

Abstract
Transcriptional repression within a complex modular promoter may play a key role in determining the action of enhancer elements. In human cytomegalovirus, the major immediate-early promoter (MIEP) locus contains a highly potent and complex modular enhancer. Evidence is presented suggesting that sequences of the MIEP between nucleotide positions −556 and −673 function to prevent transcription activation by enhancer elements from the UL127 open reading frame divergent promoter. Transient transfection assays of reporter plasmids revealed repressor sequences located between nucleotides −556 and −638. The ability of these sequences to confer repression in the context of an infection was shown using recombinant viruses generated from a bacterial artificial chromosome containing an infectious human cytomegalovirus genome. In addition to repressor sequences between −556 and −638, infection experiments using recombinant virus mutants indicated that sequences between −638 and −673 also contribute to repression of the UL127 promoter. On the basis of in vitro transcription and transient transfection assays, we further show that interposed viral repressor sequences completely inhibit enhancer-mediated activation of not only the homologous but also heterologous promoters. These and other experiments suggest that repression involves an interaction of host-encoded regulatory factors with defined promoter sequences that have the property of proximally interfering with upstream enhancer elements in a chromatin-independent manner. Altogether, our findings establish the presence of a boundary domain that efficiently blocks enhancer-promoter interactions, thus explaining how the enhancer can work to selectively activate the MIEP.
The major immediate-early promoter (MIEP) enhancer region of cytomegalovirus (CMV) is required for optimal infection (3) and plays an important role in determining the cell type tropism and state of activation in vivo (4, 5, 24, 28). The MIEP is highly complex and contains one of the most potent transcriptional enhancers known to date (7; reviewed in reference 20). Previous studies have demonstrated the importance of a wide variety of positive cis-acting enhancer modules in promoting high levels of transcription from the human CMV (HCMV) MIEP. In addition, the viral IE2 (IE86 and L40) proteins act as autorepressors of the HCMV MIEP by binding the cis repression sequence (14, 30, 34, 39, 41, 42). The location of this sequence, between the initiation site of transcription and the TATA box sequence, enables IE2 proteins to competitively block the recruitment of RNA polymerase II at the MIEP (31, 42). Repression is also predicted to be essential for establishing stringent regulation of the MIEP enhancer, which is likely to be an important feature in determining temporal and cell-type-specific patterns of viral gene expression as well as preventing promiscuous promoter interactions.
In the species-specific mouse CMV (MCMV), a similarly complex but distinct enhancer/promoter region is involved in controlling expression from its major immediate-early (MIE) genes (15). A divergent promoter upstream from its MIEP drives the expression of the MCMV ie2 gene (Fig. 1) (37). This gene is dispensable for growth in tissue culture and in vivo and comes under control of the nearby enhancer, thereby displaying IE kinetics of expression (9, 35). The observation that the MCMV enhancer plays a dual role in regulating ie1 and ie2 expression is not unexpected since by definition enhancers function equally well in either orientation. Similar to the organization of MCMV MIE locus, the genome sequence from HCMV (11) identified a divergent open reading frame (ORF) (UL127) in close proximity to the enhancer (Fig. 1) and which overlaps the modulator domain of the human MIEP (Fig. 2A). While the HCMV UL127 ORF is dispensable for replication in tissue culture, it does not appear to have any significant homology with the mouse viral ie2 gene (36, 37). In contrast to the MCMV ie2 gene, expression of UL127-encoding transcript is not detected upon infection of human foreskin fibroblasts (HFF cells) by HCMV (10). The UL127 promoter region contains an excellent TATA box (10 out of 11 nucleotides [nt] match with the MIEP TATA box [Fig. 1]) and binds CTF-1/NF-1 at the expected CAAT box position (18). Thus, the observation for inactivity of this divergent promoter is unexpected due to the strong potency of the CMV enhancer and raises the question as to why the enhancer is unable to influence expression of the UL127 promoter site. It is conceivable that repressors stringently regulate the UL127 promoter. To investigate this issue, we have examined the requirement of cis-acting sequences in the promoter-proximal region of UL127 for limiting enhancer action.
FIG. 1.

Schematic diagrams of the HCMV and MCMV genomes. Fragments containing the MIE regions are expanded below the corresponding regions of the genomes. Locations and direction of transcription of the ORFs for the MIE genes ie1, ie2, and ie3 and the potential UL127 ORF are indicated. The gray box depicts the enhancer. The TATA box sequences of the different ORFs are shown. The diagram is not drawn to scale.
FIG. 2.


NF-1 binding sites flanking the UL127 TATA box do not mediate transcriptional repression. (A) Binding of various transcription factors (marked by open boxes) to HCMV MIEP sequences between −244 and −781 (based on DNase I footprinting data [reviewed in reference 20]). Locations of the enhancer, usr, NF-1 cluster, modulator, and UL127 TATA box are shown. Numbers refer to nucleotide positions relative to the transcription start site (+1) of the MIEP. CAT reporter constructs [pSnab-BamCAT, pSnab-Bam(HI)CAT, pGACC(−673), and pGACC(−531] with various 5′ and 3′ deletion endpoints are shown below. (B) HFF, HeLa, U373-MG, and NT-2/D1 cells were transfected with 5 μg of the various promoter-CAT deletion constructs shown in panel A together with 5 μg of the control plasmid pRSV-β-gal. Cell lysates were prepared 30 h after transfection and assayed for β-galactosidase and CAT activity. For the CAT assays, cell extracts containing the same amount of β-galactosidase activity were used. A plot of the normalized percentage of CAT activity calculated for each construct taking as 1 the activity presented by pSnaB-BamCAT is shown. The CAT values shown represent the average ± standard deviation (bars) of three determinations.
Here, we present evidence that sequences located between −556 and −673 of the MIEP serve to completely block enhancer interactions with the UL127 promoter site. This region therefore plays an important role in defining the boundary of action of the MIEP enhancer in HCMV.
MATERIALS AND METHODS
Cells.
The cell lines U373-MG, HeLa, NT-2/D1, and MRC-5, as well as the HFF cells, were grown in Dulbecco's modified Eagle medium supplemented with 2 mM glutamine, 100 U of penicillin per ml, 100 μg of gentamicin per ml, and 10% (vol/vol) fetal bovine serum.
Plasmid constructions and transfections.
Plasmid pSnaB-Bam(HI)CAT was constructed by inserting a BamHI/SpeI fragment from pMIEP(HIB)CAT (17) into BamHI/SpeI-digested pSnaB-BamCAT. Plasmid pSnaB-BamCAT contains regulatory sequences of the HCMV MIE gene from nt −243 to −741 linked to the chloramphenicol acetyltransferase (CAT) gene. To construct pGACC(−531) and pGACC(−673), recombinant plasmid pGACC was generated by inserting annealed oligonucleotides G53 (5′-GATCCATTGGTTATATAGCATAACTAGTAAGCTTCTACGTAC-3′) and G52 (5′-AGCTGTACGTAGAAGCTTACTAGTTATGCTATATAACCAATG-3′), containing SnaBI, HindIII, and SpeI sites, into BamHI/HindIII-digested pMIEP(−66/+55)CAT (42). A HindIII/SnaBI fragment from Δ1 and pR55 (kindly provided by B. Fleckenstein) was cloned into pGACC digested with HindIII/SnaBI to create pGACC(−531) and pGACC(−673), respectively. Plasmids pGACC(−531) and pGACC(−673) were digested with SnaBI and BamHI and ligated into SmaI/BglII-digested pGL3-Basic (Promega, Madison, Wis.) to create pGL3(−531) and pGL3(−673), respectively. To construct pE(−66/+112)CAT and pEusr(−66/+112)CAT, a 190-bp BamHI/HindIII fragment from pMIEP(−66/+112) (44) that contains HCMV MIEP sequences from −66 to +112 was inserted into BamHI/HindIII-digested pGACC(−531) and pGACC(−598), respectively. Plasmids pGL3(−634), pGL3(−581), pGL3(−556), and pGL3(−531) were constructed using the PCR primers promExt-1 (5′-TGTACGTAGATGTACTGCCAAGT-3′), promExt-2 (5′-TGAAGCTTGGTCATTAGTTCATAGCCAT-3′), promExt-3 (5′-TGAAGCTTAGTTATTAATAGTAATCAATTAC-3′), promExt-4 (5′-TGAAGCTTCATGTTGACATTGATTATTGA-3′), and promExt-5 (5′-TGAAGCTTTTTATATTGGCTCATGTCCAAC-3′). All PCRs were performed with pMIEP(−1145/+112)CAT (19) and primer promExt-1. Primers promExt-2, promExt-3, promExt-4, and promExt-5 were used to generate pGL3(−634), pGL3(−581), pGL3(−556), and pGL3(−531), respectively. Primer promExt-1 contains a 5′ SnaBI linker, while primers promExt-2, promExt-3, promExt-4, and promExt-5 contain a 5′ HindIII linker. The corresponding PCR products were digested with SnaBI and HindIII and inserted into SnaBI/HindIII pGACC(−531). The SnaBI/BamHI fragments excised from the recombinant plasmids generated were then inserted into SmaI/BglII-digested pGL3-Basic to generate plasmids pGL3(−634), pGL3(−581), pGL3(−556), and pGL3(−531). Plasmids pGL3(Δ−531/−638) and pGL3(Δ−531/−609) were made by utilizing PCR primers promRev-1 (5′-TGGGATCCCATTGGTTATATA-3′), promRev-2 (5′-TGTGATCACATATTATGATATGG-3′), and promRev-3 (5′-TGTGATCAGTCAACATGGCGGT-3′). All PCRs were performed with pMIEP(−1145/+112)CAT and primer promRev-1. Primer promRev-2 was used to create pGL3(Δ−531/−638), and promRev-3 was used to generate pGL3(Δ−531/−609). Primer promRev-1 contains a 5′ BamHI linker, while primers promRev-2 and promRev-3 contain a 5′ BclI linker. The two PCR products generated were cloned between the SpeI and the BamHI sites of pGACC(−531). SnaBI/BamHI fragments were excised from the two recombinant plasmids constructed and inserted into SmaI/BglII-digested pGL3-Basic. The β-galactosidase expression vector pRSV-β-gal and the luciferase expression vector pRL-tk (Promega) were used as internal controls in transfection assays.
Transfections were performed by the calcium phosphate precipitation method as described previously (19). Cell lysates were prepared 30 h after transfection. β-Galactosidase and CAT assays were performed as previously described (2). Luciferase activity was determined according to the Promega's Dual-Luciferase reporter assay system technical manual. The activity of the experimental luciferase reporters was normalized to the activity of the internal control pRL-tk. For CAT assays, cell extracts containing the same amount of β-galactosidase were used. CAT activity was quantitated by using a Molecular Dynamics PhosphorImager system with ImageQuant software.
BAC mutagenesis and virus construction.
For generation of recombinant viruses UL127-GFP1, UL127-GFP10, and UL127-GFP7, three plasmids were constructed that contained a green fluorescent protein (GFP) reporter gene from pEGFP-C1 (Clontech, Palo Alto, Calif.) downstream of the UL127 sequences which were isolated from reporter plasmids pGACC(−673), pGACC(−531/−638), and pGACC(−531), respectively (Fig. 2 and 6). Note that in these constructs the GFP gene was inserted as a separate ORF upstream of the UL127 ORF but downstream of the UL127 promoter sequences. Briefly, enhanced GFP (EGFP) primers 5′-GGCCTGCAGATCTGCTAGCGCTACCGGTCGCCA-3′ and 5′-GGCCTGCAGTTTAAACTCACTTGTACAGCTCGTCCATGCC-3′, containing BglII and PmeI sites, respectively, were used to PCR amplify the EGFP gene. The resultant PCR fragment was digested with PstI and inserted into the NsiI site at position −755 of pMIEP(−1145/+112)CAT. Next a BglII site (introduced from the EGFP primer) and the SnaBI site of the wild-type (wt) MIEP sequence (at position −243) were used to replace the wt MIEP sequence with BamHI/SnaBI fragments containing the deletion mutations of pGACC(−673), pGACC(−531/−638), and pGACC(−531), respectively. A tetracycline resistance gene flanked with FRT sites was excised from plasmid pCP16 (13) and inserted into a unique PmeI site downstream of the EGFP gene. The resulting constructs were digested with PstI (−1145) and EagI (+78), generating fragments that contained one of the three different MIEP sequences each, the GFP gene, the tetracycline resistance gene, and 385 bp of downstream sequences comprising the UL127 ORF. Recombination between the linear fragments and the HCMV bacterial artificial chromosome (BAC) plasmid pHB5 (6) was performed in the recombination-proficient Escherichia coli strain JC8679 according to a recently described mutagenesis procedure (44). Minipreparations of BAC DNA isolated from 10-ml bacterial cultures were prepared and characterized by digestion with restriction enzymes SpeI, SalI, and SnaBI. HCMV BACs that had received the mutation were then transformed in the recombination-deficient E. coli strain DH10B. Excision of the tetracycline resistance cassette with Flp recombinase was performed essentially as described elsewhere (13) and confirmed by restriction enzyme analysis. In addition, the regions of interest were sequenced in the different BACs. Sequences were as expected for all recombinants except in BAC UL127-GFP10 between nt −688 and −676 relative to the transcription start site of the MIEP that contain 10 mismatched nucleotides, disrupting the NF-1 site 5′ proximal to the UL127 TATA box. Midipreparations of BACs were used to transfect MRC-5 cells for generation of infectious recombinant virus as described previously (6). Infection of HFF cells was used to generate viral stocks, and titers of infectious virus were determined by standard methods.
FIG. 6.

Construction of UL127-GFP HCMV BAC recombinant genomes. (A) The top line represents the parental (wt) HCMV genome with the SalI fragment (nt 169746 to 177147 [11]) containing the MIE region expanded below. Sequences corresponding to the enhancer and usr (open rectangle) and the UL127 ORF (black box) are indicated. Recombinant HCMV BAC genomes, UL127-GFP1, UL127-GFP10, and UL127-GFP7, containing the GFP ORF (open rectangle) under the control of the various UL127 promoter deletion mutants are shown. UL127 promoter deletions are indicated by black boxes, and the coordinates for these deletions relative to the HCMV ie1/ie2 transcription start site are shown. The diagram is not drawn to scale. (B) Ethidium bromide-stained agarose gels of SalI-digested BAC plasmids UL127-GFP1, UL127-GFP10, UL127-GFP7, and parental HCMV BAC (pHB5) after separation on a 0.5% agarose gel. Positions of size markers are shown at the right; sizes of the natural and new SalI fragments for each virus are shown with arrows.
In vitro runoff transcription assay.
Transcription reactions (25 μl) were performed with nuclear extracts prepared from HeLa cells as described previously (18). The templates pMIEP(−66/+112), pE(−66/+112), and pEusr(−66/+112) were linearized with EcoRI and used in the transcription reactions at a concentration of 25 μg/ml. Transcript products were analyzed by electrophoresis on a denaturing (8.3 M urea) 6% polyacrylamide gel. Relative levels of transcription were quantitated using a PhosphorImager (Molecular Dynamics). Poly(U) polymerase activity present in the nuclear extract was used as an internal standard to account for variability between samples.
Reverse transcriptase (RT)-mediated PCR (RT-PCR).
HFF cells were infected with the different recombinant viruses at a multiplicity of infection (MOI) of 0.1 PFU/cell. Total RNA was isolated at 13 h postinfection (hpi) by using the RNAzol B method (Tel-Test, Inc., Friendswood, Tex.) according to the manufacturer's protocol. RNA samples were treated with RNase-free DNase I for 15 min at room temperature, and the DNase was inactivated at 65°C for 15 min. The RNA was reverse transcribed using oligo(dT) primers at 42°C for 50 min, and reactions were terminated by heating at 70°C for 15 min. The reverse-transcribed products were treated with RNase H for 20 min at 37°C and amplified using the following primer sets. Primers TF-F (5′TCCTGCTCGGCTGGGTCTTCGCCCAG3′) and TF-R (5′TGTTCGGGAGGGAATCACTGCTTGAACAGT3′) were designed to amplify a 601-bp product within the human tissue factor gene. Primer GFP-R (5′TCGCCCTCGCCGGACACGCTGAACT3′) within the EGFP gene and primer GFP-F (5′CCACCATGGTGAGCAAGGGCGAGGAGC3′) located 29 to 55 nt downstream of the predicted UL127 TATA box sequence were designed to yield a 714-bp product. Primer GFP-R and primer UL127-TATA (5′CTATATAACCAATGGATCTGCTAGCGC3′), located within the predicted UL127 TATA box sequence, were designed to either fail to detect a product if the predicted TATA box was used or yield a 650-bp product in the case that an alternative TATA box upstream the predicted UL127 TATA box was used. PCRs were optimized using the HCMV recombinant BAC genomes as templates. Similar levels of PCR products were obtained for all the HCMV recombinant BACs when primer sets GFP-F/GFP-R and GFP-R/UL127 TATA were used (data not shown). PCRs were performed under the following conditions: 1 cycle at 94°C for 3 min, 30 cycles of 1 min at 94°C, 1 min at 58°C, and 1 min at 72°C, and 1 cycle at 72°C for 10 min. Amplified products were separated on a 1% agarose gel and visualized by ethidium bromide staining.
Flow cytometry.
HFF cells were infected with different HCMV recombinants at an MOI of 0.5 PFU/cell. On day 3 after infection, cultures were trypsinized, washed twice with phosphate-buffered saline (PBS) and fixed with 1% formaldehyde in PBS. Fluorescence was measured by flow cytofluorometry (fluorescence-activated cell sorting [FACS]) using a FACScan (Becton Dickinson, Mountain View, Calif.). A total of 10,000 events were collected per sample. Uncompensated fluorescence values from FL1 (GFP fluorescence, 530-nm band pass filter) and FL2 (autofluorescence; a 585-nm band pass filter) were collected along with forward angle light scatter (FSC) and side scatter (SSC) for every event. The data files were subsequently gated employing a logical gate drawn in a dot plot display of FSC versus SSC to exclude dead cells, debris, and clumps. For each data file, the resultant gated data set was displayed as a dot plot of FL2 versus GFP fluorescence and a static second region drawn around the GFP-positive (GFP+) cells, with a static third region drawn around the negative cells for the purpose of calculating statistics among the data files. The spectral differences among GFP+ and negative cells make them easy to distinguish in the bivariate display, whereas the population overlap as viewed from a single parameter histogram makes it difficult to distinguish the dull GFP+ from high autofluorescence.
Confocal laser microscopy.
HFF cells seeded onto coverslips were infected with the different HCMV recombinants at an MOI of 0.5 PFU/cell. On day 3 after infection, infected cells were fixed and permeabilized with 4% paraformaldehyde, 0.2% glutaraldehyde, and 0.2% Triton X-100 in PBS. Cells were incubated with HCMV IE monoclonal antibody 810 (dilution 1:60) for 1 h at 37°C. The coverslips were extensively washed with PBS containing 1% bovine serum albumin and incubated with a secondary antibody, tetramethyl rhodamine isothiocyanate-(TRITC)-conjugated goat anti-mouse (dilution 1:400; Sigma Immunochemicals). After a further 60-min incubation at 37°C, the coverslips were washed again with PBS containing 1% bovine serum albumin, rinsed with water, and mounted using Permount. Samples were examined using a Zeiss Axioplan confocal microscope and a 63× oil immersion objective lens. Data were collected at a resolution of 512 by 512 pixels. Data sets were processed using the MRC 1024 software and then exported for preparation for printing using Adobe Photoshop.
RESULTS
Sequences upstream of the NF-1 cluster mediate transcriptional repression of the UL127 promoter site.
Evidence for direct repression of the MIEP by its own IE2 gene product has been provided to account for the restricted expression of the IE genes in the later stages of infection (14, 30, 31, 34, 38, 39, 42). This repression system is defined by the competitive binding of the IE repressor with the RNA polymerase II recruitment step (31, 42). Our previous studies indicated that a strict requirement for this type of repression is that the position of the repressor binding site should be in the vicinity of the transcription start site (31). In other studies, binding sites 5′ proximal to a TATA box have also been shown to block the recruitment of an RNA polymerase II complex (reviewed in reference 1). The UL127 promoter is located in the center of the NF-1 cluster in which one NF-1 binding site exists 14 bp downstream of the UL127 TATA box in the immediate vicinity of the start site and another lies 10 bp upstream of the TATA box (25, 27). It is possible that these particular TATA proximal NF-1 sites may play a role in maintaining a repressed state. To investigate whether these NF-1 sites are involved in repression, we examined constructs in which promoter-proximal downstream and upstream NF-1 binding sites have been eliminated. Different cell types were cotransfected with an internal control standard and reporter plasmids with the CAT gene under the control of the UL127 promoter with or without the NF-1 binding sites. In the transient transfection assays, the wt UL127 promoter construct pSnaB-BamCAT (containing sequences between nt −243 and −741 of the MIEP) did not show any significant CAT activity in a variety of cell types (Fig. 2) despite the presence of the MIEP enhancer. The construct pSnaB-Bam(HI)CAT lacking the downstream NF-1 binding site also failed to develop significant promoter activity [Fig. 2; compare CAT responses for pSnaB-BamCAT and pSnaB-Bam(HI)CAT]. In addition, pGACC(−673), a reporter plasmid that contains neither downstream nor upstream NF-1 sites, was found to be equally ineffective in developing detectable levels of reporter gene expression [Fig. 2; compare CAT responses for pSnaB-BamCAT and pGACC(−673)]. While these experiments do not unequivocally exclude the possible involvement of NF-1, it appears that repression is not due to promoter-proximal NF-1 binding sites, indicating that additional cis-acting repression sequences might be involved. In agreement with this suggestion and in marked contrast to deletion of the NF-1 sites described above, deletion of sequences further upstream (to nt −531 relative to the MIEP transcription start site) resulted in high levels of reporter gene expression in a variety of different cell types [Fig. 2; compare CAT responses for pSnaB-BamCAT and pGACC(−531)]. It is noteworthy that the level of reporter gene activity is less than that observed with the MIEP enhancer constructs (A. Angulo and P. Ghazal, unpublished data). The reason for this lower activity is not known, but it may be due to removal of positive elements in the deletion constructs and/or presence of further repressor elements. Alternatively, the UL127 promoter may require viral factors for maximal activity. Nevertheless, the results of these experiments raise the possibility that sequences located 160-bp upstream of the UL127 promoter mediate strong repression of the UL127 promoter site.
UL127 promoter-proximal upstream sequences block enhancer activation.
We next examined in more detail the UL127 promoter-proximal upstream sequences involved in mediating transcriptional repression. Accordingly, a series of 5′ and 3′ deletion mutants (from nucleotide positions between −531 and −691) were constructed and tested for their effects on reporter (luciferase) activity in transient transfection experiments. The locations of the deletion endpoints are shown in Fig. 3A in nucleotide positions relative to the initiation site (+1) of the MIEP. In transient transfection assays, deletion of sequences between −691 and −634, in pGL3(−634), did not alter reporter activity in these cell lines [Fig. 3B; compare luciferase responses for pGL3(−673) and pGL3(−634)]. When additional sequences between −634 and −604 were deleted, an approximately 10-fold increase in reporter activity was detected [Fig. 3B; compare luciferase responses for pGL3(−673) and pGL3(−604)]. Significantly, when the construct pGL3(−581), which has an additional 23-bp region deleted, was analyzed in transient transfection assays, a further threefold increase in luciferase activity was observed [Fig. 3B; compare pGL(−581) and pGL(−604)]. Deletion of sequences between −581 and −556, in pGL3(−556), resulted in levels of reporter gene activity comparable to the ones exhibited by pGL3(−531), in which sequences from −691 to −531 were absent (Fig. 3). Altogether, these results indicate that a region between nt −556 and −634 of the MIEP mediates transcriptional repression of the UL127 promoter in a cell-type-independent manner.
FIG. 3.

Identification of the sequences that mediate transcriptional repression of the UL127 promoter. (A) Binding of various transcription factors (marked by open boxes) to HCMV-MIEP sequences between −244 and −710 (based on DNase I footprinting data [reviewed in reference 20]). The location of the UL127 TATA box is shown. Numbers refer to nucleotide positions relative to the transcription start site (+1) of the MIEP. Luciferase (LUC) reporter constructs with various 5′ and 3′ deletion endpoints are shown below. (B) HeLa and NT-2/D1 cells were transfected with 5 μg of the various promoter deletion luciferase constructs shown in panel A together with 5 μg of the control plasmid pRL-tk. Cell lysates were prepared 30 h after transfection and assayed for luciferase activity. The activity of each promoter deletion reporter construct was normalized to the activity of the internal control plasmid. A plot of the normalized percentage of luciferase activity calculated for each construct, taking as 100 the activity presented by pGL3(−531), is shown. The luciferase values shown represent the average ± standard deviation (bars) of three determinations.
In good agreement with these results, a 107-bp internal deletion mutant that eliminated sequences between nt −531 and −638, pGL3(Δ−531/−638), exhibited a 25- to 50-fold enhancement in the transcriptional activity compared with pGL3(−673) (Fig. 3). To verify further the boundary of these cis-acting repressor sequences, a mutant construct containing a shorter internal deletion of 78 bp (from nt −531 to −609) was generated (Fig. 3A). Consistent with the observed activity of pGL3(−604), this construct [pGL3(Δ−531/−609)] presented a lower reporter activity than pGL3(Δ−531/−638), indicating that sequences between −609 and −638 play a role in maintenance of the repression state (Fig. 3B). We note that these constructs collectively accommodate spacing considerations. For instance, pGL(−604) has deleted 87 bp from the promoter region, and the spacing difference between the enhancer and promoter in pGL(−531) and pGL(−531/−609) is 82 bp. In all cases, in the absence of repressor sequence, the enhancer is capable of efficiently activating the UL127 promoter in a spacing-independent manner. Altogether, we conclude from these transient transfection results that sequences between nt −556 and −634 cooperate in mediating transcriptional repression of the UL127 promoter site. These experiments thus identify within the unique sequence region (usr) a boundary domain as the unit of transcriptional repression.
To investigate whether these sequences could block enhancer activation on a heterologous promoter, the UL127 TATA box region was substituted with a core promoter fragment (from nt −66 to +112) containing the MIEP TATA box. For these experiments, three plasmids, pMIEP(−66/+112)CAT, pE(−66/+112)CAT, and pEusr(−66/+112)CAT, were constructed (Fig. 4A) and analyzed in transient transfection assays in a variety of different cells. Figure 4 shows that the pE(−66/+112)CAT reporter construct containing the enhancer region (from nt −240 to −531) upstream of the core promoter resulted in a marked increase (from 38- to 88-fold) in CAT reporter activity in comparison with the construct without the enhancer [pMIEP(−66/+122)CAT]. Strikingly, the construct pEusr(−66/+112)CAT, containing 67 bp of additional sequence from the usr (nt −531 to −598) between the enhancer and the MIEP TATA box, resulted in approximately 90% inhibition of enhancer-mediated activation of transcription [Fig. 4B; compare CAT responses for pE(−66/+112)CAT and pEusr(−66/+112)CAT]. These results support the conclusion that boundary element sequences are capable of mediating repression of enhancer function on a heterologous promoter.
FIG. 4.

Boundary domain sequences within the UL127 promoter confer repression on a heterologous promoter. (A) Schematic representation of constructs pMIEP(−66/+112)CAT, pE(−66/+112)CAT, and pEusr(−66/+112)CAT. Numbers refer to nucleotide positions relative to the MIEP transcription start site (+1, indicated by an arrow). The enhancer region, the boundary segment, and the core promoter containing the MIEP TATA box are shown. (B) HFF, U373-MG, and NT-2/D1 cells were transfected with 5 μg of either pMIEP(−66/+112)CAT, pE(−66/+112)CAT, or pEusr(−66/+112)CAT along with 5 μg of the control plasmid pRSV-β-gal. Transfections and CAT assays were performed as described in the legend to Fig. 2 and in Materials and Methods. A plot of the normalized percentage of CAT activity calculated for each construct, taking as 1 the activity presented by pMIEP(−66/+112), is shown. The CAT values shown represent the average ± standard deviation (bars) of three determinations.
We next sought to evaluate whether this mode of repression could also be observed in in vitro transcription assays. In these experiments, reporter plasmids pMIEP(−66/+112)CAT, pE(−66/+112)CAT, and pEusr(−66/+112)CAT were resected with EcoRI and assayed in transcription runoff assays using HeLa cell nuclear extracts. Transcription reactions were normalized to an internal control standard, and the amount of specific initiation of transcription was quantified by PhosphorImager analysis. As shown in Fig. 5B, the template with the MIEP core promoter [pMIEP(−66/+112)CAT] (lane 1) was transcribed at a much (fivefold) reduced level in comparison with the enhancer-containing template, pE(−66/+112)CAT (lane 2), indicating the ability of the CMV enhancer to stimulate transcription in vitro, albeit at a level much lower than observed in vivo. This level of enhancer-mediated activation was significantly lower than for transcription reactions using the pEusr(−66/+112)CAT template (compare lanes 2 and 3 in Fig. 5B). Similar inhibition results were obtained with different concentrations of reporter template (data not shown). These results suggest that cis-acting sequences within the usr repress enhancer activated transcription but do not appear to influence basal promoter activity. This is consistent with the HeLa cell transfection assay, which indicated a reduction of transcriptional activity by as much as 90% (Fig. 5A). These experiments indicate that it is possible to recapitulate, in part, repression by the boundary domain with the use of an in vitro transcription system, supporting the notion that these sequences may interact with repressor proteins to promote the process of transcriptional repression.
FIG. 5.

Boundary domain sequences within the UL127 promoter mediate repression in an in vitro transcription system. (A) HeLa cells were transfected with 5 μg of either pMIEP(−66/+112)CAT (lane 1), pE(−66/+112)CAT (lane 2), or pEusr(−66/+112)CAT (lane 3) along with 5 μg of the control plasmid pRSV-β-gal. Transfections and CAT assays were performed as described in the legend to Fig. 2 and in Materials and Methods. A plot of the normalized percentage of CAT activity calculated for each construct, taking as 1 the activity presented by pMIEP(−66/+112), is shown. (B) In vitro runoff transcription assays with different DNA templates, pMIEP(−66/+112)CAT (lane 1), pE(−66/+112)CAT (lane 2), and pEusr(−66/+112)CAT (lane 3), linearized with EcoRI. The transcription reactions were performed, processed, and subjected to polyacrylamide gel electrophoresis (18). The 360-nt specific runoff transcript is indicated by an arrow. The specific runoff transcripts were normalized to an internal control, and the amount of transcript was determined by PhosphorImager analysis (42). A plot of the normalized percentage of specific transcription for each DNA template, taking as 100 the activity developed by pE(−66/+112)CAT, is shown.
UL127 promoter-proximal upstream sequences mediate repression of transcription in the context of an HCMV infection.
To determine whether this boundary domain is responsible for mediating repression of the UL127 promoter in an HCMV-infected cell, we constructed a series of HCMV recombinants containing coding sequences for the GFP under the control of various UL127 promoter deletion mutants. To generate these HCMV recombinants, we used the recently described HCMV BAC system (6). In this system, the HCMV genome has been cloned and maintained as a BAC in E. coli, whereby viral progeny can be reconstituted after transfection of the HCMV BAC into eukaryotic cells permissive for HCMV. For the purpose of these studies, we constructed three independent HCMV BAC recombinant genomes, UL127-GFP1, UL127-GFP7, and UL127-GFP10. A schematic representation of parental HCMV and the HCMV BAC recombinants generated is shown in Fig. 6A. The recombinant virus, UL127-GFP1, contains the GFP ORF under the control of the UL127 promoter lacking sequences between nt −673 and −691, removing the 5′ proximal NF-1 site. Note that in transient transfection assays, deletion of these sequences did not result in any significant promoter activity. The second recombinant HCMV BAC, UL127-GFP10, contains the GFP ORF under the control of the UL127 promoter lacking sequences between nt −531 and −638 (Fig. 6A). Note that UL127-GFP10 also contains a disrupted 5′-proximal NF-1 site between nt −688 and −676 (see Materials and Methods). The third HCMV recombinant, UL127-GFP7, contains the UL127 promoter lacking sequences between −531 and −691 (Fig. 6A). The structure of the recombinant BACs generated was verified by extensive restriction analysis (SalI, SnaBI, and SpeI) and sequencing. Figure 6B shows the SalI restriction patterns. The DNAs from the recombinant HCMV BACs were identical to the DNA from the parental HCMV BAC pHB5 except for the presence of a new SalI fragment (7.9 to 8.1 kb) in the place of the natural SalI 7.4-kb fragment. This new fragment was generated, as predicted, from the introduction of the GFP ORF downstream of the mutant UL127 promoter sequences (Fig. 6A). These results indicate that the expected recombination events occurred within the UL127 region of the viral genome. MRC-5 cells were subsequently transfected with the HCMV BAC recombinants. Plaques developed in the cultures, and progeny mutant viruses were recovered from infected cells.
We next sought to examine whether deletion of repressor sequences within the UL127 promoter, identified in transient transfection assays, could also lead to an induction of transcription from the UL127 promoter in the context of a viral infection. For these experiments, RT-PCR was performed using RNA from cells infected with the recombinant viruses under study. HFF cells were infected at an equivalent MOI (0.1 PFU/cell) with the different recombinant viruses and parental HCMV RVHB5 reconstituted from the BAC plasmid pHB5 as a negative control (6). RNA from infected cells was harvested at 13 hpi, treated with DNase, and reverse transcribed. Two primer sets were designed to detect the presence of the GFP transcript. Both primer sets contained a common 3′ primer within the GFP gene. In the first primer set, the 5′ primer was chosen to anneal with sequences 29 to 55 bp downstream the predicted UL127 TATA box. This primer pair should detect a 714-bp product if GFP transcripts were present in infected cells. In the second primer set, the 5′ primer was designed to anneal with the predicted UL127 TATA box. This second primer set should either fail to detect a product in infected cells if the predicted UL127 TATA box was used to transcribe the GFP gene or yield a 750-bp product in the case of using an alternative TATA box upstream the predicted UL127 TATA box. As expected, when cells were infected with the parental virus RVHB5, which does not contain the GFP gene, a specific PCR-amplified product was not detected with any of the two primer sets used (Fig. 7A and B, lane 2). Similarly, when cells were infected with UL127-GFP1, the recombinant virus in which sequences from −673 to −691 of the UL127 promoter were absent, amplified PCR products were not obtained (Fig. 7A and B, lanes 4). In marked contrast, in cells infected with UL127-GFP7 and −10, the expected 714-bp PCR product resulting from amplification of GFP sequences using the first primer set was easily detected (Fig. 7A, lanes 6 and 8). However, RT-PCR performed on HFF cells infected with these two viral recombinants using the second primer set that contained the primer designed to anneal within the predicted UL127 TATA box did not yield significant levels of an amplified product (Fig. 7B, lanes 6 and 8). These data show that sequences between −531 and −638 (relative to the MIEP) play an important role in the blockage of the UL127 promoter in the context of a viral infection. In addition, these results indicate that transcription in cells infected with recombinant viruses lacking the boundary repressor sequences predominantly originates approximately 30 bp downstream the UL127 promoter TATA box. Interestingly, transcripts derived from the UL127 promoter are cycloheximide sensitive, indicating early expression kinetics instead of the anticipated IE expression (data not shown).
FIG. 7.

Detection of GFP transcripts in HFF cells infected with UL127-GFP HCMV recombinants. HFF cells were infected at an MOI of 0.1 with parental HCMV (RVHB5; lanes 1 and 2), UL127-GFP1 (lanes 3 and 4), UL127-GFP10 (lanes 5 and 6), and UL127-GFP7 (lanes 7 and 8). Total RNA was harvested at 13 hpi, treated with DNase, and reverse transcribed by using oligo(dT). PCR was performed using three different primer sets: one containing a 3′ primer that anneals within the GFP gene, GFP-R, and a 5′ primer situated 29 to 55 bp downstream the predicted UL127 TATA box, GFP-F (A); one containing primer GFP-R and a 5′ primer located within the predicted UL127 TATA box, UL127-TATA (B), and one specific for the human tissue factor gene (C). Amplified products were separated on a 1% agarose gel and visualized by ethidium bromide staining. Shown are products obtained in reactions containing RT (lanes 2, 4, 6, and 8) and in control reactions in which RT was not added (lanes 1, 3, 5, and 7). Position of size markers are shown at the left; sizes of the amplified products are indicated by arrows.
To further corroborate whether sequences within the UL127 promoter repress transcription in the context of a viral infection, we examined GFP reporter activity using confocal microscopy and FACS. In the first experiments, HFF cells were infected at an MOI of 0.5 PFU/cell with recombinant viruses UL127-GFP1, UL127-GFP7, and UL127-GFP10 and parental HCMV RVHB5. On day 3 postinfection, cells were fixed, prepared for immunofluorescence analysis using a monoclonal antibody specific for the MIE proteins and a secondary antibody conjugated to TRITC, and visualized by confocal microscopy. As expected, when cells were infected with the parental virus RVHB5, only nuclear staining due to the expression of the MIE proteins could be observed (Fig. 8, panels 1A and 2A). When cells were infected with UL127-GFP1, with sequences from −673 to −691 of the UL127 promoter deleted, GFP fluorescence was not detectable, while cells were clearly positive for expression of the MIE proteins (compare panels 1B and 2B in Fig. 8). In marked contrast, infection of HFF with UL127-GFP7 and UL127-GFP10 resulted in significant amounts of GFP expression. Confocal microscopy images showed that infected HFF cells simultaneously exhibited nuclear staining due to the MIE proteins and expressed GFP in the nucleus and cytoplasm (Fig. 8, panels C1 to -3 and D1 to -3). Therefore, sequences between −531 and −638 (relative to the MIEP) are sufficient in shutting off the UL127 promoter in the context of a viral infection in cells where the divergent MIE promoter is strongly active.
FIG. 8.
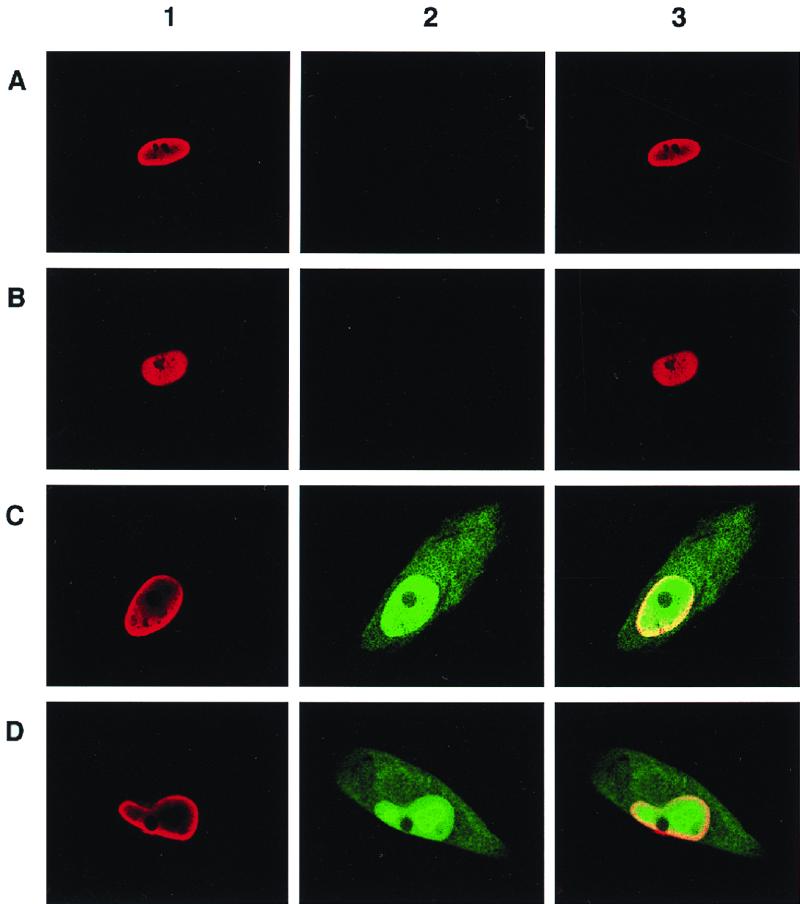
Confocal microscopy of HFF cells infected with UL127-GFP HCMV recombinants. HFF cells were infected with parental HCMV (RVHB5; A), UL127-GFP1 (B), UL127-GFP10 (C), and UL127-GFP7 (D) at an MOI of 0.5 PFU/cell. Three days after infection, cells were fixed, permeabilized, and subjected to immunofluorescence using an HCMV IE monoclonal antibody and a TRITC–anti-mouse secondary conjugate. Expression of MIE proteins can be visualized in panels 1 (red), expression of GFP is evident in panels 2 (green), and panels 3 show the merge of panels 1 and 2. Magnification, ×63.
In the next set of experiments, we quantified the levels of reporter activity by directly measuring GFP levels. For these experiments, we infected HFF cells at an MOI of 0.5 PFU/cell with the different recombinant viruses and measured fluorescence on day 3 postinfection by flow cytofluorometry. Figure 9 shows that while all cells were GFP negative after infection with UL127-GFP1 (mean fluorescence intensity with a signal-to-noise ratio of 1.3), HFF cells infected with UL127-GFP7 exhibited approximately twofold-higher levels of GFP in comparison with UL127GFP10-infected cells (compare mean fluorescence intensities with signal-to-noise ratios of 7.2 and 14.1 for UL127-GFP10 and UL127-GFP7, respectively). These results indicate that maximal derepression of the UL127 promoter in the context of the viral infection requires the removal of sequences from −531 to −673. Thus, these data are consistent with the role of sequences between −556 and −638 of the UL127 promoter as a repressor of gene expression in transient transfection assays. However, sequences between −638 and −673 also appear to contribute to the level of repression of the UL127 promoter but only in the context of an HCMV infection.
FIG. 9.

GFP expression from UL127 GFP HCMV recombinants. HFF cells were infected with UL127-GFP1, UL127-GFP10, and UL127-GFP7 at an MOI of 0.5 PFU/cell. On day 3 postinfection, cells were trypsinized and fixed with 1% formaldehyde. Fluorescence was measured by flow cytofluorometry as described in Materials and Methods. A histogram of FL1 versus the log fluorescence intensity (detected at 530 nm) is shown. A total of 10,000 events were collected for each sample. Mean fluorescence intensities with signal-to-noise ratios of 25.4/19.8, 134.6/18.8, and 222.1/15.8 were obtained for UL127-GFP1, UL127-GFP10, and UL127-GFP7, respectively.
DISCUSSION
We have presented evidence for the presence of a boundary domain located within the usr of the HCMV MIEP, whereby the putative UL127 promoter is prevented from being activated by the potent CMV enhancer. The borders of the boundary domain are positioned between the UL127 TATA box and the enhancer. Our experiments define a region between −556 and −638 (relative to the MIEP start site) that functions to dramatically repress enhancer activation of transcription at the UL127 promoter site both in transient transfection assays and in the context of virus infection. In addition, sequences between −638 and −673 were found to contribute to the level of repression only in the context of virus infection but not in transient transfection experiments.
Previously, DNase I protection experiments with DNA fragments encompassing usr sequences demonstrated (18), on the basis of different chromatographic behavior, the interaction of multiple distinct factors. Later experiments identified one of these binding components as composed of a retinoic acid receptor-retinoid X receptor (RAR-RXR) heterodimer (2). While DNA-bound RAR-RXR complexes are known to recruit transcriptional corepressors in the absence of bound ligand (12, 29), our mapping studies show that this site is not required for mediating repression. Sequences immediately adjacent to the RAR-RXR site, between nt −558 and −602 (relative to the MIEP), interact with a complex of proteins that have similar chromatographic behavior (18) (Fig. 10). These sequences resemble binding sites for the forkhead/winged helix family of transcription factors (8, 16, 32). Many members of this family are known to be potent repressors of transcription. These observations suggest that one of the candidate repressor elements may bind a cluster of winged helix repressors, although our results indicate that the repression domain is not limited to these sites alone (Fig. 10). For instance, the binding site between −604 and −632 participates in mediating repression and is known to interact with a factor that shares characteristics of the CTF family but is distinct from NF-1 (18) (Fig. 10). In addition, sequences (−638 to −673) between the CTF-like binding site and the NF-1 site proximal to the UL127 TATA box also appear to contribute to repression only in the context of the virus (Fig. 10). This region is not known to interact strongly with any nuclear factors but contains weak binding sites for YY1 (from nucleotide positions −675 to −663 and −655 to −642), a factor also known to mediate transcriptional repression (T. Stamminger, personal communication).
FIG. 10.

Sites of protein-DNA interaction on MIEP sequences between −538 and −738. Sequences between −556 and −638, corresponding to the repressor region defined in transient transfection assays, are marked by reverse print. Sequences from −638 to −673, shown to contribute to repression of the UL127 promoter in the context of the infection, are marked by a gray box. Black underlining represents the extent of strong protection observed using the phosphocellulose (p11) gel chromatography fractions P11 0.3, P11 0.6, and P11 1 (18). The cellular factors known to interact with the protected sequences are indicated. The UL127 TATA box is shown. Numbers refer to nucleotide positions relative to the transcription start site (+1) of the MIEP.
Recently, Lundquist and coworkers presented work on the characterization of a repressor region located upstream of the UL127 promoter (33). They found that sequences between −640 and −694 contribute to strong repression, while sequences between −583 and −640 effected repression to a much lesser extent in the context of the viral infection. The results of our study are in agreement with theirs regarding the presence of a repressor region but differ markedly in the precise sequences shown to be important in mediating the shutoff of UL127 expression. We believe that the conclusions drawn from these two independent studies are complementary. In particular, it is likely that the reason for the weaker repression observed by Lundquist et al. (33) for the distal repression sequences (−583 to −640) is that repression sequences between nt −556 and −583 were not removed. Indeed, resection of sequences between −556 and −638 in the context of an infection leads to a dramatic relief of repression (this study). Also in agreement with the work by Lundquist et al. (33), we found that sequences closer to the UL127 TATA box contribute to repression but only in the context of the virus. However, we note that removal of sequences between nucleotide positions −673 and −691 alone was not sufficient, in our study, to effect activation of the UL127 promoter. Therefore, while our results are different and agree only in part, they are highly complementary.
We note that the inability of the boundary region to repress the MIEP over a relatively large distance is compatible with a short-range repression mechanism (21). In this case, binding of repressors to promoter-proximal elements results in the dominant inhibition of a promoter site by upstream activators. Boundary elements have been identified in numerous drosophila and yeast genes (references 22, 26, and 40 and references therein), but few if any have been formally characterized for mammalian genes. In drosophila, short-range repressors have been elegantly shown to provide flexibility in controlling complex genetic loci. Indeed, Gray and coworkers (23) proposed that short-range repression is central for the evolution of complex promoters that are composed of multiple, autonomous regulatory elements as is prominently exhibited by the CMV MIEPs. Although the nature of the enhancer-repressor-promoter interaction is unclear, the results of this study indicate that the viral boundary domain blocks enhancer activation in a disproportional manner. In other words, there are far more activators bound to the enhancer region than there are potential repressor binding sites in the boundary region. There are several possible explanations for the ability of a limited repressor complex to block many activators, some of which challenge our view of how enhancers work. Perhaps the boundary domain recruits by protein-protein interaction a multifactor repression complex that is able to interfere with many activators. Alternatively, it is possible that multiple activators present in the enhancer coordinate communications with the basal transcription machinery by interacting with a central switching (adapter/nodal) complex. In this scenario, the boundary domain might disrupt the ability of the adapter/node to interact with the promoter. It should be noted that there is a precedent for a central switching unit in coordinating interactions with all other cis-acting regulatory modules, as demonstrated by the Endo16 system of the sea urchin embryo (43). In this example, module A of the Endo16 cis-regulatory system communicates the status of the whole regulatory system directly to the basal transcription machinery. All upstream sites work through module A and in the endogenous arrangement do not themselves interact directly with the basal transcription apparatus.
In conclusion, we have shown that repression can play an important role in determining the ability of the enhancer to activate transcription. Until now the usr was characterized as a positive control region for the MIEP (2, 18). We now show that the ability of the HCMV enhancer to bidirectionally activate transcription is stringently regulated in a negative manner by a boundary domain. This work thus provides a molecular explanation for limiting the action of the enhancer to the MIEP. Restricting the boundary of action of the CMV enhancer is likely to have important biological implications for ensuring sequential and coordinate regulation of transcription. It may not be advantageous or may even be deleterious to the virus to have genes, other than select IE genes, influenced by the proximal and long-range effects of an innately strong enhancer. The identification of the repressor proteins and target sequences, the precise mechanism for effecting inhibition, and the functional consequences of altered physiological states on UL127 promoter activation are topics that remain to be explored.
ACKNOWLEDGMENTS
We thank Fátima García del Rey for technical assistance, Susanne Etteldorf for help with the confocal microscopy, and Joe Trotter for help with the FACS analysis. We also thank Mark Stinski for making the results of his group's study available prior to publication.
This work was supported by grants from the National Institutes for Health and Novartis to P.G. A.A. was a fellow of the Ministerio de Educación y Ciencia (Spain) and is currently supported by a fellowship from the University of California Universitywide AIDS Research Program. P.G. is a Scholar of the Leukemia Society of America.
Footnotes
Publication 10492-IMM from The Scripps Research Institute.
REFERENCES
- 1.Adhya S, Geanacopoulos M, Lewis D E, Roy S, Aki T. Transcription regulation by repressosome and by RNA polymerase contact. Cold Spring Harbor Symp Quant Biol. 1998;63:1–9. doi: 10.1101/sqb.1998.63.1. [DOI] [PubMed] [Google Scholar]
- 2.Angulo A, Suto C, Heyman R A, Ghazal P. Characterization of the sequences of the human cytomegalovirus enhancer that mediate differential regulation by natural and synthetic retinoids. Mol Endocrinol. 1996;10:781–793. doi: 10.1210/mend.10.7.8813719. [DOI] [PubMed] [Google Scholar]
- 3.Angulo A, Messerle M, Koszinowski U H, Ghazal P. Enhancer requirement for murine cytomegalovirus growth and genetic complementation by the human cytomegalovirus enhancer. J Virol. 1998;72:8502–8509. doi: 10.1128/jvi.72.11.8502-8509.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskar J F, Smith P P, Climent G S, Hoffman S, Tucker C, Tenney D J, Colberg-Poley A M, Nelson J A, Ghazal P. Developmental analysis of the cytomegalovirus enhancer in transgenic animals. J Virol. 1996;70:3215–3226. doi: 10.1128/jvi.70.5.3215-3226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baskar J F, Smith P P, Nilaver G, Jupp R A, Hoffmann S, Peffer N J, Tenney D J, Colberg-Poley A M, Ghazal P, Nelson J. The enhancer domain of the human cytomegalovirus major immediate-early promoter determines cell-type-specific expression in transgenic mice. J Virol. 1996;70:3207–3214. doi: 10.1128/jvi.70.5.3207-3214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borst E, Hahn G, Koszinowski U H, Messerle M. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J Virol. 1999;73:8320–8329. doi: 10.1128/jvi.73.10.8320-8329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boshart M, Weber F, Jahn G, Dorsch-Hasler K, Fleckenstein B, Shaffner W. A very strong enhancer is located upstream of an immediate-early gene of human cytomegalovirus. Cell. 1985;41:521–530. doi: 10.1016/s0092-8674(85)80025-8. [DOI] [PubMed] [Google Scholar]
- 8.Bravieri R, Shiyanova T, Chen T H, Overdier D, Liao S. Different DNA contact schemes are used by two winged helix proteins to recognize a DNA binding sequence. Nucleic Acids Res. 1997;25:2888–2896. doi: 10.1093/nar/25.14.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardin R D, Abenes G B, Stoddart C A, Mocarski E S. Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology. 1995;209:236–241. doi: 10.1006/viro.1995.1249. [DOI] [PubMed] [Google Scholar]
- 10.Chambers J, Angulo A, Amaratunga D, Guo H, Jiang Y, Wan J S, Bittner A, Frueh K, Jackson M R, Peterson P A, Erlander M G, Ghazal P. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J Virol. 1999;73:5757–5766. doi: 10.1128/jvi.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chee M S, Bankier A T, Beck S, Bohni R, Brown C M, Cerny R, Hornsnell T, Hutchinson III C A, Kouzarides T, Martignetti J A, Preddie E, Satchwell S C, Tomlinson P, Weston K M, Barrell B G. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr Top Microbiol Immunol. 1990;154:125–169. doi: 10.1007/978-3-642-74980-3_6. [DOI] [PubMed] [Google Scholar]
- 12.Chen J D, Evans R M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 13.Cherepanov P P, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 14.Cherrington J M, Khoury E L, Mocarski E S. Human cytomegalovirus ie2 negatively regulates α gene expression via a short target sequence near the transcription start site. J Virol. 1991;65:887–896. doi: 10.1128/jvi.65.2.887-896.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorsch-Hasler K, Keil G M, Weber F, Jasin M, Schaffner W, Koszinowski U H. A long and complex enhancer activates transcription of the gene encoding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc Natl Acad Sci USA. 1985;82:8325–8329. doi: 10.1073/pnas.82.24.8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freyaldenhoven B S, Freyaldenhoven M P, Iacovoni J S, Vogt P K. Avian winged helix proteins CWH-1, CWH-2, and CWH-3 repress transcription from Qin binding sites. Oncogene. 1997;15:483–488. doi: 10.1038/sj.onc.1201189. [DOI] [PubMed] [Google Scholar]
- 17.Ghazal P. A rapid and selective method for generating deletions, insertions and clustered point mutations. BioTechniques. 1989;7:8325–8329. [PubMed] [Google Scholar]
- 18.Ghazal P, Lubon H, Reynolds-Kohler C, Hennighausen L, Nelson J A. Interactions between cellular regulatory proteins and a unique sequence region in the human cytomegalovirus major immediate-early promoter. Virology. 1990;174:18–25. doi: 10.1016/0042-6822(90)90049-w. [DOI] [PubMed] [Google Scholar]
- 19.Ghazal P, Nelson J A. Enhancement of RNA polymerase II initiation complexes by a novel DNA control domain downstream from the cap site of the cytomegalovirus major immediate-early promoter. J Virol. 1991;65:2299–2307. doi: 10.1128/jvi.65.5.2299-2307.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghazal P, Nelson J. Transcription factors and viral regulatory proteins as potential mediators of human cytomegalovirus pathogenesis. In: Becker Y, Darai G, Huang E-S, editors. Molecular aspects of human cytomegalovirus diseases. Heidelberg, Germany: Springer-Verlag Publishers; 1993. pp. 360–383. [Google Scholar]
- 21.Gray S, Cai H, Barolo S, Levine M. Transcriptional repression in the Drosophila embryo. Philos Trans R Soc Lond B. 1995;349:257–262. doi: 10.1098/rstb.1995.0111. [DOI] [PubMed] [Google Scholar]
- 22.Gray S, Levine M. Short-range transcriptional repressors mediate both quenching and direct repression within complex loci in Drosophila. Genes Dev. 1996;10:700–710. doi: 10.1101/gad.10.6.700. [DOI] [PubMed] [Google Scholar]
- 23.Gray S, Szymanski P, Levine M. Short-range repression permits multiple enhancers to function autonomously within a complex promoter. Genes Dev. 1994;8:1829–1838. doi: 10.1101/gad.8.15.1829. [DOI] [PubMed] [Google Scholar]
- 24.Grzimek N K, Podlech J, Steffens H P, Holtappels R, Schmalz S, Reddehase M J. In vivo replication of recombinant murine cytomegalovirus driven by the paralogous major immediate-early promoter-enhancer of human cytomegalovirus. J Virol. 1999;73:5043–5055. doi: 10.1128/jvi.73.6.5043-5055.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hennighausen L, Fleckenstein B. Nuclear factor 1 interacts with five DNA elements in the promoter region of the human cytomegalovirus major immediate-early gene. EMBO J. 1986;5:1367–1371. doi: 10.1002/j.1460-2075.1986.tb04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J D, Schwyter D H, Shirokawa J M, Courey A J. The interplay between multiple enhancer and silencer elements defines the pattern of decapentaplegic expression. Genes Dev. 1993;7:694–704. doi: 10.1101/gad.7.4.694. [DOI] [PubMed] [Google Scholar]
- 27.Jeang K T, Rawlins D R, Rosenfeld P J, Shero J H, Kelly T J, Hayward G S. Multiple tandemly repeated binding sites for cellular nuclear factor 1 that surround the major immediate-early promoters of simian and human cytomegalovirus. J Virol. 1987;50:1559–1570. doi: 10.1128/jvi.61.5.1559-1570.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koedood M, Fichtel A, Meier P, Mitchell P J. Human cytomegalovirus (HCMV) immediate-early enhancer/promoter specificity during embryogenesis defines target tissues of congenital HCMV infection. J Virol. 1995;69:2194–2207. doi: 10.1128/jvi.69.4.2194-2207.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld M G, Glass C K. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature. 1995;377:451–454. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 30.Lang D, Stamminger T. The 86-kilodalton IE-2 protein of human cytomegalovirus is a sequence-specific DNA-binding protein that interacts directly with the negative autoregulatory response element located near the cap site of the IE-1/2 transcriptional repressor. Nucleic Acids Res. 1994;22:3331–3338. [Google Scholar]
- 31.Lee G, Wu J, Luu P, Ghazal P, Flores O. Inhibition of the association of RNA polymerase II with the preinitiation complex by a viral transcriptional repressor. Proc Natl Acad Sci USA. 1996;67:323–331. doi: 10.1073/pnas.93.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Thurm H, Chang H W, Iacovoni J S, Vogt P K. Oncogenic transformation induced by the Qin protein is correlated with transcriptional repression. Proc Natl Acad Sci USA. 1997;94:10885–10888. doi: 10.1073/pnas.94.20.10885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lundquist C A, Meier J L, Stinski M F. A strong negative transcriptional regulatory region between the human cytomegalovirus UL127 gene and the major immediate-early enhancer. J Virol. 1999;73:9039–9052. doi: 10.1128/jvi.73.11.9039-9052.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macias M, Stinski M F. An in vitro system for human cytomegalovirus immediate early 2 protein (IE2) mediated site dependent repression of transcription and direct binding of IE2 to the major immediate early promoter. Proc Natl Acad Sci USA. 1993;90:707–711. doi: 10.1073/pnas.90.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manning C M, Mocarski E. Insertional mutagenesis of the murine cytomegalovirus genome: one prominent α gene (ie2) is dispensable for growth. Virology. 1988;167:477–484. [PubMed] [Google Scholar]
- 36.Meier J L, Stinski M F. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J Virol. 1997;71:1246–1255. doi: 10.1128/jvi.71.2.1246-1255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Messerle M, Keil G M, Koszinowski U H. Structure and expression of murine cytomegalovirus immediate-early gene 2. J Virol. 1991;65:1638–1643. doi: 10.1128/jvi.65.3.1638-1643.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson J A, Reyonods-Kohler C, Smith C. Negative and positive regulation by a short segment in the 5′-flanking region of the human cytomegalovirus major immediate-early gene. Mol Cell Biol. 1987;7:4125–4129. doi: 10.1128/mcb.7.11.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pizzorno M C, Hayward G S. The IE2 gene products of human cytomegalovirus specifically down regulate expression from the major immediate-early promoter through a target sequence located near the cap site. J Virol. 1990;64:6154–6165. doi: 10.1128/jvi.64.12.6154-6165.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Small S, Kraut R, Hoey T, Warrior T, Levine M. Transcriptional regulation of a pair-rule stripe in Drosophila. Genes Dev. 1991;5:827–839. doi: 10.1101/gad.5.5.827. [DOI] [PubMed] [Google Scholar]
- 41.Stenberg R M, Fortney J, Barlow S W, Magrane B P, Nelson J A, Ghazal P. Promoter-specific trans activation and repression by human cytomegalovirus immediate-early proteins involves common and unique protein domains. J Virol. 1990;64:1556–1565. doi: 10.1128/jvi.64.4.1556-1565.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu J, Jupp R, Stenberg R M, Nelson J A, Ghazal P. Site-specific inhibition of RNA polymerase II preinitiation complex assembly by human cytomegalovirus IE86 protein. J Virol. 1993;67:7547–7555. doi: 10.1128/jvi.67.12.7547-7555.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuh C H, Bolouri H, Davidson E H. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998;279:1896–1902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Buchholz F, Muyrers J P, Stewart A F. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]