

Abstract
Synthesis and antimalarial activity of 94 novel bipyrrole tambjamines (TAs) and a library of B-ring functionalized tripyrrole prodiginines (PGs) against a panel of Plasmodium falciparum strains are described. The activity and structure-activity relationships demonstrate that the ring-C of PGs can be replaced by an alkylamine, providing for TAs with retained/enhanced potency. Furthermore, ring-B of PGs/TAs can be substituted with short alkyl substitutions either at 4-position (replacement of OMe) or 3- and 4-positions without impacting potency. Eight representative TAs and two PGs have been evaluated for antimalarial activity against multidrug-resistant P. yoelii in mice in the dose range of 5–100 mg/kg × 4 days by oral administration. The KAR425 TA offered greater efficacy that previously observed for any PG, providing 100% protection to malaria-infected mice until day 28 at doses of 25 and 50 mg/kg × 4 days, and was also curative in this model in a single oral dose (80 mg/kg). This study presents the first account of antimalarial activity in tambjamines.
Graphical Abstract
INTRODUCTION
Malaria is a global parasitic infectious disease caused by Plasmodium parasites, among which Plasmodium falciparum (Pf) is the most dangerous one, with the highest rates of complications and mortality. It has been estimated that there are 584,000 people died from this disease in 2013 and the burden is heaviest in the African Region, where an estimated 90% of all malaria deaths occur, and in children aged under 5 years, who account for 78% of all deaths.1 On the heels of the global spread of chloroquine-resistant P. falciparum (CQRPf), resistance has also quickly developed to a variety of quinoline analogues, to antifolates, to inhibitors of electron transport, and perhaps most ominously, now to artemisinin.2,3 Therefore, novel medicinal agents are urgently needed to overcome the emergence and spread of resistance.
Prodiginines (PGs, 1a–c), tambjamines (TAs, 2a–b), and modified prodiginines (streptorubin B (3a), metacycloprodiginine (3b) and marineosins (4 and 5)) belong to a family of pyrrolylpyrromethene (PPM) alkaloids (Figure 1) derived from bacterial and marine sources.4–7 These structurally distinctive natural products can be envisioned to arise via a bifurcated process from a common precursor, 4-methoxy-2,2′-bipyrrole-5-carboxaldehyde (MBC; 6, Figure 1) and the corresponding alkylpyrrole and/or alkylamine.7–10 The natural and synthetic PPM products are undergoing intense scrutiny in the medicinal chemistry because of both their wide range of biological activities and modes of action (antimicrobial,11–14 immunosuppressive,15–22 antitumor,11,12,23 anticancer,6,24–30 antimalarial7,31–38 transmembrane anion transport,28–30,39–45 and DNA intercalation46,47). Certain PGs and TAs have also been observed to bind duplex DNA and can cleave this biomolecule in the presence of Cu(II).4,48 Some of these compounds have shown clinical potential, and in particular, PG analogue, GX15–070 has completed phase II clinical trials for the treatment of small cell lung cancer and is engaged in multiple clinical trials for the treatment of other cancer conditions.49,50
Figure 1.

Structures of PPM natural products (1–5) and their common biosynthetic precursor (6)
As a part of an ongoing interest in developing new antiparasitic agents, we reisolated the natural PGs 1a, and 3a from Streptomyces coelicolor M511, and 3b from S. longisporusruber (Figure 1).51,52 These natural PGs exhibited great potency with very low IC50 values against P. falciparum strains, a potency only slightly more than chloroquine (CQ).37 The natural PG 3b provided an excellent in vivo efficacy against multidrug-resistant P. yoelii in mice by oral route, and it was curative in this model at 100 mg/kg/day, and three of four mice were cured. This data provided the first demonstration of oral effectiveness of PGs.37 Recently we also have isolated the modified prodiginines, marineosins (5) and their pathway intermediates 23-hydroxyundecylprodiginine (1b), 23-ketoundecylprodiginine (1c) and premarineosin (4) through heterologous expression of the entire mar gene cluster and/or gene replacement mutants in a heterologous host, S. venezuelae.7 Of these, the compound 4 antimalarial activity compares favorably with the most potent naturally occurring PGs and CQ.
The structural and functional diversity and promising antimalarial activity of these natural PGs and marineosins spurred us to synthesize various analogues of these lead molecules to obtain more active compounds. We recently reported the antimalarial activity of a large library of synthetic PGs.37,38 This work has shown that a terminal nonalkylated pyrrole (ring-A), and 3,5-dialkyl substitutions on the other terminal alkylated pyrrole (ring-C) of a natural tripyrrole PGs core structure are crucial for the potent antimalarial activity. A number of the synthetic PGs were effective at lower concentrations (IC50 = 0.9–16.0 nM) against P. falciparum strains and their potency was more than the natural PGs and CQ. However, preliminary in vitro assays indicate concerns associated with the toxicity of PGs.
Our work on the potent antimalarial activity of PGs,37,38 to date have been limited to SAR studies of A- and C-ring functionalized PGs. With a few exceptions,20,24,25,30,38 there have been no reports of a comprehensive series of TAs and B-ring functionalized PGs being prepared and evaluated for biological activities. In particular, the antimalarial activities of the TAs have not been reported to the best of our knowledge. These toxicity concerns for PGs and the intriguing biological activities of these PPM scaffolds have spurred us to expand the structural and functional diversity. Therefore, we have undertaken syntheses of novel TAs and B-ring functionalized PGs for enhanced antimalarial activity and reduced toxicity. To that end, we have developed new methods for the synthesis of various 2,2′-bipyrrole-5-carboxaldehydes,53 and utilized in the generation of the novel TAs and B-ring functionalized PGs. Here we report the synthesis, and structure–activity relationships (SARs) of TAs and B-ring functionalized PGs. The results show TAs with impressive in vitro potency and low toxicity, and demonstrate that a tripyrrole structure is not required for activity. Furthermore evidence of in vivo efficacy with TAs, including curative efficacy in mice after oral administration is reported.
RESULTS AND DISCUSSION
Chemistry.
The key precursors 6–43, which are involved in the synthesis of prodiginines (PGs) and tambjamines (TAs) (Scheme 10), are depicted in Figures 2 and 3. By use of literature methodologies, MBC (6) and analogue 21 were prepared from readily available 4-methoxy-3-pyrrolin-2-one in two steps54 and 2,2′-bipyrrole-5-carboxaldehydes 7, 8, and 10–18 were synthesized by our recent methods.53 The syntheses of various new pyrrole carboxaldehydes 9, 19, 20, and 22–39 are outlined in Schemes 1, 2, 3, 4, 5, 6, 7, 8, and 9.
Scheme-10.

Synthesis of novel PGs (85–98) and TAs (99–187)
Figure 2.

Key precursors (6–20) for the synthesis of B-ring functionalized PGs and TAs
Figure 3.

Key precursors (21–43) for the synthesis of A- and B-ring functionalized PGs and TAs
Scheme 1.

Synthesis of 4-(4-chlorophenyl)-[2,2′-bipyrrole]-5-carboxaldehyde (9)
Scheme 2.

Synthesis of 2,2′-bipyrrole-5-carboxaldehyde (19)
Scheme 3.

Synthesis of 3-(pyrrol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (20)
Scheme-4.

Synthesis of isomeric [2,3′-bipyrrole]-5′-carboxaldehydes (22–25)
Scheme-5.

Synthesis of MBC′s analogues containing heteroaryl/aryl groups in the place of ring-A (26–31)
Scheme-6.

Synthesis of 3,4-dimethyl-[2,2′-bipyrrole]-5-carboxaldehydes where the ring-A contains C-alkyl groups (32–35)
Scheme 7.

Synthesis of 4′-ethyl-3,4-dimethyl-[2,2′-bipyrrole]-5-carboxaldehyde (36)
Scheme 8.

Synthesis of 3-(imidazol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (37)
Scheme 9.

Synthesis of 3-methyl-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (38) and 5,5’-methylenebis(4-ethyl-3-methyl-pyrrole-2-carboxaldehyde) (39)
Synthesis of 4-(4-chlorophenyl)-[2,2′-bipyrrole]-5-carboxaldehyde (9).
Synthesis of the aryl substituted 3-pyrrolin-2-one 48, a key synthon in the synthesis of bipyrrole-carboxaldehyde 9, was began with the coupling of Boc-glycine (44) with 2,2-dimethyl-1,3-dioxane-4,6-dione (meldrum′s acid) to afford the acylated meldrum′s acid, which was further converted into the desired intermediate 45, by an intramolecular cyclization and a subsequent decarboxylation (Scheme 1).55 The compound 45 was treated with p-toluenesulfonyl chloride in the presence of N,N-diisopropylethylamine (DIPEA) to give the tosylated product 46, in 89% yield, which was further subjected to Suzuki-coupling reaction with 4-chlorophenylboronic acid to give the N-Boc-4-aryl-3-pyrrolin-2-one 47. The desired 4-aryl-3-pyrrolin-2-one 48 was obtained in excellent yield by deprotection of the N-Boc group of 47 with trifluoroacetic acid.56 Using the reported Vilsmeier formylation method,54 48 was then smoothly transformed to 5-bromo-3-(4-chlorophenyl)-pyrrole-2-carboxaldehyde 49, which when further subjected to Suzuki coupling with N-Boc-2-pyrroleboronic acid followed by deprotection of the N-Boc group gave the desired 2,2′-bipyrrole-5-carboxaldehyde 9, in 59% yield (Scheme 1).53
Synthesis of 2,2′-bipyrrole-5-carboxaldehyde (19).
In 1988, Borger and Patel synthesized the 2,2′-bipyrrole-5-carboxaldehyde (19) in seven steps.12 In this work, we successfully accomplished 19 in two one-pot sequences from easily available pyrrole (50), as shown in Scheme 2. To that end, compound 50 was consecutively treated with N-chlorosuccinimide (NCS) and Vilsmeier reagent (POCl3/DMF, in situ generation) under controlled temperatures to obtain the 5-chloro-pyrrole-2-carboxaldehyde (51) in good yield.57 The Suzuki cross-coupling of 51 with N-Boc-2-pyrroleboronic acid followed by deprotection of the N-Boc group, provided the desired bipyrrole-carboxaldehyde 19 in 45% isolated yield (Scheme 2).
Synthesis of 3-(pyrrol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (20).
The key intermediate 53 was prepared via BartoneZard’s method, using 1-nitro-1-cyclohexene (52) as a starting material (Scheme 3).53,58,59 Upon treating with NaOH in ethylene glycol under reflux, 53 was smoothly converted to 4,5,6,7-tetrahydro-isoindole (54) in 90% yield by successive hydrolysis and decarboxylation of the ester group.53 Using the standard Vilsmeier formylation method, 54 was then transformed to 4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (55), which when further treated with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH)53 in THF at −78 °C to room temperature provided the 3-bromo-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (56). Subsequently, Suzuki cross-coupling reaction between 56 and N-Boc-2-pyrroleboronic acid and further deprotection of the N-Boc group led to the desired bipyrrole-carboxaldehyde 20 in good yield (Scheme 3).
Synthesis of isomeric [2,3′-bipyrrole]-5′-carboxaldehydes (22–25).
To investigate the ring-A positional effect on antimalarial activity, the isomeric bipyrrole-carboxaldehydes 22–25 were prepared, as shown in Scheme 4. Pyrrole-2-carboxaldehyde (40) and 3,5-dimethyl-pyrrole-2-carboxaldehyde (42) were obtained from commercial sources, and the 3-methyl-pyrrole-2-carboxaldehyde (57) and 3-ethyl-pyrrole-2-carboxaldehyde (58) were prepared according to our reported procedures.53 These pyrrole-2-carboxaldehydes were then converted into the corresponding 4-bromo-pyrrole-2-carboxaldehydes 59–62, via a regioselective bromination at 4-position using DBDMH in THF in good yields (Scheme 4).53 These 4-bromo-pyrrole-2-carboxaldehydes 59–62, were further subjected to Suzuki-coupling reaction with N-Boc-2-pyrroleboronic acid, and a subsequent treatment with LiOH in THF/MeOH (1:1) at 60 °C, resulted in the desired isomeric bipyrrole-caraboxaldehydes 22–25 (Scheme 4).
Synthesis of MBC′s analogues (26–31) containing herteroaryl/aryl groups in the place of ring-A.
To probe the exact role of the 2-pyrrolyl moiety (ring-A) on activity, we have prepared various key carboxaldehyde precursors 26–31, in which the ring-A is completely replaced by various heterocycles and/or aryl moieties and the ring-B is substituted with short alkyl groups (Scheme 5). The 5-bromo-3,4-dimethyl-pyrrole-2-carboxaldehyde (65) was prepared in 6 steps according to the literature methods from acetaldehyde (63) and nitroethane (64),53 and it was subsequently subjected to Suzuki-coupling reaction with various boronic acids and further deprotection of the Boc/TIPS group led to the corresponding carboxaldehydes 26–31 (Scheme 5).
Synthesis of 3,4-dimethyl-[2,2′-bipyrrole]-5-carboxaldehydes where the ring-A contains C-alkyl groups (32–36).
To investigate the effect of the ring-A alkyl substituents pattern on potency, we have prepared various alkylated bipyrrole-carboxaldehyde precursors 32–36, as shown in Schemes 6 and 7. The 2-acetyl-pyrrole (66a), 2,4-dimethylpyrrole (67c), and 3-ethyl-2,4-dimethylpyrrole (67d) were obtained from commercial sources, and the 2-isobutyryl-pyrrole (66b) was prepared according to the literature methods.60 The compounds 66a and 66b were then converted into the corresponding 2-alkyl-pyrroles 67a and 67b, respectively, using LiAlH4 in THF under reflux (Scheme 6).61 By using standard procedures, the N-Boc-protected pyrroles 68a–68d were prepared in excellent yields from 67a–67d using di-tert-butyl dicarbonate (Boc2O) in the presence of 4-(dimethyl amino)pyridine (DMAP), and subsequently these were converted into the corresponding 5-alkyl-(1-tert-butoxycarbonylpyrrol-2-yl)boronic acids 69a–69d.62 The resultant boronic acids 69a–69d were carried forward into the Suzuki-coupling reaction with 65 without further purification to afford their corresponding [2,2′-bipyrrole]-5-carboxaldehydes 32–35 in good yields (Scheme 6).
We have also developed a simple and convenient method for the synthesis of N-Boc-4-ethyl-2-pyrrolboronic acid (71) via a regioselective boronylation of N-Boc-3-ethyl-pyrrole (70),53 using n-BuLi/2,2,6,6-tetramethylpiperidine, and trimethyl borate (Scheme 7, Experimental Section). Further investigations to expand the substrate scope of the regioselective boronylation as well as mechanistic studies are underway in our laboratory. Finally the 4′-ethyl-3,4-dimethyl-[2,2′-bipyrrole]-5-carboxaldehyde (36) was prepared in good yield via Suzuki-coupling of 65 with boronic acid 71, followed by the deprotection of N-Boc group with LiOH (Scheme 7). The final compound 36 was fully characterized by extensive 2D NMR analysis (see Supplementary Information).
Synthesis of 3-(imidazol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (37).
To investigate the role of ring-A with an extra nitrogen atom on potency, we have replaced the ring-A by imidazole moiety, as in 37 (Scheme 8). The N-Boc-pyrrole 72 was prepared in 95% yield from compound 55 using Boc2O/DMAP, and subsequently the aldehyde group was protected by trimethyl orthoformate under acidic conditions to obtain the desired intermediate 73. The compound 73 was further reacted with triisopropyl borate/LDA in THF, and followed by aqueous solution of KHSO4/NH4Cl at room temperature to provide the desired boronic acid 74 in excellent yield.53 Finally, the Suzuki cross-coupling reaction between 74 and 2-bromo-imidazole (75), and subsequent deprotection of the N-Boc group led to the desired carboxaldehyde 37 in 65% isolated yield (Scheme 8).
Synthesis of 3-methyl-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (38) and 5,5′-methylenebis(4-ethyl-3-methyl-pyrrole-2-carboxaldehyde) (39).
We wanted to study the analogues of TAs without ring-A, therefore, two representative pyrrole aldehydes 38 and 39 (Scheme 9) were synthesized. Initially, 1-methyl-4,5,6,7-tetrahydro-isoindole (77) was synthesized from ethyl-4,5,6,7-tetrahydro-isoindole-1-carboxylate (53) via an unstable intermediate 76, using LiAlH4 in THF at 0 °C to room temperature in 85% isolated yield. The resultant alkyl-pyrrole 77 was further converted to 3-methyl-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (38) by Vilsmeier reagent (POCl3/DMF) (Scheme 9). Conversely, the bis(3-ethyl-4-methyl-pyrrol-2-yl)methane (79) was prepared from diethyl-5,5′-methylenebis(4-ethyl-3-methyl-2-pyrrolecarboxylate) (78) in excellent yields via a successive hydrolysis and a decarboxylation of the ester groups. Further Vilsmeier formylation of 79 provided the desired dicarboxaldehyde 39 in 73% isolated yield (Scheme 9).
Synthesis of novel PGs (85–98) and TAs (99–187).
By using our standardized procedures, the mono- and dialkyl/alkylaryl pyrroles 80–84 were synthesized (Figure 4).37 The acid-catalyzed condensation of either the alkyl pyrroles 80–84 or the commercially available alkyl/arylamines with various bipyrrole-carboxaldehydes and analogues 6–43, provided the desired PGs 85, 86, 88–98, and TAs 99–187, respectively, in good to excellent isolated yields (Scheme 10). The PG 85 was further treated with MeI/NaH in DMF to provide the N,N-dimethyl PG 87 in 85% isolated yield (Scheme 10).
Figure 4.

Potential substrates (80–84) for the synthesis of PGs
Synthesis of TA like Analogues (190, 191 and 194–196).
Distinct syntheses were designed and executed to obtain a different class of TA like analogues 190, 191 and 194–196, in which the crucial ring-B of TAs is completely replaced by an alkylamide/amine linkage (Scheme 11). To that end, compound 188 was synthesized via a standard condensation method (EDCl/DMAP) from 44 and 1-adamantylamine in 85% yield. Removal of the Boc group of 188 by trifluoroacetic acid:water (1:1) provided the intermediate 189 in good yield,63 which was further utilized in a condensation reaction with pyrrole-2-carboxylic acid to furnish the desired product 190. Treatment of 190 with LiAlH4 in THF at 0 °C to reflux conditions gave the 191 in 82% isolated yield (Scheme 11). Conversely, analogues 194–196, were also synthesized, as shown in Scheme 11. The pyrrole-2-carboxaldehyde (40) was subjected to Horner-Wadsworth-Emmons (HWE) reaction with methyl diethylphosphonoacetate in the presence of NaH to obtain the methyl-3-(pyrrol-2-yl)acrylate (192),64,65 which when hydrolyzed under basic (LiOH.H2O) conditions, furnished the 2-pyrrolyl acrylic acid 193. Condensation of 193 with 189 in the presence of EDCl/DMAP led to the corresponding condensed product 194, which was further treated with NaBH4/NiCl2.6H2O to give the saturated product 195. Treatment of 195 with LiAlH4 in THF at 0 °C to reflux conditions provided the desired product 196 in 78% yields (Scheme 11).
Scheme 11.

Synthesis of novel analogues (190, 191 and 194–196)
Biological Activity.
In this work, the structure-activity relationships (SARs) focused on various substitutions and positions of the ring-A, and -B and the nature of the alkylamines of TAs, and ring-B of PGs. Specifically, the modifications to the ring-B of TAs and PGs were designed in order to understand the structural requirements, as well as the necessity of the ring-B for the potent antimalarial activity. We have synthesized various series of novel TAs and B-ring functionalized PGs, and evaluated for antimalarial activity against the chloroquine-sensitive (CQS) D6, and the chloroquine-resistant (CQR) Dd2 and 7G8 strains of Pf with chloroquine (CQ) as a reference drug.66,67 In parallel, the cytotoxicity of the most potent antimalarial PGs and TAs (IC50 < 250 nM) was tested against hepatocellular HepG2 cancer cell line using mefloquine (MQ) as a control drug (see Tables 1, 2, 3, 4, 5, and 6).68,69
Table 1.
In Vitro Antimalarial Activity and Cytotoxicity of PGs (85–98)
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
||||||||||
| compd | R1 | R2 | R3 | R4 | D6 | Dd2 | 7G8 | cytotoxicity (IC50 in nM)a HepG2 | SIb (D6) | cLogPc |
|
| ||||||||||
| 85 | H | OMe | H | - | 6.1 | 4.8 | 5.5 | > 250000 | > 40983 | 4.8 |
| 86 | Me | OMe | H | - | 2250 | > 2500 | > 2500 | ntd | - | 5.1 |
| 87 | Me | OMe | Me | - | > 2500 | > 2500 | > 2500 | nt | - | 5.3 |
| 88 | H | 4-ClC6H4 | H | - | > 2500 | > 2500 | > 2500 | nt | - | 7.7 |
| 89 | n-C11H23 | H | H | H | > 2500 | > 2500 | > 2500 | nt | - | 5.2 |
| 90 | n-C11H23 | H | Et | H | 101 | 66 | 51 | 18939 | 187 | 5.7 |
| 91 | n-C11H23 | H | H | i-Pr | 1586 | 1500 | > 2500 | nt | - | 6.3 |
| 92 | n-C11H23 | H | H | t-Bu | > 2500 | > 2500 | > 2500 | nt | - | 6.7 |
| 93 | n-C11H23 | H | Et | Cl | > 2500 | > 2500 | > 2500 | nt | - | 5.8 |
| 94 | n-C11H23 | H | Me | Et | 162 | 190 | 145 | 62000 | 383 | 6.1 |
| 95 | n-C8H17 | H | Me | Et | 127 | 216 | 132 | 71000 | 559 | 4.8 |
| 96 | H | n-C8H17 | Me | Et | 41 | 53 | 61 | 57200 | 1395 | 4.9 |
| 97 | n-C7H15 | 4-FC6H4CH2 | Me | Et | 6.5 | 7.0 | 5.9 | 82024 | 12619 | 6.7 |
| 98 | 4-ClC6H4CH2 | 4-ClC6H4CH2 | Me | Et | 28 | 42 | 42 | 30600 | 1093 | 6.7 |
| 1a | 7.2 | 7.5 | 7.0 | nt | - | 4.2 | ||||
| CQ | 13 | 115 | 130 | nt | - | 3.7 | ||||
| MQ | nt | nt | nt | 21800 | - | 5.3 | ||||
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
SI (selectivity index) = IC50 (cytotoxicity)/IC50 (D6)
cLogP values were calculated using ChemBioDraw Ultra software (version 14),
nt = not tested
Table 2.
In Vitro Antimalarial Activity and Cytotoxicity of 4-Substituted B-Ring Functionalized TAs (99–129)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
||||||||
| compd | R1 | R2 | D6 | Dd2 | 7G8 | cytotoxicity (IC50 in nM)a HepG2 | SIb (D6) | cLogPc |
|
| ||||||||
| 99 | OMe | n-C4H9 | 210 | 159 | 74.6 | 23000 | 109 | 0.08 |
| 100 | OMe | n-C6H13 | 34 | 37 | 25 | 26700 | 785 | 0.9 |
| 101 | OMe | n-C8H17 | 345 | 177 | 69 | ntd | - | 1.7 |
| 102 | OMe | n-C11H23 | 55 | 53 | 23 | 9800 | 178 | 3.0 |
| 103 | OMe |
|
2400 | 2500 | 946 | nt | - | − 0.9 |
| 104 | OMe |
|
591 | 497 | 156 | nt | - | − 0.4 |
| 105 | OMe |
|
68 | 84 | 45 | 30500 | 448 | − 0.03 |
| 106 | OMe |
|
49 | 71 | 30 | 15000 | 306 | 0.4 |
| 107 | OMe |
|
23 | 34 | 15 | 10100 | 439 | 0.8 |
| 108 | OMe |
|
4.8 | 7.1 | 7.5 | 9700 | 2021 | 1.2 |
| 109 | OMe |
|
3.1 | 2.6 | 3.8 | 3300 | 1064 | 0.7 |
| 110 | OMe |
|
> 2500 | > 2500 | > 2500 | nt | - | − 0.05 |
| 111 | OMe |
|
127 | 244 | 207 | > 250000 | > 1968 | 0.5 |
| 112 | OMe | 4-ClC6H4 | 255 | 368 | 314 | nt | - | 1.1 |
| 113 | 4-ClC6H4 | n-C6H13 | 1129 | > 2500 | 564 | nt | - | 3.8 |
| 114 | 4-ClC6H4 | n-C11H23 | 664 | > 2500 | 663 | nt | - | 5.9 |
| 115 | 4-ClC6H4 |
|
1218 | > 2500 | 510 | nt | - | 2.9 |
| 116 | 4-ClC6H4 |
|
1025 | > 2500 | 415 | nt | - | 3.3 |
| 117 | 4-ClC6H4 |
|
963 | 1250 | 348 | nt | - | 3.7 |
| 118 | 4-ClC6H4 |
|
832 | 1135 | 316 | nt | - | 4.1 |
| 119 | 4-ClC6H4 |
|
> 250 | > 250 | 126 | nt | - | 3.6 |
| 120 | Me | n-C11H23 | 1167 | 1469 | 515 | nt | - | 4.2 |
| 121 | Me |
|
> 250 | > 250 | > 250 | nt | - | 2.0 |
| 122 | Me |
|
> 250 | > 250 | > 250 | nt | - | 2.4 |
| 123 | Me |
|
1.3 | 15 | 4.3 | 6900 | 5308 | 1.8 |
| 124 | Et |
|
> 250 | > 250 | > 250 | nt | - | 2.4 |
| 125 | Et |
|
> 250 | > 250 | > 250 | nt | - | 2.8 |
| 126 | Et |
|
2.5 | 16 | 7.7 | 6100 | 2440 | 2.2 |
| 127 | H |
|
> 2500 | > 2500 | > 2500 | nt | - | 1.8 |
| 128 | H |
|
> 2500 | > 2500 | > 2500 | nt | - | 2.2 |
| 129 | H |
|
341 | 295 | 235 | 70000 | 205 | 1.6 |
| CQ | 13 | 115 | 130 | nt | - | 3.7 | ||
| MQ | nt | nt | nt | 21800 | - | 5.3 | ||
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
SI (selectivity index) = IC50 (cytotoxicity)/IC50 (D6)
cLogP values were calculated using ChemBioDraw Ultra software (version 14),
nt = not tested
Table-3.
In Vitro Antimalarial Activity and Cytotoxicity of 3-Substituted B-Ring Functionalized TAs (130–141)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
||||||||
| compd | R1 | R2 | D6 | Dd2 | 7G8 | cytotoxicity (IC50 in nM)a HepG2 | SIb (D6) | cLogPc |
|
| ||||||||
| 130 | Me |
|
2107 | > 2500 | 2147 | ntd | - | 2.1 |
| 131 | Me |
|
1376 | > 250 | 1778 | nt | - | 2.5 |
| 132 | Me |
|
106 | 170 | 95 | 30000 | 283 | 2.0 |
| 133 | Et |
|
1305 | 523 | 1456 | nt | - | 2.5 |
| 134 | Et |
|
1276 | > 250 | 1326 | nt | - | 3.0 |
| 135 | Et |
|
117 | 45 | 90 | 15200 | 130 | 2.4 |
| 136 | i-Pr |
|
> 2500 | 1968 | 1980 | nt | - | 2.9 |
| 137 | i-Pr |
|
1079 | 665 | 1480 | nt | - | 3.3 |
| 138 | i-Pr |
|
26 | 20 | 31 | 18500 | 711 | 2.7 |
| 139 | t-Bu |
|
> 2500 | > 2500 | > 2500 | nt | - | 3.3 |
| 140 | t-Bu |
|
> 2500 | > 2500 | > 2500 | nt | - | 3.8 |
| 141 | t-Bu |
|
> 2500 | > 2500 | > 2500 | nt | - | 3.2 |
| CQ | 13 | 115 | 130 | nt | - | 3.7 | ||
| MQ | nt | nt | nt | 21800 | - | 5.3 | ||
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
SI (selectivity index) = IC50 (cytotoxicity)/IC50 (D6)
cLogP values were calculated using ChemBioDraw Ultra software (version 14),
nt = not tested
Table-4.
In Vitro Antimalarial Activity and Cytotoxicity of 3,4-Disubstituted B-Ring Functionalized TAs (142–165)
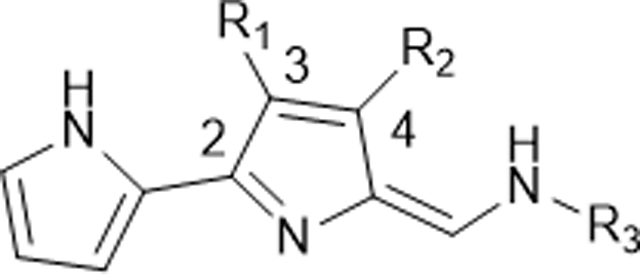
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
|||||||||
| compd | R1 | R2 | R3 | D6 | Dd2 | 7G8 | cytotoxicity (IC50 in nM)a HepG2 | SIb (D6) | cLogPc |
|
| |||||||||
| 142 | Et | Me | n-C4H9 | 883 | 680 | 260 | ntd | - | 2.0 |
| 143 | Et | Me | n-C8H17 | 1166 | 633 | 244 | nt | - | 3.7 |
| 144 | Et | Me |
|
> 2500 | 2047 | 2500 | nt | - | 1.1 |
| 145 | Et | Me |
|
62 | 55 | 60 | 19200 | 310 | 2.7 |
| 146 | Et | Me |
|
56 | 60 | 75 | 18900 | 337 | 3.1 |
| 147 | Et | Me |
|
5.5 | 4.3 | 3.6 | 3300 | 600 | 2.6 |
| 148 | Et | Me |
|
> 2500 | 1576 | 855 | nt | - | 2.4 |
| 149 | Et | Me |
|
> 2500 | > 2500 | > 2500 | nt | - | 0.3 |
| 150 | Me | Et |
|
150 | 200 | 117 | 15800 | 105 | 2.7 |
| 151 | Me | Et |
|
111 | 201 | 128 | 23900 | 215 | 3.1 |
| 152 | Me | Et |
|
19 | 14 | 14 | 4500 | 237 | 2.6 |
| 153 | Me | Me |
|
60 | 38 | 47 | 21300 | 355 | 2.3 |
| 154 | Me | Me |
|
56 | 31 | 45 | 18100 | 323 | 2.7 |
| 155 | Me | Me |
|
2.4 | 1.7 | 1.5 | 6400 | 2667 | 2.2 |
| 156 | Et | Et |
|
54 | 30 | 88 | 16900 | 313 | 3.1 |
| 157 | Et | Et |
|
39 | 26 | 58 | 13000 | 333 | 3.6 |
| 158 | Et | Et |
|
1.6 | 1.0 | 2.5 | 3900 | 2437 | 3.0 |
| 159 | –(CH2-CH2)2– |
|
35 | 39 | 23 | 6200 | 177 | 2.6 | |
| 160 | –(CH2-CH2)2– |
|
32 | 37 | 22 | 4600 | 144 | 3.1 | |
| 161 | –(CH2-CH2)2– |
|
6.1 | 7.5 | 2.8 | 2700 | 442 | 2.5 | |
| 162 | Cl | Et | t-Bu | 1217 | > 2500 | > 2500 | nt | - | 1.7 |
| 163 | Cl | Et |
|
> 2500 | > 2500 | > 2500 | nt | - | 2.4 |
| 164 | Cl | Et |
|
> 2500 | > 2500 | > 2500 | nt | - | 2.8 |
| 165 | Cl | Et |
|
2300 | > 2500 | 2250 | nt | - | 2.3 |
| CQ | 13 | 115 | 130 | nt | - | 3.7 | |||
| MQ | nt | nt | nt | 21800 | - | 5.3 | |||
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
SI (selectivity index) = IC50 (cytotoxicity)/IC50 (D6)
cLogP values were calculated using ChemBioDraw Ultra software (version 14),
nt = not tested
Table 5.
In Vitro Antimalarial Activity and Cytotoxicity of A- and B-Ring Functionalized TAs (166–187)
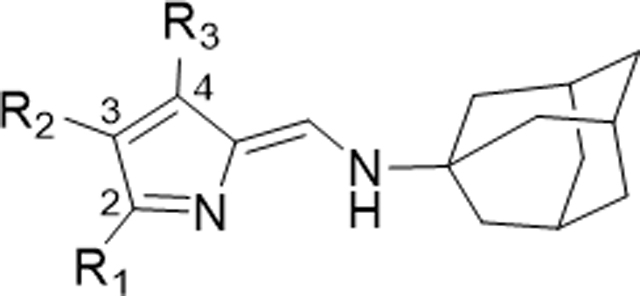
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
|||||||||
| compd | R1 | R2 | R3 | D6 | Dd2 | 7G8 | cytotoxicity (IC50 in nM)a HepG2 | SIb (D6) | cLogPc |
|
| |||||||||
| 166 | H |
|
H | > 2500 | > 2500 | > 2500 | ntd | - | 1.3 |
| 167 | H |
|
Me | > 2500 | > 2500 | > 2500 | nt | - | 1.4 |
| 168 | H |
|
Et | > 2500 | 1233 | > 2500 | nt | - | 1.9 |
| 169 | Me |
|
Me | 1418 | 1736 | 2005 | nt | - | 1.6 |
| 170 |
|
Me | Me | 250 | 328 | 215 | nt | - | 2.1 |
| 171 |
|
Me | Me | 647 | 1716 | 415 | nt | - | 2.2 |
| 172 |
|
Me | Me | 415 | 273 | 2282 | nt | - | 3.6 |
| 173 |
|
Me | Me | 1141 | 831 | >2500 | nt | - | 3.6 |
| 174 |
|
Me | Me | 318 | 388 | 161 | nt | - | 3.2 |
| 175 |
|
–(CH2-CH2)2– | 1335 | 1103 | 946 | nt | - | 1.9 | |
| 176 |
|
Me | Me | > 2500 | > 2500 | > 2500 | nt | - | 2.4 |
| 177 |
|
Me | Me | 2.1 | 2.3 | 0.5 | 3600 | 1714 | 3.0 |
| 178 |
|
Me | Me | < 2.5 | < 2.5 | < 2.5 | 1235 | > 494 | 3.7 |
| 179 |
|
Me | Me | 4.8 | 4.0 | 2.8 | 3825 | 797 | 3.1 |
| 180 |
|
Me | Me | 27 | 75 | 12 | 17920 | 664 | 3.0 |
| 181 |
|
Me | Me | 58 | 92 | 48 | 21323 | 368 | 3.9 |
| 182 | H | H | H | > 2500 | > 2500 | > 2500 | nt | - | 1.2 |
| 183 | Me | H | H | 2100 | 1682 | > 2500 | nt | - | 1.3 |
| 184 | Me | H | Me | 315 | 268 | 399 | nt | - | 1.5 |
| 185 | Me | Et | Me | 33 | 80 | 33 | 29900 | 906 | 2.2 |
| 186 | Me | –(CH2-CH2)2– | 61 | 64 | 60 | 5430 | 89 | 2.2 | |
| 187 |
|
Et | Me | > 2500 | > 2500 | > 2500 | nt | - | 4.5 |
| CQ | 13 | 115 | 130 | nt | - | 3.7 | |||
| MQ | nt | nt | nt | 21000 | - | 5.3 | |||
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
SI (selectivity index) = IC50 (cytotoxicity)/IC50 (D6)
cLogP values were calculated using ChemBioDraw Ultra software (version 14),
nt = not tested
Table 6.
In Vitro Antimalarial Activity of TA like Analogues (190, 191 and 194−196)
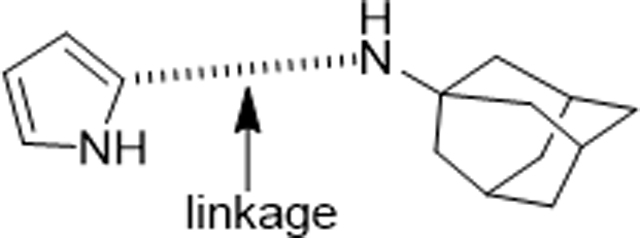
| |||||
|---|---|---|---|---|---|
| antimalarial activity (IC50 in nM)a |
|||||
| compd | linkage | D6 | Dd2 | 7G8 | cLogPb |
|
| |||||
| 190 |
|
> 2500 | > 2500 | > 2500 | 0.4 |
| 191 |
|
> 2500 | > 2500 | > 2500 | 1.6 |
| 194 |
|
> 2500 | > 2500 | > 2500 | 0.7 |
| 195 |
|
> 2500 | > 2500 | > 2500 | 0.6 |
| 196 |
|
> 2500 | > 2500 | > 2500 | 2.1 |
| CQ | - | 13 | 115 | 130 | 3.7 |
IC50 values are the average of at least three determinations, each carried out in triplicate (± 10%). In order to compare results run on different days, and with different batches of each stain; CQ was run as a positive control. All results obtained were ‘normalized’ to the CQ values of 13 nM for D6, 115 nM for Dd2 and 130 nM for 7G8.
cLogP values were calculated using ChemBioDraw Ultra software (version 14)
In Vitro Antimalarial Activity of PGs (85–98).
In our previous work, synthetic PG 85 had shown an excellent potency against Pf strains D6 (CQS) and Dd2 (CQR) with great IC50 values (Table 1), and had the most favorable profile: 92% parasite reduction at 5 mg/kg/day, 100% reduction at 25 mg/kg/day in a P. yoelii murine patent infection without any evident weight loss or clinical overt toxicity.37 To explore the N-alkyl effect on potency, initially we synthesized two N-methylated analogues 86 and 87 of the 85 (Table 1). These compounds 86 and 87 led to a large decrease in the antimalarial activity (IC50 > 2250 nM) against three Pf strains D6, Dd2 and 7G8, demonstrating that both pyrrole NH groups (ring-A and -C) of the PGs are required for potent antimalarial activity and that support our previous findings.38 To investigate the importance of the methoxy group (OMe) on ring-B, two analogues 88 and 89, in which the OMe group is replaced by 4-chlorophenyl moiety and hydrogen (complete removal of OMe), respectively, were prepared and examined for in vitro antimalarial activity. A dramatic loss of potency was observed for both compounds 88 and 89, which have an IC50 of > 2500 nM against all tested Pf strains (Table 1). Interestingly, while replacing the OMe group by ethyl unit as in 90 also led to the reduced potency (90: IC50 =101 nM versus 1a: IC50 = 7.2 nM against D6), the reduction was modest (14-fold). This result demonstrated that a short aliphatic substitution at 4-position on the ring-B could replace the OMe group and retain activity. Together, these results highlighted the importance of the OMe or short alkyl group on the ring-B of PGs for potent antimalarial activity, support our previous findings.38
We next investigated whether substitutions at 2 and 3 positions of the ring B are tolerated. A series of novel B-ring functionalized PGs 91–98, in which the ring-B is substituted with either mono- and/or di-substituents at 3- and 4-positions, were generated and examined for their in vitro antimalarial activity (Table 1). A significant loss of potency (IC50 >1500 nM) was observed for 91 and 92, containing an isopropyl, and tert-butyl groups, respectively, at 3-position on the ring-B. The adverse effect of the substitutions at 3-position on the ring-B was further confirmed by the introduction of the chloro (Cl) substitution at 3-position of 90, as with the analogue 93, which had an IC50 of > 2500 nM against all strains (90: IC50 =101 nM versus 93: IC50 > 2500 nM), suggesting that the rigid bulky substitutions or chlorine moiety (EWG) at 3-position are not preferred (Table 1). To further investigate the impact of the short alkyl substituents at both the 3- and 4-positions on ring-B, a set of mixed analogues 94–98, which contain the 3-ethyl/4-methyl groups on the ring-B, was examined. Analogues 94 and 95, which have mono-alkyl groups at 5-position of the ring-C, showed a roughly 20-fold drop in activity as compared to undecylprodiginine (1a) (Table 1). Conversely, the analogue 96 containing a monoalkyl group at 3-position on the ring-C, showed higher potency (3-fold) than 95 against all tested Pf strains, while it had 9-fold lesser potency than the corresponding OMe group containing analogue (IC50 = 4.6 nM against D637). Interestingly, the analogue 97, which has 3-alkyl and 5-alkylaryl substituents on the ring-C, showed equipotent to the 85. While the analogue 98, which has 3,5-dialkylaryl substituents on ring-C, showed ~5-fold lower potency when compared to the corresponding OMe group containing analogue 85 (Table 1), again these results are consistent and support the findings that the 3,5-disubstitutions on ring-C are very important for potent activity.37 In summary, these SAR analyses of the ring-B functionalized PGs demonstrate that the short alkyl substitutions are well tolerated at 3/4-positions on the ring-B.
In Vitro Antimalarial Activity of 4-Substituted B-Ring Functionalized TAs (99–129).
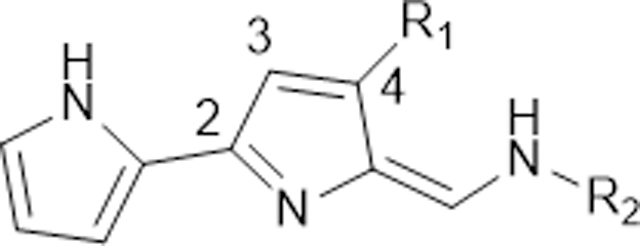
Having determined the substituents impact on the antimalarial activity of the PGs (through this and previous work37,38), we subsequently tested a hypothesis that the complete replacement of the right-hand side alkylated pyrrole (ring-C) of PGs by alkylamines, providing the TAs, might represent an opportunity to make potent and selective antimalarials with the desired “druglike” properties. Specifically, lower molecular weight (MWT) and lipophilic properties (LogP) are the two key characteristics that determine adsorption, distribution, metabolism, excretion and toxicity (ADMET) liabilities, with some ADMET parameters depending more on MWT and some on LogP.70 Subsequent TA analogues 99–129 (Table 2), which have lower MWT (< 400) and cLogP (< 4.2, except 114), were generated to obtain a SAR for the alkylamines in the place of ring-C and substituents at 4-position on the ring-B.
Initially, a series of new TAs 99–113, which have various alkyl/arylamines in the place of ring-C and the OMe group at the 4-position on the ring-B (as in natural products), were synthesized and evaluated for their in vitro antimalarial activity against Pf strains and the results are shown in Table 2. TAs 99–102 containing the n-alkylamines in the place of ring-C, exhibited good activity against all Pf strains, specifically, analogues 100 and 102 showed the highest potencies (IC50 < 50 nM) (Table 2). To probe the effect of cycloalkylamines in the place of ring-C/n-alkylamines on activity, we synthesized another set of TAs 103–109 (Table 2). Of these cycloalkylated TAs, analogues 108 and 109, which have the cyclooctylamine and 1-adamantylamine moieties, respectively, were the most potent antimalarial candidates (108: IC50 < 7.1 nM, and 109: IC50 < 3.8 nM against all tested Pf strains, see Table 2) with good selectivity and these results are more comparable to the potent PG 85 (IC50 < 4.5 nM), and the natural PG 1a (IC50 < 7.0 nM). These results, clearly demonstrated that the elongation of the cycloalkyl ring size (from cyclopropyl, 103: IC50 = 2500 nM to 1-adamantyl, 109: IC50 < 3.1 nM) lead to an increase in activity (Table 2 and Figure 5). The greatest loss of potency (IC50 > 2500 nM) was observed in 110, in which ring-C is replaced by piperidine moiety, suggesting that the free NH is required for the potent antimalarial activity. Replacement of cyclohexyl moiety with benzylpiperidine as with 111 led to slightly reduced potency (106: IC50 = 49 nM versus 111: IC50 = 127 nM against D6). The analogue 112, which contain a 4-chloroaniline in the place of ring-C showed the moderate activity (Table 2). These results unequivocal demonstrate that the ring-C of PGs can be replaced by alkylamines, providing the novel TAs with retained and/or enhanced antimalarial and cytotoxic properties.
Figure 5.

SAR of TAs (103–109) containing various cycloalkyl groups and in vitro antimalarial activity against Pf strains D6, Dd2, and 7G8
To investigate the importance of the OMe group on ring-B of TAs, another set of TAs 113–119, in which the OMe group is replaced by 4-chlorophenyl moiety, was generated and examined for their in vitro antimalarial activity (Table 2). In vitro analysis of the activity of these compounds 113–119 against Pf, demonstrated activity (IC50 > 250 nM) significantly diminished when compared to the corresponding OMe group containing TAs (100, 102, and 105–109). This work suggested that the bulky aromatic substitution at 4-position on the ring-B had an adverse effect on antimalarial activity. Interestingly the replacement of the OMe group with short alkyl substituents (methyl/ethyl) also reduced the potency of the compounds 120–122, 124 and 125 (IC50 > 250 nM) (Table 2). Conversely, the adamantly analogues 123 and 126, in which the OMe group is replaced by methyl and ethyl groups on the ring-B, respectively, showed a substantially higher potency against D6 strain (109: IC50 = 3.1 nM, versus 123: IC50 = 1.3 nM, 126: IC50 = 2.5 nM) with great selectivity. Complete removal of the OMe group on ring-B as with the analogues 127–129, resulted in the total loss of activity (127, 128: IC50 > 2500 nM vs 107: IC50 = 23 nM, 108: IC50 = 4.8 nM, and 129: IC50 = 341 nM vs 109: IC50 = 3.1 nM, 123: IC50 = 1.3 nM, 126: IC50 = 2.5 nM against D6). Together, these results again demonstrate that the substituents at 4-position on the ring-B have an important role in potent antimalarial activity, and the OMe group can be replaced by short alkyl substituents (methyl/ethyl), when 1-adamantylamine exists in the place of ring-C.
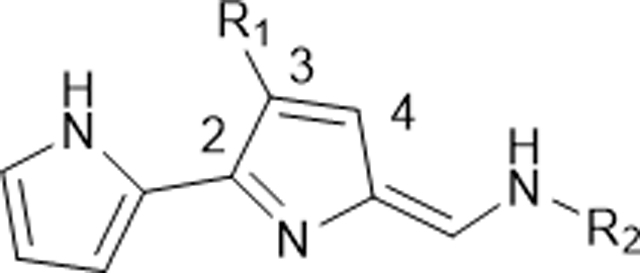
In Vitro Antimalarial Activity of 3-Substituted B-Ring Functionalized TAs (130–141).
Having established the substitution pattern at 4-position on the ring-B and the terminal alkylamines (cycloheptyl-, cyclooctyl-, and 1-adamantylamines) as optimal, we then examined the effects of substitution pattern at 3-position, where the 4-position is vacant on the ring-B of the TAs (Table 3). To that end, we generated a series of novel TAs 130–141, in which the 3-position on the ring-B is occupied with alkyl groups and screened for their antimalarial activity against Pf strains (Table 3). The greatest loss of potency was observed when the short alkyl (methyl/ethyl) groups moving from 4-position (121–126, Table 2) to the 3-position (130–141, Table 3). Moreover, the adamantly analogues 132 and 135, showed a significant decline in activity (132: IC50 = 106 nM vs 123: IC50 = 1.3 nM; and 135: IC50 = 117 nM, vs 126: IC50 = 2.5 nM against D6), and the analogue 141, had an almost total loss of activity (IC50 > 2500 nM). The one exception is the adamantly analogue 138, containing an isopropyl group at 3-position on the ring-B, which showed the better potency (IC50 < 30 nM) against all tested Pf strains with good selectivity. These results show that generally alkyl substitutions at 3-position versus the 4-position, adversely affects the potency irrespective of the terminal alkylamines.
In Vitro Antimalarial Activity of 3,4-Disubstituted B-Ring Functionalized TAs (142–165).
Exploration of the SARs around the ring-B of TAs indicated that the substitutions at 4-position were greatly favored compared to the 3-position (Tables 2 and 3). This finding is exemplified by the poor activity of the 3-substituted analogues (130–141) with the exception of 138. We next investigated whether substitutions at both the 3- and 4-positions are tolerated. We synthesized a series of 3,4-disubstituted B-ring functionalized TAs 142–149, which have 3-ethyl, and 4-methyl groups on the ring-B (Table 4). Of these 3,-4-disubstituted TAs, analogues 142–144, 148, and 149 with an n-alkyl, cyclopropyl, benzylpiperidine and morpholine moieties, respectively, showed the diminished activity (Table 4). Conversely, the analogues 145 and 146, which have cycloheptyl and cyclooctyl moieties, respectively, showed the highest potencies (Table 4) than those of the corresponding 3- and 4-monoalkyl substituted analogues (see Tables 2 and 3). Significantly, the adamantly analogue 147, showed comparable potency to that of the corresponding 4-alkyl/methoxy substituted analogues (147: IC50 = 5.5 nM versus 109: IC50 = 3.1 nM, 123: IC50 = 1.3 nM, 126: IC50 = 2.5 nM against D6), and this potency is 5–20-fold greater than the corresponding 3-alkyl substituted analogues (147: IC50 = 5.5 nM versus 132: IC50 = 106 nM, 135: IC50 = 117 nM, 138: IC50 = 26 nM against D6). Interchange of the methyl and ethyl groups between 3- and 4-positions on the ring-B as in 150–152 resulted in a ~2-fold decrease in potency (IC50 of 150–152 vs IC50 of 145–147). We were encouraged that the short alkyl substitutions at both the 3- and 4-positions on the ring-B were well tolerated with comparable and/or enhanced activities. This allowed for a variety of different analogues to be synthesized with representative examples (153–165, Table 4). The analogues 153, 154, 156, 157, 159, and 160, which contain the same alkyl groups (methyl/ethyl/-(CH2-CH2)2-) at both 3- and 4-positions on the ring-B, and cycloheptyl/cyclooctylamines in the place of ring-C, were shown comparable and/or greater potency to the dissimilar alkyl groups at both 3- and 4-positions containing TAs. Significantly, the adamantly analogues 155 (IC50 < 2.4 nM), 158 (IC50 < 2.5 nM), and 161 (IC50 < 7.5 nM) showed enhanced (2–8-fold) or comparable potency against all tested Pf strains when compared to 147 (IC50 < 5.5 nM) and 152 (IC50 < 19 nM). The biggest potency loss occurred (IC50 > 2250 nM) when we introduced a chlorine atom at 3-position on the ring-B as in 162–165 (IC50 of 156–158 vs 163–165, Table 4), and it is consistent with the observation that the chlorine atom (EWG) has an adverse effect at 3-position on the ring-B of PGs. Collectively, from the monoalkylated (Tables 2 and 3) and 3,4-dialkylated TAs (Table 4) the data clearly showed that the 3,4-disubstituted TAs containing cycloheptyl/cyclooctyl groups have significantly improved potency than the corresponding monoalkylated TAs (Tables 2 and 3), and these potencies were comparable to the corresponding OMe group containing analogues (Table 2). Notably, all the adamantyl analogues, which have short (alkyl/methoxy) groups at 4-position (Table 2) and dialkyl groups at 3/4-positions (Table 4) on ring-B, showed the greatest activity with good selectivity.
In Vitro Antimalarial Activity of A- and B-Ring Functionalized TAs (166–187).
After establishing the substitutions pattern at 3- and 4-positions on the ring-B of TAs, we investigated the importance of positioning of the ring-A at 2-position on the ring-B of TAs (Table 5), by keeping the 1-adamantlyamine as an active pharmacophore for all analogues. The TAs 166–169, in which the ring-A (2-pyrrolyl moiety) is shifted from 2- to 3-position on the ring-B and are isomeric to 129, 123, 126, and 155 (Tables 2 and 4), respectively, were synthesized and tested against Pf strains (Table 5). It is noteworthy that the potency was significantly declined against all tested Pf strains after shifting the ring-A from 2- to 3-position (166–168: IC50 > 2500 nM vs 123: IC50 = 1.3 nM, 126: IC50 = 2.5 nM, 129: IC50 = 341 nM, and 169: IC50 = 1418 nM vs 155: IC50 < 2.5 nM, against D6, Tables 2, 4 and 5). The importance of the location of nitrogen within ring-A was analyzed by moving from the 2′-position to the 3′-position (Figure 1, and Table 5), where compound 170 showed a roughly 100-fold drop in activity (170: IC50 = 250 nM vs 155: IC50 < 2.5 nM, against D6, Tables 4 and 5). We also looked at the alternatives to the ring-A at 2 position of the ring-B. Replacement of the ring-A (2-pyrrolyl) by various 2-heteroaryl/phenyl moieties (compounds, 171–175) resulted in a decrease in antimalarial activity (IC50 of 171–175 vs 155 and 161). Notably, our previous SAR investigations revealed that the ring-A (2-pyrrolyl moiety) of PGs provides optimal activity,37,38 and the current results also suggest the importance of the ring-A of TAs for the potent activity. Alkylation (methylation) on the NH group of the ring-A as in 176, resulted in a large decrease in potency (176: IC50 > 2500 nM vs 155: IC50 < 2.5 nM), suggest that the pyrrole NH (ring-A) of the TAs is important for potent antimalarial activity. Conversely, the analogues 177–181, which contain C-alkyl moieties on the ring-A, retained the potency against all tested Pf strains, suggesting that the alkyl groups are well tolerated on the ring-A.
To further investigate the exact role of the ring-A of TAs on potency, a set of mixed alkylated analogues 182–186, in which the ring-A is completely removed from the core moiety of TAs, were examined. Complete removal of the substitutions on the ring-B, dramatically reduced the potency of the compound 182 (IC50 > 2500 nM). Incorporation of the substitutions into the ring-B as in 183–186 (from mono- to tri-alkyl) resulted in a large increase in potency (Table 5), whereas the dimer 187 of the 185 showed the poorest activity. It is noteworthy that the analogues 185 and 186, which contain a monopyrrole with trialkyl substituents and an enamine moiety, showed the comparable potency to that of the corresponding bipyrrole TAs. These results demonstrated that the ring-A is not essential for the antimalarial activity, but both the trialkylated monopyrrole and enamine moiety are important. In summary, structure pruning of PGs has shown that in vitro potency can be retained and/or enhanced when moving from a tripyrrole (PGs) to bipyrrole (TAs) and even to a monopyrrole as shown in Figure 6.
Figure 6.

Structure pruning approach of the lead PG compounds (98)
In Vitro Antimalarial Activity of TA like Analogues (190, 191 and 194–196), in which the ring-B is replaced by an alkylamide/amine linkage.
Our detailed SAR explorations around the ring-A and –B and nature of alkylamines of TAs led to a robust understanding of the structural features that are required for potent antimalarial activity. We also sought to explore whether any linkage (total replacement of ring-B) between two of the most active pharmacophores (i.e. 2-pyrrolyl, and 1-adamantyl moieties) is tolerated. A set of novel TA like analogues 190, 191 and 194–196, in which ring-B is completely replaced by an alkylamide/amine linkage, were generated and screened for their antimalarial activity against Pf strains (Table 6). None of these analogues showed activity (IC50 > 2500 nM, Table 6). This data confirmed that the ring-B between ring-A and alkylamine plays an important role in the antimalarial activity of TAs and PGs as well.
In Vivo Efficacy Studies in Mice Models.
Given the attractive antiplasmodial activity of several PGs and TAs against CQS-D6, CQR-Dd2, and 7G8 strains of P. falciparum along with favourable toxicological properties against hepatocellular HepG2 cancer cell line and lower MWT and lipophilic properties, an in vivo proof of concept study in a murine P. yoelii model was undertaken with the most potent and selective analogues 98, 100, 105, 108, 109, 123, 145, 177, and 185, using side by side comparison with our previous lead PG 8537 and CQ as a reference drug (Table 7). In vivo efficacy was determined in a murine P. yoelii model,71,72 in which animals were randomly placed in groups of four and administered test drugs range of 5 mg/kg to 100 mg/kg by oral gavage on four sequential days following the day of inoculation. The in vivo data are expressed as ED50 values and reflect the dose (estimated from dose–response curves) for suppression of parasitemia by 50% relative to vehicle-only controls as assessed on day 5 of each study. In these experiments, the animals with parasitemia either on day 5 or latter were euthanized and the parasitemia free animals were kept in observation until day 28. Drug treated animals that were parasitemia free on day 28 of the experiment are defined as “cures”, and the amount of drug that was needed to achieve a cure is referred to as the “nonrecrudescence dose” (NRD).
Table 7.
In Vivo Antimalarial Efficacy of PGs and TAs in a Murine P. yoelii
| dose (mg/kg × 4 days) | % suppression of parasitaemia on day 5b | ||||
|---|---|---|---|---|---|
| compd | compd code names | structure | ED50 (mg/kg/day) | ||
|
| |||||
| control | - | - | PEG-400 | - | - |
|
| |||||
| 85 a | KAR71 |
|
5 | 90 | 2.8 |
| 25 | 100 | ||||
| 50 | 100 | ||||
| 100 | 100 | ||||
|
| |||||
| 98 | KAR276 |
|
5 | 66 | < 5 |
| 25 | 100 | ||||
| 50 | 100 | ||||
|
| |||||
| 100 | KAR458 |
|
5 | 90 | < 5 |
| 25 | 93 | ||||
| 50 | 96 | ||||
| 100 | 100 | ||||
|
| |||||
| 105 | KAR383 |
|
5 | 92 | < 5 |
| 25 | 94 | ||||
| 50 | 96 | ||||
| 100 | 100 | ||||
|
| |||||
| 108 | KAR457 |
|
5 | 93 | < 5 |
| 25 | 94 | ||||
| 50 | 99 | ||||
| 100 | 100 | ||||
|
| |||||
| 109 | KAR422 |
|
5 | 0 | 84 |
| 25 | 27 | ||||
| 50 | 38 | ||||
| 100 | 97 | ||||
|
| |||||
| 123 | KAR790 |
|
5 | 30 | 20 |
| 25 | 77 | ||||
| 50 | 100 | ||||
|
| |||||
| 145 | KAR425 |
|
5 | 100 | < 2.5 |
| 25 | 100c | ||||
| 50 | 100c | ||||
| 100 | 100 | ||||
|
| |||||
| 177 | KAR767 |
|
5 | 7 | 5.5 |
| 25 | 100 | ||||
| 50 | 100 | ||||
| 100 | 100 | ||||
|
| |||||
| 185 | KAR765 |
|
5 | 24 | 45 |
| 25 | 17 | ||||
| 50 | 67 | ||||
| 100 | 100 | ||||
|
| |||||
| CQ | - | - | 1 | 65 | 2.2 |
| 4 | 94 | ||||
| 16 | 100 | ||||
| 64 | 100 | ||||
previous lead compound,37
% suppression of parasitemia = 100 × parasitemia control group−parasitemia treated group/parasitemia control group,
provided cures (100% protection to malaria-infected mice)
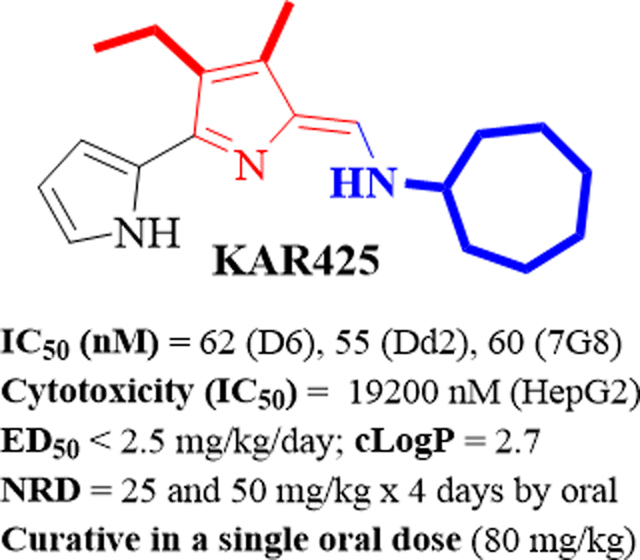
Following four once-daily doses of PGs 85 and 98 at 5 mg/kg, each reduced parasitemia by a 90% and 66% on day 5, respectively, and parasitemia free animals were observed at 25, and 50–100 mg/kg however, none of these animals were cured, while the CQ was also not curative in this model even at doses as high as 64 mg/kg/day (Table 7). The TA analogues 100, 105, and 108, each reduced parasitemia > 90% after 5, 25 and 50 mg/kg × 4 days dosing, and at the higher dose (100 mg/kg × 4 days) these reduced parasitemia 100% on day 5. Intriguingly, the TA 109 with good in vitro potency, showed much less efficacy with an ED50 value of 84 mg/kg/day, which may relate to low aqueous solubility and/or poor oral bioavailability (Table 7). Interestingly the analogue 123, in which the methyl group of ring-B is replaced the OMe group of 109, showed improved efficacy at all doses, specifically 100% reduction was observed at 50 mg/kg × 4 days on day 5. Of these TAs, the analogue 145 with 3-ethyl/4-methyl substitution pattern on the ring-B and the cycloheptylamine in the place of ring-C, provided an excellent in vivo efficacy against P. yoelii in mice with an ED50 value of < 2.5 mg/kg/day, and it cleared all parasitemia on day 5 after dosing 5 mg/kg to 100 mg/kg × 4 days. Indeed, the compound 145 provided parasite-free cures on day 28 (100% protection to malaria-infected mice) at 25 and 50 mg/kg/day, without evident weight loss and toxicity. In separate experiments, a single oral dose (80 mg/kg) of KAR425 (145) was also used. The preliminary experiments demonstrated that the KAR425 is also curative in this model and two of four mice were cured with no obvious signs of toxicity or behavior change and further higher dose studies are underway in our laboratories. The analogues 177 and 185 showed 100% parasitemia reduction on day 5 after 25–100 mg/kg and 100 mg/kg dosing, respectively, however these were not curative in this model.
CONCLUSIONS
We report here the synthesis and antimalarial activity of the novel class of potent tambjamines (TAs) and B-ring functionalized prodiginines (PGs). The compounds were synthesized via simple and inexpensive chemical procedures using easily available building blocks to respond to the demand for low-cost novel antimalarial agents. When compared to tripyrrole PGs,37,38 these bipyrrole TAs exhibited marked improvements with regard to the color properties, in vitro potency, selectivity, and in vivo efficacy. Several key findings emerged from these studies: i) the alkylated pyrrole (ring-C) can be replaced by an alkyl/cycloalkylamine, providing for TAs with retained and/or enhanced antimalarial activity, ii) the OMe group at the 4-position on the ring-B, between ring-A and ring-C/alkylamine of PGs/TAs, can be replaced with short alkyl substitutions either at 4-position or 3- and 4-positions without impacting in vitro potency, iii) the 2-pyrrolyl moiety (ring-A) must be linked at 2-position on the ring-B for potency, and it can be substituted with alkyl groups (see Figure 7). In addition, these analogues are equally effective against P. falciparum pansensitive D6 and MDR Dd2 and 7G8 strains. Some of these analogues have shown very promising in vivo efficacy in mice, specifically, the KAR425 (145) TA offered greater efficacy that previously observed for any tripyrrole PG, providing 100% protection to malaria-infected mice until day 28 at doses of 25 and 50 mg/kg × 4 days and was also curative in this model in a single oral dose (80 mg/kg). In our overall study, the KAR425 stands out as an excellent lead compound, with low molecular weight (< 300), good lipophilic profile (cLogP < 2.7), oral efficacy, and no obvious signs of toxicity or behavior change. Detailed lead optimization, pharmacology, safety, and modes of action studies of the KAR425 will be studied in our laboratories in due course to produce the antimalarial candidates for full preclinical studies.
Figure 7.

Summary of SAR analysis of PGs and TAs
EXPERIMENTAL SECTION
General.
NMR spectra were recorded on Bruker AMX-400, and AMX-600, spectrometers at 400, 600 MHz (1H), and 100, 150 MHz (13C). Experiments were recorded in CDCl3, CD3OD, acetone-d6 and DMSO-d6 at 25 °C. Chemical shifts are given in parts per million (ppm) downfield from internal standard Me4Si (TMS). HRMS (ESI) were recorded on a high-resolution (30000) thermo LTQ-Orbitrap Discovery hybrid mass spectrometer (San Jose, CA). Unless otherwise stated, all reagents and solvents were purchased from commercial suppliers and used without further purification. Reactions which required the use of anhydrous, inert atmosphere techniques were carried out under an atmosphere of argon/nitrogen. Chromatography was executed on CombiFlash® Rf 200 instrument, using silica gel (230‒400 mesh) and/or neutral alumina as the stationary phase and mixtures of ethyl acetate and hexane as eluents. Analytical HPLC analyses were performed on a Supelco Discovery HS C18 column (4.6 × 250 mm) with a linear elution gradient ranging from CH3OH/CH3CN/H2O (40%/10%/50%) to CH3OH (100%) in 0.15% trifluoroacetic acid at a flow rate of 1 mL/min. A purity of ≥ 95% has been established for all tested compounds.
Synthesis of 4-Hydroxy-2-oxo-2,5-dihydro-pyrrole-1-carboxylic acid tert-butyl ester (45).
To a stirred solution of N-(tert-butoxycarbonyl)-glycine (44; 5.0 g, 28.57 mmol) in 90 mL of anhydrous CH2Cl2 (DCM) were added meldrum′s acid (4.93 g, 34.28 mmol), and 4-dimethylaminopyridine (DMAP; 8.71 g, 71.42 mmol) under an argon atmosphere at 0 °C. A solution of isopropyl chloroformate (42.85 mL, 42.85 mmol, 1 N in toluene) was added dropwise, and the reaction mixture was stirred for 4 h at 0 °C. The reaction mixture was diluted with DCM (100 mL), washed with 15% KHSO4 (2 × 70 mL), and organic layer was dried over Na2SO4, and the solvent was evaporated under reduced pressure to give the acylated meldrum′s acid. This material was then refluxed in ethyl acetate (600 mL) for 1 h and the solvent was evaporated under reduced pressure and the product was recrystallized from ethyl acetate to give the desired product 45 (3.46 g, 61%) as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 12.13 (br s, 1H), 4.88 (s, 1H), 4.14 (s, 2H), 1.44 (s, 9H); HRMS (ESI) calcd for C9H13NaNO4 (M + Na)+ 222.0737, found 222.0740.
Synthessis of 2-Oxo-4-(toluene-4-sulfonyloxy)-2,5-dihydropyrrole-1-carboxylic acid tert-butyl ester (46).
To a stirred solution of 45 (3.4 g, 17.08 mmol) in anhydrous CH2Cl2 (150 mL) were added p-toluenesulfonyl chloride (3.24 g, 17.08 mmol), and DIPEA (4.4 g, 34.17 mmol). The resulting reaction mixture was stirred for 6 h at 25 °C. Then the reaction mixture was washed with 5% HCl (2 × 25 mL), brine and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the 46 (5.37 g, 89%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.86 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 5.75 (s, 1H), 4.22 (d, J = 1.2 Hz, 2H), 2.50 (s, 3H), 1.52 (s, 9H); HRMS (ESI) calcd for C16H19NaNO6S (M + Na)+ 376.0825, found 376.0830.
Synthesis of 4-(4-Chloro-phenyl)-2-oxo-2,5-dihydro-pyrrole-1-carboxylic acid tert-butyl ester (47).
To a degassed stirred solution of 46 (4.0 g, 11.33 mmol) and 4-chlorophenylboronic acid (2.65 g, 17.0 mmol) in 100 mL of THF at room temperature were added Pd(dppf)Cl2 (410 mg, 0.56 mmol) and a solution of cesium carbonate (11.05 g, 34.0 mmol) in water (15 mL). The reaction mixture was stirred at 25 °C for 1 h and then heated to reflux for 16 h. The reaction mixture was filtered through Celite and washed with ethyl acetate (400 mL). The organic layer was washed with saturated sodium bicarbonate (2 × 75 mL), and brine and dried over anhydrous Na2SO4. Then the organic solution was concentrated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 47 (1.82 g, 55%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 7.50 (d, J = 8.7 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 6.42 (t, J = 1.5 Hz, 1H), 4.68 (d, J = 1.5 Hz, 2H), 1.61 (s, 9H); HRMS (ESI) calcd for C15H16NaClNO3 (M + Na)+ 316.0711, found 316.0713.
Synthesis of 4-(4-Chloro-phenyl)-1,5-dihydro-pyrrol-2-one (48).
To a stirred solution of 47 (1.8 g, 6.14 mmol) in anhydrous CH2Cl2 (25 mL) was added dropwise TFA (2.8 g, 24.57 mmol). The reaction mixture was stirred for an additional hour at 25 °C. The solvent was evaporated under reduced pressure and the crude material was then dissolved in ethyl acetate (200 mL). The organic layer was washed with 5% NaHCO3, and brine and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure and the solid material was washed with CH2Cl2, to afford the pure product 48 (1.14 g, 94%) as a white solid. 1H NMR (DMSO-d6, 400 MHz) δ 8.15 (br s, 1H), 7.61 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 6.50 (t, J = 1.5 Hz, 1H), 4.30 (s, 2H); HRMS (ESI) calcd for C10H9ClNO (M + H)+ 194.0367, found 194.0372.
Synthesis of 5-Bromo-3-(4-chloro-phenyl)-pyrrole-2-carboxaldehyde (49).
To a stirred solution of diethylformamide (DEF; 1.57 g, 15.54 mmol) in anhydrous chloroform (10 mL) at 0 °C was added dropwise a solution of phosphorus oxybromide (POBr3; 3.62 g, 12.95 mmol) in chloroform (10 mL). The resulting thick suspension was stirred at 0 °C for 30 min to obtain the Vilsmeier complex as a solid. After the sample was dried in vacuo for 20 min, chloroform (50 mL) was added to the solid and the reaction mixture was cooled to 0 °C. The compound 48 (1.0 g, 5.18 mmol) was added portionwise, and the reaction mixture was warmed to room temperature and then heated at 70 °C for 16 h. The reaction mixture was poured onto ice‒water (75 mL) and the pH of the aqueous solution was adjusted to pH 9‒10 by treatment with 5 N NaOH. Dichloromethane (100 mL) was added to the resulting precipitate and the mixture was filtered through Celite. The two layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 100 mL). The organic layers were combined, washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the product was passed through a silica gel, with ethyl acetate/hexanes as eluent, to afford the pure 49 (806 mg, 55%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 10.05 (br s, 1H), 9.49 (s, 1H), 7.49‒7.40 (m, 4H), 6.42 (d, J = 2.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 178.2, 137.1, 134.5, 131.5, 130.9, 130.3, 129.9, 129.1, 128.9, 113.6, 113.1; HRMS (ESI) calcd for C11H8BrClNO (M + H)+ 283.9472, found 283.9484.
Representative Procedure for the Synthesis of 4-(4-Chloro-phenyl)-[2,2’]bipyrrolyl-5-carboxaldehyde (9).
To a degassed stirred solution of 49 (1.0 g, 3.53 mmol), and N-Boc-2-pyrroleboronic acid (1.11 g, 5.30 mmol) in 10% water/dioxane (50 mL) were added Pd(PPh3)4 (204 mg, 0.17 mmol) and Na2CO3 (749 mg, 7.06 mmol). The reaction mixture was stirred for 3 h at 100 °C and poured onto water (100 mL). The pH of the solution was lowered to pH 7 with 2 N HCl and extracted with ethyl acetate (3 × 75 mL). The combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the residue was dissolved in methanol (25 mL) and evaporated the solvent to remove the volatile B(OMe)3. This was then dissolved in THF (10 mL) and LiOH (850 mg, 35.33 mmol) in methanol (10 mL) was added dropwise under an argon atmosphere at room temperature. The resulting reaction mixture was stirred at room temperature for 30 min. On completion of the reaction, the solvent was removed under reduced pressure. The resulting solid was picked up with ethyl acetate (200 mL), washed with water and brine and dried over anhydrous Na2SO4. The organic solvent was removed under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure 9 (562 mg, 59%). 1H NMR (DMSO-d6, 400 MHz) δ 12.09 (br s, 1H), 11.31 (br s, 1H), 9.46 (s, 1H), 7.58 (d, J = 8.7 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 6.93 (m, 1H), 6.81 (m, 1H), 6.72 (d, J = 2.5 Hz, 1H), 6.14 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 177.1, 135.6, 133.6, 132.6, 132.3, 130.5 (2C), 128.7 (2C), 127.5, 123.0, 120.2, 109.3, 108.1, 106.4; HRMS (ESI) calcd for C15H12ClN2O (M + H)+ 271.0633, found 271.0639.
Synthesis of 5-Chloro-pyrrole-2-carboxaldehyde (51).
To a stirred solution of pyrrole (50; 5.0 g, 74.62 mmol) in 200 mL of dry THF was added N-chlorosuccinimide (NCS; 9.92 g, 74.62 mmol) under an argon atmosphere at −78 °C. The reaction mixture was stirred for an additional 4 h at the same temperature and placed at −20 °C for overnight. To the reaction mixture was added dropwise Vilsmeier reagent (149.25 mmol, in-situ generation from POCl3/DMF, 0 °C, 1 h) in 100 mL of DCM at −20 °C. The reaction mixture was stirred for 10 h while it was allowed to warm to room temperature. The solvent was removed under reduced pressure and added 100 mL of water. To the stirred mixture, sodium hydroxide (2 N, 100 mL) was added slowly and the reaction mixture was allowed to stir for 1 h at room temperature. Ethyl acetate (300 mL) was added to the resulting precipitate, the two layers were separated and the aqueous layer was further extracted with ethyl acetate (2 × 100 mL). The organic layers were combined, washed with brine and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 51 (3.46 g, 36%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 12.28 (br s, 1H), 9.31 (s, 1H), 6.85 (dd, J = 2.3, 4.0 Hz, 1H), 6.14 (dd, J = 2.3, 4.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 178.2, 131.9, 126.0, 122.4, 110.0; HRMS (ESI) calcd for C5H5ClNO (M + H)+ 130.0054, found 130.0055.
Synthesis of [2,2’-Bipyrrole]-5-carboxaldehyde (19).
Compound 19 (558 mg, 45%) was synthesized by the same procedure as described for 9. 1H NMR (CDCl3, 400 MHz) δ 11.98 (br s, 1H), 11.24 (br s, 1H), 9.35 (s, 1H), 7.00 (dd, J = 2.3, 3.9 Hz, 1H), 6.89 (m, 1H), 6.73 (m, 1H), 6.54 (dd, J = 2.3, 3.9 Hz, 1H), 6.12 (m, 1H); HRMS (ESI) calcd for C9H9N2O (M + H)+ 161.0709, found 161.0713.
Synthesis of Ethyl 4,5,6,7-tetrahydro-isoindole-1-carboxylate (53).
To a stirred solution of 52 (5.0 g, 39.37 mmol) and ethyl isocyanoacetate (5.33 g, 47.24 mmol) in 1:1 mixture of THF and ethanol (100 mL) was added portion-wise anhydrous potassium carbonate (10.86 g, 78.74 mmol). The reaction mixture was then stirred at room temperature for 3 days. The mixture was poured into water (100 mL), acidified to pH 5 with 2 N HCl, and extracted with diethyl ether (3 × 100 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 53 (4.93 g, 65%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 9.28 (br s, 1H), 6.67 (d, J = 2.9 Hz, 1H), 4.33 (q, J = 7.1 Hz, 2H), 2.85 (t, J = 5.8 Hz, 2H), 2.57 (t, J = 6.0 Hz, 2H), 1.77 (m, 4H), 1.38 (t, J = 7.1 Hz, 3H); HRMS (ESI) calcd for C11H16NO2 (M + H)+ 194.1176, found 194.1184.
Synthesis of 4,5,6,7-Tetrahydro-isoindole (54).
Sodium hydroxide (1.47 g, 36.71 mmol) was added to a solution of 53 (3.8 g, 18.35 mmol) in anhydrous ethylene glycol (20 mL) under an argon atmosphere at room temperature, and the reaction mixture was heated to reflux and stirred at refluxing temperature for an hour. After cooling to room temperature, the reaction mixture was taken up in n-hexane, washed with water and dried over anhydrous Na2SO4. Evaporation of the solvent under reduced pressure afforded the 54 (2.0 g, 90%) as a white solid that was directly used in the next step without further purification. 1H NMR (CDCl3, 400 MHz) δ 7.92 (br s, 1H), 6.53 (d, J = 2.6 Hz, 2H), 2.67 (m, 4H), 1.80 (m, 4H); HRMS (ESI) calcd for C8H12N (M + H)+ 122.0964, found 122.0969.
Representative Procedure for the Synthesis of 4,5,6,7-Tetrahydro-isoindole-1-carboxaldehyde (55) by Standard Vilsmeier Conditions.
Phosphorus oxychloride (POCl3; 5.05 g, 33.05 mmol) was added dropwise to dimethylformamide (DMF; 2.41 g, 33.05 mmol) at 0 °C. The resulting solution was stirred at 0 °C until the formation of the Vilsmeier complex as a solid. After the solid was dried in vacuo for 20 min, dichloromethane (50 mL) was added to the solid and the reaction mixture was cooled to 0 °C. A solution of 54 (2.0 g, 16.52 mmol) in DCM (50 mL) was added dropwise, and the reaction mixture was warmed to room temperature and then stirred for 10 h. After removing all solvent under vacuo, the residue was mixed with water (100 mL). To the stirred mixture, sodium hydroxide (5.28 g, 132.23 mmol) was added slowly and the reaction mixture was allowed to stir for 1 h at room temperature. Ethyl acetate (200 mL) was added to the resulting precipitate, the two layers were separated, and the aqueous layer was further extracted with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with brine and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 55 (1.84 g, 75%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 10.23 (br s, 1H), 9.51 (s, 1H), 6.87 (d, J = 2.8 Hz, 1H), 2.86 (t, J = 5.9 Hz, 2H), 2.55 (t, J = 6.0 Hz, 2H), 1.80 (m, 4H); HRMS (ESI) calcd for C9H12NO (M + H)+ 150.0913, found 150.0920.
Representative Procedure for the Synthesis of 3-Bromo-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (56).
To a stirred solution of 55 (2.0 g, 13.42 mmol) in THF (100 mL) was added portion-wise DBDMH (1.90 g, 6.71 mmol) in a period of 10 min at −78 °C. Then the reaction mixture was stirred for 5 h while it was allowed to warm to room temperature. The reaction was quenched with 5% aqueous KHSO4 solution, and extracted with ethyl acetate (3 × 75 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 56 (2.48 g, 82%). 1H NMR (CDCl3, 400 MHz) δ 10.60 (br s, 1H), 9.41 (s, 1H), 2.83 (m, 2H), 2.42 (m, 2H), 1.77 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 175.7, 134.7, 128.9, 122.7, 110.5, 22.8, 22.6, 21.3, 21.0; HRMS (ESI) calcd for C9H11BrNO (M + H)+ 228.0019, found 228.0031.
Synthesis of 3-(Pyrrol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (20).
Compound 20 (682 mg, 72%) was synthesized by the same procedure as described for 9. 1H NMR (DMSO-d6 + CDCl3, 400 MHz) δ 10.64 (br s, 1H), 10.35 (br s, 1H), 8.77 (s, 1H), 6.21 (s, 1H), 5.81 (s, 1H), 5.56 (s, 1H), 2.19 (s, 2H), 1.98 (s, 2H), 1.16 (m, 4H); 13C NMR (DMSO-d6 + CDCl3, 100 MHz) δ 173.0, 133.1, 128.8, 125.6, 122.5, 117.9, 116.7, 108.2, 107.5, 21.9, 21.5, 21.1, 19.6; HRMS (ESI) calcd for C13H15N2O (M + H)+ 215.1179, found 215.1188.
Synthesis of Compounds 59–62.
Compounds 59 (1.36 g, 75%), 60 (1.27 g, 78%), 61 (1.35 g, 79%), and 62 (1.35 g, 83%) were synthesized by the same procedure as described for 56.
4-Bromo-pyrrole-2-carboxaldehyde (59).
1H NMR (CDCl3, 400 MHz) δ 10.16 (br s, 1H), 9.49 (d, J = 1.0 Hz, 1H), 7.15 (m, 1H), 7.00 (m, 1H); HRMS (ESI) calcd for C5H5BrNO (M + H)+ 173.9549, found 173.9555.
4-Bromo-3,5-dimethyl-pyrrole-2-carboxaldehyde (60).
1H NMR (CDCl3, 400 MHz) δ 10.82 (br s, 1H), 9.45 (s, 1H), 2.36 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 176.2, 137.0, 133.0, 127.7, 101.5, 12.2, 10.0; HRMS (ESI) calcd for C7H9BrNO (M + H)+ 201.9862, found 201.9871.
4-Bromo-3-methyl-pyrrole-2-carboxaldehyde (61).
1H NMR (acetone-d6, 400 MHz) δ 11.17 (br s, 1H), 9.69 (d, J = 0.7 Hz, 1H), 7.25 (d, J = 3.2 Hz, 1H), 2.32 (s, 3H); HRMS (ESI) calcd for C6H7BrNO (M + H)+ 187.9705, found 187.9711.
4-Bromo-3-ethyl-pyrrole-2-carboxaldehyde (62).
1H NMR (CDCl3, 400 MHz) δ 10.08 (br s, 1H), 9.34 (s, 1H), 7.20 (d, J = 2.6 Hz, 1H), 2.47 (q, J = 7.6 Hz, 2H), 1.25 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C7H9BrNO (M + H)+ 201.9862, found 201.9869.
Synthesis of 22–25.
Compounds 22 (647 mg, 70%), 23 (673 mg, 72%), 24 (623 mg, 67%), and 25 (608 mg, 65%) were synthesized by the same procedure as described for 9, with modifying the reaction conditions for deprotection of N-Boc group. The crude material was dissolved in THF (10 mL) and LiOH (10 equiv.) in methanol (10 mL) was added dropwise under an argon atmosphere. The resulting mixture was stirred at 60 °C for 2 h.
[2,3’-Bipyrrole]-5’-carboxaldehyde (22).
1H NMR (CD3OD, 400 MHz) δ 9.43 (d, J = 1.0 Hz, 1H), 7.37 (dd, J = 1.6, 2.5 Hz, 1H), 7.15 (d, J = 1.6 Hz, 1H), 6.70 (dd, J = 1.5, 2.7 Hz, 1H), 6.23 (dd, J = 1.5, 3.4 Hz, 1H), 6.10 (dd, J = 2.7, 3.4 Hz, 1H); 13C NMR (CD3OD, 100 MHz) δ 180.7, 134.4, 127.7, 123.8, 122.6, 118.1, 117.2, 109.4, 104.7; HRMS (ESI) calcd for C9H9N2O (M + H)+ 161.0709, found 161.0713. Note. Two NH protons are not appering under these conditions.
2’,4’-Dimethyl-[2,3’-bipyrrole]-5’-carboxaldehyde (23).
1H NMR (DMSO-d6, 400 MHz) δ 11.68 (br s, 1H), 10.59 (br s, 1H), 9.51 (s, 1H), 6,76 (br s, 1H), 6.08 (br s, 1H), 5.94 (br s, 1H), 2.28 (s, 3H), 2.24 (s, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 176.4, 135.2, 129.8, 127.7, 124.2, 117.3, 117.1, 107.9, 107.0, 12.1, 9.5; HRMS (ESI) calcd for C11H13N2O (M + H)+ 189.1022, found 189.1026.
4’-Methyl-[2,3’-bipyrrole]-5’-carboxaldehyde (24).
1H NMR (DMSO-d6, 600 MHz) δ 11.78 (br s, 1H), 10.78 (br s, 1H), 9.64 (s, 1H), 7.33 (d, J = 3.0 Hz, 1H), 6.71 (dd, J = 1.8, 2.4 Hz, 1H), 6.10 (dd, J = 1.8, 3.0 Hz, 1H), 6.07 (dd, J = 2.4, 3.0 Hz, 1H), 2.41 (s, 3H); 13C NMR (DMSO-d6 + CDCl3, 100 MHz) δ 176.2, 128.7, 125.8, 124.5, 122.1, 118.1, 115.6, 107.1, 104.0, 8.9; HRMS (ESI) calcd for C10H11N2O (M + H)+ 175.0866, found 175.0871.
4’-Ethyl-[2,3’-bipyrrole]-5’-carboxaldehyde (25).
1H NMR (DMSO-d6, 600 MHz) δ 9.69 (s, 1H), 7.40 (d, J = 2.7 Hz, 1H), 6.70 (dd, J = 1.7, 2.7 Hz, 1H), 6.25 (d, J = 3.2 Hz, 1H), 6.16 (dd, J = 2.7, 3.2 Hz, 1H), 2.70 (q, J = 7.3 Hz, 2H), 1.12 (t, J = 7.3 Hz, 3H); HRMS (ESI) calcd for C11H13N2O (M + H)+ 189.1022, found 189.1027. Note. Two NH protons are not appering under these conditions.
Synthesis of 26–31.
Compounds 26 (276 mg, 55%), 27 (266 mg, 57%), 28 (296 mg, 63%), 29 (346 mg, 68%), 30 (346 mg, 67%), and 31 (385 mg, 65%) were synthesized by the same procedure as described for 9 with modifying the reaction conditions for the deprotection of N- triisopropylsilyl group. The crude material was dissolved in THF (10 mL) and TBAF (2 equiv.) was added dropwise under an argon atmosphere. The resulting mixture was stirred at room temperature for 15 min.
1’,3,4-Trimethyl-[2,2’-bipyrrole]-5-carboxaldehyde (26).
1H NMR (CDCl3, 400 MHz) δ 9.62 (s, 1H), 8.84 (br s, 1H), 6.77 (dd, J = 1.8, 2.4 Hz, 1H), 6.28 (dd, J = 1.8, 3.7 Hz, 1H), 6.23 (dd, J = 2.4, 3.7 Hz, 1H), 3.61 (s, 3H), 2.33 (s, 3H), 2.02 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 176.8, 131.8, 129.6, 129.0, 124.3, 124.2, 120.4, 111.3, 108.4, 34.8, 9.6, 9.0; HRMS (ESI) calcd for C12H14NaN2O (M + Na)+ 225.0998, found 225.1006.
3,4-Dimethyl-[2,3’-bipyrrole]-5-carboxaldehyde (27).
1H NMR (CDCl3, 400 MHz) δ 9.52 (s, 1H), 9.10 (br s, 1H), 8.64 (br s, 1H), 7.12 (m, 1H), 6.89 (m, 1H), 6.48 (m, 1H), 2.31 (s, 3H), 2.15 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 175.4, 134.3, 133.3, 127.9, 119.1, 117.1, 116.6, 115.5, 106.9, 9.8, 8.9; HRMS (ESI) calcd for C11H12NaN2O (M + Na)+ 189.1022, found 189.1028.
5-(Furan-2-yl)-3,4-dimethyl-pyrrole-2-carboxaldehyde (28).
1H NMR (CDCl3, 400 MHz) δ 9.63 (s, 1H), 9.52 (br s, 1H), 7.48 (dd, J = 1.6, 2.8 Hz, 1H), 6.64 (dd, J = 1.6, 3.6 Hz, 1H), 6.52 (dd, J = 2.8, 3.6 Hz, 1H), 2.30 (s, 3H), 2.17 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 176.8, 146.5, 142.1, 132.1, 128.6, 128.0, 117.9, 111.9, 107.8, 9.6, 8.6; HRMS (ESI) calcd for C11H11NaN2O (M + Na)+ 212.0682, found 212.0689.
3,4-Dimethyl-5-(thiophen-2-yl)-pyrrole-2-carboxaldehyde (29).
1H NMR (CDCl3, 400 MHz) δ 9.63 (s, 1H), 9.53 (br s, 1H), 7.37 (dd, J = 1.6, 2.7 Hz, 1H), 7.34 (dd, J = 1.6, 3.5 Hz, 1H), 7.13 (dd, J = 2.7, 3.5 Hz, 1H), 2.33 (s, 3H), 2.21 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 176.8, 133.6, 132.6, 131.4, 128.9, 127.8, 125.7, 124.2, 118.9, 9.9, 8.9; HRMS (ESI) calcd for C11H11NaNOS (M + Na)+ 228.0454, found 228.0459.
3,4-Dimethyl-5-phenyl-pyrrole-2-carboxaldehyde (30).
1H NMR (CDCl3, 400 MHz) δ 9.64 (s, 1H), 9.49 (br s, 1H), 7.52 (m, 2H), 7.46 (m, 2H), 7.39 (m, 1H), 2.34 (s, 3H), 2.17 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 176.9, 137.0, 132.6, 131.7, 129,0, 128.9 (2C), 128.2, 127.8 (2C), 118.6, 9.8, 9.0; HRMS (ESI) calcd for C13H13NaNO (M + Na)+ 222.0889, found 222.0897.
5-(Indol-2-yl)-3,4-dimethyl-pyrrole-2-carboxaldehyde (31).
1H NMR (DMSO-d6, 400 MHz) δ 11.43 (br s, 2H), 9.62 (s, 1H), 7.44 (m, 2H), 7.09 (m, 2H), 6.82 (s, 1H), 2.29 (s, 3H), 2.21 (s, 3H); 13C NMR (CDCl3 + DMSO-d6, 100 MHz) δ 177.1, 136.1, 130.9, 129.6, 129.2, 128.8, 128.3, 122.1, 120.2, 119.6, 118.4, 111.2, 101.5, 10.0, 8.5; HRMS (ESI) calcd for C15H15N2O (M + H)+ 239.1179, found 239.1188.
Representative Procedure for the Synthesis of 2-Ethyl-pyrrole (67a).
To a stirred suspension of LiAlH4 (3.49 g, 91.74 mmol) in dry THF (50 mL) was added dropwise 66a (5.0 g, 45.87 mmol) in THF (50 mL) at 0 °C. Then the resulting solution was heated to reflux overnight. The reaction was quenched with saturated solution of sodium sulfate. The insoluble solid was filtrated off, and washed with DCM (100 mL). Then the combined organic solution was concentrated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 67a (4.0 g, 92%).
2-Isobutyl-pyrrole (67b).
(4.26 g, 95%); HRMS (ESI) calcd for C8H14N (M + H)+ 124.1121, found 124.1126.
Representative Procedure for the Synthesis of tert-Butyl 2-ethyl-pyrrole-1-carboxylate (68a).
4-(Dimethyl-amino)pyridine (DMAP; 257 mg, 2.10 mmol) was added to a stirred solution of 67a (2.0 g, 21.05 mmol), and di-tert-butyl dicarbonate (Boc2O; 6.23 g, 27.36 mmol) in acetonitrile (50 mL) and the reaction left to stir for 1 h at room temperature. Dichloromethane (150 mL) was added and the solution was washed with water and brine and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure 68a (3.90 g, 95%). HRMS (ESI) calcd for C11H18NO2 (M + H)+ 196.1332, found 196.1335.
tert-Butyl 2-isobutyl-pyrrole-1-carboxylate (68b).
(3.40 g, 94%), 1H NMR (CDCl3, 400 MHz) δ 7.21 (dd, J = 1.6, 2.4 Hz, 1H), 6.09 (dd, J = 1.6, 3.6 Hz, 1H), 5.95 (dd, J = 2.4, 3.6 Hz, 1H), 2.73 (d, J = 7.0 Hz, 2H), 1.93 (m, 1H), 1.61 (s, 9H), 0.94 (d, J = 6.3 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 149.6, 135.1, 120.9, 112.3, 109.7, 83.1, 37.8, 28.0 (3C), 27.7, 22.5 (2C): HRMS (ESI) calcd for C13H22NO2 (M + H)+ 224.1645, found 224.1649.
tert-Butyl 2,4-dimethyl-pyrrole-1-carboxylate (68c).
(3.77 g, 92%), 1H NMR (CDCl3, 400 MHz) δ 6.94 (s, 1H), 5.80 (s, 1H), 2.41 (s, 3H), 2.02 (s, 3H), 1.60 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 149.6, 131.6, 120.4, 117.5, 114.2, 82.8, 28.1 (3C), 15.4, 11.7; HRMS (ESI) calcd for C11H18NO2 (M + H)+ 196.1332, found 196.1339.
tert-Butyl 3-ethyl-2,4-dimethyl-pyrrole-1-carboxylate (68d).
(3.26 g, 90%), 1H NMR (CDCl3, 400 MHz) δ 6.96 (s, 1H), 2.38 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.01 (s, 3H), 1.61 (s, 9H), 1.07 (t, J = 7.6 Hz, 3H); HRMS (ESI) calcd for C13H22NO2 (M + H)+ 224.1645, found 224.1653.
Representative Procedure for the Synthesis of (1-(tert-Butoxycarbonyl)-5-ethyl-pyrrol-2-yl)boronic acid (69a).
To a stirred solution of 2,2,6,6-tetramethylpiperidine (2.60 g, 18.46 mmol) in dry THF (50 mL) was added dropwise n-BuLi (1.6 M in pentane, 12.5 mL, 20.0 mmol) under an argon atmosphere at −78 °C. The reaction mixture was allowed to warm to 0 °C and maintained at that temperature for 30 min. After cooling again to −78 °C, a solution of 68a (3.0 g, 15.38 mmol) in THF (10 mL) was added. The reaction mixture was stirred for 2 h at −78 °C prior to the addition of trimethyl borate (7.92 g, 76.92 mmol). The solution was allowed to react at ambient temperature overnight. The reaction mixture was diluted with EtOAc (200 mL), washed with water, and brine solution and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation to furnish the desired product 69a (3.12 g, 85%) as a brown solid. The product 69a was carried forward into the next reaction without further purification. The products 69b (1.82 g, 76%), 69c (1.59 g, 65%), and 69d (1.62 g, 68%) were also carried forward into the next reaction without further purification.
Synthesis of 32–35.
Compounds 32 (403 mg, 75%), 33 (467 mg, 77%), 34 (1.59 g, 57%), and 35 (1.62 g, 55%) were synthesized by the same procedure as described for 9.
5’-Ethyl-3,4-dimethyl-[2,2’-bipyrrole]-5-carboxaldehyde (32).
1H NMR (DMSO-d6, 600 MHz) δ 10.99 (s, 1H), 10.94 (br s, 1H), 9.46 (s, 1H), 6.35 (br s, 1H), 5.89 (br s, 1H), 2.61 (br s, 2H), 2.22 (s, 3H), 2.06 (s, 3H), 1.21 (br s, 3H); 13C NMR (CDCl3 + DMSO-d6, 150 MHz) δ 175.0, 135.6, 131.8, 130.9, 127.5, 122.0, 115.3, 108.9, 106.0, 20.3, 13.6, 9.9, 8.9; HRMS (ESI) calcd for C13H17N2O (M + H)+ 217.1335, found 217.1348.
5’-Isobutyl-3,4-dimethyl-[2,2’-bipyrrole]-5-carboxaldehyde (33).
1H NMR (CDCl3 + DMSO-d6, 400 MHz) δ 11.18 (br s, 1H), 10.92 (br s, 1H), 9.24 (s, 1H), 6.40 (br s, 1H), 5.85 (s, 1H), 2.41 (d, J = 7.1 Hz, 2H), 2.17 (s, 3H), 2.04 (s, 3H), 1.86 (m, 1H), 0.85 (d, J = 6.4 Hz, 6H); 13C NMR (CDCl3 + DMSO-d6, 150 MHz) δ 173.6, 134.4, 134.2, 133.3, 127.5, 122.3, 116.8, 110.2, 107.9, 37.3, 29.1, 22.4 (2C), 10.3, 8.7; HRMS (ESI) calcd for C15H21N2O (M + H)+ 245.1648, found 245.1660.
3,3’,4,5’-Tetramethyl-[2,2’-bipyrrole]-5-carbaldehyde (34).
1H NMR (CDCl3, 400 MHz) δ 9.52 (s, 1H), 9.24 (br s, 1H), 8.40 (br s, 1H), 5.84 (d, J = 2.6 Hz, 1H), 2.31 (s, 6H), 2.16 (s, 3H), 2.09 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 174.2, 130.4, 127.4, 127.2, 117.0, 116.7, 116.6, 107.7 (2C), 11.6, 11.1, 8.6, 7.8; HRMS (ESI) calcd for C13H17N2O (M + H)+ 217.1335, found 217.1348.
4’-Ethyl-3,3’,4,5’-tetramethyl-[2,2’-bipyrrole]-5-carboxaldehyde (35).
HRMS (ESI) calcd for C15H21N2O (M + H)+ 245.1648, found 245.1656.
Synthesis of (1-(tert-Butoxycarbonyl)-4-ethyl-pyrrol-2-yl)boronic acid (71).
Compound 71 (1.81 g, 74%) was synthesized by the same procedure as described for 69a. The product 71 was carried forward into the next reaction without further purification.
Synthesis of 4’-Ethyl-3,4-dimethyl-[2,2’-bipyrrole]-5-carboxaldehyde (36).
Compound 36 (413 mg, 77%) was synthesized by the same procedure as described for 9. 1H NMR (DMSO-d6, 400 MHz) δ 11.26 (br s, 1H), 10.75 (br s, 1H), 9.48 (s, 1H), 6.70 (s, 1H), 6.35 (s, 1H), 2.45 (q, J = 7.5 Hz, 2H), 2.22 (s, 3H), 2.07 (s, 3H), 1.15 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 173.4, 130.8, 129.8, 126.3, 125.6, 122.2, 122.0, 114.6, 107.4, 18.6, 14.0, 8.9, 7.3; HRMS (ESI) calcd for C13H17N2O (M + H)+ 217.1335, found 217.1346.
Synthesis of tert-Butyl 1-formyl-4,5,6,7-tetrahydro-isoindole-2-carboxylate (72).
Compound 72 (3.17 g, 95%) was synthesized by the same procedure as described for 68a. 1H NMR (CDCl3, 400 MHz) δ 10.38 (s, 1H), 7.14 (s, 1H), 2.88 (t, J = 5.8 Hz, 2H), 2.52 (t, J = 5.6 Hz, 2H), 1.74 (m, 4H), 1.69 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 183.4, 148.7, 137.3, 129.1, 123.8, 122.7, 84.8, 28.0 (3C), 24.2, 22.7, 22.6, 21.5; HRMS (ESI) calcd for C14H19NaNO3 (M + Na)+ 272.1257, found 272.1263.
Synthesis of tert-Butyl 1-(dimethoxymethyl)-4,5,6,7-tetrahydro-isoindole-2-carboxylate (73).
A solution of aldehyde 72 (2.0 g, 8.03 mmol), trimethyl orthoformate (1.70 g, 16.06 mmol) and a catalytic amount (30 mg) of p-toluenesulfonic acid (PTSA) in MeOH (20 mL) was stirred at room temperature for 1 h. The reaction mixture was diluted with Et2O (200 mL) and washed with a solution of NaHCO3. The organic layer was washed with water and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 73 (2.01 g, 85%). HRMS (ESI) calcd for C16H26NO4 (M + H)+ 296.1856, found 296.1863.
Synthesis of (3-Formyl-4,5,6,7-tetrahydro-isoindol-1-yl)boronic acid (74).
To a stirred solution of 73 (1.2 g, 4.06 mmol) in THF (10 mL) was added triisopropyl borate (1.14 g, 6.10 mmol). The solution was cooled to 0‒5 °C in an ice bath, and lithium diisopropylamide (LDA; 2 N, 4 mL, 8.13 mmol) was added over 20 min and stirring was continued for an additional hour. The saturated ammonium chloride (5 mL) and 10% aqueous potassium bisulfate solution (50 mL) were added to adjust the pH 2, followed by stirring at room temperature for 2 h. The reaction mixture was diluted with EtOAc (200 mL), washed with brine solution and dried over anhydrous Na2SO4. The solvent was removed by rotary evaporation to furnish the desired product 74 (738 mg, 94%) as an orange solid. The product 74 was carried forward into the next reaction without further purification.
Synthesis of 3-(Imidazol-2-yl)-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (37).
Compound 37 (478 mg, 65%) was synthesized by the same procedure as described for 9. 1H NMR (DMSO-d6, 400 MHz) δ 12.02 (s, 1H), 11.68 (br s, 1H), 9.53 (s, 1H), 7.26 (br s, 1H), 7.07 (br s, 1H), 2.79 (m, 4H), 1.72 (m, 4H); 13C NMR (CDCl3 + DMSO-d6, 100 MHz) δ 176.9, 139.6, 132.5, 129.3, 127.3, 126.2, 120.7, 117.2, 22.8, 22.6, 22.3, 20.9; HRMS (ESI) calcd for C12H14N3O (M + H)+ 216.1131, found 216.1136.
Synthesis of 1-Methyl-4,5,6,7-tetrahydro-isoindole (77).
To a stirred suspension of LiAlH4 (1.57 g, 41.45 mmol) in dry THF (50 mL) was added dropwise 53 (2.0 g, 10.36 mmol) in THF (50 mL) at 0 °C. Then the resulting solution was stirred at same temperature for additional 3 h and heated to reflux overnight. The reaction was quenched with saturated solution of sodium sulfate. The insoluble solid was filtrated off, and washed with DCM (100 mL). Then the combined organic solution was concentrated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 77 (1.19 g, 85%). 1H NMR (CDCl3, 400 MHz) δ 7.61 (br s, 1H), 6.47 (d, J = 2.8 Hz, 1H), 2.71 (t, J = 5.8 Hz, 2H), 2.60 (t, J = 6.0 Hz, 2H), 1.90 (s, 3H), 1.87 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 121.9, 120.1, 115.5, 110.7, 24.2 (2C), 22.3, 21.5, 10.9; HRMS (ESI) calcd for C9H14N (M + H)+ 136.1121, found 136.1117.
Synthesis of 3-Methyl-4,5,6,7-tetrahydro-isoindole-1-carboxaldehyde (38).
Compound 38 (917 mg, 76%) was synthesized by the same procedure as described for 55. 1H NMR (CDCl3, 400 MHz) δ 10.10 (br s, 1H), 9.37 (s, 1H), 2.82 (t, J = 5.8 Hz, 2H), 2.41 (t, J = 6.0 Hz, 2H), 2.23 (s, 3H), 1.78 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 175.0, 135.4, 135.1, 126.5, 120.2, 23.3, 22.8, 21.0, 20.8, 11.3; HRMS (ESI) calcd for C10H14NO (M + H)+ 164.1070, found 164.1065.
Synthesis of Bis(3-ethyl-4-methyl-1H-pyrrol-2-yl)methane (79).
Compound 79 (1.13 g, 92%) was synthesized by the same procedure as described for 54. 1H NMR (CDCl3, 400 MHz) δ 7.25 (br s, 2H), 6.26 (t, J = 1.2 Hz, 2H), 3.73 (s, 2H), 2.36 (q, J = 7.5 Hz, 4H), 1.97 (s, 6H), 1.02 (t, J = 7.5 Hz, 6H); HRMS (ESI) calcd for C15H23N2 (M + H)+ 231.1856, found 231.1861.
Synthesis of 5,5’-Methylenebis(4-ethyl-3-methyl-1H-pyrrole-2-carbaldehyde) (39).
Compound 39 (907 mg, 73%) was synthesized by the same procedure as described for 55. 1H NMR (CDCl3, 400 MHz) δ 11.46 (br s, 2H), 9.48 (s, 2H), 3.86 (s, 2H), 2.31 (q, J = 7.4 Hz, 4H), 2.18 (s, 6H), 0.81 (t, J = 7.5 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 176.5, 134.2, 130.0, 127.9, 123.7, 22.4, 16.3, 14.9, 8.4. (Dimer); HRMS (ESI) calcd for C17H23N2O2 (M + H)+ 287.1786, found 287.1782.
Representative Procedure for the Synthesis of Prodiginine (85).
To a stirred solution of 6 (250 mg, 1.31 mmol) and 2,4-dialkylpyrrole (80; 829 mg, 2.63 mmol) in anhydrous methanol (50 mL) was added methanolic 2 N HCl (catalytic amount). The resulting brightly colored solution was stirred for 5 h at room temperature. The methanol was removed under reduced pressure and the product was chromatographed on neutral alumina, with ethyl acetate/hexanes as eluent, to afford the desired prodiginine analogue 85.HCl (468 mg, 68%) as a bright red colored compound. 1H NMR (CDCl3, 400 MHz) δ 12.85 (br s, 1H), 12.81 (br s, 1H), 12.65 (br s, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.26 (m, 5H), 7.06 (d, J = 8.1 Hz, 2H), 7.01 (s, 1H), 6.97 (m, 1H), 6.38 (m, 1H), 6.09 (d, J = 1.9 Hz, 1H), 5.87 (d, J = 1.6 Hz, 1H), 4.23 (s, 2H), 4.00 (s, 3H), 3.93 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 166.2, 149.1, 148.8, 141.0, 138.3, 136.4, 132.5, 132.2, 130.5 (2C), 129.8 (2C), 128.7 (4C), 127.9, 123.9, 122.1, 121.7, 118.3, 113.4, 112.9, 112.2, 93.1, 58.9, 33.8, 31.9; HRMS (ESI) calcd for C28H24Cl2N3O (M + H)+ 488.1291, found 488.1284; IR (KBr) vmax 3320, 3010, 2845, 1510, 1045, 742 cm−1.
Synthesis of 5’-((3,5-Bis(4-chlorobenzyl)-1-methyl-pyrrol-2-yl)methylene)-4’-methoxy-1-methyl-2,2’-bipyrrole (87).
To a stirred solution of prodiginine 85 (50 mg, 0.10 mmol) in DMF (10 mL) was added NaH (10 mg, 0.41 mmol) at 0 °C. The resulting bright red suspension was stirred for 10 min, and methyl iodide (58 mg, 0.41 mmol) was added at 0 °C and stirred for additional 30 min. The reaction mixture was warmed to room temperature, and gradually poured into ice cold water and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with water and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was chromatographed on neutral alumina, with ethyl acetate/hexanes as eluent, to afford the desired prodiginine 87 (46 mg, 85%). 1H NMR (CDCl3, 400 MHz) δ 7.24 (d, J = 8.3 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.3 Hz, 2H), 6.84 (s, 1H), 6.74 (br s, 1H), 6.68 (dd, J = 1.5, 3.8 Hz, 1H), 6.17 (dd, J = 2.6, 3.7 Hz, 1H), 5.92 (s, 1H), 5.75 (s, 1H), 4.25 (s, 2H), 3.96 (s, 3H), 3.90 (s, 3H), 3.89 (s, 2H), 3.63 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 167.8, 161.2, 142.1, 140.7, 136.9, 132.4, 131.6, 130.1 (3C), 129.8 (2C), 129.3, 128.8, 128.7 (3C), 127.8 (2C), 127.2, 115.3, 113.5, 111.7, 108.4, 96.9, 58.4, 37.5, 33.0, 32.7, 29.7; HRMS (ESI) calcd for C30H28Cl2N3O (M + H)+ 516.1604, found 516.1607.
Representative Procedure for the Synthesis of Tambjamine (99).
To a stirred solution of 6 (100 mg, 0.52 mmol) and n-butylamine (77 mg, 1.05 mmol) in anhydrous methanol (10 mL) was added methanolic 2 N HCl (catalytic amount). The resulting pale yellow colored solution was stirred at refluxing temperature for 5 h and the solvent was removed under reduced pressure. The crude solid was dissolved in EtOAc (50 mL) and washed with 2 N HCl (2 × 10 mL). The organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the product was chromatographed on neutral alumina, with ethyl acetate/hexanes as eluent, to afford the desired tambjamine 99 (117 mg, 91%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ 7.26 (s, 1H), 6.98 (dd, J = 1.3, 2.7 Hz, 1H), 6.67 (dd, J = 1.3, 3.6 Hz, 1H), 6.20 (dd, J = 2.7, 3.6 Hz, 1H), 5.87 (s, 1H), 3.84 (s, 3H), 3.41 (t, J = 7.1 Hz, 2H), 1.67 (m, 2H), 1.37 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 163.7, 142.2, 140.3, 124.0, 122.7, 113.1, 110.8, 110.7, 91.1, 58.5, 50.7, 32.2, 19.7, 13.6; HRMS (ESI) calcd for C14H20N3O (M + H)+ 246.1601, found 246.1605. IR (KBr) vmax 3299, 2936, 1420, 1175, 722 cm−1.
Representative Procedure for the Synthesis of N-(adamantan-1-yl)-2-(((oxoboranyl)methylene)amino)acetamide (188).
To a stirred solution of 44 (2.0 g, 11.43 mmol) in a mixture of THF (25 mL) and CH2Cl2 (25 mL) were added 1-adamantylamine (2.07 g, 13.71 mmol), DMAP (348 mg, 2.85 mmol), and N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDCl, 2.62 g, 13.71 mmol). The reaction mixture was stirred at room temperature for 4 h. The reaction was quenched with saturated aqueous NH4Cl solution (50 mL) and extracted with ethyl acetate (3 × 100 mL). The combined organic phases were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the product was chromatographed on neutral alumina, with ethyl acetate/hexanes as eluent, to afford the desired product 188 (2.99 g, 85%).1H NMR (CDCl3, 400 MHz) δ 6.01 (br s, 1H), 5.52 (br s, 1H), 3.64 (d, J = 4.7 Hz, 2H), 2.03 (m, 3H), 1.96 (d, J = 2.9 Hz, 6H), 1.64 (m, 6H), 1.41 (s, 9H); HRMS (ESI) calcd for C17H29N2O3 (M + H)+ 309.2173, found 309.2180.
Representative Procedure for the Synthesis of N-(Adamantan-1-yl)-2-aminoacetamide (189).
Compound 188 (2.5 g, 8.11 mmol) was dissolved in 20 mL of trifluoroacetic acid/water (1:1) and stirred at room temperature for 3 h. The reaction mixture was neutralized with 2 N NaOH and extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with brine, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to obtain the pure product 189 (1.60 g, 95%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 6.88 (br s, 1H), 3.20 (s, 2H), 2.06 (br s, 3H), 1.97 (d, J = 2.8 Hz, 6H), 1.70 (m, 6H); HRMS (ESI) calcd for C12H21N2O (M + H)+ 209.1648, found 209.1646. Note. Two NH protons are not appering under these conditions.
Synthesis of N-(2-((Adamantan-1-yl)amino)-2-oxoethyl)-pyrrole-2-carboxamide (190).
Compound 190 (1.09 g, 81%) was synthesized by the same procedure as described for 188. 1H NMR (CDCl3, 400 MHz) δ 9.83 (br s, 1H), 7.21 (t, J = 5.3 Hz 1H), 6.85 (m, 1H), 6.69 (m, 1H), 6.16 (m, 1H), 6.08 (s, 1H), 3.90 (d, J = 5.3 Hz, 2H), 1.99 (br s, 3H), 1.92 (d, J = 2.6 Hz, 6H), 1.59 (m, 6H); 13C NMR (CDCl3 + CD3OD, 100 MHz) δ 169.1, 162.4, 124.9, 122.2, 111.2, 109.5, 51.0, 43.2, 42.2 (3C), 36.2 (3C), 29.4 (3C); HRMS (ESI) calcd for C17H23NaN3O2 (M + Na)+ 324.1682, found 324.1693.
Synthesis of N1-((Pyrrol-2-yl)methyl)-N2-(adamantan-1-yl)ethane-1,2-diamine (191).
Compound 191 (186 mg, 82%) was synthesized by the same procedure as described for 67a. 1H NMR (CDCl3, 400 MHz) δ 9.52 (br s, 1H), 6.74 (dd, J = 1.9, 2.7 Hz, 1H), 6.11 (dd, J = 3.0, 5.6 Hz, 1H), 6.02 (d, J = 1.9 Hz, 1H), 3.80 (s, 2H), 3.00 (br s, 2H), 2.76 (m, 4H), 2.08 (br s, 3H), 1.69–1.60 (m, 12H); 13C NMR (CDCl3, 100 MHz) δ 130.1, 117.5, 107.8, 106.4, 51.5, 48.9, 46.1, 42.1 (3C), 39.4, 36.5 (3C), 29.5 (3C); HRMS (ESI) calcd for C17H28N3 (M + H)+ 274.2278, found 274.2287.
Synthesis of 3-(Pyrrol-2-yl)-acrylic acid methyl ester (192).
To a stirred suspension of NaH (910 mg, 37.89 mmol) in 50 mL of anhydrous dimethoxyethane at 0 °C was added dropwise a methyl diethylphosphnoacetate (7.96 g, 37.89 mmol). The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to room temperature. Pyrrole-2-carboxaldehyde (40; 3.0 g, 31.58 mmol) was added and the reaction mixture was stirred for additional 4 h. The reaction was quenched with ice-water and extracted with ethyl acetate (3 × 50 mL). The combined organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 192 (3.72 g, 78%). Rf value of the product 192 is similar to the starting material 40, the visualization of the product was black spot on TLC after applying the iodine vapor. 1H NMR (CDCl3, 600 MHz) δ 9.26 (br s, 1H), 7.61 (d, J = 15.9 Hz, 1H), 6.94 (d, J = 1.4 Hz, 1H), 6.58 (s, 1H), 6.29 (d, J = 3.6 Hz, 1H), 6.10 (d, J = 15.9 Hz, 1H), 3.80 (s, 3H). HRMS (ESI) calcd for C8H10NO2 (M + H)+ 152.0706, found 152.0710.
Synthesis of 3-(Pyrrol-2-yl)-acrylic acid (193).
To a stirred suspension of 192 (2.0 g, 13.24 mmol) in a mixture of THF (50 mL) and water (60 mL) was added LiOH.H2O (1.66 g, 39.73 mmol). The reaction mixture was stirred at 60 °C for 12 h after which it was cooled to 0 °C and washed with ethyl acetate (3 × 30 mL). The aqueous layer was carefully acidified to pH 2 with 2 N HCl and extracted with ethyl acetate (3 × 50 mL). The combined extracts were dried over anhydrous Na2SO4, and concentrated under reduced pressure to give the pure product 193 (1.74 g 96%). 1H NMR (CD3OD, 600 MHz) δ 7.51 (d, J = 15.8 Hz, 1H), 6.91 (dd, J = 1.1, 2.3 Hz, 1H), 6.49 (dd, J = 1.1, 3.5 Hz, 1H), 6.18 (dd, J = 2.2, 4.9 Hz, 1H), 6.04 (d, J = 15.8 Hz, 1H); HRMS (ESI) calcd for C7H7NaNO2 (M + Na)+ 160.0369, found 160.0363. Note. NH and COOH protons are not appearing under these conditions.
Synthesis of N-(2-Adamantan-1-yl)amino)-2-oxoethyl)-3-(pyrrol-2-yl)acrylamide (194).
Compound 194 (2.02 g, 85%) was synthesized by the same procedure as described for 188. 1H NMR (DMSO-d6, 600 MHz) δ 11.34 (s, 1H), 7.95 (t, J = 5.8 Hz, 1H), 7.27 (d, J = 15.7 Hz, 1H), 7.26 (s, 1H), 6.91 (dd, J = 2.4, 3.7 Hz, 1H), 6.41 (s, 1H), 6.26 (d, J = 15.7 Hz, 1H), 6.12 (dd, J = 2.4, 5.6 Hz, 1H), 3.71 (d, J = 5.8 Hz, 2H), 1.99 (s, 3H), 1.92 (d, J = 2.7 Hz, 6H), 1.61 (m, 6H); 13C NMR (DMSO-d6, 100 MHz) δ 167.9, 165.9, 129.8, 128.5, 121.6, 114.9, 111.5, 109.5, 50.7, 42.5, 41.0 (3C), 36.0 (3C), 28.8 (3C); HRMS (ESI) calcd for C19H25NaN3O2 (M + Na)+ 350.1839, found 350.1853.
Representative Procedure for the Synthesis of 195.
To a stirred solution of 194 (500 mg, 1.52 mmol) in methanol (10 mL) at room temperature was added NiCl2.6H2O (180 mg, 0.76 mmol). When the clear solution acquired a greenish color, the whole reaction mixture was brought to 0 °C and NaBH4 (85 mg, 2.29 mmol) was added portion-wise. The black colored reaction mixture was stirred for 30 min at 0 °C, and the solvent was removed under reduced pressure. The crude product was dissolved in ethyl acetate (50 mL), and treated with aqueous NH4Cl (2 × 10 mL). The organic layer was washed with brine and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 195 (473 mg, 94%) as a white solid. 1H NMR (DMSO-d6, 600 MHz) δ 11.50 (s, 1H), 7.97 (t, J = 5.8 Hz, 1H), 7.24 (s, 1H), 6.55 (dd, J = 2.3, 3.9 Hz, 1H), 5.86 (dd, J = 2.6, 5.4 Hz, 1H), 5.72 (s, 1H), 3.62 (d, J = 5.8 Hz, 2H), 2.75 (t, J = 7.4 Hz, 2H), 2.41 (t, J = 7.4 Hz, 2H), 2.00 (br s, 3H), 1.91 (d, J = 2.6 Hz, 6H), 1.60 (br s, 6H); 13C NMR (DMSO-d6, 100 MHz) δ 171.8, 167.9, 130.8, 116.0, 107.0, 104.2, 50.7, 42.3, 41.0 (3C), 35.9 (3C), 35.3, 28.8 (3C), 23.2; HRMS (ESI) calcd for C19H28N3O2 (M + H)+ 330.2176, found 330.2169.
Synthesis of N1-(3-(Pyrrol-2-yl)propyl)-N2-(adamantan-1-yl)ethane-1,2-diamine (196).
Compound 196 (178 mg, 78%) was synthesized by the same procedure as described for 67a. 1H NMR (CDCl3, 400 MHz) δ 9.28 (br s, 1H), 6.68 (s, 1H), 6.12 (t, J = 2.8 Hz, 1H), 5.92 (m, 1H), 2.78–2.66 (m, 8H), 2.16 (br s, 2H), 2.09 (br s, 3H), 1.83 (m, 2H), 1.86–1.67 (m, 12H); 13C NMR (CDCl3, 100 MHz) δ 132.1, 116.2, 107.9, 104.9, 50.1, 50.0, 48.9, 42.6 (3C), 39.6, 36.7 (3C), 29.6 (4C), 25.6; HRMS (ESI) calcd for C19H32N3 (M + H)+ 302.2591, found 302.2587.
In Vitro Antimalarial Activity: P. falciparum Growth Inhibition:
In vitro antimalarial activity was determined by the Malaria SYBR Green I-based Fluorescence (MSF) assay described previously66 with minor modifications as previously described,67 and expressed as the compound concentration inhibiting growth by 50% (IC50).
HepG2 Cytotoxicity Assay.
Drugs were dissolved in DMSO to make 10 mM stock solutions. Human hepatocarcinoma cells (HepG2) were maintained on RPMI-1640 medium supplemented with 10% fetal bovine serum at 37 °C in a humidified 5% CO2 atmosphere. Cells were seeded at a density of 2 × 104 per well in 96-well flat-bottom tissue culture plates containing complete medium in a total volume of 160 μL/well. The cells were allowed to attach at 37 °C overnight. On the following day, drug solutions (40 μL/well) were serially diluted with complete culture medium across the plate. The plates were then incubated at 37 °C and 5% CO2 for another 24–36 h. Afterward, the medium was aspirated and replaced with complete RPMI medium (200 μL/well), and the plates were incubated for an additional 24 h at 37 °C and 5% CO2. An aliquot of a stock solution of resazurin (Alamar Blue, prepared in 1 × PBS) was then added at 20 μL per well (final concentration 10 μM), and the plates were returned to the incubator for 3 h. After this period, fluorescence in each well, indicative of cellular redox activity was measured in a Gemini EM plate reader with excitation wavelength at 560 nm and emission wavelength at 590 nm.68,69 IC50 values were determined by nonlinear regression analysis of logistic concentration–fluorescence intensity curves (GraphPad Prism software).
In Vivo Efficacy Against Murine Malaria:
The in vivo activity of selected PGs and TAs was assessed against the blood stages using a modified 4-day test.71,72 A 4- to 5-week-old female CF1 mice (Charles River Laboratories) were infected intravenously with 2.5 × 105 P. yoelii (Kenya strain, MR4 MRA-428) parasitized erythrocytes from a donor animal. Drug administration commenced the day after the animals were inoculated (day 1). The test compounds were dissolved in PEG-400 and administered by oral gavage once daily for four successive days; chloroquine phosphate was used as a positive control. Blood for blood film analysis and body weights were obtained on the day following the last dose and then at weekly intervals through day 28. Blood films were Giemsa stained and examined microscopically to determine the levels of parasitemia. These blood samples were collected from the tail vein with the aid of a syringe-needle. All mice were observed daily to assess their clinical signs, which were recorded. Animals with observable parasitemia following the experiment were euthanized; animals cleared of parasites from their bloodstream were observed daily with assessment of parasitemia performed weekly until day 28 at which point we score the animal(s) as cured of infection, and the animals were euthanized. All treated mice with a negative smear on day 28 were considered cured (100% protection). ED50 values (mg/kg/day) were derived graphically from the dose required to reduce parasite burden by 50% relative to drug-free controls.
Supplementary Material
ACKNOWLEDGMENT.
This work was supported by a grant from the National Institutes for Health (GM077147).
ABBREVIATIONS USED.
- CQRPf
chloroquine-resistant P. falciparum
- CQR
chloroquine-resistant
- CQS
chloroquine-sensitive
- Pf
Plasmodium falciparum
- PGs
prodiginines: TAs, tambjamines
- PPM
pyrrolylpyrromethene
- SAR
structure-activity relationship
- CQ
chloroquine
- MQ
mefloquine
- IC50
half maximal inhibitory concentration
- nM
nanomolar
- MDR
multidrug-resistant
- ADMET
adsorption, distribution, metabolism, excretion and toxicity
- ED50
median effective dose
- NRD
non-recrudescence dose (the amount of drug needed for 100% protection to malaria-infected mice until day 28)
Footnotes
The authors declare no competing financial interest.
United States provisional patent application has been filed by the Portland State University to protect the intellectual property described in this report.
ASSOCIATED CONTENT
Supporting Information. Structural characterization data and spectra (NMR, and HRMS) of all final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.WHO. World Malaria Report 2014. http://www.who.int/malaria/publications/world_malaria_report_2014/en/
- 2.Hyde JE Drug-resistant malaria. Trends Parasitol. 2005, 21, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; An SS; Yeung S; Singhasivanon P; Day NP; Lindegardh N; Socheat D; White NJ Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manderville RA Synthesis, proton-affinity and anti-cancer properties of the prodigiosin-group natural products. Curr. Med. Chem.: Anti-Cancer Agents 2001, 1, 195–218. [DOI] [PubMed] [Google Scholar]
- 5.Fürstner A Chemistry and biology of roseophilin and the prodigiosin alkaloids: a survey of the last 2500 years. Angew. Chem. Int. Ed. 2003, 42, 3582–3603. [DOI] [PubMed] [Google Scholar]
- 6.Boonlarppraadab C; Kauffman CA; Jensen PR; Fenical W Marineosins A and B, cytotoxic spiroaminals from a marine-derived actinomycete. Org. Lett. 2008, 10, 5505–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salem SM; Kancharla P; Florova G; Gupta S; Lu W; Reynolds KA Elucidation of final steps of the marineosins biosynthetic pathway through identification and characterization of the corresponding gene cluster. J. Am. Chem. Soc. 2014, 136, 4565–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haynes SW; Sydor PK; Stanley AE; Song L; Challis GL Role and substrate specificity of the Streptomyces coelicolor RedH enzyme in undecylprodiginine biosynthesis. Chem. Commun. 2008, 1865–1867. [DOI] [PubMed] [Google Scholar]
- 9.Sydor PK; Barry SM; Odulate OM; Barona-Gomez F; Haynes SW; Corre C; Song L; Challis GL Regio- and stereodivergent antibiotic oxidative carbocyclizations catalysed by Rieske oxygenase-like enzymes. Nat. Chem. 2011, 3, 388–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burke C; Thomas T; Egan S; Kjelleberg S The use of functional genomics for the identification of a gene cluster encoding for the biosynthesis of an antifungal tambjamine in the marine bacterium Pseudoalteromonas tunicate. Environ. Microbiol. 2007, 9, 814–818. [DOI] [PubMed] [Google Scholar]
- 11.Kojiri K; Nakajima S; Suzuki H; Okura A; Suda H A new antitumor substance, BE-18591, produced by a streptomycete. I. Fermentation, isolation, physico-chemical and biological properties. J. Antibiot. 1993, 46, 1799–1803. [DOI] [PubMed] [Google Scholar]
- 12.Boger DL; Patel MJ Total synthesis of prodigiosin, prodigiosene, and desmethoxyprodigiosin: Diels-Alder reactions of heterocyclic azadienes and development of an effective palladium(II)-promoted 2,2’-bipyrrole coupling procedure. J. Org. Chem. 1988, 53, 1405–1415. [Google Scholar]
- 13.Alihosseini F; Ju KS; Lango J; Hammock BD; Sun G Antibacterial colorants: characterization of prodiginines and their applications on textile materials. Biotechnol. Prog. 2008, 24, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchal E; Uddin MI; Smithen DA; Hawco CLA; Lanteigne M; Overy DP; Kerr RG; Thompson A Antimicrobial activity of non-natural prodigiosenes. RSC Adv. 2013, 3, 22967–22971. [Google Scholar]
- 15.Nakamura A; Nagai K; Ando K; Tamura G Selective suppression by prodigiosin of the mitogenic response of murine splenocytes. J. Antibiot. 1985, 39, 1155–1159. [DOI] [PubMed] [Google Scholar]
- 16.Tsuji RF; Yamamoto M; Nakamura A; Katoka T; Magae J; Nagai K; Jamasaki M Selective immunosuppression of prodigiosin 25-C and FK 506 in the murine immune system. J. Antibiot. 1990, 43, 1293–1301. [DOI] [PubMed] [Google Scholar]
- 17.Stepkowski SM; Erwin-Cohen RA; Behbod F; Wang M-E; Qu X; Tejpal N; Nagy ZS; Kahan BD; Kirken RA Selective inhibitor of Janus tyrosine kinase 3, PNU 156804, prolongs allograft survival and acts synergistically with cyclosporine but additively with rapamycin. Blood 2002, 99, 680–689. [DOI] [PubMed] [Google Scholar]
- 18.Stepkowski SM; Nagy ZS; Wang M-E; Behbod F; Erwin-Cohen R; Kahan BD; Kirken RA The role of stat5 in the induction of regulatory T cells in transplantation tolerance. Transplant. Proc. 2001, 33, 3835–3836. [DOI] [PubMed] [Google Scholar]
- 19.Magae J; Miller JW; Nagai K; Shearer GM Effect of metacycloprodigiosin, an inhibitor of killer T cells, on murine skin and heart transplants. J. Antibiot.1996, 49, 86–90. [DOI] [PubMed] [Google Scholar]
- 20.D’Alessio R; Bargiotti A; Carlini O; Colotta F; Ferrari M; Gnocchi P; Isetta A; Mongelli N; Motta P; Rossi A; Rossi M; Tibolla M; Vanotti E Synthesis and immunosuppressive activity of novel prodigiosin derivatives. J. Med. Chem. 2000, 43, 2557–2565. [DOI] [PubMed] [Google Scholar]
- 21.Tanigaki K; Sato T; Tanaka Y; Nishikawa A; Nagai K; Kawashima H; Ohkuma S BE-18591 as a new H+/Cl− symport ionophore that inhibits immunoproliferation and gastritis. FEBS Lett. 2002, 524, 37–42. [DOI] [PubMed] [Google Scholar]
- 22.Han SB; Kim HM; Kim YH; Lee CW; Jang ES; Son KH; Kim SU; Kim YK T-cell specific immunosuppression by prodigiosin isolated from Serratia marcescens. Int. J. Immunopharmacol. 1998, 20, 1–13. [DOI] [PubMed] [Google Scholar]
- 23.Williams RP; Hearn WR Prodigiosin. Antibiotics 1967, 2, 410–432. [Google Scholar]
- 24.Regourd J; Al-Sheikh Ali A; Thompson A Synthesis and anticancer activity of C-ring-functionalized prodigiosin analogues. J. Med. Chem. 2007, 50, 1528–1536. [DOI] [PubMed] [Google Scholar]
- 25.Aldrich LN; Stoops SL; Crews BC; Marnett LJ; Lindsley CW Total synthesis and biological evaluation of tambjamine K and a library of unnatural analogs. Bioorg. Med. Chem. Lett. 2010, 20, 5207–5211. [DOI] [PubMed] [Google Scholar]
- 26.Smithen DA; Forrester AM; Corkery DP; Dellaire G; Colpitts J; McFarland SA; Berman JN Thompson A Investigations regarding the utility of prodigiosenes to treat leukemia. Org. Biomol. Chem. 2013, 11, 62–68. [DOI] [PubMed] [Google Scholar]
- 27.Hawco CLA; Marchal E; Uddin MI; Baker AEG; Corkery DP; Dellaire G; Thompson A Synthesis and biological evaluation of prodigiosene conjugates of porphyrin, estrone and 4-hydroxytamoxifen. Bioorg. Med. Chem. 2013, 21, 5995–6002. [DOI] [PubMed] [Google Scholar]
- 28.Sessler JL; Eller LR; Cho W-S; Nicolaou S; Aguilar A; Lee JT; Lynch VM; Magda DJ Synthesis, anion-binding properties, and in vitro anticancer activity of prodigiosin analogues. Angew. Chem. Int. Ed. 2005, 44, 5989–5992. [DOI] [PubMed] [Google Scholar]
- 29.Díaz de Greñu B; Hernández PI; Espona M; Quiñonero D; Light ME; Torroba T; Pérez-Tomás R; Quesada R Synthetic prodiginine obatoclax (GX15–070) and related analogues: anion binding, transmembrane transport, and cytotoxicity properties. Chem. –Eur. J 2011, 17, 14074–14083. [DOI] [PubMed] [Google Scholar]
- 30.Marchal E; Rastogi S; Thompson A; Davis JT Influence of B-ring modifications on proton affinity, transmembrane anion transport and anti-cancer properties of synthetic prodigiosenes. Org. Biomol. Chem. 2014, 12, 7515–7522. [DOI] [PubMed] [Google Scholar]
- 31.Castro AJ Antimalarial activity of prodigiosin. Nature 1967, 213, 903–904. [DOI] [PubMed] [Google Scholar]
- 32.Gerber NN A new prodiginine (prodigiosin-like) pigment from streptomyces. Antimalarial activity of several prodiginines. J. Antibiot. 1975, 28, 194–199. [DOI] [PubMed] [Google Scholar]
- 33.Davidson DE Jr.; Johnsen DO; Tanticharoenyos P; Hickman RL; Kinnamon KE Evaluating new antimalarial drugs against trophozoite induced Plasmodium cynomolgi malaria in rhesus monkeys. Am. J. Trop. Med. Hyg. 1976, 25, 26–33. [DOI] [PubMed] [Google Scholar]
- 34.Isaka M; Jaturapat A; Kramyu J; Tanticharoen M; Thebtaranonth Y Potent in vitro antimalarial activity of metacycloprodigiosin isolated from Streptomyces spectabilis BCC 4785. Antimicrob. Agents Chemother. 2002, 46, 1112–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazaro JEH; Nitcheu J; Predicala RZ; Mangalindan GC; Nesslany F; Marzin D; Concepcion GP; Diquet B Heptyl prodigiosin, a bacterial metabolite, is antimalarial in vivo and non-mutagenic in vitro. J. Nat. Toxins 2002, 11, 367–377. [PubMed] [Google Scholar]
- 36.Marchal E; Smithen DA; Uddin M, I.; Robertson AW; Jakeman DL; Mollard V; Goodman CD; MacDougall KS; McFarland SA; McFadden GI; Thompson A Synthesis and antimalarial activity of prodigiosenes. Org. Biomol. Chem. 2014, 12, 4132–4142. [DOI] [PubMed] [Google Scholar]
- 37.Papireddy K; Smilkstein M; Kelly JX; Shweta.; Salem SM; Alhamadsheh M; Haynes SW; Challis GL; Reynolds KA Antimalarial activity of natural and synthetic prodiginines. J. Med. Chem. 2011, 54, 5296–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kancharla P; Lu W; Salem SM; Kelly JX Reynolds KA Stereospecific synthesis of 23-hydroxyundecylprodiginines and analogues, and conversion to antimalarial premarineosins via a novel Rieske oxygenase catalyzed bicyclization. J. Org. Chem. 2014, 79, 11674–11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saes Dias RI; Regourd J; Santacroce PV; Davis JT; Jakeman DL; Thompson A Chloride anion transport and copper-mediated DNA cleavage by C-ring functionalized prodigiosenes. Chem. Commun. 2007, 2701–2703. [DOI] [PubMed] [Google Scholar]
- 40.Seganish JL; Davis JT Prodigiosin is a chloride carrier that can function as an anion exchanger. Chem. Commun. 2005, 5781–5783. [DOI] [PubMed] [Google Scholar]
- 41.Melvin MS; Tomlinson JT; Park G; Day CS; Saluta GS; Kucera GL; Manderville RA Influence of the A-ring on the proton affinity and anticancer properties of the prodigiosins. Chem. Res. Toxicol. 2002, 15, 734–741. [DOI] [PubMed] [Google Scholar]
- 42.Matsuya H; Okamoto M; Ochi T; Nishikawa A; Shimizu S; Kataoka T; Nagai K; Wasserman HH; Ohkuma S Prodigiosins uncouple lysosomal vacuolar-type ATPase through promotion of H+/Cl− symport. Biochem. J. 1998, 334, 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gale PA; Light ME; McNally B; Navakhun K; Sliwinski KE; Smith BD Co-transport of H+/Cl– by a synthetic prodigiosin mimic. Chem. Commun. 2005, 3773–3775. [DOI] [PubMed] [Google Scholar]
- 44.Davis JT; Gale PA; Okunola OA; Prados P; Iglesias-Sanchez JC; Torroba T; Quesada R Using small molecules to facilitate exchange of bicarbonate and chloride anions across liposomal membranes. Nat. Chem. 2009, 1, 138–144. [DOI] [PubMed] [Google Scholar]
- 45.Rastogi S; Marchal E; Uddin I; Groves B; Colpitts J; McFarland SA; Davis JT; Thompson A Synthetic prodigiosenes and the influence of C-ring substitution on DNA cleavage, transmembrane chloride transport and basicity. Org. Biomol. Chem. 2013, 11, 3834–3845. [DOI] [PubMed] [Google Scholar]
- 46.Melvin MS; Ferguson DC; Lindquist N; Manderville RA DNA Binding by 4-methoxypyrrolic natural products. Preference for intercalation at AT sites by tambjamine E and prodigiosin. J. Org. Chem. 1999, 64, 6861–6869. [DOI] [PubMed] [Google Scholar]
- 47.Cavalcanti BC; Júnior HVN; Seleghim MHR; Berlinck RGS; Cunha GMA; Moraes MO; Pessoa C Cytotoxic and genotoxic effects of tambjamine D, an alkaloid isolated from the nudibranch Tambja eliora, on Chinese hamster lung fibroblasts. Chem. Biol. Interact. 2008, 174, 155–162. [DOI] [PubMed] [Google Scholar]
- 48.Melvin MS; Tomlinson JT; Saluta GR; Kucera GL;Lindquist N; Manderville RA Double-strand DNA cleavage by copper·prodigiosin. J. Am. Chem. Soc. 2000, 122, 6333–6334. [Google Scholar]
- 49.Borthakur G; O’Brien S; Ravandi-Kashani F; Giles F; Schimmer AD; Viallet J; Kantarjian H A Phase I trial of the small molecule Pan-Bcl-2 family inhibitor obatoclax mesylate (GX15–070) administered by 24 hour infusion every 2 weeks to patients with myeloid malignancies and chronic lymphocytic leukemia (CLL). Blood (ASH Annual meeting Abstracts) 2006, 108, 2654. [Google Scholar]
- 50.Nguyen M; Marcellus RC; Roulston A; Watson M; Serfass L; Madiraju SRM; Goulet D; Viallet J; Bélec L; Billot X; Acoca S; Purisima E; Wiegmans A; Cluse L; Johnstone RW; Beauparlant P; Shore GC Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 19512–19517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen K; Rannulu NS; Cai Y; Lane P; Liebl AL; Rees BB; Corre C; Challis GL; Cole RB Unusual odd-electron fragments from even-electron protonated prodiginine precursors using positive-ion electrospray tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1856–1866. [DOI] [PubMed] [Google Scholar]
- 52.Wasserman HH; Rodgers GC; Keith DD Metacycloprodigiosin, a tripyrrole pigment from Streptomyces longisporus ruber. J. Am. Chem. Soc. 1969, 91, 1263–1264. [DOI] [PubMed] [Google Scholar]
- 53.Kancharla P; Reynolds KA Synthesis of 2,2′-bipyrrole-5-carboxaldehydes and their application in the synthesis of B-ring functionalized prodiginines and tambjamines. Tetrahedron 2013, 69, 8375–8385. [Google Scholar]
- 54.Dairi K; Tripathy S; Attardo G; Lavallee J-F Two-step synthesis of the bipyrrole precursor of prodigiosins. Tetrahedron Lett. 2006, 47, 2605–2606. [Google Scholar]
- 55.Li W-R; Lin ST; Hsu N-M; Chern M-S Efficient total synthesis of pulchellalactam, a CD45 protein tyrosine phosphatase inhibitor. J. Org. Chem. 2002, 67, 4702–4706. [DOI] [PubMed] [Google Scholar]
- 56.Yoon-Miller SJP; Opalka SM; Pelkey ET Short synthesis of 4-aryl-3-pyrrolin-2-ones. Tetrahedron Lett. 2007, 48, 827–830. [Google Scholar]
- 57.Leen V; Braeken E; Luckermans K; Jackers C; Van der Auweraer M; Boens N; Dehaen W A versatile, modular synthesis of monofunctionalized BODIPY dyes. Chem. Commun. 2009, 4515–4517. [DOI] [PubMed] [Google Scholar]
- 58.Roth SD; Shkindel T; Lightner DA Intermolecularly hydrogen-bonded dimeric helices: tripyrrindiones. Tetrahedron 2007, 63, 11030–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ono N; Kawamura H; Bougauchi M; Maruyama K Intermolecularly hydrogen-bonded dimeric helices: tripyrrindiones. Tetrahedron 1990, 46, 7483–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yadav JS; Reddy BVS; Kondaji G; Rao RS; Kumar SP Zinc-mediated acylation and sulfonation of pyrrole and its derivative. Tetrahedron Lett. 2002, 43, 8133–8135. [Google Scholar]
- 61.He Y; Lin M; Li Z; Liang X; Li G; Antilla JC Direct synthesis of chiral 1,2,3,4-tetrahydropyrrolo[1,2-a]pyrazines via a catalytic asymmetric intramolecular aza-Friedel–Crafts reaction. Org. Lett. 2011, 13, 4490–4493. [DOI] [PubMed] [Google Scholar]
- 62.Fürstner A; Grabowski J; Lehmann CW Total synthesis and structural refinement of the cyclic tripyrrole pigment nonylprodigiosin. J. Org. Chem. 1999, 64, 8275–8280. [DOI] [PubMed] [Google Scholar]
- 63.Schnölzer M; Alewood P; Jones A; Alewood D; Kent SBH In situ neutralization in Boc-chemistry solid phase peptide synthesis. Int. J. Peptide Res. Therap. 2007, 13, 31–44. [DOI] [PubMed] [Google Scholar]
- 64.Wadsworth W Synthetic applications of phosphoryl-stabilized anions. Org. React. 1977, 25, 73–253. [Google Scholar]
- 65.Wadsworth WS Jr.; Emmons WD Ethyl cyclohexylideneacetate. Org. Synth.1965, 45, 44. [Google Scholar]
- 66.Smilkstein MJ; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly JX; Smilkstein MJ; Cooper RA; Lane KD; Johnson RA; Janowsky A; Dodean RA; Hinrichs DJ; Winter R; Riscoe M Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitizers in Plasmodium falciparum. Antimicrob. Agents Chemother. 2007, 51, 4133–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhi-Jun Y; Sriranganathan N; Vaught T; Arastu SK; Ahmed SA A dye-based lymphocyte proliferation assay that permits multiple immunological analyses: mRNA, cytogenetic, apoptosis, and immunophenotyping studies. J. Immunol. Methods 1997, 210, 25–39. [DOI] [PubMed] [Google Scholar]
- 69.Nilsen A; Miley GP; Forquer IP; Mather MW; Katneni K; Li Y; Pou S; Pershing AM; Stickles AM; Ryan E; Kelly JX; Doggett JS; White KL; Hinrichs DJ; Winter RW; Charman SA; Zakharov LN; Bathurst I; Burrows JN; Vaidya AB; Riscoe MK Discovery, synthesis, and optimization of antimalarial 4(1H)-quinolone-3-diarylethers. J. Med. Chem. 2014, 57, 3818–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gleeson MP Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- 71.Peters W; Davies EE; Robinson BL The chemotherapy of rodent malaria, XXIII Causal prophylaxis, part II: Practical experience with Plasmodium yoelii nigeriensis in drug screening. Ann. Trop. Med. Parasitol. 1975, 69, 311–328. [PubMed] [Google Scholar]
- 72.Ager AJ Rodent malaria models; Springer-Verlag: New York, 1984, vol. 68, pp 225–264. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.