

Abstract
Background
Coronary artery disease (CAD) is a leading cause of death among the 38.4 million people with HIV globally. The extent to which cardiovascular polygenic risk scores (PRSs) derived in non‐HIV populations generalize to people with HIV is not well understood.
Methods and Results
PRSs for CAD (GPSMult) and lipid traits were calculated in a global cohort of people with HIV treated with antiretroviral therapy with low‐to‐moderate atherosclerotic cardiovascular disease risk enrolled in REPRIEVE (Randomized Trial to Prevent Vascular Events in HIV). The PRSs were associated with baseline lipid traits in 4495 genotyped participants, and with subclinical CAD in a subset of 662 who underwent coronary computed tomography angiography. Among participants who underwent coronary computed tomography angiography (mean age, 50.9 [SD, 5.8] years; 16.1% women; 41.8% African, 57.3% European, 1.1% Asian), GPSMult was associated with plaque presence with odds ratio (OR) per SD in GPSMult of 1.42 (95% CI, 1.20–1.68; P=3.8×10−5), stenosis >50% (OR, 2.39 [95% CI, 1.48–3.85]; P=3.4×10−4), and noncalcified/vulnerable plaque (OR, 1.45 [95% CI, 1.23–1.72]; P=9.6×10−6). Effects were consistent in subgroups of age, sex, 10‐year atherosclerotic cardiovascular disease risk, ancestry, and CD4 count. Adding GPSMult to established risk factors increased the C‐statistic for predicting plaque presence from 0.718 to 0.734 (P=0.02). Furthermore, a PRS for low‐density lipoprotein cholesterol was associated with plaque presence with OR of 1.21 (95% CI, 1.01–1.44; P=0.04), and partially calcified plaque with OR of 1.21 (95% CI, 1.01–1.45; P=0.04) per SD.
Conclusions
Among people with HIV treated with antiretroviral therapy without documented atherosclerotic cardiovascular disease and at low‐to‐moderate calculated risk in REPRIEVE, an externally developed CAD PRS was predictive of subclinical atherosclerosis. PRS for low‐density lipoprotein cholesterol was also associated with subclinical atherosclerosis, supporting a role for low‐density lipoprotein cholesterol in HIV‐associated CAD.
Registration
URL: https://www.reprievetrial.org; Unique identifier: NCT02344290.
Keywords: coronary CT angiography, people with HIV, polygenic risk scores, subclinical atherosclerosis
Subject Categories: Precision Medicine, Lipids and Cholesterol
Nonstandard Abbreviations and Acronyms
- ARIC
Atherosclerosis Risk in Communities
- PCE
pooled cohort equation
- PRS
polygenic risk score
- PWH
people with HIV
- REPRIEVE
Randomized Trial to Prevent Vascular Events in HIV
Clinical Perspective.
What Is New?
Cardiovascular disease such as coronary artery disease is a leading cause of morbidity and death in people with HIV (PWH); quantification of cumulative genetic risk using polygenic risk scores (PRSs) has potential as a complementary modality for predicting coronary artery disease in PWH.
An externally developed PRS for coronary artery disease was predictive of subclinical coronary artery disease in a diverse cohort of PWH treated with antiretroviral therapy without documented cardiovascular disease at low‐to‐moderate traditional risk and improves upon prediction by traditional risk estimators.
PRSs for other cardiometabolic traits such as lipid levels and blood pressure were also predictive in this cohort of PWH.
What Are the Clinical Implications?
Cardiovascular PRSs derived in the general population may further guide risk stratification among a vulnerable cohort of PWH without documented cardiovascular disease and at low‐to‐moderate estimated risk, who may be otherwise missed by traditional risk predictors.
By using existing PRSs to effectively identify individuals with the greatest subclinical atherosclerosis, this study directly supports clinical use of PRSs for primary prevention of cardiovascular disease in this vulnerable population of PWH.
The genetic association of low‐density lipoprotein cholesterol with coronary artery plaque among PWH supports the role of low‐density lipoprotein cholesterol in HIV‐associated coronary atherosclerosis.
There are approximately 38.4 million people with HIV (PWH) globally. 1 With the widespread use and effectiveness of antiretroviral therapy (ART) in preventing morbidity and death from AIDS, chronic diseases, most notably atherosclerotic cardiovascular disease (ASCVD), are becoming more prevalent in this population. 2 , 3 Mortality from ASCVD such as coronary artery disease (CAD) has now risen to be one of the leading non‐AIDS causes of death in PWH, outpacing rates in the general population. 4
PWH remain at outsized risk for ASCVD despite effective viral suppression with ART. 5 , 6 Proposed causes include increased inflammation, thrombosis, vulnerable noncalcified plaques, and atherogenic lipid profiles with notably elevated oxidized low‐density lipoprotein cholesterol (LDL‐C). 5 , 6 Indeed, studies comparing participants with or without HIV infection found that traditional risk models for CAD, such as pooled cohort equations (PCEs) or Framingham Risk Scores, 7 are frequently inaccurate among PWH compared with the general population. 8 , 9 The mechanisms driving the excess risk are poorly understood. Therefore, there is a critical need to develop improved strategies to predict ASCVD in this population, and to identify asymptomatic PWH at greatest risk for future ASCVD to guide preventive measures.
CAD has a strong genetic component, 10 and corresponding polygenic risk scores (PRSs) have emerged as a tool to stratify lifetime genetic risk for ASCVD in the general population. 11 , 12 , 13 , 14 , 15 Risk mitigation interventions such as lifestyle adjustments and cholesterol‐lowering medications may be targeted to individuals found to be at increased polygenic risk on the basis of epidemiological and post hoc analyses of clinical trials. 14 , 16 While it is well appreciated that PRSs perform differentially by genetic ancestry, 17 recent analyses have shown that age and sex also influence CAD PRS performance. 18 , 19 Therefore, there is a need to assess generalizability of CAD PRS, and its role in stratifying HIV‐associated CAD is not well understood. Coronary computed tomography angiography (CCTA) has emerged as a powerful tool to extract surrogate indices for clinical CAD risk to assess such novel biomarkers. 8 , 20 , 21
In this study, we demonstrated the ability of polygenic scores to predict subclinical CAD, atherosclerotic plaque phenotypes, and related cardiometabolic traits among the largest and most diverse cohort of PWH without prior evidence of ASCVD and at low‐to‐moderate traditional ASCVD risk, enrolled in a primary prevention trial. 22 Our findings offer new insights in risk stratification and mechanisms of HIV‐associated atherosclerosis.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Participants
Our study consisted of 4495 genotyped PWH without clinical cardiovascular disease (CVD) with low‐to‐moderate estimated cardiovascular risk, out of 7770 total who were enrolled in REPRIEVE (Randomized Trial to Prevent Vascular Events in HIV) (Figure S1A). 23 Data were derived from a subset of the REPRIEVE cohort (N=4837) enrolling from 52 AIDS Clinical Trial Group sites participating in REPRIEVE, for whom genotyping was performed. REPRIEVE is a prospective, randomized, placebo‐controlled trial assessing the efficacy of pitavastatin calcium versus placebo for the prevention of major adverse cardiovascular events. 22 Participants were enrolled in >100 sites across 12 countries during 2015 to 2019. Inclusion criteria included age ≥40 and ≤75 years, on any ART regimen for at least 6 months before study entry, CD4+ T‐cell count >100 cells/mm3, and fasting LDL‐C meeting specific thresholds depending on the 10‐year ASCVD risk score estimated by the 2013 American College of Cardiology/American Heart Association PCEs (Figure S1B). 22 Exclusion criteria included history of ASCVD and prior diagnoses of stable or unstable angina. Further details regarding inclusion and exclusion criteria and the design of REPRIEVE have been previously published. 20 , 22 As final data collection is ongoing in REPRIEVE, baseline data were used for these analyses.
Our CCTA analyses included a subset of 662 genotyped participants, of 805 total from the United States, who were enrolled into the REPRIEVE Mechanistic Substudy (Figure S1A). 20 , 24 In the Mechanistic Substudy, each participant received CCTA that quantified baseline subclinical CAD at 31 US sites. Enrollment was screened on the basis of willingness and absence of contraindications to CCTA (eg, known contrast reaction).
Each clinical research site obtained institutional review board/ethics committee approval and any other applicable regulatory entity approvals. Participants were provided with study information, including discussion of risks and benefits, and signed the approved declaration of informed consent. Secondary use of the data for the present analyses were approved by the Massachusetts General Hospital Institutional Review Board (protocol no. 2020P003693).
Genotyping and Polygenic Risk Score Calculation
Genotyping was performed using Illumina Infinium HTS (Illumina, Inc., San Diego, CA). From the original 729 804 variants, 625 869 variants (85.8%) remained after removing variants that were duplicates, multiallelic, >2% variant missingness, minor allele count <3, and located on the Y chromosome or mitochondrial genome. Twelve of the 4495 (0.27%) participants were noted to have either undefined self‐reported sex assigned at birth, >5% missingness, or heterozygosity > ± 4 SD from the mean; these participants were not excluded from analysis.
Genotype data were imputed using TOPMed via an online server (https://imputation.biodatacatalyst.nhlbi.nih.gov/) using the parameters: TOPMed r2 Reference Panel, GrCh37/hg19 Array Build, 0.001 rsq filter, and Eagle v2.4 Phasing. 25
A recently developed PRS for CAD, GPSMult, 26 was constructed using LDPred2, 27 incorporating the weighted effects of over 1.2 million single‐nucleotide polymorphisms from 58 genome‐wide association studies for CAD (>222 000 cases and >914 000 controls), other atherosclerotic diseases, and their risk factors from multiancestry cohorts external to REPRIEVE, as previously described. 26 The scoring weights were applied to the TOPMed 25 imputed genotype of each REPRIEVE participant using the Plink software 28 to obtain raw polygenic scores for CAD.
The PRS for lipid profiles and blood pressure for each REPRIEVE participant was constructed by applying publicly available scoring weights (https://www.pgscatalog.org/publication/PGP000332/), 29 externally derived from the UK Biobank, 30 to the TOPMed imputed genotype of each REPRIEVE participant using the Plink software to obtain raw polygenic scores. The specific weights for each trait are: LDL‐C (PGS002654), high‐density lipoprotein cholesterol (HDL‐C; PGS002646), total cholesterol (PGS002669), triglyceride (PGS002670) concentrations, and systolic blood pressure (PGS002666) and diastolic blood pressure (PGS002639).
Final PRS values were residualized, population structure adjusted, and standardized, as is standard for the field. 31 , 32 This is accomplished by calculating the residuals from multidimensional linear regression with raw PRS as the outcome, and covariates using 10 principal components derived from principal component analysis 13 of the genotype from the entire study population. The rationale is that adjustment for these principal components minimizes genetic association test confounding caused by population stratification. 33 , 34
Study Outcomes
Baseline characteristics included age, sex (assigned at birth), self‐reported ancestry, body mass index, smoking history, physical activity, and diet (physical activity and diet assessments were conducted using the Rapid Eating and Activity Assessment for Patients Questionnaire). 35 Laboratory measurements included CD4 count 36 and serum concentrations of total cholesterol, LDL‐C, HDL‐C, oxidized LDL‐C, triglycerides, and glucose. Ancestry was defined to be the estimated genetic similarity based on self‐report or computationally derived values, determined using the following strategy: participants were assigned to African, European, South Asian, or East Asian ancestry on the basis of self‐report, and others (ie, “Unknown,” “Other,” “American Indian,” “More Than One Race,” or “Asian” without specifying either South or East Asian), assigned to 1 of the 4 ancestries using the nearest neighbor classification algorithm to the principal component of genetic ancestry 13 from the 1000 Genomes Project. 37
A subset of participants included in our analyses had undergone CCTA as part of the REPRIEVE Mechanistic Substudy. The computed tomographic imaging was performed using a 64‐slice or greater computed tomographic scanner at the enrolled sites. 20 CAD was assessed in a standardized fashion by a central core laboratory with expert readers blinded to clinical and treatment information with excellent reproducibility. 20 Measurements of subclinical CAD included coronary artery calcium scores, and the presence of plaque, stenosis >50%, noncalcified plaque, and vulnerable plaque features defined by the presence of any 1 of 3 phenotypes: positive remodeling (remodeling index >1.1), low attenuation (<30 Hounsfield units), and napkin‐ring sign (low central attenuation with ringlike peripheral high attenuation). 24 , 38 Measurements also include computed tomographic Leaman scores, which account for the degree of stenosis, coronary dominance, plaque location, and composition. 39 All measurements were compared in the entire cohort and in subgroups on the basis of demographics, 10‐year ASCVD risk score, and other baseline features. 20
Statistical Analysis
Distribution of GPSMult percentiles were determined among cases and controls for each subclinical CAD phenotype and compared using the 2‐sided Student's t test. Participants were also subgrouped by low (<5%), borderline (5%–7.5%), or intermediate (>7.5%) ASCVD risk on the basis of the PCE 10‐year estimated risk. Within each subgroup, Leaman scores were compared between participants in the top and bottom PRS tertile using the 2‐sided Wilcoxon rank‐sum test. Odds ratios (ORs) based on GPSMult for subclinical CAD, along with associated 95% CIs and P values, were computed using logistic regression; covariates were age, sex, and the first 10 principal components of ancestry. 12 , 40 All reported ORs are per standard deviation in PRS; all PRSs are standardized (centered around mean with a unit SD) in this study. GPSMult was also associated with plaque presence by subgroups distinguished by sex, age, 10‐year ASCVD risk, CD4 nadir, 36 and ancestry. Comparisons between GPSMult and Leaman score or natural logarithm of the coronary artery calcium score+1 were performed using linear regression analysis (adjusting for age, sex, and top 10 principal components derived from genotype), and by comparing the average concentration per quintile of GPSMult. Statistical testing for differences in OR between subgroups was determined by a multiple‐degrees‐of‐freedom test for heterogeneity using metagen in R (R Foundation for Statistical Computing, Vienna, Austria). The variance explained by each putative CVD risk factor (GPSMult, sex, age, ancestry, use of antihypertensive medication, smoking status, systolic blood pressure [SBP], and baseline concentrations of LDL‐C, HDL‐C, and glucose) for predicting plaque presence was determined using Nagelkerke pseudo R 2 and associated P values, using the Nagelkerke function in R. Area under the receiver operating characteristic curve, or C‐statistic, for logistic regression to predict subclinical CAD phenotypes was determined using the Cstat function in R, with 95% CIs calculated with bootstrapping using the boot function. Comparisons between LDL‐C PRS and LDL‐C or oxidized LDL‐C concentrations were performed using linear regression analysis (adjusting for age, sex, and top 10 principal components derived from genotype), and by comparing the average concentration per percentile of LDL‐C PRS. Similar comparisons were conducted for HDL‐C, total cholesterol, triglycerides, SBP, and diastolic blood pressure.
Results
Overall participant characteristics among participants enrolling from AIDS Clinical Trial Group sites in REPRIEVE are shown in Table S1. Among the cohort of 4495 participants in REPRIEVE with polygenic risk data available (Figure S1A), the mean age at study enrollment was 49.9 [SD, 6.4] years. A total of 1652 (36.8%) participants were women, and 2327 (51.8%) were of African ancestry, 1118 (24.9%) of European ancestry, 600 (13.3%) of East Asian ancestry, and 450 (10.0%) of South Asian ancestry (Table S2). The median estimated 10‐year ASCVD risk was 3.9% (interquartile range, 1.7–6.6). At the time of enrollment, there were several differences in baseline characteristics between participants among different traditional 10‐year ASCVD risk categories (low, 0%–<5% [n=2644, 58.8%], borderline, 5%–7.5% [n=1040, 23.1%], and intermediate, >7.5% [n=804, 17.9%]) by the PCEs. 41 Patients with higher ASCVD risk were more likely to be older; men; smokers; hypertensive; of African ancestry; and have higher triglyceride and glucose concentrations and lower HDL‐C, CD4 count, and estimated glomerular filtration rate. 42 There were no differences in physical activity, body mass index, and LDL‐C concentration at the time of enrollment among patients in different ASCVD risk tertiles.
Among our studied subset of 662 participants with both genetic and CCTA data (Figure S1A), the mean age at study enrollment was 50.9 [SD, 5.8] years. A total of 106 (16.1%) participants were women, 277 (41.8%) were of African ancestry, 379 (57.3%) of European ancestry, 6 (0.9%) of East Asian ancestry, and 1 (0.2%) of South Asian ancestry (Table). The median estimated 10‐year ASCVD risk was 4.5% interquartile range, 2.6–6.8). Similar to the general cohort, there were several differences in baseline characteristics when categorized by ASCVD risk tertiles. With respect to plaque characteristics, 325 (49.2%) had evidence of plaque, 268 (40.6%) had visible noncalcified plaque segments, 24 (3.7%) had stenosis >50%, and 153 (23.2%) had plaque with vulnerable features, including 149 (22.6%) with positive remodeling, 40 (6.1%) with low attenuation, and 22 (3.3%) with a napkin‐ring sign on CCTA. As expected, participants with higher ASCVD risk exhibited more evidence of subclinical atherosclerosis in this genotyped subset (Table). 24 , 41
Table .
Baseline Characteristics for Participants With CCTA Measurements Stratified by ASCVD Category
| Variable | All participants (N=662), mean (SD) | ASCVD risk 0% to <5% (n=368), mean (SD) | ASCVD risk 5%–7.5% (n=162), mean (SD) | ASCVD risk >7.5% (n=130), mean (SD) | P value |
|---|---|---|---|---|---|
| Age, y | 50.9 (5.8) | 48.6 (4.7) | 52.5 (5.5) | 55.1 (5.8) | <0.0001 |
| Body mass index, kg/m2 | 27.2 (4.4) | 27.3 (4.4) | 27.1 (4.0) | 27.4 (4.7) | 0.515 |
| Total years smoked | 21.5 (12.2) | 19.1 (11.0) | 21.1 (11.7) | 26.3 (13.7) | <0.0001 |
| CD4 count, cells/mm3 | 628.9 (268.9) | 633.1 (278.5) | 624.1 (259.8) | 623.2 (253.9) | 0.984 |
| eGFR, mL/min | 88.7 (16.4) | 89.4 (16.1) | 88.5 (16.3) | 86.8 (17.0) | 0.123 |
| LDL‐C concentration, mg/dL | 107.4 (30.3) | 106.2 (30.1) | 111.8 (32.5) | 105.8 (27.8) | 0.977 |
| HDL‐C concentration, mg/dL | 50.1 (17.7) | 52.4 (17.7) | 48.0 (19.4) | 46.2 (14.6) | 0.0008 |
| Triglyceride concentration, mg/dL | 134.3 (83.4) | 124.3 (73.5) | 146.5 (97.5) | 147.7 (87.4) | 0.0037 |
| Glucose concentration, mg/dL | 93.0 (12.1) | 92.2 (12.2) | 92.8 (11.6) | 95.4 (12.4) | 0.0078 |
| Variable | All participants, n/N (%) | ASCVD risk 0% to <5%, n/N (%) | ASCVD risk 5%–7.5%, n/N (%) | ASCVD risk >7.5%, n/N (%) |
|---|---|---|---|---|
| Sex | ||||
| Female | 106/660 (16.1) | 90/368 (24.5) | 13/162 (8) | 3/130 (2.3) |
| Male | 554/660 (83.9) | 278/368 (75.5) | 149/162 (92) | 127/130 (97.7) |
| Ancestry | ||||
| African | 277/662 (41.8) | 129/368 (35.1) | 70/162 (43.2) | 77/130 (59.2) |
| European | 379/662 (57.3) | 234/368 (63.6) | 92/162 (56.8) | 52/130 (40) |
| East Asian | 6/662 (0.9) | 5/368 (1.4) | 0/162 (0) | 1/130 (0.8) |
| South Asian | 1/662 (0.2) | 0/368 (0) | 0/162 (0) | 0/130 (0) |
| Hypertension | 206/662 (31.1) | 79/368 (21.5) | 52/162 (32.1) | 75/130 (57.7) |
| Physical activity | ||||
| Poor | 220/655 (33.6) | 117/364 (32.1) | 54/161 (33.5) | 49/130 (37.7) |
| Intermediate | 357/655 (54.5) | 205/364 (56.3) | 84/161 (52.2) | 68/130 (52.3) |
| Ideal | 78/655 (11.9) | 42/364 (11.5) | 23/161 (14.3) | 13/130 (10) |
| Diet | ||||
| Poor | 179/659 (27.2) | 92/368 (25) | 49/162 (30.2) | 38/129 (29.5) |
| Intermediate | 355/659 (53.9) | 192/368 (52.2) | 89/162 (54.9) | 74/129 (57.4) |
| Ideal | 125/659 (19) | 84/368 (22.8) | 24/162 (14.8) | 17/129 (13.2) |
| Plaque characteristics | ||||
| Any plaque | 325/660 (49.2) | 147/368 (39.9) | 91/162 (56.2) | 87/130 (66.9) |
| Stenosis >50% | 24/650 (3.7) | 7/366 (1.9) | 8/159 (5) | 9/125 (7.2) |
| Calcified plaque | 117/660 (17.7) | 45/368 (12.2) | 34/162 (21) | 38/130 (29.2) |
| Plaque with vulnerable features | 153/660 (23.2) | 66/368 (17.9) | 38/162 (23.5) | 49/130 (37.7) |
| Low‐attenuation plaque | 40/660 (6.1) | 13/368 (3.5) | 10/162 (6.2) | 17/130 (13.1) |
| Partially calcified plaque | 229/660 (34.7) | 104/368 (28.3) | 66/162 (40.7) | 59/130 (45.4) |
| Noncalcified plaque | 73/660 (11.1) | 35/368 (9.5) | 20/162 (12.3) | 18/130 (13.8) |
| Visible noncalcified plaque segments | 268/660 (40.6) | 123/368 (33.4) | 77/162 (47.5) | 68/130 (52.3) |
| Visible noncalcified plaque or plaque with vulnerable features | 282/660 (42.7) | 132/368 (35.9) | 79/162 (48.8) | 71/130 (54.6) |
| Napkin‐ring sign | 22/660 (3.3) | 6/368 (1.6) | 5/162 (3.1) | 11/130 (8.5) |
| Plaque present | 325/660 (49.2) | 147/368 (39.9) | 91/162 (56.2) | 87/130 (66.9) |
| Positive remodeling | 149/660 (22.6) | 65/368 (17.7) | 37/162 (22.8) | 47/130 (36.2) |
Table of baseline characteristics for 662 participants with CCTA measurements, including stratification, into ASCVD tertiles (0% to <5%, 5%–7.5%, and >7.5%). The top section of baseline characteristics (rows Age to Triglyceride concentration) lists the mean (SD) value among all participants, in addition to stratification by ASCVD tertiles. P value corresponds to Student's t test between <5% and >7.5% ASCVD risk cohorts. The bottom section of baseline characteristics (rows Sex to Plaque characteristics) counts the number of participants in each variable subcategory among all participants, in addition to stratification by ASCVD tertiles. ASCVD indicates atherosclerotic cardiovascular disease; CCTA, coronary computed tomography angiography; eGFR, estimated glomerular filtration rate; HDL‐C, high‐density lipoprotein cholesterol; and LDL‐C, low‐density lipoprotein cholesterol.
We calculated the PRS for CAD, GPSMult, 26 for 662 participants in the Mechanistic Substudy. Across multiple measures of subclinical CAD, participants with evidence of CAD had a significantly higher PRS percentile compared with participants without evidence of CAD (Figure 1A). The average PRS percentile among participants with plaque present on CCTA was 53.2 compared with 45.0 among participants without plaque (P=2.9×10−4), and similarly 56.8 versus 48.1 for noncalcified plaque (P=0.017), 55.2 versus 47.2 (P=0.0027) for plaque with vulnerable features, 54.2 versus 45.2 for visible noncalcified plaque or plaque with vulnerable features (P=8.4×10−5), and 66.1 versus 48.2 for stenosis >50% (P=0.0033) (Figure 1A; Table S3). While high CAD PRS was significantly associated with the average Leaman score only among those with low traditional clinical risk (0%–<5%) ASCVD category, enrichment was not significantly different across borderline and intermediate risk categories (Figure 1B). We used logistic regression to calculate the association between plaque presence and GPSMult (Table S4). Overall, GPSMult was associated with plaque presence on CCTA with an OR of 1.42 per SD in GPSMult (95% CI, 1.20–1.68; P=3.8×10−5; Figure 2). The significant association between GPSMult and plaque presence was consistent across multiple important demographic and clinical subgroups, including those of male sex at birth, ages <50 and ≥50 years, ASCVD risk <5%, CD4 nadirs of 50 to 199 cells/μL and 200 to 249 cells/μL, and European and African ancestries (Figure 2).
Figure 1. Associations between GPSMult and measures of subclinical CAD.
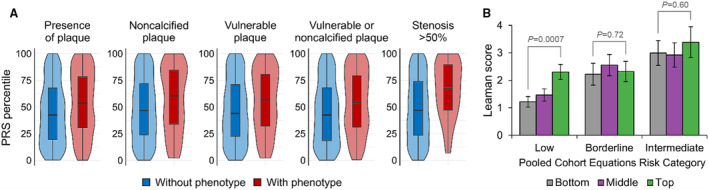
A, Violin plots with box and whiskers showing the distribution of GPSMult percentiles among participants either negative (blue) or positive (red) for 5 measures of subclinical CAD. The whiskers represent maximum and minimum, box span from first to third quartiles, and the middle line represents median. B, Among participants stratified at low (<5%), borderline (5%–7.5%), or intermediate (>7.5%) risk by PCEs, the plot of the average Leaman score between participants was determined to be in the bottom (<33.3%), middle (33.3%–66.7%), and top (>66.7%) tertiles of CAD PRS, respectively. The difference between CAD tertiles was significant (P=7.2×10−4) for the low PCE risk category (n=365) but not significant between tertiles in the borderline (n=159) and intermediate (n=125) categories (P=0.72 and P=0.60, respectively). CAD indicates coronary artery disease; and PCEs, pooled cohort equations.
Figure 2. Associations between GPSMult and plaque presence, by participant subgroups.
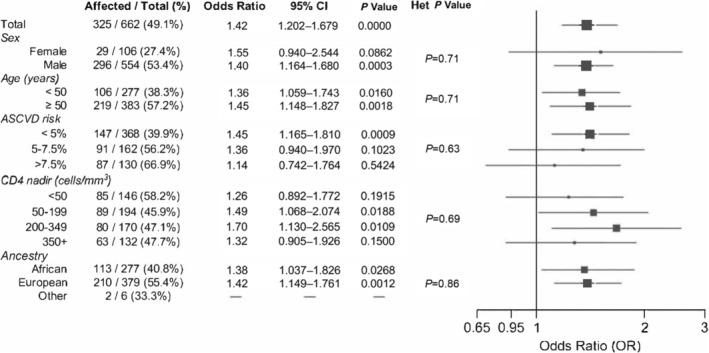
Among participants with CCTA measurements, the table (left) and forest plot (right) show the OR and associated P value for the association between CAD PRS and presence of plaque. Plot includes the OR after categorizing participants on the basis of sex, age, 10‐year ASCVD risk, nadir CD4 count, and ancestry. Het P Value lists the P values from the multiple‐degrees‐of‐freedom test for heterogeneity for different subgroup ORs of each category. ASCVD indicates atherosclerotic cardiovascular disease; CAD, coronary artery disease; CCTA, coronary computed tomography angiography; OR, odds ratio; and PRS, polygenic risk score.
Among the participants of the Mechanistic Substudy, GPSMult was significantly associated with multiple subclinical CAD features detected by CCTA, particularly certain high‐risk features (Figure 3A). Such features included stenosis >50% with an OR of 2.39 per SD in GPSMult (95% CI, 1.48–3.85; P=3.4×10−4) and visible noncalcified plaque or plaque with vulnerable features (OR, 1.45 [95% CI, 1.23–1.72]; P=9.6×10−6). Furthermore, GPSMult was linearly associated with Leaman scores (0.402 per SD in GPSMult; P=1.2×10−4) and the natural logarithm of coronary artery calcium scores (0.308 per SD in GPSMult; P=4.0×10−5; Table S5). This was consistent with significant differences in Leaman or natural logarithm coronary artery calcium scores (P=0.0088 and P=0.012, respectively) between those in the top and bottom quintiles in GPSMult (Figure 3B and 3C).
Figure 3. Associations between GPSMult and different measures of subclinical CAD.
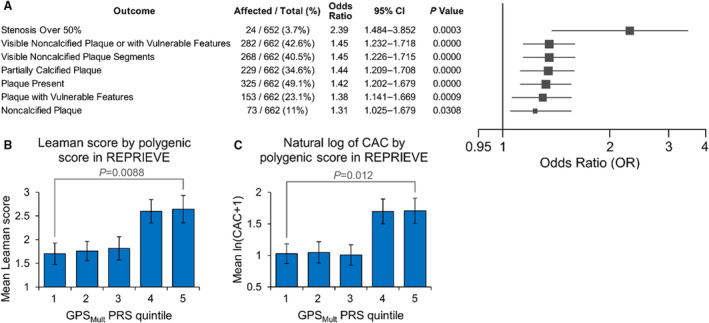
A, Among participants with CCTA measurements, the table (left) and forest plot (right) show the OR and associated P value for the association between CAD PRS and different measures of subclinical CAD along each row. Vulnerable plaque features include napkin‐ring sign, low attenuation plaques, and positive remodeling. The mean of the Leaman score (B) and natural logarithm (ln) of theCAC score+1 (C), by quintile of GPSMult. Error bars correspond to the standard error. Significant difference in Leaman score and ln(CAC+1) between the top and bottom quintiles of PRS (P=0.0088 and P=0.012, respectively), with the Wilcoxon rank‐sum test. CAC indicates coronary artery calcium; CAD, coronary artery disease; CCTA, coronary computed tomography angiography; OR, odds ratio; PRS, polygenic risk score; and REPRIEVE, Randomized Trial to Prevent Vascular Events in HIV.
In addition to the aforementioned metrics for association, the variance explained by GPSMult was significant for multiple measures of subclinical CAD, with Nagelkerke “pseudo” R 2=0.027 (P=4.3×10−4) for plaque presence (Figure S2A through S2C). Variance explained by GPSMult was especially strong for high‐risk CAD phenotypes, often comparable with many established ASCVD risk factors. For predicting noncalcified or vulnerable plaques, GPSMult had a variance explained of R 2=0.029 (P=2.4×10−4), compared with 0.056 (P=2.8×10−7) for age, 0.031 (P=1.5×10−4) for sex, 0.038 (P=2.6×10−5) for LDL‐C, 0.016 (P=0.0065) for use of antihypertensive medication, and 0.032 (P=0.0020) for smoking status. For predicting stenosis >50%, GPSMult had a variance explained of R 2=0.099 (P=1.0×10−4), compared with 0.049 (P=0.0075) for age, 0.071 (P=0.0011) for sex, and 0.062 (P=0.028) for smoking status.
Next, we determined the ability of GPSMult to discriminate for subclinical CAD over PCE, the standard for ASCVD risk prediction, using the area under the receiver operating characteristic curve, or C‐statistic. 43 GPSMult (with age and sex) performed better than PCEs, and combining GPSMult with PCEs led to an improved C‐statistic for presence of plaque of 0.734, a significant increase of 0.016 (95% CI, 0.003–0.033) from 0.718 with PCE alone (Figure 4A and 4B). The combined C‐statistic was 0.700 for visible noncalcified plaque or plaque with vulnerable features, and 0.819 for stenosis >50%, an increase of 0.024 (95% CI, 0.0045–0.045) and 0.065 (95% CI, 0.0076–0.113), respectively, from C‐statistics with PCEs alone. Similar findings were obtained for noncalcified or vulnerable plaque and stenosis >50% (Figure 4A and 4B). Finally, the change in C‐statistic with leave‐one‐out of either GPSMult or each component of the PCE exhibited comparable results with that of Nagelkerke pseudo R 2 (Figure 4C through 4E, S2D). 36
Figure 4. Importance of different CAD risk factors in predicting subclinical CAD.
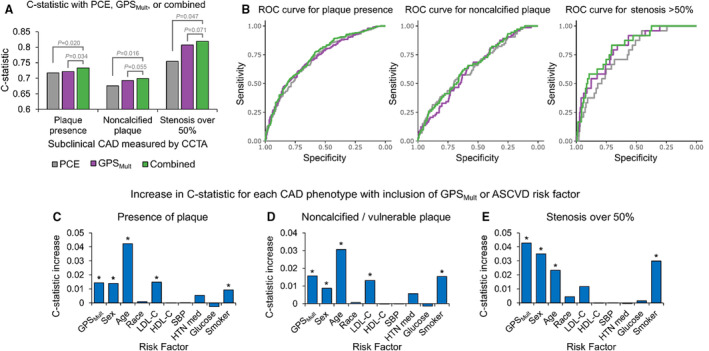
A, Plot of C‐statistics (y axis) for 3 different subclinical CAD phenotypes (x axis) with GPSMult (with age and sex) alone, PCE alone, or GPSMult with PCE. Significance values determined using bootstrapping. B, ROC curves corresponding to the same outcome and color labels shown in (A) (which report on area under the ROC, or C‐statistic). C through E, Plots of increase in C‐statistic (y axis), from a model with leave‐one‐out of GPSMult or each nongenetic ASCVD risk factor (x axis), compared with a combined model with both GPSMult and nongenetic factors, for 3 different subclinical CAD phenotypes (each graph). An asterisk above the bar indicates significant increase at P<0.05, determined using bootstrapping. Figure S2D and S2E lists the corresponding plot values and P values, respectively. CAD indicates coronary artery disease; HDL‐C, high‐density lipoprotein cholesterol; HTN med, antihypertensive medication; LDL‐C, low‐density lipoprotein cholesterol; PCE, pooled cohort equation; ROC, receiver operating characteristic; and SBP, systolic blood pressure.
Next, we evaluated the performance of polygenic scores in predicting other cardiometabolic traits in this cohort of PWH. We calculated the PRS for LDL‐C using weights previously derived from PolyPred, 29 then compared them with fasting LDL‐C and oxidized LDL‐C concentrations obtained at baseline. LDL‐C PRS was linearly associated with fasting LDL‐C (8.86 mg/dL per SD in PRS, P=1.04×10−86) and oxidized LDL‐C concentrations (2.97 mg/dL per SD, P=1.76×10−4) (Figure 5A and 5B; Table S6). The difference in average LDL‐C concentrations between the top and bottom percentiles of PRS was 44.8 mg/dL, and 19.9 mg/dL for oxidized LDL‐C concentrations (Figure 5C and 5D). In addition, LDL‐C PRS was associated with presence of plaque 24 with an OR of 1.21 (95% CI, 1.01–1.44; P=0.039) and partially calcified plaque with OR of 1.21 (95% CI, 1.01–1.45; P=0.041) per SD in PRS (Figure S3A and S3B).
Figure 5. Associations between LDL‐C PRS and LDL‐C or oxidized LDL‐C concentrations.
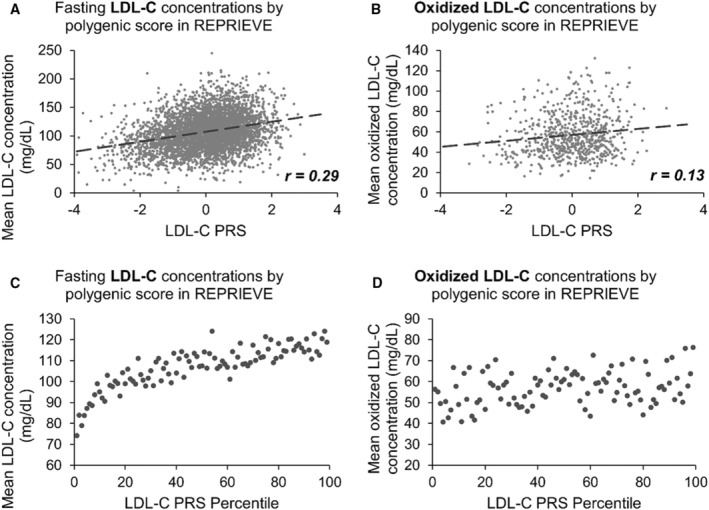
A, Plot of the baseline LDL‐C concentration (mg/dL) with corresponding LDL‐C PRS for each participant (x axis). Correlation coefficient of 0.29. B, Plot of the baseline oxidized LDL‐C concentration (mg/dL) with corresponding LDL‐C PRS for each participant (x axis). Correlation coefficient of 0.13. C, Plot of the mean baseline LDL‐C concentration (mg/dL), among participants in each LDL‐C PRS percentile (x axis). D, Plot of the mean baseline oxidized LDL‐C concentration (mg/dL), among participants in each LDL‐C PRS percentile (x axis). HDL‐C indicates high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; PRS, polygenic risk score; and REPRIEVE, Randomized Trial to Prevent Vascular Events in HIV.
We calculated PRS for total cholesterol, total triglycerides, and HDL‐C, and compared them with the respective baseline values for each participant. We found linear associations between PRSs for these 3 traits and the measured baseline values, with 9.23 mg/dL (P=7.76×10−69) for total cholesterol, 14.77 mg/dL for triglycerides (P=4.06×10−36), and 4.52 mg/dL (P=3.06×10−79) for HDL‐C per SD of their respective PRS (Table S6; Figure S4A through S4C). In addition, we performed the same analysis on SBP and diastolic blood pressure, finding linear associations between PRSs and measured baseline values for both SBP and diastolic blood pressure, with 2.06 mmHg (P=3.70×10−22) and 1.26 mm Hg (P=2.84×10−18) per SD of their respective PRS (Figure S4D and S4E). Unlike for LDL‐C PRS, there were no significant associations between PRS for total cholesterol, triglycerides, HDL‐C, SBP, or diastolic blood pressure, and subclinical CAD phenotypes (Table S7).
Discussion
Individuals with HIV remain at higher risk for ASCVD despite effective viral suppression using ART. Improved prediction of ASCVD risk in this vulnerable population, such as by accounting for cumulative genetic risk through PRS, may lead to better clinical care. In this study, we demonstrated that PRS for CAD and other cardiometabolic traits, which have been optimized for the general population, are applicable to a global cohort of participants with HIV without previously documented ASCVD and at low‐to‐moderate traditional estimated risk. Our findings support the applicability and potential clinical use of polygenic scores for cardiometabolic traits in individuals with HIV in several ways.
First, cardiometabolic PRSs derived from individuals without HIV identify PWH who have subclinical CAD, and this risk was consistent across subgroups by key sociodemographics, including age, sex, 10‐year ASCVD risk, ancestry, and HIV‐specific features. This association between PRS and CAD was particularly strong with more severe CAD phenotypes such as stenosis >50% and noncalcified plaque or plaque with vulnerable features, which are each strongly associated with incident major adverse cardiovascular events. 39 , 44 Together, these results demonstrate that, while HIV provides strong acquired stimuli for atherogenesis, genetic predisposition still also plays a role in atherogenesis among PWH.
Second, our population of PWH, those without documented evidence of ASCVD and calculated to have low‐to‐medium traditional risk for future clinical events, is unique and warrants specialized study. PWH exhibits excess and unique ASCVD characteristics, such as increased noncalcified plaque, suggesting additional pathophysiology at play in driving excess cardiovascular risk. 5 , 6 Because they are categorized as low‐to‐medium ASCVD risk, combined with increased discrimination and disparities in PWH accessing medical care, this population is highly vulnerable to be missed by screening measures for primary prevention. 45 , 46 By showing that existing polygenic scores effectively identify those with the greatest subclinical atherosclerosis in this population, our study supports clinical use of PRS for primary prevention of CVD in this vulnerable population.
Third, this is the most diverse population evaluated for cardiometabolic PRS in PWH and the largest study to directly measure subclinical CAD in a subset of participants. The 4495 genotyped participants enrolled in REPRIEVE are of majority non‐European ancestry, of whom 662 also have subclinical CAD measurements from CCTA, enabling comparison between PRS and direct measures of CAD. A related study of 6284 PWH from the Centers for AIDS Research Network of Integrated Clinical Systems found correlations between PRSs for lipid traits and corresponding concentrations in serum, but we now extend insights to CAD PRSs and for subclinical CAD. 47 Another study in 345 PWH from the Swiss HIV cohort study found CAD PRSs to be associated with subclinical CAD presence alone from CCTA but restricted to a cohort of exclusively European ancestry. 48 , 49 To our knowledge, our study is the first to demonstrate that a new multiancestry PRS 26 can reliably predict multiple measures of subclinical CAD, especially high‐risk plaque phenotypes, among PWH from diverse ancestries.
Fourth, our results highlight the importance of PRSs as an orthogonal and additive modality to traditional risk factors and algorithms such as PCEs in predicting ASCVD risk among asymptomatic PWH. In our cohort, individuals of African ancestry had higher estimated traditional 10‐year ASCVD risk on the basis of PCEs, which are predominantly composed of nongenetic factors that would in theory dampen the strength of polygenic association. However, we found that GPSMult achieved comparable relative effects in estimating polygenic risk for CAD between participants of African and European ancestries, supporting its generalizability as an orthogonal modality for cardiovascular risk stratification in PWH. Furthermore, GPSMult was comparable with sex, age, LDL‐C concentration, and smoking status at predicting subclinical CAD presence. Notably, including GPSMult significantly increased the C‐statistic compared with PCEs alone for assessing ASCVD risk, attaining 0.73 for plaque presence and 0.82 for high‐risk plaque phenotypes. This is comparable with studies in cohorts without HIV; genome‐wide CAD PRSs with known CVD risk factors in a French‐Canadian cohort of over 11 000 participants yielded C‐statistic of 0.72–0.89 for prevalent CAD, 50 and in the ARIC (Atherosclerosis Risk in Communities) and MESA (Multiethnic Study of Atherosclerosis) cohorts (7237 total participants), yielded C‐statistics of 0.701 and 0.660, respectively. 51 Our results were also comparable with studies in PWH; genome‐wide CAD PRSs in the aforementioned Swiss HIV Cohort Study 48 , 49 yielded a C‐statistic of 0.75 for subclinical CAD plaques using nongenetic risk factors alone, which increased to 0.78 with inclusion of CAD PRSs. 48
Fifth, this study aids in identifying potential mechanisms for the elevated CVD risk in PWH, which can guide new therapeutic approaches. We found that genetic risk for elevated LDL‐C, quantified as LDL‐C PRS, was associated with presence of subclinical CAD. This finding supports a role for LDL‐C in elevating risk for CAD in PWH, and that lowering LDL‐C, such as with a statin, 52 may be a reasonable strategy for primary prevention. This observation is consistent with the recently released topline results of REPRIEVE, which showed that LDL‐C‐lowering from pitavastatin calcium versus placebo led to a reduction in major adverse cardiovascular events among PWH. 53 , 54
PRSs have traditionally struggled in performance in non‐European ancestry groups, likely because currently available genome‐wide association study data sets to train PRSs are of predominantly European ancestry. 55 , 56 , 57 This study benefited from the use of GPSMult, which was recently invented to excel at multiancestry predictive performance, 26 and using it we were able to achieve comparable relative effects between participants of European and African ancestries. To further improve the applicability of PRSs, urgent efforts are still needed to further diversify genome‐wide association study data sets by recruiting a diverse population of individuals, particularly those of African ancestry and other populations currently underrepresented in genomics data sets.
This study is not without limitations. While the entire genotyped cohort was large and included >4000 global participants, GPSMult was studied only among those with measurements of subclinical CAD using CCTA, which is a smaller number of participants from the United States with more limited racial and ethnic diversity. Future studies with a greater number and more diverse participants receiving CCTA would improve generalizability and power in risk prediction for subclinical CAD. In addition, as the REPRIEVE 22 trial follow‐up is ongoing with final data collection under way, we are not yet able to examine associations with incident clinical cardiovascular outcomes. While we focus on polygenic risk from traditional ASCVD risk factors on cardiometabolic traits, this does not preclude the presence of other mechanisms that act to exacerbate CAD in PWH, such as increased inflammation, which has been shown to be associated with plaque in prior studies in the REPRIEVE cohort. 24 Future studies that evaluate the applicability of CAD PRSs to predict risk for major adverse cardiovascular events in PWH, as well as whether this risk is ameliorated with pharmacological, dietary, and lifestyle interventions, remains to be determined.
Conclusions
In a diverse cohort of patients with HIV without known CVD, high predicted polygenic risk for CAD was associated with increased prevalence of subclinical CAD. An LDL‐C PRS was also predictive of subclinical CAD, which is consistent with the role of LDL‐C in HIV‐associated coronary atherosclerosis. PRSs for other cardiometabolic traits including lipid profiles and blood pressure were also predictive of corresponding traits in this cohort. Altogether, our findings support the validity and potential clinical use of polygenic scores for cardiometabolic traits among PWH.
Sources of Funding
This study is supported through National Institutes of Health grants U01HL1233369 to the Clinical Coordinating Center, U01HL123339 to the Data Coordinating Center, as well as funding from Kowa Pharmaceuticals America, Inc, Gilead Sciences, and ViiV Healthcare. The National Institute of Allergy and Infectious Diseases supported this study through grants UM1 AI068636, which supports the AIDS Clinical Trials Group Leadership and Operations Center; and UM1 AI106701, which supports the AIDS Clinical Trial Group Laboratory Center. This work was supported by the KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst (to Dr Patel); grants K08HL168238 (to Dr Patel), R01HL151283 (to Dr Natarajan), R01HL148565 (to Dr Natarajan), R01HL148050 (to Dr Natarajan), from the National Heart, Lung, and Blood Institute; and grant 1U01HG011719 from the National Human Genome Research Institute (to Drs Patel and Natarajan). The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the the National Institutes of Health, National Heart, Lung, and Blood Institute, the National Institute of Allergy and Infectious Diseases, or the US Department of Health and Human Services.
Disclosures
Dr Bhattacharya reports support from the John S LaDue Fellowship in Cardiovascular Research and consulting fees from Casana Care Inc, unrelated to the submitted work. Dr Lu reports funding to his institution for the REPRIEVE trial from the National Heart, Lung, and Blood Institute (NHLBI) and Kowa, as well as unrelated funding to his institution from AstraZeneca, Ionis, Johnson & Johnson Innovation, and MedImmune. Dr Ribaudo reports grants from the National Institutes of Health (NIH)/NHLBI and Kowa Pharmaceuticals during the conduct of the study, as well as grants from NIH/National Institute of Allergy and Infectious Diseases; NIH/NHLBI; NIH/National Institute of Diabetes and Digestive and Kidney Diseases, and NIH/National Institute on Aging, outside the submitted work. Dr Zanni reports grant support through her institution from NIH/National Institute of Allergy and Infectious Diseases and Gilead Sciences, Inc., relevant to the conduct of the study, as well as grants from the NIH/National Institute of Allergy and Infectious Diseases andNIH/NHLBI outside the submitted work. Dr Grinspoon reports grant support through his institution from the NIH, Kowa Pharmaceuticals America, Inc., Gilead Sciences, Inc., and ViiV Healthcare for the conduct of the study; personal fees from Theratechnologies and ViiV; and service on the Scientific Advisory Board of Marathon Asset Management, all outside the submitted work. Dr Natarajan reports investigator‐initiated grants from Allelica, Amgen, Apple, AstraZeneca, Boston Scientific, Roche/Genentech, and Novartis; personal fees from Allelica, Apple, AstraZeneca, Blackstone Life Sciences, GV, HeartFlow, Foresite Labs, Novartis, Roche/Genentech, and Novartis; is a co‐founder of TenSixteen Bio, and a scientific advisory board member of Esperion Therapeutics, Preciseli, and TenSixteen Bio; and spousal employment at Vertex Pharmaceuticals, all unrelated to the present work. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S7.
Figures S1–S4.
Acknowledgments
The study investigators thank the study participants, site staff, and study‐associated personnel for their ongoing participation in the trial. In addition, the study investigators thank the following: the AIDS Clinical Trial Group for clinical site support; AIDS Clinical Trial Group Clinical Trials Specialists (Laura Moran, MPH and Jhoanna Roa, MD) for regulatory support; the data management center, Frontier Science Foundation, for data support; the Center for Biostatistics in AIDS Research for statistical support; and the Community Advisory Board for input for the community.
This manuscript was sent to Samuel S. Gidding, MD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.033413
For Sources of Funding and Disclosures, see page 11.
Contributor Information
Aniruddh P. Patel, Email: apatel32@mgh.harvard.edu.
Pradeep Natarajan, Email: pnatarajan@mgh.harvard.edu.
References
- 1. Global HIV & AIDS statistics — fact sheet. UNAIDS. 2024. Accessed February 15, 2024. https://www.unaids.org/en/resources/fact‐sheet
- 2. Shah ASV, Stelzle D, Lee KK, Beck EJ, Alam S, Clifford S, Longenecker CT, Strachan F, Bagchi S, Whiteley W, et al. Global burden of atherosclerotic cardiovascular disease in people living with HIV: systematic review and meta‐analysis. Circulation. 2018;138:1100–1112. doi: 10.1161/CIRCULATIONAHA.117.033369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sudano I, Spieker LE, Noll G, Corti R, Weber R, Lüscher TF. Cardiovascular disease in HIV infection. Am Heart J. 2006;151:1147–1155. doi: 10.1016/j.ahj.2005.07.030 [DOI] [PubMed] [Google Scholar]
- 4. Feinstein MJ, Hsue PY, Benjamin LA, Bloomfield GS, Currier JS, Freiberg MS, Grinspoon SK, Levin J, Longenecker CT, Post WS, et al. Characteristics, prevention, and management of cardiovascular disease in people living with HIV: a scientific statement from the American Heart Association. Circulation. 2019;140:e98–e124. doi: 10.1161/CIR.0000000000000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Longenecker CT, Sullivan C, Baker JV. Immune activation and cardiovascular disease in chronic HIV infection. Curr Opin HIV and AIDS. 2016;11:216–225. doi: 10.1097/COH.0000000000000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zanni MV, Schouten J, Grinspoon SK, Reiss P. Risk of coronary heart disease in patients with HIV infection. Nat Rev Cardiol. 2014;11:728–741. doi: 10.1038/nrcardio.2014.167 [DOI] [PubMed] [Google Scholar]
- 7. Topel ML, Shen J, Morris AA, Al Mheid I, Sher S, Dunbar SB, Vaccarino V, Sperling LS, Gibbons GH, Martin GS, et al. Comparisons of the Framingham and pooled cohort equation risk scores for detecting subclinical vascular disease in blacks versus whites. Am J Cardiol. 2018;121:564–569. doi: 10.1016/j.amjcard.2017.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaikh K, Bhondoekhan F, Haberlen S, Nakanishi R, Roy SK, Alla VM, Brown TT, Lee J, Osawa K, Almeida S, et al. Coronary artery plaque progression and cardiovascular risk scores in men with and without HIV‐infection. AIDS. 2022;36:215–224. doi: 10.1097/QAD.0000000000003093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soares C, Kwok M, Boucher K‐A, Haji M, Echouffo‐Tcheugui JB, Longenecker CT, Bloomfield GS, Ross D, Jutkowtiz E, Sullivan JL, et al. Performance of cardiovascular risk prediction models among people living with HIV: a systematic review and meta‐analysis. JAMA Cardiol. 2023;8:139–149. doi: 10.1001/jamacardio.2022.4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangino M, Spector T. Understanding coronary artery disease using twin studies. Heart. 2013;99:373–375. doi: 10.1136/heartjnl-2012-303001 [DOI] [PubMed] [Google Scholar]
- 11. Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018;19:581–590. doi: 10.1038/s41576-018-0018-x [DOI] [PubMed] [Google Scholar]
- 12. Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang M, Menon R, Mishra S, Patel AP, Chaffin M, Tanneeru D, Deshmukh M, Mathew O, Apte S, Devanboo CS, et al. Validation of a genome‐wide polygenic score for coronary artery disease in South Asians. J Am Coll Cardiol. 2020;76:703–714. doi: 10.1016/j.jacc.2020.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klarin D, Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nat Rev Cardiol. 2022;19:291–301. doi: 10.1038/s41569-021-00638-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Sullivan JW, Ashley EA, Elliott PM. Polygenic risk scores for the prediction of cardiometabolic disease. Eur Heart J. 2023;44:89–99. doi: 10.1093/eurheartj/ehac648 [DOI] [PubMed] [Google Scholar]
- 16. Patel AP, Khera AV. Advances and applications of polygenic scores for coronary artery disease. Ann Rev Med. 2023;74:141–154. doi: 10.1146/annurev-med-042921-112629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Surakka I, Wolford BN, Ritchie SC, Hornsby WE, Sutton NR, Elvenstad Gabrielsen M, Skogholt AH, Thomas L, Inouye M, Hveem K, et al. Sex‐specific survival bias and interaction modeling in coronary artery disease risk prediction. Circ Genom Precis Med. 2023;16:e003542. doi: 10.1161/CIRCGEN.121.003542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marston NA, Pirruccello JP, Melloni GEM, Koyama S, Kamanu FK, Weng L‐C, Roselli C, Kamatani Y, Komuro I, Aragam KG, et al. Predictive utility of a coronary artery disease polygenic risk score in primary prevention. JAMA Cardiol. 2023;8:130–137. doi: 10.1001/jamacardio.2022.4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffmann U, Lu MT, Olalere D, Adami EC, Osborne MT, Ivanov A, Aluru JS, Lee S, Arifovic N, Overton ET, et al. Rationale and design of the Mechanistic Substudy of the Randomized Trial to Prevent Vascular Events in HIV (REPRIEVE): effects of pitavastatin on coronary artery disease and inflammatory biomarkers. Am Heart J. 2019;212:1–12. doi: 10.1016/j.ahj.2019.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Post WS, Budoff M, Kingsley L, Palella FJ, Witt MD, Li X, George RT, Brown TT, Jacobson LP. Associations between HIV infection and subclinical coronary atherosclerosis. Ann Intern Med. 2014;160:458–467. doi: 10.7326/M13-1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grinspoon SK, Fitch KV, Overton ET, Fichtenbaum CJ, Zanni MV, Aberg JA, Malvestutto C, Lu MT, Currier JS, Sponseller CA, et al. Rationale and design of the Randomized Trial to Prevent Vascular Events in HIV (REPRIEVE). Am Heart J. 2019;212:23–35. doi: 10.1016/j.ahj.2018.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grinspoon SK, Douglas PS, Hoffmann U, Ribaudo HJ. Leveraging a landmark trial of primary cardiovascular disease prevention in human immunodeficiency virus: introduction from the REPRIEVE Coprincipal investigators. J Infect Dis. 2020;222:S1–S7. doi: 10.1093/infdis/jiaa098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoffmann U, Lu MT, Foldyna B, Zanni MV, Karady J, Taron J, Zhai BK, Burdo T, Fitch KV, Kileel EM, et al. Assessment of coronary artery disease with computed tomography angiography and inflammatory and immune activation biomarkers among adults with HIV eligible for primary cardiovascular prevention. JAMA Network Open. 2021;4:e2114923. doi: 10.1001/jamanetworkopen.2021.14923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel AP, Wang M, Ruan Y, Koyama S, Clarke SL, Yang X, Tcheandjieu C, Agrawal S, Fahed AC, Ellinor PT, et al. A multi‐ancestry polygenic risk score improves risk prediction for coronary artery disease. Nat Med. 2023;29:1793–1803. doi: 10.1038/s41591-023-02429-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics. 2021;36:5424–5431. doi: 10.1093/bioinformatics/btaa1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot WJ, Khera AV, Okada Y, Martin AR, Finucane HK, Price AL. Leveraging fine‐mapping and multipopulation training data to improve cross‐population polygenic risk scores. Nat Genet. 2022;54:450–458. doi: 10.1038/s41588-022-01036-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khera AV, Chaffin M, Zekavat SM, Collins RL, Roselli C, Natarajan P, Lichtman JH, D'Onofrio G, Mattera J, Dreyer R, et al. Whole‐genome sequencing to characterize monogenic and polygenic contributions in patients hospitalized with early‐onset myocardial infarction. Circulation. 2019;139:1593–1602. doi: 10.1161/CIRCULATIONAHA.118.035658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hao L, Kraft P, Berriz GF, Hynes ED, Koch C, Kumar P KV, Parpattedar SS, Steeves M, Yu W, Antwi AA, et al. Development of a clinical polygenic risk score assay and reporting workflow. Nat Med. 2022;28:1006–1013. doi: 10.1038/s41591-022-01767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 34. Patel AP, Fahed AC. Pragmatic approach to applying polygenic risk scores to diverse populations. Curr Protoc. 2023;3:e911. doi: 10.1002/cpz1.911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Douglas PS, Umbleja T, Bloomfield GS, Fichtenbaum CJ, Zanni MV, Overton ET, Fitch KV, Kileel EM, Aberg JA, Currier J, et al. Cardiovascular risk and health among people with human immunodeficiency virus (HIV) eligible for primary prevention: insights from the REPRIEVE trial. Clin Infect Dis. 2021;73:2009–2022. doi: 10.1093/cid/ciab552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ellis RJ, Badiee J, Vaida F, Letendre S, Heaton RK, Clifford D, Collier AC, Gelman B, McArthur J, Morgello S, et al. CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS. 2011;25:1747–1751. doi: 10.1097/QAD.0b013e32834a40cd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi‐Yang Fritz M, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferencik M, Mayrhofer T, Bittner DO, Emami H, Puchner SB, Lu MT, Meyersohn NM, Ivanov AV, Adami EC, Patel MR, et al. Use of high‐risk coronary atherosclerotic plaque detection for risk stratification of patients with stable chest pain: a secondary analysis of the PROMISE randomized clinical trial. JAMA Cardiol. 2018;3:144–152. doi: 10.1001/jamacardio.2017.4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Araújo GP, Garcia‐Garcia HM, Dores H, Carvalho MS, Jerónimo Sousa P, Marques H, Ferreira A, Cardim N, Campante Teles R, Raposo L, et al. Coronary computed tomography angiography‐adapted Leaman score as a tool to noninvasively quantify total coronary atherosclerotic burden. Int J Cardiovasc Imaging. 2013;29:1575–1584. doi: 10.1007/s10554-013-0232-8 [DOI] [PubMed] [Google Scholar]
- 40. Aragam KG, Jiang T, Goel A, Kanoni S, Wolford BN, Atri DS, Weeks EM, Wang M, Hindy G, Zhou W, et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat Genet. 2022;54:1803–1815. doi: 10.1038/s41588-022-01233-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goff DC, Lloyd‐Jones DM, Bennett G, Coady S, D'Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O'Donnell CJ, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. Circulation. 2014;129:S49–S73. doi: 10.1161/01.cir.0000437741.48606.98 [DOI] [PubMed] [Google Scholar]
- 42. Fitch KV, McCallum SA, Erlandson KM, Overton ET, Zanni MV, Fichtenbaum C, Aberg JA, Fulda ES, Kileel EM, Moran LE, et al. Diet in a global cohort of adults with HIV at low‐to‐moderate traditional cardiovascular disease risk. AIDS. 2022;36:1997–2003. doi: 10.1097/QAD.0000000000003344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aragam KG, Natarajan P. Polygenic scores to assess atherosclerotic cardiovascular disease risk: clinical perspectives and basic implications. Circ Res. 2020;126:1159–1177. doi: 10.1161/CIRCRESAHA.120.315928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carrigan TP, Nair D, Schoenhagen P, Curtin RJ, Popovic ZB, Halliburton S, Kuzmiak S, White RD, Flamm SD, Desai MY. Prognostic utility of 64‐slice computed tomography in patients with suspected but no documented coronary artery disease. Eur Heart J. 2008;30:362–371. doi: 10.1093/eurheartj/ehn605 [DOI] [PubMed] [Google Scholar]
- 45. Sullivan PS, Satcher Johnson A, Pembleton ES, Stephenson R, Justice AC, Althoff KN, Bradley H, Castel AD, Oster AM, Rosenberg ES, et al. Epidemiology of HIV in the USA: epidemic burden, inequities, contexts, and responses. Lancet. 2021;397:1095–1106. doi: 10.1016/S0140-6736(21)00395-0 [DOI] [PubMed] [Google Scholar]
- 46. Ballocca F, Gili S, D'Ascenzo F, Marra WG, Cannillo M, Calcagno A, Bonora S, Flammer A, Coppola J, Moretti C, et al. HIV infection and primary prevention of cardiovascular disease: lights and shadows in the HAART era. Prog Cardiovasc Dis. 2016;58:565–576. doi: 10.1016/j.pcad.2016.02.008 [DOI] [PubMed] [Google Scholar]
- 47. Cheng H, Sewda A, Marquez‐Luna C, White SR, Whitney BM, Williams‐Nguyen J, Nance RM, Lee WJ, Kitahata MM, Saag MS, et al. Genetic architecture of cardiometabolic risks in people living with HIV. BMC Med. 2020;18:288. doi: 10.1186/s12916-020-01762-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schoepf IC, Thorball CW, Kovari H, Ledergerber B, Buechel RR, Calmy A, Weber R, Kaufmann PA, Nkoulou R, Schwenke JM, et al. Polygenic risk scores for prediction of subclinical coronary artery disease in persons with human immunodeficiency virus (HIV): the Swiss HIV cohort study. Clin Infect Dis. 2023;76:48–56. doi: 10.1093/cid/ciac758 [DOI] [PubMed] [Google Scholar]
- 49. Schoepf IC, Thorball CW, Ledergerber B, Engel T, Raffenberg M, Kootstra NA, Reiss P, Hasse B, Marzolini C, Thurnheer C, et al. Coronary artery disease–associated and longevity‐associated polygenic risk scores for prediction of coronary artery disease events in persons living with human immunodeficiency virus: the Swiss HIV cohort study. Clin Infect Dis. 2021;73:1597–1604. doi: 10.1093/cid/ciab521 [DOI] [PubMed] [Google Scholar]
- 50. Wünnemann F, Sin Lo K, Langford‐Avelar A, Busseuil D, Dubé M‐P, Tardif J‐C, Lettre G. Validation of genome‐wide polygenic risk scores for coronary artery disease in French Canadians. Circ Genom Precis Med. 2019;12:e002481. doi: 10.1161/CIRCGEN.119.002481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, Shaffer CM, Kundu S, Robinson‐Cohen C, Psaty BM, Rich SS, et al. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA. 2020;323:627–635. doi: 10.1001/jama.2019.21782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilt TJ, Bloomfield HE, MacDonald R, Nelson D, Rutks I, Ho M, Larsen G, McCall A, Pineros S, Sales A. Effectiveness of statin therapy in adults with coronary heart disease. Arch Intern Med. 2004;164:164. doi: 10.1001/archinte.164.13.1427 [DOI] [PubMed] [Google Scholar]
- 53. Daily statin reduces the risk of cardiovascular disease in people living with HIV, large NIH study finds. National Institutes of Health. April 11, 2023. Accessed February 15, 2024. https://www.nih.gov/news‐events/news‐releases/daily‐statin‐reduces‐risk‐cardiovascular‐disease‐people‐living‐hiv‐large‐nih‐study‐finds#:~:text=A%20planned%20interim%20analysis%20of,with%20those%20receiving%20a%20placebo
- 54. Grinspoon SK, Fitch KV, Zanni MV, Fichtenbaum CJ, Umbleja T, Aberg JA, Overton ET, Malvestutto CD, Bloomfield GS, Currier JS, et al. Pitavastatin to prevent cardiovascular disease in HIV infection. N Engl J Med. 2023;389:687–699. doi: 10.1056/NEJMoa2304146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kamiza AB, Toure SM, Vujkovic M, Machipisa T, Soremekun OS, Kintu C, Corpas M, Pirie F, Young E, Gill D, et al. Transferability of genetic risk scores in African populations. Nat Med. 2022;28:1163–1166. doi: 10.1038/s41591-022-01835-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, Feldman M, Peterson R, Domingue B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dikilitas O, Schaid DJ, Tcheandjieu C, Clarke SL, Assimes TL, Kullo IJ. Use of polygenic risk scores for coronary heart disease in ancestrally diverse populations. Curr Cardiol Rep. 2022;24:1169–1177. doi: 10.1007/s11886-022-01734-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S7.
Figures S1–S4.