

Abstract
Esters of kynurenic acid, a known neuroprotective agent were reacted with cyclic amino acids to yield novel alkoxymethylated products under optimized reaction conditions. The importance of amino acid based (primary, secondary, biogenic and synthetic) organic additives was proven by the conduction of numerous test reactions. Thoroughly extended investigations, directly focusing on amino acid catalysis, which is an emerging and up-to-date field of catalysis and green chemical processes, have been conducted. The mechanism of the alkoxymethylation reaction was proposed and later the findings supported the hypothesis of the first retro-Mannich step (formation of the ortho-quinone methide intermediate) and subsequent formation of the alkoxymethylated derivatives. As a preparative result, two novel kynurenic acid derivatives bearing an alkoxymethyl moiety and two additional derivatives having amino acid residues at the site C-3 were synthesized, thus setting the scope and limitations of the modified Mannich reaction of kynurenic acid derivatives using amino acid nucleophiles. The mechanistic investigations highlighted the significant physicochemical effects of used nucleophiles on the amino-acid driven one-pot retro-Mannich initiated alkoxylation of kynurenic acid.
Keywords: Kynurenic acid, Amino acid catalysis, Mannich reaction, Alkoxyalkylation
1. Introduction
Kynurenic acid (KYNA) (1) is an endogenous quinoline derivative, biosynthesized from l-triptophane via the kynurenine pathway. The compound is of great importance due to its neuroprotective property as aphysiological levels are measured in a broad range of neurological disorders such as Parkinson and Huntington disease, epilepsy, and migraine [[1], [2], [3], [4], [5], [6], [7], [8], [9]].
Recent studies focused on the modification of kynurenic acid in order to enhance its penetrability through the blood–brain barrier. Amidation is a versatile means of structural fine tuning to form salts via substitution with diamines yielding compounds (2a–b) bearing tertiary amino moieties [[10], [11], [12]]. Another major method of structural fine tuning is the modified Mannich reaction on the analogy of 1-naphthol. In the presence of formaldehyde and a secondary amine, numerous C-3 aminoalkylated compounds were synthesized (3a–c) [[12], [13], [14], [15], [16]]. Compounds with both amide and aminoalkyl moieties were also successfully prepared (4a–b) (Fig. 1.) [11].
Fig. 1.

Kynurenic acid and its chemically fine-tuned derivatives.
Amino acids are in focus regarding drug research due to their zwitterionic characteristics. Derivatives of widely known compounds, such as zidovudine [17], camptothecine [18], melphalan [19], and docetaxel [20] have been synthesized [21] (Fig. 2.)
Fig. 2.

Derivatives of zidovudine (5a–d), camptothecine (6a–d), melphalan (7), and docetaxel (8a–d) incorporating amino acid moieties.
The Mannich reaction is one of the known procedures to incorporate amino acid residues in compounds [[22], [23], [24], [25], [26]]. Performing the modified Mannich reaction, using kynurenic acid ethyl ester (9), paraformaldehyde, and l-proline (10) as an N-nucleophile, was attempted to synthesize a novel, zwitterionic kynurenic acid–amino acid hybrid (11).
2. Results
In our preliminary research, the reaction of compound 9 with paraformaldehyde and l-proline 1.0 equiv. each was conducted in absolute ethanol at 80 °C in a pressure-resistant vessel. The solvent was chosen on the basis of the ability of dissolving each reactant. After reaching a maximal conversion and then purification of the reaction mixture, the analysis of the product proved that the amino acid moiety is not present in the molecule. Instead, an unexpected product (12) bearing an ethoxymethylene moiety at the C-3 position was formed. Solvent ethanol acted as an O-nucleophile under the examined conditions, thus yielding an ether-type compound (Scheme 1.).
Scheme 1.

Modified Mannich reaction of 9 with l-proline and paraformaldehyde.
Product 12 attracted our attention, since it is a novel C-3 modified kynurenic acid derivative, able to broaden the spectrum of the possible neuroactive KYNA analogues.
In the literature a wide palette of alkoxyalkylation reactions of aromatic compounds bearing active hydrogen were published [27,28]. Reactions were carried out using catalysts such as boron- or phosphorous-modified HZSM-5 [29], ZBM-10 [30], HCl/HOAc/H2O [31] or HCl/MeOH [32] systems or bis(trifluoromethanesulfonyl)imide [33]. Results of alkoxyalkylations via the usage of N-nucleophiles were disclosed starting with (−)-cannabidiol and (−)-cannabigerol [34] as well as resorcinarenes [35]. Iminodiacetic acid, an amino diacid was also applied as catalyst for alkoxyalkylation reactions of resorcinarene derivatives [36,37].
Having results of the preliminary reaction at hand, test reactions were conducted using common strong or weak acid or base additives. NaOEt, Et3N, acetic acid or p-toluenesulfonic acid (1.0 equiv.) and 1.0 equiv. paraformaldehyde in abs. ethanol was added to 12. Monitoring the reaction after 60 h indicated no conversion. Similar observation was made in microwave-assisted reactions at 100 and 120 °C temperatures. These findings led to the conclusion that for the formation of compound 12 a secondary N-nucleophile is necessary.
In the next step, 9 was reacted with 1.0 equiv. paraformaldehyde and 1.0 equiv. cyclic amines (13–15) in abs. ethanol in a pressure-resistant vessel for 60 h to reach maximum conversion [11]. Upon monitoring the reaction, crude NMR spectra and TLC-analysis showed that using pyrrolidine (13) and piperidine (14), no trace of 12 was observed. The only reaction products were aminoalkylated derivatives (16, 17). When morpholine (15), a significantly weaker base containing an O-heteroatom was used, ethoxymethylated compound 12 was detectable along with aminoalkylated product 18 (Scheme 2., Table 1.).
Scheme 2.

Reaction of 9 with cyclic amines 13–15 or cyclic ammonium acetates 19–21 and paraformaldehyde in ethanol.
Table 1.
Maximal conversion per cents measured in the reaction of 9 with cyclic amines 13–15 and paraformaldehyde in ethanol.
| Used nucleophile |
Maximal conversion |
|||
|---|---|---|---|---|
| # | pKa | Mannich base | Compound 12 | |
| 13 | 11.00 | 16 | 75 %a | – |
| 14 | 11.00 | 17 | 98 %a | – |
| 15 | 8.51 | 18 | 92 %a | 4 %b |
Measured in 22.5-h reactions at plateau.
Measured in 8-h reaction (12 decomposed upon longer treatment).
Based on this information, a series of reactions was conducted using acetate salts of 13–15 as N-nucleophiles (19–21). Compounds 19–21 are able to mimic the corresponding amino acids on the grounds of acidity as pyrrolidine acetate mimicked l-proline used previously. Compound 9 was reacted with 1.0 equiv. paraformaldehyde and acetates (19–21) in abs. ethanol until no change was detectable in conversion (Scheme 2.). Both crude NMR spectra and TLC-analysis showed that the conversion per cents of Mannich bases 16–18 dropped significantly in each reaction and compound 12 was detectable (Table 2.).
Table 2.
Maximal conversion per cents in the reaction of 9 with cyclic ammonium acetates 19–21 and paraformaldehyde in ethanol.
| Used nucleophile |
Maximal conversion (at reaction time) |
||||
|---|---|---|---|---|---|
| # | pKa | Mannich bases | Compound 12 | ||
| 19 | HOAc: 4.54 | 13: 11.00 | 16 | 56 % (28 h) | 3 % (1–3 h) |
| 20 | 14: 11.00 | 17 | 34 % (20 h) | 5 % (1–5 h) | |
| 21 | 15: 8.51 | 18 | 60 % (12 h) | 20 % (9 h) | |
Full conversion per cents of 12 in the three test reactions were in the order of pyrrolidine acetate < piperidine acetate < morpholine acetate. These findings can be explained by either the electron-withdrawing ability of the oxygen atom in morpholine (thus making it the weakest base and strongest conjugate acid of the three compounds) or the presence of oxygen itself as a hydrogen-bond acceptor, able to coordinate an ethanol molecule.
Additional test reactions were run with l-proline (pKa = 1.94, 10.33) (10), d-pipecolic acid (pKa = 2.06, 10.39) (22), and (S)-morpholine-3-carboxylic acid (pKa = 1.61, 8.52) (23) as N-nucleophiles. Compound 9 was reacted with 1.0 equiv. paraformaldehyde and amino acids (10, 22, 23) in abs. ethanol until no change in conversion was detectable. Having the crude NMR spectra at hand, it was found that C-3 aminoalkylated products could not be detected in either reaction (Scheme 3.).
Scheme 3.

Reaction of 9 with cyclic amino acids 10, 22, 23 and paraformaldehyde in ethanol.
In accordance with our previous results, maximal conversion per cents of 12 in the three test reactions were in the order of nucleophiles 10 < 22 < 23 used. These findings support the hypothesis of the importance of the oxygen heteroatom and prove that the acidic condition is necessary but it is not a sufficient factor regarding the formation of the ethoxymethylated product (Fig. 3.). The differences between conversions while using nucleophiles 10 or 19 and 20 or 22 can be explained with the steric effects regarding the ability of the nucleophilic attack between the 5-membered and 6-membered rings.
Fig. 3.

Conversion curves of 12 depending of the used amino acid.
In order to clarify the previous results, eight more zwitterionic N-nucleophiles (24–31) were utilized in the test reactions (Table 3). Compound 9 was reacted with 1.0 equiv. paraformaldehyde and nucleophiles 24–31 in abs. ethanol in a pressure-resistant vessel for 60 h to reach maximum conversion.
Table 3.
Conversion per cents in the reactions of 9 with additional zwitterionic nucleophiles 24–31 and paraformaldehyde in ethanol.
| Compound # | Structure | pKa | Amine character | Acid character | Outcome |
|---|---|---|---|---|---|
| 24 | 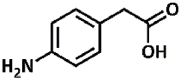 |
3.39, 5.51 | Primary aromatic | Aliphatic | No conversion |
| 25 | 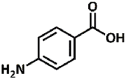 |
2.69, 4.77 | Primary aromatic | Aromatic | No conversion |
| 26 | 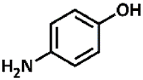 |
5.43, 10.40 | Primary aromatic | Weak phenolic | No conversion |
| 27 | 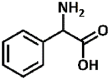 |
2.29, 8.64 | Primary aliphatic, at benzylic site | Aliphatic | No conversion |
| 28 | 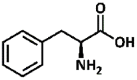 |
2.47, 9.45 | Primary aliphatic | Aliphatic, large side-chain | No conversion |
| 29 | 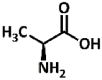 |
2.47, 9.48 | Primary aliphatic | Aliphatic, short side-chain | Aminoalkylated product (32) |
| 30 | 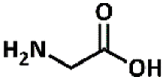 |
2.31, 9.24 | Primary aliphatic | Aliphatic, no alkyl side-chain | Aminoalkylated product (33) |
| 31 | 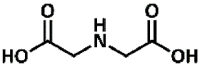 |
2.12, 2.90, 9.63 | Secondary aliphatic | Aliphatic dicarboxylic acid | 15 % conversion (12) |
Both crude NMR spectra and TLC-analysis showed no conversion of 9 in the case of nucleophiles 24–28. Utilizing nucleophile 31, the maximal conversion per cent of 12 was found to be 15 %, which is significantly lower than that achieved with additive 23. This information led to the deduction that only secondary N-nucleophiles facilitate the formation of compound 12. Compound 31 being a potent acidic secondary amine nucleophile, the hypothesis is verified that for the ethoxymethylation reaction, an acid functional group on the nucleophile is necessary but not sufficient. Furthermore, the hydrogen-bond acceptor O-heteroatom is proposed to coordinate ethanol.
In the case of primary amino acid nucleophiles with side chains of negligible steric hindrance (29, 30), stable Mannich bases 32, 33 formed. It should be emphasized that in the case of compound 31 fair conversion and yield could be achieved (44 %), although 32 formed in a significantly lower conversion and yield (9 %) thus setting the scope and limitations of the original aim of the modified Mannich reaction of KYNA esters with amino acids (Scheme 4.).
Scheme 4.

Reaction of 9 with primary amino acids 29 and 30 and paraformaldehyde in ethanol.
In addition, we aimed to explore the mechanism of the ethoxymethylation reaction. It was hypothesized that first an unstable Mannich base forms (I and zwitterionic I-enol and I-oxo) with the secondary amino acids. Next, upon the elimination of the amino acid, an ortho-quinone methide (II) forms which, via Michael addition, leads to compound 12 (Scheme 5.).
Scheme 5.

Proposed mechanism of formation of 12 starting from compound 9, using secondary amino acids 10, 22 or 34 and paraformaldehyde in ethanol.
In order to elaborate the previous mechanism, esterification of compound 18 was conducted. Since utilizing thionyl chloride yielded complex reaction mixture, other coupling reagents (DIC, DCC, EDAC.HCl, CDI, HCTU, Ac2O) were tested. Among these, N,N′-diisopropylcarbodiimide (DIC) in abs. ethanol at reflux temperature gave the highest conversion (Scheme 6.).
Scheme 6.

Esterification of Mannich base 18 with coupling agent DIC in ethanol.
TLC and crude NMR analysis showed full converison of compound 18 (Fig. 4./A and B). Besides the formation of the aminoester (34), 12 also started to form as a side-product with approximately 1–7.7 M ratio (Fig. 4./B). Characteristic signals could be assigned to compound 34. Note, however, that the signals of compound 12 are also detectable. The reaction was quenched and worked-up at that point. Surprisingly, after the work-up process and chromatographic purification, 12 was found to be present in significantly higher (2–3) molar ratio in comparison to 34 (Fig. 4./C) thus proving that compound 34 is highly prone to transformation to 12 also encumbering its isolation.
Fig. 4.

Crude NMR spectra of the esterification reaction of 18 with coupling agent DIC in ethanol.
Formation of 12 indirectly proved the unstable amino acid Mannich ester (I) → ortho-quinone methide (II) → 12 route shown in Scheme 6. According to our proposed explanation, first the unisolable Mannich product I forms. Thanks to its zwitterionic trait, autoprotonation occurs yielding a quaternary ammonium compound at the benzylic site with good leaving group property. After the retro-Mannich type elimination of the amino acid, ortho-quinone methide (II), an excellent Michael acceptor forms, which readily reacts with the O-nucleophile solvent ethanol to form 12.
As a next step the optimal equivalence of 23 was investigated in the reaction. Compound 23 was chosen for equivalence optimization as the highest conversion towards 12 could be achieved with this additive. In order to clarify the role of the catalyst, additive or reagent in the reaction of 23, four test reactions were conducted with different equivalences. Compound 9 was reacted with paraformaldehyde and 23 (0.1, 0.5, 1.0, and 2.0 equiv.) in abs. ethanol until maximum conversions were observed. Crude NMR spectra of samples taken from the reaction chambers were analyzed and conversion curves were drawn (Fig. 5.).
Fig. 5.

Conversion curves (12 assigned) of the C-3 ethoxymethylation reaction of 9 using different equivalents of 23.
The curves showed that a significant decrease in conversion occurred when applying 0.1 equiv. 23 in contrast to the utilization of 1.0 equivalent. When using 2.0 equiv. 23 the conversion of 12 measurably increased thanks to the higher molar ratio of the nucleophile. Interestingly, the optimal equivalent of 23 was found to be 0.5, an additive-range equivalent. Analyzing crude NMR spectra it can be hypothesized that lower nucleophile concentration subdues certain side reactions thus leading to higher conversion. These findings are supported by crude NMR-spectra since, in the case of the reaction with 0.5 equiv. 23, less unidentifiable signals can be observed in contrast to using 1.0 or 2.0 equivalents of additive.
Our next objective was to expand the scope of the reaction with diverse aldehyde or alcohol components. First, we changed the aldehyde. Compound 9 was reacted with 1.0 equiv. of the corresponding aldehyde (benzaldehyde, butyraldehyde, or phenylacetaldehyde) and 0.5 equiv. additive 23 in abs. ethanol. Both TLC and crude NMR analyses showed no conversion. A possible explanation of the failure of the reaction is the steric hindrance of the corresponding aldehydes [38]. Next, we modified the alcohol component in the reaction. We started with methanol the simplest alcohol followed by using bulkier primary, secondary, and tertiary alcohols. Kynurenic acid methyl ester [39] was reacted with 1.0 equiv. paraformaldehyde and 0.5 equiv. 23 in methanol for 60 h at 80 °C in a closed vessel. TLC and crude NMR analyses showed the formation of a complex reaction mixture from which the desired product was not possible to isolate.
Henceforth, compound 9 was reacted with 1.0 equiv. paraformaldehyde and 0.5 equiv. 23 in isopropyl alcohol for 60 h at 80 °C in a pressure-resistant vessel in order to produce the alkoxymethylated product. It was found that transesterification to the isopropyl ester occurred simultaneously to isopropoxymethylation. Only compound 35 formed (21 % maximal conversion in 26-h reaction) (Scheme 7.).
Scheme 7.

Reaction of 9 with paraformaldehyde in isopropyl alcohol.
Conducting the reaction in tBuOH, a bulky tertiary alcohol, no conversion was observed after 60 h under the previously set conditions, thus setting the scope and limitations of the secondary aminoacid-mediated alkoxyalkylation reaction of kynurenic acid esters.
3. Materials and methods
For TLC analyses Merck Kieselgel 60 F254 plates were used. Melting points were measured on a Hinotek X-4 melting point apparatus with heating velocity of 4 °C/min. The 1H and 13C NMR spectra were recorded in DMSO‑d6 solutions in 5 mm tubes at room temperature (RT), on a Bruker DRX-500 spectrometer with a 5 mm BBO Prodigy Probe (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H, 13C). Optical rotation values were measured on a Jasco P-2000 Polarimeter. The HRMS flow injection analysis was performed with Thermo Scientific Q Exactive Plus hybrid quadrupole-Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled to a Waters Acquity I-Class UPLC™ (Waters, Manchester, UK). FTIR spectra were measured on a Bruker Alpha II FTIR device in transmittance mode.
All chemicals were purchased from BLDPharm with the minimal purity of 95 %. The used solvents were absolute solvents. Chemicalize was used for pKa calculations, 05.2024., https://chemicalize.com, developed by ChemAxon.
3.1. General procedure of the alkoxyalkylation and/or aminomethylation test reactions
4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (9, 50 mg, 0.23 mmol) was dissolved in 3.0 mL absolute ethanol and then 7.0 mg paraformaldehyde (1.0 equivalent, 0.23 mmol), 1.0 equivalent (in case of 23 0.1, 0.5, 1.0 or 2.0 equivalents) of the corresponding N-nucleophile (10, 13–15, 19–31) was added in a sealable tubular flask. The reaction chamber was sealed and heated in silicone oil bath at 80 °C for 60 h. Samples were taken at least four times. Before analysis, the solvent was evaporated followed by using the aforementioned NMR device.
3.1.1. Ethyl 3-(ethoxymethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylate (12)
Synthesis was carried out according to the general procedure, then the solvent was evaporated and column chromatography was used (n-hexane: acetone 3 : 1) to purify compound 12 and then crystallized with n-hexane. Yield: 21 mg (33 %). M.p.: 98–102 °C. 1H NMR (DMSO‑d6): 1.08 (t, 3H, J = 7.15 Hz); 1.39 (t, 3H, J = 6.69 Hz); 3.43 (q, 2H, J = 7.21 Hz); 4.42 (q, 2H, J = 6.60 Hz); 4.58 (s, 2H); 7.37 (t, 1H, J = 6.70 Hz); 7.70 (t, 1H, J = 6.88 Hz); 7.73 (d, 1H, J = 7.95 Hz); 8.12 (d, 1H, J = 7.58 Hz); 12.00 (brs, 1H). 13C NMR (DMSO‑d6): 14.25; 15.54; 62.25; 62.92; 65.49; 117.25; 119.30; 124.46; 124.96; 125.63; 132.94; 139.48; 140.16; 163.89; 176.58 (Figs. S1 and S2). HRMS calcd for [M + H]+ m/z = 276.1230, found m/z = 276.1228 (Fig. S3). FTIR spectrum in Fig. S4.
3.1.2. Isopropyl 3-(isopropoxymethyl)-4-oxo-1,4-dihydroquinoline-2-carboxylate (35)
4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (9, 50 mg, 0.23 mmol) was dissolved in 3.0 mL absolute isopropyl alcohol, and then 7.0 mg paraformaldehyde (1.0 equivalent, 0.23 mmol), 16 mg 23 (0.5 equivalent, 0.115 mmol) was added in a sealable tubular flask. The reaction chamber was sealed and heated in silicone oil bath at 80 °C. For the test reaction, 60-h reaction was carried out, whereas for the highest conversion and yield, 26-h reaction was used. Solvent was evaporated, followed by column chromatogprahy (n-hexane: acetone 3 : 1) to purify compound 35, crystallized with n-hexane. Yield: 11 mg (16 %). M.p.: 142–144 °C. 1H NMR (DMSO‑d6): 1.06 (d, 6H, J = 6.28 Hz); 1.39 (d, 6H, J = 6.28 Hz); 3.51–3.58 (m, 1H, J = 6.23 Hz); 4.57 (s, 2H); 5.15–5.24 (m, 1H, J = 6.53 Hz); 7.37 (t, 1H, J = 7.60 Hz); 7.70 (t, 1H, J = 6.93 Hz); 7.73 (d, 1H, J = 8.05 Hz); 8.10 (d, 1H, J = 8.58 Hz); 11.98 (brs, 1H). 13C NMR (DMSO‑d6): 21.81; 22.47; 59.57; 70.55; 71.18; 117.47; 119.25; 124.40; 124.94; 125.65; 132.90; 139.46; 139.48; 155.00; 172.33 (Figs. S5 and S6). HRMS calcd for [M + H]+ m/z = 304.1543, found m/z = 304.1540 (Fig. S7). FTIR spectrum in Fig. S8.
3.2. (S)-3-(((1-Carboxyethyl)amino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (32)
4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (9, 50 mg, 0.23 mmol) was dissolved in 3.0 mL ethanol and then 7 mg paraformaldehyde (1.0 equivalent, 0.23 mmol) and 1.0 equivalent of 29 (21 mg, 0.23 mmol) was added in a round-bottom flask. The reaction chamber was heated in silicone oil bath at reflux temperature for 95 h. Compound 32 crystallized from the reaction mixture. Crystals were filtered from the warm mixture and then washed with ethanol. Yield: 6 mg (9 %). M.p.: decomposes at 257 °C, [α]D25 −26.45° (c 0.0128, H2O). 1H NMR (DMSO‑d6): 1.54 (d, 3H, J = 7.48 Hz) 4.35–4.45 (dd, 2H, J1 = 17.96 Hz, J2 = 25.73 Hz); 4.80 (q, 1H, J = 7.24 Hz); 7.40 (t, 1H, J = 7.57 Hz); 7.73 (t, 1H, J = 7.60 Hz); 7.81 (d, 1H, J = 8.39 Hz); 8.20 (d, 1H, J = 8.18 Hz). 13C NMR (DMSO‑d6): 15.05; 44.27; 50.00; 119.32; 120.47; 121.77; 123.53; 124.87; 126.10; 132.18; 140.30; 163.82; 176.14; 178.16 (Figs. S9 and S10). HRMS calcd for of [M − H2O - H]- m/z = 271.0727, found m/z = 271.0729 (Fig. S11). FTIR spectrum in Fig. S12.
3.3. (((Carboxymethyl)amino)methyl)-4-oxo-1,4-dihydroquinoline-2-carboxylic acid (33)
4-Oxo-1,4-dihydroquinoline-2-carboxylic acid ethyl ester (9, 50 mg, 0.23 mmol) was dissolved in 3.0 mL ethanol and then 7 mg paraformaldehyde (1.0 equivalent, 0.23 mmol), and 1.0 equivalent of 30 (18 mg, 0.23 mmol) was added in a round-bottom flask. The reaction chamber was heated in silicone oil bath at reflux temperature for 88 h. Compound 33 crystallized from the reaction mixture. Crystals filtered from the warm mixture were washed with ethanol. Yield: 28 mg (44 %). M.p.: decomposes at 231 °C. 1H NMR (DMSO‑d6): 4.32 (s, 2H); 4.40 (s, 2H); 7.40 (t, 1H, J = 7.50 Hz); 7.73 (t, 1H, J = 7.77 Hz); 7.81 (d, 1H, J = 8.70 Hz); 8.20 (d, 1H, J = 8.04 Hz). 13C NMR (DMSO‑d6): 43.99; 47.46; 119.33; 120.42; 123.55; 124.86; 126.13; 128.14; 132.20; 140.27; 163.95; 169.82; 173.75 (Figs. S13 and S14). HRMS calcd for [M − H2O - H]- m/z = 257.0568, found m/z = 257.0569 (Fig. S15). FTIR spectrum in Fig. S16.
4. Conclusions
Novel C-3 alkoxymethylated derivatives of kynurenic acid were synthesized with cyclic secondary alpha amino acid additives. The importance of a secondary acidic N-nucleophile was proven. Test reactions were conducted using common organic additives, including cyclic amines and their acetate salts, as well as using other acidic N-nucleophiles. Result showed the importance of the local acidic conditions, particularly when using amino acids. Moreover, the importance of the corresponding heterocycle and its ability to coordinate the O-nucleophile were also highlighted. A mechanism of the alkoxyalkylation of kynurenic acid ethyl esters was proposed. Optimal equivalence of the amino acid was set. Two novel kynurenic acid derivatives, containing glycine or l-alanine fragments were synthesized, thus proving the scope and limitations of the aminoalkylation reaction of kynurenic acid ethyl ester using alpha amino acids. Limitations were set of alkoxyalkylation reactions and two novel kynurenic acid derivatives were synthesized bearing ethoxymethyl or isopropoxymethyl moieties at the site C-3.
CRediT authorship contribution statement
Péter Simon: Writing – review & editing, Writing – original draft, Investigation. Bálint Lőrinczi: Writing – review & editing, Writing – original draft, Investigation. István Szatmári: Writing – review & editing, Conceptualization.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgements
The authors thank the Hungarian Research Foundation (OTKA No. K-138871), the Ministry of Human Capacities, Hungary grant, TKP-2021-EGA-32. P. S. was supported by the ÚNKP-23-3 -SZTE-185 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development and Innovation Fund. The authors also thank Dr. Róbert Berkecz (Institute of Pharmaceutical Analysis, University of Szeged) for the HRMS investigations and Dr. Éva Anna Enyedy (Department of Molecular and Analytical Chemistry, University of Szeged) for the FTIR investigations.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e32188.
Abbreviations
- CDI
1,1′-Carbonyldiimidazole
- DCC
N,N′-Dicyclohexylcarbodiimide
- DIC
N,N′-Diisopropylcarbodiimide
- DMSO‑d6
Deuterated dimethylsulfoxide
- EDAC.HCl
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- HCTU
O-(1H-6-Chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HZSM-5
protonated form of Zeolite Socony Mobil-5
- KYNA
Kynurenic acid
- NMR
Nuclear Magnetic Resonance (spectroscopy or spectrum)
- TLC
Thin Layer Chromatography
- TMS
Tetramethylsilane
- ZBM
Zeolite-bentonite Mixture(s)
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zádori D., Veres G., Szalárdy L., Klivényi P., Toldi J., Vécsei L. Glutamatergic dysfunctioning in alzheimer's disease and related therapeutic targets. J. Alzheimers Dis. 2014;42:177–187. doi: 10.3233/JAD-132621. [DOI] [PubMed] [Google Scholar]
- 2.Zádori D., Nyiri G., Szőnyi A., Szatmári I., Fülöp F., Toldi J., Freund T.F., Vécsei L., Klivényi P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington's disease. J. Neural. Transm. 2011;118:865–875. doi: 10.1007/s00702-010-0573-6. [DOI] [PubMed] [Google Scholar]
- 3.Rózsa É., Robotka H., Vécsei L., Toldi J. The Janus-face kynurenic acid. J. Neural. Transm. 2008;115:1087–1091. doi: 10.1007/s00702-008-0052-5. [DOI] [PubMed] [Google Scholar]
- 4.Sas K., Robotka H., Toldi J., Vécsei L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007;257:221–239. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 5.Gigler G., Szénási G., Simó A., Lévay G., Hársing L.G., Sas K., Vécsei L., Toldi J. Neuroprotective effect of L-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur. J. Pharmacol. 2007;564:116–122. doi: 10.1016/j.ejphar.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 6.Kiss C., Shepard P.D., Bari F., Schwarcz R. Cortical spreading depression augments kynurenate levels and reduces malonate toxicity in the rat cortex. Brain Res. 2004;1002:129–135. doi: 10.1016/j.brainres.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Luchowska E., Luchowski P., Sarnowska A., Wielosz M., Turski W.A. Pol. J. Pharmacol. 2003;55:443–447. [PubMed] [Google Scholar]
- 8.Nilsson L.K., Linderholm K.R., Engberg G., Paulson L., Blennow K., Lindström L.H., Nordin C., Karanti A., Persson P., Erhardt S. Elevated levels of kynurenic acid in the cerebrospinal fluid of male patients with schizophrenia. Schizophr. Res. 2005;80:315–322. doi: 10.1016/j.schres.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Fejes A., Párdutz A., Toldi J., Vécsei L. Kynurenine metabolites and migraine: experimental studies and therapeutic perspectives. Curr. Neuropharmacol. 2011;9:376–387. doi: 10.2174/157015911795596621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fülöp F., Szatmári I., Vámos E., Zádori D., Toldi J., Vécsei L. Syntheses, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr. Med. Chem. 2009;16:4828–4842. doi: 10.2174/092986709789909602. [DOI] [PubMed] [Google Scholar]
- 11.Lőrinczi B., Csámpai A., Fülöp F., Szatmári I. Synthesis of new C-3 substituted kynurenic acid derivatives. Molecules. 2020;25:937–951. doi: 10.3390/molecules25040937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy K., Plangár I., Tuka B., Gellért L., Varga D., Demeter I., Farkas T., Kis Zs, Marosi M., Zádori D., Klivényi P., Fülöp F., Szatmári I., Vécsei L., Toldi J. Synthesis and biological effects of some kynurenic acid analogs. Bioorg. Med. Chem. 2011;19:7590–7596. doi: 10.1016/j.bmc.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Tiszlavicz Z., Németh B., Fülöp F., Vécsei L., Tápai K., Ocsovszky I., Mándi Y. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-α (TNF-α) production by mononuclear cells, HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011;383:447–455. doi: 10.1007/s00210-011-0605-2. [DOI] [PubMed] [Google Scholar]
- 14.Gellért L., Fuzik J., Göblös A., Sárközi K., Marosi M., Kis Z., Farkas T., Szatmári I., Fülöp F., Vécsei L., Toldi J. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur. J. Pharmacol. 2011;667:182–187. doi: 10.1016/j.ejphar.2011.05.069. [DOI] [PubMed] [Google Scholar]
- 15.Marosi M., Nagy D., Farkas T., Kis Z., Rózsa É., Robotka H., Fülöp F., Vécsei L., Toldi J. A novel kynurenic acid analogue: a comparison with kynurenic acid. An in vitro electrophysiological study. J. Neural. Transm. 2010;117:183–188. doi: 10.1007/s00702-009-0346-2. [DOI] [PubMed] [Google Scholar]
- 16.Vámos E., Párdutz Á., Varga H., Bohár Z., Tajti J., Fülöp F., Toldi J., Vécsei L. l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology. 2009;57:425–429. doi: 10.1016/j.neuropharm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 17.Turk G., Moroni G., Pampuro S., Briñón M.C., Salomón H. Antiretroviral activity and cytotoxicity of novel zidovudine (AZT) derivatives and the relation to their chemical structure. Int. J. Antimicrob. Agents. 2002;20:282–288. doi: 10.1016/s0924-8579(02)00191-7. [DOI] [PubMed] [Google Scholar]
- 18.Deshmukh M., Chao P., Kutscher H.L., Gao D., Sinko P.J. A series of α-amino acid ester prodrugs of camptothecin: in vitro hydrolysis and A549 human lung carcinoma cell cytotoxicity. J. Med. Chem. 2010;53:1038–1047. doi: 10.1021/jm901029n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chrzanowski K., Bielawska A., Pałka J. Proline analogue of melphalan as a prodrug susceptible to the action of prolidase in breast cancer MDA-MB 231 cells. Il Farmaco. 2003;58:1113–1119. doi: 10.1016/S0014-827X(03)00164-2. [DOI] [PubMed] [Google Scholar]
- 20.Ma H., Chen G., Wang T., Li Q., Liu Y. Design, synthesis, and biological evaluation of a novel water‐soluble prodrug of docetaxel with amino acid as a linker. Chem. Biol. Drug Des. 2016;88:363–369. doi: 10.1111/cbdd.12762. [DOI] [PubMed] [Google Scholar]
- 21.Vale N., Ferreira A., Matos J., Fresco P., Gouveia M. Amino acids in the development of prodrugs. Molecules. 2018;23:2318–2378. doi: 10.3390/molecules23092318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Currie G.S., Drew M.G.B., Harwood L.M., Hughes D.J., Luke R.W.A., Vickers R.J. Chirally templated boronic acid Mannich reaction in the synthesis of optically active α-amino acids. J. Chem. Soc. Perkin Trans. 2000;1:2982–2990. [Google Scholar]
- 23.Müller R., Röttele H., Henke H., Waldmann H. Asymmetric steering of the mannich reaction with phthaloyl amino acids. Chem. Eur J. 2000;6:2032–2043. doi: 10.1002/1521-3765(20000602)6:11<2032::aid-chem2032>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 24.Csütörtöki R., Szatmári I., Mándi A., Kurtán T., Fülöp F. Synthesis of hydroxynaphthyl-substituted α-amino acid derivatives via a modified mannich reaction. Synlett. 2011;13:1940–1946. [Google Scholar]
- 25.Short J.H., Ours C.W. The use of amino acids in the mannich reaction. J. Heterocycl. Chem. 1975;12:869–876. [Google Scholar]
- 26.Pivarcsik T., Dömötör O., Mészáros J.P., May N.V., Spengler G., Csuvik O., Szatmári I., Enyedy É.A. 8-Hydroxyquinoline-Amino acid hybrids and their half-sandwich Rh and Ru complexes: synthesis, anticancer activities, solution Chemistry and interaction with biomolecules. Int. J. Mol. Sci. 2021;22:11281–11309. doi: 10.3390/ijms222011281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keshipour S., Shaabani A., Pedarpour M., Sarvary A. An oxy-Michael addition: 2,5-dihydroxy-1,4-benzoquinone-assisted synthesis of 1-[ethoxy(phenyl)methyl]-2-naphthol and 5-[ethoxy(phenyl)methyl]-6-hydroxyquinoline derivatives. Res. Chem. Intermed. 2014;40:149–156. [Google Scholar]
- 28.Boeckman R.K., Miller J.R. Direct enantioselective organocatalytic hydroxymethylation of aldehydes catalyzed by α,α-diphenylprolinol trimethylsilyl ether. Org. Lett. 2009;11:4544–4547. doi: 10.1021/ol9017479. [DOI] [PubMed] [Google Scholar]
- 29.Wu X., Shan X., Dong Q., Chang H., Li Z. New synthetic route for benzyl methyl ether and benzyl alcohol from benzene and formaldehyde on shape-selective. catalysts Jingxi Huangong. 2001;18:114–116. [Google Scholar]
- 30.U. Griesbach, U. Mueller, K. Ebel, J. Botzem, B. Yilmaz, C. Stock, A Process for the Regioselective Preparation of Benzyl Methyl Ethers, WO/2009/059941.
- 31.Goswami J., Borthakur N., Goswami A. A water based method for hydroxymethylation of phenols and phenolic ketones. J. Chem. Res., Synop. 2003;4:200–203. [Google Scholar]
- 32.S. Jackson P., A.D. Robertson, V. Kenche, P. Thompson, H. Prabaharan, K. Anderson, B. Abbott, I. Goncalves, W. Nesbitt, S. Schoenwalder, D. Saylik, Preparation of Morpholinyl- and Pyridinyl-Substituted Heterobicyclic Ketones as Selective Inhibitors of Phosphoinositide 3-kinase β for Use against Thrombosis, WO/2004/016607.
- 33.Cossy J., Lutz F., Alauze V., Meyer C. Carbon-carbon bond forming reactions by using bistrifluoromethanesulfonimide. Synlett. 2002;1:45–48. [Google Scholar]
- 34.Lőrincz E.B., Tóth G., Spolárics J., Herczeg M., Hodek J., Zupkó I., Minorics R., Ádám D., Oláh A., Zouboulis C.C., Weber J., Nagy L., Ostorházi E., Bácskay I., Borbás A., Herczegh P., Bereczki I. Mannich-type modifications of (-)-Cannabidiol and (-)-Cannabigerol leading to new, bioactive derivatives. Sci. Rep. 2023;13:19618–19637. doi: 10.1038/s41598-023-45565-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nummelin S., Falabu D., Shivanyuk A., Rissanen K. Alkoxy-, acyloxy-, and bromomethylation of resorcinarenes. Org. Lett. 2004;6:2869–2872. doi: 10.1021/ol049179z. [DOI] [PubMed] [Google Scholar]
- 36.Urbaniak M., Pedrycz A., Gawdzik B., Wzorek A. Preparation of partially functionalised resorcinarene derivatives. Supramol. Chem. 2013;25:777–781. [Google Scholar]
- 37.Urbaniak M., Iwanek W. Synthesis of alkoxymethyl derivatives of resorcinarene via the Mannich reaction catalysed with iminodiacetic acid. Tetrahedron. 2006;62:1508–1511. [Google Scholar]
- 38.Lőrinczi B., Simon P., Szatmári I. Synthesis of indole-coupled KYNA derivatives via C–N bond cleavage of mannich bases. Int. J. Mol. Sci. 2022;23:7152–7164. doi: 10.3390/ijms23137152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borza I., Kolok S., Galgóczy K., Gere A., Horváth C., Farkas S., Greiner I., Domány G. Kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorg. Med. Chem. Lett. 2007;17:406–409. doi: 10.1016/j.bmcl.2006.10.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.