rabot S, Canoui-Poitrine F, Mestivier D. et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures,” (in eng. Proc Natl Acad Sci U S A. [2019 Nov 26]. 116(48):24285–24295. doi: 10.1073/pnas.1912129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao L, Cho WC, Nicolls MR. Colorectal cancer-associated microbiome patterns and signatures,” (in eng. Front Genet. 2021;12:787176. doi: 10.3389/fgene.2021.787176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peters BA, Hayes RB, Goparaju C, Reid C, Pass HI, Ahn J. The microbiome in lung cancer tissue and recurrence-free survival. (In Eng), Cancer Epidemiol Biomarkers Prev. 2019. Apr. 28(4):731–740. doi: 10.1158/1055-9965.EPI-18-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. JNCI J Nat Cancer Inst. 2013;105(24):1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, Ng SC, Tsoi H, Dong Y, Zhang N. et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6(1):8727. doi: 10.1038/ncomms9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Liu F, Ling Z, Tong X, Xiang C, Moschetta A. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7(6):e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takashima Y, Kawamura H, Okadome K, Ugai S, Haruki K, Arima K, Mima K, Akimoto N, Nowak JA, Giannakis M. et al. Enrichment of bacteroides fragilis and enterotoxigenic bacteroides fragilis in CpG island methylator phenotype-high colorectal carcinoma. (In Eng), Clin Microbiol Infect. [2024 Jan 22]. 30(5):630–636. doi: 10.1016/j.cmi.2024.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorrie CL, Mirčeta M, Wick RR, Edwards DJ, Thomson NR, Strugnell RA, Pratt NF, Garlick JS, Watson KM, Pilcher DV. et al. Gastrointestinal carriage is a major reservoir of Klebsiella pneumoniae infection in intensive care patients. Clin Infect Dis. 2017;65(2):208–215. doi: 10.1093/cid/cix270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lau HY, Huffnagle GB, Moore TA. Host and microbiota factors that control Klebsiella pneumoniae mucosal colonization in mice. (In Eng), Microbes Infect. 2008. Oct. 10(12–13):1283–1290. doi: 10.1016/j.micinf.2008.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Q, Su X, Zhang C, Chen W, Wang Y, Yang X, Liu D, Zhang Y, Yang R. Klebsiella pneumoniae Induces inflammatory bowel disease through caspase-11–Mediated IL18 in the gut epithelial cells. Cell Mol Gastroenterol Hepatol. 2023;15(3):613–632. doi: 10.1016/j.jcmgh.2022.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB. et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature. 2020;580(7802):269–273. doi: 10.1038/s41586-020-2080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rashid A, Shen L, Morris JS, Issa J-PJ, Hamilton SR. CpG Island methylation in colorectal adenomas. Am J Pathol. 2001;159(3):1129–1135. doi: 10.1016/s0002-9440(10)61789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toyota M, Ohe-Toyota M, Ahuja N, Issa J-PJ. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97(2):710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gihawi A, Ge Y, Lu J, Puiu D, Xu A, Cooper CS, Brewer DS, Pertea M, Salzberg SL. et al. Major data analysis errors invalidate cancer microbiome findings,” (in eng. mBio. [2023 Oct 31]. 14(5):e0160723. doi: 10.1128/mbio.01607-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hermida LC, Gertz EM, Ruppin E. Predicting cancer prognosis and drug response from the tumor microbiome. (In Eng), Nat Commun. [2022 May 24]. 13(1):2896. doi: 10.1038/s41467-022-30512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.von Rosenvinge EC, O’May GA, Macfarlane S, Macfarlane GT, Shirtliff ME. Microbial biofilms and gastrointestinal diseases,” (in eng. Pathog Dis. 2013. Feb. 67(1):25–38. doi: 10.1111/2049-632X.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dejea CM, Sears CL. Do biofilms confer a pro-carcinogenic state? Gut Microbes. 2016;7(1):54–57. doi: 10.1080/19490976.2015.1121363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, Roslani AC, Wick EC, Mongodin EF, Loke MF. et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. Npj Biofilms Microbiomes. 2017;3(1):2017–11–29. doi: 10.1038/s41522-017-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tricarico R, Madzo J, Scher G, Cohen M, Jelinek J, Maegawa S, Nagarathinam R, Scher C, Chang W-C, Nicolas E. et al. TET1 and TDG suppress inflammatory response in intestinal tumorigenesis: implications for colorectal tumors with the cpg island methylator phenotype. Gastroenterology. 2023;164(6):921–936.e1. doi: 10.1053/j.gastro.2023.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Puccini A, Berger MD, Naseem M, Tokunaga R, Battaglin F, Cao S, Hanna DL, McSkane M, Soni S, Zhang W. et al. Colorectal cancer: epigenetic alterations and their clinical implications. Biochimica Et Biophysica Acta (BBA) - Rev Cancer. 2017;1868(2):439–448. doi: 10.1016/j.bbcan.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16(6):341–352. doi: 10.1038/nri.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaul A, Mandal S, Davidov O, Peddada SD. Analysis of Microbiome Data in the Presence of Excess Zeros. Front Microbiol. 2017;8. doi: 10.3389/fmicb.2017.02114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.