

Abstract
Diphenyl ether derivatives inhibit mycobacterial cell wall synthesis by inhibiting an enzyme, enoyl-acyl carrier protein reductase (InhA), which catalyzes the last step in the fatty acid synthesis cycle of genus Mycobacterium. To select and validate a protein crystal structure of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis for designing inhibitors using molecular modelling, a cross-docking and correlation study was performed. A series of novel 1-(3-(3-hydroxy-4-phenoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl) ethan-1-ones were synthesized from this model and screened for their antitubercular activity against M. tuberculosis H37Rv. Compound PYN-8 showed good antitubercular activity on M. tuberculosis H37Rv (MIC=4–7μM) and Mycobacterium bovis (% inhibition at 10μM=95.91%). Cytotoxicity of all the synthesized derivatives was assessed using various cell lines and they were found to be safe. Structure of PYN-8 was also confirmed by single crystal X–ray diffraction. The molecular modelling studies also corroborated the biological activity of the compounds. Further, in-silico findings revealed that all these tested compounds exhibited good ADME properties and drug-likeness and thus may be considered as potential candidates for further drug development.
Keywords: TB, Diphenylether, InhA, molecular docking, Correlation study, antitubercular
1. Introduction:
Tuberculosis (TB) is one of the top 10 causes of death and the leading cause from a single infectious agent (above HIV/AIDS).(1) In 2017, TB caused an estimated 1.3 million deaths (range, 1.2–1.4 million) among HIV-negative people and there were an additional 300 000 deaths from TB (range,266 000–335 000) among HIV-positive people. There were cases in all countries and age groups, but overall 90% were adults (aged ≥15 years), in the most productive years of their life. Diagnosis and successful treatment of people with TB averts millions of deaths each year (an estimated 54 million over the period 2000–2017).(2) The latest treatment outcome data for new cases show a global treatment success rate of 82% in 2016. Today’s TB treatments take too long to cure and can be toxic. They are also complicated to administer which results in poor patient compliance.(3) New treatments that are faster, simpler, and affordable are urgently needed.
Enoyl carrier protein reductase is a validated target for Mycobacterium.(4) It is an enzyme in the type II fatty acid synthase system, which is involved in the biosynthesis of mycolic acids. Mycolic acids are a major component of mycobacterial cell wall and are responsible for virulence of the microorganism.(5) InhA catalyzes the NADH-dependent reduction of the double bond of 2-trans-enoyl-[acyl-carrier protein], an essential step in the fatty acid elongation cycle of the FAS-II pathway. Isoniazid (INH), a first line antitubercular drug, targets this enzyme. InhA is inhibited by an active form of isoniazid which is formed by reacting with the cofactor NAD(H), bound to the active site of the enzyme. It forms a covalent adduct (isonicotinic acyl NADH), which binds to InhA with high affinity.(6)The activation is carried out by the catalase peroxidase enzyme encoded by katG, and mutation in this gene is the cause of resistance to isoniazid in 64% of clinical isolates.(7) Therefore, molecules which directly inhibit InhA without any bio-activation circumvent this mechanism of resistance and are promising candidates. Such a direct inhibitor of InhA is Triclosan(a diphenyl ether derivative).(8) However, it is a weak inhibitor of InhA.(9) Therefore, in the present work, a series of diphenyl ether derivatives were designed and synthesized by studying the nature of the substrate binding pocket and the interactions of potent inhibitors with the amino acid residues of the active site. The design used in this work is shown the Figure 1.
Figure 1.
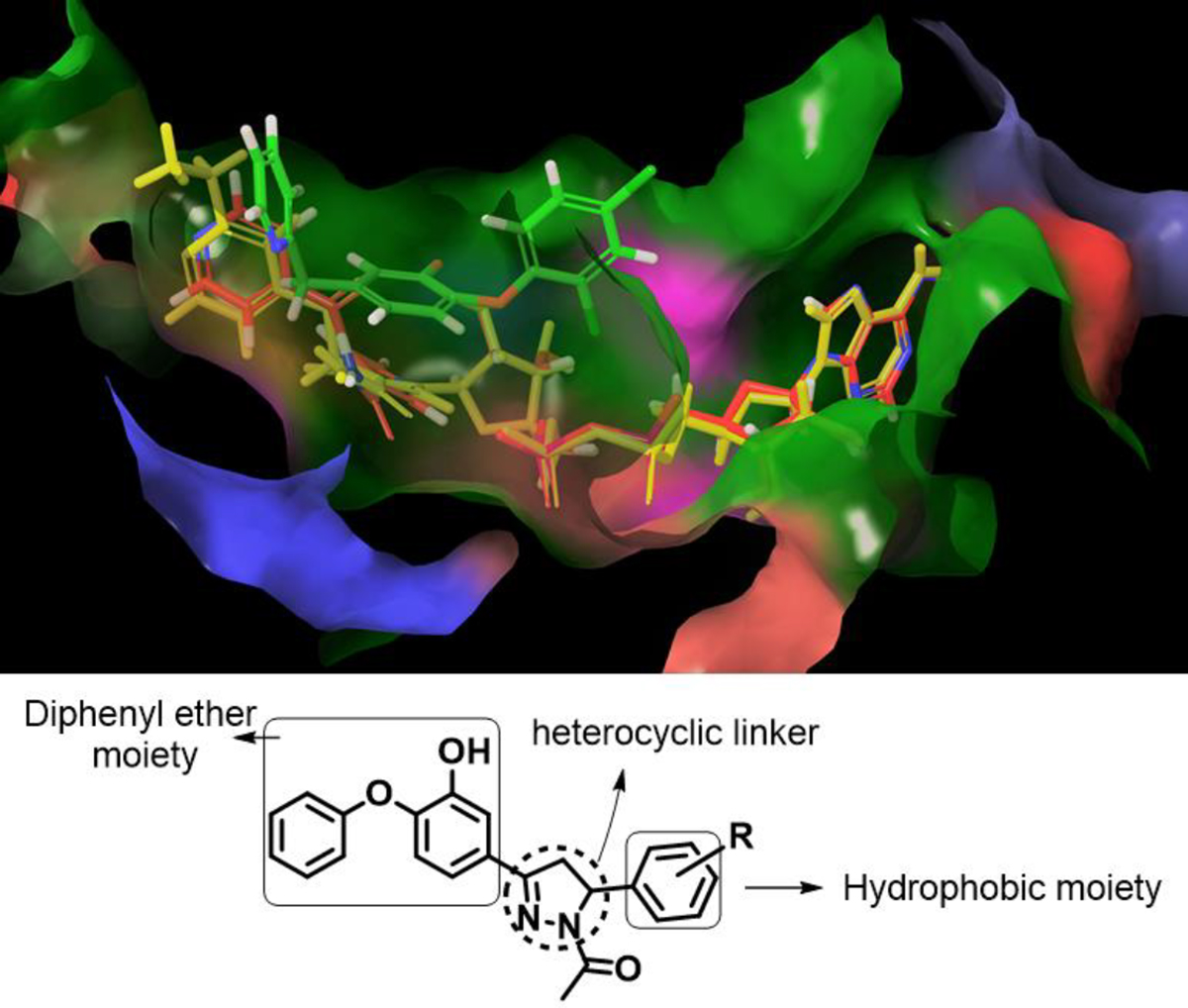
Design strategy and active site of InhA showing diphenyl ether derivative substituted at 5th chlorine position protruding into the isonicotinoyl binding pocket containing InhA complexes of isoniazid (red colour) and ethionamide (yellow colour)
Physicochemical properties of the inhibitor molecules are a major factor for drug efficacy. Therefore, Lipinski’s rule was followed while designing the molecules to fulfil these requirements. The synthesized compounds were screened against H37Rv strain of Mycobacterium tuberculosis and Mycobacterium bovis. The safety profiles of the molecules were also determined using various cell lines. To study the plausible mechanism of inhibition and for determination of ADME profile of the synthesized molecules, molecular modelling study was carried out.
2. Result and Discussion:
2.1. Drug Design Strategy
To select and validate a protein crystal structure of enoyl-acyl carrier protein reductase of Mycobacterium tuberculosis for designing of the inhibitor molecules using molecular modelling, a cross-docking and correlation study was performed. Molecules were designed by studying the nature of the substrate binding pocket of InhA. Also, the interactions of various potent direct inhibitors (diphenyl ether analogues) with the amino acid residues, as exhibited in their protein crystallographic structures, was studied. Sullivan et al reported the crystal structures of diphenyl ether derivatives complexed with InhA, giving detailed insight into the binding pocket interactions with the direct inhibitors. A basic scaffold was then assigned which had the attributes required for interactions in a favourable way with the binding pocket. Hydrogen bonding interaction between amino acid residue Tyrosine 158 and the hydroxyl group of the diphenyl moiety was considered essential for activity.(10) Also, the π-π stacking interaction of the phenyl ring of the diphenyl ether moiety with the pyridyl moiety of the NAD 300 cofactor InhA protein was observed in potent inhibitors.(11) Comparison of the crystal structures of InhA complexes of isoniazid-NAD and ethionamide-NAD adducts with InhA-triclosan complex gives clues for structural modifications in diphenyl ether scaffold for increasing affinity towards InhA.(12) Hydrophobic substitutions in place of the chloro group at 5 position of A ring in diphenyl ether scaffold might help in occupying and increasing interactions with isonicotinoyl binding pocket which is important for improved activity.(13) Additionally for the better positioning of the aryl group(hydrophobic substituent) in to the target INH binding pocket a heterocyclic linker was designed between the hydrophobic moiety and the 5 position carbon on A ring.
There are 87 crystal structures available hitherto in the protein data bank for the protein, enoyl-(acyl-carrier-protein) reductase, of Mycobacterium tuberculosis of strain H37Rv. It was thus essential to select a right protein crystal structure, for the design and docking studies of the inhibitor molecules. The primary prerequisite for selection of the protein crystal structure is, the presence of a co-crystallised ligand in it, which is similar to the inhibitor molecule scaffold. The protein crystal structure should be able to accommodate all the analogues of the scaffold molecule without much deviation, so as to corroborate the in-vitro activity results. Thus, a cross docking study involving docking of the co-crystallised ligands, extracted from the selected protein crystal structures on each of the protein crystal structure was performed. The protein crystal structures were selected based on screening parameters mentioned in the experimental section.
2.1.1. Cross Docking Study
All the ligands extracted from the 11 proteins were docked on every protein. Root-mean-square deviation (RMSD) of every ligand with its docked pose in all the proteins, relative to its pose in the aligned ligand set, was calculated. The values of the RMSDs are listed in the table 1. Ligands with RMSD values more than 2 Å were considered as outliers. The protein crystal with the least number of outliers and the least average RMSD value was thought to be appropriate for use to study the docking of a variety of diphenyl ether derivatives. The protein with PDB entry code 3FNE had the least average RMSD value and no outliers. It had a resolution of 1.98 Å and a triclosan derivative, 2-(2,4-dichlorophenoxy)-5-(pyridin-2-ylmethyl) phenol as its co-crystallized ligand.
Table 1.
RMSD of the extracted ligands after docking on the respective crystal structures with poses in their native crystal structure.
| Co-crystallized ligand |
2X23 | 4OYR | 4OXY | 4OXK | 4OHU | 2X22 | 3FNH | 3FNG | 3FNF | 3FNE | 1P45 | Average RMSD (Å) | Outliers | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein crystal structure | ||||||||||||||
|
| ||||||||||||||
| 2X23 with water | 0.893 | 0.955 | 0.631 | 1.164 | 1.118 | 0.301 | 10.313 | 2.043 | 1.784 | 1.591 | 1.004 | 1.982 | 2 | |
| 2X23 without water | 0.849 | 0.954 | 0.595 | 1.637 | 1.09 | 0.921 | 10.443 | 1.382 | 1.785 | 1.589 | 1.001 | 2.022 | 1 | |
| 4OYR with water | 1.393 | 0.254 | 1.516 | 10.689 | 1.298 | 1.365 | 10.415 | 2.2 | 1.941 | 1.715 | 9.422 | 3.837 | 4 | |
| 4OYR without water | 1.391 | 0.226 | 0.817 | 2.011 | 1.336 | 1.363 | 1.979 | 8.909 | 1.941 | 1.715 | 10.116 | 2.891 | 3 | |
| 4OXY with water | 1.192 | 1.334 | 1.237 | 1.985 | 1.512 | 1.111 | 2.028 | 2.021 | 1.725 | 1.678 | 0.76 | 1.508 | 2 | |
| 4OXY without water | 1.166 | 1.066 | 1.08 | 2.362 | 1.367 | 1.069 | 2.125 | 1.212 | 1.059 | 1.696 | 0.726 | 1.357 | 2 | |
| 4OXK with water | 0.97 | 1.195 | 1.232 | 1.324 | 1.401 | 1.151 | 1.79 | 1.891 | 0.622 | 0.637 | 0.599 | 1.165 | 0 | |
| 4OXK without water | 0.971 | 0.778 | 1.232 | 1.328 | 1.453 | 1.169 | 1.739 | 0.985 | 0.75 | 0.713 | 0.649 | 1.070 | 0 | |
| 4OHU with water | 1.72 | 0.847 | 1.109 | 11.336 | 0.452 | 1.669 | 9.724 | 2.181 | 9.467 | 8.857 | 6.168 | 4.866 | 6 | |
| 4OHU without water | 1.046 | 0.866 | 1.108 | 11.757 | 0.306 | 1.009 | 9.723 | 2.186 | 9.197 | 8.857 | 6.168 | 4.748 | 6 | |
| 2X22 with water | 0.893 | 1.271 | 0.629 | 9.51 | 1.306 | 0.848 | 9.866 | 8.854 | 8.816 | 9.038 | 9.091 | 5.466 | 6 | |
| 2X22 without water | 1.011 | 0.959 | 0.984 | 11 | 1.308 | 0.896 | 9.872 | 8.851 | 8.828 | 9.054 | 8.988 | 5.614 | 6 | |
| 3FNH with water | 2.321 | 1.703 | 1.709 | 1.428 | 2.398 | 2.375 | 1.667 | 1.645 | 0.663 | 1.318 | 0.571 | 1.618 | 3 | |
| 3FNH without water | 2.321 | 1.703 | 1.709 | 1.428 | 2.398 | 2.375 | 1.667 | 1.645 | 0.663 | 1.318 | 0.571 | 1.618 | 3 | |
| 3FNG with water | 1.179 | 1.435 | 1.178 | 1.427 | 1.648 | 1.262 | 1.451 | 1.744 | 0.611 | 0.534 | 0.592 | 1.187 | 0 | |
| 3FNG without water | 1.273 | 1.483 | 0.918 | 1.209 | 1.705 | 1.345 | 1.481 | 1.735 | 0.515 | 1.911 | 0.591 | 1.288 | 0 | |
| 3FNF with water | 1.128 | 1.582 | 1.144 | 1.164 | 1.789 | 1.272 | 1.91 | 0.737 | 0.763 | 0.943 | 0.486 | 1.174 | 0 | |
| 3FNF without water | 1.411 | 1.538 | 1.51 | 1.452 | 1.835 | 1.557 | 2.233 | 0.672 | 0.713 | 0.706 | 0.659 | 1.299 | 0 | |
| 3FNE with water | 1.126 | 1.197 | 1.292 | 1.501 | 1.325 | 1.183 | 1.095 | 0.962 | 0.608 | 0.302 | 0.442 | 1.003 | 0 | |
| 3FNE without water | 1.549 | 1.239 | 1.045 | 1.445 | 1.315 | 1.647 | 1.604 | 0.508 | 0.351 | 0.298 | 0.45 | 1.041 | 0 | |
| 1P45 with water | 2.001 | 1.356 | 1.643 | 1.987 | 1.512 | 2.04 | 2.178 | 1.879 | 0.839 | 1.576 | 0.572 | 1.598 | 3 | |
| 1P45 without water | 1.207 | 1.369 | 1.641 | 2.248 | 1.418 | 1.214 | 2.331 | 0.697 | 0.839 | 1.592 | 0.609 | 1.379 | 2 | |
2.1.2. Correlation Study
To validate the outcome of the cross-docking study and therefore the applicability of the protein 3FNE, a correlation study was done. Twenty-five molecules from the literature with their experimental IC50 values were collected.(14)(15)(16) These molecules were then docked on the protein 3FNE. Molecular mechanics/generalized born surface area (MM/GBSA) was used to calculate the binding free energy of the ligand-protein complex. Prime MM/GBSA calculations were performed for the complexes obtained after docking of the molecules on 3FNE and the pIC50 values were correlated with the free binding energy of the ligand-protein complexes. The MM/GBSA values and the pIC50 values as listed in the Table 2. A graph was plotted between the MM/GBSA values and the pIC50 values and a Pearson’s correlation coefficient value of 0.73 was obtained (Figure 2). This validates the protein crystal structure 3FNE chosen after the screening of the available crystal structures of InhA. It also hints that the mechanism of action of the synthesized diphenyl ether derivatives could plausibly be the inhibition of InhA.
Table 2.
Binding free energy of diphenyl ether analogues (with respective codes as in the reference) from literature with their IC50 and pIC50 values.
| Molecules | IC50 | pIC50 | ΔG Bind (Kcal/mol) |
|---|---|---|---|
|
| |||
| 8PP | 5 | 8.30103 | −90.4033 |
| PT092 | 10 | 8 | −91.3701 |
| PT113 | 12.1 | 7.917215 | −86.8914 |
| 5PP | 17 | 7.769551 | −82.6221 |
| PT095 | 29.7 | 7.527244 | −82.4981 |
| PT096 | 44.6 | 7.350665 | −86.2859 |
| PT114 | 48 | 7.318759 | −90.9673 |
| 13b | 48 | 7.318759 | −86.7372 |
| PT070 | 50.7 | 7.294992 | −90.5046 |
| 14c | 55 | 7.259637 | −88.0505 |
| PT013 | 61.9 | 7.208309 | −88.3477 |
| PT133 | 79.7 | 7.098542 | −86.73 |
| 4PP | 80 | 7.09691 | −80.3218 |
| PT109 | 86 | 7.065502 | −83.2069 |
| PT111 | 100 | 7 | −87.3732 |
| 14PP | 150 | 6.823909 | −73.6168 |
| 3b | 236 | 6.627088 | −80.5168 |
| PT134 | 238.8 | 6.621966 | −80.0489 |
| 3e | 650 | 6.187087 | −81.2368 |
| 10a | 1550 | 5.809668 | −67.8625 |
| PT108 | 1570 | 5.8041 | −77.4535 |
| 2PP | 2000 | 5.69897 | −71.675 |
| 11a | 2360 | 5.627088 | −58.3266 |
| 12a | 3200 | 5.49485 | −64.9083 |
| 2d | 100000 | 4 | −62.3432 |
Figure 2.
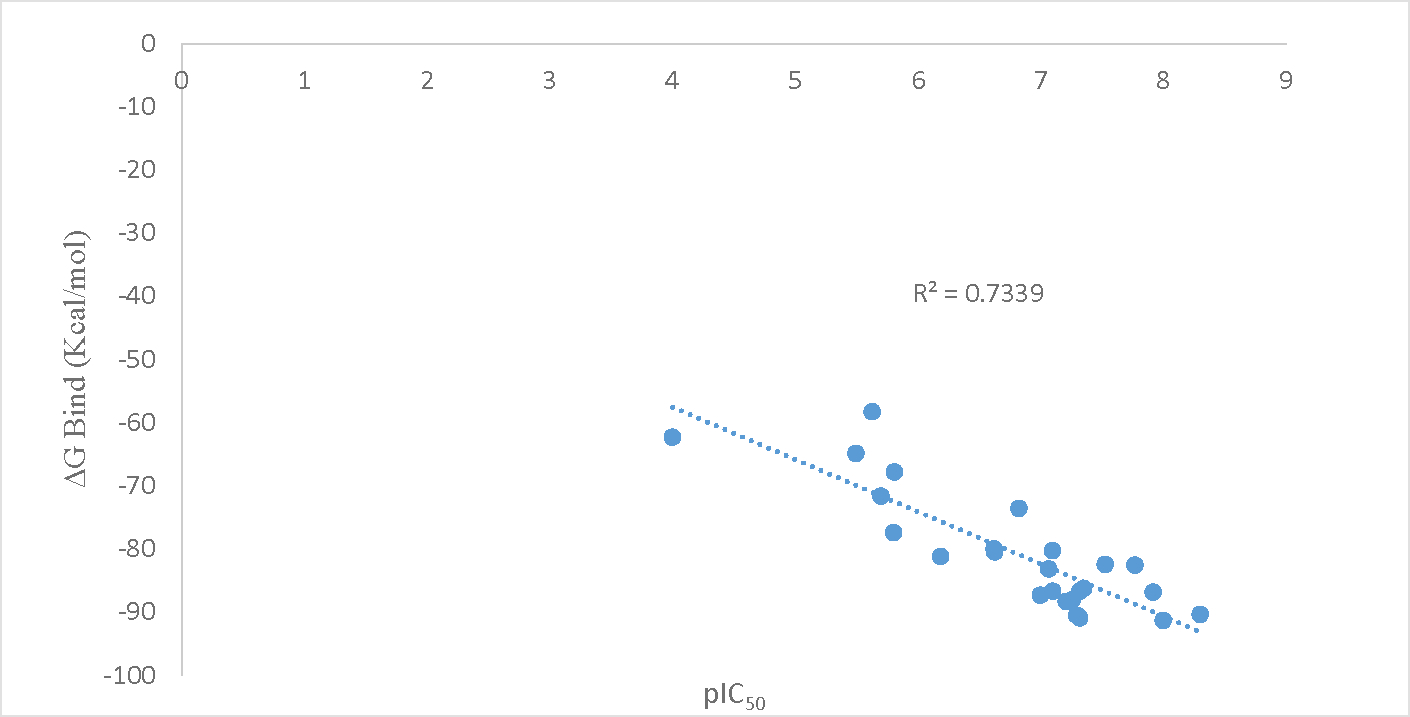
Binding free energy vs pIC50 correlation graph with correlation value R2
2.2. Induced Fit Docking
Induced fit docking (IFD) of the synthesized PYN-8 and Triclosan generated good poses with RMSD of 0.093 A°. PYN-8 showed hydrogen bonding interaction indicated by yellow dotted lines, with the Tyrosine 158 residue. A π-π stacking interaction with the pyridyl ring of NAD 300 was observed (indicated by blue dotted line). A cation- π interaction (indicated by green dotted line) with the pyridine nitrogen of NAD 300 was also observed. The induced fit docking score of synthesized PYN-8 (−541.09) was better than triclosan (−536.48) supporting its better activity. The interactions are shown in the Figure 3 and Figure 4.
Figure 3.
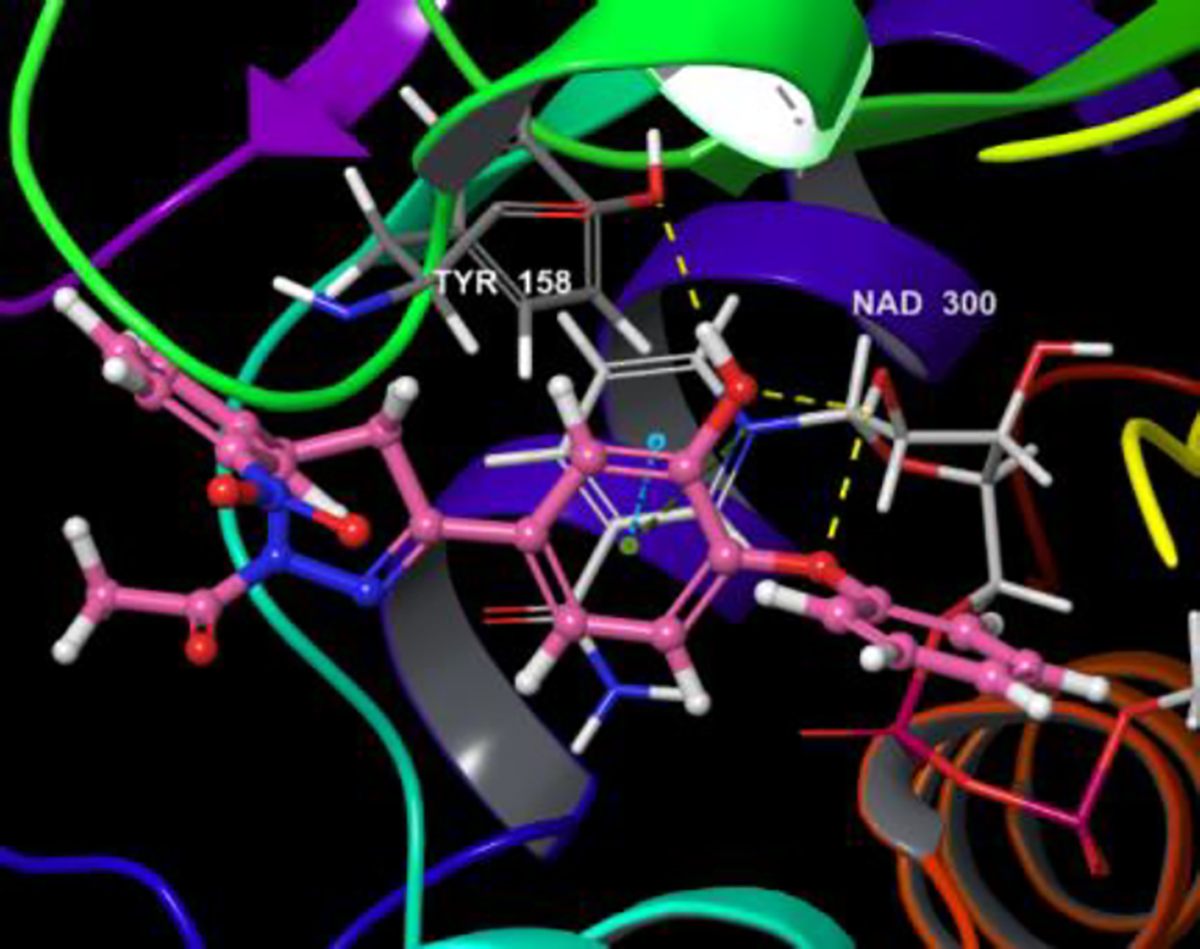
Induced fit docked pose of PYN-8 on 3FNE
Figure 4.
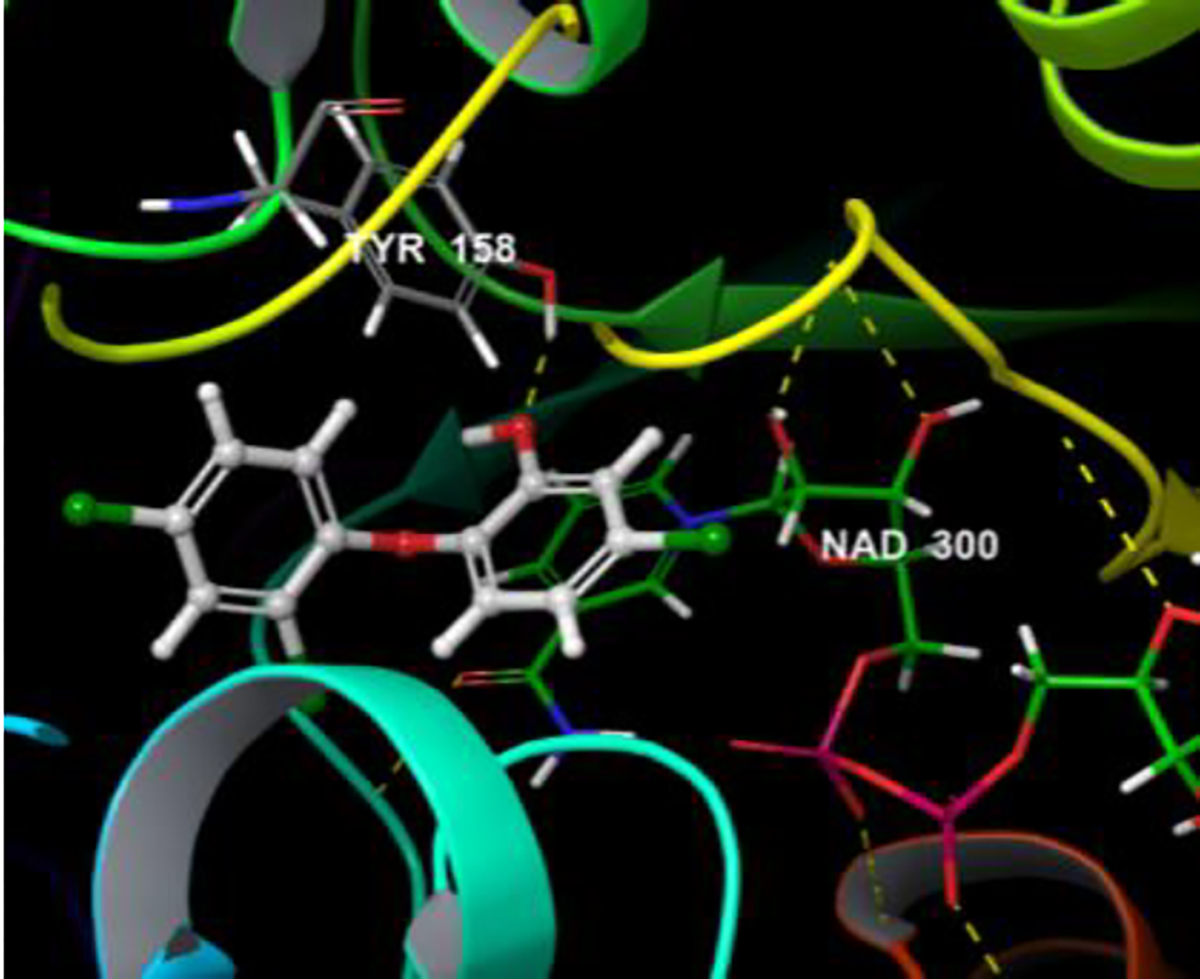
Induced fit docked pose of Triclosan on 3FNE
2.3. Water Map Analysis
To understand the better activity of meta nitro substituted derivative (PYN-8) compared to other analogues, a water map analysis was performed. Water map shows the hydration-sites in a protein along with their thermodynamic properties. Based on the disorderliness of the water molecules in the hydration-sites it can be assessed if a hydration site has to be displaced or eschewed.(17) It was observed that molecule PYN-8 displaced a hydration-site with site number 68 as shown in the Figure 5. This hydration-site has an enthalpy (ΔH) of 2.24 kcal/mol, entropy (−TΔS) 1.61 kcal/mol and Gibbs free energy(ΔG) 3.85 kcal/mol. These values indicate that it is favourable to displace this hydration site. The meta nitro group on the phenyl ring of the PYN-8 helps to increase the hydrophobicity of the phenyl ring. The displacement of the unstable water by this hydrophobic group might be the reason for better activity of PYN-8.
Figure 5.
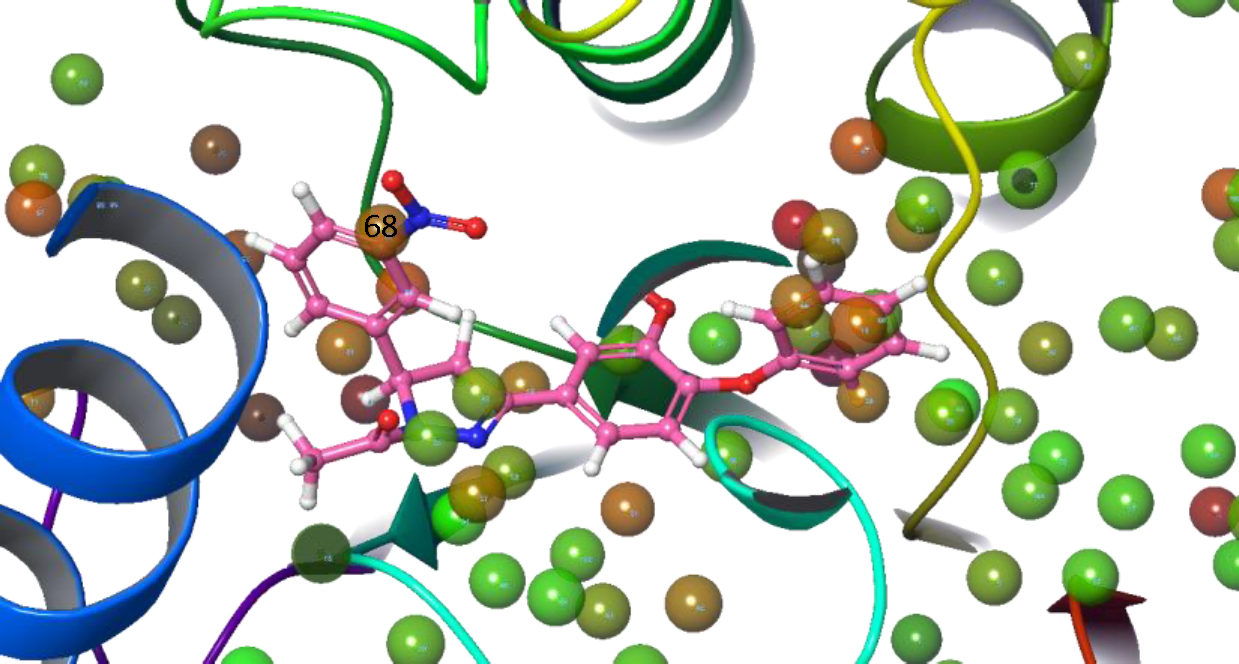
Water map of 3FNE protein showing PYN-8 displacing water site number 68
2.4. Drug-likeness Properties
Many hits often fail in the advanced drug discovery phases.(18) Therefore, it is vital to evaluate lead likeness and drug-likeness of hits obtained after a comprehensive screening of molecules. Lipinski’s rule of five was formulated to ascertain drug likeness of a chemical compound with properties that would probably make it an orally active drug. In the drug discovery process, molecules that abide these rules face lesser attrition rates for failure in clinical trials. The probability of reaching the market for such molecules increases because, the parameters of this rule were set after observing that most of the orally administered drugs are moderately lipophilic and small molecules.(19) Thus, we also determined the ADME properties of the synthesized compounds in-silico and all the compounds showed desirable ADME profile.
2.5. Chemistry
The diphenyl ether pyrazoline derivatives were synthesized as depicted in the Scheme-1. The synthesis of 1-(3-methoxy-4-phenoxyphenyl)ethanone was done via Chan-Lam coupling. Phenyl boronic acid and acetovanillone were reacted in the presence of copper acetate monohydrate, pyridine in anhydrous dichloromethane with activated molecular sieves at room temperature. 1-(3-methoxy-4-phenoxyphenyl)ethanone was then deprotected using BBr3 in anhydrous dichloromethane at −78°C.(20) 1-(3-hydroxy-4-phenoxyphenyl)ethan-1-one thus obtained was reacted with various substituted benzaldehydes using lithium hydroxide monohydrate as catalyst for Claisen-Schmidt condensation in ethanol to give various substituted 1-(3-hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-ones.(21)
Scheme 1. Synthesis of pyrazoline derivatives of diphenyl ethers (PYN series).
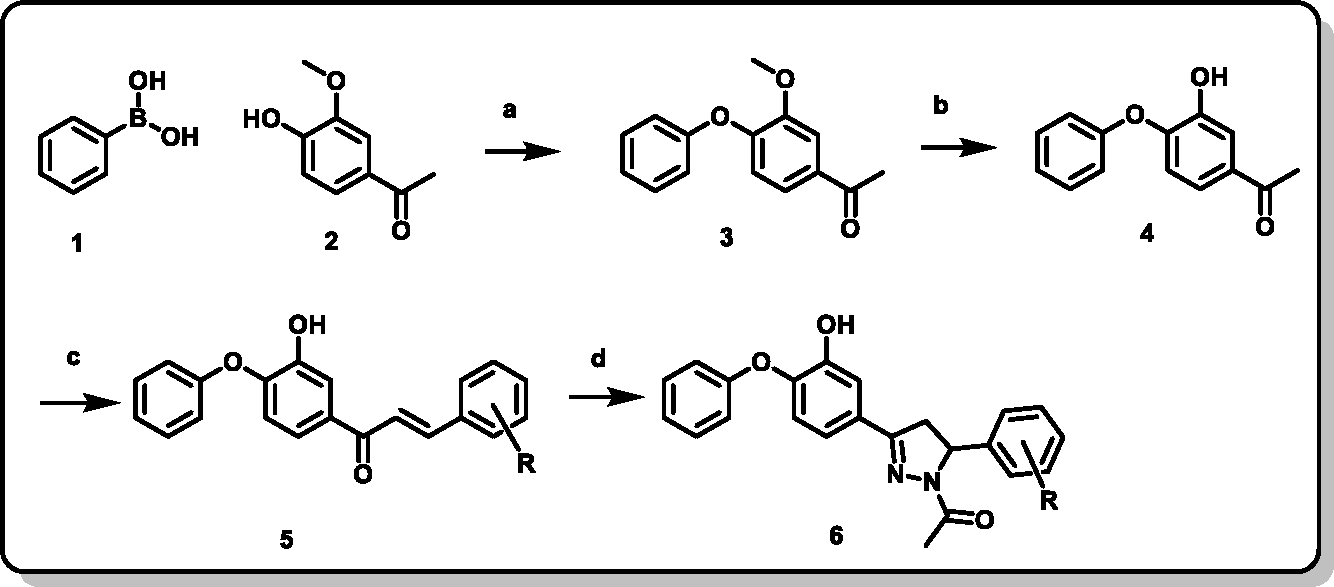
1) Phenyl boronic acid 2) 3-hydroxy-4-phenoxy 3) 1-(3-methoxy-4-phenoxyphenyl)ethan-1-one 4) 1-(3-hydroxy-4-phenoxyphenyl)ethan-1-one 5) (E)-1-(3-hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-one 6) 1-(3-(3-hydroxy-4-phenoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one acetophenone a) Copper acetate monohydrate, Pyridine, anhydrous dichloromethane, 4Å Molecular Sieves b) BBr3, anhydrous dichloromethane, −78°C c) Substituted benzaldehydes, LiOH.H2O, Ethanol d) Hydrazine hydrate, Glacial acetic acid
The 1-(3-hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-ones were then converted to 1-(3-(3-hydroxy-4-phenoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-ones by refluxing them with hydrazine hydrate in acetic acid for a duration of 3 hours. The synthesized compounds were purified by crystallization and column chromatography. Structures of all the synthesized final derivatives were confirmed by 1H, 13C NMR and HRMS.
A typical 1H NMR spectrum of 1-(3-(3-hydroxy-4-phenoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-ones showed protons of the pyrazoline moiety at three different positions. The methylene protons in the pyrazoline ring were observed at δ 3.01–3.11 ppm and δ 3.65–3.73 ppm both with a coupling constant of 17.6–17.8 Hz. The stereogenic proton appeared between δ 5.46–5.59 ppm with a coupling constant of 11.8–12.0 Hz. A singlet or a small hump at δ 5.7–5.8 ppm indicated the OH protons. The methyl protons of the acetyl group on the nitrogen of pyrazoline ring were observed at δ 2.3–2.42 ppm as a singlet. The HRMS (ESI/Q-TOF) showed [M + H]+ peaks corresponding to their molecular formula weight.
Further, the structure of compound PYN-8 was confirmed by single crystal X-ray diffraction study as shown in the Figure 6.
Figure 6.
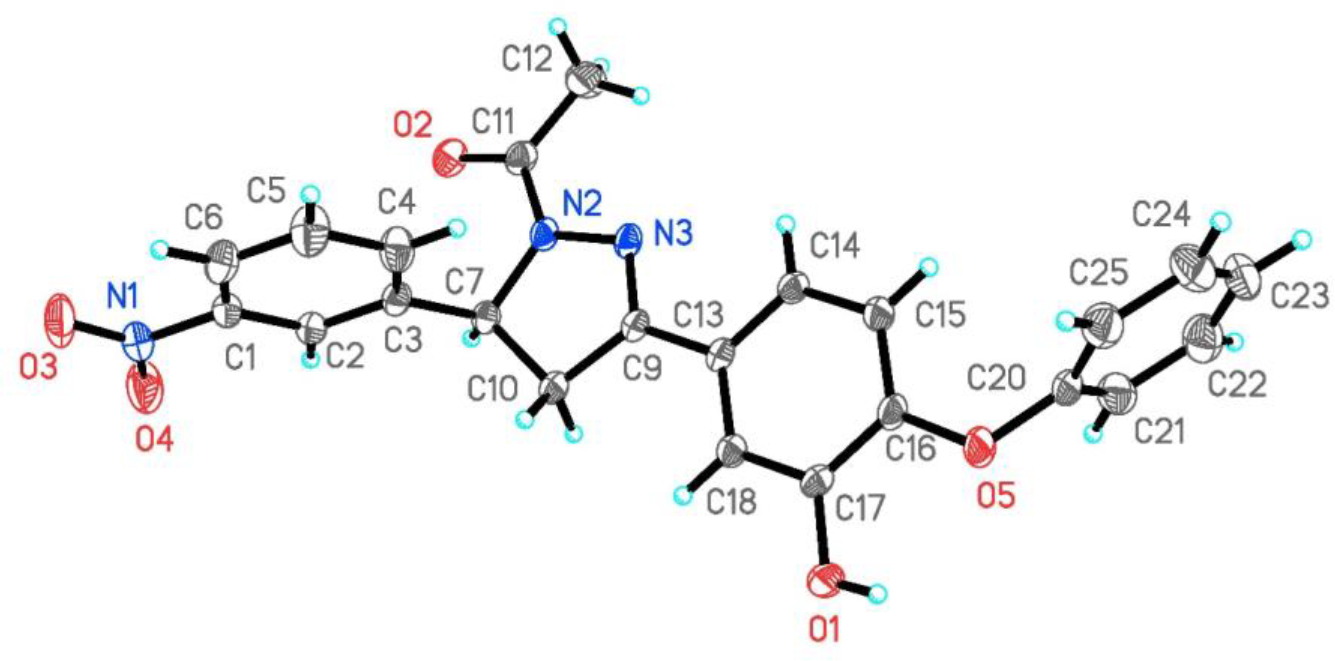
ORTEP diagram of PYN-8. Displacement ellipsoids are drawn at the 30% probability level.
2.6. Anti-tubercular Study
All the synthesized final derivatives (1–11) were screened for their anti-tubercular activity against Mycobacterium tuberculosis, strain H37Rv. They were also tested for their inhibitory effect against Mycobacterium bovis. The derivative with meta nitro substitution PYN-8, showed better activity than the other analogues. Similar trend of activity in both Mycobacterium tuberculosis and Mycobacterium bovis signifies the activity values. The activity values are as reported in the Table 4.
Table 4.
In-vitro MIC values of synthesized derivatives determined against Mycobacterium tuberculosis H37Rv strain in two different media.
| Compound Code | (7H9/ADC/Tw) MIC | (GAST-Fe) MIC | ||
|---|---|---|---|---|
|
| ||||
| Day 7 | Day 14 | Day 7 | Day 14 | |
| (μM) | (μM) | (μM) | (μM) | |
|
| ||||
| PYN-1 | 123 | >246 | 61 | 123 |
| PYN-2 | 240 | >240 | 120 | >239 |
| PYN-3 | 28–55 | >222 | 28 | >222 |
| PYN-4 | 128 | 128 | 128 | 128 |
| PYN-5 | >259 | >239 | 32 | 32 |
| PYN-6 | 31 | >249 | 31 | 62 |
| PYN-7 | 61 | 61 | 61 | 61 |
| PYN-8 | 3 | 4–7 | 15 | 15 |
| PYN-9 | 111–222 | 111–222 | 111 | 111 |
| PYN-10 | 64 | 64 | 64 | 64 |
| PYN-11 | 259 | 259 | 129 | 259 |
| Triclosan | 21 | 43 | 43 | 43 |
| Isoniazid | 0.8 | 1 | 0.2 | 0.2–0.4 |
2.7. Cytotoxicity Assay
For the treatment of tuberculosis, it is essential to take anti-tubercular drugs for a long period of time (at least 6 months). Thus, it is necessary to study cytotoxic effects of the drug candidates. The compounds were therefore evaluated at 10μM concentration for their cytotoxic effect on MCF-7, PANC-1, PC-3, SKOV-3 and HeLa cell lines. Additionally, they were screened against BD-11 and HEK cell lines where their cytotoxic concentration was greater than 100μM. Therefore, these studies indicate that these compounds do not have any cytotoxic effect. The cytotoxicity data is shown in the Table 5.
Table 5.
Percentage inhibition values of cancer cells and Mycobacterium bovis at 10μM concentration of the compounds
| Compound Code | % inhibition of cells at 10μM concentration |
|||||
|---|---|---|---|---|---|---|
| MCF7 | PANC-1 | PC-3 | SKOV-3 | HeLa | M. bovis | |
|
| ||||||
| PYN-1 | 6.77 | 4.29 | 17.29 | 2.71 | 4.45 | 10.15 |
| PYN-2 | 4.75 | 5.42 | 18.10 | 3.05 | 4.78 | 23.00 |
| PYN-3 | 6.16 | 5.17 | −2.67 | 4.12 | 4.10 | −22.14 |
| PYN-4 | 4.07 | 3.20 | 11.41 | 4.32 | 3.31 | −19.60 |
| PYN-5 | 4.04 | 1.07 | −9.88 | 4.79 | 3.06 | 11.04 |
| PYN-6 | 1.65 | 2.17 | 12.01 | 0.97 | 4.40 | 38.00 |
| PYN-7 | 5.15 | 3.27 | 0.04 | 4.50 | 3.66 | −35.36 |
| PYN-8 | 4.88 | 4.06 | 5.06 | 4.32 | 3.52 | 95.91 |
| PYN-9 | 4.68 | 2.69 | −4.23 | 3.92 | 3.32 | 29.55 |
| PYN-10 | 4.80 | 2.68 | −4.02 | 4.92 | 3.25 | 32.17 |
| PYN-11 | 5.65 | 2.27 | 0.82 | 3.67 | 2.99 | −35.31 |
| Doxorubicin(2μM) | 81.84 | 60.82 | 76.52 | 77.37 | 47.91 | - |
| Rifampicin | - | - | - | - | - | 99.3 |
| Isoniazid | - | - | - | - | - | 99.1 |
3. Conclusion
A protein crystal structure, 3FNE, was selected after screening all the available protein X-ray crystal structures from protein data bank. The selection was made after cross docking and validation study of the protein crystal structures. The molecules were designed using insights from the interaction patterns of known diphenyl ether derivatives. The synthesized compounds were then evaluated for their antitubercular activity against Mycobacterium tuberculosis H37Rv strain and Mycobacterium bovis. Compound PYN-8 was found to be most active among the synthesized derivatives with MIC=1.5–3.12 μg/mL in 7H9/ADC/Tw medium and showed a 95.91% inhibition of Mycobacterium bovis at 10μM concentration. All the synthesized compounds were subjected to cytotoxicity evaluation against various cell lines and were found to be noncytotoxic. The ADME properties calculated in-silico predicted good pharmacokinetic profile of the synthesized molecules. These analogues can be further developed to improve their InhA inhibitory activity.
4. Experimental:
4.1. General Chemistry
All chemicals and reagents for synthesis were procured from Sigma Aldrich (St. Louis, MO, USA) and Spectrochem Pvt. Ltd. (Bengaluru, India). LR grade solvent were used in the reaction. Distilled solvents were used for column chromatography (CC). Silica gel 60 F254 precoated TLC (Thin layered chromatography) plates were used for monitoring the reactions and visualized in UV chamber. The compounds were purified by CC or recrystallization. Column chromatography was performed on silica gel 60120 (100–200 mesh). Silica gel for CC and TLC plate both were purchased from Merck (Germany). The synthesized compounds were characterized using 1H and 13C NMR and mass spectrometry. 1H- and 13C-NMR spectra were recorded using Bruker Ascend 400 MHz, Chemical shift values (δ) are expressed in ppm relative to internal standard, tetramethylsilane (TMS). The mass spectra were recorded on Agilent Technologies 6545 Q-TOF LC/MS.
4.1.1. Synthesis of 1-(3-methoxy-4-phenoxyphenyl)ethan-1-one
1-(3-methoxy-4-phenoxyphenyl)ethan-1-one was synthesized as described in the previously reported procedure(22).The compound was used without further purification in the next step with a crude yield of 81%.
4.1.2. Synthesis of 1-(3-hydroxy-4-phenoxyphenyl)ethan-1-one
1-(3-hydroxy-4-phenoxyphenyl)ethan-1-one was synthesized from 1-(3-methoxy-4-phenoxyphenyl)ethanone as described in the previously reported procedure(22). The crude compound was purified using column chromatography using ethyl acetate: hexane (3:7) to give 76% yield.
4.1.3. Synthesis of 1-(3-hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-ones
1-(3-hydroxy-4-phenoxyphenyl)ethan-1-one (0.66 mmol) in EtOH (1mL) and LiOH·H2O (1.4 mmol) was stirred for 10 min at room temperature. Then various substituted benzaldehydes (0.79 mmol) were added and stirred at room temperature until complete consumption of the starting materials. A yellow precipitate was usually obtained after completion of the reaction. EtOH was removed under reduced pressure. The residue was diluted with water (5 mL) and neutralized with 2% dilute HCl. It was then extracted with Ethyl acetate (10 mL). The organic layer was washed with saturated NaCl solution (5 mL), dried with MgSO4 and concentrated under reduced pressure. It was then recrystallized from ethanol to afford various phenylprop-2-en-1-ones in 80–89% yield.
4.1.4. General method for the synthesis of substituted 1-(3-(3-hydroxy-4-phenoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one
1-(3-hydroxy-4-phenoxyphenyl)-3-phenylprop-2-en-1-ones (0.47 mmol) and hydrazine hydrate (1.41 mmol) in glacial acetic acid (2 mL) were refluxed for 2 h. Upon completion of the reaction, the mixture was poured into ice cold water (20 mL). The precipitate was then filtered and washed with cold water (10mL) and re-crystallized from ethanol to give pure product.
1-(5-(4-chlorophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-1); %Yield:
88%, 1H NMR (400 MHz, Chloroform-d) δ 8.16 – 7.95 (m, 2H), 7.52 (d, J = 7.6 Hz, 1H), 7.49 – 7.37 (m, 2H), 7.32 (t, J = 7.7 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 6.79 (d, J = 8.4 Hz, 1H), 5.77 (s, 1H), 5.59 (dd, J = 12.0, 5.0 Hz, 1H), 3.73 (dd, J = 17.8, 12.0 Hz, 1H), 3.05 (dd, J = 17.8, 5.0 Hz, 1H), 2.36 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 168.92, 155.89, 153.16, 147.36, 145.98, 140.35, 133.48, 130.11, 129.09, 127.53, 127.13, 124.44, 119.34, 118.74, 117.94, 114.15, 59.46, 42.28, 21.92. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19ClN2O3+ 407.1157; found 407.1148.
1-(3-(3-hydroxy-4-phenoxyphenyl)-5-(4-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-2); %Yield:
91%, 1H NMR (400 MHz, Chloroform-d) δ 8.16 – 8.10 (m, 2H), 7.40 (d, J = 2.0 Hz, 1H), 7.37 – 7.28 (m, 4H), 7.19 (s, 1H), 7.17 – 7.07 (m, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.79 (d, J = 8.5 Hz, 1H), 5.73 (s, 1H), 5.58 (dd, J = 12.0, 5.0 Hz, 1H), 3.72 (dd, J = 17.7, 12.0 Hz, 1H), 3.03 (dd, J = 17.7, 5.0 Hz, 1H), 2.35 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 155.76, 153.07, 148.82, 147.44, 147.37, 146.24, 130.14, 127.09, 126.70, 124.55, 124.34, 119.38, 118.83, 117.85, 114.15, 59.53, 42.12, 21.84, 1.02. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19N3O5+ 418.1397; found 418.1391.
1-(5-(4-bromophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-3); %Yield:
87%, 1H NMR (400 MHz, Chloroform-d) δ 7.42 – 7.35 (m, 3H), 7.32 (t, J = 7.8 Hz, 2H), 7.11 (t, J = 9.3 Hz, 2H), 7.04 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 6.79 (d, J = 8.5 Hz, 1H), 5.73 (s, 1H), 5.46 (dd, J = 12.5, 4.5 Hz, 1H), 3.65 (dd, J = 17.7, 11.9 Hz, 1H), 3.01 (dd, J = 17.3, 4.7 Hz, 1H), 2.33 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 168.94, 155.91, 153.20, 147.39, 146.00, 140.85, 132.03, 130.09, 127.49, 127.46, 124.41, 121.58, 119.33, 118.73, 117.98, 114.18, 59.52, 42.23, 21.90. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19BrN2O3+ 451.0652; found 451.0636.
1-(5-(4-fluorophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-4); %Yield:
85%, 1H NMR (400 MHz, Chloroform-d) δ 7.48 (d, J = 2.1 Hz, 1H), 7.43 – 7.34 (m, 2H), 7.25 – 7.14 (m, 4H), 7.06 (dd, J = 7.7, 1.5 Hz, 2H), 7.04 – 6.96 (m, 2H), 6.86 (d, J = 8.5 Hz, 1H), 5.77 (s, 1H), 5.56 (dd, J = 11.8, 4.6 Hz, 1H), 3.72 (dd, J = 17.6, 11.8 Hz, 1H), 3.10 (dd, J = 17.7, 4.6 Hz, 1H), 2.40 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 167.88, 154.89, 152.16, 146.34, 144.91, 129.08, 126.59, 126.43, 126.35, 123.39, 118.30, 117.70, 116.94, 114.86, 114.64, 113.13, 58.36, 41.34, 20.92, −1.02. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19FN2O3+ 391.1452; found 391.1439.
1-(3-(3-hydroxy-4-phenoxyphenyl)-5-(p-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-5); %Yield:
84%, 1H NMR (400 MHz, Chloroform-d) δ 7.47 (d, J = 2.0 Hz, 1H), 7.43 – 7.34 (m, 2H), 7.22 – 7.13 (m, 2H), 7.06 (d, J = 7.7 Hz, 2H), 6.86 (d, J = 8.4 Hz, 1H), 5.55 (dd, J = 11.8, 4.5 Hz, 1H), 3.69 (dd, J = 17.6, 11.8 Hz, 1H), 3.11 (dd, J = 17.6, 4.5 Hz, 1H), 2.40 (s, 3H), 2.30 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 167.02, 154.20, 151.54, 145.61, 143.96, 137.05, 135.47, 128.16, 127.67, 125.94, 123.68, 122.35, 117.38, 116.74, 116.30, 112.41, 57.95, 40.55, 20.06, 19.22. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C24H22N2O3+ 387.1703; found 387.1701.
1-(3-(3-hydroxy-4-phenoxyphenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-6); %Yield:
87%, 1H NMR (400 MHz, Chloroform-d) δ 7.48 (d, J = 2.0 Hz, 1H), 7.43 – 7.34 (m, 2H), 7.22 – 7.12 (m, 4H), 7.10 – 7.02 (m, 2H), 6.85 (t, J = 8.1 Hz, 3H), 5.82 (s, 1H), 5.54 (dd, J = 11.7, 4.5 Hz, 1H), 3.77 (s, 3H), 3.68 (dd, J = 17.6, 11.7 Hz, 1H), 3.11 (dd, J = 17.6, 4.5 Hz, 1H), 2.39 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 168.87, 159.04, 156.04, 153.35, 147.44, 145.82, 134.09, 130.06, 127.85, 126.96, 124.29, 119.28, 118.64, 118.11, 114.24, 59.52, 55.29, 42.34, 21.97. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C24H22N2O4+ 403.1652; found 403.1641.
1-(5-(3-chlorophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-7); %Yield:
82%, 1H NMR (400 MHz, Chloroform-d) δ 7.47 (d, J = 2.0 Hz, 1H), 7.38 (t, J = 7.9 Hz, 2H), 7.30 – 7.03 (m, 8H), 6.86 (d, J = 8.5 Hz, 1H), 5.54 (dd, J = 11.9, 4.7 Hz, 1H), 3.71 (dd, J = 17.9, 11.6 Hz, 1H), 3.09 (dd, J = 17.7, 4.7 Hz, 1H), 2.42 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 168.98, 155.93, 153.20, 147.41, 146.00, 143.81, 134.80, 130.23, 130.09, 127.93, 127.47, 125.73, 124.39, 123.92, 119.35, 118.72, 118.01, 114.21, 59.54, 42.32, 21.91. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19ClN2O3+ 407.1157; found 407.1141.
1-(3-(3-hydroxy-4-phenoxyphenyl)-5-(3-nitrophenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-8); %Yield:
88%, 1H NMR (400 MHz, Chloroform-d) δ 8.09 – 7.99 (m, 2H), 7.52 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.40 (d, J = 2.0 Hz, 1H), 7.32 (t, J = 7.7 Hz, 2H), 7.16 – 7.08 (m, 2H), 7.00 (d, J = 8.1 Hz, 2H), 6.79 (d, J = 8.4 Hz, 1H), 5.77 (s, 1H), 5.59 (dd, J = 12.0, 5.0 Hz, 1H), 3.73 (dd, J = 17.8, 12.0 Hz, 1H), 3.05 (dd, J = 17.8, 5.0 Hz, 1H), 2.36 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 169.16, 155.85, 153.09, 148.71, 147.42, 146.21, 143.84, 132.08, 130.11, 129.97, 127.12, 124.46, 122.81, 120.84, 119.41, 118.77, 117.97, 114.24, 59.44, 42.22, 21.88. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19N3O5+ 418.1397; found 418.1386.
1-(5-(3-bromophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-9); %Yield:
79%, 1H NMR (400 MHz, Chloroform-d) δ 7.47 (d, J = 2.1 Hz, 1H), 7.44 – 7.32 (m, 4H), 7.24 – 7.13 (m, 4H), 7.10 – 7.02 (m, 2H), 6.86 (d, J = 8.5 Hz, 1H), 5.53 (dd, J = 11.9, 4.7 Hz, 1H), 3.72 (dd, J = 17.7, 11.9 Hz, 1H), 3.09 (dd, J = 17.7, 4.7 Hz, 1H), 2.42 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 169.00, 155.93, 153.22, 147.41, 146.02, 144.07, 130.87, 130.52, 130.09, 128.63, 127.45, 124.39, 123.01, 119.36, 118.71, 118.02, 114.23, 59.49, 42.34, 21.91. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19BrN2O3+ 451.0652; found 451.0634.
1-(5-(3-fluorophenyl)-3-(3-hydroxy-4-phenoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-10); %Yield:
81%, 1H NMR (400 MHz, Chloroform-d) δ 7.46 (d, J = 2.0 Hz, 1H), 7.38 (t, J = 7.9 Hz, 2H), 7.29 (td, J = 7.6, 5.5 Hz, 1H), 7.22 – 7.14 (m, 2H), 7.09 – 6.99 (m, 3H), 6.99 – 6.88 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 5.57 (dd, J = 11.8, 4.6 Hz, 1H), 3.71 (dd, J = 17.7, 11.9 Hz, 1H), 3.09 (dd, J = 17.7, 4.6 Hz, 1H), 2.42 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 167.97, 154.95, 152.26, 146.42, 144.98, 129.55, 129.47, 129.05, 126.47, 123.32, 120.26, 120.24, 118.29, 117.66, 117.05, 113.73, 113.52, 113.22, 111.67, 111.45, 58.53, 41.29, 20.86. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C23H19FN2O3+ 391.1452; found 391.1439.
1-(3-(3-hydroxy-4-phenoxyphenyl)-5-(m-tolyl)-4,5-dihydro-1H-pyrazol-1-yl)ethan-1-one (PYN-11); %Yield:
84%, 1H NMR (400 MHz, Chloroform-d) δ 7.48 (d, J = 2.0 Hz, 1H), 7.38 (t, J = 7.8 Hz, 2H), 7.19 (ddd, J = 8.6, 5.9, 2.6 Hz, 3H), 7.04 (dd, J = 17.6, 6.5 Hz, 5H), 6.86 (d, J = 8.4 Hz, 1H), 5.55 (dd, J = 11.8, 4.5 Hz, 1H), 4.78 (s, 8H), 3.70 (dd, J = 17.6, 11.8 Hz, 1H), 3.11 (dd, J = 17.7, 4.6 Hz, 1H), 2.42 (s, 3H), 2.32 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 168.98, 156.03, 153.47, 147.44, 145.86, 141.79, 138.59, 130.06, 128.82, 128.48, 127.79, 126.17, 124.29, 122.55, 119.31, 118.64, 118.11, 114.27, 60.04, 42.52, 21.94, 21.47. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C24H22N2O3+ 387.1703; found 387.1691.
4.2. Anti-tubercular Activity
4.2.1. Anti-mycobacterial Assay
Mycobacterium bovis (BCG) Pasteur is a surrogate model for high throughput screening of chemical compounds to identify newer antimycobacterial agents.(23) Primary screening at single concentration (10 μM) was performed against Mycobacterium bovis in 96-well flat-bottomed polystyrene microtiter sterile plates (Nunc). Test compounds prepared in DMSO were dispensed into triplicate wells before addition of the assay components. Ninety-eight μl of inoculum (overnight culture with 0.6 OD diluted at 1:1000 in 7H9 broth) was distributed into sterile micro titre plates. This dilution of test compounds with inoculums provided 10 μM of final concentration of the compounds in screening medium. To better ascertain the activity of the compounds, controls like DMSO as a solvent control, media control (Blank) as well as Rifampicin and Isoniazid were added as positive controls for inhibition of Mycobacterium bovis growth in every plate. The peripheral wells of assay plates were filled with sterile distilled water to avoid evaporation in assay wells. Inoculated plates were stacked in groups of 7–8 plates. Plates were carefully allowed to incubate at 37°C and at 80% relative humidity. The plates were incubated for four days. Growth of the bacteria was studied after the incubation period using turbidometry, by measuring the absorbance at 600 nm using a Multi-Mode Reader (Perkin Elmer). Percentage growth inhibition was determined against DMSO control. The growth inhibition effect of compounds was calculated as:Percentage Inhibition= 100 x [OD with compound- OD of Negative control] / [OD of Positive control-OD of Negative control].
Every assay plate had DMSO in one column as negative control which corresponds to 100% growth and positive controls (Rifampicin and Isoniazid) in one column.
4.2.3. M. tuberculosis (H37Rv) inhibition assay
All the final synthesized substituted diphenyl ether derivatives were evaluated for in vitro antitubercular activity against Mycobacterium tuberculosis H37Rv strain. Minimum inhibitory concentrations were determined in triplicate in iron-supplemented GAST (pH 6.6) or in Middlebrook 7H9 broth base supplemented with 0.2% glycerol, 0.4% glucose, 0.5% BSA fraction V, 0.08% NaCl and 0.05% Tween 80 in 96w plates according to the broth microdilution method using drugs from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. Briefly, two-fold serial dilutions of compounds in DMSO were added to 50 mL of medium in round-bottom 96-well plates (Nunc). An equal volume of Mycobacterium tuberculosis H37Rv ATCC27294 diluted to 10,000 cells/mL in the respective medium was added to each well. Positive controls included isoniazid and triclosan. Plates were incubated for 1 and 2 weeks at 37°C after which growth was determined using an inverted enlarging mirror. The MIC was determined as the lowest concentration of compound that completely inhibited growth of the bacterium.(24)(25)(26)
4.3. Cytotoxicity
4.3.1. Resazurin Assay
All cell lines used in this study were purchased from the American Type Culture Collection (ATCC). MCF7, PANC-1, PC-3, SK-OV-3 and HeLa were grown in Dulbecco’s modified Eagle’s medium (containing 10% FBS in a humidified atmosphere of 5% CO2 at 37°C). PC-3 cells were cultured in RPMI-1640 medium containing non-essential amino acids, 1 mM sodium pyruvate, 10 mg/mL bovine insulin, and 10% FBS. Cells were trypsinized when sub-confluent from T75 flasks/90 mm dishes and seeded in 96-well plates. The synthesized test compounds were evaluated for their in vitro antiproliferative activity in five different human cancer cell lines by Resazurin assay. The cell lines were grown in their respective media and seeded into 96-well microtiter plates in 100 μL aliquots at plating densities depending on the doubling time of individual cell lines. The microtiter plates were incubated at 37°C, 5% CO2, 95% air, and 100% relative humidity for 24 h prior to addition of drugs. Aliquots of 1 μL of the test compounds were added to the wells in triplicates already containing 100 μL of cells, resulting in the required final drug concentrations, plates were incubated further for 48 h. The assay was terminated by the addition of 50 μL of Resazurin solution and incubated for 60 min at 37°C. The Fluorescence Intensity was read on a multimode plate reader (Tecan M200) at a wavelength of 560 nm Excitation / 590nm Emission. The cell growth is directly proportional to the measure of absorbance and is used to calculate the IC50 values.
Percentage Inhibition= 100 × [OD with compound- OD of Negative control] / [OD of Positive control- OD of Negative control].
4.3.2. Sulforhodamine B Assay
The cytotoxic activity of compounds was also tested on BD-11 and HEK using the sulforhodamine-B (SRB) (Sigma Aldrich, Germany) microculture colorimetric assay. Cells were seeded in 96-well plates on day zero, at appropriate cell densities. After 24 hours, the cells were treated with serial dilutions of the compounds (6.25 to 100 lM). The plates were incubated at 37°C in a humidified incubator with 5% CO2 for 96 hours. The final concentration of DMSO never exceeded 0.5%, which was shown to be non-toxic to the cells. After 96 hours of drug exposure percentages of surviving cells compared to untreated controls were determined. The cells were fixed with 10% TCA after discarding the supernatant medium from the 96-well plates. The plates were then allowed to stand at 4°C. Then the cells were washed in a strip washer four times with water using alternate dispensing and aspiration procedures. 100 μL of 0.05% SRB and incubated at room temperature for about 30 min. After dying, the plates were washed with 1% acetic acid to remove the dye and allowed to air dry overnight. Then, 50 μL of 10 mM Tris base solution (pH10) was added to each well and absorbance was measured at 520 nm using a 96-well plate reader (Tecan Spectra, Crailsheim, Germany). IC50 was calculated using GraphPad Prism software.
4.4. Drug Design Strategy
4.4.1. Data Set for Cross Docking Study
A total of 87 crystal structures of Enoyl-acyl-carrier-protein reductase of Mycobacterium tuberculosis available in RCSB protein data bank were selected for the present study. However, only the crystal structures containing co-crystallized inhibitors similar to triclosan were selected for the study. Of the numerous chains of InhA, only the A-chain was considered for the comparative study of the crystal structures. Out of 15 selected crystal structures, 4 were not considered appropriate for the study because they either had a missing inhibitor ligand in the A-chain or had missing coordinates for the substrate binding loop.
4.4.2. Protein Preparation
Selected 11 protein crystal structures were prepared using protein preparation module of Schrodinger. Bond orders of the protein structure were assigned, and hydrogens were added. The hydroxyl, Asparagine, Glutamine and Histidine states were optimized using ProtAssign. The complex was subjected to restrained minimization using OPLS_2005 force field, which was terminated when the root-mean-square deviation of the heavy atoms relative to their initial location reached a maximum value of 0.3Å.
The inhibitor ligands from respective crystal structures were extracted out and were aligned using flexible shape-based alignment in the Flexible ligand alignment module as shown in Figure 9. The prepared protein structures were aligned on the protein PDB entry code 2X23(27) according to the protein back bone using protein structure alignment as shown in Figure 8. In order to test the significance of water molecules in the study another set of protein structures were created by removing all the water molecules from the crystal structures.
Figure 9.
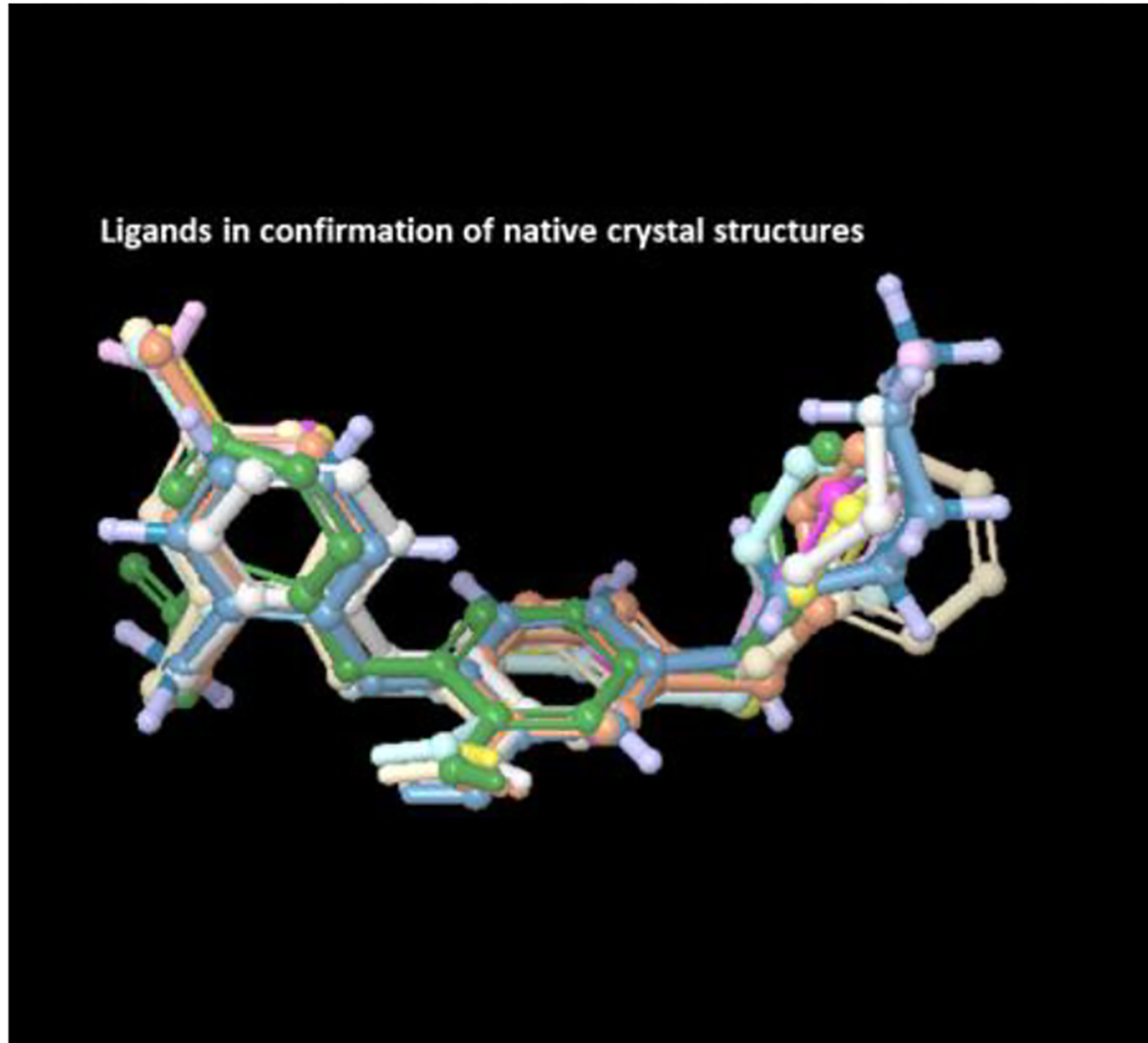
All the extracted ligands aligned using flexible ligand alignment
Figure 8.
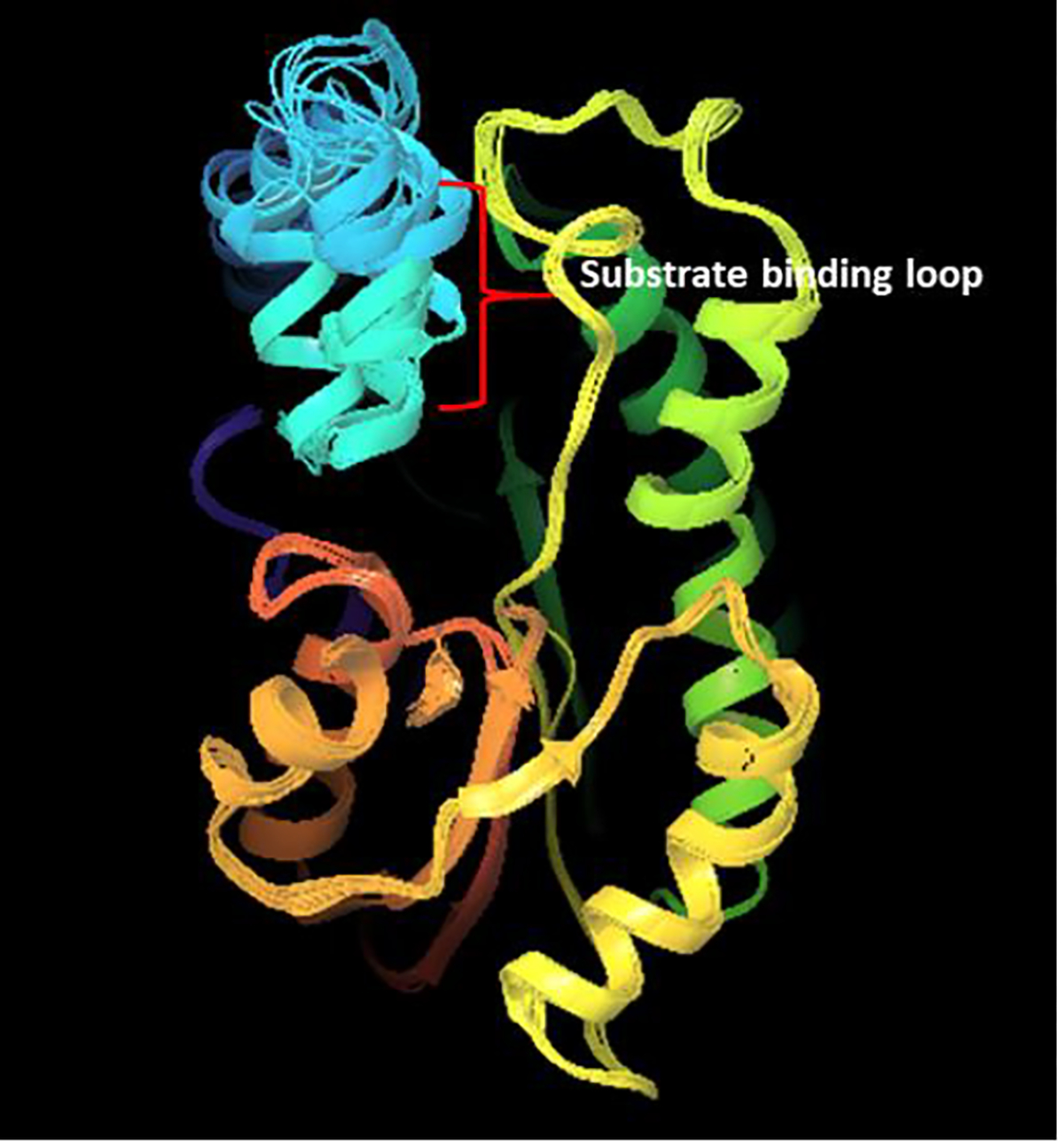
All 11 crystal structures aligned over 2X23 crystal structure
4.4.3. Receptor Grid Generation
Receptor grid was then generated on the chain-A of all the prepared protein structures using glide grid generation with centre of the grid as centroid of the co-crystallized ligand.
4.4.4. Docking
Docking was performed using glide dock on each of the protein crystal structure using all the extracted ligands. The docking was performed in extra precision (XP) mode. Every ligand was docked on all the 11 protein crystal structures.
4.4.5. RMSD Calculation
The aligned ligands were chosen individually and super positioned over their docked poses in different protein structures. Root-mean-square deviations (RMSD) of all the docked poses on proteins containing crystal waters and the ones without the crystal water were then determined with respect to the aligned ligand structures
4.4.6. Data Set for Correlation Study
Twenty-five diphenyl ether derivatives collected from literature (28) were used for this correlation study. The molecules had wide variation in their range of biological activity. The IC50 values were converted to −log IC50 (pIC50).
4.4.7. Molecular Docking
All the 25 inhibitors were docked on protein 3FNE which was prepared by using protein preparation wizard module keeping all the default settings and by removing explicitly all the water molecules from the crystal structure. The selected molecules were drawn using build panel in Maestro, Schrödinger. Bond orders of all the structures were corrected and structures were energy minimized using LigPrep, Schrödinger. Grid was generated by using centroid of the co-crystallized ligand in the protein-ligand complex crystal as centre. Extra precision (XP) mode (Glide, Schrödinger) was applied for analysing the binding modes of the inhibitors.
4.4.8. Prime MM/GBSA Calculations
The MM/GBSA binding free energy was calculated (Prime, Schrödinger) using the receptor ligand complex obtained from molecular docking done in extra precision (XP) mode. VSGB2.0 solvation model and OPLS_2005 force field were used for the simulation.
4.5. Induced Fit Docking
The receptor protein and the ligands were prepared as per the protocol mentioned above. Triclosan and PYN-8 were docked into the active site of 3FNE using the Induced Fit Docking Protocol (IFD) (Glide, Schrödinger)(29). First the van der Waals radii of protein and ligand atoms are scaled down by a factor of 0.5, and ligands are then docked into the fixed receptor using the Glide SP docking protocol. Then optimal orientation of the side chains of the binding site residues is predicted by Prime. Finally, the optimised binding site was used for re-docking of the ligand and poses were scored with Glide XP. Following IFD twenty top-scoring poses were obtained.
4.6. Water Map
Water map (WaterMap, Schrödinger) was generated using the protein 3FNE after preparation of the protein using protein preparation wizard (Epik, Schrödinger)(30). The calculation was run for 9ns (300K,1atm) which is a three-step process. It involves running a molecular dynamics simulation of protein-ligand complex with explicit water molecules. Clustering algorithm then locates the hydration sites and finally thermodynamic properties of the waters are calculated.
4.7. X-ray Diffraction
X-ray data of compound PYN-8 was collected at room temperature using a Bruker Smart Apex CCD diffractometer with graphite monochromated MoKα radiation (λ=0.71073Å) with ω-scan method(31). Preliminary lattice parameters and orientation matrices were obtained from four sets of frames.
Integration and scaling of intensity data was accomplished using SAINT program(31). The structure was solved by direct methods using SHELXS(32) and refinement was carried out by full-matrix least-squares technique using SHELXL(32).Anisotropic displacement parameters were included for all non-hydrogen atoms. Oxygen bound hydrogen atoms were located in different Fourier maps and their positions and isotropic displacement parameters were refined. All hydrogen atoms were positioned geometrically and treated as riding on their parent carbon atoms [C-H = 0.93–0.97 Å and Uiso(H) = 1.2Ueq(C) for other hydrogen atoms]. All attempts to model a disordered ethylacetate solvent (used for crystallization) failed. Therefore, the solvent contributions have been removed using the SQUEEZE procedure in PLATON(33). SQUEEZE calculated a void volume of approximately 334 Å3 occupied by 73 electrons per unit cell.
Crystal Data for PYN-8: C23H19N3O5 (M =417.41 g/mol): triclinic, space group P-1 (no. 2), a = 11.2865(7) Å, b = 11.4236(7) Å, c = 11.9381(7) Å, α = 115.8430(10)°, β = 93.3520(10)°, γ = 111.2500(10)°, V = 1247.36(13) Å3, Z = 2, T = 294.15 K, μ(MoKα) = 0.080 mm-1, Dcalc = 1.111 g/cm3, 15426 reflections measured (3.924° ≤ 2Θ ≤ 56.71°), 6041 unique (Rint = 0.0259, Rsigma = 0.0341) which were used in all calculations. The final R1 was 0.0573 (I > 2σ(I)) and wR2 was 0.1671 (all data). CCDC 1883097 contains supplementary Crystallographic data for the structure. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0) 1223 336 033; deposit@ccdc.cam.ac.uk].
Table 3.
In-silico ADME Properties as Predicted by Qikprop
| Compound | Mol MW | HD | HA | QPlogPo/w | QPPCaco | QPlog BB | %Human Oral Absorption | Rule Of Five | PSA |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| PYN-1 | 406.8 | 1 | 4.75 | 5.291 | 1605.4 | −0.305 | 100 | 1 | 69.6 |
| PYN-2 | 417.4 | 1 | 5.75 | 4.084 | 192.5 | −1.564 | 91.747 | 0 | 114.7 |
| PYN-3 | 451.3 | 1 | 4.75 | 5.367 | 1609.9 | −0.293 | 100 | 1 | 70.4 |
| PYN-4 | 390.4 | 1 | 4.75 | 5.036 | 1596.5 | −0.358 | 100 | 1 | 69.7 |
| PYN-5 | 386.4 | 1 | 4.75 | 5.107 | 1609.4 | −0.484 | 100 | 1 | 70.3 |
| PYN-6 | 402.4 | 1 | 5.5 | 4.928 | 1782.8 | −0.5 | 100 | 0 | 78.1 |
| PYN-7 | 406.8 | 1 | 4.75 | 5.335 | 1802.7 | −0.254 | 100 | 1 | 69.6 |
| PYN-8 | 417.4 | 1 | 5.75 | 4.127 | 217.1 | −1.516 | 92.936 | 0 | 113.1 |
| PYN-9 | 451.3 | 1 | 4.75 | 5.412 | 1803.4 | −0.244 | 100 | 1 | 69.6 |
| PYN-10 | 390.4 | 1 | 4.75 | 5.075 | 1801.3 | −0.304 | 100 | 1 | 69.9 |
| PYN-11 | 386.4 | 1 | 4.75 | 5.148 | 1814.6 | −0.431 | 100 | 1 | 69.8 |
HD – hydrogen bond donor, HA – hydrogen bond acceptor, PSA- Polar Surface Area
Acknowledgment
The authors are thankful to Manipal Academy of Higher Education and Manipal College of Pharmaceutical Sciences for providing necessary supports and facilities to carry out the present research work. Authors are thankful to D’IICT and National Mol Bank (NMB) facility of CSIR-IICT, Hyderabad, India for the help in biological evaluation of compounds. Authors also thank Manipal-Schrödinger Centre for Molecular Simulations. This work was funded in part by the Intramural Research Program of the NIH, NIAID.
Footnotes
Conflict of Interest
The authors confirm that this article content has no conflict of interest.
References:
- 1.WHO | Tuberculosis (TB). WHO [Internet]. World Health Organization; 2018. [cited 2019 Mar 1]; Available from: https://www.who.int/gho/tb/en/ [Google Scholar]
- 2.World Health Organization. BCG vaccine: WHO position paper, February 2018 – Recommendations. Vaccine. 2018. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Guidelines for treatment of drug-susceptible tuberculosis and patient care. World Health Organization. 2017. [Google Scholar]
- 4.Rožman K, Sosič I, Fernandez R, Young RJ, Mendoza A, Gobec S, et al. A new ‘golden age’ for the antitubercular target InhA. Drug Discovery Today. 2017. [DOI] [PubMed] [Google Scholar]
- 5.Forrellad MA, Klepp LI, Gioffré A, Sabio y García J, Morbidoni HR, Santangelo M de la P, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992; [DOI] [PubMed] [Google Scholar]
- 7.Narang A, Giri A, Gupta S, Garima K, Bose M, Varma-Basil M. Contribution of putative efflux pump genes to isoniazid resistance in clinical isolates of Mycobacterium tuberculosis. Int J Mycobacteriology [Internet]. Medknow Publications and Media Pvt. Ltd.; 2017. [cited 2019 Apr 29];6(2):177. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28559521 [DOI] [PubMed] [Google Scholar]
- 8.McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis [4]. Nature. 1998. [DOI] [PubMed] [Google Scholar]
- 9.Parikh SL, Xiao G, Tonge PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry. 2000;39(26):7645–50. [DOI] [PubMed] [Google Scholar]
- 10.Luckner SR, Liu N, Am Ende CW, Tonge PJ, Kisker C. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem. 2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J Mol Biol. 1999; [DOI] [PubMed] [Google Scholar]
- 12.Freundlich JS, Wang F, Vilchèze C, Gulten G, Langley R, Schiehser GA, et al. Triclosan derivatives: Towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem. 2009;4(2):241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chollet A, Maveyraud L, Lherbet C, Bernardes-Génisson V. An overview on crystal structures of InhA protein: Apo-form, in complex with its natural ligands and inhibitors. Eur J Med Chem. 2018;146:318–43. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, et al. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem Biol. 2006; [DOI] [PubMed] [Google Scholar]
- 15.Pan P, Knudson SE, Bommineni GR, Li HJ, Lai CT, Liu N, et al. Time-dependent diaryl ether inhibitors of InhA: Structure-activity relationship studies of enzyme inhibition, antibacterial activity, and in vivo efficacy. ChemMedChem. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.am Ende CW, Knudson SE, Liu N, Childs J, Sullivan TJ, Boyne M, et al. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorganic Med Chem Lett. 2008;18(10):3029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cappel D, Sherman W, Beuming T. Calculating Water Thermodynamics in the Binding Site of Proteins – Applications of WaterMap to Drug Discovery. Curr Top Med Chem. 2017; [DOI] [PubMed] [Google Scholar]
- 18.Wei E, Liu B, Lin S, Liang F. Multicomponent reaction of chalcones, malononitrile and DMF leading to γ-ketoamides. Org Biomol Chem. Royal Society of Chemistry; 2014;12(33):6389–92. [DOI] [PubMed] [Google Scholar]
- 19.Lipinski CA. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discovery Today: Technologies. 2004. [DOI] [PubMed] [Google Scholar]
- 20.Cheraïti N, Brik ME. Synthesis of a new triprotonated ligand and selective O-demethylation of methyl aryl ether by boron tribromide. Tetrahedron Lett. 1999;40(23):4327–30. [Google Scholar]
- 21.Bhagat S, Sharma R, Sawant DM, Sharma L, Chakraborti AK. LiOH · H 2 O as a novel dual activation catalyst for highly efficient and easy synthesis of 1, 3-diaryl-2-propenones by Claisen – Schmidt condensation under mild conditions. 2006;244:20–4. [Google Scholar]
- 22.Kar SS, Bhat VG, Shenoy VP, Bairy I, Shenoy GG. Design, synthesis and evaluation of novel diphenyl ether derivatives against drug susceptible and resistant strains of Mycobacterium tuberculosis. Chem Biol Drug Des. 2018; [DOI] [PubMed] [Google Scholar]
- 23.Khan A, Sarkar D. A simple whole cell based high throughput screening protocol using Mycobacterium bovis BCG for inhibitors against dormant and active tubercle bacilli. J Microbiol Methods. 2008; [DOI] [PubMed] [Google Scholar]
- 24.Barot KP, Jain SV., Gupta N, Kremer L, Singh S, Takale VB, et al. Design, synthesis and docking studies of some novel (R)-2-(4′- chlorophenyl)-3-(4′-nitrophenyl)-1,2,3,5-tetrahydrobenzo[4,5] imidazo [1,2-c]pyrimidin-4-ol derivatives as antitubercular agents. Eur J Med Chem [Internet]. Elsevier Masson SAS; 2014;83:245–55. Available from: 10.1016/j.ejmech.2014.06.019 [DOI] [PubMed] [Google Scholar]
- 25.Kaniga K, Cirillo DM, Hoffner S, Ismail NA, Kaur D, Lounis N, et al. A multilaboratory, multicountry study to determine MIC quality control ranges for phenotypic drug susceptibility testing of selected First-Line Antituberculosis Drugs, Second-Line Injectables, Fluoroquinolones, Clofazimine, and Linezolid. J Clin Microbiol. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Candice SDM, Feng TS, Van Der Westhuyzen R, Gessner RK, Street LJ, Morgans GL, et al. Aminopyrazolo[1,5-a]pyrimidines as potential inhibitors of Mycobacterium tuberculosis: Structure activity relationships and ADME characterization. Bioorganic Med Chem [Internet]. Elsevier Ltd; 2015;23(22):7240–50. Available from: 10.1016/j.bmc.2015.10.021 [DOI] [PubMed] [Google Scholar]
- 27.Kumar V, Sobhia ME. Insights into the bonding pattern for characterizing the open and closed state of the substrate-binding loop in Mycobacterium tuberculosis InhA. Future Med Chem. 2014; [DOI] [PubMed] [Google Scholar]
- 28.Pan P, J. Tonge P, Tonge P. Targeting InhA, the FASII Enoyl-ACP Reductase: SAR Studies on Novel Inhibitor Scaffolds. Curr Top Med Chem [Internet]. 2012;12:672–93. Available from: http://www.ingentaconnect.com/content/ben/ctmc/2012/00000012/00000007/art00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand / Receptor Induced Fit Effects. 2006;534–53. [DOI] [PubMed] [Google Scholar]
- 30.Robinson D, Bertrand T, Carry JC, Halley F, Karlsson A, Mathieu M, et al. Differential Water Thermodynamics Determine PI3K-Beta/Delta Selectivity for Solvent-Exposed Ligand Modifications. J Chem Inf Model. 2016;56(5):886–94. [DOI] [PubMed] [Google Scholar]
- 31.Bruker (2001). SAINT (Version 6.28a) & SMART (Version 5.625). Bruker AXS Inc., Madison, Wisconsin, USA. [Google Scholar]
- 32.Sheldrick GM. Crystal structure refinement with {\it SHELXL}. Acta Crystallogr Sect C [Internet]. 2015. Jan;71(1):3–8. Available from: 10.1107/S2053229614024218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spek AL. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr Sect C Struct Chem. 2015; [DOI] [PubMed] [Google Scholar]