

Abstract
The influenza virus remains a significant cause of mortality worldwide due to the limited effectiveness of currently available vaccines. A key challenge to the development of universal influenza vaccines is high antigenic diversity resulting from antigenic drift. Overcoming this challenge requires novel research tools to measure the breadth of serum antibodies directed against many virus strains across different antigenic subtypes. Here, we present a protocol for analyzing the breadth of serum antibodies against diverse influenza virus strains using a protein microarray of influenza antigens.
This influenza antigen microarray is constructed by printing purified hemagglutinin and neuraminidase antigens onto a nitrocellulose-coated membrane using a microarray printer. Human sera are incubated on the microarray to bind antibodies against the influenza antigens. Quantum-dot-conjugated secondary antibodies are used to simultaneously detect IgG and IgA antibodies binding to each antigen on the microarray. Quantitative antibody binding is measured as fluorescence intensity using a portable imager. Representative results are shown to demonstrate assay reproducibility in measuring subtype-specific and cross-reactive influenza antibodies in human sera.
Compared to traditional methods such as ELISA, the influenza antigen microarray provides a high throughput multiplexed approach capable of testing hundreds of sera for multiple antibody isotypes against hundreds of antigens in a short time frame, and thus has applications in serosurveillance and vaccine development. A limitation is the inability to distinguish binding antibodies from neutralizing antibodies.
Keywords: Immunology and Infection, Issue 149, Protein microarray, Influenza virus, Antigen, Antibody, Hemagglutinin, Immunity
Introduction
The influenza virus is responsible for a loss of 20 million life-years annually by death or disability, including 1% of all deaths worldwide each year, with disproportionate impacts on the elderly and populations in the tropics and developing world1,2,3. In addition to the disease burden of seasonal epidemics, the emergence of novel influenza strains via genetic re-assortment either naturally in common hosts or artificially for bioterrorism could lead to worldwide pandemics with rapid spread and high lethality4,5. While numerous influenza vaccines are currently available, their effectiveness is limited by subtype specificity6, creating the need to develop universal influenza vaccines that confer long-lasting immunity against multiple virus strains7.
A key challenge to the development of universal influenza vaccines is high antigenic diversity across strains. The antigenic specificity of current vaccines combined with antigenic variation of circulating viruses creates a mismatch between vaccine strains and circulating strains. This confers an evolutionary advantage favoring further genetic drift away from vaccine strains during an epidemic, limiting vaccine efficacy often to less than 50%8,9. An additional source of antigenic mismatch is egg-adaptive viral mutations generated during vaccine manufacture, which lead to antibodies that bind poorly to circulating viruses10,11.
Overcoming this challenge of high antigenic diversity will require novel research tools to characterize the breadth of pre-existing and elicited immune responses across clinically relevant antigenic variants in serum and mucosal specimens. Currently available methods, including hemagglutination inhibition (HAI), microneutralization (MN), and traditional ELISA, are limited to detecting antibodies against a single virus strain at a time, so their use for detection of multiple antibody isotypes against multiple virus strains quickly exhausts available clinical specimen and laboratory resources. Furthermore, HAI and MN require live virus culture that is only available in specialized laboratories.
Protein microarrays, potentially consisting of up to thousands of antigens printed onto nitrocellulose-coated slides as shown in Figure 1, can fill this need12. These microarrays can be produced and probed in a high throughput manner while consuming small quantities of clinical specimen to determine quantitative antibody isotype/subtype levels against each individual antigen on the array. This approach to antigen discovery has been applied to diagnostic and vaccine development against multiple infectious pathogens13. To date, we have produced protein microarrays for over 35 pathogens including over 60,000 total expressed proteins and used them to probe over 30,000 human sera from infected and control individuals. A recently developed portable imaging platform for microarray slides has made this methodology more accessible to the end user14.
Figure 1: Schematic of protein microarray.

Each slide contains multiple pads each with a single array, which consists of hundreds of antigens printed onto spots arranged in a grid, with each spot containing one antigen adsorbed onto the 3-dimensional topography of the nitrocellulose surface to which antibodies from serum are bound.
Building on extensive previous work by multiple contributors in the field15,16,17,18,19, an influenza protein microarray was recently developed that contains over 250 purified hemagglutinin (HA) antigenic variants with representation of all 18 subtypes12,20. Using this methodology, a natural influenza infection was demonstrated to generate broadly reactive IgG and IgA antibodies against phylogenetically related HA subtypes, while an intramuscular influenza vaccination generated only subtype-specific IgG antibodies21. However, adding an adjuvant that activates toll-like receptors to influenza vaccines was shown to broaden the elicited IgG antibody response across HA subtypes in animal studies22.
This microarray is currently being used to probe sera collected from a prospective cohort study of college students who were followed for influenza infection. Here, the methodology of the influenza antigen microarray with demonstration of assay reproducibility to detect subtype-specific and cross-reactive antibodies in a subset of specimens from this study is reported.
Protocol
All human sera are handled and disposed according to approved institutional protocols for biosafety with use of protective personal equipment. All laboratory personnel participating in this protocol have received training in biosafety and research ethics.
1. Produce influenza antigen microarrays
- Design and obtain antigen set for microarray
- Obtain expressed and purified protein antigens as lyophilized powder.
- Print antigens onto microarray slides
- Reconstitute each lyophilized antigen to a concentration of 0.1 mg/mL in phosphate-buffered saline (PBS) with 0.001% Tween-20 (T-PBS). Transfer 10 μL of each reconstituted antigen to individual wells of an untreated 384-well flat-bottom plate.
- Print antigens using a microarray printer (the microarray printer used in this study is no longer commercially available, see Discussion) with low-volume microarray spotting pins that aspirate antigen into the sample channel and deposit via direct contact and capillary action onto 16-pad nitrocellulose-coated glass slides.
- Program the printing software (e.g., Gridder) with the source plate configuration and printing parameters.
- Use the pull-down menu to select the name of the plate type that will be used with this printing method. For this study, use an untreated 384-well flat-bottom plate.
- Select the text box next to Number of Plates and type the number of sample plates that will be used in this printing protocol. For this study, use 1 plate.
- Select a pin configuration to use with this method. For this study, use an 8-pin configuration.
- Ensure the origin offsets are the distances (in the X- and Y-directions) between the slide origin (which is calibrated in the Administrative section) and the location where the printing pins will start printing on the slides.
- Using the Array Design tab, define the size and shape of the arrays (dot spacing and number of dots per subarray). For this study, print 324 spots (180 μm diameter with 300 μm spacing) onto 16-pad slides in an 18×18 format using 8 pins.
- Select the parameters for how the printing pins pick up and dispense samples. For this study, each pin aspirates 250 nL of antigen solution and prints 1 nL onto each of 40 spots (total of 20 slides for all 8 pins)
- Configure the pin cleaning protocol and blotting protocol. For this study, each pin prints antigen, is dipped in sterile ddH2O in sonicated wash container, and then aspirates next antigen.
-
Define the sequence in which the sample blocks are printed onto the slides. The array software will construct an annotated grid index (.gal) file to describe the arrangement of antigens within each microarray.NOTE: The top row is used for fiducials (with fluorescence at all wavelengths used in imaging, e.g., mix of Qdot 585 nm streptavidin conjugate and Qdot 800 nm streptavidin conjugate in this study) to orient grids during imaging. For long printing runs, antigens in source plate can be periodically re-suspended by pipetting up and down and then centrifuging, or new source plate can be prepared and used.
- Place un-probed microarray slides in a lightproof box and keep in a desiccator cabinet at room temperature for long-term storage. NOTE: The protocol may be paused indefinitely at this point.
- Perform quality control check (requires poly-histidine tags)
- Attach the slide to probing chambers and rehydrate with blocking buffer as described in step 2.1.1–2.1.2 and shown in Figure 2.
-
Dilute the mouse monoclonal poly-His antibody 1:100 in filtered 1x blocking buffer.NOTE: If non-purified protein antigens (e.g., expressed in in vitro transcription and translation system) are directly used to print microarrays, add components of the protein expression system (e.g., E. coli lysate) in a ratio of 1:10 with the blocking buffer used for serum dilution in order to block any antibodies directed against these components.
- Add 100 μL of diluted poly-His antibody to each slide chamber containing array pad after aspiration and incubate for 2 h at room temperature or overnight at 4 °C on shaker. Wash 3x with T-TBS buffer as described in step 2.2.1. Dilute biotin-conjugated goat anti-mouse IgG secondary antibody 1:200 in blocking buffer, add 100 μL per well after aspiration, and incubate for 1 h at room temperature on shaker. Wash 3x with T-TBS buffer.
- Dilute Qdot 585 nm streptavidin conjugate to 4 nM in blocking buffer, add 100 μL per well after aspiration, and incubate for 1 h at room temperature on shaker. Wash 3x with T-TBS buffer, and then once with TBS buffer (without Tween).
-
Dissemble and quantify slides as described in step 2.2.5 – 3.1.2.NOTE: Protein antigens must contain His10 tags to use this quality control protocol. Alternatively, if a different tag is included, quality control check can be performed with antibody or ligand for that tag.
Figure 2: Schematic of influenza antigen microarray printing and probing protocol.

From left to right, microarray is printed using onto nitrocellulose-coated slides, which are used to probe sera for IgG and IgA antibodies using quantum-dot-conjugated secondary antibodies, with slides imaged using a portable imager, and results analyzed to generate a heat map.
2. Probe sera for influenza antibodies using microarrays
- Incubate sera on microarrays for antibody binding
-
Attach microarray slides to chambers using clips and place in frames as shown in Figure 3.NOTE: Always avoid touching the microarray pad with hands and instruments. Ensure that slides are oriented with the pad side up and the small notch in upper right corner.
-
Rehydrate microarray slides with 100 μL per well of filtered 1x blocking buffer, and dilute serum 1:100 in 100 μL of blocking buffer (can use untreated 96-well plates or 2 mL tubes). Incubate both rehydrated microarray slides in covered frames and diluted sera separately for 30 minutes at room temperature on orbital shaker at 100–250 rpm. Perform all subsequent incubation steps similarly on the shaker.NOTE: Sera should be aliquoted and frozen at −80 °C for long-term storage to minimize freeze-thaw cycles and should be vortexed to mix and centrifuged to remove particulates prior to use. Observe slide chambers carefully during and after this step to detect any leakage that requires re-assembling slide chambers.
- Using pipette tips connected to a vacuum line with secondary collection flask, carefully aspirate blocking buffer from corner of each chamber without touching pads. Perform all subsequent aspiration steps similarly. Add diluted sera to pads quickly after aspiration in order to not allow pads to dry.
- Place covered frames in trays inside a secondary container surrounded by moist paper towels and sealed to prevent evaporation. Incubate overnight at 4 °C on rocking shaker (alternatively, can incubate for 2 h at room temperature on orbital shaker at 100–250 rpm).
-
- Label bound serum antibodies with quantum-dot-conjugated secondary antibodies
- Aspirate sera from chambers carefully as described above, add 100 μL per well of T-TBS buffer (20 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20 in ddH2O adjusted to pH 7.5 and filtered, can be obtained commercially), and incubate for 5 min on orbital shaker at 100–250 rpm. Repeat this wash step a total of 3x (all subsequent wash steps are performed similarly).
-
Prepare mixture of secondary antibodies diluted to 1 μM in blocking buffer and mix thoroughly by pipetting prior to and during use to maintain homogeneity.NOTE: To maintain assay reproducibility, the same batch of each secondary antibody should be used for all probing experiments for which quantitative comparison of data across experiments is planned. The specific concentration of secondary antibody may need to be varied depending on the affinity; follow manufacturer’s protocols whenever available.
- Aspirate buffer from chambers after final wash, add 100 μL per well of secondary antibody mixture, and incubate for 2 h at room temperature on shaker.
- Aspirate secondary antibody mixture wash 3x with T-TBS buffer, and then wash once with TBS buffer (without Tween).
- Dissemble microarray slides from chambers carefully to avoid touching pads, rinse gently with filtered ddH2O, and dry by placing in 50 mL tubes and centrifuging at 500 x g for 10 min.
-
Place probed microarray slides in a lightproof box and keep in a desiccator cabinet at room temperature for long-term storage.NOTE: The protocol may be paused for up to 1 week at this point.
Figure 3: Procedure for attaching probing chamber to microarray slide.

From A to F, the probing chamber is placed on top of slide in correct orientation, attached to the slide using horizontal clips on the sides, and placed in the probing tray.
Quantify antibody binding to antigens within microarray
- Visualize microarray slides and quantify spot fluorescence intensity to measure antibody binding
- Acquire images of microarray slides using the portable imager with built-in software.
- In the Configure Imager tab, select the proper slide configuration. For this study, use 16-pad slides.
- In the Image Control tab, select the proper fluorescent channel, and adjust the gain, exposure time, and acquisition time depending on the reactivity of the sera to obtain optimal images. For this study, the fluorescent channels for IgA and IgG were 585 nm and 800 nm respectively, and imaging settings were gain of 50, exposure time of 500 ms, and acquisition time of 1 s.
-
Click on Capture to start the process of acquiring the image.NOTE: Microarray slides can be re-imaged at multiple settings without degradation of signal as long as they are stored dark and dry. Other imaging systems can be used if compatible with the slides.
- Detect array spots using grids oriented based on the fiducial markers and measure spot intensity as median of pixel intensity minus background measured around spots. Perform this quantification algorithm in batch using the built-in imager software, which utilizes the .gal file constructed in step 1.2.2 to connect spot intensities to individual antigens on each microarray.
- In the File Info panel, upload the .gal file by selecting from its folder on the computer, and specify the folder where the analysis output files are to be saved in the Analysis Options section.
- In the Image Control tab, open one of the acquired images to be quantified and select the Auto button in the upper right corner.
- In the Array Analysis section in the lower right corner, create a fiducial template as instructed by the software.
-
Click on the Batch Analysis, select the folder that contains the images to be quantified, and select the fiducial template that was created in the previous step. The software analyzes each image and quantifies the spot intensity.NOTE: This step will generate a .csv file containing spot intensities quantifying antibodies within each serum specimen that bind to each individual antigen on the microarray that can subsequently be manipulated in spreadsheet manipulation or analysis software.
-
Analyze raw data to compare antibody binding across antigens and across serum specimens. For this study, IgA and IgG antibodies measured as 585 nm and 800 nm fluorescent spot intensities were compared across all antigens between 2 independent runs of the experiment using different slides on different days, and correlation analysis was performed to measure assay reproducibility.NOTE: For non-purified proteins printed as expression mixture, data analysis should begin with background subtraction of a no DNA control.
Representative Results
As a demonstration of the protocol, baseline sera were assayed from 16 individuals within a prospective cohort study of college students followed for influenza infection on the influenza antigen microarray. To demonstrate assay reproducibility, these specimens were probed twice, on different slides and different days.
For this study, purified influenza antigens containing His10 tags were obtained from commercial vendor (see Table of Materials) and collaborators. These antigens include 251 total HA antigens, with 63 globular head domains (HA1) and 186 full-length proteins (HA0), including 96 monomeric HA0 proteins and 90 trimerized HA0 proteins containing fused trimerization (“foldon”) domain23. A full list of antigens and controls used in this study is included as a Supplementary File. For this study, secondary antibodies used were goat anti-human IgG conjugated to quantum dot emitting at 800 nm (GAH-IgG-Q800) and goat anti-human IgA conjugated to quantum dot emitting at 585 nm (GAH-IgA-Q585) for multiplex detection of IgG and IgA antibodies as shown in Figure 3. IgG and IgA antibody binding to different subtypes and molecular forms of influenza HA antigens were compared for sera obtained from the clinical cohort described above.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| 16-pad nitrocellulose-coated glass slides | Grace Bio Labs | 305016 | |
| 1x GVS FAST blocking buffer | Fischer Scientific | 10485356 | |
| ArrayCam portable imager | Grace Bio Labs | 400S | Other imaging devices can be used to visualize slides if capable of achieving the resolution of the microarray spots and the excitation and emission wavelengths of the quantum dots. |
| Biotin-conjugated goat anti-mouse-IgG antibody | Thermo Fischer | 31800 | |
| HiBase 384-well plate | Greiner Bio-One | T-3037–11 | |
| Microarray pins | ArrayIt | GMP2 | Each different microarray printer may require its own custom microarray pins. |
| Mouse monoclonal poly-His antibody | Sigma-Aldrich | H1029 | |
| OmniGrid 100 microarray printer | GeneMachines | The version of the microarray printer used in this work is no longer commercially available, but the updated similar equipment is the OmniGrid Accent microarray printer from Digilab (Hopkinton, MA), and the same protocol can be carried out with most commercially available microarray printers. | |
| ProPlate slide chambers | Grace Bio Labs | 246890 | |
| ProPlate slide clips | Grace Bio Labs | 204838 | |
| ProPlate slide frames | Grace Bio Labs | 246879 | |
| Quantum dot 585 nm conjugated goat anti-human-IgA antibody | Grace Bio Labs | 110620 | |
| Quantum dot 585 nm streptavidin conjugate | Thermo Fischer | Q10111MP | |
| Quantum dot 800 nm conjugated goat anti-human-IgG antibody | Grace Bio Labs | 110610 |
The resulting heat map is shown in Figure 4 with graphical representation in Figure 5. In these figures, only the clinically relevant subtypes with high representation on the array are labeled to save space, with “+” denoting all remaining subtypes of higher number, and minor or less well represented subtypes included as ordered (e.g., H2 in between H1 and H3). A full list of strains and subtypes on the array is included as Supplementary File. These data demonstrate that antibodies to the head group of HA are subtype-specific with expected high quantity of antibodies to clinically prevalent strains (H1N1, H3N2, and B) and low quantity of antibodies to other strains. However, antibodies to the whole HA, which includes the stalk domain, are more cross-reactive across subtypes, and this effect appears to be augmented when the whole HA is trimerized. This result is not unexpected, as the whole HA includes the stem region, which is highly conserved across subtypes. Therefore, antibodies to whole HA molecules from non-clinical subtypes (e.g., H5 and H7) likely represent anti-stem antibodies originally elicited against clinical subtypes (e.g., H1 and H3) that are cross-reacting with the stem regions of the other HA subtypes on the array. This point illustrates the importance of including both head HA and whole molecule HA on the array to distinguish between antibodies to the head and the stem regions which show different reactivity profiles.
Figure 4: Representative results of influenza antigen microarray.

Heat maps represent antigen-specific antibody responses, with each row representing a single antigen arranged by molecule, subtype, and strain, and each column representing a probing run of a single specimen, arranged by antibody isotype and run (A, white = 0, black = 20000, red = 40000 fluorescence intensity). The antigen subtypes including all hemagglutinin subtypes from 1 to 18 and all neuraminidase subtypes from 1 to 10 are arranged vertically and labeled on the left. A comparison of the fluorescence intensity between two runs demonstrates good assay reproducibility by linear regression for IgA (B) and IgG(C).
Figure 5: Breadth of serum antibodies measured on influenza antigen microarray.
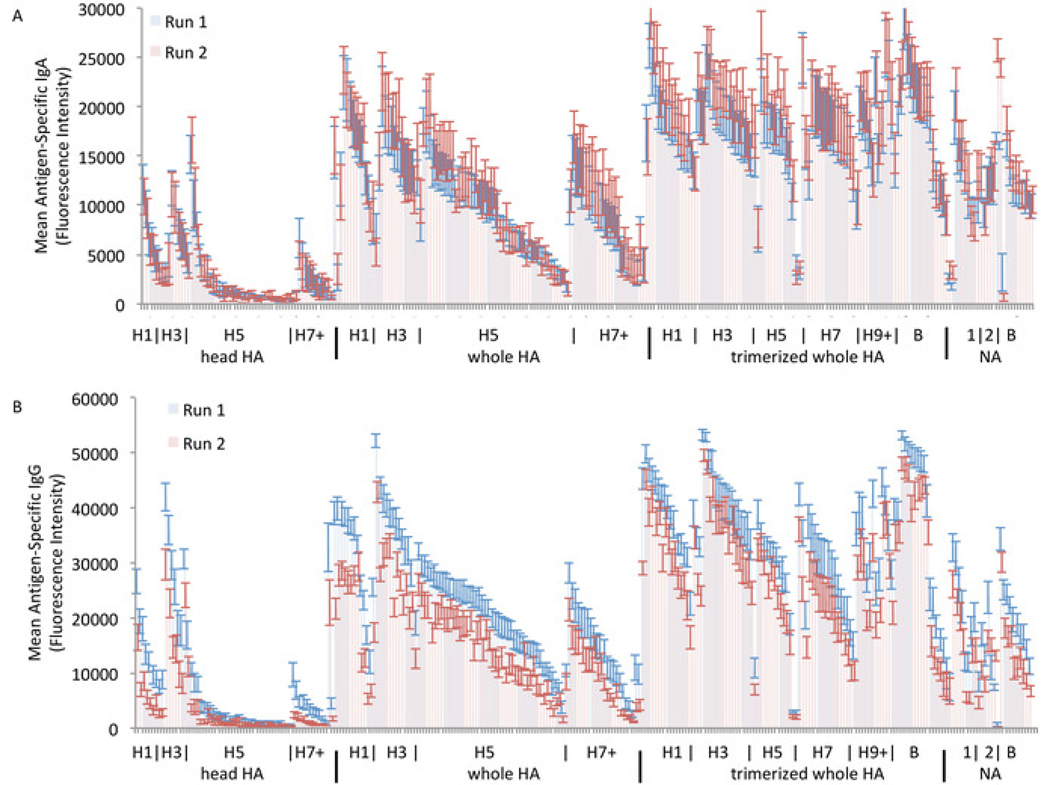
Serum IgA (A) and IgG (B) are grouped by HA and NA molecular forms and subtypes to demonstrate high specificity of HA head group antibodies for clinical subtypes and high cross-reactivity of whole HA and trimerized whole HA antibodies with inclusion of stalk region.
The assay demonstrates good reproducibility across probing runs. The second run does show slightly lower IgG antibodies across all strains, although the pattern between the strains is consistent. This across-the-board slight decrease is likely due to batch-to-batch variability in the secondary antibody, which was changed between runs for IgG but not for IgA. Thus, as noted in the protocol, it is recommended to use the same batch of each secondary antibody for any experiments between which quantitative comparison is planned. If different batches of antibody are necessary due to a high number of samples to be tested, we recommended including shared samples between experimental runs with different antibody batches to allow for quantitative comparison with correction.
Discussion
The influenza antigen microarray protocol described here is adaptable to any project that requires analyzing antibody responses to many antigens. The microarray platform can be used with any desired set of protein antigens expressed in any system that can achieve 0.1 mg/mL or higher yield with or without purification as previously described12. If non-purified protein antigens (e.g., expressed in in vitro transcription and translation system) are directly used to print microarrays, components of protein expression system (e.g., E. coli lysate) should be added 1:10 to blocking buffer used for dilution of sera and quality control antibodies in order to block any antibodies directed against these components. Slide configurations are available with a lower number of larger pads on each slide to accommodate a higher number of antigens per array. For this study, we used a GeneMachines OmniGrid 100 microarray printer that utilizes pins that directly contact the nitrocellulose surface to deposit spots on the array. While this microarray printer is no longer commercially available, other commercially available microarray printers can be used in this protocol but may require custom pins (either contact or non-contact) and software and should have sufficient spot resolution and compatibility with slides and imager. Depending on reactivity of sera, dilutions from 1:50 to 1:400 can be used. Serum-free buffer can be used as a negative control, while monoclonal antibodies known to bind to antigen can be used as a positive control.
Users should be aware of a few troubleshooting issues. The purpose of the quality control check using antibodies to the His tag is to check for any antigens for which printing was not successful. Any spots that consistently give low signal in quality control check of multiple arrays likely represent insufficient antigen printed. Possible reasons include aggregation or precipitation or antigen in source plate or poor contact between printing pin and microarray slide due to variable thickness of nitrocellulose pad. At this point, we do not perform assay normalization based on quantitative results of the quality control check, given that the detected binding of anti-His antibodies can be influenced by availability of this tag, which depends on the 3-dimensional conformation specific to each antigen.
The influenza antigen microarray provides several advantages over traditional methods such as ELISA and is complementary to functional assays such as HAI and MN. The 16-pad protein microarray is a sample-sparing technology with capacity to measure antibodies of multiple isotypes simultaneously against approximately 300 antigens from 1 μL of serum. The number of antigens can be increased to the thousands by decreasing the number of pads per slides. The multiplexed assay also spares personnel time and consumable resources, given that hundreds of sera can be probed for antibodies in 2 days, and all materials other than slides and reagents are re-usable so do not generate large amounts of plastic waste.
While microarray printers may not be widely distributed, microarray slides can be printed at a centralized location and then transported to the end user for probing. The only equipment required for probing is the low-cost and portable imager. The goal of disseminating this protocol is to make utilization of this technique more widespread.
The main limitations of the influenza antigen microarray are the inability to characterize the function and kinetics of the detected antibodies. The microarray is detecting a polyclonal set of binding antibodies for each antigen. These antibodies may or may not be functional in neutralizing virus in HAI and MN assays. However, HAI and MN assays require live virus culture with associated need for specialized facilities with high-level biosafety cabinets to test for antibodies against avian subtypes of influenza, whereas the protein microarray does not involve live virus components so can be utilized in any basic laboratory. With respect to binding kinetics, a single dilution of serum probed with an array containing a single concentration of each antigen yields a single data point, which represents a composite of quantity and affinity summated over all antibodies that bind to the antigen. To fully resolve the antigen-antibody binding kinetics, multiple antigen concentrations and/or serial dilutions of sera are required.
Despite these limitations, the influenza antigen microarray is a useful tool to characterize breadth of influenza antibodies across the antigenic landscape that can complement functional assays that are more limited in throughput and availability.
Supplementary Material
Content is shown for all 324 spots on the array, including blanks, fiducials, controls (human IgG and IgA and anti-human IgG and IgA at 0.1 mg/mL and 0.3 mg/mL), and antigens with information on source, molecular form, subtype, and strain. Abbreviations are as follows: for source, Sino = Sino Biological Inc., FKL = Florian Krammer Laboratory; for molecular form, HA1 = head HA, HA0 = whole HA, HA2 = stalk HA, NP = nucleoprotein. For antigens sourced from Sino Biological Inc., catalog numbers are shown; for antigens sourced from Krammer Laboratory, antigen IDs are listed.
Acknowledgments
The authors would like to acknowledge Prof. Don Milton (Institute of Applied Public Health, University of Maryland, College Park, MD, USA) for collecting the human sera under University of Maryland IRB protocol #313842 funded by DARPA N66001-18-2-4015 P00001. The authors would also like to acknowledge Prof. Florian Krammer (Icahn School of Medicine, Mt. Sinai, NY, USA) for providing trimerized HA and NA antigens funded by ODNI IARPA DJF-15-1200-K-0001725. S. Khan is partially supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through grant KL2 TR001416. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/59973/
Disclosures
The authors have no disclosures.
References
- 1.Murray CJ et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 380 (9859), 2197–2223 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Renegar KB, Crouse D, Floyd RA, Krueger J. Progression of influenza viral infection through the murine respiratory tract: the protective role of sleep deprivation. Sleep. 23 (7), 859–863 (2000). [PubMed] [Google Scholar]
- 3.Li L. et al. Heterogeneity in Estimates of the Impact of Influenza on Population Mortality: A Systematic Review. American Journal of Epidemiology. 187 (2), 378–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fauci AS Seasonal and pandemic influenza preparedness: science and countermeasures. Journal of Infectious Diseases. 194 Suppl 2, S73–76 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Duggal A, Pinto R, Rubenfeld G, Fowler RA Global Variability in Reported Mortality for Critical Illness during the 2009–10 Influenza A(H1N1) Pandemic: A Systematic Review and Meta-Regression to Guide Reporting of Outcomes during Disease Outbreaks. PLoS One. 11 (5), e0155044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demicheli V, Jefferson T, Ferroni E, Rivetti A, Di Pietrantonj C. Vaccines for preventing influenza in healthy adults. Cochrane Database Systematic Reviews. 2, CD001269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erbelding EJ et al. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. Journal of Infectious Diseases. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie H. et al. H3N2 Mismatch of 2014–15 Northern Hemisphere Influenza Vaccines and Head-to-head Comparison between Human and Ferret Antisera derived Antigenic Maps. Scientific Reports. 5, 15279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tricco AC et al. Comparing influenza vaccine efficacy against mismatched and matched strains: a systematic review and meta-analysis. BMC Medicine. 11, 153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katz JM, Webster RG Efficacy of inactivated influenza A virus (H3N2) vaccines grown in mammalian cells or embryonated eggs. Journal of Infectious Disease. 160 (2), 191–198 (1989). [DOI] [PubMed] [Google Scholar]
- 11.Zost SJ et al. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proceedings of the National Academy of Science U. S. A. 114 (47), 12578–12583 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies DH et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proceedings of the National Academy of Science U. S. A. 102 (3), 547–552 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang L, Felgner PL A systems biology approach for diagnostic and vaccine antigen discovery in tropical infectious diseases. Current Opinion in Infectious Diseases. 28 (5), 438–445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain A. et al. Evaluation of quantum dot immunofluorescence and a digital CMOS imaging system as an alternative to conventional organic fluorescence dyes and laser scanning for quantifying protein microarrays. Proteomics. 16 (8), 1271–1279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desbien AL et al. Development of a high density hemagglutinin protein microarray to determine the breadth of influenza antibody responses. Biotechniques. 54 (6), 345–348 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Mace CR et al. Label-free, arrayed sensing of immune response to influenza antigens. Talanta. 83 (3), 1000–1005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koopmans M. et al. Profiling of humoral immune responses to influenza viruses by using protein microarray. Clinical and Microbiology Infections. 18 (8), 797–807 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Bucukovski J, Latorre-Margalef N, Stallknecht DE, Miller BL A Multiplex Label-Free Approach to Avian Influenza Surveillance and Serology. PLoS One. 10 (8), e0134484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meade P, Latorre-Margalef N, Stallknecht DE, Krammer F. Development of an influenza virus protein microarray to measure the humoral response to influenza virus infection in mallards. Emerging Microbes and Infection. 6 (12), e110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vigil A, Davies DH, Felgner PL Defining the humoral immune response to infectious agents using high-density protein microarrays. Future Microbiology. 5 (2), 241–251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakajima R. et al. Protein Microarray Analysis of the Specificity and Cross-Reactivity of Influenza Virus Hemagglutinin-Specific Antibodies. mSphere. 3 (6) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Hoeven N. et al. A Formulated TLR7/8 Agonist is a Flexible, Highly Potent and Effective Adjuvant for Pandemic Influenza Vaccines. Scientific Reports. 7, 46426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krammer F. et al. A carboxy-terminal trimerization domain stabilizes conformational epitopes on the stalk domain of soluble recombinant hemagglutinin substrates. PLoS One. 7 (8), e43603 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Content is shown for all 324 spots on the array, including blanks, fiducials, controls (human IgG and IgA and anti-human IgG and IgA at 0.1 mg/mL and 0.3 mg/mL), and antigens with information on source, molecular form, subtype, and strain. Abbreviations are as follows: for source, Sino = Sino Biological Inc., FKL = Florian Krammer Laboratory; for molecular form, HA1 = head HA, HA0 = whole HA, HA2 = stalk HA, NP = nucleoprotein. For antigens sourced from Sino Biological Inc., catalog numbers are shown; for antigens sourced from Krammer Laboratory, antigen IDs are listed.