

Abstract
BACKGROUND
Alzheimer's disease (AD) prevalence increases with age, yet a small fraction of the population reaches ages > 100 years without cognitive decline. We studied the genetic factors associated with such resilience against AD.
METHODS
Genome‐wide association studies identified 86 single nucleotide polymorphisms (SNPs) associated with AD risk. We estimated SNP frequency in 2281 AD cases, 3165 age‐matched controls, and 346 cognitively healthy centenarians. We calculated a polygenic risk score (PRS) for each individual and investigated the functional properties of SNPs enriched/depleted in centenarians.
RESULTS
Cognitively healthy centenarians were enriched with the protective alleles of the SNPs associated with AD risk. The protective effect concentrated on the alleles in/near ANKH, GRN, TMEM106B, SORT1, PLCG2, RIN3, and APOE genes. This translated to >5‐fold lower PRS in centenarians compared to AD cases (P = 7.69 × 10−71), and 2‐fold lower compared to age‐matched controls (P = 5.83 × 10−17).
DISCUSSION
Maintaining cognitive health until extreme ages requires complex genetic protection against AD, which concentrates on the genes associated with the endolysosomal and immune systems.
Highlights
Cognitively healthy cent enarians are enriched with the protective alleles of genetic variants associated with Alzheimer's disease (AD).
The protective effect is concentrated on variants involved in the immune and endolysosomal systems.
Combining variants into a polygenic risk score (PRS) translated to > 5‐fold lower PRS in centenarians compared to AD cases, and ≈ 2‐fold lower compared to middle‐aged healthy controls.
Keywords: Aging, Alzheimer's disease, Cognitively healthy centenarians, Endolysosomal, Genes, Genome‐wide association studies, Heritability, Immunity, Protection, Resilience
1. BACKGROUND
The average human life expectancy continues to grow and by 2050 there will be 3.2 million centenarians in the world. 1 At old ages, a major contributor to poor health is cognitive decline and dementia, of which Alzheimer's disease (AD) is the most common type. 2 , 3 However, AD is not an inevitable consequence of aging, as testified by a small proportion of the population that reaches at least 100 years while maintaining a high level of cognitive and physical functions. 4 , 5 This raises the question of whether these cognitively healthy centenarians have exceptional features that protect or delay the onset of dementia, and whether such mechanisms may be genetically encoded.
AD is a progressive disorder characterized by loss of cognitive functions, ultimately leading to loss of independence and death, for which an effective treatment is lacking. 3 , 6 The greatest risk factor for AD is age: the disease is rare at 60 years, and the incidence of AD reaches ≈ 40% per year at 100 years of age. 7 Next to aging, heritability plays an important role that changes dramatically with age. While the heritability of AD with age at onset < 65 years is estimated to be 90% to 100%, mostly due to autosomal dominant or strong risk‐increasing genetic variants, 8 it decreases to 60% to 80% for ages at AD onset of ≈ 75 years (determined by twin studies), based on a unique mix of rare and common risk factors, and further declines with later ages at AD onset. 9 Approximately 30% of the genetic risk of AD is attributable to the ε4 allele of the apolipoprotein E (APOE) gene. Large collaborative genome‐wide association studies (GWAS) have collectively identified 86 single nucleotide polymorphisms (SNPs) that are associated with a slight modification of the risk of AD. 10 , 11
Intriguingly, the reverse is also true, as ≈ 60% of the chance to survive to 100 years in good cognitive health depends on inheriting favorable genetic factors, 12 comprising a relative depletion of risk‐increasing variants and an enrichment of advantageous genetic variants that associate with a prolonged (brain) health. 13 , 14 , 15 In fact, in 2018 we reported that the effect size of 29 SNPs that were associated with AD risk was increased on average 2‐fold when using cognitively healthy centenarians as controls rather than controls age‐matched with the AD cases. 16 Consequently, cognitively healthy centenarians had a significantly lower polygenic risk score (PRS), compared to AD cases and age‐matched controls.
In the current study, we aimed to further expand on these findings by investigating the prevalence in cognitively healthy centenarians of the 86 SNPs that are currently associated with AD risk, based on the most recent GWAS for AD. 10 We studied the effect of individual AD‐associated SNPs as well as their combined effect (PRS) on prolonged cognitive health. Furthermore, we identified risk‐increasing and protective SNPs that were, respectively, most depleted or enriched in a cohort of cognitively healthy centenarians, which allowed us to highlight the biological mechanisms most strongly involved with resilience against AD.
2. METHODS
2.1. Cohort description
We included 6747 individuals in this study. Of these, 2542 were AD cases, either clinically diagnosed with probable AD from the Amsterdam Dementia Cohort (ADC, N = 2060) 17 , 18 , 19 or pathologically confirmed from the Netherlands Brain Bank (N = 482). 20 The diagnosis of probable AD in the ADC cohort was based on the clinical criteria formulated by the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) and based on the National Institute of Aging–Alzheimer Association (NIA‐AA). All subjects underwent a standard diagnostic assessment including neurological examination, blood tests, magnetic resonance imaging, electroencephalogram, and cerebrospinal fluid (CSF) analysis (available for most patients). Together, this diagnostic procedure reduces the chance of misdiagnosis. 17 As age‐matched controls, we used (1) a sample of 1776 Dutch older adults from the Longitudinal Aging Study of Amsterdam (LASA), 21 (2) a sample of 1524 older adults with subjective cognitive decline who visited the memory clinic of the Alzheimer Center Amsterdam and SCIENCe project and were labeled cognitively normal after the extensive examination, 17 (3) a sample of 62 healthy controls from the Netherlands Brain Bank, 20 (4) a sample of 196 individuals from the twin study, 22 and (5) a sample of 85 older adults from the 100‐plus Study (partners of centenarians’ offspring). 5 All age‐matched controls were cognitively healthy at the time of inclusion in this study. Individuals with subjective cognitive decline were followed over time in the SCIENCe project, and only individuals who did not convert to mild cognitive impairment or dementia during follow‐up were included in this study. Additional information about inclusion criteria for these cohorts is available in Supplementary Material. As alternative (extreme) healthy controls, we used 360 cognitively healthy centenarians from the 100‐plus Study cohort. 5 This study includes Dutch‐speaking individuals who (1) can provide official evidence for being aged ≥ 100 years; (2) self‐report to be cognitively healthy, which is confirmed by a proxy; (3) consent to the donation of a blood sample; (4) consent to (at least) two home visits from a researcher including an interview and neuropsychological test battery. 5 The medical ethics committee of the Amsterdam UMC approved all studies. All participants and/or their legal representatives provided written informed consent for participation in clinical and genetic studies.
2.2. Genotyping and imputation of 86 selected SNPs
We included 85 SNPs that were significantly associated with AD in the latest GWAS by Bellenguez et al., 10 plus SNP rs12459419 near CD33 (Table S1 in supporting information). 23 , 24 After quality control and genotype imputation of the genetic data (see Supplementary Methods: Genotyping and Imputation), all individuals passed quality control. Before analysis, we excluded individuals with a family relation (identity‐by‐descent ≥ 0.2), 25 and we kept only individuals of European ancestry (based on 1000Genomes clustering), 26 leaving 2281 AD cases, 3165 age‐matched controls, and 346 cognitively healthy centenarians for the analyses.
2.3. Single variant analyses
As reference effect size for each SNP, we used the effect sizes resulting from the comparison of 39,106 clinically diagnosed AD cases and 401,577 age‐matched controls used in the discovery phase by Bellenguez et al. (Table S1). 10 We excluded the proxy phenotypes which Bellenguez et al. included in their multi‐stage meta‐analysis, as these are based on paternal and maternal disease status rather than clinical diagnosis, which typically leads to a dilution of the SNP effect sizes. For each AD‐associated SNP, we calculated the change in effect size relative to the reference effect size comparing (1) AD cases versus cognitively healthy centenarians, (2) AD cases versus age‐matched controls, and (3) age‐matched controls versus cognitively healthy centenarians (see Supplementary Methods: Change in effect size). In a sensitivity analysis, we compared the frequency of each SNP between early‐onset (age at onset ≤ 65 years) and late‐onset AD cases, to highlight potential genetic modifiers.
RESEARCH IN CONTEXT
Systematic review: Genetic studies of Alzheimer's disease (AD) have identified common genetic variants that influence the risk of AD. By contrast, the genetic factors associated with long‐term resilience against AD are mostly elusive. We studied the genetic variants associated with AD in cognitively healthy centenarians, that is, individuals > 100 years with maintained cognitive health.
Interpretation: Cognitively healthy centenarians were enriched with the protective alleles of AD‐associated variants. The variants with the largest effect size in centenarians functionally map to endolysosomal and immunological/clearance mechanisms. A polygenic risk score combining all AD variants was > 5‐fold lower in the cognitive healthy centenarians compared to AD cases and almost 2‐fold lower compared to AD age‐matched controls.
Future directions: Our article highlights the importance of further investigation of protective genetic variants and their effects on maintaining health. The prioritization of the biological processes associated with the strongest protection pinpoints those mechanisms involved in the resilience against AD.
2.4. Polygenic risk score
We combined all 86 SNPs into a PRS, resembling an individual's net genetic risk of AD. As weights for the PRS, we conventionally used the effect sizes of the meta‐analysis including both clinically diagnosed AD cases and by‐proxy phenotypes, reflecting the final results of Bellenguez et al. (Table S1). Given the large effect size associated with the two APOE SNPs (rs429358 and rs7412), we calculated PRS including and excluding these two SNPs. We assessed the association between PRS and AD risk by comparing the scaled PRS distributions (μ = 0, σ = 1) between AD cases, age‐matched controls, and cognitively healthy centenarians, in a pairwise manner and splitting by sex. We also tested for any difference in the PRS between early‐onset AD and late‐onset AD samples. For the associations, we used logistic regression models adjusting for population stratification (PC1‐5). The resulting effect sizes (log of odds ratio) can be interpreted as the odds ratio difference per one standard deviation increase in the PRS, with the corresponding 95% confidence intervals.
2.5. The contribution of a centenarian
In genetic studies, the power to detect a significant SNP association is affected by both the effect size and the sample size, which includes the number of cases and controls in the comparison. This can be approximated using power analyses. The greater the effect size, the smaller the required number of cases and controls to achieve statistical significance in an association. Through a power analysis, we determined the potential additional statistical power offered by cognitively healthy centenarians compared to typical controls in a case‐control study of AD. To do so, for each SNP identified by Bellenguez et al. we calculated the number of normal (age‐matched) controls and cognitively healthy centenarians necessary to obtain 80% power to find a SNP association at P value = 0.05. We assumed (1) 8000 AD cases, (2) the minor allele frequency as reported in the reference GWAS (Table S1), and (3) the observed effect size from our comparisons (AD cases versus age‐matched controls, and AD cases versus cognitively healthy centenarians). Because the direction of effect must be consistent with the direction reported in Bellenguez et al. we excluded SNPs for which we observed an opposite direction of effect in both AD cases versus age‐matched controls and AD cases versus cognitively healthy centenarians. Then, for each SNP, we compared 8000 AD cases to 200 controls, and recursively increased the number of controls by 200 until a power of at least 80% was found or the number of controls was twice the number of AD cases (i.e., 16,000). When a SNP association reached at least 80% power, we regarded it as converging. The ratio between the number of age‐matched controls and cognitively healthy centenarians, for each SNP, indicates the increase in statistical power of a single centenarian relative to age‐matched controls. We simulated the analysis using several thresholds for the number of AD cases to use (2500, 5000, 8000, and 10,000) and found that after 8000 no additional SNPs converged.
2.6. In silico functional analysis
We investigated the biological pathways associated with the SNPs with the largest effect‐size differences between cognitively healthy centenarians and age‐matched controls. We selected SNPs for which, based on our power analysis, the number of cognitively healthy centenarians was at least half of the number of age‐matched controls to achieve the same power. For the functional analysis, we used the functional annotation section of snpXplorer web server with default settings. 27 This tool performs (1) variant‐to‐gene mapping using integrating variant consequences (coding, intronic, intergenic) and quantitative trait loci (eQTLs and sQTLs), followed by (2) gene‐set enrichment analysis, and (3) clustering of the enriched terms. 27 The clusters of enriched terms were compared to clusters obtained from a previous study including all AD‐associated SNPs based on the same method. 27
2.7. Implementation
Quality control of the genotype data, population stratification analysis, and relatedness analyses were performed with PLINK (v1.90 and v2.0). Association analyses, downstream analyses, and plots were performed with R (v4.2). For the power analyses, we adapted the likelihood ratio test framework implemented in the R package genpwr. 28 The scripts are publicly available at https://github.com/TesiNicco/Centenarians_AD.
3. RESULTS
3.1. Quality control of genetic data and SNPs
The mean age at study inclusion of the 2281 AD cases was 67.96 ± 9.84 (interquartile range [IQR] = [61–74], 55% females, of which 971 were early‐onset AD with age at diagnosis ≤ 65 years), the mean age of the 3165 age‐matched controls was 62.57 ± 8.66 (IQR = [57–66], 48% females), and the mean age of the 346 cognitively healthy centenarians was 101.05 ± 2.51 (IQR = [100–102], 71% females; Table 1, Figure S1 in supporting information). The median quality of the imputed SNPs was r 2 = 0.95 and ranged from 0.45 to 0.99 (Table S2 in Supplementary Tables); all imputed SNPs were included in the analyses.
TABLE 1.
Population characteristics.
| AD cases | Age‐matched controls | Cognitively healthy centenarians | |
|---|---|---|---|
| Sample size | 2281 | 3165 | 346 |
| Age | 67.96 ± 9.84 | 62.57 ± 8.66 | 101.05 ± 2.51 |
| Females (%) | 1265 (55%) | 1507 (48%) | 247 (71%) |
| APOE ε2 (%) | 3% | 9% | 13% |
| APOE ε4 (%) | 43% | 17% | 7% |
Note: Additional information about the cohorts used is available in Methods and Supplementary Methods (in supporting information) sections. Reference to the cohorts described in this table are: Holstege et al., 5 van der Flier and Scheltens, 17 Rademaker et al., 20 Hoogendijk et al.,21 Willemsen et al., 22 and Slot et al. 29 Age = age at onset for AD cases, age at study inclusion for age‐matched controls, and cognitively healthy centenarians.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E.
3.2. AD cases versus cognitively healthy centenarians
Comparing AD cases to cognitively healthy centenarians, the effect size across all 86 tested SNPs increased by a median 1.78‐fold (IQR: 0.51–2.85) relative to the published effect sizes; Figure 1; Figure S2, Table S3 and Table S4 in Supplementary Tables). 10 Overall, a relatively small difference in allele frequency may lead to a large increase in effect size (Figure 1). For 59 SNPs the change in effect size was > 1 (P = 3.6 × 10−4 based on a one‐tailed binomial test, Figure 1) and ranged from 1.07 (rs785129 near HS3ST5 gene) to 5.91 (rs112403360 in ANKH gene, Table S3). Cognitively healthy centenarians did not include carriers of the rare rs60755019 (in TREML2), while the carriers frequency in AD cases was 0.18% and 0.14% in age‐matched controls (Table S4). For nine SNPs (in or near the genes EPDR1, MAF, PLCG2, RIN3, ANKH, TMEM106B, SORT1, GRN, and WDR12), the effect size was increased more than 4‐fold compared to previously published effect sizes (change > 4). The effect of 16 SNPs was not increased compared to the reference effect sizes (0 < change < 1, Figure 1 and Table S3), and the effect of 11 SNPs was opposite compared to the reference effects (change < 0, Figure 1, Figure S2 and Table S3). Despite the small sample size of cognitively healthy centenarians, the association for 8 out of 85 SNPs with AD reached significance after correction for multiple testing (false discovery rate [FDR] < 5%): ANKH, GRN, PLCG2, RIN3, ABCA7, BIN1, and the two APOE SNPs, Figure 1 and Table S3). We note that in a sensitivity analysis comparing early‐onset AD to late‐onset AD, we observed one significant association after multiple testing correction (rs7384878 in/near ZCWPW1, FDR < 5%, Table S5 in Supplementary Tables).
FIGURE 1.
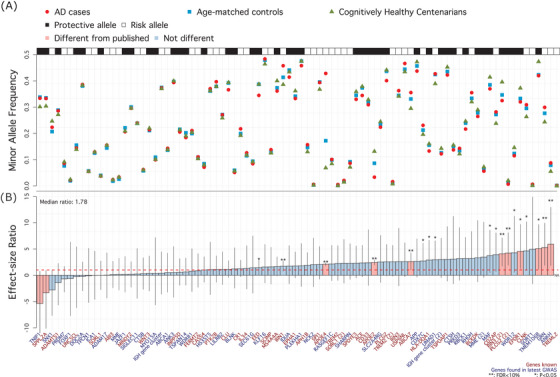
Single variant associations summary. A (top), Raw minor allele frequency in AD patients (red circles), age‐matched controls (blue squares), and cognitively healthy centenarians (green triangles). Black and white annotation squares refer to whether the plotted allele (the minor allele) was associated with an increased risk of AD (risk allele, white) or a decreased risk of AD (protective allele, black). B (bottom), Change in effect size comparing observed effect sizes (AD cases vs. cognitively healthy centenarians) to the reference effect sizes (Bellenguez et al. 10 ). Blue genes refer to novel SNP–AD associations discovered by Bellenguez et al. for the first time, while red genes were known before Bellenguez et al. The dashed red line at 1 indicates the published effect size from the literature. *: P value of association < 0.05; **: FDR‐corrected P value of association < 0.05; pink bars indicate SNPs for which observed effect size is significantly different from published effect size. AD, Alzheimer's disease; FDR, false discovery rate; SNP, single nucleotide polymorphism.
3.3. AD cases versus age‐matched controls
The 2281 AD patients have mainly early‐onset AD, such that they are likely enriched with risk‐increasing genetic variants relative to the predominantly late‐onset AD cases included in Bellenguez et al. 10 Therefore, our AD dataset may explain part of the change in effect sizes observed in our AD versus cognitively healthy centenarians analysis. To investigate the contribution of the AD cases, we compared them to 3165 age‐matched controls. We observed a 1.16‐fold increased effect size relative to the published effect sizes (IQR: 0.60–1.76), which is significantly lower than the 1.78‐fold increased effect size in the comparison of AD cases versus cognitively healthy centenarians (P = 0.004 comparing the distributions of effect size change, Figure S3 and Table S6 in Supplementary Tables). The change in effect size was > 1 for 48 SNPs and ranged from 1.01 (rs73223431 near PTK2B gene) to 4.47 (rs141749679 near SORT1 gene). In total, a significant association after multiple test corrections (FDR < 5%) was identified for 11 SNPs, in or near SORT1, RHOH, PLCG2, HLA‐DQA1, EED, RIN3, APH1B, TREM2, BIN1, and the two APOE SNPs, Table S6).
3.4. Age‐matched controls versus cognitively healthy centenarians
Next, we investigated whether the AD‐associated SNPs differentially contribute to maintaining cognitive health at old age compared to maintaining cognitive health at younger ages. For this, we compared the effect sizes of age‐matched controls to cognitively healthy centenarians and found that they were increased by a median 0.58‐fold (IQR: −0.23 to 1.45) relative to the published effect sizes in Bellenguez et al. (Figure S4 and Table S7 in Supplementary Tables). 10 The change in effect size was > 2‐fold for 17 SNPs, and 1‐ to 2‐fold for 13 SNPs. The effect sizes of 29 SNPs were not increased compared to the reference effects, and the effect of 27 SNPs was opposite. Altogether, a significant association after multiple test corrections (FDR < 5%) was identified only for the two APOE SNPs, Table S7).
3.5. Polygenic risk score
We assigned two PRSs to each subject, one including the weighted effect of all 86 SNPs, and a second excluding the effect of the two APOE SNPs (Figure 2). Then, we compared the distribution of the PRSs among AD cases, age‐matched controls, and cognitively healthy centenarians (Figure 2 and Table S8 in Supplementary Tables). In all comparisons, the PRSs in AD cases was significantly higher. AD patients versus age‐matched controls, excluding the two APOE SNPs: odds ratio [OR] = 1.54, 95% confidence interval [CI] = [1.45–1.63], P = 1.55 × 10−47; including APOE SNPs: OR = 2.55, 95% CI = [2.39–2.72], P = 2.09 × 10−176). AD patients versus cognitively healthy centenarians, excluding the two APOE SNPs: OR = 1.97, 95% CI = [1.74–2.23], P = 2.75 × 10−26; including APOE SNPs, OR = 5.07, 95% CI = [4.25–6.06], P = 1.54 × 10−71). We found a significantly lower PRS in centenarians compared to age‐matched controls, excluding APOE SNPs: OR = 0.77, 95% CI = [0.69–0.88], P = 2.57 × 10−5; including APOE SNPs, OR = 0.53, 95% CI = [0.46–0.62], P = 2.92 × 10−17. Notably, all analyses remained significant after splitting by sex. The only exception was that the PRS between cognitively healthy centenarian males and healthy control males (PRS without APOE) lost its statistical significance, likely attributable to the limited number of male centenarians (N = 99, OR = 1.14, 95% CI = [0.92–1.41], P = 2.44 × 10‐1; Table S8). Also, in this sample, the distribution of PRSs of the early‐onset AD cases was not different from the late‐onset AD cases (P > 0.05, Table S8).
FIGURE 2.
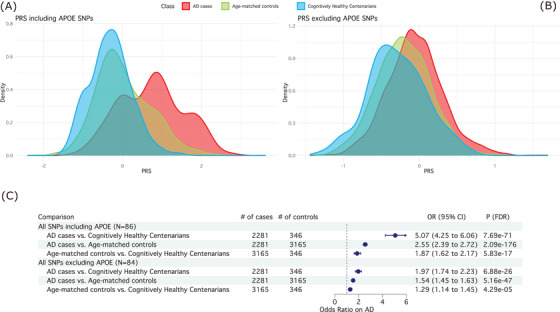
Summary of PRS analyses. A (top left), Distribution of the PRS including the two APOE SNPs (86 SNPs in total) in AD cases (red), age‐matched controls (blue), and cognitively healthy centenarians (green). B (top right), Distribution of the PRS excluding the two APOE SNPs (84 SNPs in total). C (bottom), Association statistics (OR, 95% CI, and corrected P value) and forest plot of the PRS including and excluding APOE SNPs. For the comparisons, we used logistic regression models in a pairwise manner (i.e., AD cases vs. cognitively healthy centenarians, AD cases vs. age‐matched controls, and age‐matched controls vs. cognitively healthy centenarians), controlling for population substructure. AD, Alzheimer's disease; APOE, apolipoprotein E; CI, confidence interval; OR, odds ratio; PRS, polygenic risk score; SNP, single nucleotide polymorphism.
3.6. The contribution of a centenarian
In a simulation, we estimated the number of age‐matched controls and cognitively healthy centenarians required to reach 80% power to find an association at P = 0.05, we used a subset of 67 common SNPs for which the direction of effect in our analyses matched that of Bellenguez et al. (see section 2.5: The contribution of a centenarian; Table S3, and Table S9 in Supplementary Tables). For eight SNPs, a total of 16,000 controls did not guarantee the power of 80% (i.e., no convergence) using both age‐matched controls and cognitively healthy centenarians, which is likely due to the small effect sizes associated with these SNPs (Figure 3A and Table S9). For the remaining 59 SNPs, an association at P = 0.05 (convergence) was observed comparing 8000 AD cases with on average 6183 ± 5680 age‐matched controls (median = 3600, IQR = 2300–8800) or 3745 ± 5436 cognitively healthy centenarians (median = 1200, IQR = 600–3300; Figure 3 and Table S9). On average, based on 59 AD SNPs, and specifically within our cohort of individuals, the power of a single cognitively healthy centenarian in a GWAS of AD is equivalent to that of 5.86 typical age‐matched controls (median = 2.4, IQR = [1.00–6.58], Figure 3 and Table S9).
FIGURE 3.
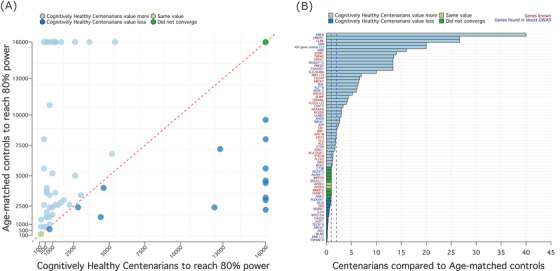
Relationship between cognitively healthy centenarians and age‐matched controls. A (left), Number of individuals (age‐matched controls on the y axis and centenarians on x axis) necessary to achieve 80% power for a SNP association at P = 0.05, assuming 8000 AD cases. We restricted this analysis to common variants (MAF > 1%) with expected direction of effect in our comparisons (N = 67 SNPs, see Methods). Note that, for this reason, some variants enriched in cognitively healthy centenarians such as rs13237518 (TMEM106B) and rs13237518 (SORT1) could not be represented here. Each dot represents a SNP: dark green dots identify the eight SNPs that did not converge using both age‐matched controls and centenarians (i.e., the power did not reach 80%). Light green dots indicate the two APOE SNPs, for which N = 200 individuals (age‐matched controls and centenarians) were enough to guarantee 80% power. Light blue dots identify SNPs for which the number of cognitively healthy centenarians (to achieve 80% power) was lower than the number of age‐matched controls. Of these, N = 13 SNPs did not converge using age‐matched controls. Conversely, dark blue dots identify SNPs for which the number of age‐matched controls was lower than the number of cognitively healthy centenarians. Of these, N = 8 SNPs did not converge using cognitively healthy centenarians. B (right), Ratio between the number of age‐matched controls and the number of cognitively healthy centenarians, for each SNP. Color code is the same as (A). SNPs larger than the blue dotted line (N = 31, ratio > 2) were used for functional annotation and gene‐set enrichment analysis. AD, Alzheimer's disease; APOE, apolipoprotein E; GWAS, genome‐wide association study; MAF, minor allele frequency; SNP, single nucleotide polymorphism.
3.7. Functional implications
We then functionally annotated and performed gene‐set enrichment analysis using 31 SNPs for which the power of a single centenarian was > 2‐fold increased than age‐matched controls (Figure 3B). Of 31 SNPs, only 2 were coding (rs143332484 in TREM2 and rs72824905 in PLCG2); 23 were annotated to their likely affected gene(s) using eQTL, sQTL, and Combined Annotation Dependent Depletion (CADD) information; and 6 SNPs were annotated solely based on their genomic position (Table S10 in Supplementary Tables). The resulting genes were used as input for gene‐set enrichment analysis. After clustering the enriched Gene Ontology (GO) terms based on a semantic similarity measure, we found two clusters of pathways, pointing toward the immune system and endo‐lysosomal trafficking (Figure 4A‐B and Table S11 in Supplementary Tables). The immune system cluster of pathways included activation and regulation of immune response (genes CR1, MS4A6A, IGH‐cluster, RIN3, KAT8, GRN, SCIMP, RBCK1, APP, RHOH, OTULIN, MAPK9, PLCG2, and TREM2), leukocyte activation and differentiation (genes CD55, CR1, IGH‐cluster, APP, GRN, PLCG2, and TREM2), macrophage activation (genes GRN, APP, PLCG2, and TREM2), and neuroinflammatory response (genes GRN, LILRA5, PLCG2, KAT8, and TREM2). The endo‐lysosomal trafficking cluster of pathways included marked immunological aspects: endocytosis and phagocytosis (genes IGH‐cluster, RIN3, ABCA7, LILRB4, APP, RHOH, PLCG2, and TREM2), interleukin‐6 metabolism (genes SCIMP, LILRA5, APP, PLCG2, and TREM2), and amyloid clearance (genes ABCA7, MME, APP, and TREM2) (Figure 4C, Table S11). We compared these clusters to five clusters from a previous study including all AD‐associated SNPs. 27 A significant overlap was found only between the endolysosomal trafficking cluster (this analysis) and (1) the amyloid clearance cluster (previous study, chi‐square P = 3.38 × 10−5), and (2) immune trafficking and migration cluster (previous study, chi‐square P = 2.07 × 10−4). Conversely, no significant overlap was found regarding clusters of pathways pointing to activation of immune response (P = 0.49), organization and metabolic processes, and amyloid beta (Aβ) and tau formation.
FIGURE 4.
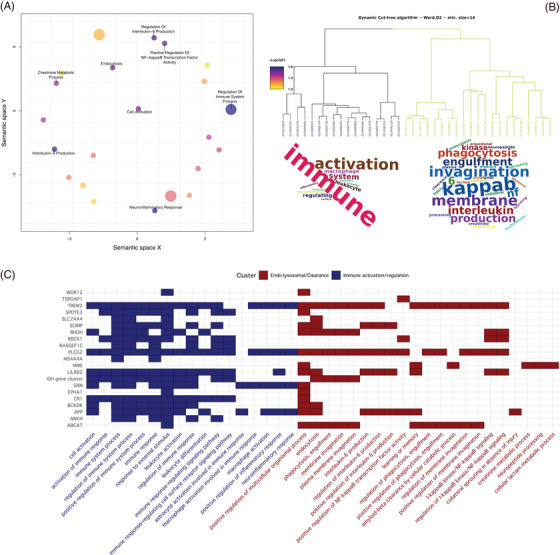
Functional annotation of SNPs with the largest effect in cognitively healthy centenarians. The figure shows the result of the functional annotation of 31 SNPs for which the number of cognitively healthy centenarians required to achieve 80% power was at least half of the number of age‐matched controls required to achieve the same power. Functional annotation analysis was performed using snpXplorer. 27 A, Result of the gene‐set enrichment analysis followed by REVIGO analysis, which clusters enriched pathways based on a semantic similarity measure. B, Dendrogram of the main enriched pathways along with their cluster (branches color code for cluster assignment) and word clouds showing the main terms enriched in the underlying pathways. C, Mapping between significant pathways (x axis), AD‐associated SNPs (y axis, labeled with the name of the gene as provided by Bellenguez et al. 10 ), and the relative gene‐set enrichment cluster. AD, Alzheimer's disease; SNP, single nucleotide polymorphism.
4. DISCUSSION
Based on common AD‐associated SNPs as identified by GWAS, self‐reported cognitively healthy centenarians from the 100‐plus Study are genetically protected against AD. Cognitively healthy centenarians have a lower frequency of almost all risk‐increasing alleles and a higher frequency of protective alleles, which indicates that maintaining cognitive health depends on having an advantageous function across all genetically associated mechanisms. However, the centenarians are most strongly depleted with the risk alleles in ANKH, GRN, and SORT1, and most strongly enriched with the protective alleles in TMEM106B, EPDR1, PLCG2 (rs72824905), and RIN3 (rs12590654). For these alleles, the effect sizes were > 4‐fold increased comparing AD cases to cognitively healthy centenarians rather than age‐matched controls. Together, our findings suggest that prolonged cognitive health depends on maintaining specific aspects of the endolysosomal and immune system, and on the resistance of accumulating neuropathology.
The centenarians had a significantly lower PRS for AD compared to middle‐aged healthy individuals, both including and excluding the effect of the two APOE alleles. The effect size across risk alleles was increased by an average 1.78‐fold comparing AD cases to cognitively healthy centenarians as controls rather than age‐matched controls. These effects were confirmed in both males and females. However, the increase in effect size concentrates on specific alleles, indicating that prolonged cognitive health especially depends on the maintenance of the associated cellular processes.
Cognitively healthy centenarians are most strongly enriched with the protective allele of the ANKH gene (rs112403360), which is associated with hippocampal sclerosis and Braak neurofibrillary tangles stages. 30 Impairment of the ANKH gene leads to excessive mineralization, including calcification of arteries leading to joint pain, arthritis, atherosclerosis, and diabetes. 31 , 32 Together, this suggests that the prolonged cognitive health in centenarians may be supported by maintained vasculature and low pathology load in the brain.
Furthermore, it is intriguing that the protective alleles of the GRN‐, TMEM106B‐, and the SORT1‐associated loci are among the strongest enriched in cognitively healthy centenarians, as these three genes all contribute to endolysosomal trafficking. 33 , 34 , 35 It is notable that these loci were previously identified in context of frontotemporal lobar degeneration (FTLD) risk. 36 , 37 This might suggest that these FTLD risk alleles also influence the risk of AD; that some AD patients may have FTLD as a comorbidity; or that FTLD patients were misdiagnosed as AD patients, influencing the GWAS. 30 Regardless of rationale, the strong enrichment of these three alleles underlines the importance of a functional endolysosomal trafficking mechanism in maintained cognitive health during aging. This is further supported by a strong enrichment of the protective allele of RIN3 (rs12590654 and rs7401792), the function of which is also associated with endolysosomal function and axonal trafficking. 38
EPDR1 (mammalian ependymin‐related protein 1) is a transmembrane protein that plays a crucial role in adhesion of neural cells. 39 Although its role in AD is currently not clear, EPDR1 was shown to be downregulated in AD patients compared to controls, 40 and has been implicated in dopaminergic regulation of neurogenesis and neuroendocrine function in goldfish. 41 While speculative, our finding that cognitively healthy centenarians are enriched with a protective EPDR1 allele may confirm a role for prolonged neurogenesis in maintaining cognitive health. 42
Protective alleles in genes modulating immune and neuroinflammatory response (PLCG2, CR1, TREM2, OTULIN, MS4A‐cluster) were strongly enriched in cognitively healthy centenarians, suggesting that maintaining an efficient regulation of neuro‐immune response during aging is an important aspect of cognitive health. Notably, the protective coding SNP rs72824905, leading to the gain‐of‐function p.P522R substitution in PLCG2, provides proof of concept that only a limited increase in immune activation translates to a beneficial effect, as stronger gain‐of‐function mutations in PLCG2 (e.g., p.S707Y and p.L848P) are associated with autoimmune disorders such as PLCγ2‐associated antibody deficiency and immune dysregulation syndrome (PLAID) and autoinflammation, antibody deficiency, and immune dysregulation syndrome (APLAID). 13 , 43 , 44
TREM2 is well known to be involved in microglial activation and phagocytosis in the same pathway as PLCG2. The protective allele of the rs75932628 coding SNP in TREM2 (i.e., the arginine at residue 74), was enriched in the centenarians and was shown to increase microglial activation and expression of proinflammatory cytokines. 45 Altogether, a slightly more responsive immune and neuroinflammatory response in cognitively healthy centenarians seems to better cope with the physiological accumulation of pathology over time and promote a long‐term maintenance of cognitive health. 46
The protective alleles of SNPs near ABCA7 (rs12151021), SORL1 (rs74685827), APP (rs2154481), and APOE (rs429358 and rs7412) were all enriched in cognitively healthy centenarians. These genes are involved in immune–lipid signaling pathways that lead to the clearance of amyloid peptides in the brain. 47 Specifically, the ABCA7 gene is involved in Aβ processing and clearance, while the SORL1 gene codes for a retromer receptor involved in the trafficking of amyloid precursor protein (APP), thereby preventing Aβ secretion. 13 , 48 Interestingly, in the brains donated by cognitively healthy centenarians we observed Aβ deposits across many regions; however, the load of Aβ neuropathology remained limited. 49 This suggests that enrichment of protective alleles may support the resistance of the accumulation of amyloid pathology.
Finally, we expect that the genetically driven enhancements of conserved molecular mechanisms will have limited functional effects, as impactful changes are likely to have damaging effects. This is exemplified by the limited functional effects of strongly protective coding variants in PLCG2, APP, and APOE. 13 , 46 , 50 , 51
While the main aim of this study was to identify the AD‐associated genetic loci that most prominently associated with escaping AD, our study also suggests that genetic comparisons of diseased individuals to those who are resilient to the disease maximize the identified effect sizes. The comparison of AD patients and age‐matched controls yielded effect sizes comparable to published effect sizes, highlighting that the observed increased effects were mostly due to the cognitively healthy centenarians. In fact, 84% (37/44) of the SNPs that were associated with AD for the first time in Bellenguez et al. had the same direction of effects, despite their very small effect sizes. We were able to replicate the association of 8 SNPs at FDR < 5%, while a comparison of these AD cases with more than 10 times the number of age‐matched controls allows for the replication of 11 SNPs. Together, we estimated that the contribution of one centenarian to a case–control analysis is equivalent to on average six age‐matched controls. While this highlights the power of analyzing extreme phenotypes, we acknowledge that assembling a sufficiently large cohort of cognitively healthy centenarians for the discovery of novel disease loci in a case–control comparison is challenging. 5 Furthermore, because maintained cognitive health concurs with extreme longevity in our centenarians, the effect sizes on pure AD risk of such newly identified loci would have to be determined in a (targeted) age‐matched comparison between AD cases and age‐matched controls. Indeed, prior analyses have demonstrated that cognitively healthy centenarians carry an abundance of longevity‐related genetic variations, some of which might even alleviate the adverse effect of APOE ε4 allele. 42 , 51 We acknowledge that, given the association between maintained cognitive health and maintained physical health, 52 genetic differences associated with resilience against AD may also be representative of overall survival until extreme age. However, the 86 SNPs studied here were all discovered in an age‐matched GWAS of AD and, except for APOE‐ and HLA‐associated SNPs, these are not detected by previous GWASs of survival, longevity, and/or other age‐related diseases. 14 , 53 , 54 , 55 , 56 This suggests that the tested alleles may be in large part representative of resilience to AD rather than overall decline, and that the differential effect sizes when using cognitively healthy centenarians as controls points toward the most important mechanisms associated with escaping AD and other dementias until old age. In addition, to disentangle the genetics underlying AD resilience, it would be ideal to compare cognitively healthy centenarians to centenarians who are affected with AD, representing an estimated 75% of all centenarians in the population. 57 However, ethical considerations precluded the inclusion of centenarians affected with AD in the 100‐plus Study. Therefore, for this analysis, we had to refrain to comparisons to unaffected and affected younger individuals.
Our study was conducted in a genetically homogeneous population: cognitively healthy centenarians as well as AD cases and age‐matched controls are all from the same Dutch (White) population, and we are aware that genetic associations with AD differ among individuals from different ancestries, 58 which likely extends to the genetics associated with the long‐term maintenance of cognitive health. We acknowledge that AD patients, age‐matched controls, and cognitively healthy centenarians were from different studies, each with their own inclusion criteria: individuals with subjective cognitive decline (included as age‐matched controls) and the AD patients presented at the clinic with complaints, while the participants of the 100‐plus and LASA studies were actively approached for study inclusion. Therefore, we cannot exclude that comparisons were affected by inclusion biases introduced by differences between individuals willing to contribute to research and those seeking care for clinical complaints. Last, we acknowledge that part of the individuals used in this study was also included in the GWAS study we used as a reference. However, these individuals represent < 2% of all AD cases included in the GWAS, and < 0.5% of all controls included, a negligible fraction.
In summary, we find that cognitively healthy centenarians are genetically protected against AD and that the alleles with the largest effects are involved in sustaining specific aspects of the immune and endolysosomal systems, which may prevent accumulation of amyloid and other neuropathological hallmarks of AD.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
The medical ethics committee of the Amsterdam UMC approved all studies. All participants and/or their legal representatives provided written informed consent for participation in clinical and genetic studies.
Supporting information
Supplementary Methods and Figures
Supplementary Tables
Disclosure Form
ACKNOWLEDGMENTS
Niccolò Tesi is appointed at ABOARD. Sven van der Lee, Henne Holstege, Marcel Reinders and Wiesje van der Flier are recipients of ABOARD. ABOARD is a public–private partnership receiving funding from ZonMW National Dementia program (#73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; #LSHM20106). More than 30 partners, including de Hersenstichting (Dutch Brain Foundation) participate in ABOARD (https://www.alzheimer‐nederland.nl/onderzoek/projecten/aboard).
Tesi N, van der Lee S, Hulsman M, et al. Cognitively healthy centenarians are genetically protected against Alzheimer's disease. Alzheimer's Dement. 2024;20:3864–3875. 10.1002/alz.13810
REFERENCES
- 1. United Nations, Department of Economic and Social Affair 2019. Retrieved from Profiles of Ageing 2019. 2019.
- 2. Nichols E, Szoeke CEI, Vollset SE, et al. Global, regional, and national burden of Alzheimer's disease and other dementias, 1990‐2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18:88‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. 2012 Alzheimer's disease facts and figures. Alzheimer's & Dementia. 2012;8:131‐168. [DOI] [PubMed] [Google Scholar]
- 4. Perls T. Dementia‐free centenarians. Exp Gerontol. 2004;39:1587‐1593. [DOI] [PubMed] [Google Scholar]
- 5. Holstege H, Beker N, Dijkstra T, et al. The 100‐plus Study of cognitively healthy centenarians: rationale, design and cohort description. Eur J Epidemiol. 2018;33:1229‐1249. doi: 10.1007/s10654-018-0451-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winblad B, Amouyel P, Andrieu S, et al. Defeating Alzheimer's disease and other dementias: a priority for European science and society. The Lancet Neurology. 2016;15:455‐532. [DOI] [PubMed] [Google Scholar]
- 7. Corrada MM, Brookmeyer R, Paganini‐Hill A, Berlau D, Kawas CH. Dementia incidence continues to increase with age in the oldest old: the 90+ study. Ann Neurol. 2010;67:114‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holstege H, Hulsman M, Charbonnier C, et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer's disease. Nat Genet. 2022;54:1786‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168‐174. [DOI] [PubMed] [Google Scholar]
- 10. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer's risk‐altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 2013;33:13320‐13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sebastiani P, Perls TT. The genetics of extreme longevity: lessons from the new England centenarian study. Front Genet. 2012;3:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seto M, Weiner RL, Dumitrescu L, Hohman TJ. Protective genes and pathways in Alzheimer's disease: moving towards precision interventions. Mol Neurodegeneration. 2021;16:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joshi PK, Pirastu N, Kentistou KA, et al. Genome‐wide meta‐analysis associates HLA‐DQA1/DRB1 and LPA and lifestyle factors with human longevity. Nat Commun. 2017;8(1):910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DESGESCO (Dementia Genetics Spanish Consortium), EADB (Alzheimer Disease European DNA biobank), EADB (Alzheimer Disease European DNA biobank), IFGC (International FTD‐Genomics Consortium), IPDGC (The International Parkinson Disease Genomics Consortium), IPDGC (The International Parkinson Disease Genomics Consortium), RiMod‐FTD (Risk and Modifying factors in Fronto‐Temporal Dementia), Netherlands Brain Bank (NBB) , et al. A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer's disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol. 2019;138:237‐250. doi: 10.1007/s00401-019-02026-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tesi N, van der Lee SJ, Hulsman M, et al. Centenarian controls increase variant effect sizes by an average twofold in an extreme case–extreme control analysis of Alzheimer's disease. Eur J Hum Genet. 2018;27(2):244‐253. doi: 10.1038/s41431-018-0273-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Flier WM, Scheltens P. Amsterdam dementia cohort: performing research to optimize care. Journal of Alzheimer's Disease. 2018;62:1091‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dreves MAE, van Harten AC, Visser LNC, et al. Rationale and design of the ABOARD project (A Personalized Medicine Approach for Alzheimer's Disease). A&D Transl Res & Clin Interv. 2023;9:e12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Flier WM, de Vugt ME, Smets EMA, Blom M, Teunissen CE. Towards a future where Alzheimer's disease pathology is stopped before the onset of dementia. Nat Aging. 2023;3:494‐505. [DOI] [PubMed] [Google Scholar]
- 20. Rademaker MC, de Lange GM, Palmen SJMC. The Netherlands brain bank for psychiatry. Handbook of Clinical Neurology. Elsevier; 2018:3‐16. [DOI] [PubMed] [Google Scholar]
- 21. Hoogendijk EO, Deeg DJH, Poppelaars J, et al. The Longitudinal Aging Study Amsterdam: cohort update 2016 and major findings. Eur J Epidemiol. 2016;31:927‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willemsen G, de Geus EJC, Bartels M, et al. The Netherlands twin register biobank: a resource for genetic epidemiological studies. Twin Research and Human Genetics. 2010;13:231‐245. [DOI] [PubMed] [Google Scholar]
- 23. de Rojas I, Moreno‐Grau S, Tesi N, et al. Common variants in Alzheimer's disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12:3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43:436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson CA, Pettersson FH, Clarke GM, Cardon LR, Morris AP, Zondervan KT. Data quality control in genetic case‐control association studies. Nat Protoc. 2010;5:1564‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. 1000 Genomes Project Consortium , Auton A, Brooks LD, et al, 1000 Genomes Project Consortium . A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tesi N, van der Lee S, Hulsman M, Holstege H, Reinders MJT. snpXplorer: a web application to explore human SNP‐associations and annotate SNP‐sets. Nucleic Acids Res. 2021;49:W603‐W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore CM, Jacobson SA, Fingerlin TE. Power and sample size calculations for genetic association studies in the presence of genetic model misspecification. Hum Hered. 2019;84:256‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Slot RER, Verfaillie SCJ, Overbeek JM, et al. Subjective Cognitive Impairment Cohort (SCIENCe): study design and first results. Alzheimer's Research & Therapy. 2018;10(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Katsumata Y, Shade LM, Hohman TJ, et al. Multiple gene variants linked to Alzheimer's‐type clinical dementia via GWAS are also associated with non‐Alzheimer's neuropathologic entities. Neurobiol Dis. 2022;174:105880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nugent S, Potvin O, Cunnane SC, Chen T‐H, Duchesne S. Associating Type 2 diabetes risk factor genes and FDG‐PET brain metabolism in normal aging and Alzheimer's disease. Front Aging Neurosci. 2020;12:580633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pendleton A, Johnson MD, Hughes A, et al. Mutations in ANKH cause chondrocalcinosis. Am Hum Genet. 2002;71:933‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rhinn H, Abeliovich A. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that Regulate Aging Phenotypes. Cell Syst. 2017;4:404‐415. .e5. [DOI] [PubMed] [Google Scholar]
- 34. Finch N, Carrasquillo MM, Baker M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simon MJ, Logan T, DeVos SL, Di Paolo G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. 2023;33:324‐339. [DOI] [PubMed] [Google Scholar]
- 36. Van Deerlin VM, Sleiman PMA, Martinez‐Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP‐43 inclusions. Nat Genet. 2010;42:234‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baker M, Mackenzie IR, Pickering‐Brown SM, et al. Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916‐919. [DOI] [PubMed] [Google Scholar]
- 38. Shen R, Zhao X, He L, et al. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer's disease. Transl Neurodegener. 2020;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shashoua VE. Ependymin, a brain extracellular glycoprotein, and CNS plasticity. Ann NY Acad Sci. 1991;627:94‐114. [DOI] [PubMed] [Google Scholar]
- 40. Li M, Geng R, Li C, et al. Dysregulated gene‐associated biomarkers for Alzheimer's disease and aging. Translational Neuroscience. 2021;12:83‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xing L, Martyniuk CJ, Esau C, Da Fonte DF, Trudeau VL. Proteomic profiling reveals dopaminergic regulation of progenitor cell functions of goldfish radial glial cells in vitro. J Proteomics. 2016;144:123‐132. [DOI] [PubMed] [Google Scholar]
- 42. Tesi N, van der Lee SJ, Hulsman M, et al. Polygenic risk score of longevity predicts longer survival across an age continuum. The Journals of Gerontology: Series A. 2020;76(5):750‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koss H, Bunney TD, Behjati S, Katan M. Dysfunction of phospholipase Cγ in immune disorders and cancer. Trends Biochem Sci. 2014;39:603‐611. [DOI] [PubMed] [Google Scholar]
- 44. Kutukculer N, Topyildiz E, Berdeli A, et al. Four diseases, PLAID, APLAID, FCAS3 and CVID and one gene (PHOSPHOLIPASE C, GAMMA‐2; PLCG2): striking clinical phenotypic overlap and difference. Clin Case Rep. 2021;9:2023‐2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong L, Chen X‐F, Wang T, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Diks AM, Teodosio C, De Mooij B, et al. Carriers of the p.P522R variant in PLCγ2 have a slightly more responsive immune system. Mol Neurodegeneration. 2023;18:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Deming Y, Filipello F, Cignarella F, et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer's disease risk. Sci Transl Med. 2019;11:eaau2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mishra S, Knupp A, Szabo MP, et al. The Alzheimer's gene SORL1 is a regulator of endosomal traffic and recycling in human neurons. Cell Mol Life Sci. 2022;79:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang M, Ganz AB, Rohde S, et al. The correlation between neuropathology levels and cognitive performance in centenarians. Alzheimer's & Dementia. 2023;19(11):5036‐5047. [DOI] [PubMed] [Google Scholar]
- 50. Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature. 2012;488:96‐99. [DOI] [PubMed] [Google Scholar]
- 51. Arboleda‐Velasquez JF, Lopera F, O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beker N, Sikkes SAM, Hulsman M, et al. Longitudinal maintenance of cognitive health in centenarians in the 100‐plus Study. JAMA Netw Open. 2020;3:e200094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deelen J, Beekman M, Uh H‐W, et al. Genome‐wide association meta‐analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum Mol Genet. 2014;23:4420‐4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Deelen J, Evans DS, Arking DE, et al. A meta‐analysis of genome‐wide association studies identifies multiple longevity genes. Nat Commun. 2019;10(1):3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Timmers PR, Mounier N, Lall K, et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. eLife. 2019;8:e39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Timmers PRHJ, Wilson JF, Joshi PK, Deelen J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nat Commun. 2020;11:3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Poon LW, Woodard JL, Stephen Miller L, et al. Understanding dementia prevalence among centenarians. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2012;67A:358‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sherva R, Zhang R, Sahelijo N, et al. African ancestry GWAS of dementia in a large military cohort identifies significant risk loci. Mol Psychiatry. 2023;28:1293‐1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods and Figures
Supplementary Tables
Disclosure Form