

Abstract
We present a detailed and quantitative analysis of the functional characteristics of the 1,000-nucleotide segment at the 5′ end of the human immunodeficiency virus type 1 (HIV-1) RNA genome. This segment of the viral genome contains several important cis-acting sequences, including the TAR, polyadenylation, viral att site, minus-strand primer-binding site, and 5′ splice donor sequences, as well as coding sequences for the matrix protein and the N-terminal half of the capsid protein. The genetic footprinting technique was used to determine quantitatively the abilities of 134 independent insertion mutations to (i) make stable viral RNA, (ii) assemble and release viral RNA-containing viral particles, and (iii) enter host cells, complete reverse transcription, enter the nuclei of host cells, and generate proviruses in the host genome by integration. All of the mutants were constructed and analyzed en masse, greatly decreasing the labor typically involved in mutagenesis studies. The results confirmed the presence of several previously known functional features in this region of the HIV genome and provided evidence for several novel features, including newly identified cis-acting sequences that appeared to contribute to (i) the formation of stable viral transcripts, (ii) viral RNA packaging, and (iii) an early step in viral replication. The results also pointed to an unanticipated trans-acting role for the N-terminal portion of matrix in the formation of stable viral RNA transcripts. Finally, in contrast to previous reports, the results of this study suggested that detrimental mutations in the matrix and capsid proteins principally interfered with viral assembly.
As a step toward a high-resolution functional map of the complete human immunodeficiency virus type 1 (HIV-1) genome, we carried out a detailed study of a 1-kb segment at the 5′ end of the viral RNA genome. This portion of the viral genome was chosen for two reasons. First, it contains many previously identified functional elements (see Fig. 2), including several cis-acting elements and sequences encoding the matrix and capsid proteins. Comparison of our results to previous studies of this region served as a means to validate our approach. Second, several of the functions attributed to this region have not been mapped to particular sequences, suggesting that much remains to be discovered in this segment of the genome.
FIG. 2.

Map of the mutations evaluated in this study. Previously described features, indicated on the map, are TAR, the polyadenylation signal (poly-A), the att site (att), the primer-binding site (PBS), the kissing loop domain (KLD), the major splice donor (sd), two Gag-binding stem-loop structures (SL), alpha helices 1 to 5 (H1 to H5) of matrix, the basic region of matrix (BR), the beta hairpin of capsid (β), helices 1 to 4 (H1 to H4) of capsid, and the cyclophilin A-binding region of capsid (CyPA). Numbers indicate nucleotide positions at the borders of major regions in the HIV genome. Arrows indicate positions of insertional mutations evaluated in this study.
We used a genetic footprinting (26) method to construct and analyze a large number of mutants distributed in a relatively random fashion over the selected segment of the genome. A library of 15-bp insertion mutants was constructed in vitro using MuA transposase and then selected en masse for the ability to carry out various phases of the viral life cycle. Each mutant contained a single insertion, which included a restriction endonuclease recognition sequence.
Nucleic acid samples of the library taken before and after each functional test were analyzed to assess the fitness and recovery of each mutant. Mutants defective for a given phase of the viral life cycle were relatively depleted at that step. This mutagenesis approach permitted the efficient functional classification of defects in viral replication caused by individual mutations. Although our findings are for the most part in accordance with results of previous studies, we were able to identify several novel functional features of the HIV genome.
MATERIALS AND METHODS
Plasmids.
The replication-defective HIV-1 proviral clone mutagenized in this report (HIVpuro) was derived from pHIV-APΔenvΔVifΔVpr (27) and subcloned into either Bluescript KS+ (Stratagene) or pBS-Kan (a Bluescript KS+-derived vector in which the ampicillin resistance gene was replaced by the kanamycin resistance gene). pHIV-APΔenvΔVifΔVpr was constructed from HIV-AP, an HIV-1 proviral clone containing the human placental alkaline phosphatase gene in place of nef (13), by making multiple deletions to eliminate most of env, vif, and vpr. To construct HIVpuro, the human placental alkaline phosphatase gene was replaced by the puromycin acetyltransferase gene driven by the simian virus 40 promoter (19). In addition, host DNA sequences flanking the proviral sequences were eliminated. PCR mutagenesis was used to eliminate the five BsgI sites originally present in the plasmid (G→C at position 1186, C→G at position 2538, A→C at position 4820, A→T at position 5719, and A→C at position 5848). These changes did not detectably affect viral replication as measured by endpoint titration (data not shown). A fragment of HIVpuro was subcloned into Bluescript KS+, mutagenized (see below), and subsequently cloned back into HIVpuro to generate a library of mutant proviruses.
Mutagenesis.
The mutagenesis procedure was a modification of the method described by Singh et al. (26). MuA transposase was purified as described elsewhere (1). The double-stranded oligonucleotide (Not15) used for mutagenesis was made by annealing Not15A (5′-TGCGGCCGCGCACGAAAAACGCGAAAGCGTTTCACGATAAATGCGAAAAC-3′) and Not15B (5′-GTTTTCGCATTTATCGTGAAACGCTTTCGCGTTTTTCGTGCGCGGCCGCA-3′) in 50 mM NaCl. This oligonucleotide contains recognition sequences for both MuA transposase and the NotI restriction endonuclease. The integration reaction was performed by incubating the Not15 duplex oligonucleotide, target plasmid, and MuA transposase at 30°C for 1 h (25). The five-nucleotide gaps resulting from the integration events were repaired by Taq DNA polymerase-mediated nick translation. The products of these nick translation reactions were then digested with NotI and recircularized by ligation. A detailed description of reaction conditions for the mutagenesis can be found at our website (http://cmgm.stanford.edu/pbrown/footprint.html).
Cell culture.
293, 293T, and HOS cells were grown in Dulbecco's modified Eagle's medium containing 4.5 g of glucose/liter and 10% defined fetal calf serum (HyClone). 293T cells were used for all transient transfection experiments. 293 cells were used for all stable transfection experiments and transductions by virions produced by transient transfection. HOS cells were used for transductions by virions produced from transduced or stably transfected cells. Cells were grown at 37°C in 5% CO2. Puromycin selection was performed with puromycin (Sigma) at 2.5 μg/ml for 293 cells and 5 μg/ml for HOS cells.
Transfections and transductions.
Transient and stable transfections using a Lipofectamine Plus kit (Gibco/BRL) were performed according to the recommended protocol; 30 μg of total plasmid DNA, 60 μl of Plus reagent, and 40 μl of Lipofectamine were used for each 15-cm-diameter tissue culture dish. For stable transfections, puromycin selection was initiated 48 h posttransfection. For transient transfections, the medium was changed 48 h posttransfection and virus was harvested 72 h posttransfection. Viral stocks were diluted to the desired titer in medium containing Polybrene (4 μg/ml; Sigma) and used to transduce cells for 2 h at 37°C. Puromycin selection was initiated 48 h after transduction.
Nucleic acid preparation and manipulation.
Plasmid DNA was purified using a Qiagen plasmid DNA kit and subsequently banded in a cesium chloride gradient (24). A Qiagen blood and cell culture genomic DNA kit was used to prepare genomic DNA from tissue culture samples. Total cellular RNA was prepared using a Qiagen RNeasy total RNA kit. Viral RNA was prepared by pelleting virions by ultracentrifugation (28,000 rpm for 2 h at 4°C in a Beckman SW28 rotor), decanting the supernatant, resuspending the viral pellet in the residual medium, and using a Qiagen Oligotex direct mRNA kit.
Sequencing reactions were performed by the dideoxy termination technique using Sequenase 2.0 (United States Biologicals). Reverse transcription (RT) of cellular RNA and virion RNA samples was performed with 100 ng of template RNA (quantitated by UV spectrophotometry) with the HIV-specific oligonucleotides HIV521 (5′-GGGAGCTCTCTGGCTAACTAGGG-3′) and HIV1573r (5′-CATCCTATTTGTTCCTGAAGGG-3′) according to the manufacturer's instructions (Titan reverse transcription kit; Boehringer-Mannheim).
PCR was performed in a mixture containing 20 mM Tris-HCl (pH 8.55), 150 ng of bovine serum albumin/ml, 16 mM (NH4)2SO4, 3.5 mM MgCl2, 625 μM each deoxynucleoside triphosphate, 0.25 μM each primer, and 1 U of Taq DNA polymerase (AmpliTaq; Perkin-Elmer) per 50-μl reaction. Nonradioactive (cold) PCR conditions consisted of 2 min at 94°C followed by 30 cycles of 30 s at 94°C, 30 s at 55°C, and 2 min at 72°C. Radioactive (hot) PCR conditions consisted of 2 min at 94°C followed by 25 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C.
Pretreatment of streptavidin-agarose beads.
Streptavidin-agarose beads (Sigma) were incubated for 1 h in the presence of poly(dl-dC) (200 μg per ml of streptavidin agarose slurry) in 1× binding buffer (12% [vol/vol] glycerol, 12 mM HEPES [pH 7.9], 4 mM Tris-HCl [pH 8.0], 60 mM KCl, 1 mM EDTA, 1 mM dithiothreitol), washed extensively, and resuspended in 1× binding buffer as a 50% slurry.
Footprinting.
Initial amplification of nucleic acid samples was performed using the cold PCR protocol with HIV-specific primers HIV37 (5′-TGGAAGGGCTAATTCACTCCCAAAG-3′), HIV493 (5′-TCTCTCTGGTTAGACCAGATCTG-3′), HIV521 (5′-GGGAGCTCTCTGGCTAACTAGGG-3′), and HIV1573r (5′-CATCCTATTTGTTCCTGAAGGG-3′); 10 ng of plasmid samples, 1/10 of the products of RT reactions (equivalent to 10 ng of input RNA), or 0.5 μg of genomic DNA samples was used as the template; 10 ng of cold PCR products was used for hot PCRs. For hot PCRs, one HIV-specific primer was 5′ end labeled with [γ-32P]ATP and T4 DNA polynucleotide kinase (New England Biolabs), while the other primer was 5′ biotinylated (Operon). High-specific-activity [γ-32P]ATP (160 μCi/mmol, 23 pmol/μl; ICN) was used for radiolabeling at a stoichiometry of 1 pmol of ATP/1 pmol of oligonucleotide. Hot PCR products were treated with single-stranded affinity matrix (Clontech), purified, and adsorbed to 50 μl of pretreated streptavidin-agarose beads in 1× binding buffer for 1 h at 25°C. The beads were then washed twice with 0.5 ml of 1× binding buffer for 15 min at 25°C, washed once with 0.5 ml of 1× restriction enzyme buffer 3 (New England Biolabs), and incubated in 50 μl of 1× restriction enzyme buffer 3 with 20 U of restriction enzyme NotI for 1 h at 37°C. The supernatant from this digestion step was separated from the beads by centrifugation through a Micro Bio-spin column (Bio-Rad) and ethanol precipitated. Samples were analyzed by denaturing polyacrylamide-urea electrophoresis.
Sources of variability in the data.
One kilobase of the HIV genome was analyzed in 10 overlapping intervals of 200 to 300 bp by using 10 primer pairs. Each mutant was thus examined using at least two primer pairs for amplification. Moreover, each series of transfection-transduction experiments was carried out in its entirety in triplicate. It was therefore necessary to develop a normalization procedure so that data from separate gels and different replicates could be combined to determine the quantitative effect of each mutation.
Within each triplicate, data for a given nucleic acid sample from different footprinting reactions and different gels were normalized using an algorithm that finds a scalar factor for each experiment, minimizing the sum of the weighted coefficients of variance for each mutant (15). The coefficient of variance for each mutant was weighted by the number of measurements for that mutant. The normalized data were then averaged. Data from triplicate experiments were normalized using the same algorithm and averaged. Details about the normalization algorithm are available online (http://cmgm.stanford.edu/pbrown/footprint.html).
After data from each individual nucleic acid sample were normalized and averaged between experiments and between triplicates, data for different nucleic acid samples were normalized based on previous findings that certain areas of the viral genome, such as the C terminus of matrix, are consistently tolerant to small, in-frame insertions (7, 10).
Most of the variability in the data arose from differences between gels or primers rather than sampling error incurred during the selection procedure, variability between PCRs, or inconsistencies in other nucleic acid manipulations (data not shown).
Quantitation.
Autoradiographs were scanned using a flatbed scanner (Hewlett-Packard ScanJet IIc) at a resolution of 300 dots/inch resolution, with brightness and contrast set at 125 (50%), using the DeskScan II utility (Hewlett-Packard). Scanned images were read into a Matlab-based application (15; http: //cmgm.stanford.edu/pbrown/footprint.html) by which individual bands were selected and quantitated for peak intensity values.
RESULTS
Constructing a library of insertion mutants.
The objective was to make a large number of mutations of the same type at diverse positions in a 1-kb segment of the HIV genome and to assess the performance of each mutant at several points in the viral replication cycle. An in vitro transposition reaction was used to introduce a 15-bp insertion mutation into a replication-defective HIV-1 proviral genome in which the env gene had been replaced by the puromycin acetyltransferase gene. The mutations were made in the segment of the HIV proviral genome extending from nucleotide positions 1 to 1514. This segment included the 5′ long terminal repeat (LTR), the 5′ untranslated region, the complete matrix gene, and the 5′ half of the capsid gene. Mutants and mutations are numbered according to the nucleotide position immediately 5′ to the insertion.
The MuA transposase was used to perform a concerted in vitro transposition reaction, introducing a pair of identical double-stranded DNA oligonucleotides into a double-stranded circular target DNA molecule (Fig. 1) (R. A. Crowley, L. C. Laurent, and P. O. Brown, unpublished data). Each oligonucleotide contained recognition sequences for both MuA transposase and the NotI restriction endonuclease. MuA transposase inserted this pair of oligonucleotides into the target DNA in a staggered fashion, which eventually results in a 5-bp duplication of the target DNA sequence flanking the insertion sequence. The products of the transposition reaction were gapped linear double-stranded DNA molecules with oligonucleotides attached at either end in an inverted orientation. After the gaps were filled in by nick translation, the reaction products were digested with NotI, which both cleaves the oligonucleotide such that only the desired insert sequences remain and generates compatible cohesive ends. Finally, a ligation reaction was performed at low DNA concentration to favor intramolecular ligation events. The final products were circular DNA molecules containing the palindromic insert sequence derived from the oligonucleotides, 5′-TGCGGCCGCA-3′, flanked by 5-bp tandem duplications of the target sequence. The insertions retained the NotI recognition sequence, which was used during the analysis procedure. The mutant library constructed in this way contained 157,000 independent clones, each with an insertion mutation in a 1,500 bp segment of the HIV genome. Sixteen individual mutant clones, with insertions at positions 150, 202, 232, 322, 521, 586, 740, 890, 976, 1009, 1031, 1139, 1228, 1231, 1241, and 1363, were isolated and sequenced. These clones were used as markers to facilitate the mapping of all the other insertions during analysis.
FIG. 1.

Mutagenesis scheme using MuA tranposase. Oligonucleotides used for mutagenesis are represented by bold lines ( ), the target DNA plasmid is represented by thin lines, and the 5-bp sequences in the target DNA that were duplicated during mutagenesis are represented by open boxes (
), the target DNA plasmid is represented by thin lines, and the 5-bp sequences in the target DNA that were duplicated during mutagenesis are represented by open boxes ( ).
).
The inserted oligonucleotide was designed so that mutations in coding sequences would be in-frame insertions of five codons. The identity of the amino acids encoded by the insertions depended on both the reading frame and the sequences in the target DNA adjacent to the insertion site.
The positions of the insertion mutations that were analyzed are indicated in Fig. 2. Although the mutants were diverse, the mutagenized segment was not saturated, since the Mu transposase does not make insertions at the same frequency at all sites. Moreover, since transcription starts at nucleotide 456 in the 5′ LTR, the effects of mutations in U3 (nucleotides 1 to 456) could not be assessed. Comparison of the transfected DNA with mRNA samples isolated from the transfected cells, viral RNA, and progeny proviruses confirmed the expected eliminations of mutants with insertions in U3, indicating that those nucleic acid samples were not contaminated with the original transfected plasmid DNA (data not shown).
Sampling populations of mutants at different steps in the viral replication cycle.
To identify insertions that disrupted cis-acting elements (e.g., transcriptional modulators, the packaging sequence, and the viral att site), the library was either transiently or stably transfected into producer cells. A plasmid encoding vesicular stomatitis virus G protein (VSV-G) was transiently transfected into the producer cells to pseudotype the env-defective mutant virions. Virions were harvested from these cells and used to transduce fresh HOS cells. Nucleic acid samples were collected at various steps during this experiment (Fig. 3). Depletion of mutants at each of these steps in the viral replication cycle was followed by analysis of these nucleic acid samples by genetic footprinting.
FIG. 3.

Relationship of nucleic acid samples analyzed by genetic footprinting to specific steps in the life cycle. A decrease in the abundance of a given mutant between two of the indicated nucleic acid samples implies that the mutant is defective in one of the intervening steps of the viral life cycle.
A similar strategy was used to determine the effects of insertions in trans-acting sequences. Since more than one DNA often enters a given cell during transfection, complementation could occur between trans-acting elements in a transfection experiment (Fig. 4). To minimize the confounding effect of complementation on analysis of sequences that can function in trans, a first round of transient transfection was conducted, cotransfecting the mutant library with a VSV-G expression construct. The goal was to produce a VSV-G pseudotyped, phenotypically mixed population in which mutants with defective trans-acting functions were rescued by complementation. Since approximately half of the clones in the library were wild type (i.e., did not contain an insertion), this complementation was easy to achieve. These virions were then used to transduce fresh host cells at a multiplicity of transduction of 0.05. The transduced cells were selected using puromycin, and this pool of cells was used as the starting population of producer cells for a subsequent round of transduction in which defects in trans-acting sequences could be analyzed in the absence of complementation. Nucleic acids representing stages in the replication cycle were isolated and analyzed by genetic footprinting.
FIG. 4.

Design of transfection and transduction experiments. The library of mutagenized proviruses was either transiently or stably transfected into cells to produce populations of mutant virions. In our experiments, the viral genomes of mutants defective in trans-acting factors were efficiently rescued during the transient transfection by phenotypic mixing but were not detectably rescued during the stable transfection. The virus produced in the transient transfection experiment was used to transduce fresh, untransduced cells at a low multiplicity, resulting in a population of producer cells that contained a single provirus per cell. Symbols represent wild-type viral genome ( ), replication-defective viral genome with mutation in trans-acting factor (
), replication-defective viral genome with mutation in trans-acting factor ( ), wild-type viral protein (
), wild-type viral protein ( ), and mutant viral protein (
), and mutant viral protein ( ).
).
Although we did not directly measure the number of proviruses per producer cell, the results described below show that complementation of trans-acting sequences occurred very efficiently during the first round of transduction in our transient transfection experiments but not to any appreciable degree in our stable transfection experiments.
Footprinting analysis procedure.
Samples of genomic DNA from producer cells or transduced cells, mRNA from producer cells, or genomic RNA from virions were subjected to an initial round of amplification by either PCR (for DNA samples) or RT-PCR (for RNA samples). PCR was then performed on these preamplified samples, using one 32P-labeled DNA primer and one biotinylated DNA primer (Fig. 5). The primers were complementary to HIV sequences and flanked the region to be analyzed. The products of this second PCR were treated with a single-stranded nucleic acid-binding resin to remove incomplete extension products, bound to streptavidin-agarose beads, and digested with NotI. The released radioactive products were concentrated and subjected to electrophoresis on denaturing polyacrylamide-urea gels. This assay generates a radioactive product of unique length for each mutation; the length depends only on the position of the insertion in the HIV sequence. Figure 6 shows a typical genetic footprinting gel. Adjacent to each band, the position of the insertion sequence, quantitative measurements of recovery in several nucleic acid samples, and local nucleic acid sequences for the corresponding mutant are indicated.
FIG. 5.

Genetic footprinting scheme using flanking PCR and restriction digestion. A collection of insertion mutants is subjected to PCR using one radioactively labeled primer ( ) and one biotinylated primer (
) and one biotinylated primer ( ). The PCR products are captured by streptavidin-agarose resin (
). The PCR products are captured by streptavidin-agarose resin ( ) and digested with a restriction enzyme that recognizes a site in the insertion sequence. The radioactively labeled ends of the PCR products are released.
) and digested with a restriction enzyme that recognizes a site in the insertion sequence. The radioactively labeled ends of the PCR products are released.
FIG. 6.

Genetic footprinting of library of mutagenized proviruses and nucleic acid samples from the transient transfection experiment, second round (the uncomplemented round) of viral production and transduction (cellular RNA, viral RNA, and transduced cell genomic DNA). Numbers directly to the left of the gel indicate exact positions of insertions. In this numbering convention, the first nucleotide of the HIV provirus is at position 1. Quantitative values for recovery of the mutants represented by each band, in each sample, averaged from normalized measurements are also given to the left of the gel. The nucleic acid sequences of the mutants represented by each band are written to the right of the gel. The sequences derived from the insertion oligonucleotide are boxed, and the target sequence duplications are underlined. Eighty-nine percent of 19 individually sequenced mutants contained the expected precise 5-bp duplication flanking the 9-bp insertion.
cis-acting versus trans-acting elements.
Mutations that disrupt cis-acting sequences can be distinguished from those that disrupt trans-acting sequences by their inability to be complemented by wild-type viral genomes.
The fitness of individual mutants in single-cycle transductions are summarized in Fig. 7. Figure 7A shows the relative fitness of mutants in completing one round of transduction initiated with proviruses introduced into producer cells by first-round transient transfection, thus allowing complementation by wild-type proviruses. Figure 7B shows the relative fitness of mutants in completing one round of transduction initiated with proviruses introduced into producer cells by low-multiplicity transduction (minimizing the possibility of complementation). Mutations that significantly impaired replication in both complemented (Fig. 7A) and uncomplemented (Fig. 7B) transductions all mapped between positions 457 and 811. Mutations in sequences between positions 811 and 1488 all appeared to be complemented by wild-type viral genomes in transductions initiated from transfected cells, but many of these insertions impaired fitness in the absence of complementation.
FIG. 7.

Percent recovery of mutants through single-cycle transductions. Data are not shown for mutants for which the coefficient of variation between triplicate experiments was greater than 0.5, except in cases where the observed phenotypes were confirmed by reanalysis. Ordinate values are plotted on a logarithmic scale. Tick marks indicate mutants that display severe depletions (<45% recovery). Mutations that compromise replication in both the presence and the absence of complementation are considered to be located in cis-acting sequences, while those that affect only uncomplemented replication cycles are considered to be located in trans-acting sequences. (A) Percent recovery of mutants after a single round of transduction in the presence of complementation. Data are from the transient transfection experiment, first round of transduction. (B) Percent recovery of mutants after a single round of transduction in the absence of complementation. Data are from the transient transfection experiment, second round of transduction. Data are not given for mutants whose abundance after the first round of infection was too low to allow accurate quantitation of further depletion.
Mutations that impair viral RNA transcription or stability.
The four mutations in the TAR region (positions 456 to 506) severely compromised replication (Fig. 8). These mutants are probably defective for Tat-cyclin T1 binding, which would result in diminished transcription.
FIG. 8.
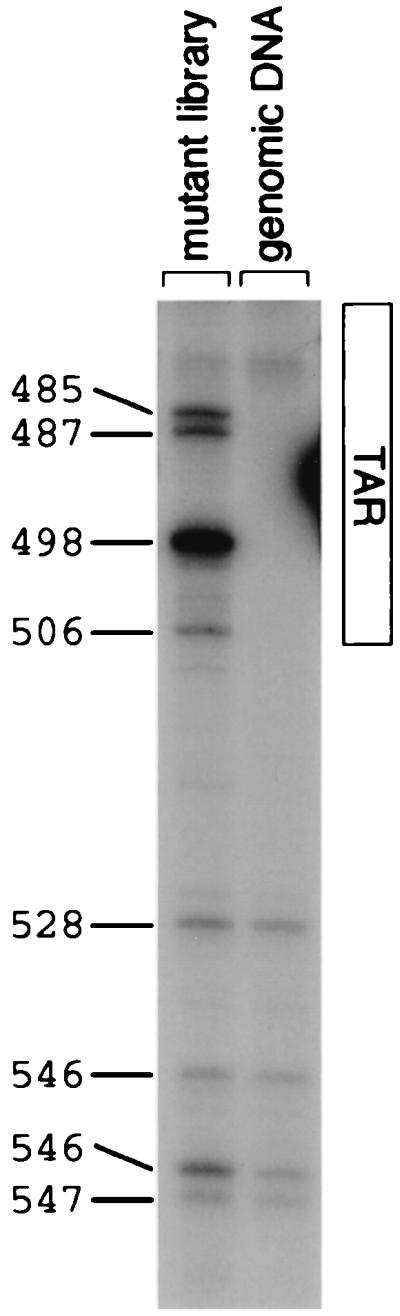
Genetic footprinting of library of mutagenized proviruses and infected cell genomic DNA sample from the transient transfection experiment, first round (the complemented round) of viral production and transduction. Numbers directly to the left of the gel indicate exact positions of insertions. In this numbering convention, the first nucleotide of the HIV provirus is at position 1. The extent of the TAR region is indicated to the right of the gel.
In the transient transfection experiment, insertions at positions 528, 537, and 547 had detrimental effects on the second, but not the first, round of transduction (Fig. 7). In the second round of the transient transfection experiment, the most pronounced effects of the insertions at these positions were on transcript abundance (Fig. 9). However, in the stable transfection experiment, the mutations at positions 528 and 547 resulted in decreases in fitness in the phase of the life cycle reflected by the transition from viral RNA in the producer cells to viral RNA in extracellular virions (Fig. 10). The most probable explanation for these observations is that all three mutations, which are in or near the polyadenylation consensus sequence (positions 527 to 532), interfered with polyadenylation. This defect would not be observed in the first round of transduction since only the 5′ LTR was mutagenized and it was not until the first round of reverse transcription that mutations were transferred to the 3′ LTR, where the operative polyadenylation signal lies. In addition, mutations at positions 528 and 547 may result in packaging defects that are complementable in trans (Fig. 7 and 10).
FIG. 9.

Effects of selected mutations in cis-acting sequences in the transient transfection experiment. These mutants were replication competent in the first round of transduction but defective for transcript formation in the second round, possibly indicating that it is the copy of these cis-acting sequences in the 3′ LTR which is active.
FIG. 10.

Recovery of genomes carrying mutations in cis-acting sequences, at several steps of a single-cycle uncomplemented transduction. Samples were collected from the stable transfection experiment. Percent recovery was calculated by dividing the abundance of a mutant in a given nucleic acid sample by the abundance of that mutant in the genomic DNA sample from the stable transfection. Data are not shown for points where the abundance of a particular mutant was very low in the preceding nucleic acid sample or where the coefficient of variation between triplicate experiments was greater than 0.5. Graphs are plotted on a log scale. Tick marks indicate mutants that display severe depletions (<50% of preceding nucleic acid sample). Mutants which were depleted in the cellular RNA sample are considered to be defective in transcript formation, mutants which were depleted in the virion RNA sample are considered to be defective in packaging, and mutants which were depleted in the transduced cell genomic DNA sample are considered to be defective in an early step in viral transduction.
Six other cis-acting mutations (at positions 542, 691, 692, 694, 722, and 755) resulted in moderately to severely decreased transcript levels in producer cells under all conditions tested (Fig. 10), suggesting that these mutations may impair transcriptional enhancer elements or reduce transcript stability.
Mutations in cis-acting sequences that affect viral assembly.
A cis-acting RNA packaging signal has previously been mapped to the few hundred base pairs around the 5′ splice donor site and the 5′ end of gag. This region includes the “kissing loop” dimerization and packaging signal, the 5′ splice donor site, and two stem-loop structures which have been found to bind in vitro to Gag and nucleocapsid proteins (2, 3, 6, 23). We found that in all experiments, most genomes with mutations in the interval between positions 666 and 810 were underrepresented in virions compared to the abundance of the corresponding RNA in the producer cells (Fig. 10).
The existence of a supplementary packaging signal is suggested in a report by Vicenzi et al. (28), describing a deletion of the 5′ one-third of U5 that results in a 10-fold decrease in RNA packaging. Indeed, we found that some mutations in U5 (at positions 528, 547, 571, 585, and 604) appeared to impair viral RNA packaging (Fig. 10).
The packaging defects observed for these mutants appeared to be partially relieved by complementation (compare Fig. 10 and 7A). If viral genomes with insertions at these positions are still able to form dimers, dimerization with wild-type viral genomes might partially relieve the packaging defect of these mutant genomes.
cis-acting mutations that impair steps between virus production and integration.
Mutations at positions 571 to 618 and 691 to 712 resulted in a reduction in fitness for the portion of the replication cycle that includes viral entry, uncoating, RT, nuclear entry, and integration (Fig. 10). The segment between positions 571 and 618 is in U5, just 5′ to the att site. Although no specific function for this region of U5 has previously been defined, its proximity to the att site raises the possibility that sequences in this region might affect recognition of the viral genome by integrase. Alternatively, the proximity of this interval to the primer-binding site suggests a possible role in initiation of RT (16). The second group of mutations, between positions 691 and 712, is in the kissing loop motif. A role for sequences in the 5′ stem-loop of the kissing loop motif (nucleotides 692 to 738) in the synthesis of proviral DNA (in addition to the expected role in genomic RNA packaging) has been described elsewhere (20).
The paucity of mutants for which we could recognize significant and specific defects in early steps of viral infection is probably due to the design of our experimental system. It is likely that elimination of mutants at steps in the viral life cycle occurring earlier in our series of experiments (e.g., transcription or assembly) prevents our recognition of additional effects these mutations might have on viral entry, RT, or integration. For example, two mutations (at positions 647 and 648) located in the primer-binding site were severely depleted in the virion RNA sample (transient transfection experiment [data not shown]), making it impossible to see further reductions in the transduced cell genomic DNA sample that would reflect the expected defect in RT. Our pool of insertion mutants did not include any detectable mutations that destroyed the att site in U5, the one feature in this segment of the genome known to be essential for integration.
Mutations in matrix.
Sequences at the 5′ end of the matrix coding sequence (positions 791 to 802) appeared to contribute to viral RNA packaging in cis (see above). A few mutations near the 5′ end of the matrix gene (positions 840 to 893) appeared to impair the production of stable transcripts (Fig. 11). These trans-acting mutations, which could be rescued by complementation, were located in the sequences that encode the C-terminal end of helix 1, a loop between helix 1 and helix 2, and the N-terminal half of helix 2. This region contains many basic residues and is at the edge of the globular domain of matrix that faces away from the trimer interfaces. The phenotype of these mutants suggests that the matrix domain of the Gag polyprotein might have a role in enhancing transcription or stabilizing the viral RNA genome in the producer cell prior to budding. Supporting this possibility, it was first proposed (5) and subsequently demonstrated in vitro (17) that the matrix protein has RNA-binding activity.
FIG. 11.

Recovery of mutations in matrix and capsid at several steps of a single-cycle uncomplemented transduction. Samples were collected from the transient transfection experiment, second round of viral production and transduction. Percent recovery was calculated by dividing the abundance of a mutant in a given nucleic acid sample by the abundance of that mutant in the transduced cell genomic DNA sample from the first round of transduction. Data are not shown for points where the abundance of a particular mutant was very low in the preceding nucleic acid sample or where the coefficient of variation between triplicate experiments was greater than 0.5. Graphs are plotted on a log scale. Tick marks indicate mutants that display severe depletions (<45% of preceding nucleic acid sample). Mutants which were depleted in the cellular RNA sample are considered to be defective in transcript formation, mutants which were depleted in the virion RNA sample are considered to be defective in assembly, and mutants which were depleted in the transduced cell genomic DNA sample are considered to be defective in an early step in viral replication. All mutations in capsid that affected viral replication showed their effects in trans.
Most of the insertions between positions 901 and 1095, the region encoding the core of the globular domain of matrix, primarily impaired the steps in the life cycle between transcript accumulation and budding (Fig. 11 and 12). These mutations could be complemented in trans. The observed phenotype is consistent with a defect in viral assembly due to impaired folding of the matrix domain of the Gag polyprotein or defective Gag polyprotein processing.
FIG. 12.

Percent recovery of matrix and capsid mutants through the viral assembly process and the early steps of the viral life cycle. Data are derived from single-round uncomplemented viral production and transduction cycles. A schematic of the phases of the life cycle tested is drawn above the graphs. Numbers to the right of each point indicate the number of mutants from which data were averaged. Below each data point is a schematic of the region of matrix or capsid evaluated. Error bars indicate 95% confidence intervals. (A) Data for the matrix protein. Data are shown for insertions in the complete matrix protein ( [amino acids 1 to 132; nucleotides 792 to 1187]), the N-terminal region (
[amino acids 1 to 132; nucleotides 792 to 1187]), the N-terminal region ( [amino acids 1 to 35; nucleotides 792 to 896]), the central region (
[amino acids 1 to 35; nucleotides 792 to 896]), the central region ( [amino acids 36 to 102; nucleotides 897 to 1097]), and the C-terminal region (
[amino acids 36 to 102; nucleotides 897 to 1097]), and the C-terminal region ( [amino acids 104 to 130; nucleotides 1101 to 1181]). (B) Data for the N-terminal half of the capsid protein. Data are shown for insertions in the N-terminal half (
[amino acids 104 to 130; nucleotides 1101 to 1181]). (B) Data for the N-terminal half of the capsid protein. Data are shown for insertions in the N-terminal half ( [amino acids 1 to 101; nucleotides 1188 to 1490]), the N-terminal beta hairpin (
[amino acids 1 to 101; nucleotides 1188 to 1490]), the N-terminal beta hairpin ( [amino acids 1 to 15; nucleotides 1188 to 1232]), the central helical region (
[amino acids 1 to 15; nucleotides 1188 to 1232]), the central helical region ( [amino acids 17 to 81, nucleotides 1236 to 1430]), and the cyclophilin A-binding region (
[amino acids 17 to 81, nucleotides 1236 to 1430]), and the cyclophilin A-binding region ( [amino acids 85 to 96; nucleotides 1440 to 1475]).
[amino acids 85 to 96; nucleotides 1440 to 1475]).
Consistent with previous studies (7, 10), most of the mutations in the C-terminal domain of matrix (positions 1105 to 1175 in this study), which consists of a long alpha-helical tail that extends away from the globular domain, have little apparent effect on any of steps in viral replication (Fig. 11 and 12).
Mutations in capsid.
In agreement with other reports (8, 18), we found that mutations in the sequence encoding the N-terminal half of the capsid protein severely impaired viral replication (Fig. 7B). Capsid mutants with insertions between positions 1240 and 1428 were defective both at a step in viral production (assembly or budding) and at an early step in transduction of new host cells (Fig. 11 and 12). This result is particularly notable for the contrast to most of the matrix mutants, which appeared to be specifically defective at the assembly/budding step (Fig. 12).
Mutants with insertions in, and immediately adjacent to, the N-terminal β hairpin (positions 1208 to 1228) and the cyclophilin A-binding region (1443 to 1472) of capsid were able to form viral particles but were defective in a step in early infection (Fig. 11 and 12). X-ray crystallographic and biochemical studies (12, 30, 32) support the theory that the β-hairpin structure forms only after proteolytic maturation of the viral particle. An extended, relatively disordered conformation during assembly may account for the fact that insertions in this region do not impair viral particle formation. The β hairpin and the cyclophilin A-binding regions are the only regions in the N-terminal domain of capsid that protrude from a tightly packed helical core. These structural differences might explain the differential effects of insertions in these regions on assembly. It has been suggested that the disassembly of the viral core (uncoating) that occurs after viral entry and before the initiation of RT depends on an interaction between capsid and cyclophilin A (4, 11). Therefore, insertions in the cyclophilin A-binding region that interfere with this interaction might affect uncoating.
DISCUSSION
In the experiments described here, a large number of mutants with insertions at diverse positions in the 5′ 1-kb segment of the HIV-1 genome were followed en masse through two rounds of replication. This strategy permitted a comprehensive, comparative examination of the effects of each mutation on viral replication. Selection and analysis of the mutants in parallel provided built-in internal controls for variables such as sample recovery and efficiency of analysis procedures. Retroviruses, particularly HIV, have been extensively studied using a variety of techniques. As a result, the basic mechanisms of major steps in the retroviral life cycle have been elucidated and many functional features of the retroviral genome have been mapped. Yet numerous important questions about retroviral replication remain to be answered. There is, therefore, still much to be learned from a systematic dissection of the HIV genome.
Mutations of any type can produce unpredictable consequences. For example, results from a previous genetic footprinting experiment (26) suggest that 36-bp insertion mutations may in some cases be less disruptive to the structure of a small RNA molecule than 12-bp substitution mutations. The mutation introduced in this study, a 15-bp insertion mutation, was designed to be disruptive enough to affect reproductive fitness of mutant viruses yet subtle enough to produce partially active mutants. The observation that many mutants with insertions at diverse locations had partially defective phenotypes demonstrates the usefulness of this type of mutagenesis strategy. Interestingly, these partially active mutants contained insertions in known essential sequences, as well as in sequences not previously noted to be important for viral replication.
The three-dimensional structures of matrix, capsid, and many HIV RNA elements have revealed much about the properties of these proteins and RNAs, and much directed mutagenesis has been done based on these structures. However, in our studies, the severity of the effects of mutations does not predictably correlate with the locations of insertions with respect to known secondary or tertiary structures. This observation suggests that important functional data can still be derived by thorough, nondirected mutagenesis approaches, at least until we know better how to predict function based on structural information.
Expected results and novel observations.
The experiments defined several functional features in the HIV-1 genome, some of which have not previously been described. We mapped three kinds of cis-acting sequences: sequences that affect transcription or transcript stability, sequences involved in viral RNA packaging, and sequences important for an early step in viral transduction. Some of these sequences were found in intervals previously implicated in these functions (e.g., TAR and the kissing loop motif), and others were found in previously unrecognized locations.
The phenotypes of several mutations near the N terminus of matrix suggested an unforeseen trans-acting function for matrix in transcript formation or stabilization. In contrast to previous reports, we observed that many mutations that map to the globular core of matrix had marked effects on assembly and that mutations in the helical core of the N-terminal domain of capsid caused defects in both assembly and an early step in infection (perhaps disassembly).
Mutations in the β-hairpin and cyclophilin A-binding regions of capsid primarily resulted in early replication defects. The behavior of the mutations in the cyclophilin A-binding region was consistent with its previously postulated role in uncoating.
How might the same mutation in capsid cause defects in both assembly and disassembly? Assembly involves the aggregation of Gag and Gag-Pol polyproteins, whereas disassembly normally occurs after proteolysis of these polyproteins into smaller proteins. A mutation that impairs assembly might also cause the viral particles that do form to be aberrant. Viruses with mutations in the N-terminal half of capsid have previously been noted to have abnormal core morphologies (8, 22). Perhaps this abnormal structure compromises steps essential for uncoating, such as proteolytic maturation or cyclophilin A incorporation.
Discrepancies with previously published results.
The inconsistencies between our results and previous reports can be grouped into two classes. First, mutations at 25 positions in the matrix gene resulted in severe defects in replication in our study, while mutations disrupting some of the same sites were reportedly tolerated in a previous study (10). Second, we found that many mutations in the N-terminal half of capsid impaired viral assembly. Previous studies have mapped the residues important for Gag multimerization and viral assembly to the C-terminal domain of capsid (8, 14, 21, 29), while viruses with mutations in the N-terminal domain of capsid were found to be competent for viral assembly, although sometimes abnormal in core morphology (8, 9, 21, 22, 31).
These discrepancies may result from differences in experimental method or interpretation. First, the precise locations and types of mutations differ. Second, the methods used to assess replication competence differ between reports. Third, in the previous work, viral assembly was measured in a variety of ways, including exogenous RT assays, RNase protection, Western blotting for viral proteins, and electron micrography. These assays implicitly define viral assembly in ways that reflect different features of the assembly process and can reasonably be expected to be differentially affected by mutations. In addition, some of the assays in previous reports were inherently qualitative, whereas we measured quantitative incorporation of viral RNA into extracellular particles.
For mutations that severely compromise viral replication, the experiments presented here are likely to have led to an overestimate of the ability of these mutants to replicate. In the selection strategy, the uncomplemented transduction cycles were carried out using either stably transfected cells or cells that had been transduced at low multiplicity of infection (MOI) as producer cells. It is likely, however, that complementation occurred in both populations of cells. For the virus-producing cells transduced at low MOI, the measured MOI was approximately 0.05. Assuming that the number of proviruses per cell followed a Poisson distribution, approximately 2.5% of the cells that were transduced by at least one virus were actually transduced by more than one virus, allowing complementation to occur in those cells. Hence, for viruses carrying complete recessive loss-of-function mutations in trans-acting factors, one would expect a background survival of approximately 2.5%.
Possible extensions of this work.
In the original report describing the genetic footprinting technique, this method was used to generate a high-resolution functional map of a small (200-bp) gene encoding an RNA molecule (26). Modifications to the genetic footprinting procedure permitted us to analyze a much larger (1,000-bp) stretch of nucleic acid, including both cis-acting and protein-coding sequences. The experiments presented here involved the isolation and analysis of complex nucleic acid samples, including cellular RNA, virion RNA, and genomic DNA samples from a selection scheme in eukaryotic cells. Thus, genetic footprinting could be used to map the functional features in any DNA sequence if an appropriate selection scheme were available.
By examining only three stages of the replication cycle, we could only make a rough assignment of the stages of the HIV life cycle affected by each mutation. Refinement of our picture of viral replication can be achieved by footprinting nucleic acid samples representing more finely differentiated steps. In addition, a more diverse set of phenotypes could be characterized by using a variation on the method used here. Insertion mutations with different insert sequences and substitution mutations could be introduced and analyzed (reference 26 and unpublished results). Extending this genetic footprinting analysis to the entire HIV genome is likely to provide new insights into the functional organization of the HIV genome.
ACKNOWLEDGMENTS
We thank Marc Laurent for extensive software support, Kiyoshi Mizuuchi for MuA transposase, and Richard Sutton for helpful discussion.
Funds for this work were provided by NIH grant HG00983 and the Howard Hughes Medical Institute. Patrick O. Brown is an associate investigator of the Howard Hughes Medical Institute. Louise C. Laurent was supported in part by a Medical Scientist Training Program training grant.
Footnotes
Present address: University of California at San Francisco Medical School, San Francisco, CA 94143.
REFERENCES
- 1.Baker T A, Mizuuchi M, Savilahti H, Mizuuchi K. Division of labor among monomers within the Mu transposase tetramer. Cell. 1993;74:723–733. doi: 10.1016/0092-8674(93)90519-v. [DOI] [PubMed] [Google Scholar]
- 2.Berkowitz R D, Luban J, Goff S P. Specific binding of human immunodeficiency virus type 1 Gag polyprotein and nucleocapsid protein to viral RNAs detected by RNA mobility shift assays. J Virol. 1993;67:7190–7200. doi: 10.1128/jvi.67.12.7190-7200.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berkowitz R D, Goff S P. Analysis of binding elements in the human immunodeficiency virus type 1 genomic RNA and nucleocapsid protein. Virology. 1994;202:233–246. doi: 10.1006/viro.1994.1339. [DOI] [PubMed] [Google Scholar]
- 4.Braaten D, Franke E K, Luban J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol. 1996;70:3551–3560. doi: 10.1128/jvi.70.6.3551-3560.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bukrinskaya A G, Vorkunova G K, Tentsov Y. HIV-1 matrix protein p17 resides in cell nuclei in association with genomic RNA. AIDS Res Hum Retroviruses. 1992;8:1795–1801. doi: 10.1089/aid.1992.8.1795. [DOI] [PubMed] [Google Scholar]
- 6.Clever J, Sassetti C, Parslow T G. RNA secondary structure and binding sites for gag gene products in the 5′ packaging signal of human immunodeficiency virus type 1. J Virol. 1995;69:2101–2109. doi: 10.1128/jvi.69.4.2101-2109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorfman T, Mammano F, Haseltine W A, Göttlinger H G. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1994;68:1689–1696. doi: 10.1128/jvi.68.3.1689-1696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorfman T, Bukovsky A, Ohagen A, Höglund S, Göttlinger H G. Functional domains of the capsid protein of human immunodeficiency virus type 1. J Virol. 1994;68:8180–8187. doi: 10.1128/jvi.68.12.8180-8187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke E K, Yuan H E, Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 10.Freed E O, Orenstein J M, Buckler-White A J, Martin M A. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol. 1994;68:5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gamble T R, Vajdos F F, Yoo S, Worthylake D K, Houseweart M, Sundquist W I, Hill C P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 12.Gitti R K, Lee B M, Walker J, Summers M F, Yoo S, Sundquist W I. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- 13.He J, Landau N R. Use of a novel human immunodeficiency virus type 1 reporter virus expressing human placental alkaline phosphatase to detect an alternative viral receptor. J Virol. 1995;69:4587–4592. doi: 10.1128/jvi.69.7.4587-4592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jowett J B, Hockley D J, Nermut M V, Jones I M. Distinct signals in human immunodeficiency virus type 1 Pr55 necessary for RNA binding and particle formation. J Gen Virol. 1992;73:3079–3086. doi: 10.1099/0022-1317-73-12-3079. . (Erratum, 74:943, 1993.) [DOI] [PubMed] [Google Scholar]
- 15.Laurent L C. Functional characterization of the HIV genome by genetic footprinting. Ph.D. dissertation. University of California at San Francisco; 1998. [Google Scholar]
- 16.Leis J, Aiyar A, Cobrinik D. Regulation of initiation of reverse transcription in retroviruses. In: Skalka A M, Goff S P, editors. Reverse transcriptase. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1993. pp. 33–48. [Google Scholar]
- 17.Lochrie, M. A., S. Waugh, D. G. Pratt, Jr., J. Clever, T. G. Parslow, and B. Polisky. 1997. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 gag polyprotein. Nucleic Acids Res. 25:2902–2910. [DOI] [PMC free article] [PubMed]
- 18.Mammano F, Ohagen A, Höglund S, Göttlinger H G. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J Virol. 1994;68:4927–4936. doi: 10.1128/jvi.68.8.4927-4936.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgenstern J P, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paillart J C, Berthoux L, Ottmann M, Darlix J L, Marquet R, Ehresmann B, Ehresmann C. A dual role of the putative RNA dimerization initiation site of human immunodeficiency virus type 1 in genomic RNA packaging and proviral DNA synthesis. J Virol. 1996;70:8348–8354. doi: 10.1128/jvi.70.12.8348-8354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reicin A S, Paik S, Berkowitz R D, Luban J, Lowy I, Goff S P. Linker insertion mutations in the human immunodeficiency virus type 1 gag gene: effects on virion particle assembly, release, and infectivity. J Virol. 1995;69:642–650. doi: 10.1128/jvi.69.2.642-650.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reicin A S, Ohagen A, Yin L, Hoglund S, Goff S P. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J Virol. 1996;70:8645–8652. doi: 10.1128/jvi.70.12.8645-8652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakaguchi K, Zambrano N, Baldwin E T, Shapiro B A, Erickson J W, Omichinski J G, Clore G M, Gronenborn A M, Appella E. Identification of a binding site for the human immunodeficiency virus type 1 nucleocapsid protein. Proc Natl Acad Sci USA. 1993;90:5219–5223. doi: 10.1073/pnas.90.11.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J, Maniatis T, Fritsch E F. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 25.Savilahti H, Rice P A, Mizuuchi K. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J. 1995;14:4893–4903. doi: 10.1002/j.1460-2075.1995.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh I R, Crowley R A, Brown P O. High-resolution functional mapping of a cloned gene by genetic footprinting. Proc Natl Acad Sci USA. 1997;94:1304–1309. doi: 10.1073/pnas.94.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutton R E, Wu H T, Rigg R, Böhnlein E, Brown P O. Human immunodeficiency virus type 1 vectors efficiently transduce human hematopoietic stem cells. J Virol. 1998;72:5781–5788. doi: 10.1128/jvi.72.7.5781-5788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicenzi E, Dimitrov D S, Engelman A, Migone T S, Purcell D F, Leonard J, Englund G, Martin M A. An integration-defective U5 deletion mutant of human immunodeficiency virus type 1 reverts by eliminating additional long terminal repeat sequences. J Virol. 1994;68:7879–7890. doi: 10.1128/jvi.68.12.7879-7890.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Poblotzki A, Wagner R, Niedrig M, Wanner G, Wolf H, Modrow S. Identification of a region in the Pr55gag-polyprotein essential for HIV-1 particle formation. Virology. 1993;193:981–985. doi: 10.1006/viro.1993.1210. [DOI] [PubMed] [Google Scholar]
- 30.von Schwedler U K, Stemmler T L, Klishko V Y, Li S, Albertine K H, Davis D R, Sundquist W I. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998;17:1555–1568. doi: 10.1093/emboj/17.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C T, Barklis E. Assembly, processing, and infectivity of human immunodeficiency virus type 1 gag mutants. J Virol. 1993;67:4264–4273. doi: 10.1128/jvi.67.7.4264-4273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wlodawer A, Erickson J W. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. doi: 10.1146/annurev.bi.62.070193.002551. [DOI] [PubMed] [Google Scholar]