

Abstract
INTRODUCTION
Amyloid beta and tau pathology are the hallmarks of sporadic Alzheimer's disease (AD) and autosomal dominant AD (ADAD). However, Lewy body pathology (LBP) is found in ≈ 50% of AD and ADAD brains.
METHODS
Using an α‐synuclein seed amplification assay (SAA) in cerebrospinal fluid (CSF) from asymptomatic (n = 26) and symptomatic (n = 27) ADAD mutation carriers, including 12 with known neuropathology, we investigated the timing of occurrence and prevalence of SAA positive reactivity in ADAD in vivo.
RESULTS
No asymptomatic participant and only 11% (3/27) of the symptomatic patients tested SAA positive. Neuropathology revealed LBP in 10/12 cases, primarily affecting the amygdala or the olfactory areas. In the latter group, only the individual with diffuse LBP reaching the neocortex showed α‐synuclein seeding activity in CSF in vivo.
DISCUSSION
Results suggest that in ADAD LBP occurs later than AD pathology and often as amygdala‐ or olfactory‐predominant LBP, for which CSF α‐synuclein SAA has low sensitivity.
Highlights
Cerebrospinal fluid (CSF) real‐time quaking‐induced conversion (RT‐QuIC) detects misfolded α‐synuclein in ≈ 10% of symptomatic autosomal dominant Alzheimer's disease (ADAD) patients.
CSF RT‐QuIC does not detect α‐synuclein seeding activity in asymptomatic mutation carriers.
Lewy body pathology (LBP) in ADAD mainly occurs as olfactory only or amygdala‐predominant variants.
LBP develops late in the disease course in ADAD.
CSF α‐synuclein RT‐QuIC has low sensitivity for focal, low‐burden LBP.
Keywords: alpha‐synuclein seed amplification assay, Dominantly Inherited Alzheimer Network, Lewy body pathology, real‐time quaking‐induced conversion
1. INTRODUCTION
Autosomal dominant Alzheimer's disease (ADAD) is a rare form of Alzheimer's disease (AD) that arises from mutations in the genes encoding presenilin 1 (PSEN1), presenilin 2 (PSEN2), or amyloid precursor protein (APP), all affecting APP processing. 1 The hallmarks of sporadic AD (sAD) and ADAD are the accumulation of extracellular amyloid beta plaques and the aggregation of hyperphosphorylated tau proteins inside neurons, leading to the progressive loss of synapses and neurons. 2 However, AD brains often exhibit co‐pathologies, including other abnormal protein aggregates, especially misfolded α‐synuclein (α‐syn), forming intraneuronal Lewy bodies (LBs) and Lewy neurites. 3 With increased frequency compared to age‐matched non‐AD individuals, LB pathology (LBP) has been documented post mortem in 31% to 54% of sAD and 27% to 85% of ADAD patients’ brains, with significant variability among studies. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12
In AD brains, especially those with ADAD 8 and early onset sAD, α‐syn immunoreactivity is often predominantly or exclusively detected in the amygdala with limited associated accumulation in other limbic areas and in the brainstem. 4 , 6 , 7 , 11 These findings have been referred to as the amygdala‐predominant variant of LBP (Amg‐LBP) in which α‐syn accumulation does not follow the typical pattern of topographic distribution associated with Parkinson's disease (PD) and dementia with Lewy bodies (DLB) described by the Braak staging. 13 The strong link with AD and the particular anatomical distribution suggest that Amg‐LBP might occur secondarily to AD pathology, the latter modulating the susceptibility of different brain regions to LBP.
Until recently, our knowledge of LBP in AD came from post mortem studies. Consequently, the mechanisms underlying the connection between AD and LB pathologies, the timing of α‐syn accumulation, and the specificities of α‐syn–related pathology in ADAD compared to sAD are poorly understood.
The development of seed amplification assays (SAAs) of misfolded α‐syn has recently provided a robust in vivo biomarker for LBP. 14 , 15 SAAs have demonstrated high specificity and sensitivity in detecting pathological α‐syn seeds in the cerebrospinal fluid (CSF) of patients with PD and DLB, even during the prodromal (i.e., isolated rapid eye movement sleep behavior disorder or mild cognitive impairment) or preclinical stage. 16 , 17 , 18 , 19 , 20
This study aimed to investigate the presence and timing of SAA reactivity to detect LBP in ADAD in vivo. Initially, we investigated the presence of LBP via SAA examinations of CSF in local cohorts of the Dominantly Inherited Alzheimer Network (DIAN) in Germany, including living asymptomatic and symptomatic ADAD patients as well as healthy controls. Next, we tested the CSF of ADAD cases in a cohort with post mortem semiquantitative assessment of LBP.
2. METHODS
2.1. Study participants
The first part of this study involved participants from Munich and Tübingen DIAN study sites, including mutation non‐carriers (n = 29), asymptomatic (n = 26), and symptomatic (n = 15) mutation carriers. Asymptomatic individuals were defined by having a global Clinical Dementia Rating (CDR) score = 0, whereas symptomatic by a CDR score > 0. The second part of the study included 12 symptomatic individuals with neuropathological information available in addition to the core DIAN dataset. All these cases were evaluated by the DIAN observational study (DIAN‐Obs, data freeze number 16), and their CSF was provided by the DIAN‐Obs biorepository (Washington University, St. Louis, Missouri, USA). All cases with neuropathology were evaluated by the DIAN‐Obs Neuropathology Core (Washington University, St. Louis, Missouri, USA). Among the symptomatic individuals, two patients, labelled “converters,” were asymptomatic at baseline DIAN assessment and became symptomatic (CDR > 0) during follow‐up. In this context, baseline is defined as the first clinical assessment with CSF sampling.
2.2. Ethics approval
DIAN received approval from the ethics committees of Ludwig–Maximilians–University Munich and Eberhardt Karls University Tübingen (371‐13; 535/2011BO1, respectively), as well as the institutional review board committee of Washington University in St. Louis, Missouri, USA (201106339). In addition, permission to perform these measurements was obtained from the ethics committee of Ludwig–Maximilians‐University (371‐13). All participants provided written informed consent for CSF donation for research purposes; similarly, brain donations were obtained after acquiring written informed consent from participants and/or their legal representatives in accordance with applicable local laws and practices.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using online resources such as PubMed, Web of Science, and Scopus. We found several studies applying the α‐synuclein (α‐syn) seed amplification assay (SAA) to detect Lewy body pathology (LBP) in vivo in patients with Parkinson's disease, dementia with Lewy bodies and sporadic Alzheimer's disease (sAD), including prodromal syndromes. However, we did not find publications on autosomal dominant Alzheimer's disease (ADAD).
Interpretation: In this study, we investigated the timing of occurrence and prevalence of misfolded α‐syn positivity as evidence of LBP, which is a frequent post mortem finding in AD. Our findings suggest that in ADAD, LBP occurs later than AD pathology and often as amygdala‐ or olfactory‐predominant LBP, for which cerebrospinal fluid α‐syn SAA has low sensitivity.
Future directions: Future studies should expand the in vivo analysis of LBP by α‐syn SAA to larger cohorts of ADAD patients, stratified for the type of mutation. Moreover, the prevalence of LBP detected by α‐syn SAA should be determined in sporadic early‐onset AD. Finally, the kinetic properties of brain misfolded α‐syn should be compared between individuals with typical LBD and those with the amygdala‐predominant variant.
2.3. CSF collection and analysis
CSF samples were collected by lumbar puncture (LP) from living individuals and processed as previously described. 1 CSF α‐syn SAA (i.e., real‐time quaking‐induced conversion assay [RT‐QuIC]), including the recombinant wild‐type human α‐syn purification, was performed as previously described, 16 with minor , modifications. We ran the same positive and negative control samples throughout all experiments to optimize the comparison among fluorescent signal responses in different plates. As positive controls, we used samples from two patients with normal pressure hydrocephalus for whom a large CSF volume was available, who consistently showed a 4 of 4 positive wells response over at least 10 consecutive runs, indicating a high α‐syn seeding activity. Conversely, the negative controls were CSF from individuals with no clinical evidence of neurodegenerative disease consistently displaying a negative RT‐QuIC response (0 of 4). To overcome batch‐to‐batch variations and intrinsic plate‐to‐plate variability, we normalized the relative fluorescent units for every time point to the median of the maximum intensity (Imax) reached by four positive control replicates within each plate and expressed it as a percentage. We then set the threshold at 20% of the above parameter and the cut‐off at 30 hours. The rationale of the 20% choice stems from the observation that the samples expected to be negative (e.g., CSF from patients lacking LB pathology at neuropathologic examination) sometimes show a slight increase in the fluorescence signal toward the end of the run (between 20 and 30 hours). Therefore, reducing this threshold below the value of 20% would have increased the risk of having false positive readings. CSF samples were deemed positive when at least two of four replicates crossed the threshold as described. 16
TABLE 1.
Characteristics of the asymptomatic study participants from the Munich & Tübingen DIAN cohorts at baseline α‐syn SAA assessments and numbers of consecutive α‐syn SAA assessments per participant.
| Asymptomatic mutation carriers (n = 26) | Mutation non‐carriers (n = 29) | P value | |
|---|---|---|---|
| Age, years (SD) | 35.2 (10.1) | 34.6 (9.3) | 0.82 |
| Parental AAO, median years (interquartile range) | 53 (47.0–56.3) | 48 (40.5–52.5) | 0.14 |
| EYO (SD) | −16.6 (9.0) | N/A | N/A |
| Participants with 1/2/3/4 consecutive α‐syn SAA assessments, n | 8/9/4/5 | 11/13/4/1 | 0.29 |
Abbreviations: AAO, age at onset; DIAN, Dominantly Inherited Alzheimer Network; EYO, estimated years from symptom onset; N/A, not applicable; SD, standard deviation; α‐syn SAA, α‐synuclein seed amplification assay.
TABLE 2.
Characteristics of the symptomatic study participants and converters at baseline α‐syn SAA assessment, numbers of consecutive α‐syn SAA assessments per participant, and information if an autopsy was performed.
| Participant # | α‐syn SAA result | Consecutive α‐syn SAA assessments, [n] | Sex | Time point of first and last LP relative to symptom onset, [years] | Affected gene | Autopsy |
|---|---|---|---|---|---|---|
| Symptomatic participants | ||||||
| 1 | Positive | 1 | m | 7 | PSEN1 | no |
| 2 | Positive | 4 | m | 5, 7 | APP | no |
| 3 | Positive | 1 | m | 7 | PSEN1 | yes |
| 4 | Negative | 1 | m | 2 | PSEN1 | no |
| 5 | Negative | 1 | m | 7 | PSEN1 | no |
| 6 | Negative | 1 | m | 4 | PSEN1 | no |
| 7 | Negative | 1 | f | 3 | PSEN1 | no |
| 8 | Negative | 3 | m | 3, 4 | APP | no |
| 9 | Negative | 4 | f | 3, 8 | PSEN1 | no |
| 10 | Negative | 4 | m | 3, 8 | PSEN1 | no |
| 11 | Negative | 3 | m | 0, 3 | APP | no |
| 12 | Negative | 2 | f | 2, 3 | PSEN1 | no |
| 13 | Negative | 2 | f | 5, 6 | APP | no |
| 14 | Negative | 2 | f | 5, 6 | PSEN1 | no |
| 15 | Negative | 2 | m | 4, 6 | APP | no |
| 16 | Negative | 1 | m | 6 | PSEN1 | yes |
| 17 | Negative | 4 | m | 1, 4 | PSEN1 | yes |
| 18 | Negative | 2 | f | 2, 3 | PSEN1 | yes |
| 19 | Negative | 4 | f | 2, 7 | PSEN1 | yes |
| 20 | Negative | 2 | f | 1, 2 | PSEN1 | yes |
| 21 | Negative | 1 | m | 3 | PSEN1 | yes |
| 22 | Negative | 2 | m | 7, 8 | APP | yes |
| 23 | Negative | 2 | f | 2,3 | PSEN1 | yes |
| 24 | Negative | 1 | f | 8 | PSEN1 | yes |
| 25 | Negative | 1 | m | 5 | PSEN1 | yes |
| Converters | ||||||
| 26 | Negative | 2 | f | –1, 1 | APP | no |
| 27 | Negative | 2 | f | –1, 2 | PSEN1 | yes |
Abbreviations: APP, gene encoding the amyloid precursor protein; f, female; LP, lumbar puncture; m, male; PSEN1, gene encoding presenilin 1; α‐syn SAA, α‐synuclein seed amplification assay.
All CSF α‐syn SAA analyses were carried out at the Neuropathology Laboratory of the IRCCS Institute of Neurological Science of Bologna, Italy, by personnel blinded to the participants’ clinical or genetic status.
2.4. Post mortem neuropathology
Neuropathologic assessment included a systematic evaluation of the left hemibrain by experienced neuropathologists (RJP and NJC) according to an established protocol 8 and National Institute on Aging–Alzheimer's Association consensus criteria for AD neuropathological changes. 21 LBP was assessed by immunohistochemistry using an antibody against phosphorylated α‐syn (Phospho‐α‐synuclein[Ser129], Cell Applications).
TABLE 3.
Characteristics of Lewy body pathology in the most representative areas of the autopsied study participants.
| Participant, # | Time CSF collection—autopsy (years) a | α‐syn immuno‐reactivity | Distribution of Lewy body pathology | Burden of Lewy body pathology by area | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OLF ctx | ME/LC | SN | CA1/subic | ENT ctx | Amg | aC | NEO ctx | ||||
| 3 | 0.7 | Yes | Diffuse, mild in brainstem | ++++ | + | + | +++ | +++ | ++++ | + | + ˟ |
| 16 | 2.4 | Yes | Olfactory only | +++ | – | – | – | – | – | – | – |
| 17 | 3.1 | No | – | – | – | – | – | – | – | – | – |
| 18 | 0.6 | Yes | Brainstem/limbic | +/‐ | – | +++ | – | +/‐ | ++ | +/‐ | – |
| 19 | 1.1 | Yes | Amygdala predominant | ++ | – | – | ++ | – | +++ | – | – |
| 20 | 5.7 | No | – | n.a. | – | – | – | – | – | – | – |
| 21 | 2.4 | Yes | Amygdala/limbic predominant | ++ | – | + | + | +++ | +++ | + | – |
| 22 | 2.2 | Yes | Amygdala predominant | +++ | – | – | – | ++ | +++ | – | – |
| 23 | 2.9 | Yes | Olfactory only | [+] | – | – | – | – | – | – | – |
| 24 | 3.1 | Yes | Olfactory only | ++ | – | – | – | – | – | – | – |
| 25 | 4.9 | Yes | Amygdala only | – | – | – | – | – | +/++ | – | – |
| 27 | 1.4 | Yes | Rare sparse Lewy pathology | n.a. | – | [+] | – | – | – | + | – |
Note: α‐syn immunoreactivity burden was scored as follows :— = none; +/– = very rare (< 3 LB in the area); + = rare/sparse (< 1 LB inclusion per x10 field); ++ = moderate (1–3 LBs); +++ = frequent (4–10 LBs); ++++ = numerous (> 10 LBs).
Abbreviations: aC, anterior cingulate cortex; Amg, amygdala; CA1, cornus ammonis sector 1 (hippocampus); CSF, cerebrospinal fluid; EC, entorhinal cortex; LBs, Lewy bodies; LC, locus coeruleus; ME, medulla oblongata; n.a., not available; NEO ctx, neocortex; OLF ctx, olfactory cortex; SN, substantia nigra; subic, subiculum.
We indicated the last lumbar puncture in cases undergoing multiple assessments.
Represents an average score (from—or + in most cortical areas to +++ in superior temporal gyrus). Score in square brackets indicates that only Lewy neurites were detected.
LBs were scored semi‐quantitatively according to McKeith et al.’s 22 criteria (—= none; + = < 1 LB inclusion per 10x objective field; ++ = 1–3 LBs; +++ = 4–10 LBs; ++++ = > 10 or numerous LBs) in the following regions: medulla oblongata, locus coeruleus, pontine tegmentum, substantia nigra, basal forebrain including nucleus basalis, olfactory cortex (with olfactory tract and peduncle when available), striatum, thalamus, pallidum, dentate gyrus, hippocampal areas CA4‐CA2, CA1, subiculum, parahippocampal gyrus, fusiform gyrus, amygdala, anterior entorhinal cortex, anterior cingulate gyrus, superior and middle temporal gyri, middle frontal gyrus, precentral gyrus (when available), inferior parietal lobule, and occipital cortex. Where LBs were rare or absent, Lewy neurites/grains were scored separately from LBs.
For each case, the stage of LBP was classified according to Braak criteria for PD 13 and McKeith criteria for DLB only in cases with brainstem involvement. 23 Other cases, as appropriate, were classified as Amg‐LBP 6 or simply described by area(s) of involvement.
2.5. Statistical analyses
Values for continuous parameters were expressed as means ± standard deviations; categorical parameters, as absolute values (%) or as median and interquartile range. The Student t test or Mann–Whitney U test were used to compare continuous parameters. The chi‐squared test was used for the number of consecutive SAA assessments per participant.
3. RESULTS
There were no significant differences in demographic features between asymptomatic mutation carriers and non‐carriers (Table 1). All CSF α‐syn SAA assessments in the asymptomatic cohort were negative.
Details on the symptomatic ADAD participants, including the mutated genes, are reported in Table 2. The results obtained in the symptomatic/converter cohorts are shown together (n = 27, of which n = 15 were DIAN participants from Munich and Tübingen, and n = 12 were ADAD individuals who underwent neuropathological assessment). The mean age of symptom onset was 43 ± 7.8 years. The range between age at onset and age at first LP was 1 to 8 years (Table 2), and the interval from the last LP to death in the autopsy‐confirmed cases ranged from 0.7 to 5.7 years (Table 3, Figure S1 in supporting information).
The CSF α‐syn SAA was positive in 3/27 (11.1%) symptomatic participants. In one of them (participant #2), each of four consecutive CSF samples taken longitudinally during 4 years of follow‐up starting from 4 years after symptom onset gave a positive result. All six SAA‐positive CSF samples (i.e., three patients at baseline plus three longitudinal analyses in one individual) showed α‐syn seeding activity in all tested replicates (i.e., 4 of 4). Compared to the positive controls, the positive ADAD patients showed a longer time to the threshold (Lag phase) and a tendency toward a lower Imax, suggesting a lower seeding activity (Figure S2 in supporting information). Both clinical converters to mild cognitive impairment tested negative by CSF α‐syn SAA at baseline (asymptomatic stage) and at 2 and 1 years after the development of cognitive decline.
Of the 12 brain donors, all of whom had developed high levels of AD neuropathologic changes, 10 (83%) also displayed LBP (Table 3, Figure 1). However, only one of them (10%) showed positive seeding activity by CSF α‐syn SAA (Table 3). The brain of the α‐syn SAA positive donor (participant #3) showed the highest burden of LBP, and was the only one with neocortical LBP (i.e., Braak stage 6; diffuse neocortical stage according to McKeith et al., 23 although with an atypical distribution due to the mild involvement of the brainstem). Time between LP and death in this individual was relatively short, that is, 0.7 years. Among the other 11 participants with neuropathologic data, all with negative α‐syn SAA results, one had substantial LBP within the amygdala and substantia nigra (participant #18), four (33.3%) showed the Amg‐LBP variant pattern (#19, 21, 22, 25), three (25%) had immunoreactivity restricted to the olfactory area (piriform cortex, olfactory tract, and peduncle; participants #16, 23, 24), two were negative (#17 and 20) and one virtually negative (#27).
FIGURE 1.
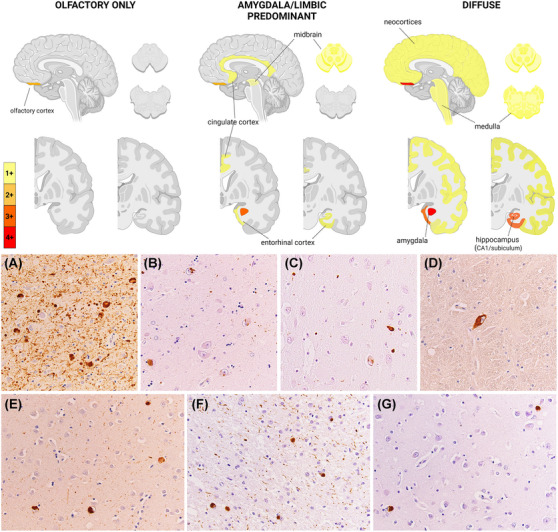
Representative LB pathology features in ADAD. The upper panel shows brain maps of LB pathology distribution in the three representative profiles (i.e., olfactory only, amygdala/limbic predominant, and diffuse) observed in the ADAD individuals at neuropathologic examination. Color scale of the brain maps represents the mean semiquantitative scores in each profile. The lower panel shows LBP features in the ADAD brains. A–C, Severe to moderate/mild LB and neuritic pathology in the amygdala (A, LBP ++++, case #3; B, LBP +++, case #21; C, LBP ++, case #18). D, Isolated syn immunoreactivity in a neuron of the medullary raphe, and (E) moderate LBP in the neocortex of the superior temporal gyrus in the only brain with diffuse LBP (case #3). F, Abundant Lewy neurites and LB in the olfactory tract, and (G) scattered LBs in the olfactory cortex of case #18. Magnification is x40 for all micrographs. ADAD, autosomal dominant Alzheimer's disease; LB, Lewy bodies; LBP, Lewy body pathology.
The four individuals with Amg‐LBP showed mild to moderate LBP involving only the amygdala (n = 1) or the amygdala together with other limbic structures, such as the entorhinal cortex and the hippocampus, with sparse or no brainstem involvement (Table 3). The time between LP and death in these four participants was 1.4, 2.4, 3.1, and 5.7 years, respectively.
Notably, 53 out of 82 participants (64.6%) underwent at least two consecutive α‐syn SAA assessments using material of independent LPs. All intraindividual consecutive α‐syn SAA assessments showed the same result as at baseline, highlighting the complete consistency of results within each participant.
4. DISCUSSION
The results of the present study, combined with those recently obtained in sAD, 20 , 24 shed new light on the association and pathogenetic interaction between LBP and AD pathology in the symptomatic phase of AD. The significantly lower percentage of LBP we detected via SAA in CSF of symptomatic patients with ADAD compared to sAD patients (11% vs. 21%) suggests a lower prevalence of “typical” LBP (i.e., following Braak staging) in ADAD compared to sAD most likely due to the significantly lower mean age of patients with symptomatic ADAD compared to those with sAD. Indeed, a positive association between age and “typical” LBP has been documented in both non‐AD and AD‐affected brains, especially between the presenile and senile age. 11 , 25
In contrast to “typical” LBP, the Amg‐LBP variant, which almost only affects patients with AD pathology, suggesting a secondary synucleinopathy, is more common in patients with early‐onset AD than in patients with late‐onset AD. 11 Consistent with these previously published data, 4 of the 12 ADAD participants in our neuropathologically verified cohort showed LBP compatible with Amg‐LBP, whereas only one had diffuse LBP reaching the neocortical areas. The low overall detection sensitivity of the α‐syn SAA for LBP in this ADAD cohort is in line with the previously reported reduced sensitivity of these assays for the Amg‐LBP variants and other conditions in which the LBP is only focally present (e.g., limited to the lower brainstem or the olfactory areas) compared to the virtually full sensitivity in cases with a neocortical or limbic stage of LB disease pathology. 26 , 27 , 28 In line with these findings, by applying the same SAA protocol to a cohort of 59 CSF and brain pairs, we have recently confirmed the 100% sensitivity of our assay in detecting the neocortical and limbic LBP stages. 28 One might argue that the 100% sensitivity in “typical” LBD at the limbic stage compared to the negative finding we obtained in this study in the two participants with involvement of the limbic areas might represent a discrepancy. However, there is a profound difference in the LBP load/burden between the limbic stage of typical LBD, characterized by prominent involvement (corresponding to a score +++ / ++++ of the present study) of the brainstem (and hypothalamus/basal forebrain as well) and the ADAD participant with limbic predominant pathology showing minimal or only focal pathology in those areas. Consequently, the different overall burden of LB pathology might be sufficient to explain the different sensitivity of our α‐syn SAA between the two patient groups. However, we cannot entirely dismiss the hypothesis that Amg‐LBP in ADAD could be due to a different strain of α‐syn showing a lower SAA reactivity as it was demonstrated for multiple system atrophy. 16 , 28 , 29
The range of intervals between CSF assessment and death in our cohort may also have contributed to the low positivity rate of α‐syn SAA in this study. Indeed, given the focal and sparse nature of the LBP in many of our patients with post mortem evaluation, it is plausible to believe that some patients were free of misfolded α‐syn deposition at the time of LP. The secondary nature of the Amg‐LBP variant, likely triggered or strongly modulated by AD pathology, also fits the idea of LBP being a relatively late event in most ADAD brains.
A limitation of the present study concerns the low number of individuals with positive α‐syn SAA not allowing a stratification according to mutated gene (e.g., PSEN1 vs. APP). Moreover, the mean estimated years to symptom onset (EYO) of −16.6 years in the cohort of asymptomatic ADAD individuals suggests that in many cases, the amyloid deposition was in very early stages as the amyloid deposition typically starts ≈ −20 EYO. 30 Finally, the timing between LP and death was heterogeneous in the cohort with post mortem neuropathology.
In summary, in this relatively small but comprehensive cohort of patients with ADAD, including asymptomatic carriers and symptomatic patients with or without post mortem neuropathology, we show that LBP can be detected as a CSF biomarker in symptomatic mutation carriers but at a lower rate than in sAD. This is likely due to the lower incidence of “typical” transitional/limbic and neocortical/diffuse LBP stages in presenile patients and the focal and late occurring nature of Amg‐LBP in ADAD.
While the clinical consequences of LBP in ADAD remain to be fully understood, analyzing CSF biomarkers of LBP in vivo could provide a better precision‐medicine approach for the clinical management of ADAD patients and for designing and interpreting data from disease‐modifying drug trials.
CONFLICT OF INTEREST STATEMENT
JL reports speaker fees from Bayer Vital, Biogen, EISAI, Merck, Roche, TEVA, and Zambon; consulting fees from Axon Neuroscience, EISAI, and Biogen; author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers; and is inventor in a patent “Oral Phenylbutyrate for Treatment of Human 4‐Repeat Tauopathies” (EP 23 156 122.6) filed by LMU Munich. In addition, he reports compensation for serving as chief medical officer for MODAG GmbH; is beneficiary of the phantom share program of MODAG GmbH; and is inventor in a patent “Pharmaceutical Composition and Methods of Use” (EP 22 159 408.8) filed by MODAG GmbH, all activities outside the submitted work. NCF reports consulting fees from Biogen, Eisai, Ionis, Lilly, Roche/Genentech, and Siemens all paid to UCL; he has also served on a Data Safety Monitoring Board for Biogen. OH has acquired research support (for the institution) from ADx, AVID Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer, and Roche. In the past 2 years, he has received consultancy/speaker fees from AC Immune, Amylyx, Alzpath, BioArctic, Biogen, Cerveau, Eisai, Eli Lilly, Fujirebio, Merck, Novartis, Novo Nordisk, Roche, Sanofi, and Siemens. FL has research support from NIA, NIH, Biogen, Tau‐Consortium, Roche and is a consultant of Viewmind and Biogen. JCM is funded by NIH grants # P30 AG066444; P01AG003991; P01AG026276. Neither Dr. Morris nor his family owns stock or has equity interest (outside of mutual funds or other externally directed accounts) in any pharmaceutical or biotechnology company. AMG serves on SABs for Genentech and Muna Therapeutics and has received consultancy/speaker fees from Biogen. RSV reports consultancy or speaker fees from Ionis, AviadoBio, NovoNordisk, Pfizer, Neuraxpharm, and Roche diagnosis. GH has ongoing research collaborations with Roche, UCB, Abbvie; serves as a consultant for Abbvie, Alzprotect, Amylyx, Aprineua, Asceneuron, Bayer, Bial, Biogen, Biohaven, Epidarex, Ferrer, Kyowa Kirin, Lundbeck, Novartis, Retrotope, Roche, Sanofi, Servier, Takeda, Teva, UCB; received honoraria for scientific presentations from Abbvie, Bayer, Bial, Biogen, Bristol Myers Squibb, Kyowa Kirin, Pfizer, Roche, Teva, UCB, Zambon; holds a patent on Treatment of Synucleinopathies (US 10,918,628 B2; EP 17 787 904.6‐1109 / 3 525 788); received publication royalties from Academic Press, Kohlhammer, and Thieme. All other authors have no conflicts to report. Author disclosures are available in the supporting information.
DIAN consortium member list
| Last name | First name | Institution | Affiliation |
|---|---|---|---|
| Bateman | Randall | Washington University | Washington University School of Medicine in St. Louis |
| Daniels | Alisha J. | Washington University | Washington University in St. Louis |
| Courtney | Laura | Washington University | Washington University School of Medicine in St. Louis |
| McDade | Eric | Washington University | Washington University School of Medicine in St. Louis, Department of Neurology |
| Llibre‐Guerra | Jorge J. | Washington University | Dominantly Inherited Alzheimer's Network, Department of Neurology, Washington University School of Medicine in St.Louis |
| Supnet‐Bell | Charlene | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Xiong | Chengie | Washington University | Washington University in St. Louis, School of Medicine |
| Xu | Xiong | Washington University | Washington University in St. Louis, School of Medicine |
| Lu | Ruijin | Washington University | Washington University in St. Louis, School of Medicine |
| Wang | Guoqiao | Washington University | Washington University in St. Louis, School of Medicine |
| Li | Yan | Washington University | Washington University in St. Louis, School of Medicine |
| Gremminger | Emily | Washington University | Washington University in St. Louis, School of Medicine |
| Perrin | Richard J. | Washington University | Department of Pathology and Immunology, Department of Neurology, Knight Alzheimer Disease Research Center, Washington University School of Medicine, St. Louis, MO, USA |
| Franklin | Erin | Washington University | Department of Pathology and Immunology, Washington University in St. Louis |
| Ibanez | Laura | Washington University | Department of Psychiatry, Department of Neurology, and NeuroGenomics and Informatics Center |
| Jerome | Gina | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Herries | Elizabeth | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Stauber | Jennifer | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Baker | Bryce | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Minton | Matthew | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Cruchaga | Carlos | Washington University |
1. Department of Psychiatry, Washington University School of Medicine, St. Louis, MO, USA 2. NeuroGenomics and Informatics Center, Washington University School of Medicine, St. Louis, MO, USA |
| Goate | Alison M. | Mount Sinai | Dept. of Genetics & Genomic Sciences, Dept. of Neuroscience, Ronald M. Loeb Center for Alzheimer's Disease, Icahn School of Medicine at Mount Sinai, New York, NY |
| Renton | Alan E. | Mount Sinai | Ronald M. Loeb Center for Alzheimer's Disease, Dept of Genetics and Genomic Sciences and Nash Family Dept of Neuroscience, Icahn School of Medicine at Mount Sinai |
| Picarello | Danielle M. | Mount Sinai | Ronald M. Loeb Center for Alzheimer's Disease, Dept of Genetics and Genomic Sciences and Nash Family Dept of Neuroscience, Icahn School of Medicine at Mount Sinai |
| Benzinger | Tammie | Washington University | Washington University in St. Louis, Department Radiology |
| Gordon | Brian A. | Washington University | Washington University in St. Louis, Department Radiology |
| Hornbeck | Russ | Washington University | Washington University in St. Louis, Department Radiology |
| Chen | Allison | Washington University | Washington University School of Medicine, St. Louis |
| Chen | Charles | Washington University | Washington University School of Medicine, St. Louis |
| Flores | Shaney | Washington University | Washington University School of Medicine, St. Louis |
| Joseph‐Mathurin | Nelly | Washington University | Washington University School of Medicine, St. Louis |
| Jarman | Steve | Washington University | Washington University School of Medicine, St. Louis |
| Jackson | Kelley | Washington University | Washington University School of Medicine, St. Louis |
| Keefe | Sarah | Washington University | Washington University School of Medicine, St. Louis |
| Koudelis | Deborah | Washington University | Washington University School of Medicine, St. Louis |
| Massoumzadeh | Parinaz | Washington University | Washington University School of Medicine, St. Louis |
| McCullough | Austin | Washington University | Washington University School of Medicine, St. Louis |
| McKay | Nicole | Washington University | Washington University School of Medicine, St. Louis |
| Nicklaus | Joyce | Washington University | Washington University School of Medicine, St. Louis |
| Pulizos | Christine | Washington University | Washington University School of Medicine, St. Louis |
| Wang | Qing | Washington University | Washington University School of Medicine, St. Louis |
| Sabaredzovic | Edita | Washington University | Washington University School of Medicine, St. Louis |
| Smith | Hunter | Washington University | Washington University School of Medicine, St. Louis |
| Scott | Jalen | Washington University | Washington University School of Medicine, St. Louis |
| Simmons | Ashlee | Washington University | Washington University School of Medicine, St. Louis |
| Rizzo | Jacqueline | Washington University | Washington University School of Medicine, St. Louis |
| Hassenstab | Jason | Washington University | Associate Professor of Neurology and of Psychological & Brain Sciences, Washington University in St. Louis |
| Smith | Jennifer | Washington University | Department of Neurology, Washington University in St. Louis |
| Stout | Sarah | Washington University | Department of Neurology, Washington University in St. Louis |
| Aschenbrenner | Andrew J. | Washington University |
Assistant Professor of Neurology Washington University in St. Louis |
| Karch | Celeste M. | Washington University | Department of Psychiatry, Washington University in St Louis |
| Marsh | Jacob | Washington University | DIAN Fibroblast and Stem Cell Bank, Washington University in St. Louis |
| Morris | John C. | Washington University | Washington University in St. Louis, Department of Neurology and the Knight Alzheimer Disease Research Center |
| Holtzman | David M. | Washington University | Department of Neurology, Hope Center for Neurological Disorders, Knight Alzheimer's Disease Research Center, Washington University in St. Louis |
| Barthelemy | Nicolas | Washington University | Washington University in St. Louis, School of Medicine, Department of Neurology |
| Xu | Jinbin | Washington University | Department of Radiology, Washington University in St. Louis |
| Noble | James M. | Columbia University | Taub Institute for Research on Alzheimer's Disease and the Aging Brain, G.H. Sergievsky Center, Department of Neurology, Columbia University Irving Medical Center |
| Berman | Sarah B. | University of Pittsburgh | University of Pittsburgh Departments of Neurology and Clinical & Translational Science |
| Ikonomovic | Snezana | University of Pittsburgh | University of Pittsburgh, Department of Neurology |
| Nadkarni | Neelesh K. | University of Pittsburgh | University of Pittsburgh, Departments of Medicine (Geriatric Medicine) and Neurology |
| Day | Gregory | Mayo | Department of Neurology, Mayo Clinic in Florida; Jacksonville, FL |
| Graff‐Radford | Neill R. | Mayo | Department of Neurology, Mayo Clinic in Florida; Jacksonville, FL |
| Farlow | Martin | Indiana University | Indiana University School of Medicine |
| Chhatwal | Jasmeer P. | BWH | Massachusetts General Hospital, Brigham and Women's Hospital, Harvard Medical School |
| Ikeuchi | Takeshi | Niigata | Brain Research Institute, Niigata University |
| Kasuga | Kensaku | Niigata | Brain Research Institute, Niigata University |
| Niimi | Yoshiki | Tokyo | Specially appointed lecturer, Unit for Early and Exploratory Clinical Development, The University of Tokyo |
| Huey | Edward D. | Butler | Memory and Aging Program, Butler Hospital, Professor, Department of Psychiatry and Human Behavior, Alpert Medical School, Brown University |
| Salloway | Stephen | Butler | Memory and Aging Program, Butler Hospital, Departments of Psychiatry and Human Behavior and Neurology, Alpert Medical School, Brown University |
| Schofield | Peter R. | Sydney |
1. Neuroscience Research Australia, Sydney, Australia 2. School of Medical Sciences, University of New South Wales, Sydney, Australia |
| Brooks | William S. | Sydney |
1. Neuroscience Research Australia, Sydney, Australia; and 2. School of Medical Sciences, University of New South Wales, Sydney, Australia |
| Bechara | Jacob A. | Sydney | Neuroscience Research Australia, Sydney, Australia |
| Martins | Ralph | Perth | Edith Cowan University |
| Fox | Nick C. | UCL |
1. Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom 2. UK Dementia Research Institute at UCL, London, United Kingdom |
| Cash | David M. | UCL |
1. Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom 2. UK Dementia Research Institute at UCL, London, United Kingdom |
| Ryan | Natalie S. | UCL |
1. Dementia Research Centre, UCL Queen Square Institute of Neurology, London, United Kingdom 2. UK Dementia Research Institute at UCL, London, United Kingdom |
| Jucker | Mathias | Tubingen |
1. German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany 2. Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Laske | Christoph | Tubingen |
1. German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany 2. Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Hofmann | Anna | Tubingen |
1. German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany 2. Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Kuder‐Buletta | Elke | Tubingen | German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany |
| Graber‐Sultan | Susanne | Tubingen | German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany |
| Obermueller | Ulrike | Tubingen |
1. German Center for Neurodegenerative Diseases (DZNE) Tübingen, Tübingen, Germany 2. Hertie‐Institute for Clinical Brain Research, University of Tübingen, Tübingen, Germany |
| Levin | Johannes | Munich | 1. German Center for Neurodegenerative Diseases, site Munich; 2. Department of Neurology, Ludwig‐Maximilians‐Universität München, Munich, Germany; 3. Munich Cluster for Systems Neurology (SyNergy), Munich, Germany |
| Roedenbeck | Yvonne | Munich | 1. German Center for Neurodegenerative Diseases, site Munich; 2. Department of Neurology, Ludwig‐Maximilians‐Universität München, Munich, Germany; 3. Munich Cluster for Systems Neurology (SyNergy), Munich, Germany |
| Vöglein | Jonathan | Munich |
1. Department of Neurology, LMU University Hospital, LMU Munich, Munich, Germany 2. German Center for Neurodegenerative Diseases (DZNE), Munich, Germany |
| Lee | Jae‐Hong | Seoul | Asian Medical Center, Seoul, South Korea |
| Roh | Jee Hoon | Seoul | Korea University College of Medicine, Seoul, South Korea |
| Sanchez‐Valle | Raquel | Barcelona | Alzheimer's Disease and Other Cognitive Disorders Group, Neurology Service, Hospital Clínic de Barcelona, FRCB‐IDIBAPS, University of Barcelona, Barcelona (Spain) |
| Rosa‐Neto | Pedro | McGill | Translational Neuroimaging Laboratory, McGill University Research Centre for Studies in Aging, Department of Neurology and Neurosurgery, Psychiatry and Pharmacology and Therapeutics, McGill University, Montreal, Canada |
| Allegri | Ricardo F. | FLENI/Salta | Department of Cognitive Neurology, Instituto Neurológico Fleni, Buenos Aires, Argentina |
| Chrem Mendez | Patricio | FLENI | Department of Cognitive Neurology, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Surace | Ezequiel | FLENI | Department of Molecular Biology and Neuropathology, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Vazquez | Silvia | FLENI | Center of Molecular Imaging, Institute for Neurological Research Fleni, Buenos Aires, Argentina |
| Lopera | Francisco | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia |
| Leon | Yudy Milena | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia |
| Ramirez | Laura | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia |
| Aguillon | David | Medellin | Grupo de Neurociencias de Antioquia (GNA), Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia |
| Levey | Allan I. | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA, USA |
| Johnson | Erik C.B | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA, USA |
| Seyfried | Nicholas T. | Emory | Goizueta Alzheimer's Disease Research Center, Emory University, Atlanta, GA, USA |
| Ringman | John | University of Southern California | Department of Neurology, Keck School of Medicine of USC, Universty of Southern California |
| Fagan | Anne M. | Washington University | Department of Neurology, Washington University in St. Louis |
| Mori | Hiroshi | Osaka Metropolitan University | |
| Masters | Colin | University of Melbourne | Florey Institute, The University of Melbourne |
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. The authors acknowledge the altruism of the participants and their families and the contributions of the DIAN research and support staff at each participating site for their contributions to this study. The study was supported by the German research foundation under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy ‐ ID 390857198, the German Ministry of Research and Education (Förderkennzeichen: FKZ161L0214B, FKZ161L0214C CLINSPECT‐M), the grant Ricerca Finalizzata‐2021‐12374386, funded by the Ministry of Health, and the #NextGenerationEU (NGEU), funded by the Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006). Data collection and sharing for this project was supported by the Dominantly Inherited Alzheimer Network (DIAN, U19AG032438), funded by the National Institute on Aging (NIA), the Alzheimer's Association (SG‐20‐690363‐DIAN LATAM), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Dementia Research Center (KDRC—HU21C0066), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec – Santé.
Open access funding enabled and organized by Projekt DEAL.
Levin J, Baiardi S, Quadalti C, et al. α‐Synuclein seed amplification assay detects Lewy body co‐pathology in autosomal dominant Alzheimer's disease late in the disease course and dependent on Lewy pathology burden. Alzheimer's Dement. 2024;20:4351–4365. 10.1002/alz.13818
Contributor Information
Johannes Levin, Email: jlevin@med.uni-muenchen.de.
Piero Parchi, Email: piero.parchi@unibo.it.
REFERENCES
- 1. Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795‐804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morris JC, Weiner M, Xiong C, et al. Autosomal dominant and sporadic late onset Alzheimer's disease share a common in vivo pathophysiology. Brain. 2022;145:3594‐3607. doi: 10.1093/brain/awac181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson JL, Xie SX, Baer DR, et al. Pathological combinations in neurodegenerative disease are heterogeneous and disease‐associated. Brain. 2023;146:2557‐2569. doi: 10.1093/brain/awad059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha‐synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365‐1370. doi: 10.1016/s0002-9440(10)65722-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parkkinen L, Soininen H, Alafuzoff I. Regional distribution of alpha‐synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:363‐367. doi: 10.1093/jnen/62.4.363 [DOI] [PubMed] [Google Scholar]
- 6. Uchikado H, Lin W‐L, DeLucia MW, Dickson DW. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha‐synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685‐697. doi: 10.1097/01.jnen.0000225908.90052.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leverenz JB, Fishel MA, Peskind ER, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease‐ and mutation‐specific pathologic phenotype. Arch Neurol. 2006;63:370‐376. doi: 10.1001/archneur.63.3.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi‐center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology. 2015;35:390‐400. doi: 10.1111/neup.12205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ringman JM, Monsell S, Ng DW, et al. Neuropathology of autosomal dominant Alzheimer disease in the national Alzheimer coordinating center database. J Neuropathol Exp Neurol. 2016;75:284‐290. doi: 10.1093/jnen/nlv028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144:2186‐2198. doi: 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sepulveda‐Falla D, Villegas Lanau CA, White C, et al. Comorbidities in Early‐Onset Sporadic versus Presenilin‐1 Mutation‐Associated Alzheimer's Disease Dementia: Evidence for Dependency on Alzheimer's Disease Neuropathological Changes. medRxiv. 2023.08.14.23294081. doi: 10.1101/2023.08.14.23294081 medRxiv [DOI]
- 13. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197‐211. doi: 10.1016/s0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- 14. Fairfoul G, McGuire LI, Pal S, et al. Alpha‐synuclein RT‐QuIC in the CSF of patients with alpha‐synucleinopathies. Ann Clin Transl Neurol. 2016;3:812‐818. doi: 10.1002/acn3.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Groveman BR, Orrù CD, Hughson AG, et al. Rapid and ultra‐sensitive quantitation of disease‐associated α‐synuclein seeds in brain and cerebrospinal fluid by αSyn RT‐QuIC. Acta Neuropathol Commun. 2018;6:7. doi: 10.1186/s40478-018-0508-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rossi M, Candelise N, Baiardi S, et al. Ultrasensitive RT‐QuIC assay with high sensitivity and specificity for Lewy body‐associated synucleinopathies. Acta Neuropathol. 2020;140:49‐62. doi: 10.1007/s00401-020-02160-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rossi M, Baiardi S, Teunissen CE, et al. Diagnostic value of the CSF α‐Synuclein real‐time quaking‐induced conversion assay at the prodromal MCI stage of dementia with Lewy bodies. Neurology. 2021;97:e930‐e940. doi: 10.1212/WNL.0000000000012438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iranzo A, Mammana A, Muñoz‐Lopetegi A, et al. Misfolded α‐synuclein assessment in the skin and CSF by RT‐QuIC in isolated REM sleep behavior disorder. Neurology. 2023;100:e1944‐e1954. doi: 10.1212/WNL.0000000000207147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siderowf A, Concha‐Marambio L, Lafontant D‐E, et al. Assessment of heterogeneity among participants in the Parkinson's Progression Markers Initiative cohort using α‐synuclein seed amplification: a cross‐sectional study. Lancet Neurol. 2023;22:407‐417. doi: 10.1016/S1474-4422(23)00109-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmqvist S, Rossi M, Hall S, et al. Cognitive effects of Lewy body pathology in clinically unimpaired individuals. Nat Med. 2023;29:1971‐1978. doi: 10.1038/s41591-023-02450-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863‐1872. doi: 10.1212/01.wnl.0000187889.17253.b1 [DOI] [PubMed] [Google Scholar]
- 23. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89:88‐100. doi: 10.1212/WNL.0000000000004058 [DOI] [PubMed] [Google Scholar]
- 24. Quadalti C, Palmqvist S, Hall S, et al. Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med. 2023;29:1964‐1970. doi: 10.1038/s41591-023-02449-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 1988;51:745‐752. doi: 10.1136/jnnp.51.6.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall S, Orrù CD, Serrano GE, et al. Performance of αSynuclein RT‐QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun. 2022;10:90. doi: 10.1186/s40478-022-01388-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α‐synuclein in the context of co‐pathology and Non‐LBD Diagnoses. Ann Neurol. 2022;92:650‐662. doi: 10.1002/ana.26453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bentivenga GM, Mammana A, Baiardi S, et al. Performance of a seed amplification assay for misfolded alpha‐synuclein in cerebrospinal fluid and brain tissue in relation to Lewy body disease stage and pathology burden. Acta Neuropathol.. 2024;147(1):18. doi: 10.1007/s00401-023-02663-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shahnawaz M, Mukherjee A, Pritzkow S, et al. Discriminating α‐synuclein strains in Parkinson's disease and multiple system atrophy. Nature. 2020;578(7794):273‐277. doi: 10.1038/s41586-020-1984-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91:e1295‐e1306. doi: 10.1212/WNL.0000000000006277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information