

Summary
Early modern humans lived as hunter-gatherers for millennia before agriculture, yet the genetic adaptations of these populations remain a mystery. Here, we investigate selection in the ancient hunter-gatherer-fisher Jomon and contrast pre- and post-agricultural adaptation in the Japanese archipelago. Building on the successful validation of imputation with ancient Asian genomes, we identify selection signatures in the Jomon, particularly robust signals from KITLG variants, which may have influenced dark pigmentation evolution. The Jomon lacks well-known adaptive variants (EDAR, ADH1B, and ALDH2), marking their emergence after the advent of farming in the archipelago. Notably, the EDAR and ADH1B variants were prevalent in the archipelago 1,300 years ago, whereas the ALDH2 variant could have emerged later due to its absence in other ancient genomes. Overall, our study underpins local adaptation unique to the Jomon population, which in turn sheds light on post-farming selection that continues to shape contemporary Asian populations.
Subject areas: Genomics, Anthropology
Graphical abstract
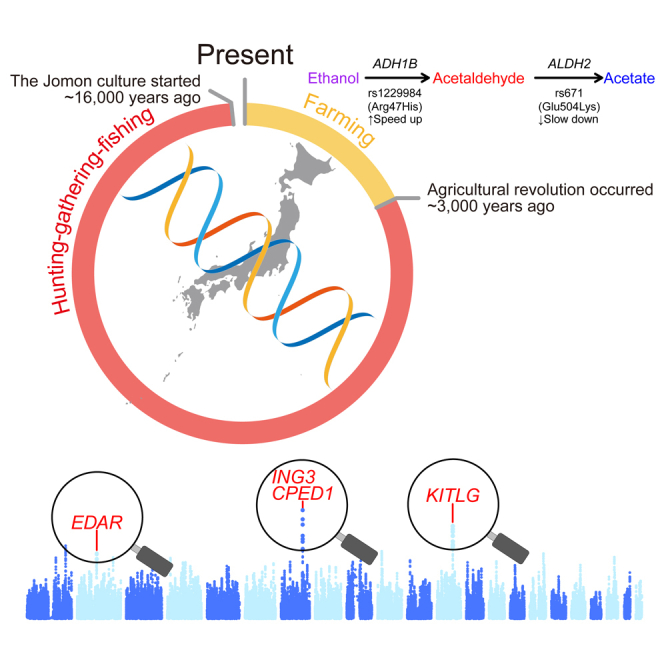
Highlights
-
•
Genomic imputation accurately brings ancient Asian genomes to higher coverages
-
•
Selection scans of imputed genomes reveal adaptive evolution in the ancient Jomon
-
•
Well-known adaptive variants in Asia (EDAR/ADH1B/ALDH2) emerged post-agriculture
Genomics; Anthropology
Introduction
For tens of thousands of years, human populations were exclusively reliant on hunting and foraging for sustenance, before the advent of agriculture during the Holocene period.1,2,3 While this shift is a relative blip on the evolutionary timescale, it had an enormous impact on global lifeways.4,5,6 Much effort has been devoted to uncovering the population dynamics that accompanied the spread of farming, which include migration, admixture, turnover, and growth,7,8,9,10,11 as well as adaptation to novel selective pressures that emerged from the change of dietary habits and the increase of pathogen loads following this transition.12,13,14,15,16,17,18,19 However, it is equally critical to understand the evolutionary trajectories of pre-agricultural humans, including selective pressures that characterized these early hunter-gatherer populations.
Extant hunter-gatherers often inhabit isolated or extreme environments that are not suited to agriculture,20 offering an accessible model for studying the genetic basis of human adaptation, such as metabolic pathways specialized for high-fat diet21 and short stature potentially beneficial for efficient hunting.22,23 It is unknown whether or not similar patterns of phenotypic adaptation would have also been present or even advantageous in pre-agricultural populations living in less hostile terrain. A population-scale dataset of ancient genomes from pre-agricultural humans makes it possible to identify the adaptive traits that are present or absent in these populations,24,25 which in turn provides critical insights in the distinctive natures of pre- and post-farming evolution.
The indigenous Jomon population of prehistoric Japan, known as one of the most deeply diverged populations in East Asia,8,26,27 provides an unparalleled opportunity to explore adaptive history throughout lifeway transitions from pre- and post-agriculture. The Japanese archipelago is marked by many thousands of years of insular isolation for the Jomon, prior to more recent shifts to rice farming and then to state formation.8,28,29 This history makes the region an ideal contained system in which to contrast pre- and post-agricultural populations.
In this study, we leverage genomic imputation30,31,32,33,34 to increase the genomic resolution of low-coverage ancient genomes. This approach statistically infers the likely identity of missing genomic data based on available sequence information and common haplotypic patterns, usually determined through the use of a phased reference panel (e.g., 1000 Genome phase 330,35). However, it remains still unclear how effectively imputation performs for populations like the Jomon, whose ancestry is only represented in present-day reference panels by Japanese populations.8 Here, we assess and implement the imputation of ancient Japanese genomes and those from continental Asia, uncovering the footprints of natural selection that are uniquely characteristic of pre-agricultural hunter-gatherers and the origins of adaptive traits that continue to characterize modern populations.
Results
Assessing the performance of imputation in ancient Asian individuals
The applicability of imputation to ancient Asian individuals remains relatively unexplored compared to those from various geographic regions within West Eurasia.30,31,32,33,34 In particular, haplotypic patterns or variation in ancient populations may not be necessarily well represented in large present-day reference panels, which may result in inaccurate calls or biases toward included populations.36 It is therefore important to establish how well deeply diverged ancestral lineages—which often have no or a very little genetic contribution to present-day populations—can be imputed to higher coverage (Figures 1 and S1).
Figure 1.

Performance of imputing heterozygotes in five ancient individuals
(A and B) The ability to impute ancient Asian individuals is assessed by two metrics: (A) the concordance measures how many of heterozygous sites in the original diploid genomes are successfully recovered from imputation, while (B) the accuracy quantifies how many of the sites that are imputed as heterozygotes are validated with the diploid data. These metrics are calculated for seven different depths of lower coverage data downsampled from the original data (i.e., 0.01, 0.05, 0.1, 0.5, 1, 2, and 5 ). The lines with open circles represent transition mutations, while those with closed circles represent transversions. The ancient genomes included are as follows: two Jomon individuals (JpKa6904 and F23, represented by red and pink respectively), a 31,600-year-old Upper Palaeolithic North Siberian (Yana1 represented by green), and two Siberian individuals from the Baikal Region and Krasnoyarsk Krai (irk034 and kra001, represented by blue and orange, respectively).
Here, we took an approach in which high coverage ancient individuals were downsampled, put through an imputation pipeline implemented by GLIMPSE,37 and compared to their original diploid genotypes. This approach was applied to two individuals of Jomon who have been sequenced to relatively high-coverage (8,819-year-old “JpKa6904” and 3,755-year-old “F23”; coverages of approximately 7.5 and 35 respectively)8,38 and three additional individuals that have different temporal, spatial, and archaeological contexts (31,600-year-old Upper Palaeolithic “Yana1” representing the Ancient North Siberian lineage, a 5,533-year-old individual from the Baikal region “irk034”, and a 4,186-year-old individual from Krasnoyarsk Krai “kra001”).39,40
Our analysis shows that high proportions of diploid genotypes were successfully recovered from imputation (>98.9%), which we define as “concordance rates,” with genome coverage of >0.5 (Figures 1A, S1A, and S1B). The concordance rates are extremely high for homozygous sites (>99.7%) for both transitions and transversions. The concordance rates for transition sites are marginally lower than transversions when focusing on heterozygous sites. However, they become comparable once the coverage reaches 0.5 (Figure 1A). The accuracy per site is nevertheless high in heterozygous sites for both transitions and transversions (>97%) (Figures 1B, S1C, and S1D), supporting an idea that alleles produced through this imputation pipeline are reliable, even for the coverage as low as 0.05 . However, the number of imputed heterozygotes significantly decreases when the coverage drops below 0.5 primarily due to misclassifications of heterozygotes as homozygotes (Figures 1A and S2). We thus defined a coverage cut-off as 0.5 for excluding ancient individuals from haplotypic-based analyses, which is consistent with findings from the previous studies involving ancient samples from other geographic contexts.30,31 Given the high accuracy rates observed regardless of coverage and mutation type, we included all available individuals in frequency-based analyses, i.e., selection scans. This is strong evidence that imputed data provides a firm foundation upon which to conduct further demographic and selection analysis.
Genetic uniqueness of hunter-gatherer-fisher Jomon in the broader Asian context
Leveraging IBDSeq41 with 126 imputed ancient genomes from various parts of East Eurasia with coverage of >0.5 (Table S1), we provide a comprehensive picture of identity-by-descent (IBD) sharing across all pairs of individuals (Figure 2A). Our principal component analysis (PCA) based on the IBD patterns captures the positioning and clustering of samples that broadly mirror their geographic or cultural affiliations in the continent, such as the individuals from Yellow River42 or the Baikal populations split into the Neolithic and Bronze Age groups.43 The most striking observation is the degree to which the Jomon stand out from all continental populations in the second principal component (PC2). Regardless of temporal and spatial variation (Figure S3), as well as a lack of kinship (Figure S4), the twelve Jomon individuals display a uniquely high amount of internal IBD compared to the rest of the dataset (Figure S5). This pattern is still observable even if analysis is restricted to shorter or longer lengths (1–5 Mb, 5–10 Mb, or >10 Mb) of shared IBD (Figure S6), although the tightness of the cluster decreases somewhat as when only longer fragments are included. In line with previous studies, the survival of Jomon ancestry is evident in a historical individual from the Ryukyu Islands,44,45 three individuals from the Kofun period and in individuals from the historic Korean Three Kingdoms period. However, Jomon ancestry is not evenly distributed, with one of the Kofun samples sharing more IBD with the Yellow River population, which supports the previously reported genetic affinity between the Kofun and people with widespread East Asian ancestry8 and an uneven level of sharing within the Historic Korean population in line with differing levels of reported Jomon ancestry.
Figure 2.

Haplotypic analysis of 126 imputed ancient Asian individuals
(A) Principal component analysis based on a matrix of identity-by-descent (IBD) sharing between all pairs of imputed ancient individuals. Different colors show different geographic or cultural backgrounds.
(B) ROH profiles stratified by their total lengths of short (<1.6 Mb) and long (>1.6 Mb) fragments. Individuals with the coverage <0.5 in their original data (i.e., pre-imputed data) are excluded due to insufficient numbers of imputed sites available for this analysis.
The Jomon also stand out from other Asian populations with their highest total levels of runs-of-homozygosity (ROH) in Asia (Figures 2B and S7), with an excessive amount of short ROH fragments (<1.6 Mb in size). This pattern supports a strong bottleneck and small population size in the Jomon, as shown in a previous study.8 In particular, the highest levels of ROH are observed in the two Jomon individuals (F5 and F23) from the same site (i.e., Funadomari site of Rebun Island, north of Hokkaido); they also share excessively high amount of IBD (>1,000 Mb) compared with any pairs of the other Jomon individuals (Figure S8). This enhanced resolution of genomic data identifies a degree of geographic substructure within the otherwise largely uniform Jomon, which has been previously undetectable with pseudo-haploid data.8 Additionally, the lack of evidence on inbreeding in this set of ancient East Eurasians highlights the role of demography including population size and admixture, rather than cultural processes, in shaping the elevated levels of autozygosity as is the case of ancient West Eurasians46
Detecting genetic signatures of local adaptation in pre-agricultural hunter-gatherers
The long-lasting isolation of Jomon in the Japanese archipelago prior to the arrival of agriculture may make it possible to genomically identify adaptive traits that are unique to a hunter-gatherer lifeway. To scan for signatures of natural selection at a genome-wide scale, we applied the population branch statistic (PBS)47 to a set of 19 Jomon individuals (Figure S3) by using Utah residents (CEPH) with Northern and Western European ancestry (CEU) and Han Chinese in Beijing, China (CHB) as an outgroup and a reference, respectively. This allele frequency-based approach can mitigate confounding effects of the strong linkage disequilibrium found throughout the Jomon genomes on the selection scan (Figure S9). Our sliding window analysis identified 12 regions with high PBS values ranked in the top 0.1% of the empirical distribution (Figures 3, S10, and S11), highlighting selective pressures unique to the Jomon lineage and those absent in the Jomon but characteristic of the broader East Asian populations. The strongest signals of selection in the Jomon, ranking within the top 0.01%, are observed in two regions on chromosomes 7 and 12 (Figure 3) and are robust to the choice of different reference populations in the scans (Figure S10). Furthermore, these two regions are marked by a significant reduction in genetic diversity entirely or locally within each window as expected from a recent selective sweep48 (Figures 3 and S11). In contrast, although PBS values exceed the top 0.1% around the EDAR locus, a well-known adaptive gene in East Asia, the region exhibits a high level, rather than a low level, of genetic diversity in the Jomon compared with the other populations (Figures 3 and S11).
Figure 3.

A genome-wide scan for positive selection in the Jomon
Top: Transformed PBS represents rankings of windows (size: 80 SNPs; step between windows: 10 SNPs) by their PBS values. The dashed horizontal lines show the 99.9th (magenta) and 99.99th (purple) percentiles of the empirical distribution. Middle: Heterozygosity of Jomon (red), Han Chinese in Beijing (CHB; blue), and Utah Residents with Northern and Western European ancestry (CEU; orange) in the regions on chromosomes 2, 7, and 12. The protein-coding genes present in each region are shown by solid lines with magenta. The dashed lines mark the locations of focal SNPs in each region. Bottom: Frequencies of focal alleles in ancient populations and modern-day references. The bar plots represent the frequencies of ancestral (blue) and derived (orange) alleles for SNPs of the region on chromosomes 2, 7, and 12, respectively. The frequencies of other SNPs (rs150038188 and rs77567846 on chromosome 7; rs11495049 on chromosome 7) are shown in Figure S16.
EDAR has strong associations with a variety of traits (Figure S12A) including hair thickness,49 earlobe and chin shape,50 and a suite of morphological variants on teeth.51,52 A focal mutation at Val370Ala (rs3827760) is generally at extremely high frequencies across the ancient as well as modern datasets (Figure 3). Intriguingly, this allele is completely absent from all Jomon individuals included in this analysis. This finding is further validated by counting raw reads carrying the ancestral or derived alleles (Table S2), as well as by estimating allele frequencies based on these read counts16,25 (Figure S13). These results suggest that this adaptive mutation was almost absent in the population ancestral to the Jomon. The oldest ancient individual reported to have this mutation is an Upper Pleistocene individual dated to 19,587–19,175 cal BP, the end of Last Glacial Maximum (LGM), from the Amur River Basin, northern China (AR19K),53 a finding we also observe (Figure S14). Given that the Jomon lineage has been estimated to have emerged 15,000–20,000 years ago,8 our analysis provides an upper limit of the temporal origin of ∼20,000 years ago for this adaptive mutation, which in turn suggests that this mutation emerged in response to the environmental conditions observed during the LGM.
The highest peak observed in the selection scan analysis comes from a region on chromosome 7, with a window including four protein-coding genes: KCND2, TSPAN12, ING3, and CPED1 (Figures 3 and S11). Compared with the present-day or ancient population, the Jomon display particularly low heterozygosity in the region, in which several single nucleotide polymorphisms (SNPs) show significant associations with bone health (Figure S12B). Quantifying differences in allele frequencies between the Jomon and each of the continental populations, we were able to further narrow potential targets of natural selection down to three SNPs, whose frequencies are among the most differentiated sites in the region (rs150038188, rs143809091, and rs77567846; Figure S15). These SNPs have derived allele frequencies of >94% in Jomon, with phenotypic impacts on heel bone mineral density, an indicator of physical activity level54 or of osteoporosis55 (Table S3). These frequencies contrast with those observed in modern Africans and European populations (<5%) and in ancient and modern East Asian populations (<50%) (Figures 3, S13, and S16A).
We additionally found strong signals of selection in a region on chromosome 12 (Figure 3), which also encompasses several protein-coding genes. This includes KITLG, a gene well-known for its functional roles in pigmentation. The heterozygosity in Jomon is extremely low downstream of this gene when compared to other ancient and modern populations (Figures 3 and S12C): within this region, two SNP sites (rs74381527 and rs11495049) are highly differentiated in their frequencies between Jomon and ancient or modern continental populations (Figure S17). The derived alleles are almost exclusively observed in Jomon (60%) and are rarely seen in other populations (Figures 3, S13, and S16B). Notably, the UK Biobank dataset56,57 reveals a clear phenotypic impact associated with these alleles, manifesting as darker hair and skin pigmentation (see Table S4). This observed phenotypic effect may be facilitated by alterations in KITLG expression, as indicated in the GTEx data.58
Tracing selection following the introduction of agriculture
The presence or absence of adaptive variants identified from modern populations in the Jomon is also informative as to whether selection occurred before or after the agricultural revolution. One of the traits specifically associated with modern people of East Asian descent is a physiological reaction to the consumption of alcohol in which the skin of affected individuals turns a pinkish-red color, a phenomenon known as “Asian Flush.”59,60 This response is caused by an accumulation of excessive acetaldehyde in the blood due to atypical forms of enzymes involved in alcohol metabolism.61 Variants in genes that encode proteins associated with different stages of the alcohol breakdown pathway, such as rs1229984 at ADH1B and rs671 at ALDH2 (Figure 4A), are present at elevated frequencies in East Asian populations (Figure S18)62,63 and appear to have undergone recent selection in modern Japanese populations.13,63,64 The temporal patterns of allele frequencies are consistent across SNPs in strong linkage disequilibrium (r2 > 0.8) with those two loci (Table S5), supporting the accuracy of imputation regardless of transitions or transversions (as shown in Figures 1 and S1).
Figure 4.

Tracing selection within the alcohol metabolism pathway pre- and post-farming
(A) Temporal changes in allele frequencies at rs1229984 and rs671 in Japanese and Chinese populations. The bar plots represent the frequencies of ancestral (blue) and derived (orange) alleles for those SNPs. Two enzymes are mainly involved in this pathway: first, ADH1B converts ethanol into acetaldehyde, and then ALDH2 digests acetaldehyde into acetate. The nonsynonymous mutation at ADH1B (rs1229984) speeds up the conversion process, while the mutation at ALDH2 (rs671) slows down the digestion. Natural selection on this pathway is directed toward the local accumulation of acetaldehyde.
(B) Contrasting the recent population growth with the emergence of a novel trait that facilitates the accumulation of acetaldehyde. Top: A temporal change in effective population size estimated from IBD sharing between individuals. The estimates are obtained from five different time points: four individuals of the Odake site (Jomon; 5,979 years before present, BP), two individuals of the Kosaku site (Jomon; 4,159 years BP), two individuals of the Funadomari site (Jomon; 3,755 years BP), three individuals of the Iwade site (Kofun; 1,348 years BP), and 104 individuals of modern Japanese. Bottom: Evolution of genetic potential to retain acetaldehyde. The plots show genetic scores of alcohol consumption estimated from the two SNPs (rs1229984 and rs671)65 among individuals from the Jomon (n = 19), Kofun (n = 3), and modern Japanese (n = 104). The x axis represents time with the same scale as that in the top plot.
Our imputation and allelic count approaches clearly demonstrate the complete absence of both adaptive alleles among the Jomon population (Figures 4A and S13; Table S2), supporting a possibility that they were brought by later arrivals to the archipelago from the continent. A “C-to-T” missense mutation at rs1229984 (Arg47His) results in a more active form of the alcohol dehydrogenase 1B (ADH1B) enzyme66 and is observed at high frequencies in present-day East Asian populations (Figures 4A and S18A). Our diverse set of ancient genomes from pre- and post-farming populations show that this variant is observable in individuals from the broad Yellow River cluster in ancient China, historic Southern China, historic Korea, and the imperial Kofun period of Japan (Figures S13 and S18A). The earliest occurrence of this mutation that we observe is ∼4,000 years ago during the Late Neolithic period (Figure 4A), which roughly coincides with an intensification in rice farming and an increased genetic affinity to present-day populations from China and Southeast Asia compared to the preceding Middle Neolithic period.42,67 The frequency continued to increase within China in subsequent periods of the Late Bronze Age and Iron Age and is at its highest level in present-day populations. By the historic period, the allele had become prevalent in Southern China and Korea. Given that the Kofun period saw a large influx of people from continental East Asia, including Yellow River Basin, via the Korean Peninsula,8 this allele was likely introduced through migration to the archipelago during this time and spread into Japanese populations through admixture with indigenous people. The total absence of the allele in the Jomon may also suggest that this mutation and its resulting phenotype conferred no advantage to the hunter-gatherer lifeway, and that its later rise in frequency may be due to the impact of subsistence change after the introduction to farming to the region.
In contrast to rs1229984, a “G-to-A” missense mutation at rs671 is unobservable across all ancient populations from our dataset, except for in Historic Korea (Figures 4A, S13, S18B, and S19). Although no confirmed reads carry this mutation, the majority of SNPs in strong linkage disequilibrium with rs671 support the presence of derived alleles in this population (Figure S19). This mutation changes the position of amino acid residue 504 from glutamic acid to lysine, leading to a slower breakdown of acetaldehyde to acetate via the aldehyde dehydrogenase enzyme.68 This result suggests that the two mutations directly affecting alcohol metabolism on the same pathway occurred at separate stages of history, within different cultural contexts. The emergence of adaptive mutations is often contextualized into the growth of human populations (i.e., the larger a population becomes, the more adaptive variants appear in the population.69 Due to the complete fixation of ancestral alleles at both sites, the genetic potential to retain more acetaldehyde was low in the Jomon, where the population size was constantly small over time (Figure 4B). However, this trait became more variable when the population started to increase in the Kofun period. This evolution further accelerated during which the population exploded in the last 1,300 years, resulting in a bimodality of this phenotypic variation in the modern-day population.
Discussion
Our study performs the first in-depth analysis of natural selection in hunter-gatherer-fisher Jomon. We first demonstrate high rates of accuracy and concordance for genotype imputation in ancient Asian individuals including two Jomon individuals. The vast majority of sites used for the direct comparison of diploid and imputed data are homozygous for either reference or alternative alleles; these sites are imputable with very high rates of concordance and accuracy as long as the coverage is > 0.01 (Figures 1 and S1). In contrast, the ability to recreate heterozygotes is limited by a higher genome coverage of 0.5 due to a reduction in the number of heterozygous sites correctly imputed (Figure S2). Thus, imputing low-coverage genomes (e.g., <0.5 ) comes with a caveat that it may lead to an overall lack of heterozygosity. We also design our imputation framework to minimize potential impacts of ancient DNA damage by imputing data without transition sites, which results in lower concordance rates of imputed sites with transitions than those of transversions. Still, a high rate of accuracy is achievable across all coverage, suggesting that imputed sites are useful, regardless of transitions or transversions, for allele-based analysis such as determining the presence or absence of adaptive alleles. This validation of the successful imputation of ancient Asian genomes enabled us to confidently increase the genomic resolution of individuals from this region and opened novel analytical approaches.
Taking advantage of the potential of imputed data for the deeper analysis, our study provides new insights into pre- and post-farming selection. Following their arrival and subsequent isolation on the archipelago, the population profile of the Jomon became markedly different from those of mainland Asia (Figure 2), with a high level of temporal and spatial genetic homogeneity due to a strong bottleneck and small effective population size as previously shown.8 The divergence of the Jomon lineage appears to have preceded the highly prevalent EDAR V370A mutation within Asia (Figure 3); this finding is supported by a lower rate of incisor shoveling in Jomon.70 While this variant is at near ubiquity in a diverse range of genetic clusters from Asia (Figures 3 and S14), it is completely absent in those who predate the emergence of Jomon lineage estimated to be 15,000–20,000 years ago (i.e., a 33,000-year-old Upper Pleistocene individual from Amur region, a 31,600-year-old individual from Yana River in Siberia, and 24,000-year-old Upper Paleolithic Siberian from Mal’ta).8 Given that the presence of this variant is traceable at least for the last 19,000 years (Figure S14), this mutation could have appeared during the LGM when aridity increased and biological productivity reduced dramatically.71,72
A lack of this selection on EDAR V370A in Jomon in turn raises an intriguing question as to how they coped in extreme environments. Our analysis mapped the most pronounced allele frequency differences between Jomon and other populations onto the two genomic loci including ING3 on chromosome 7 and KITLG on chromosome 12 (Figure 3). ING3 and its surrounding region have robust associations with heel bone mineral density (Figure S12B; Table S3), loss of which results in osteoporosis, a common chronic form of disability. Knockdown of this gene impairs osteoblast differentiation but enhances adipogenesis; this implies its function in determining the fate of mesenchymal progenitors.73 Thus, this selection may have been driven by the high levels of physical activity required for hunting and gathering, which has been also implicated from Mesolithic hunter-gatherers in Europe.74 The other gene, KITLG, is well-known for its associations with variation in hair and skin pigmentation75 as well as its adaptive roles in the ancestor of modern European populations.76 These phenotypic and selection signals come from the enhancer region upstream of the KITLG transcription start site; a reduction in enhancer activity and KITLG expression results in lighter coat color in mice.77,78 However, our scan of Jomon genomes identified selection signatures from its downstream region including the derived alleles that are almost exclusive to Jomon (Figures 3 and S13) and associated with the increase of KITLG expression as well as dark hair color and dark skin pigmentation (Table S4). European hunter-gatherers tend to have darker skin colors even under low environmental UV radiation compared with later farmer,31,79,80 which was potentially due to their accessibility to a diet rich in vitamin D.81 Our results highlight a new possibility that dark pigmentation may have been favored in such pre-agricultural hunter-gatherers, rather than persisting as the ancestral state.82 Given the pleiotropic effects of KITLG in melanogenesis, hematopoiesis, and gametogenesis,83 these variants may have been maintained in this population not directly for skin pigmentation but for other aspects of human physiology.
These selective pressures could have been relaxed due to population movement and admixture8 or increased sedentism and warm and stable climate when paddy field rice cultivation spread into the archipelago in the Yayoi period,84,85 as suggested in a previous study.86 Selection may have then shifted toward the emergence of novel mechanisms on alcohol digestion in this transition (Figure 4). One example of this is the ADH1B allele, which accelerates the rate of conversion from ethanol to acetaldehyde, and arrived in Japan by the time of the Kofun period. While this mutation was completely absent in the Jomon, it started to increase in its frequency in the Yellow River region of China from the Late Neolithic onward (Figure 4A). This mutation is, however, unobservable in the West Liao River, Amur River, and Neolithic Baikal populations (Figure S18A), who all represent Northeast Asian ancestry introduced into the Japanese archipelago during the Yayoi period.8 The prevalence of this adaptive allele in Korea during the period coinciding with the Kofun era supports the possibility that this allele was brought to the archipelago with the arrival of East Asian ancestry during that time. Another mutation in the same pathway subsequently emerged, which slows down the digestion of acetaldehyde into acetate by ALDH2. This mutation is only observable in Historic Korea at a very low frequency among our diverse set of ancient populations and individuals (Figures 4A and S18B), suggesting its very recent origin, possibly within the last millennium to two millennia consistent with recent findings.87 This post-agricultural selection has shaped the inter-individual variability in the alcohol metabolism and “Asian Flush” phenotype observed among the modern populations (Figure 4B).
In summary, taking advantage of the applicability and reliability of genotype imputation to increase the genomic resolution of ancient genomes and the accessibility to both hunter-gatherers and farmers, our study provides genetic evidence on the shift in selective pressures before and after the agricultural revolution in Japan, and more broadly across East Asia. Population movement and admixture appears to have played a critical role not only in transforming lifeways but also in producing phenotypic variation that continues to characterize modern populations. Our approaches illustrate the utility of rapidly growing ancient human genomes with modern bioinformatic methods for understanding regional impacts of agricultural transitions on local adaptation and phenotypic variation in human populations.
Limitations of the study
The main limitation of this study is the small sample size of the Jomon population. We included all accessible Jomon genomes, nineteen individuals in total, into our imputation framework and selection scans. However, it is important to highlight that seven of these genomes exhibit high missing rates in their imputed data, as evidenced in Figure S2. This is primarily due to their failure to meet the 0.5 coverage cut-off. While we are confident with the reliability of the imputed genotypes, it is worth emphasizing the need for additional ancient genomes to validate and strengthen the findings presented in this study.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information on materials, datasets, and protocols should be directed to and will be fulfilled by the lead contact, Shigeki Nakagome (NAKAGOMS@tcd.i.e).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The 260 ancient previously published genomes used in this study are all listed in Table S1.
-
•
This paper does not report the original code.
-
•
The full imputed database (in PLINK format) and any additional information required to reanalyse the data is available upon request from the lead author.
Method details
Data processing and imputation pipeline
260 ancient genomes (listed in Table S1) were processed using the same pipeline as those used in a previous study8 with two additional steps: aligned reads were further filtered for a higher mapping quality threshold of 25 using samtools version 1.7,93 and all reads with less than 34 bp in length were removed.
We used the 1000 Genomes Phase 3 as the reference panel for imputation,35 which is composed of 75,880,357 SNP sites. All bi-allelic variants were called from ancient data, and the corresponding genotype likelihoods were calculated using bcftools v1.1188 with the arguments “-mpileup” and “-call” respectively, and subsequently indexed with “-index”. The genotype likelihood score for all transition sites was manually lowered to “0” before imputation in order to account for potential errors caused by damaged ancient DNA that would influence the imputation output.
Ancient genomes were imputed using GLIMPSE v1.0.1.37 The computational speed of this method made it possible to impute each ancient individual independently, and not as a single batch in which the imputation would be informed by markers in the other ancient samples. For any known second-degree relatives within the dataset, only the highest coverage individual was imputed. Input genomes were first split into chunks of 2 Mb windows with a buffer size of 200 kb using GLIMPSE_chunk and subsequently imputed with GLIMPSE_phase. These chunks were then ligated using GLIMPSE_ligate. Output SNPs were further filtered based on their output genotype likelihood score, with only SNPs reaching 99% considered in further analysis.
Quantification and statistical analysis
Validation of imputation
We assessed the performance of imputation by downsampling the processed BAM file for five selected high coverage ancient individuals to lower coverages (i.e., 0.01, 0.05, 0.1, 0.5, 1, 2, and 5X), using DownsampleSam, part of Picard tools version 1.101 (https://gatk.broadinstitute.org/hc/en-us/articles/360036431292-DownsampleSam-Picard) with random seed “1234”. We independently imputed each individual at each coverage under the same pipeline, and compared the output sites to the original sequence data. The five individuals with >7.5 coverage that were included in this comparison were: two Jomon individuals “JpKa6904”8 and “F23”,38 sequenced to a depth of approx. 7.5 and 35 respectively; two individuals from the Baikal region “irk034” and “kra001”39 14.1 and 13.2 respectively; and the ∼33kya Upper Paleolithic North Siberian “Yana1”40 with a coverage of approx. 23 .
Diploid variants were called from processed bam files using HaplotypeCaller, which is part of GATK version 3.7–0.89 Sites were further filtered for a minimum allele balance of 40%, a base quality of 30, and a depth of 10 using FilterVcf in GATK version 4.3–089 and vcftools v0.1.13.94
Direct comparisons were made between sites common to the imputed and downsampled individuals. Performance was measured under two metrics: a) concordance, which was defined as rate at which diploid sequence data was correctly re-created through imputation (i.e., overlapping sites divided by the sites present in the diploid data) and b) accuracy, which was defined as how often imputed data accurately matched the sequence data (i.e., overlapping sites divided by the number of imputed sites).
Jomon kinship analysis
We further assessed kinship within the entire Jomon population (n = 19) to look for potential deeper relatives (third to fifth degree relations) using KING v2.2.5 –kinship.91 The imputed data were filtered for minor allele frequencies >1% and transversions only.
IBD analysis
Identical-by-descent (IBD) segments of the genome across imputed individuals were identified using IBDSeq vr120641 using parameters as follows: --errormax = 0.00 --ibdlod = 2 --r2max = 0.15 --r2window = 500. Analysis was confined to sites filtered for minor allele frequencies >1% and transversions only. Low-coverage individuals that showed >15% missingness in their imputed genotypes were removed from analysis, as were the lower coverage individuals of any second-degree relatives; SNPs with >10% missingness rate were subsequently removed using --geno 0.1 in PLINK v1.90b4.4.92 The IBDSeq output was further used to estimate recent population sizes using IBDne90 with default parameters. Estimated population sizes were calculated by taking a harmonic mean of the values estimated between 5 and 15 generations. Principal component analysis based on the shared IBD between each individual was calculated using the prcomp function in R.
ROH analysis
We measured the proportion of imputed genomes under runs of homozygosity (ROH) using PLINK v1.90b4.4.92 As in IBD analysis, this analysis was conducted on sites filtered for minor allele frequencies >1% and transversions only, with low-coverage individuals (>15% missingness) and poorly imputed SNPs (>10% missingness after the exclusion of low-coverage individuals) not included in the analysis. ROH was measured in each imputed genome using the following options: -homozyg --homozyg-density 50 --homozyg-gap 100 --homozyg-kb 500 --homozyg-snp 50 --homozyg-window-het 1 --homozyg-window-snp 50 --homozyg-window-threshold 0.05.
Positive selection scan
We used PLINK v1.90b4.492 to calculate allele frequencies, specifically focusing on sites filtered for minor allele frequencies >1%, limited to transversions, and containing no missing genotypes in at least 12 Jomon genomes with coverage >0.5 (see Figure S3). We then computed Fst values using an estimator as described in Weir and Cockerham (1984)95 for all pairwise comparisons including Jomon (i.e., test population), CEU (i.e., outgroup), and each of several reference populations (i.e., CHB, the other modern East Asians included in 1000 Genome Phase 3, ancient hunter-gatherers from the Baikal region, ancient farmers from Yellow River Basin, and a group of individuals from Historic Korea). These Fst values were further used to measure the Population Branch Statistic (PBS)47 between the Jomon population and each reference population, with CEU serving as the outgroup. This method has been shown to be powerful in detecting population specific selection in humans;47 it allows us to identify SNPs with a change in allele frequency specific to the branch leading to the Jomon. If this change is notably extreme (i.e., the branch length is significantly longer than the genome-wide average), it can be considered as a sign of positive selection in the Jomon population.
To detect signatures of selection, we employed a sliding window approach with an 80-SNP window size and a 10-SNP step size. This approach identified 12 genomic intervals that contained windows with mean PBS values above the 99.9th percentile of the empirical distribution (Figure S11). Two of these intervals achieved a more stringent cut-off with the values above the 99.99th percentile of the distribution (Figure 3). These selection signatures were characterised by overlapping signals of phenotypic associations retrieved from BioMart in Ensembl (Ensembl Variation 104), with the filters for a human reference genome (GRCh37.p13) and “Study type: GWAS”.
We further searched for phenotypes or molecular traits that have significant associations with SNPs uniquely differentiated in Jomon using Open Target Genetics56,57 or GTEx Portal58 (see Table S2). One of the phenotypes chosen for this analysis is skin color (Data-Field 1717), which is coded by eight different categorical values: 1 (Very fair), 2 (Fair), 3 (Light olive), 4 (Dark olive), 5 (Brown), and 6 (Black). Thus, positive values of effect sizes were considered as genetic effects on darker skin pigmentation.
Adjacent sites in linkage disequilibrium (r2 ≥ 0.8) with the focal SNPs were identified based on the modern Japanese population from the 1000 Genomes Phase 3 panel,35 with the commands in PLINK as follows: --r2 --ld-snp site_position -ld-window-kb 1000 -ld-window 99999 --ld-window-r2 0.8.
Adaptive allele frequencies based on allelic counts
To validate the differences in allele frequencies among imputed ancient populations, we estimated the frequencies based on numbers of reads carrying ancestral and derived alleles at each site of interest in the pre-imputed data. We initially retrieved aligned sequence reads (i.e., BAM files) of ancient Asian individuals from Japan (i.e., Jomon and Kofun), Korea, the Yellow River region, the West Liao River region, and the Amur River region respectively. Second, we utilised CollectAllelicCounts in GATK version 4.0.5.1 to quantify the presence of ancestral and derived alleles at a given site. Then, we estimated the allele frequency in each ancient population, taking into account individuals who possessed imputed genotypes at the site (Figure S13). This estimation was carried out using a likelihood function described in a previous study.25
Polygenic score calculation
We quantified the genetic potential to retain acetaldehyde for each individual from ancient (i.e., 19 individuals of Jomon and three individuals of Kofun) and modern Japanese (JPT; n = 104). The scores were calculated from genotypes at rs1229984 (ADH1B) and rs671 (ALDH2) weighted by the effect sizes estimated from a genome-wide association study (GWAS) on alcohol consumption in a large cohort of BioBank Japan samples.65 Due to variable coverage across ancient individuals, not all SNPs were present in a given sample. To account for missing information in this calculation, we used an equation modified from a previous study80: , where is the number of SNPs (i.e., two), is the genotype at the i-th SNP (i.e., 0, 1, or 2, depending on how many effect alleles an individual has), and is the GWAS-estimated effect size. The genetic potential was defined in a way that an individual with a higher score has a lower potential to retain acetaldehyde in the blood.
Acknowledgments
This work was supported by the Science Foundation Ireland/Health Research Board/Wellcome Trust Biomedical Research Partnership Investigator Award (no. 205072 to D.G.B.), the SAKIGAKE Project in Kanazawa University (to T.G.), Wellcome Trust ISSF Award (to S.N.), and grants from the Japan Society for the Promotion of Science KAKENHI (nos. 20H05822 and 20H05815 to T.G. and 22H02711 to S.N.). We would also like to thank the reviewers for their constructive feedback.
Author contributions
S.N. conceived and supervised the study. N.P.C and M.M. assessed and analyzed imputed data. N.P.C conducted demographic analysis. S.N. performed selection scans. N.P.C and S.N. wrote the manuscript with input from all coauthors.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110050.
Supplemental information
References
- 1.Price T.D., Bar-Yosef O. The Origins of Agriculture: New Data, New Ideas: An Introduction to Supplement 4. Curr. Anthropol. 2011;52:S163–S174. [Google Scholar]
- 2.Barker G. Oxford University Press on Demand; 2009. The Agricultural Revolution in Prehistory: Why Did Foragers Become Farmers? [Google Scholar]
- 3.Diamond J., Bellwood P. Farmers and their languages: the first expansions. Science. 2003;300:597–603. doi: 10.1126/science.1078208. [DOI] [PubMed] [Google Scholar]
- 4.Bocquet-Appel J.-P., Bar-Yosef O. Springer Science & Business Media; 2008. The Neolithic Demographic Transition and its Consequences. [Google Scholar]
- 5.Richards M.P. A brief review of the archaeological evidence for Palaeolithic and Neolithic subsistence. Eur. J. Clin. Nutr. 2002;56:16. doi: 10.1038/sj.ejcn.1601646. p following 1262. [DOI] [PubMed] [Google Scholar]
- 6.Pearce-Duvet J.M.C. The origin of human pathogens: evaluating the role of agriculture and domestic animals in the evolution of human disease. Biol. Rev. Camb. Philos. Soc. 2006;81:369–382. doi: 10.1017/S1464793106007020. [DOI] [PubMed] [Google Scholar]
- 7.Skoglund P., Malmström H., Raghavan M., Storå J., Hall P., Willerslev E., Gilbert M.T.P., Götherström A., Jakobsson M. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 8.Cooke N.P., Mattiangeli V., Cassidy L.M., Okazaki K., Stokes C.A., Onbe S., Hatakeyama S., Machida K., Kasai K., Tomioka N., et al. Ancient genomics reveals tripartite origins of Japanese populations. Sci. Adv. 2021;7:eabh2419. doi: 10.1126/sciadv.abh2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamba C., Jones E.R., Teasdale M.D., McLaughlin R.L., Gonzalez-Fortes G., Mattiangeli V., Domboróczki L., Kővári I., Pap I., Anders A., et al. Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 2014;5:5257. doi: 10.1038/ncomms6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones E.R., Zarina G., Moiseyev V., Lightfoot E., Nigst P.R., Manica A., Pinhasi R., Bradley D.G. The Neolithic Transition in the Baltic Was Not Driven by Admixture with Early European Farmers. Curr. Biol. 2017;27:576–582. doi: 10.1016/j.cub.2016.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patin E., Siddle K.J., Laval G., Quach H., Harmant C., Becker N., Froment A., Régnault B., Lemée L., Gravel S., et al. The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nat. Commun. 2014;5:3163. doi: 10.1038/ncomms4163. [DOI] [PubMed] [Google Scholar]
- 12.Field Y., Boyle E.A., Telis N., Gao Z., Gaulton K.J., Golan D., Yengo L., Rocheleau G., Froguel P., McCarthy M.I., Pritchard J.K. Detection of human adaptation during the past 2000 years. Science. 2016;354:760–764. doi: 10.1126/science.aag0776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada Y., Momozawa Y., Sakaue S., Kanai M., Ishigaki K., Akiyama M., Kishikawa T., Arai Y., Sasaki T., Kosaki K., et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018;9:1631. doi: 10.1038/s41467-018-03274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson N., Isakov M., Booth T., Büster L., Fischer C.-E., Olalde I., Ringbauer H., Akbari A., Cheronet O., Bleasdale M., et al. Large-scale migration into Britain during the Middle to Late Bronze Age. Nature. 2022;601:588–594. doi: 10.1038/s41586-021-04287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burger J., Kirchner M., Bramanti B., Haak W., Thomas M.G. Absence of the lactase-persistence-associated allele in early Neolithic Europeans. Proc. Natl. Acad. Sci. USA. 2007;104:3736–3741. doi: 10.1073/pnas.0607187104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagome S., Alkorta-Aranburu G., Amato R., Howie B., Peter B.M., Hudson R.R., Di Rienzo A. Estimating the Ages of Selection Signals from Different Epochs in Human History. Mol. Biol. Evol. 2016;33:657–669. doi: 10.1093/molbev/msv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luca F., Perry G.H., Di Rienzo A. Evolutionary adaptations to dietary changes. Annu. Rev. Nutr. 2010;30:291–314. doi: 10.1146/annurev-nutr-080508-141048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davy T., Ju D., Mathieson I., Skoglund P. Hunter-gatherer admixture facilitated natural selection in Neolithic European farmers. Curr. Biol. 2023;33:1365–1371.e3. doi: 10.1016/j.cub.2023.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerner G., Neehus A.-L., Philippot Q., Bohlen J., Rinchai D., Kerrouche N., Puel A., Zhang S.-Y., Boisson-Dupuis S., Abel L., et al. Genetic adaptation to pathogens and increased risk of inflammatory disorders in post-Neolithic Europe. Cell Genom. 2023;3:100248. doi: 10.1016/j.xgen.2022.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee R.B., Daly R.H., Daly R., Cambridge University Press . Cambridge University Press; 1999. The Cambridge Encyclopedia of Hunters and Gatherers. [Google Scholar]
- 21.Fumagalli M., Moltke I., Grarup N., Racimo F., Bjerregaard P., Jørgensen M.E., Korneliussen T.S., Gerbault P., Skotte L., Linneberg A., et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science. 2015;349:1343–1347. doi: 10.1126/science.aab2319. [DOI] [PubMed] [Google Scholar]
- 22.Jarvis J.P., Scheinfeldt L.B., Soi S., Lambert C., Omberg L., Ferwerda B., Froment A., Bodo J.-M., Beggs W., Hoffman G., et al. Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet. 2012;8:e1002641. doi: 10.1371/journal.pgen.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perry G.H., Foll M., Grenier J.-C., Patin E., Nédélec Y., Pacis A., Barakatt M., Gravel S., Zhou X., Nsobya S.L., et al. Adaptive, convergent origins of the pygmy phenotype in African rainforest hunter-gatherers. Proc. Natl. Acad. Sci. USA. 2014;111:E3596–E3603. doi: 10.1073/pnas.1402875111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quintana-Murci L. Human Immunology through the Lens of Evolutionary Genetics. Cell. 2019;177:184–199. doi: 10.1016/j.cell.2019.02.033. [DOI] [PubMed] [Google Scholar]
- 25.Mathieson I., Lazaridis I., Rohland N., Mallick S., Patterson N., Roodenberg S.A., Harney E., Stewardson K., Fernandes D., Novak M., et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gakuhari T., Nakagome S., Rasmussen S., Allentoft M.E., Sato T., Korneliussen T., Chuinneagáin B.N., Matsumae H., Koganebuchi K., Schmidt R., et al. Ancient Jomon genome sequence analysis sheds light on migration patterns of early East Asian populations. Commun. Biol. 2020;3:437. doi: 10.1038/s42003-020-01162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McColl H., Racimo F., Vinner L., Demeter F., Gakuhari T., Moreno-Mayar J.V., van Driem G., Gram Wilken U., Seguin-Orlando A., de la Fuente Castro C., et al. The prehistoric peopling of Southeast Asia. Science. 2018;361:88–92. doi: 10.1126/science.aat3628. [DOI] [PubMed] [Google Scholar]
- 28.Habu J. Cambridge University Press; 2004. Ancient Jomon of Japan. [Google Scholar]
- 29.Mizoguchi K. Cambridge University Press; 2013. The Archaeology of Japan: From the Earliest Rice Farming Villages to the Rise of the State. [Google Scholar]
- 30.Sousa da Mota B., Rubinacci S., Cruz Dávalos D.I., G Amorim C.E., Sikora M., Johannsen N.N., Szmyt M.H., Włodarczak P., Szczepanek A., Przybyła M.M., et al. Imputation of ancient human genomes. Nat. Commun. 2023;14:3660. doi: 10.1038/s41467-023-39202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cassidy L.M., Maoldúin R.Ó., Kador T., Lynch A., Jones C., Woodman P.C., Murphy E., Ramsey G., Dowd M., Noonan A., et al. A dynastic elite in monumental Neolithic society. Nature. 2020;582:384–388. doi: 10.1038/s41586-020-2378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hui R., D’Atanasio E., Cassidy L.M., Scheib C.L., Kivisild T. Evaluating genotype imputation pipeline for ultra-low coverage ancient genomes. Sci. Rep. 2020;10:18542. doi: 10.1038/s41598-020-75387-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martiniano R., Cassidy L.M., Ó'Maoldúin R., McLaughlin R., Silva N.M., Manco L., Fidalgo D., Pereira T., Coelho M.J., Serra M., et al. The population genomics of archaeological transition in west Iberia: Investigation of ancient substructure using imputation and haplotype-based methods. PLoS Genet. 2017;13:e1006852. doi: 10.1371/journal.pgen.1006852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irving-Pease E.K., Refoyo-Martínez A., Barrie W., Ingason A., Pearson A., Fischer A., Sjögren K.-G., Halgren A.S., Macleod R., Demeter F., et al. The selection landscape and genetic legacy of ancient Eurasians. Nature. 2024;625:312–320. doi: 10.1038/s41586-023-06705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Günther T., Nettelblad C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet. 2019;15:e1008302. doi: 10.1371/journal.pgen.1008302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubinacci S., Ribeiro D.M., Hofmeister R.J., Delaneau O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat. Genet. 2021;53:120–126. doi: 10.1038/s41588-020-00756-0. [DOI] [PubMed] [Google Scholar]
- 38.Kanzawa-Kiriyama H., Jinam T.A., Kawai Y., Sato T., Hosomichi K., Tajima A., Adachi N., Matsumura H., Kryukov K., Saitou N., Shinoda K.I. Late Jomon male and female genome sequences from the Funadomari site in Hokkaido, Japan. Anthropol. Sci. 2019;127:83–108. doi: 10.1537/ase.190415. [DOI] [Google Scholar]
- 39.Kılınç G.M., Kashuba N., Koptekin D., Bergfeldt N., Dönertaş H.M., Rodríguez-Varela R., Shergin D., Ivanov G., Kichigin D., Pestereva K., et al. Human population dynamics and Yersinia pestis in ancient northeast Asia. Sci. Adv. 2021;7:eabc4587. doi: 10.1126/sciadv.abc4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sikora M., Pitulko V.V., Sousa V.C., Allentoft M.E., Vinner L., Rasmussen S., Margaryan A., de Barros Damgaard P., de la Fuente C., Renaud G., et al. The population history of northeastern Siberia since the Pleistocene. Nature. 2019;570:182–188. doi: 10.1038/s41586-019-1279-z. [DOI] [PubMed] [Google Scholar]
- 41.Browning B.L., Browning S.R. Detecting identity by descent and estimating genotype error rates in sequence data. Am. J. Hum. Genet. 2013;93:840–851. doi: 10.1016/j.ajhg.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ning C., Li T., Wang K., Zhang F., Li T., Wu X., Gao S., Zhang Q., Zhang H., Hudson M.J., et al. Ancient genomes from northern China suggest links between subsistence changes and human migration. Nat. Commun. 2020;11:2700. doi: 10.1038/s41467-020-16557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Barros Damgaard P., Martiniano R., Kamm J., Moreno-Mayar J.V., Kroonen G., Peyrot M., Barjamovic G., Rasmussen S., Zacho C., Baimukhanov N., et al. The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science. 2018;360:eaar7711. doi: 10.1126/science.aar7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooke N.P., Mattiangeli V., Cassidy L.M., Okazaki K., Kasai K., Bradley D.G., Gakuhari T., Nakagome S. Genomic insights into a tripartite ancestry in the Southern Ryukyu Islands. Evol. Hum. Sci. 2023;5:e23. doi: 10.1017/ehs.2023.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robbeets M., Bouckaert R., Conte M., Savelyev A., Li T., An D.-I., Shinoda K.-I., Cui Y., Kawashima T., Kim G., et al. Triangulation supports agricultural spread of the Transeurasian languages. Nature. 2021;599:616–621. doi: 10.1038/s41586-021-04108-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ceballos F.C., Gürün K., Altınışık N.E., Gemici H.C., Karamurat C., Koptekin D., Vural K.B., Mapelli I., Sağlıcan E., Sürer E., et al. Human inbreeding has decreased in time through the Holocene. Curr. Biol. 2021;31:3925–3934.e8. doi: 10.1016/j.cub.2021.06.027. [DOI] [PubMed] [Google Scholar]
- 47.Yi X., Liang Y., Huerta-Sanchez E., Jin X., Cuo Z.X.P., Pool J.E., Xu X., Jiang H., Vinckenbosch N., Korneliussen T.S., et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R., et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujimoto A., Kimura R., Ohashi J., Omi K., Yuliwulandari R., Batubara L., Mustofa M.S., Samakkarn U., Settheetham-Ishida W., Ishida T., et al. A scan for genetic determinants of human hair morphology: EDAR is associated with Asian hair thickness. Hum. Mol. Genet. 2008;17:835–843. doi: 10.1093/hmg/ddm355. [DOI] [PubMed] [Google Scholar]
- 50.Peng Q., Li J., Tan J., Yang Y., Zhang M., Wu S., Liu Y., Zhang J., Qin P., Guan Y., et al. EDARV370A associated facial characteristics in Uyghur population revealing further pleiotropic effects. Hum. Genet. 2016;135:99–108. doi: 10.1007/s00439-015-1618-6. [DOI] [PubMed] [Google Scholar]
- 51.Park J.-H., Yamaguchi T., Watanabe C., Kawaguchi A., Haneji K., Takeda M., Kim Y.-I., Tomoyasu Y., Watanabe M., Oota H., et al. Effects of an Asian-specific nonsynonymous EDAR variant on multiple dental traits. J. Hum. Genet. 2012;57:508–514. doi: 10.1038/jhg.2012.60. [DOI] [PubMed] [Google Scholar]
- 52.Kimura R., Yamaguchi T., Takeda M., Kondo O., Toma T., Haneji K., Hanihara T., Matsukusa H., Kawamura S., Maki K., et al. A common variation in EDAR is a genetic determinant of shovel-shaped incisors. Am. J. Hum. Genet. 2009;85:528–535. doi: 10.1016/j.ajhg.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mao X., Zhang H., Qiao S., Liu Y., Chang F., Xie P., Zhang M., Wang T., Li M., Cao P., et al. The deep population history of northern East Asia from the Late Pleistocene to the Holocene. Cell. 2021;184:3256–3266.e13. doi: 10.1016/j.cell.2021.04.040. [DOI] [PubMed] [Google Scholar]
- 54.Pettersson U., Nilsson M., Sundh V., Mellström D., Lorentzon M. Physical activity is the strongest predictor of calcaneal peak bone mass in young Swedish men. Osteoporos. Int. 2010;21:447–455. doi: 10.1007/s00198-009-0982-2. [DOI] [PubMed] [Google Scholar]
- 55.Bauer D.C., Ewing S.K., Cauley J.A., Ensrud K.E., Cummings S.R., Orwoll E.S., Osteoporotic Fractures in Men (MrOS) Research Group Quantitative ultrasound predicts hip and non-spine fracture in men: the MrOS study. Osteoporos. Int. 2007;18:771–777. doi: 10.1007/s00198-006-0317-5. [DOI] [PubMed] [Google Scholar]
- 56.Ghoussaini M., Mountjoy E., Carmona M., Peat G., Schmidt E.M., Hercules A., Fumis L., Miranda A., Carvalho-Silva D., Buniello A., et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2021;49:D1311–D1320. doi: 10.1093/nar/gkaa840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mountjoy E., Schmidt E.M., Carmona M., Schwartzentruber J., Peat G., Miranda A., Fumis L., Hayhurst J., Buniello A., Karim M.A., et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 2021;53:1527–1533. doi: 10.1038/s41588-021-00945-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolff P.H. Ethnic differences in alcohol sensitivity. Science. 1972;175:449–450. doi: 10.1126/science.175.4020.449. [DOI] [PubMed] [Google Scholar]
- 60.Chan A.W. Racial differences in alcohol sensitivity. Alcohol Alcohol. 1986;21:93–104. [PubMed] [Google Scholar]
- 61.Eriksson C.J. The role of acetaldehyde in the actions of alcohol (update 2000) Alcohol Clin. Exp. Res. 2001;25:15S–32S. doi: 10.1097/00000374-200105051-00005. [DOI] [PubMed] [Google Scholar]
- 62.Oota H., Pakstis A.J., Bonne-Tamir B., Goldman D., Grigorenko E., Kajuna S.L.B., Karoma N.J., Kungulilo S., Lu R.-B., Odunsi K., et al. The evolution and population genetics of the ALDH2 locus: random genetic drift, selection, and low levels of recombination. Ann. Hum. Genet. 2004;68:93–109. doi: 10.1046/j.1529-8817.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 63.Han Y., Gu S., Oota H., Osier M.V., Pakstis A.J., Speed W.C., Kidd J.R., Kidd K.K. Evidence of positive selection on a class I ADH locus. Am. J. Hum. Genet. 2007;80:441–456. doi: 10.1086/512485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang M., Huang X., Li R., Xu H., Jin L., He Y. Detecting recent positive selection with high accuracy and reliability by conditional coalescent tree. Mol. Biol. Evol. 2014;31:3068–3080. doi: 10.1093/molbev/msu244. [DOI] [PubMed] [Google Scholar]
- 65.Matoba N., Akiyama M., Ishigaki K., Kanai M., Takahashi A., Momozawa Y., Ikegawa S., Ikeda M., Iwata N., Hirata M., et al. GWAS of 165,084 Japanese individuals identified nine loci associated with dietary habits. Nat. Hum. Behav. 2020;4:308–316. doi: 10.1038/s41562-019-0805-1. [DOI] [PubMed] [Google Scholar]
- 66.Sherva R., Rice J.P., Neuman R.J., Rochberg N., Saccone N.L., Bierut L.J. Associations and interactions between SNPs in the alcohol metabolizing genes and alcoholism phenotypes in European Americans. Alcohol Clin. Exp. Res. 2009;33:848–857. doi: 10.1111/j.1530-0277.2009.00904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong G., Zhang S., Yang Y., Chen J., Chen F. Agricultural intensification and its impact on environment during Neolithic Age in northern China. Chin. Sci. Bull. 2016;61:2913–2925. doi: 10.1360/n972016-00547. [DOI] [Google Scholar]
- 68.Luo H.-R., Wu G.-S., Pakstis A.J., Tong L., Oota H., Kidd K.K., Zhang Y.-P. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487Lys. Gene. 2009;435:96–103. doi: 10.1016/j.gene.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 69.Hawks J., Wang E.T., Cochran G.M., Harpending H.C., Moyzis R.K. Recent acceleration of human adaptive evolution. Proc. Natl. Acad. Sci. USA. 2007;104:20753–20758. doi: 10.1073/pnas.0707650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kitagawa Y., Manabe Y., Oyamada J., Rokutanda A. Deciduous dental morphology of the prehistoric Jomon people of Japan: comparison of nonmetric characters. Am. J. Phys. Anthropol. 1995;97:101–111. doi: 10.1002/ajpa.1330970203. [DOI] [PubMed] [Google Scholar]
- 71.Claussen M., Selent K., Brovkin V., Raddatz T., Gayler V. Impact of CO2 and climate on Last Glacial maximum vegetation – a factor separation. Biogeosciences. 2013;10:3593–3604. doi: 10.5194/bg-10-3593-2013. [DOI] [Google Scholar]
- 72.Hlusko L.J., Carlson J.P., Chaplin G., Elias S.A., Hoffecker J.F., Huffman M., Jablonski N.G., Monson T.A., O’Rourke D.H., Pilloud M.A., Scott G.R. Environmental selection during the last ice age on the mother-to-infant transmission of vitamin D and fatty acids through breast milk. Proc. Natl. Acad. Sci. USA. 2018;115:E4426–E4432. doi: 10.1073/pnas.1711788115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chesi A., Wagley Y., Johnson M.E., Manduchi E., Su C., Lu S., Leonard M.E., Hodge K.M., Pippin J.A., Hankenson K.D., et al. Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat. Commun. 2019;10:1260. doi: 10.1038/s41467-019-09302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cox S.L., Ruff C.B., Maier R.M., Mathieson I. Genetic contributions to variation in human stature in prehistoric Europe. Proc. Natl. Acad. Sci. USA. 2019;116:21484–21492. doi: 10.1101/690545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sulem P., Gudbjartsson D.F., Stacey S.N., Helgason A., Rafnar T., Magnusson K.P., Manolescu A., Karason A., Palsson A., Thorleifsson G., et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 2007;39:1443–1452. doi: 10.1038/ng.2007.13. [DOI] [PubMed] [Google Scholar]
- 76.Sturm R.A., Duffy D.L. Human pigmentation genes under environmental selection. Genome Biol. 2012;13:248. doi: 10.1186/gb-2012-13-9-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guenther C.A., Tasic B., Luo L., Bedell M.A., Kingsley D.M. A molecular basis for classic blond hair color in Europeans. Nat. Genet. 2014;46:748–752. doi: 10.1038/ng.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller C.T., Beleza S., Pollen A.A., Schluter D., Kittles R.A., Shriver M.D., Kingsley D.M. cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007;131:1179–1189. doi: 10.1016/j.cell.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brace S., Diekmann Y., Booth T.J., van Dorp L., Faltyskova Z., Rohland N., Mallick S., Olalde I., Ferry M., Michel M., et al. Ancient genomes indicate population replacement in Early Neolithic Britain. Nat. Ecol. Evol. 2019;3:765–771. doi: 10.1038/s41559-019-0871-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ju D., Mathieson I. The evolution of skin pigmentation-associated variation in West Eurasia. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2009227118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jablonski N.G. The evolution of human skin pigmentation involved the interactions of genetic, environmental, and cultural variables. Pigm. Cell Melanoma Res. 2021;34:707–729. doi: 10.1111/pcmr.12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jablonski N.G., Chaplin G. Colloquium paper: human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA. 2010;107:8962–8968. doi: 10.1073/pnas.0914628107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reissmann M., Ludwig A. Pleiotropic effects of coat colour-associated mutations in humans, mice and other mammals. Semin. Cell Dev. Biol. 2013;24:576–586. doi: 10.1016/j.semcdb.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 84.McMichael A.J. Insights from past millennia into climatic impacts on human health and survival. Proc. Natl. Acad. Sci. USA. 2012;109:4730–4737. doi: 10.1073/pnas.1120177109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ryan T.M., Shaw C.N. Gracility of the modern Homo sapiens skeleton is the result of decreased biomechanical loading. Proc. Natl. Acad. Sci. USA. 2015;112:372–377. doi: 10.1073/pnas.1418646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watanabe Y., Ohashi J. Modern Japanese ancestry-derived variants reveal the formation process of the current Japanese regional gradations. iScience. 2023;26:106130. doi: 10.1016/j.isci.2023.106130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cong P.-K., Bai W.-Y., Li J.-C., Yang M.-Y., Khederzadeh S., Gai S.-R., Li N., Liu Y.-H., Yu S.-H., Zhao W.-W., et al. Genomic analyses of 10,376 individuals in the Westlake BioBank for Chinese (WBBC) pilot project. Nat. Commun. 2022;13:2939. doi: 10.1038/s41467-022-30526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Browning S.R., Browning B.L. Accurate Non-parametric Estimation of Recent Effective Population Size from Segments of Identity by Descent. Am. J. Hum. Genet. 2015;97:404–418. doi: 10.1016/j.ajhg.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weir B.S., Cockerham C.C. Estimating f-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The 260 ancient previously published genomes used in this study are all listed in Table S1.
-
•
This paper does not report the original code.
-
•
The full imputed database (in PLINK format) and any additional information required to reanalyse the data is available upon request from the lead author.