

Abstract

As the chemistry that surrounds the field of strained hydrocarbons, such as bicyclo[1.1.0]butane, continues to expand, it becomes increasingly advantageous to develop alternative reactivity modes that harness their unique properties to access new regions of chemical space. Herein, we report the use of photoredox catalysis to promote the single-electron oxidation of bicyclo[1.1.0]butanes. The synthetic utility of the resulting radical cations is highlighted by their ability to undergo highly regio- and diastereoselective [2π + 2σ] cycloaddition reactions. The most notable feature of this transformation is the breadth of alkene classes that can be employed, including nonactivated alkenes, which have so far been elusive for previous strategies. A rigorous mechanistic investigation, in conjunction with DFT computation, was undertaken in order to better understand the physical nature of bicyclo[1.1.0]butyl radical cations and thus provides a platform from which further studies into the synthetic applications of these intermediates can be built upon.
Introduction
Since its first synthesis in 1959,1 bicyclo[1.1.0]butane (BCB) has captured the imagination of chemists due to its innate strain energy and relative ease of assembly and handling.2,3 The highly diverse reactivity of BCB-containing compounds, facilitated by the release of strain upon breaking the bridging C1–C3 bond, has allowed such structures to become valuable building blocks for the generation of sp3-rich carbocycles and heterocycles.2−5 Perhaps the most prominent reactivity mode that has been utilized in this context is the addition of nucleophiles and nucleophilic radicals to the bridgehead of electron-deficient BCB compounds (Figure 1A).6−9 In recent years, alternative strategies have also emerged, such as electrophilic addition,10−12 reduction13,14 or Lewis acid activation15−18 of adjacent carbonyl fragments to trigger ring-opening, pyridine-boryl radical transfer19,20 and photochemical excitation of the strained bridging bond to access the corresponding diradical.21,22 Employing these strategies has led to the development of many unique transformations, and as a consequence, BCB-containing compounds have become cemented as valuable synthetic building blocks. This is most clearly evidenced by their application in areas such as bioconjugation23,24 and in the assembly of rigid and sp3-rich arene isosteres, structures of particular interest in a medicinal chemistry context.10,11,25 However, in order to find new applications and access unexplored regions of chemical space, alternative reactivity modes that harness the unique properties of BCBs are required.
Figure 1.
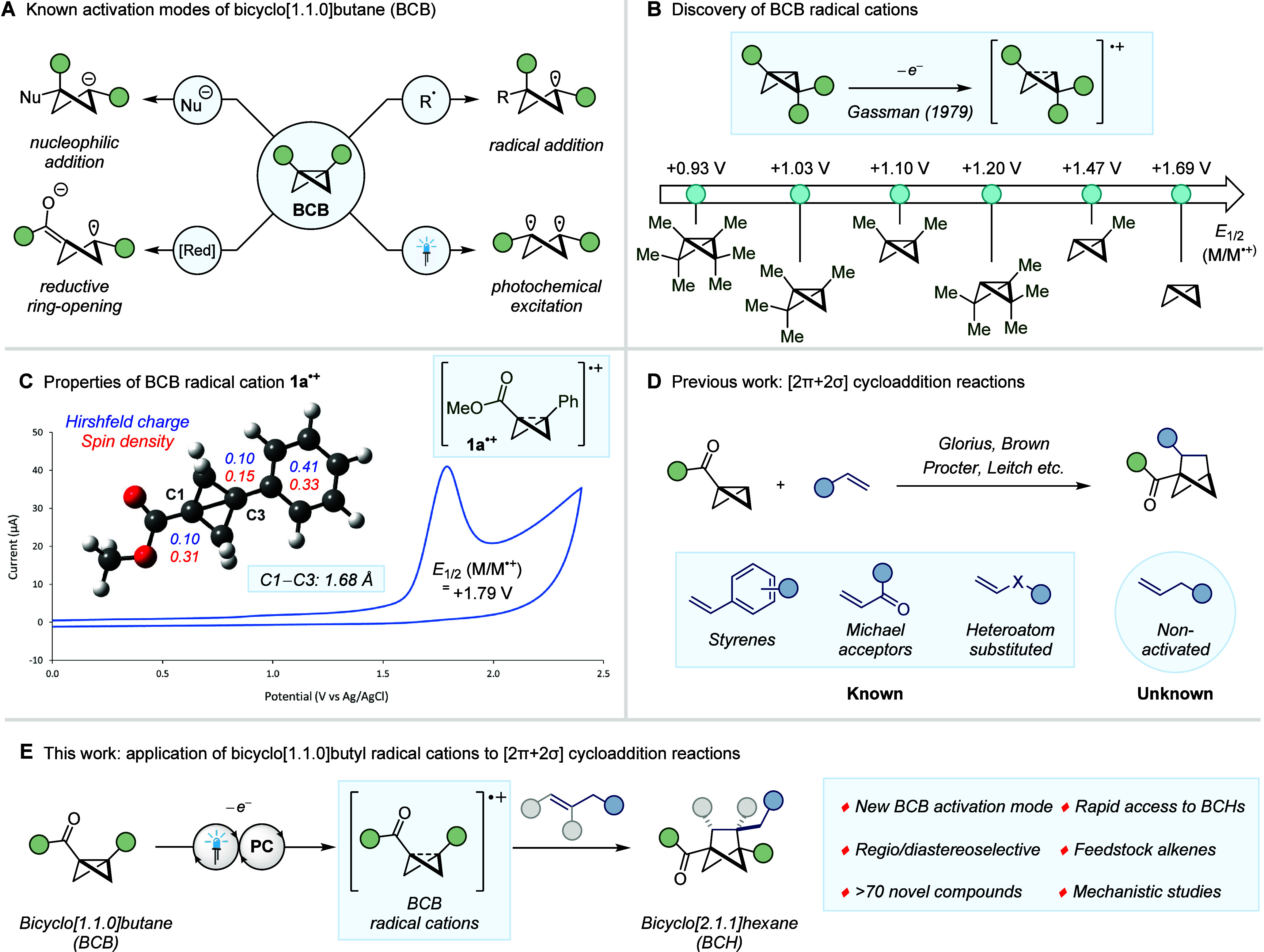
(A) Known activation modes of bicyclo[1.1.0]butane. (B) Discovery of the direct oxidation of substituted bicyclo[1.1.0]butane compounds.27 (C) Oxidation potential of BCB 1a vs Ag/AgCl (2 M LiCl in EtOH) and physical properties of bicyclo[1.1.0]butyl radical cation 1a•+. (D) Previous reports of [2π + 2σ] cycloaddition reactions. (E) This work: application of bicyclo[1.1.0]butyl radical cations to [2π + 2σ] cycloaddition reactions.
In 1979, Gassman reported a study on the relationship between alkyl substitution and the ease of oxidation of strained hydrocarbons.26 By measuring the half-wave potentials of a variety of substituted bicyclo[1.1.0]butanes, it was demonstrated that the σ-framework of the bicycle could readily undergo single-electron oxidation to the corresponding radical cation (Figure 1B).27 In addition, Hoz and co-workers postulated the existence of a radical cation intermediate, arising from single-electron oxidation, in the bromination of bicyclo[1.1.0]butane analogs.28 Despite these discoveries, the synthetic potential of bicyclo[1.1.0]butyl radical cations has been severely underexplored,29 with nonselective nucleophilic addition being the only transformation demonstrated for these intermediates.30−32 We therefore determined to assess the feasibility of employing single-electron oxidation via photoredox catalysis as a strategy to access alternative bicyclo[1.1.0]butane reactivity.
Results and Discussion
To avoid the issues associated with handling low-molecular-weight strained hydrocarbons, electron-deficient BCB 1a, known to be nonvolatile and easily synthesized, was investigated (Figure 1C). Although this species contains an electron-withdrawing group directly appended to the σ-framework, cyclic voltammetry studies clearly showed that this compound could be oxidized, with a half-wave potential of +1.79 V vs Ag/AgCl (2 M LiCl in EtOH). Density functional theory (DFT) calculations of the condensed Hirshfeld charges and spin densities of the radical cation revealed that both the overall charge and the spin were largely delocalized across the bridging bond of the BCB framework as well as the aromatic ring (Figure 1C). Analyzing these values revealed that the C1 and C3 carbon atoms have a greater contribution of the overall spin (0.31 and 0.15, respectively) compared to the positive charge (0.10 and 0.10, respectively), which is more concentrated on the aryl ring (0.41). Additionally, the bridging bond remains intact upon oxidation and shows an elongation of just 0.16 Å compared to the ground state,33 highlighting the difference between this activation strategy and energy transfer, where σ-bond cleavage occurs to form the corresponding diradical.21
With an understanding of the accessibility and physical properties of BCB radical cations, a reactivity regime that cannot be achieved using previously known strategies was pursued. Specifically, we targeted [2π + 2σ] cycloaddition reactions to access bicyclo[2.1.1]hexane (BCH) compounds, highly valuable isosteres of ortho- and meta-substituted benzene that are sp3-rich with well-defined substituent exit vectors.34−37 Although [2π + 2σ] cycloaddition reactions that harness the strained bond of BCB have been reported by our own research group,18,38−40 as well as those of Brown,21 Procter,13 Leitch25 and others,14−17,19,20,41 in all cases, alkenes that can be employed require a radical stabilizing group, electron-withdrawing group, or heteroatom directly appended to the double bond, depending on the respective mechanism (Figure 1D). Conversely, it is known that styrene-type radical cations,42−44 intermediates that display remarkably similar levels of charge and spin delocalization to 1a•+ (see Supporting Information for details), have the ability to participate in [2π + 2π] cycloaddition reactions with olefins that are not stabilized by an adjacent π-system or carbonyl unit.45−47 Therefore, we believed that harnessing BCB radical cations could provide a general method for [2π + 2σ] cycloaddition reactions, allowing the transformation to occur with multiple distinct classes of olefins, including simple feedstock alkenes that have so far been elusive.
Herein, we report the successful application of bicyclo[1.1.0]butyl radical cations to [2π + 2σ] cycloaddition reactions to generate a unique selection of BCH structures (Figure 1E). The most notable features of this strategy are the breadth of alkene classes that can be employed and the remarkable levels of regio- and diastereoselectivity that can be achieved using single-electron oxidation as the activation mode for BCB. A rigorous experiment-based mechanistic study, in conjunction with DFT computation, was undertaken in order to better understand this process and illuminate how these strained radical cations interact with alkenes. Consequently, we believe that the work described here can serve as a platform from which further studies into the potential synthetic applications of BCB radical cations can be built upon.
Reaction Development
Our investigation commenced with the exploration of suitable photocatalysts, capable of promoting the single-electron oxidation of the BCB moiety. Olefin 2a, containing no adjacent heteroatom or radical stabilizing group, was selected as the model coupling partner due to its presumed inactivity under any currently known BCB [2π + 2σ] cycloaddition conditions. Upon screening photocatalysts across a wide range of oxidation potentials, it was demonstrated that [Mes2AcrtBu2]ClO4 (E1/2 (PC*/PC•–) = +2.00 V vs SCE),48 irradiated with blue LEDs, could catalyze the desired [2π + 2σ] cycloaddition reaction with 2a (Figure 2A, entry 5).49−51 Indeed, it was observed that employing photocatalysts that display an excited state oxidation potential below +1.86 V (or significantly greater than +2.00 V) failed to deliver any observable product (entries 1–6). After an extensive exploration of the reaction conditions (see Supporting Information for full optimization details), it was found that improvements to the yield could be achieved upon increasing the equivalents of the alkene coupling partner and performing the reaction in MeNO2 (Figure 2B, entries 7–10). Although this reaction represents the first example of a bicyclo[1.1.0]butane [2π + 2σ] cycloaddition with a simple alkyl substituted alkene, the overall yield is partially limited by the side reactions that can occur from the BCB radical cation, such as dimerization.31,32 Finally, control reactions, in which the photocatalyst and light source were omitted, were performed (entries 11–12). The inability to access any cycloaddition product under these conditions clearly shows that product formation is dependent on the generation of the excited state photocatalyst and does not arise as a result of direct excitation of either the BCB or alkene substrates.
Figure 2.
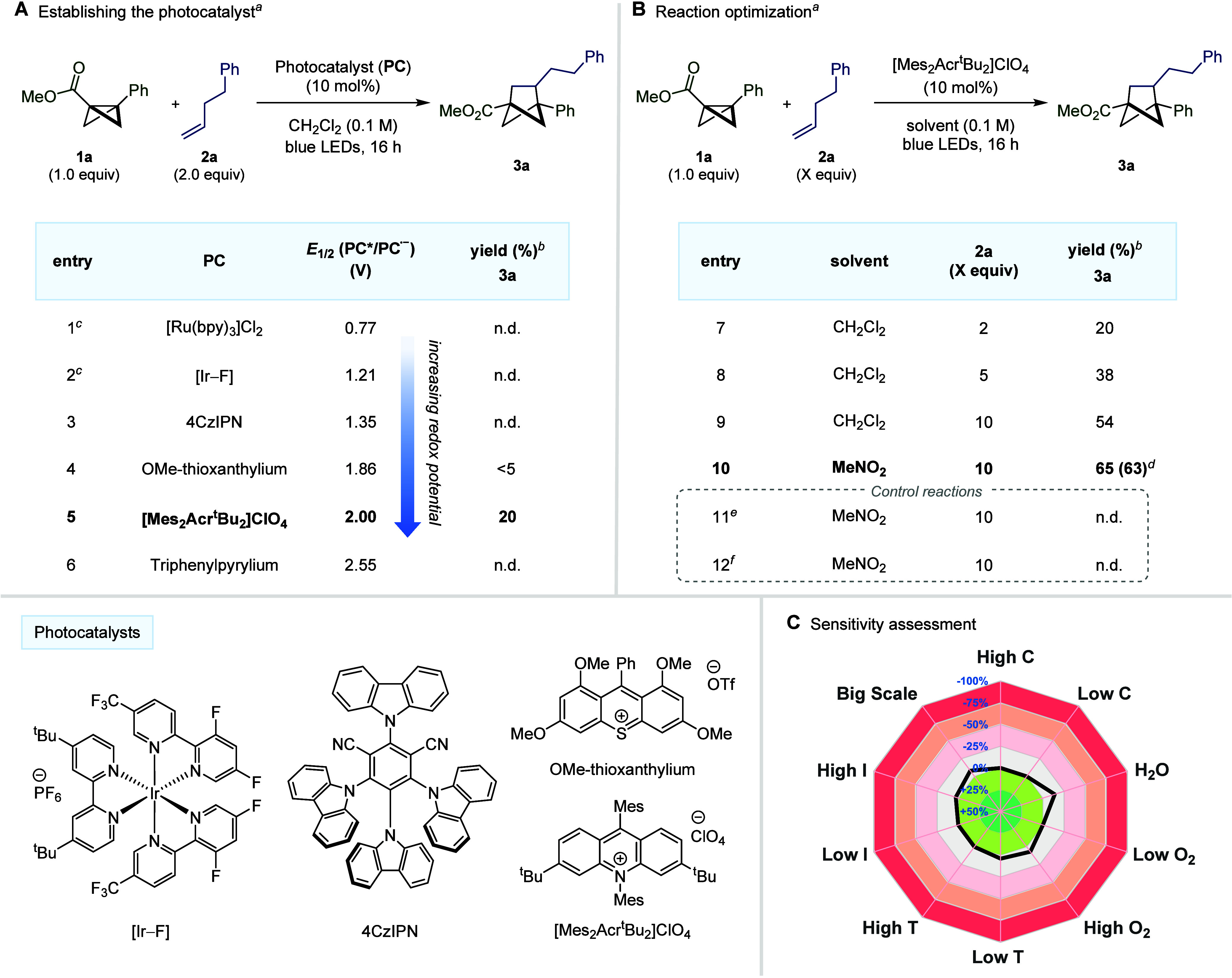
Optimization of the BCB radical cation [2π + 2σ] cycloaddition reaction. (A) Establishing the photocatalyst. (B) Optimization of the reaction conditions. (C) Sensitivity assessment of the reaction conditions. aReactions performed on 0.05 mmol scale under blue LED (λmax = 425 nm) irradiation. bYields determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard. c2 mol % of photocatalyst. dIsolated yield on 0.2 mmol scale. eNo photocatalyst. fReaction performed in absence of light.
In order to assess the robustness and reproducibility of the newly established protocol, a reaction condition-based sensitivity assessment was performed (Figure 2C).52 Interestingly, the reaction was shown to be remarkably tolerable toward perturbations in the temperature (T), concentration (c), oxygen level, and light intensity (I), with only a slight decrease in yield observed when H2O was added to the reaction mixture. Additionally, the photocatalyzed reaction could be performed on 4.0 mmol scale, showing a relatively small erosion in isolated yield compared to the standard reaction (53% vs 63%).
Reaction Scope
With the optimized reaction conditions in hand, the scope of the reaction with respect to the olefin was systematically investigated to both assess the generality of the transformation and discover the limits of reactivity (Figure 3). As well as the simple hydrocarbon 1-hexene (3b), propene gas could also be employed under the same reaction conditions to access 3c in 38% yield. Additionally, functional groups such as primary halides (3d–e), terminal alkynes (3f), ketones (3g), ethers (3h), esters (3i), internal alkynes (3j), thiophenes (3k) sulfones (3l), quinolines (3m), phthalimides (3n), and amino acid derivatives (3o) were all compatible with the transformation and provided a single regioisomer of the desired products. However, limitations to the reaction were discovered when it was observed that some nucleophilic fragments such as unprotected alcohols and amines could not be tolerated in the alkene fragment (see Supporting Information for all failed substrates). Exploring the scope of the alkene substitution pattern demonstrated that internal alkenes such as cyclopentene and cyclohexene could be used to access BCH structures 3p and 3q exclusively as the cis-diastereomer. However, increasing the ring size to cyclooctene resulted in isomerization to the trans-isomer (3r). Pleasingly, noncyclic 1,2-disubstituted alkenes such as (E)-oct-3-ene were also compatible, providing 3s in 30% yield as a single diastereomer. Unsurprisingly, with no significant electronic or steric bias, a mix of regioisomers was observed for this substrate. On the other hand, when trisubstituted alkene 2-methylpent-2-ene was utilized, only a single regioisomer (3t) was detected in the reaction mixture, demonstrating the ability of the system to clearly distinguish between mono- and disubstituted sp2 carbon atoms. Given that bicyclo[2.1.1]hexane structures are seen as potential sp3-rich isosteres for ortho- and meta-substituted benzene, we next investigated the tolerance of natural product and approved pharmaceutical derived alkenes in the newly developed [2π + 2σ] cycloaddition reaction. Promisingly, substrates derived from the anti-inflammatory drug ibuprofen (3u) and the β-lactamase inhibitor sulbactam (3v) could be tolerated. In addition, derivatives of the cholesterol lowering pharmaceutical fenofibrate (3w), the gout medication probenecid (3x), and a protected glucose analogue (3y) were all capable of accessing the desired BCH products. As complex alkene substrates could be deemed more precious than the BCB coupling partner, we also demonstrated that inverting the stoichiometry of this reaction to have the olefin as the limiting reagent could also provide access to the desired products in comparable yields (see Supporting Information for details).
Figure 3.
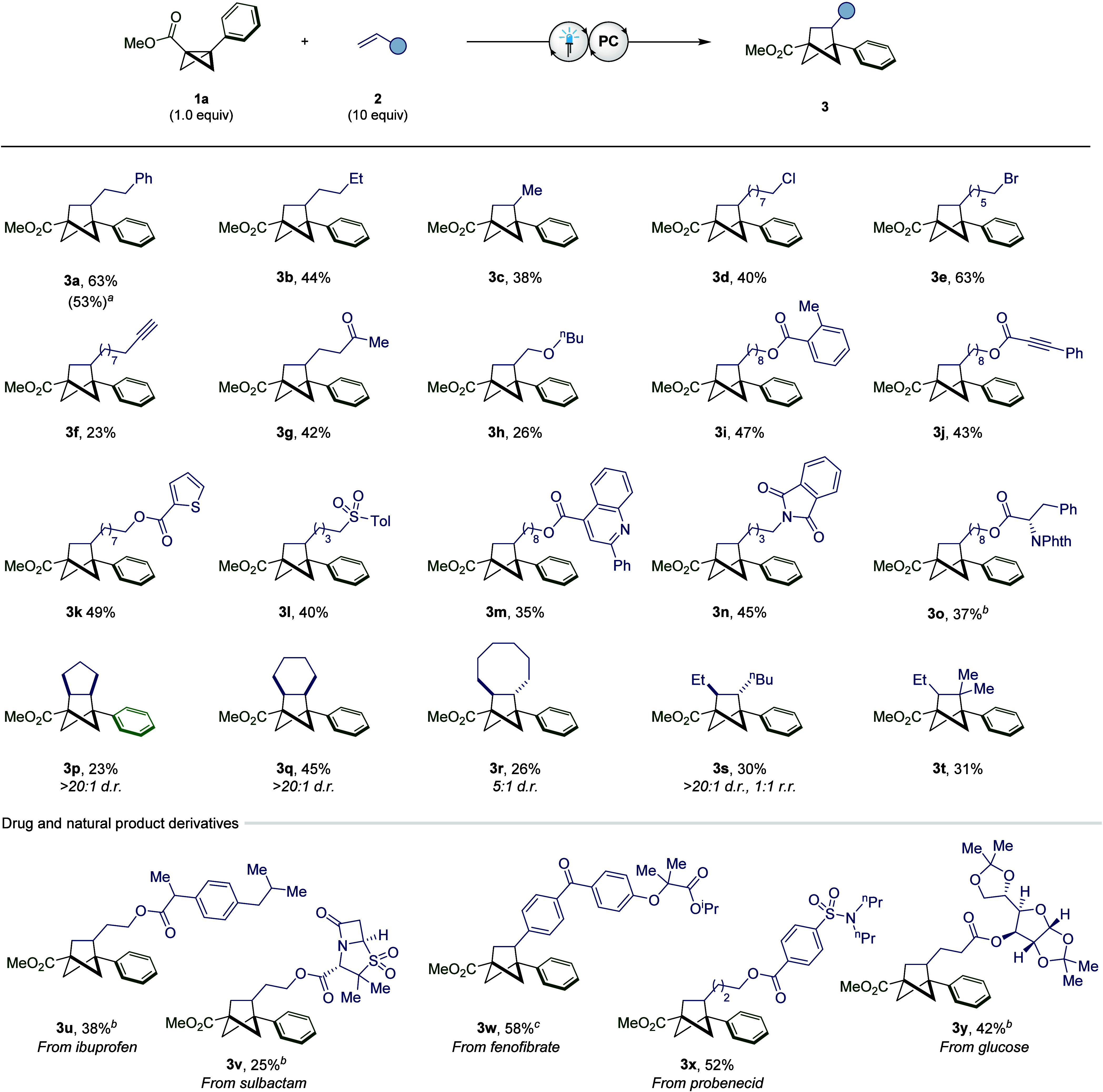
Bicyclo[1.1.0]butane [2π + 2σ] cycloaddition reaction with nonactivated alkenes. Reaction conditions: 1a (0.2 mmol), 2 (2.0 mmol), [Mes2AcrtBu2]ClO4 (10 mol %), MeNO2 (0.1 M), blue LEDs (λmax = 425 nm), 16 h. Isolated yields given. The d.r. and r.r. values were determined by 1H NMR analysis of the crude reaction mixture. aReaction performed on 4.0 mmol of 1a. bSubstrates that contain a pre-existing stereocenter were formed as a 1:1 mix of diastereomers. cUsing conditions from Figure 4 (see below).
Initially, we hypothesized that the extension of the transformation to include “activated” alkenes, such as styrenes, would be challenging, as these compounds are known to be susceptible to oxidation by the photocatalyst. Although this was indeed observed, styrene-type substrates typically exhibited an improved yield in the reaction due to their enhanced reactivity with the BCB radical cation (Figure 4).
Figure 4.
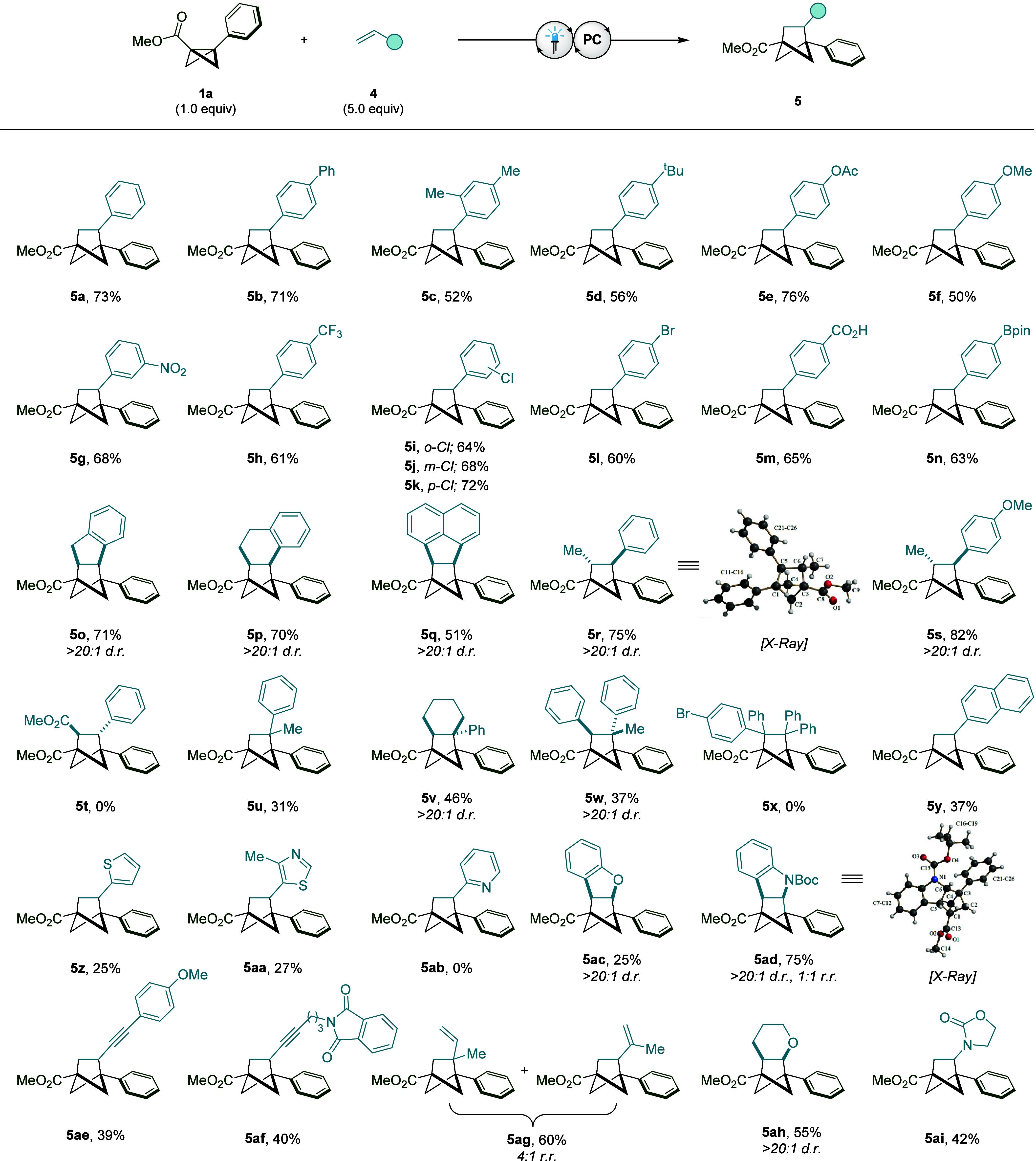
Bicyclo[1.1.0]butane [2π + 2σ] cycloaddition reaction with activated alkenes. Reaction conditions: 1a (0.2 mmol), 4 (1.0 mmol), [Mes2AcrtBu2]ClO4 (10 mol %), MeCN (0.1 M), blue LEDs (λmax = 425 nm), 16 h. Isolated yields given. The d.r. and r.r. values were determined by 1H NMR analysis of the crude reaction mixture.
Despite involving a direct interaction with an electron-deficient radical cation intermediate, styrene-type alkenes bearing both electron-donating and electron-withdrawing groups were viable in this transformation, and all delivered the desired products as single regioisomers (5a–h). Additionally, highly versatile functional handles such as halides (5i–l), carboxylic acids (5m) and boronic esters (5n) could also be tolerated under the reaction conditions, providing the potential for further derivatization of these substrates. When exploring the effect of alkene substitution, it was again observed that cyclic 1,2-disubstituted substrates are capable of accessing the desired BCH products, exclusively as the cis-diastereomer (5o–q). In the case of acyclic (E)-1,2-disubstituted olefins, only the trans-isomer is detected (5r–s), with the stereochemistry confirmed by X-ray crystallography. The limit of reactivity was located when highly electron-deficient Michael-type alkenes were observed to be unsuitable for this transformation (5t), making this approach complementary to previously reported radical-based BCB [2π + 2σ] cycloaddition reactions.13,14,19 However, subjecting 1,1-disubstituted and trisubstituted alkenes to the newly developed reaction conditions could provide access to highly substituted BCH substrates 5u, 5v, and 5w, although tetrasubstituted alkenes were too sterically hindered to deliver the desired cycloadduct (5x).
One of the key drawbacks of previously reported BCB [2π + 2σ] cycloaddition reactions is the limited generality with respect to the alkene coupling partner, and so we next turned our attention to other classes of olefin which could be employed. In addition to vinyl naphthalene (5y), heterocycles containing Lewis basic atoms such as vinyl thiophene (5z) and thiazole (5aa) were amenable to the cycloaddition reaction, although vinylpyridine was deemed unsuitable (5ab). Interestingly, this transformation could also be used to facilitate the dearomatization of heterocycles such as benzofuran (5ac) and indole (5ad), as well as being compatible with enynes (5ae–af), dienes (5ag), enol ethers (5ah), and enamine-type substrates (5ai). These results demonstrate the remarkable variety of olefins that can interact with BCB radical cations and highlight the inimitable reactivity of this synthetic intermediate.
When exploring the electronic effect of the BCB fragment, we were eager to discover whether a relationship between the aptitude for cycloaddition and the compound oxidation potential could be established (Figure 5). First, it was discovered that BCB substrates which do not bear an electron-withdrawing group possess a considerably lower oxidation potential and yield only trace product under the developed cycloaddition conditions (5aj). However, ester and amide containing BCB compounds, bearing aryl substitution that did not greatly perturb the substrate oxidation potential, could effectively deliver the desired BCH products (5ak–am). Considerably decreasing the electron density of the aromatic system, through the addition of a trifluoromethyl group, resulted in a BCB compound with an oxidation potential of +2.00 V which failed to deliver the desired cycloadduct and thus represents the upper limit of BCB oxidation by [Mes2AcrtBu2]ClO4 under these conditions (5an). It must also be stated that removal of the aryl ring entirely resulted in a drastic increase in oxidation potential (5ao), presumably due to the inability of the corresponding radical cation to delocalize into the aromatic system. From the data obtained from these cyclic voltammetry studies, a redox window for reactivity was established allowing BCB compounds to first be analyzed using this technique and then only be employed if their oxidation potential falls within this potential range. Using this guiding principle, a variety of aryl substitution patterns, different ester groups, amides, and ketones were all shown to be suitable substrates in this transformation (5ap–ax).
Figure 5.
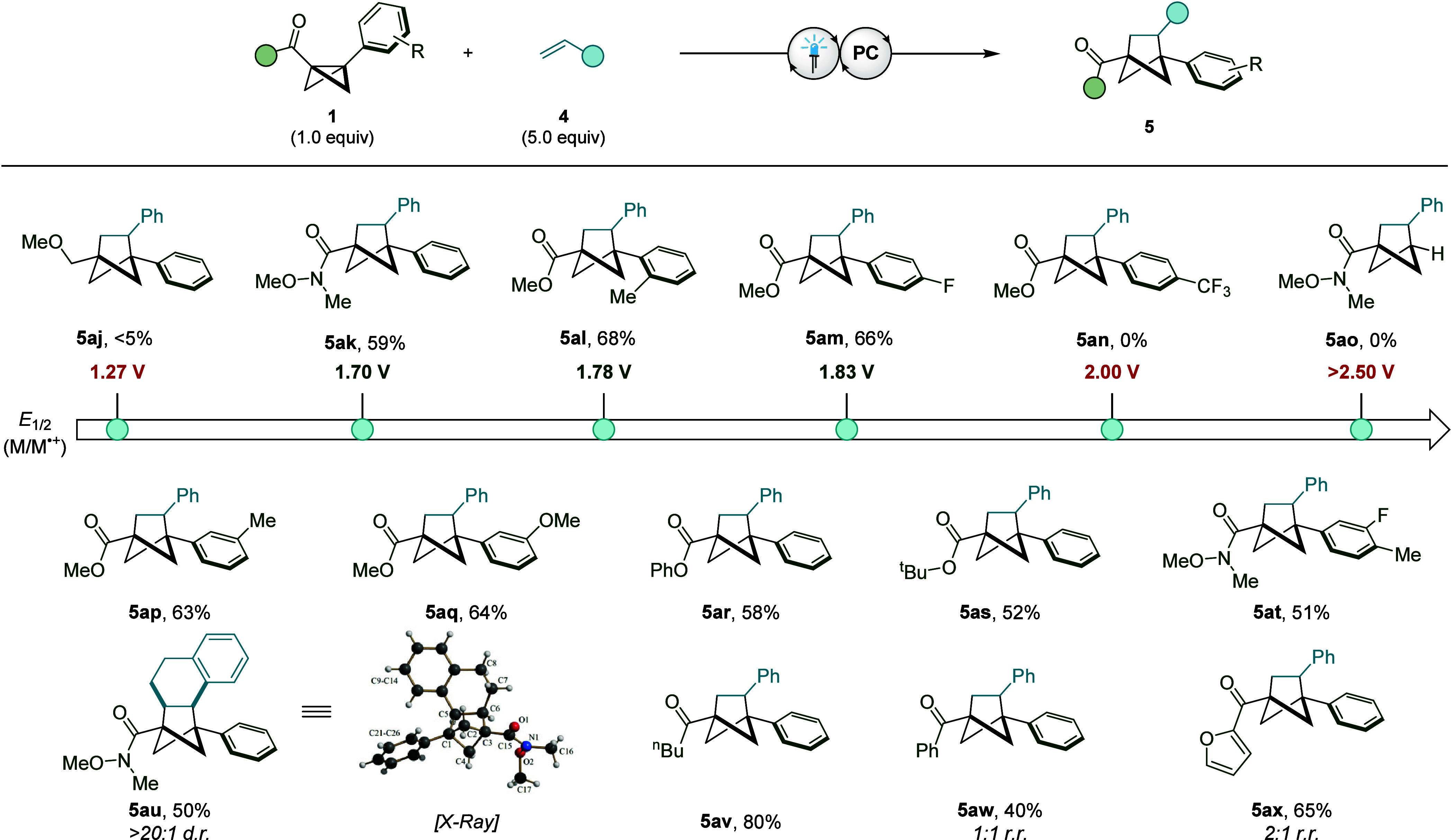
Effect of BCB oxidation potential on the [2π + 2σ] cycloaddition reaction. Reaction conditions: 1 (0.2 mmol), 4 (1.0 mmol), [Mes2AcrtBu2]ClO4 (10 mol %), MeCN (0.1 M), blue LEDs (λmax = 425 nm), 16 h. Isolated yields given. Oxidation potentials of the corresponding BCB starting materials are given in MeCN against the Ag/AgCl electrode (2 M LiCl in EtOH). The d.r. and r.r. values were determined by 1H NMR analysis of the crude reaction mixture.
Mechanistic Studies
To confirm that BCB radical cation 1•+ is indeed responsible for reactivity, and to establish the mechanism of the [2π + 2σ] cycloaddition reaction, we set about designing experiments that could provide a deeper understanding of the transformation described. First, UV/vis spectroscopy of the individual reaction components revealed that the photocatalyst [Mes2AcrtBu2]ClO4 is the only light absorbing species at λ = 425 nm, confirming that direct excitation of either BCB 1a or alkene 2a cannot be responsible for reactivity (Figure 6A). Furthermore, Stern–Volmer quenching studies clearly demonstrated that BCB 1a is an effective quencher of the photocatalyst excited state, whereas alkene 2a gave no indication that it can interact with this excited state species (Figure 6B). However, quenching was detected, albeit to a lesser extent than for 1a, upon the addition of styrene (4a). These observations are in full corroboration with the cyclic voltammetry (CV) experiments that were performed (Figure 6C). Here, both BCB 1a (+1.79 V vs Ag/AgCl) and styrene 4a (+2.03 V vs Ag/AgCl) show oxidation peaks that were deemed accessible for the photocatalyst excited state, whereas 2a was observed to have an oxidation potential well outside this range (+2.36 V vs Ag/AgCl). Additionally, the quantum yield for the standard reaction was calculated to be φ = 3.7, revealing that this cycloaddition transformation can proceed via a radical chain mechanism. Overall, these results strongly suggest that, in the case of nonactivated alkenes, BCB radical cation 1•+ acts as the key intermediate in the [2π + 2σ] cycloaddition reaction. Despite undergoing oxidation by the photocatalyst, radical cations arising from styrene-type alkenes were found, during DFT studies, to be unable to lead to product formation and so this alternative pathway could be eliminated as a possibility (see Supporting Information for details). Given that the oxidation of the bicyclobutane framework constitutes an activation mode that has been underexplored in synthesis, we performed further mechanistic experiments to determine whether the initial interaction of the BCB radical cation and the alkene proceeds via radical addition to give an intermediate of type 6, or occurs via alkene nucleophilic addition to give the corresponding “electromer” (7, Figure 6D).53 When radical trapping agent TEMPO was added to the standard reaction, product formation was not entirely suppressed, whereas the addition of MeOH resulted primarily in the formation of 9, with only trace product being observed (Figure 6E). Furthermore, in the case of the TEMPO experiment, the observation of a 1:1:1 adduct of 1a, alkene, and TEMPO (8) suggests the presence of a carbon-centered radical in the mechanism. In an attempt to establish whether a carbon-centered radical is present at the C8 position of the BCB-alkene adduct (6), cyclopropane-containing alkenes 2z and 4ay were subjected to standard reaction conditions. However, no cyclopropane ring opening could be detected in either case and cycloadducts 3z and 5ay were isolated in 40% and 41% yield, respectively (Figure 6F). Given that intramolecular cyclization may still occur at a faster rate than cyclopropane ring-opening, these results are not entirely conclusive in elucidating the nature of the initial interaction of the BCB radical cation and the alkene. Consequently, we turned to computational calculations to provide key insights into the operative mechanism.
Figure 6.
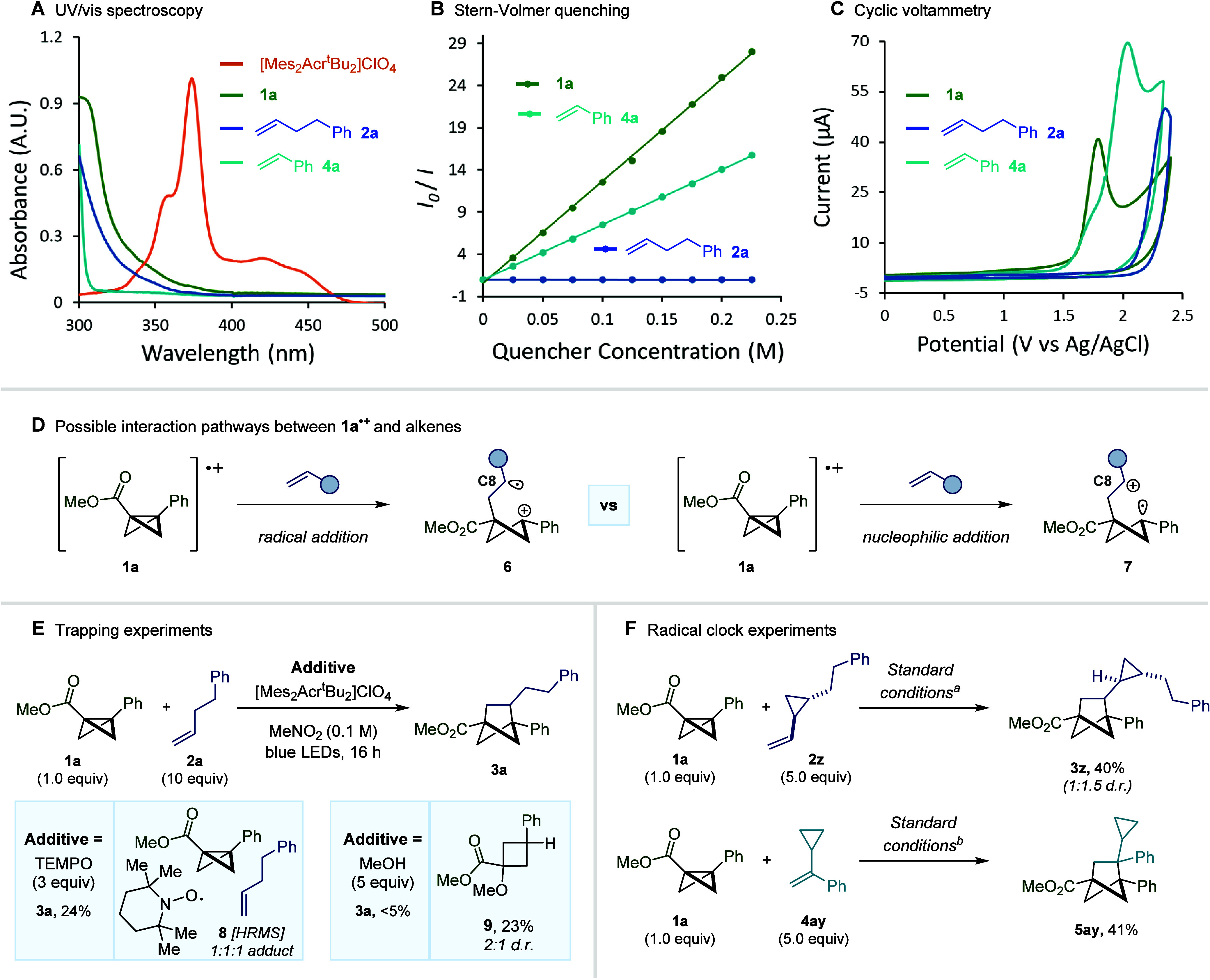
Mechanistic studies. (A) Ultraviolet–visible absorption spectra of the reaction components. (B) Stern–Volmer quenching studies. (C) Cyclic voltammetry measurements versus the Ag/AgCl reference electrode (2 M LiCl in EtOH). (D) Potential intermediates arising from the interaction of 1a•+ and an alkene. (E) Trapping experiments. (F) Radical clock experiments. aStandard conditions from Figure 3. bStandard conditions from Figure 4.
DFT Calculations
When employing density functional theory (DFT) calculations to further study the reaction pathway, it was observed that the complexation of the radical cation 1a•+ with a simple alkene (propene) to form IM-I•+ is exergonic by 1.2 kcal/mol (Figure 7A). Subsequent insertion of the alkene fragment into the BCB scaffold was found to be a kinetically facile process (TS-Ia), with a free energy barrier of 10.0 kcal/mol with respect to the preceding IM-I•+. To rationalize the regiochemistry of this initial bond forming process, all other possible transition states (TS-Ib, TS-Ic, and TS-Id) were computed and were all found to have significantly higher free energy barriers. From TS-Ia, formation of the subsequent intermediate IM-II•+ was determined to be exergonic by 5.5 kcal/mol.
Figure 7.
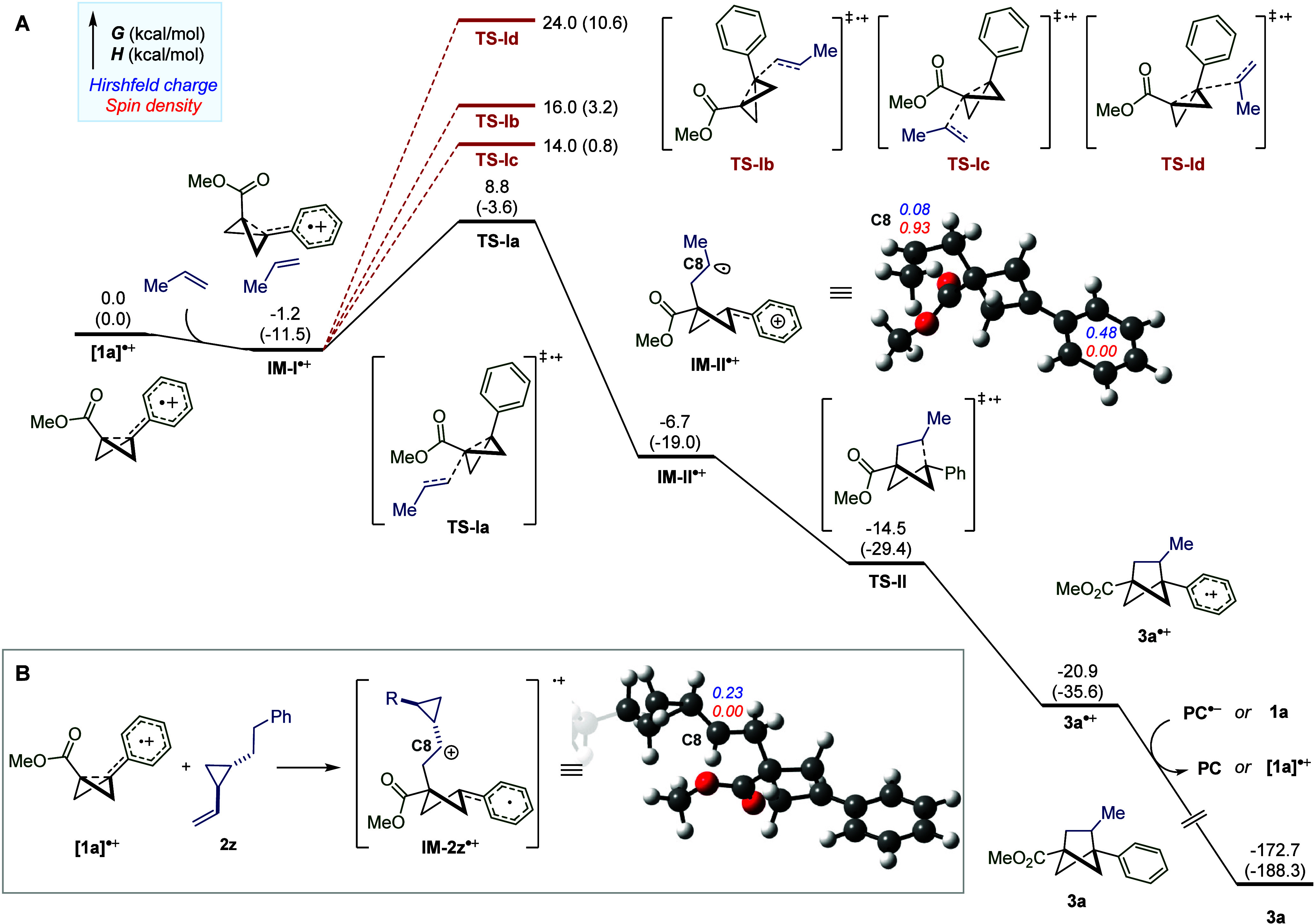
(A) Computed reaction coordinate profile of the [2π + 2σ] cycloaddition reaction between BCB radical cation 1a•+ and propene. (B) Computed spin densities and Hirshfeld charges of radical clock intermediate IM-2z•+. All DFT calculations were conducted at either ωb97xd/def-TZVPP/CPCM (solvent = MeCN) or ωb97xd/def2SVP/CPCM (solvent = MeCN) levels of theory (see Supporting Information for full details).
Upon closer analysis of IM-II•+ we identified that the spin density of the radical cation is highly localized on the propene α-carbon (C8). In order to rationalize the results of the radical clock experiments, we also calculated the spin densities for the corresponding intermediate with substrate 2z (IM-2z•+, Figure 7B) and found that, in this case, no spin density is localized on the carbon adjacent to the cyclopropane (C8). From this, we can conclude that the distribution of spin and charge density in the intermediate following the initial interaction of 1a•+ with olefins is highly dependent on the nature of the alkene substituents. The final C–C bond formation step in the mechanism (TS-II) was found to be a barrierless process, with an estimated free energy of −14.5 kcal/mol using a restrained calculation (see Supporting Information). These results also indicate that the intramolecular ring-closure step would occur at a faster rate than cyclopropane ring-opening.54 Finally, reduction of the thermodynamically stable 3a•+ (−20.9 kcal/mol) can then occur from either the reduced photocatalyst or a neutral BCB molecule to turn over the radical chain and generate BCH product 3a.
Conclusion
In conclusion, we have identified a new strategy for the single-electron oxidative activation of bicyclo[1.1.0]butane via photoredox catalysis. The synthetic utility of the resulting radical cation was highlighted by its ability to undergo [2π + 2σ] cycloaddition reactions in a highly regio- and diastereoselective fashion. The scope of the transformation with respect to the alkene coupling partner was remarkably broad, allowing the cycloaddition of styrene-type, heteroatom substituted, and, for the first time in this reaction class, nonactivated alkenes. A comprehensive experimental and computational mechanistic study was undertaken that confirmed the involvement of BCB radical cations and illuminated the nature of their interaction with olefins. We foresee that the work presented above can serve as a platform from which further studies into the potential synthetic applications of bicyclo[1.1.0]butyl radical cations can be built upon.
Acknowledgments
We sincerely thank Dr. Alessia Petti, Dr. Chetan C. Chintawar, Subhabrata Dutta, Johannes Erchinger, and Christian Gutheil (Universität Münster) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c04403.
Author Contributions
§ J.L.T. and F.S. contributed equally.
Generous financial support from the Alexander von Humboldt Foundation (J.L.T.) and the ERC Advanced Grant (Agreement No. 101098156, HighEnT, F.S.) are gratefully acknowledged. DFT calculations were performed on the IDRE Hoffman2 cluster at the University of California, Los Angeles, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI1053575). Additional funding was provided to K.N.H. by the NSF (CHE2153972).
The authors declare no competing financial interest.
Supplementary Material
References
- Wiberg K. B.; Ciula R. P. Ethyl bicyclo[1.1.0]butane-1-carboxylate. J. Am. Chem. Soc. 1959, 81, 5261–5262. 10.1021/ja01528a060. [DOI] [Google Scholar]
- Golfmann M.; Walker J. C. L. Bicyclobutanes as unusual building blocks for complexity generation in organic synthesis. Commun. Chem. 2023, 6, 9. 10.1038/s42004-022-00811-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C. B.; Milligan J. A.; Tilley L. J.; Sodano T. M. Bicyclobutanes: from curiosities to versatile reagents and covalent warheads. Chem. Sci. 2022, 13, 11721–11737. 10.1039/D2SC03948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett A. Recent advances in the chemistry of bicyclo- and 1-azabicyclo[1.1.0]butanes. Pure Appl. Chem. 2020, 92, 751–765. 10.1515/pac-2019-1007. [DOI] [Google Scholar]
- Tyler J. L.; Aggarwal V. K. Synthesis and applications of bicyclo[1.1.0]butyl and azabicyclo[1.1.0]butyl organometallics. Chem.—Eur. J. 2023, 29, e202300008 10.1002/chem.202300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkowska J.; Durka J.; Gryko D. Strain release - an old tool for new transformations. Chem. Commun. 2020, 56, 5718–5734. 10.1039/D0CC01771J. [DOI] [PubMed] [Google Scholar]
- Lopchuk J. M.; Fjelbye K.; Kawamata Y.; Malins L. R.; Pan C.-M.; Gianatassio R.; Wang J.; Prieto L.; Bradow J.; Brandt T. A.; Collins M. R.; Elleraas J.; Ewanicki J.; Farrell W.; Fadeyi O. O.; Gallego G. M.; Mousseau J. J.; Oliver R.; Sach N. W.; Smith J. K.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-release heteroatom functionalization: development, scope, and stereospecificity. J. Am. Chem. Soc. 2017, 139, 3209–3226. 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianatassio R.; Lopchuk J. M.; Wang J.; Pan C.-M.; Malins L. R.; Prieto L.; Brandt T. A.; Collins M. R.; Gallego G. M.; Sach N. W.; Spangler J. E.; Zhu H.; Zhu J.; Baran P. S. Strain-release amination. Science 2016, 351, 241–246. 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellotti P.; Glorius F. Strain-release photocatalysis. J. Am. Chem. Soc. 2023, 145, 20716–20732. 10.1021/jacs.3c08206. [DOI] [PubMed] [Google Scholar]
- Ma X.; Sloman D. L.; Han Y.; Bennett D. J. A selective synthesis of 2,2-difluorobicyclo[1.1.1]pentane analogues: ″BCP-F2″. Org. Lett. 2019, 21, 7199–7203. 10.1021/acs.orglett.9b02026. [DOI] [PubMed] [Google Scholar]
- Bychek R. M.; Hutskalova V.; Bas Y. P.; Zaporozhets O. A.; Zozulya S.; Levterov V. V.; Mykhailiuk P. K. Difluoro-substituted bicyclo[1.1.1]pentanes for medicinal chemistry: design, synthesis, and characterization. J. Org. Chem. 2019, 84, 15106–15117. 10.1021/acs.joc.9b01947. [DOI] [PubMed] [Google Scholar]
- McNamee R. E.; Thompson A. L.; Anderson E. A. Synthesis and applications of polysubstituted bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 2021, 143, 21246–21251. 10.1021/jacs.1c11244. [DOI] [PubMed] [Google Scholar]
- Agasti S.; Beltran F.; Pye E.; Kaltsoyannis N.; Crisenza G. E. M.; Procter D. J. A catalytic alkene insertion approach to bicyclo[2.1.1]hexane bioisosteres. Nat. Chem. 2023, 15, 535–541. 10.1038/s41557-023-01135-y. [DOI] [PubMed] [Google Scholar]
- Yan H.; Liu Y.; Feng X.; Shi L. Hantzsch esters enabled [2π + 2σ] cycloadditions of bicyclo [1.1.0] butanes and alkenes under photo conditions. Org. Lett. 2023, 25, 8116–8120. 10.1021/acs.orglett.3c03222. [DOI] [PubMed] [Google Scholar]
- Radhoff N.; Daniliuc C. G.; Studer A. Lewis acid catalyzed formal (3 + 2)-cycloaddition of bicyclo[1.1.0]butanes with ketenes. Angew. Chem., Int. Ed. 2023, 62, e202304771 10.1002/anie.202304771. [DOI] [PubMed] [Google Scholar]
- Ni D.; Hu S.; Tan X.; Yu Y.; Li Z.; Deng L. Intermolecular formal cycloaddition of indoles with bicyclo[1.1.0]butanes by Lewis acid catalysis. Angew. Chem., Int. Ed. 2023, 62, e202308606 10.1002/anie.202308606. [DOI] [PubMed] [Google Scholar]
- Tang L.; Xiao Y.; Wu F.; Zhou J.-L.; Xu T.-T.; Feng J.-J. Silver-catalyzed dearomative [2π + 2σ] cycloadditions of indoles with bicyclobutanes: access to indoline fused bicyclo[2.1.1]hexanes. Angew. Chem., Int. Ed. 2023, 62, e202310066 10.1002/anie.202310066. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Paulus F.; Daniliuc C. G.; Glorius F. Catalytic formal [2π + 2σ] cycloaddition of aldehydes with bicyclobutanes: expedient access to polysubstituted 2-oxabicyclo[2.1.1]hexanes. Angew. Chem., Int. Ed. 2023, 62, e202305043 10.1002/anie.202305043. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Lin S.; Li Y.; Xue J.-H.; Li Q.; Wang H. Pyridine-boryl radical-catalyzed [2π + 2σ] cycloaddition of bicyclo[1.1.0]butanes with alkenes. ACS Catal. 2023, 13, 5096–5103. 10.1021/acscatal.3c00305. [DOI] [Google Scholar]
- Xu M.; Wang Z.; Sun Z.; Ouyang Y.; Ding Z.; Yu T.; Xu L.; Li P. Diboron(4)-catalyzed remote [3 + 2] cycloaddition of cyclopropanes via dearomative/rearomative radical transmission through pyridine. Angew. Chem., Int. Ed. 2022, 61, e202214507 10.1002/anie.202214507. [DOI] [PubMed] [Google Scholar]
- Guo R.; Chang Y.-C.; Herter L.; Salome C.; Braley S. E.; Fessard T. C.; Brown M. K. Strain-release [2π + 2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes initiated by energy transfer. J. Am. Chem. Soc. 2022, 144, 7988–7994. 10.1021/jacs.2c02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A. R.; Wang Y.; Wiberg K. B. Excited states and photochemistry of bicyclo[1.1.0]butane. J. Phys. Chem. A 2009, 113, 1686–1695. 10.1021/jp807407c. [DOI] [PubMed] [Google Scholar]
- Tokunaga K.; Sato M.; Kuwata K.; Miura C.; Fuchida H.; Matsunaga N.; Koyanagi S.; Ohdo S.; Shindo N.; Ojida A. Bicyclobutane carboxylic amide as a cysteine-directed strained electrophile for selective targeting of proteins. J. Am. Chem. Soc. 2020, 142, 18522–18531. 10.1021/jacs.0c07490. [DOI] [PubMed] [Google Scholar]
- Schwartz B. D.; Smyth A. P.; Nashar P. E.; Gardiner M. G.; Malins L. R. Investigating bicyclobutane-triazolinedione cycloadditions as a tool for peptide modification. Org. Lett. 2022, 24, 1268–1273. 10.1021/acs.orglett.1c04071. [DOI] [PubMed] [Google Scholar]
- Dhake K.; Woelk K. J.; Becica J.; Un A.; Jenny S. E.; Leitch D. C. Beyond bioisosteres: divergent synthesis of azabicyclohexanes and cyclobutenyl amines from bicyclobutanes. Angew. Chem., Int. Ed. 2022, 61, e202204719 10.1002/anie.202204719. [DOI] [PubMed] [Google Scholar]
- Gassman P. G.; Yamaguchi R. Electrochemical oxidation of strained hydrocarbons. J. Am. Chem. Soc. 1979, 101, 1308–1310. 10.1021/ja00499a057. [DOI] [Google Scholar]
- Gassman P. G.; Mullins M. J.; Richtsmeier S.; Dixon D. A. Effect of alkyl substitution on the ease of oxidation of bicyclo[1.1.0]butanes. Experimental verification of PRDDO calculations for the nature of the HOMO of bicyclo[1.1.0]butane. J. Am. Chem. Soc. 1979, 101, 5793–5797. 10.1021/ja00513a054. [DOI] [Google Scholar]
- Hoz S.; Livneh M.; Cohen D. Cyclobutane-bicyclobutane system. Part 13. Bromination of bicyclobutanes: a possible case of an electron-transfer mechanism. J. Am. Chem. Soc. 1987, 109, 5149–5156. 10.1021/ja00251a018. [DOI] [Google Scholar]
- Concurrent with the work presented in this manuscript, bicyclobutyl radical cations were suggested as a possible intermediate in the dearomatizing cycloaddition reaction of bicyclobutane and phenols. See:; Dutta S.; Lee D.; Ozols K.; Daniliuc C. G.; Shintani R.; Glorius F. Photoredox-enabled dearomative [2π + 2σ] cycloaddition of phenols. J. Am. Chem. Soc. 2024, 146, 2789–2797. 10.1021/jacs.3c12894. [DOI] [PubMed] [Google Scholar]
- Gassman P. G.; Carroll G. T. The reaction of 1,2,2-trimethylbicyclo[1.1.0]butane with excited state 1-cyanonaphthalene. Tetrahedron 1986, 42, 6201–6206. 10.1016/S0040-4020(01)88081-9. [DOI] [Google Scholar]
- Gassman P. G.; Olson K. D.; Walter L.; Yamaguchi R. Photoexcitation of nonconjugated, strained, saturated hydrocarbons. Relationship between ease of oxidation and the quenching of naphthalene fluorescence by saturated hydrocarbons. J. Am. Chem. Soc. 1981, 103, 4977–4979. 10.1021/ja00406a067. [DOI] [Google Scholar]
- Gassman P. G.; Olson K. D. Photochemistry of saturated hydrocarbons. Mechanistic changes as a function of methyl substitution in the photosensitized reactions of the tricyclo[4.1.0.02, 7]heptyl system. J. Am. Chem. Soc. 1982, 104, 3740–3742. 10.1021/ja00377a042. [DOI] [Google Scholar]
- Saettel N. J.; Wiest O. Sterically crowded bicyclo[1.1.0]butane radical cations. J. Org. Chem. 2003, 68, 4549–4552. 10.1021/jo0267549. [DOI] [PubMed] [Google Scholar]
- Denisenko A.; Garbuz P.; Shishkina S. V.; Voloshchuk N. M.; Mykhailiuk P. K. Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem., Int. Ed. 2020, 59, 20515–20521. 10.1002/anie.202004183. [DOI] [PubMed] [Google Scholar]
- Herter L.; Koutsopetras I.; Turelli L.; Fessard T.; Salomé C. Preparation of new bicyclo[2.1.1]hexane compact modules: an opening towards novel sp3-rich chemical space. Org. Biomol. Chem. 2022, 20, 9108–9111. 10.1039/D2OB01669A. [DOI] [PubMed] [Google Scholar]
- Mykhailiuk P. K. Saturated bioisosteres of benzene: where to go next?. Org. Biomol. Chem. 2019, 17, 2839–2849. 10.1039/C8OB02812E. [DOI] [PubMed] [Google Scholar]
- Rigotti T.; Bach T. Bicyclo[2.1.1]hexanes by visible light-driven intramolecular crossed [2 + 2] photocycloadditions. Org. Lett. 2022, 24, 8821–8825. 10.1021/acs.orglett.2c03606. [DOI] [PubMed] [Google Scholar]
- Kleinmans R.; Pinkert T.; Dutta S.; Paulisch T. O.; Keum H.; Daniliuc C. G.; Glorius F. Intermolecular [2π + 2σ]-photocycloaddition enabled by triplet energy transfer. Nature 2022, 605, 477–482. 10.1038/s41586-022-04636-x. [DOI] [PubMed] [Google Scholar]
- Kleinmans R.; Dutta S.; Ozols K.; Shao H.; Schäfer F.; Thielemann R. E.; Chan H. T.; Daniliuc C. G.; Houk K. N.; Glorius F. ortho-Selective dearomative [2π + 2σ] photocycloadditions of bicyclic aza-arenes. J. Am. Chem. Soc. 2023, 145, 12324–12332. 10.1021/jacs.3c02961. [DOI] [PubMed] [Google Scholar]
- Dutta S.; Lu Y.-L.; Erchinger J. E.; Shao H.; Studer E.; Schäfer F.; Wang H.; Rana D.; Daniliuc C. G.; Houk K. N.; Glorius F. Double Strain-Release [2π + 2σ]-Photocycloaddition. J. Am. Chem. Soc. 2024, 146, 5232–5241. 10.1021/jacs.3c11563. [DOI] [PubMed] [Google Scholar]
- de Robichon M.; Kratz T.; Beyer F.; Zuber J.; Merten C.; Bach T. Enantioselective, intermolecular [π2+σ2] photocycloaddition reactions of 2(1H)-quinolones and bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 2023, 145, 24466–24470. 10.1021/jacs.3c08404. [DOI] [PubMed] [Google Scholar]
- Ischay M. A.; Ament M. S.; Yoon T. P. Crossed intermolecular [2 + 2] cycloaddition of styrenes by visible light photocatalysis. Chem. Sci. 2012, 3, 2807–2811. 10.1039/c2sc20658g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Lu Z.; Yoon T. P. [2 + 2] cycloadditions by oxidative visible light photocatalysis. J. Am. Chem. Soc. 2010, 132, 8572–8574. 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Ischay M. A.; Fry C. G.; Yoon T. P. Radical cation Diels-Alder cycloadditions by visible light photocatalysis. J. Am. Chem. Soc. 2011, 133, 19350–19353. 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz D. P.; Griesbeck A. G.; Alle R.; Perez-Ruiz R.; Neudörfl J. M.; Meerholz K.; Schmalz H.-G. Molecular oxygen as a redox catalyst in intramolecular photocycloadditions of coumarins. Angew. Chem., Int. Ed. 2012, 51, 6000–6004. 10.1002/anie.201201222. [DOI] [PubMed] [Google Scholar]
- Poplata S.; Tröster A.; Zou Y.-Q.; Bach T. Recent advances in the synthesis of cyclobutanes by olefin [2 + 2] photocycloaddition reactions. Chem. Rev. 2016, 116, 9748–9815. 10.1021/acs.chemrev.5b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Chen C. An approach for the synthesis of nakamuric acid. Tetrahedron 2015, 71, 3690–3693. 10.1016/j.tet.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzer L.; Sandfort F.; Strieth-Kalthoff F.; Glorius F. Carbonyl-olefin cross-metathesis through a visible-light-induced 1,3-diol formation and fragmentation sequence. Angew. Chem., Int. Ed. 2018, 57, 16219–16223. 10.1002/anie.201810221. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-transfer state of 9-mesityl-10-methylacridinium ion with a much longer lifetime and higher energy than that of the natural photosynthetic reaction center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Ohkubo K. Organic synthetic transformations using organic dyes as photoredox catalysts. Org. Biomol. Chem. 2014, 12, 6059–6071. 10.1039/C4OB00843J. [DOI] [PubMed] [Google Scholar]
- Joshi-Pangu A.; Lévesque F.; Roth H. G.; Oliver S. F.; Campeau L.-C.; Nicewicz D.; DiRocco D. A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]
- Pitzer L.; Schäfers F.; Glorius F. Rapid assessment of the reaction-condition-based sensitivity of chemical transformations. Angew. Chem., Int. Ed. 2019, 58, 8572–8576. 10.1002/anie.201901935. [DOI] [PubMed] [Google Scholar]
- Bally T. Isomerism: the same but different. Nat. Chem. 2010, 2, 165–166. 10.1038/nchem.564. [DOI] [PubMed] [Google Scholar]
- Newcomb M. Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151–1176. 10.1016/S0040-4020(01)85808-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.