

Abstract
Metabolic dysfunction‐associated steatotic liver disease (MASLD), with its steadily increasing prevalence, represents now a major problem in public health. A proper referral could benefit from tools allowing more precise risk stratification. To this end, in recent decades, several genetic variants that may help predict and refine the risk of development and progression of MASLD have been investigated. In this review, we aim to discuss the role genetics in MASLD plays in everyday clinical practice. We performed a comprehensive literature search of PubMed for relevant publications. Available evidence highlights the emergence of genetic‐based noninvasive algorithms for diagnosing fatty liver, metabolic dysfunction‐associated steatohepatitis, fibrosis progression and occurrence of liver‐related outcomes including hepatocellular carcinoma. Nevertheless, their accuracy is not optimal and application in everyday clinical practice remains challenging. Furthermore, susceptible genetic markers have recently become subjects of great scientific interest as therapeutic targets in precision medicine. In conclusion, decisional algorithms based on genetic testing in MASLD to facilitate the clinician decisions on management and treatment are under growing investigation and could benefit from artificial intelligence methodology.
Keywords: genetic, MASH, MASLD, metabolic dysfunction associated steatotic liver disease, PNPLA3, steatohepatitis

INTRODUCTION
Metabolic dysfunction‐associated steatotic liver disease (MASLD) is the most common cause of cirrhosis and by 2030 is predicted to become the leading indication for liver transplantation in the western countries among patients with or without hepatocellular carcinoma (HCC). 1 In the scenario of a constantly increasing burden of MASLD, identifying patients at higher risk of progression to justify HCC screening represents one of the greatest clinical challenges, which is also affected by the heterogeneity in MASLD pathogenesis that reflects on a wide diversification in MASLD patients, whose disease might progress slower or faster. 2 In the last decade, non‐tests have been developed for early identification of patients at higher risk of advanced fibrosis who should be referred to a secondary and/or tertiary center. In this contest, genetics could be a tool for risk stratification (Figure 1). The role of genetics in MASLD predisposition and liver fibrosis progression has been widely investigated, but little is known about the role of genetics in predicting hepatic and extra‐hepatic events, and in driving drug development.
FIGURE 1.
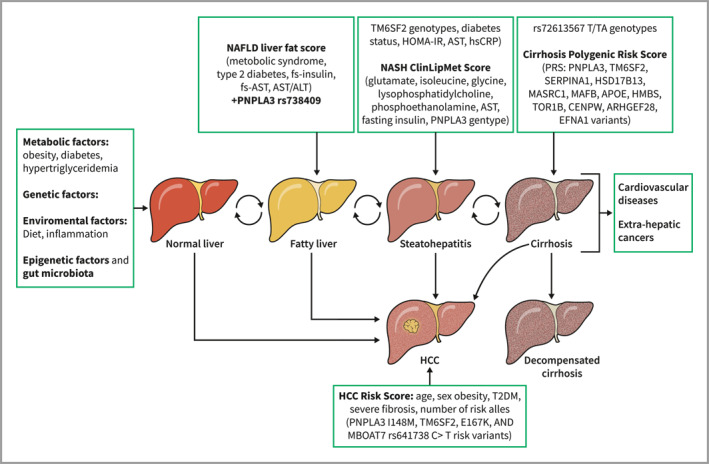
Genetic scores and risk stratification in the full spectrum of MASLD. Together with metabolic and environmental factors, the full spectrum of MASLD has been associated with genetic variants which predispose to MASLD development and progression. Several genetic scores combined with clinical and laboratory data have been developed for the early identification of subjects at higher risk of severe liver disease, who might deserve adequate surveillance. MASLD, metabolic dysfunction‐associated steatotic liver disease.
AIMS AND METHODS
In this review, we aim to discuss the role genetics in MASLD plays in everyday clinical practice. For this purpose, we performed a comprehensive literature search of PubMed for relevant publications.
Key genes in MASLD
MASLD is a systemic disease whose complex pathogenesis accounts for metabolic, genetic, environmental and microbial factors. 2 Over the last decades, several exome‐wide and genome‐wide association studies have identified inherited common variants in genes such as patatin‐like phospholipase domain‐containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane bound O‐acyltransferase domain containing 7 (MBOAT7) and hydroxysteroid 17‐beta dehydrogenase 13 (HSD17B13) associated with MASLD development and its progression 3 Romeo et colleagues first demonstrated how the PNPLA3 rs738409 C > G single nucleotide polymorphism (SNP) ‐leading to the I148M protein variant‐was strongly associated with increased hepatic fat levels and hepatic inflammation. 4 PNPLA3 encodes for a lipoprotein lipase but its catalytic activity is disrupted in the rs738409 G variant, which finally leads to fat accumulation in hepatocytes. It also confers a pro‐inflammatory and a profibrogenic phenotype in hepatic stellate cells by reducing retinol release. Besides, recent evidence highlighted that the PNPLA3 rs738409 SNP confers a dysfunctional phenotype of mitochondrial function with a reduction of de novo lipogenesis and an implementation in b‐oxidation/ketogenesis, which may play a direct role in liver disease progression. 5
This evidence was further confirmed by Anstee and colleagues, who pointed‐out that the full histological spectrum of MASLD was affected by genetic variants of PNPLA3, TM6SF2 and HSD17B13. 6 Indeed, excess lipid accumulation in lipid droplets has also been linked to the SNP rs58542926 C > T in the TM6SF2 gene: the gene variant is associated with an alteration in the assembly of triglycerides and with a reduction of very‐low‐density lipoproteins (VLDL) secretion. 7 Despite the limited knowledge on its role, opposite functions were attributed to HSD17B13 rs72613567 T > TA variant. The HSD17B13 is a liver‐specific lipid droplet associated protein; it has been observed that its overexpression promotes hepatic steatosis in mice, while the loss‐of‐function variants seems to be protective against the development of MASLD and its progression. 8 , 9
The main genetic variants associated with MASLD cause a modulation of lipid metabolism and consensual lipotoxicity, these effects in turn leading to the association between genetic variants and liver disease severity in terms of liver inflammation, ballooning, steatohepatitis and fibrosis, which is a key point to be considered when searching for a new therapeutic target for the treatment of metabolic steatohepatitis.
Notably, genetic structure seems to influence the variability in the prevalence of fatty liver disease among individuals of different ethnicities: the lower frequency of PNPLA3 rs738409 G variant 4 and of other genetic risk variants contributes to protecting African ancestry from fatty liver disease development and progression. 10
MASLD diagnosis
MASLD is defined as the presence of steatosis in >5% of hepatocytes according to histological analysis and its diagnosis requires the exclusion of secondary causes of steatosis and of a significant alcohol intake. 11 The first‐line tool recommended in clinical practice is conventional ultrasound, which may non‐invasively detect steatosis in patients with metabolic risk factors. Among non‐invasive tools, controlled attenuation parameter measurement by transient elastography with values above 275 dB/m is widely accepted to diagnose steatosis because of its high sensitivity. 12
In the last decade, numerous evidences have highlighted the role of common genetic variants in the development of MASLD. As cited before, much evidence confirmed the association between the PNPLA3 rs738409 SNP and an increased MASLD risk. 4 , 13 Kotronen et al. developed a MASLD liver fat score based on clinical and laboratory data and then tested the influence of the PNPLA3 rs738409 C > G SNP in prediction accuracy, which improved by less than 1% when the genetic information was added to the MASLD liver fat score (area under the receiver‐operating characteristic curve [AUROC] 0.872 +/− 0.02, 95% CI: 0.84–0.91 if PNPLA3 genotype added vs. AUROC 0.866 +/− 0.02, 95% CI: 0.83–0.90, without PNPLA3 genotype). In their multivariate logistic regression analyses PNPLA3 rs738409 G variant remained independently associated with MASLD. 14 Recent evidence by Stender and colleagues revealed that obesity may amplify the connection of the genetic variants with steatosis: for instance, in participants with body mass index (BMI) >35 kg/m2, homozygosity for the PNPLA3 rs738409 G‐allele was associated with higher median hepatic triglyceride content than in Iean individuals. 15 Consistently, we can argue that even if genetic variants can lead to liver fat development, they could be useful for the identification of patients at risk of developing liver fat when interacting with metabolic risk factors, but not for the diagnosis of already existing steatosis.
Metabolic dysfunction‐associated steatohepatitis (MASH) diagnosis
Liver biopsy still remains the gold standard to diagnose MASH, while proposed noninvasive scores (routine blood markers as alanine aminotransferase [ALT], adipokines as adiponectin, markers of cell death as cytokeratin 18 fragments, metabolic parameters as insulin resistance, ‐omics markers or combination scoring systems) failed to achieve good diagnostic accuracy. 12 In this contest, several risk scores including genetic information were developed in the recent years (Table 1). Hyysalo et al. added the PNPLA3 rs738409 SNP in a risk score based on aspartate aminotransferase (AST) and fasting insulin which predicted MASH with an AUROC of 0.774 in Finns and 0.759 in Italians. 16 Another scoring system based on SNPs in PNPLA3 (rs738409) and TM6SF2 (rs58542926) and clinical factors showed an acceptable accuracy (AUROC value of 0.787) to identify patients with MASLD, with or without diabetes, at risk of progression to MASH. 17 Moreover, it was demonstrated that the accuracy in risk stratification might improve when the genetic risk factors are added to models based on clinical and laboratory data. For instance, Paternostro and colleagues showed that the area under curves (AUCs) of the prediction of NAFLD activity score (NAS) ≥5 on liver biopsy were higher when PNPLA3 rs738409, HSB17B13 rs72613567 or their combination were added in clinical risk score (based on age, sex, BMI, diabetes and ALT), but not when TM6SF2 rs58542926 was added; still, a higher diagnostic accuracy in the prediction of advanced fibrosis was reached with the addition of genetic variants (baseline model: AUC 0.777; addition of PNPLA3 rs738409: AUC 0.789; addition of TM6SF2 rs58542926: AUC 0.786). 18 Previously, Zhou and colleagues developed the MASH ClinLipMet Score associating clinical variables (AST and fasting insulin), PNPLA3 rs738409 information and also metabolomics data (5 molecular metabolites identified by mass spectrometry‐based methods: glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, phosphoethanolamine 40:6): this score non‐invasively identified patients with MASH with an AUROC of 0.866 (95% CI, 0.820–0.913), reflecting a significantly higher accuracy than scores with only clinical information. 19 These noninvasive scores suggest a potential utility of genetic testing for the identification of MASH patients, but they were mostly developed in bariatric populations and need further validation before being considered worthy of introduction in clinical practice.
TABLE 1.
Genetic scores for the prediction of MASH.
| Reference | Score | Outcome | AUROC | Sensitivity | Specificity |
|---|---|---|---|---|---|
| Hyysalo et al. | NASH score: PNPLA3 genotype, AST and fasting insulin | Prediction of MASH | 0.774 (95% C.I.: 0.709, 0.839) in Finns; 0.759 (95% C.I.: 0.711, 0.807) in Italians | 71.6% in Finns; 39% in Italians | 73.5% in Finns; 89% in Italians |
| Koo et al. | NASH PT scores: PNPLA3 and TM6SF2 genotypes, diabetes status, HOMA‐IR, AST and hsCRP | Prediction of MASH | 0.859 (95% CI, 0.817–0.901) | 0.881 | 0.684 |
| Zhou et al. | NASH ClinLipMet score: AST, fasting insulin, PNPLA3 genotype, glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, phosphoethanolamine 40:6 | Prediction of MASH | 0.866 (95% CI, 0.820–0.913) | 85.5% | 72.1% |
| Paternostro et al. | Age, sex, BMI, diabetes, ALT (baseline model) + PNPLA3 genotype | Prediction of NAS ≥5 | 0.766 | ||
| Baseline model + HSB17B13 genotype | Prediction of NAS ≥5 | 0.766 | |||
| Baseline model + PNPLA3 and HSB17B13 genotype | Prediction of NAS ≥5 | 0.775 | |||
| Baseline model + TM6SF2 genotype | Prediction of NAS ≥5 | 0.762 | |||
| Age, sex, BMI, diabetes (baseline model for advanced fibrosis) + PNPLA3 genotype | Prediction of advanced fibrosis | 0.789 | |||
| Baseline model for advanced fibrosis + TM6SF2 genotype | Prediction of advanced fibrosis | 0.786 | |||
| Baseline model for advanced fibrosis + HSD17B13 genotype | Prediction of advanced fibrosis | 0.777 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUROC, area under the receiver‐operating characteristic curve; BMI, body mass index; MASH, metabolic dysfunction‐associated steatohepatitis; NAS, NAFLD activity score.
Fibrosis progression and cirrhosis development in MASLD
In patients with biopsy‐proven MASH, the fibrosis stage is the main prognostic factor. As reported by Taylor et colleagues, the risk of liver events is 2.6, 5.2 and 12.7 times higher in patients with F2, F3 and F4 fibrosis stages, respectively, compared to that of F0 fibrosis patients, and the severity of liver fibrosis also predicts a higher risk of occurrence of extrahepatic complication. 20 , 21 As mentioned before, liver biopsy result is still the gold standard not only for MASH diagnosis but also for staging fibrosis. However, its constrained by sampling errors, its cost and its limited reproducibility. This led to the recommendation of the use of NITs to stratify the risk of advanced fibrosis and also of developing cirrhosis and liver‐related events. 12 , 22 Specifically, FIB‐4, liver stiffness measurement (LSM) and the AGILE 3 + scores showed good accuracy in predicting the development of liver‐related events during follow‐up. 23 , 24 In this setting, the PNPLA3 rs738409 C > G variant was independently associated with fibrosis progression in an Italian biopsy‐proven cohort of MASLD patients with paired FIB‐4 and LSM (OR 1.65; 95% CI, 1.12–2.42; p = 0.01 and OR 1.90; 95% CI, 1.05–3.42; p = 0.03 respectively). 25 Consistently, Gellert‐Kristensen and colleagues made up a genetic risk score (GRS) for fatty liver disease based on PNPLA3 rs738409, TM6SF2 rs58542926 and HSD17B13 rs72613567 SNPs that conferred not just a 12‐fold higher risk of cirrhosis but even a 29‐fold higher risk of HCC among individuals from general population. 26 Along this line, it has been proved that polygenic risk scores may improve the discriminatory ability of the clinical risk test as FIB‐4: recent evidence shows that in subjects with diabetes and indeterminate FIB‐4 (1.3–2.67), the determination of the homozygous GG genotype of PNPLA3 rs738409 identifies with good accuracy MASLD patients at higher risk of developing cirrhosis during follow‐up 27 (Table 2). All in all, this evidence could support the use of genetic testing in clinical practice for stratifying the risk of liver fibrosis progression and cirrhosis development in at‐risk patients.
TABLE 2.
Genetic scores and association with and prediction of fibrosis progression/cirrhosis.
| Reference | Score | Outcome | OR (95% C.I.) | AUROC | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| Gellert–Kristensen | GRS (genetic risk score): PNPLA3 p.I148M, TM6SF2 p.E167K, and HSD17B13 rs72613567 T/TA genotypes | Risk of cirrhosis for GRS = 6 | 12 (95% CI, 7.7, 19) | Not available | Not available | |
| Chen et al. | Cirrhosis PRS (polygenic risk score): PNPLA3, TM6SF2, SERPINA1, HSD17B13, MARC1, MAFB, APOE, HMBS, TOR1B, CENPW, ARHGEF28, EFNA1 variants | Prediction of incident cirrhosis in patients with diabetes and FIB4 1.3–2.67 | 0.73 (95% CI, 0.65–0.81) in MGI; 0.65 (95% CI, 0.58–0.73) in UKBB | Not available | Not available |
Abbreviation: AUROC, area under the receiver‐operating characteristic curve.
Genetic and liver‐related outcomes
In the last few years, the ability of genetics to predict liver‐related outcomes has been a subject essential to early identify patients at higher risk of hepatic morbidity and mortality.
Several both cross‐sectional and cohort studies provided the evidence that genetics may also help to detect the development of HCC and/or liver decompensation itself and to stratify complication risk in MASLD patients (Table 3).
TABLE 3.
Genetic scores and prediction of liver‐related outcomes.
| Reference | Score | Outcome | AUROC | Sensitivity | Specificity |
|---|---|---|---|---|---|
| Donati et al. | HCC risk score: Age, sex, obesity, T2DM, severe fibrosi, number of risk alleles (PNPLA3 I148M, TM6SF2 E167K, and MBOAT7 rs641738 C > T risk variants | Prediction of HCC | 0.96 ± 0.04 | 96% | 89% |
| Bianco et al. | PRS‐HFC: PNPLA3, TM6SF2, MBOAT7 and GCKR risk alleles | Prediction of HCC | 0.64 | 43% | 80% |
| PRS‐5: PRS adjusted for the rs72613567 HSD17B13 variant | Prediction of HCC | 0.65 | 43% | 79% | |
| Pennisi et al. | GEMS score: Sex, age, diabetes, HDL, albumin, PLT and PNPLA3, TM6SF2, HSD17B13 genotype | Prediction of liver‐related events | 0.87 | Not available | Not available |
| De vincentis et al. | Clinical scores (NFS, FIB4, APRI, BARD Forns) + PRS‐HFC | Prediction of severe liver disease (SLD) in overall population | 0.683, 0.670, 0.648, 0.603, 0.735 for NFS, FIB‐4, APRI, BARD and Forns respectively | Not available | Not available |
| Clinical scores (NFS, FIB4, APRI, BARD Forns) + PRS‐HFC | Prediction of SLD in subgroup with diabetes | 0.722, 0.753, 0.742, 0.669, 0.791 for NFS, FIB‐4, APRI, BARD and Forns respectively | Not available | Not available | |
| Clinical scores (NFS, FIB4, APRI, BARD Forns) + PRS‐HFC | Prediction of SLD in subgroup with obesity | 0.721, 0.723, 0.683, 0.671, 0.771 for NFS, FIB‐4, APRI, BARD and Forns respectively | Not available | Not available | |
| Clinical scores (NFS, FIB4, APRI, BARD Forns) + PRS‐HFC | Prediction of SLD in subgroup with FLI ≥60 | 0.700, 0.717, 0.684, 0.655, 0.754 for NFS, FIB‐4, APRI, BARD and Forns respectively | Not available | Not available |
Abbreviations: AUROC, area under the receiver‐operating characteristic curve; FLI, fatty liver index; GCKR, glucokinase regulator; HDL, high‐density lipoprotein; NFS, NAFLD fibrosis score.
In a Caucasian cohort of patients with MASLD, the homozygous GG genotype of PNPLA3 rs738409 conferred a 5‐fold increased risk of developing HCC. 28 Consistent with this evidence, Grimaudo and colleagues found that PNPLA3 rs738409 variant was independently associated with a higher risk of developing liver decompensation and HCC in a large cohort of patients with MASLD, with HR of 2.10 and 2.68, respectively, even if all events occurred in F3 or cirrhotic patients. 29 Shao et al. also demonstrated that in a Japanese cohort, the OR of MASLD‐related cirrhosis complications significantly increased in the presence of the homozygous GG genotype of PNPLA3 rs738409 (OR = 3.165; 95% CI = 1.073–10.294; p = 0.046) when compared to the CC genotype. 30 Subsequently, similar evidence was obtained in a larger cohort of Japanese MASLD‐biopsy proven patients over a longer follow‐up period: the PNPLA3 rs738409 G allele predicted liver related event with an HR of ∼16. 31 A metanalysis by Singal et al. pooled evidence on the association between PNPLA3 rs738409 and HCC, showing that the PNPLA3 rs738409 G allele increased the risk of HCC in patients with MASH as an independent risk factor. 32
When looking at other common variants, recently, Eldafashi et al. confirmed the association between the PNPLA3 rs738409 and TM6SF2 rs58542926 SNPs with MASLD‐HCC, but they also suggested that their impact on the development of cancer may be related to the promotion of MASLD itself. 33 Conversely, Liu et al. demonstrated that the carriage of the C > T minor allele of TM6SF2 rs58542926 was significantly associated with the stage of fibrosis (β = 0.549 ± 0.135, 95% CI 0.285–0.813; p = 5.57 × 10⁻⁵), but no significant association was found with the onset of HCC in multivariate analysis including well known risk factors (age, sex, BMI, diabetes and cirrhosis), 34 Moreover, Donati et al. suggested that the risk MBOAT7 rs641738 T allele promotes HCC especially in non‐cirrhotic MASLD patients both in univariate (OR 2.18, 95% C.I. 1.30–3.63; p = 0.003) and multivariate analysis (OR 1.81, 95% C.I. 1.24–2.69; p = 0.002). Further, their HCC risk score including genetic risk variants identified patients with HCC with an AUROC of 0.96 ± 0.04 (96% sensitivity, 89% specificity). 35 The protective role of the loss‐of‐function T > TA variant of HSD17B13 rs72613567 on developing MASLD and advanced fibrosis was widely investigated. Interestingly, this protective role might vanish among patients with already advanced chronic liver disease (ACLD), as suggested by Ting et al. 36 This was further supported by Scheiner et al. and Gil‐Gomez later who showed that the variant was associated with hepatic decompensation and liver‐related mortality in those patients who already had portal hypertension and ACLD. 37 , 38
Genetic variants can also interact among them in modulating the risk of HCC and liver‐related complications. Longo et al. found out that carrying all the three variants (PNPLA3 rs738409, TM6SF2 rs58542926, and the rs641738 in TMC4/MBOAT7 locus) confers up to 2‐fold higher risk of developing HCC (OR, 1.73; 95% CI, 1.09–2.74; p = 0.01) even if adjusted for the presence of fibrosis. 39 Along this line, Bianco et colleagues first developed a polygenic risk score PRS‐HFC (Polygenic Risk Score—Hepatic Fat Content) combining the most common genetic variants (PNPLA3 rs738409 ‐TM6SF2 rs58542926 –GCKR rs1260326 ‐ MBOAT7 rs641738) further adjusted for HSD17B13 rs72613567 (PRS‐5) which predicted advanced fibrosis and HCC in MASLD cohort with robust statistical associations (OR of ∼12 and ∼9 respectively), even though with moderate diagnostic accuracy for HCC identification (AUROC 0.64 and 0.65 for PRS‐HFC e PRS‐5 respectively). Interestingly, this association between genetic predisposition to steatosis and HCC was also confirmed in patients without severe liver fibrosis or cirrhosis proving that liver fat may directly promote liver cancer. 40 Other risk scores including genetic variants were developed to predict liver‐related events, as GEMS (Genetic and Metabolic Staging) score based on clinical and metabolic parameters (age, sex, platelets, high‐density lipoprotein, diabetes) and on the common genetic variants. This score stratifies the risk of liver‐related events in patients with MASLD and FIB‐4 >1.3 with good accuracy (AUC 0.87 at 1, 3 and 5 years), which becomes suboptimal (AUC 0.70, 0.69 and 0.67 at 1, 3 and 5 years respectively) when applied in the general population. 41 Furthermore, De Vincentis et al. verified that combining a polygenic risk score based on PNPLA3 rs738409, TM6SF2 rs58542926, GCKR rs1260326 and MBOAT7 rs641738 (PRS‐HFC) with clinical fibrosis scores as NAFLD fibrosis score (NFS) and FIB‐4 improved the accuracy in risk stratification and in prediction of cirrhosis and liver events in the overall population and in subgroups with different baseline risk (diabetes, obesity, FLI ≥ 60). They also observed an increased incidence of advanced liver disease in individuals with intermediate or high clinical fibrosis scores and unfavorable genetic assessment (PRS‐HFC ≥0.396). 42
The impact of common genetic variants on mortality has also been investigated. Unalp‐Arida and Ruhl provided evidence on the capability of genetics to predict liver‐related mortality in their study based on data from the third U.S. National Nutrition and Health Examination Survey (NHANES III) database. In fact, they showed a direct association between liver disease mortality and PNPLA3 rs738409 G allele heterozygosity with HR, 2.9, and the G allele homozygosity with HR, 18.2 in multivariate analysis, independently from other relevant covariates. 43 Similarly, Grimaudo and colleagues found that PNPLA3 rs738409 C > G variant was independently associated with a higher risk of liver‐related death in a large cohort of patients with MASLD, with HR of 3.64. 29 This evidence was recently supported by Gellert–Kristensen and colleagues, who found that the genetic risk variants associated with hepatic steatosis were independently associated with liver‐related mortality in the general population, with the strongest association for individuals homozygous for the PNPLA3 rs738409 G‐allele (HR 2.77, 95% CI 1.77, 4.33). In addition, liver‐related mortality was higher in individuals with an increasing GRS (combining PNPLA3 rs738409, TM6SF2 rs58542926 and HSD17B13 rs72613567 risk alleles). Instead, none of these genetic risk variants were significantly associated with all‐cause, cardiovascular‐related and extrahepatic cancer‐related mortality. 44 Other studies confirmed the association of the PNPLA3 rs738409 GG genotype with an increased risk of developing cirrhosis in patients with MASLD, but no association was found with overall mortality when compared to advanced fibrosis at diagnosis: PNPLA3 rs738409 GG genotype effect on promoting disease was of more importance in patients who had no advanced fibrosis at baseline. 45
Overall the reported evidence suggests that genetic testing could help in identifying MASLD patients at higher risk of developing liver‐related events and of death probably worthy of more intensive prevention strategies and follow‐up but this is rendered more challenging by the evidence that, as expected, the predictive role of a genetic variant for advanced liver disease or hepatic events is higher when examined from the general population compared to at‐risk individuals.
Genetics and extra‐hepatic outcomes
Worthy of note is the evidence that the first common cause of death in MASLD patients is cardiovascular disease followed by extrahepatic cancers and then by liver‐related mortality. 46
The susceptible polygenic background is one of the players in the variability of MASLD phenotypes, and it seems to play a role even in the field of extra‐hepatic outcomes. In the last decade, conflicting findings on cardiovascular events in MASLD patients have come to light. In a biopsy‐proven Sicilian MASLD cohort, Petta et al. observed a higher severity of carotid plaques and intima‐media thickness (IMT) in MASLD patients <50 years carrying the PNPLA3 rs738409 GG genotype, that was confirmed in multivariate analyses and validated in a cohort with clinical or histological diagnosis of MASLD from Northern Italy. 47 These results are partially in line with the evidence from the NHANES data (1991–1994) that PNPLA3 rs738409 G‐allele was independently associated with increased cardiovascular disease mortality in the total population after adjustment for age and sex (HR 1.31, 95% CI 1.07–1.61, p = 0.011), but not in subjects with MASLD. 48
Opposite results were later found in a Japanese retrospective study on a biopsy‐proven MASLD cohort where there was no significant association between PNPLA3 rs738409 GG genotype and increased cardiovascular risk in those patients, suggesting its protective role in that contest, while the PNPLA3 rs738409 CC genotype was independently associated with the incidence of cardiovascular (CV) diseases with an HR 3.66, 95% CI = 1.63–8.35; p = 0.002. 49 Similar findings were shown in a large cohort of Chinese overweight/obese patients. 50 These conflicting results may be related to the racial differences, but further investigation is needed to clarify this issue. Finally, PNPLA3 rs738409 and TM6SF2 rs58542926 SNP were demonstrated to confer an antiatherogenic plasma lipid profile and protection against CV events, respectively, in obese subjects: in an European cross‐sectional cohort of biopsy‐proven MASH patients, Dongiovanni and colleagues confirmed that TM6SF2 rs58542926 T risk allele promoted progressive MASH but they also pointed out that this variant, by reducing secretion of very‐low‐density lipoproteins (VLDLs) from the liver, conferred a protective profile against carotid atherosclerosis (OR, 0.48; 95% CI: 0.25–0.94; p = 50.031). 51 , 52
Emerging evidence suggests a link between MASLD and chronic kidney disease (CKD), and that genetics may be involved in renal injury. 53 Mantovani et al. recently investigated the role of known MASLD‐risk alleles in renal dysfunction and their results highlighted that the PNPLA3 rs738409 G‐allele homozygosity confers a higher risk of CKD and lower eGFR levels among patients with diabetes. 54 Other evidence showed a protective role of the HSD17B13 rs72613567, an allele, which was associated with lower levels of albuminuria in MASLD patients. 55
Finally, little evidence suggests a role of the MASLD genetic background and the incidence of extra‐hepatic malignancies. For example, in a Japanese study cited previously, the PNPLA3 rs738409 GG genotype was significantly associated with a higher incidence of extrahepatic cancers in MASLD patients in univariate analysis (HR 3.64, CI 95% (1.41–9.44), p = 0.008) but not in multivariate analysis. 56 Conversely, no association was found between PNPLA3 rs738409 SNP and extrahepatic cancer‐related mortality in the US general population and MASLD patients. 49
All in all the presented data suggest a potential role of at risk variants for MASLD/MASH in modulating the risk of extrahepatic complications even if with contrasting results, probably affected by ethnicity and clinical setting, and consequently worthy of further investigations.
Genetics and MASLD/MASH treatment
Given its role in modulating lipid metabolism, PNPLA3 genotype has been rationally proposed as potentially affecting treatment response in patients with MASLD, but discrepancies in treatment response were observed.
Shen et al. conducted a post‐hoc analysis of a randomized control trial on lifestyle modification program in MASLD patients and their results suggest that the PNPLA3 rs738409 G allele confers a higher susceptibility to the effects of lifestyle modification, resulting to be independently associated with intra‐hepatic triglyceride content reduction: reduction of intrahepatic triglyceride content in patients with CC, CG, and GG genotypes was of 3.7 ± 5.2% (p = 0.003), 6.5 ± 3.6% (p < 0.001), and 11.3 ± 8.8% < (p < 0.001), respectively. 48 Similarly, patients homozygous for the PNPLA3 rs738409 G allele experienced a significantly greater reduction of liver fat content compared to carriers of the PNPLA3 rs738409 CC genotype after a 6‐day trial of hypocaloric low‐carbohydrate diet. 57
Nonetheless, contrasting findings suggesting that the PNPLA3 rs738409 SNP may predispose MASLD patients to a poor treatment response were also observed. The C allele of PNPLA3 rs738409 was associated with a greater reduction of body weight compared to the G allele in a cohort of Japanese patients with MASLD after 1 year of diet therapy, while the G allele was significantly associated with a greater reduction of LSM. 58 In a study evaluating the effects of a nutraceutical compound (sylimarin + vitamin E) in MASLD patients, the PNPLA3 rs738409 GG genotype group did not respond to the treatment in comparison to wild type patients who exhibited a statistically significant improvement in laboratory parameters (e.g., reduction of transaminases). 59 Moreover, the genetic background has been proved to influence the response to GLP1RA, which demonstrated great efficacy in MASLD treatment. Chen et al. showed that exenatide reduced lipid deposition in hepatocytes less significantly in PNPLA3 rs738409 GG genotype carriers than in the CC genotype and similar findings were observed in a small group of patients with diabetes mellitus treated with exenatide: carriers of the PNPLA3 rs738409 G allele had no reduction in liver fat content. 60
All the evidence of genetics affecting MASLD/MASH prognosis makes it a promising approach for the development of precision medicine.
The accumulation of PNPLA3 rs738409 I148M variant protein on lipid droplets is a required factor for a pathological phenotype in hepatocytes. This allows us to assume that silencing of PNPLA3 rs738409 could recover lipid metabolism in hepatocytes and improve liver damage. Linden et al. investigated this field using an antisense oligonucleotide (ASO) targeting the PNPLA3 gene in a model of mice fed steatogenic diets and engineered to overexpress the PNPLA3 rs738409 SNP: that PNPLA3 silencing led to a reduction in steatosis and fibrosis accumulation especially in animals homozygous for the mutated variant. 61 Consistently specific target therapy based on PNPLA3 ASO in MASH patients carrying the PNPLA3 rs738409 G variant is under investigation in phase IIb clinical trials. 62
An alternative potential targeting approach is to mimic the protective effect of the loss‐of‐function of the HSD17B13 rs72613567 SNP. In a recent phase I‐II randomized controlled trial, Mak et al. used RNA interference which resulted to be able to selectively reduce messenger RNA and protein levels of hepatic HSD17B13 rs72613567, leading to a reduction of transaminases in healthy volunteers and patients with confirmed or suspected MASH. 63 Consequently, phase I clinical trials are under development to assess knockdown of hepatic HSD17B13 mRNA. 64
These studies represent a proof of concept toward a precision medicine strategy for a personalized treatment of patients with MASLD/MASH based on their genetic background.
Genetic versus “omics” approaches to predict liver‐related outcomes
The recent development of different “omics” approaches allowed the pursuit of deeper knowledge on the pathophysiology of MASLD and the subsequent development of diagnostic and prognostic tools as well as the identification of new therapeutic targets. Even if this topic is outside the primary aim of this review, we reported as an example some preliminary interesting data that could be complementary to genetic profiling for the identification of at‐risk patients.
Several ongoing research have been focusing on the role of transcriptomic signatures, which include coding and non‐coding RNA, in affecting transcriptional activity. The upregulation of the thrombospondin 2 gene (coding RNA) in patients with steatosis has been linked with the severity of the disease and it showed a higher diagnostic accuracy for metabolic steatohepatitis compared to other scores (AUROC 0.96 vs. 0.88 and 0.84 for NFS and FIB‐4 respectively). 65
Among upregulated mRNAs, IL32 has been identified as a potential biomarker for MASH and advanced fibrosis. Baselli et al. proposed a diagnostic model of MASLD with IL32‐ALT‐AST that achieved a higher diagnostic accuracy in identifying MASLD compared with aminotransferases alone (AUROC 0.92 vs. 0.81). 66 Alterations in microRNAs (belonging to non‐coding RNA) implicated in metabolic pathways have been discovered to be involved in MASLD development and progression. Among the most widely investigated miRNAs, miR‐122 and miR‐192 showed a robust upregulation in patients with hepatic steatosis and miR‐122 and miR‐34 a have been demonstrated to have moderate diagnostic accuracy in distinguishing MASLD from healthy individuals (AUC 0.82) and MASH from MASLD (AUC 0.78), respectively. 67 However, further studies are needed to better define their potential diagnostic and prognostic value.
The proteome, due to post‐translational modifications and different splicing, is more dynamic than the genome or transcriptome, but is closer to the final phenotype, which renders it a preferred tool for MASLD identification and stratification. 68
Several studies identified proteomic signatures as potential biomarkers, even with high diagnostic accuracy. Wood et al. identified eight proteins (as aminoacylase one and sex hormone binding globulin) associated with steatosis in multivariate analysis (AUC 0.86), with a higher diagnostic accuracy when PNPLA3 rs738409 information and phenotypic variables were added (AUC 0.93). 69 Additionally, a proteomic‐based classifier, proposed by Luo et al., was able to differentiate patients with early (F0–F2) and advanced (F3–F4) fibrosis with an AUC of 0.83. 70
An improvement in understanding metabolic pathways implicated in the development and progression of MASLD has been offered by the study of the metabolome, which includes metabolic products such as cellular lipids. Thus, the lipidomics represents a major subfield of metabolomics able to capture the disease severity. Mayo et al. assessed two panels of triglycerides, which could discriminate the first between MASLD and healthy liver and the second between MASLD and MASH with good accuracy (AUC 0.88 and 0.79, respectively). 71 Similarly, other lipid metabolites were later identified to be able to differentiate MASLD from healthy controls and MASLD from MASH with high diagnostic accuracy, and with different metabolomics profiles between obese and non‐obese individuals ( 72 , 73 ).
Among the “omics”, metagenomics represents a recent field that refers to the study of the gut microbiome and its metabolites, which may play a potential pathogenetic role in the development and progression of MASLD. Emerging evidence supports their utility as biomarkers in the diagnosis and prognosis of the disease. Among all, a metagenomic diagnostic model proposed by Loomba et al., including 37 microbial species in combination with anthropometric features (age, BMI), was able to detect advanced fibrosis in a cohort of biopsy‐proven MASLD patients with high diagnostic accuracy (AUC 0.94) ( 74 ).
All in all these data support further research in the setting of “omics” as a diagnostic/prognostic tool in MASLD, but comprehensive and validated analyses considering together markers arising from different “omics” are necessary to go toward a personalized‐medicine approach. As an example, Zhou et al. combining genetic polymorphisms with lipidomics and metabolomics data improved the diagnostic accuracy compared to scores with genetic or clinical information alone. 19
CONCLUSION
In the recent years, our understanding of key genes in MASLD and their underlying pathophysiological mechanisms have allowed the development of several tools to better identify at‐risk patients who deserve proper surveillance. Nevertheless, their application in everyday clinical practice remains challenging and their inclusion in decisional algorithms to facilitate the clinician decision on management and treatment of MASLD could benefit from artificial intelligence methodology. Moreover, in the era of precision medicine, target therapies under development that can selectively silence genetic variants implicated in the promotion and progression of MASLD appear to be a promising near‐term reality, and we are waiting for ongoing industrial development and clinical trials.
AUTHOR CONTRIBUTIONS
Adele Tulone, Grazia Pennisi, Carlo Ciccioli, Giuseppe Infantino, Claudia La Mantia, Roberto Cannella, Francesco Mercurio, and Salvatore Petta had full control of the study design, data analysis, interpretation, and preparation of the article. All authors were involved in planning the analysis and drafting the article. The final draft of the article was approved by all the authors.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
ACKNOWLEDGMENT
The research leading to these results has received funding from MIUR under PNRR M4C2I1.3 Heal Italia project PE00000019 CUP B73C22001250006 to Prof. Salvatore Petta. The project is supported by the Italian PNRR‐MAD‐2022‐12375656 project. The project is supported by PRIN 2022 2022L273C9. The project is supported by RF‐2021‐12372399.
Tulone A, Pennisi G, Ciccioli C, Infantino G, La Mantia C, Cannella R, et al. Are we ready for genetic testing in metabolic dysfunction‐associated steatotic liver disease? United European Gastroenterol J. 2024;12(5):638–48. 10.1002/ueg2.12556
Adele Tulone and Grazia Pennisi contributed to the study in equal parts.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A, et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin Gastroenterol Hepatol. 2019;17(4):748.e3–755.e3. 10.1016/j.cgh.2018.05.057 [DOI] [PubMed] [Google Scholar]
- 2. Bugianesi E, Petta S. MASLD/MASH. J Hepatol. 2022;77(2):549–550. 10.1016/j.jhep.2022.02.006 [DOI] [PubMed] [Google Scholar]
- 3. Trépo E, Valenti L. Update on MASLD genetics: from new variants to the clinic. J Hepatol. 2020;72(6):1196–1209. 10.1016/j.jhep.2020.02.020 [DOI] [PubMed] [Google Scholar]
- 4. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. 10.1038/ng.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luukkonen PK, Porthan K, Ahlholm N, Rosqvist F, Dufour S, Zhang XM, et al. The PNPLA3 I148M variant increases ketogenesis and decreases hepatic de novo lipogenesis and mitochondrial function in humans. Cell Metab. 2023;35(11):1887–1896.e5. 10.1016/j.cmet.2023.10.008 [DOI] [PubMed] [Google Scholar]
- 6. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome‐wide association study of non‐alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol. 2020;73(3):505–515. 10.1016/j.jhep.2020.04.003 [DOI] [PubMed] [Google Scholar]
- 7. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–356. 10.1038/ng.2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17β‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2014;111(31):11437–11442. 10.1073/pnas.1410741111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma Y, Belyaeva OV, Brown PM, Fujita K, Valles K, Karki S, et al. 17‐Beta Hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology. 2019;69(4):1504–1519. 10.1002/hep.30350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romeo S, Valenti L. African genetic ancestry and protection against fatty liver disease. Liver Int. 2022;42(10):2122–2123. 10.1111/liv.15364 [DOI] [PubMed] [Google Scholar]
- 11. European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO) . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64:1388–1402. [DOI] [PubMed] [Google Scholar]
- 12. European Association for the Study of the Liver . Electronic address: easloffice@easloffice.eu; Clinical Practice Guideline Panel; Chair; EASL Governing Board representative; Panel members: EASL Clinical Practice Guidelines on non‐invasive tests for evaluation of liver disease severity and prognosis ‐ 2021 update. J Hepatol. 2021;75:659–689. [DOI] [PubMed] [Google Scholar]
- 13. Walker RW, Belbin GM, Sorokin EP, Van Vleck T, Wojcik GL, Moscati A, et al. A common variant in PNPLA3 is associated with age at diagnosis of MASLD in patients from a multi‐ethnic biobank. J Hepatol. 2020;72(6):1070–1081. 10.1016/j.jhep.2020.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, et al. Prediction of non‐alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. 2009;137(3):865–872. 10.1053/j.gastro.2009.06.005 [DOI] [PubMed] [Google Scholar]
- 15. Stender S, Kozlitina J, Nordestgaard BG, Tybjærg‐Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–847. 10.1038/ng.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hyysalo J, Männistö VT, Zhou Y, Arola J, Kärjä V, Leivonen M, et al. A population‐based study on the prevalence of MASH using scores validated against liver histology. J Hepatol. 2014;60(4):839–846. 10.1016/j.jhep.2013.12.009 [DOI] [PubMed] [Google Scholar]
- 17. Koo BK, Joo SK, Kim D, Lee S, Bae JM, Park JH, et al. Development and validation of a scoring system, based on genetic and clinical factors, to determine risk of steatohepatitis in Asian patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2020;18(11):2592.e10–2599.e10. 10.1016/j.cgh.2020.02.011 [DOI] [PubMed] [Google Scholar]
- 18. Paternostro R, Staufer K, Traussnigg S, Stättermayer AF, Halilbasic E, Keritam O, et al. Combined effects of PNPLA3, TM6SF2 and HSD17B13 variants on severity of biopsy‐proven non‐alcoholic fatty liver disease. Hepatol Int. 2021;15(4):922–933. 10.1007/s12072-021-10200-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Y, Orešič M, Leivonen M, Gopalacharyulu P, Hyysalo J, Arola J, et al. Noninvasive detection of nonalcoholic steatohepatitis using clinical markers and circulating levels of lipids and metabolites. Clin Gastroenterol Hepatol. 2016;14(10):1463.e6–1472.e6. 10.1016/j.cgh.2016.05.046 [DOI] [PubMed] [Google Scholar]
- 20. Taylor RS, Taylor RJ, Bayliss S, Hagström H, Nasr P, Schattenberg JM, et al. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: a systematic review and meta‐analysis. Gastroenterology. 2020;158(6):1611.e12–1625.e12. 10.1053/j.gastro.2020.01.043 [DOI] [PubMed] [Google Scholar]
- 21. Pennisi G, Enea M, Romero‐Gomez M, Viganò M, Bugianesi E, Wong VW, et al. Liver‐related and extrahepatic events in patients with non‐alcoholic fatty liver disease: a retrospective competing risks analysis. Aliment Pharmacol Ther. 2022;55(5):604–615. 10.1111/apt.16763 [DOI] [PubMed] [Google Scholar]
- 22. Martin K, Hatab A, Athwal VS, Jokl E, Piper Hanley K. Genetic contribution to non‐alcoholic fatty liver disease and prognostic implications. Curr Diab Rep. 2021;21(3):8. 10.1007/s11892-021-01377-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pennisi G, Enea M, Pandolfo A, Celsa C, Antonucci M, Ciccioli C, et al. AGILE 3+ score for the diagnosis of advanced fibrosis and for predicting liver‐related events in MASLD. Clin Gastroenterol Hepatol. 2023;21:1293.e5–1302.e5. [DOI] [PubMed] [Google Scholar]
- 24. Younes R, Caviglia GP, Govaere O, Rosso C, Armandi A, Sanavia T, et al. Long‐term outcomes and predictive ability of non‐invasive scoring systems in patients with non‐alcoholic fatty liver disease. J Hepatol. 2021;75(4):786–794. 10.1016/j.jhep.2021.05.008 [DOI] [PubMed] [Google Scholar]
- 25. Pennisi G, Pipitone RM, Cammà C, Di Marco V, Di Martino V, Spatola F, et al. PNPLA3 rs738409 C>G variant predicts fibrosis progression by noninvasive tools in nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2021;19(9):1979–1981. 10.1016/j.cgh.2020.09.009 [DOI] [PubMed] [Google Scholar]
- 26. Gellert‐Kristensen H, Richardson TG, Davey Smith G, Nordestgaard BG, Tybjærg‐Hansen A, Stender S. Combined effect of PNPLA3, TM6SF2, and HSD17B13 variants on risk of cirrhosis and hepatocellular carcinoma in the general population. Hepatology. 2020;72(3):845–856. 10.1002/hep.31238 [DOI] [PubMed] [Google Scholar]
- 27. Chen VL, Oliveri A, Miller MJ, Wijarnpreecha K, Du X, Chen Y, et al. PNPLA3 genotype and diabetes identify patients with nonalcoholic fatty liver disease at high risk of incident cirrhosis. Gastroenterology. 2023;164(6):966.e17–977.e17. 10.1053/j.gastro.2023.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu YL, Patman GL, Leathart JB, Piguet AC, Burt A, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non‐alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61(1):75–81. 10.1016/j.jhep.2014.02.030 [DOI] [PubMed] [Google Scholar]
- 29. Grimaudo S, Pipitone RM, Pennisi G, Celsa C, Cammà C, Di Marco V, et al. Association between PNPLA3 rs738409 C>G variant and liver‐related outcomes in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2020;18(4):935.e3–944.e3. 10.1016/j.cgh.2019.08.011 [DOI] [PubMed] [Google Scholar]
- 30. Shao X, Uojima H, Arai T, Ogawa Y, Setsu T, Atsukawa M, et al. The risk of cirrhosis and its complications based on PNPLA3 rs738409 G allele frequency. Dig Dis. 2022;40(5):625–634. 10.1159/000521062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seko Y, Yamaguchi K, Shima T, Iwaki M, Takahashi H, Kawanaka M, et al. The greater impact of PNPLA3 polymorphism on liver‐related events in Japanese non‐alcoholic fatty liver disease patients: a multicentre cohort study. Liver Int. 2023;43(10):2210–2219. 10.1111/liv.15678 [DOI] [PubMed] [Google Scholar]
- 32. Singal A, Manjunath H, Yopp A, Beg MS, Marrero JA, Gopal P, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta‐analysis. Am J Gastroenterol. 2014;109(3):325–334. 10.1038/ajg.2013.476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eldafashi N, Darlay R, Shukla R, McCain MV, Watson R, Liu YL, et al. A PDCD1 role in the genetic predisposition to MASLD‐HCC? Cancers (Basel). 2021;13(6):1412. 10.3390/cancers13061412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JBS, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non‐alcoholic fatty liver disease. Nat Commun. 2014;5(1):4309. 10.1038/ncomms5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non‐cirrhotic individuals. Sci Rep. 2017;7(1):4492. 10.1038/s41598-017-04991-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ting YW, Kong AS, Zain SM, Chan WK, Tan HL, Mohamed Z, et al. Loss‐of‐function HSD17B13 variants, non‐alcoholic steatohepatitis and adverse liver outcomes: results from a multi‐ethnic Asian cohort. Clin Mol Hepatol. 2021;27(3):486–498. 10.3350/cmh.2020.0162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scheiner B, Stättermayer AF, Schwabl P, Bucsics T, Paternostro R, Bauer D, et al. Impact of HSD17B13 rs72613567 genotype on hepatic decompensation and mortality in patients with portal hypertension. Liver Int. 2020;40(2):393–404. 10.1111/liv.14304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gil‐Gómez A, Rojas Á, García‐Lozano MR, Muñoz‐Hernández R, Gallego‐Durán R, Maya‐Miles D, et al. Impact of a loss‐of‐function variant in HSD17B13 on hepatic decompensation and mortality in cirrhotic patients. Int J Mol Sci. 2022;23(23):11840. 10.3390/ijms231911840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Longo M, Meroni M, Paolini E, Erconi V, Carli F, Fortunato F, et al. TM6SF2/PNPLA3/MBOAT7 loss‐of‐function genetic variants impact on MASLD development and progression both in patients and in in vitro models. Cell Mol Gastroenterol Hepatol. 2022;13(3):759–788. 10.1016/j.jcmgh.2021.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bianco C, Jamialahmadi O, Pelusi S, Baselli G, Dongiovanni P, Zanoni I, et al. Non‐invasive stratification of hepatocellular carcinoma risk in non‐alcoholic fatty liver using polygenic risk scores. J Hepatol. 2021;74(4):775–782. 10.1016/j.jhep.2020.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pennisi G, Pipitone RM, Enea M, De Vincentis A, Battaglia S, Di Marco V, et al. A genetic and metabolic staging system for predicting the outcome of nonalcoholic fatty liver disease. Hepatol Commun. 2022;6(5):1032–1044. 10.1002/hep4.1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Vincentis A, Tavaglione F, Jamialahmadi O, Picardi A, Antonelli Incalzi R, Valenti L, et al. A polygenic risk score to refine risk stratification and prediction for severe liver disease by clinical fibrosis scores. Clin Gastroenterol Hepatol. 2022;20(3):658–673. 10.1016/j.cgh.2021.05.056 [DOI] [PubMed] [Google Scholar]
- 43. Unalp‐Arida A, Ruhl CE. Patatin‐like phospholipase domain‐containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the U.S. Population. Hepatology. 2020;71:820–834. 10.1002/hep.31032 [DOI] [PubMed] [Google Scholar]
- 44. Gellert‐Kristensen H, Tybjaerg‐Hansen A, Nordestgaard BG, Ghouse J, Fuchs A, Kühl JT, et al. Genetic risk of fatty liver disease and mortality in the general population: a Mendelian randomization study. Liver Int. 2023;43(9):1955–1965. 10.1111/liv.15629 [DOI] [PubMed] [Google Scholar]
- 45. Holmer M, Ekstedt M, Nasr P, Zenlander R, Wester A, Tavaglione F, et al. Effect of common genetic variants on the risk of cirrhosis in non‐alcoholic fatty liver disease during 20 years of follow‐up. Liver Int. 2022;42(12):2769–2780. 10.1111/liv.15438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Angulo P. The natural history of MASLD. In: Farrell G, McCulloch AJ, Day C, Hoboken NJ, editors. Non‐alcoholic fatty liver disease: a practical guide: Wiley Blackwell Press; 2013. p. 37–45. [Google Scholar]
- 47. Petta S, Valenti L, Marchesini G, Di Marco V, Licata A, Cammà C, et al. PNPLA3 GG genotype and carotid atherosclerosis in patients with non‐alcoholic fatty liver disease. PLoS One. 2013;8(9):e74089. 10.1371/journal.pone.0074089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen J, Wong GL, Chan HL, Chan RS, Chan H, Chu WC, et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2015;30:139–146. [DOI] [PubMed] [Google Scholar]
- 49. Wijarnpreecha K, Scribani M, Raymond P, Harnois DM, Keaveny AP, Ahmed A, et al. PNPLA3 gene polymorphism and overall and cardiovascular mortality in the United States. J Gastroenterol Hepatol. 2020;35:1789–1794. [DOI] [PubMed] [Google Scholar]
- 50. Xia M, Ma S, Huang Q, Zeng H, Ge J, Xu W, et al. MASLD‐related gene polymorphisms and all‐cause and cause‐specific mortality in an Asian population: the Shanghai Changfeng Study. Aliment Pharmacol Ther. 2022;55:705–721. [DOI] [PubMed] [Google Scholar]
- 51. Borén J, Adiels M, Björnson E, Matikainen N, Soderlund S, Ramo J, et al. Effects of PNPLA3 I148M on hepatic lipid and very‐low‐density lipoprotein metabolism in humans. J Intern Med. 2022;291:218–223. [DOI] [PubMed] [Google Scholar]
- 52. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506–514. [DOI] [PubMed] [Google Scholar]
- 53. Byrne CD, Targher G. MASLD as a driver of chronic kidney disease. J Hepatol. 2020;72:785–801. [DOI] [PubMed] [Google Scholar]
- 54. Mantovani A, Taliento A, Zusi C, Baselli G, Prati D, Granata S, et al. PNPLA3 I148M gene variant and chronic kidney disease in type 2 diabetic patients with MASLD: clinical and experimental findings. Liver Int. 2020;40:1130–1141. [DOI] [PubMed] [Google Scholar]
- 55. Sun DQ, Wang TY, Zheng KI, Zhang H, Wang X, Targher G, et al. The HSD17B13 rs72613567 variant is associated with lower levels of albuminuria in patients with biopsy‐proven nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2021;31:1822–1831. [DOI] [PubMed] [Google Scholar]
- 56. Akuta N, Kawamura Y, Arase Y, Saitoh S, Fujiyama S, Sezaki H, et al. PNPLA3 genotype and fibrosis‐4 index predict cardiovascular diseases of Japanese patients with histopathologically‐confirmed MASLD. BMC Gastroenterol. 2021;21:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sevastianova K, Kotronen A, Gastaldelli A, Perttila J, Hakkarainen A, Lundbom J, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss‐induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94(1):104–111. 10.3945/ajcn.111.012369 [DOI] [PubMed] [Google Scholar]
- 58. Seko Y, Yamaguchi K, Tochiki N, Yano K, Takahashi A, Okishio S, et al. The effect of genetic polymorphism in response to body weight reduction in Japanese patients with nonalcoholic fatty liver disease. Genes. 2021;12(5):628. 10.3390/genes12050628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Aller R, Laserna C, Rojo MÁ, Mora N, García Sánchez C, Pina M, et al. Role of the PNPLA3 polymorphism rs738409 on silymarin + vitamin E response in subjects with non‐alcoholic fatty liver disease. Rev Esp Enferm Dig. 2018;110(10):634–640. 10.17235/reed.2018.5602/2018 [DOI] [PubMed] [Google Scholar]
- 60. Chen Y, Yan X, Xu X, Yuan S, Xu F, Liang H. PNPLA3 I148M is involved in the variability in anti‐NAFLD response to exenatide. Endocrine. 2020;70(3):517–525. 10.1007/s12020-020-02470-7 [DOI] [PubMed] [Google Scholar]
- 61. Lindén D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andréasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock‐in mice. Mol Metab. 2019;22:49–61. 10.1016/j.molmet.2019.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. A study to evaluate AZD2693 in participants who are carriers of the PNPLA3 148M risk allele with non‐cirrhotic non‐alcoholic steatohepatitis with fibrosis (FORTUNA). ClinicalTrials.gov Identifier: NCT05809934; [Google Scholar]
- 63. Mak LY, Gane E, Schwabe C, Yoon KT, Heo J, Scott R, et al. A phase I/II study of ARO‐HSD, an RNA interference therapeutic, for the treatment of non‐alcoholic steatohepatitis. J Hepatol. 2023;78(4):684–692. 10.1016/j.jhep.2022.11.025 [DOI] [PubMed] [Google Scholar]
- 64. Knockdown of HSD17B13 mRNA, pharmacokinetics, safety, and tolerability, of AZD7503 in non‐alcoholic fatty liver disease. ClinicalTrials.gov Identifier: NCT05560607; [Google Scholar]
- 65. Kozumi K, Kodama T, Murai H, Sakane S, Govaere O, Cockell S, et al. Transcriptomics identify thrombospondin‐2 as a biomarker for NASH and advanced liver fibrosis. Hepatology. 2021;74(5):2452–2466. 10.1002/hep.31995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Baselli GA, Dongiovanni P, Rametta R, Meroni M, Pelusi S, Maggioni M, et al. Liver transcriptomics highlights interleukin‐32 as novel NAFLD‐related cytokine and candidate biomarker. Gut. 2020;69(10):1855–1866. 10.1136/gutjnl-2019-319226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu CH, Ampuero J, Gil‐Gomez A, Montero‐Vallejo R, Rojas A, Munoz‐Hernandez R, et al. miRNAs in patients with non‐alcoholic fatty liver disease: a systematic review and meta‐analysis. J Hepatol. 2018;69(6):1335–1348. 10.1016/j.jhep.2018.08.008 [DOI] [PubMed] [Google Scholar]
- 68. Perakakis N, Yazdani A, Karniadakis GE, Mantzoros C. Omics, big data and machine learning as tools to propel understanding of biological mechanisms and to discover novel diagnostics and therapeutics. Metabolism. 2018;87:A1–A9. 10.1016/j.metabol.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wood GC, Chu X, Argyropoulos G, Benotti P, Rolston D, Mirshahi T, et al. A multi‐component classifier for nonalcoholic fatty liver disease (NAFLD) based on genomic, proteomic, and phenomic data domains. Sci Rep. 2017;7(1):43238. 10.1038/srep43238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Luo Y, Wadhawan S, Greenfield A, Decato BE, Oseini AM, Collen R, et al. SOMAscan proteomics identifies serum biomarkers associated with liver fibrosis in patients with NASH. Hepatol Commun. 2021;5(5):760–773. 10.1002/hep4.1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mayo R, Crespo J, Martínez‐Arranz I, Banales JM, Arias M, Mincholé I, et al. Metabolomic‐based noninvasive serum test to diagnose nonalcoholic steatohepatitis: results from discovery and validation cohorts. Hepatol Commun. 2018;2(7):807–820. 10.1002/hep4.1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jung Y, Lee MK, Puri P, Koo BK, Joo SK, Jang SY, et al. Circulating lipidomic alterations in obese and non‐obese subjects with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther. 2020;52(10):1603–1614. 10.1111/apt.16066 [DOI] [PubMed] [Google Scholar]
- 73. Masarone M, Troisi J, Aglitti A, Torre P, Colucci A, Dallio M, et al. Untargeted metabolomics as a diagnostic tool in NAFLD: discrimination of steatosis, steatohepatitis and cirrhosis. Metabolomics. 2021;17(2):12. 10.1007/s11306-020-01756-1 [DOI] [PubMed] [Google Scholar]
- 74. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome‐based metagenomic signature for non‐invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cel Metab. 2017;25(5):1054.e5–1062.e5. 10.1016/j.cmet.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.