

Abstract
INTRODUCTION:
Comparative epigenomics is an emerging field that combines epigenetic signatures with phylogenetic relationships to elucidate species characteristics such as maximum life span. For this study, we generated cytosine DNA methylation (DNAm) profiles (n = 15,456) from 348 mammalian species using a methylation array platform that targets highly conserved cytosines.
RATIONALE:
Nature has evolved mammalian species of greatly differing life spans. To resolve the relationship of DNAm with maximum life span and phylogeny, we performed a large-scale cross-species unsupervised analysis. Comparative studies in many species enables the identification of epigenetic correlates of maximum life span and other traits.
RESULTS:
We first tested whether DNAm levels in highly conserved cytosines captured phylogenetic relationships among species. We constructed phyloepigenetic trees that paralleled the traditional phylogeny. To avoid potential confounding by different tissue types, we generated tissue-specific phyloepigenetic trees. The high phyloepigenetic-phylogenetic congruence is due to differences in methylation levels and is not confounded by sequence conservation.
We then interrogated the extent to which DNA methylation associates with specific biological traits. We used an unsupervised weighted correlation network analysis (WGCNA) to identify clusters of highly correlated CpGs (comethylation modules). WGCNA identified 55 distinct comethylation modules, of which 30 were significantly associated with traits including maximum life span, adult weight, age, sex, human mortality risk, or perturbations that modulate murine life span.
Both the epigenome-wide association analysis (EWAS) and eigengene-based analysis identified methylation signatures of maximum life span, and most of these were independent of aging, presumably set at birth, and could be stable predictors of life span at any point in life. Several CpGs that are more highly methylated in long-lived species are located near HOXL subclass homeoboxes and other genes that play a role in morphogenesis and development. Some of these life span–related CpGs are located next to genes that are also implicated in our analysis of upstream regulators (e.g., ASCL1 and SMAD6). CpGs with methylation levels that are inversely related to life span are enriched in transcriptional start site (TSS1) and promoter flanking (PromF4, PromF5) associated chromatin states. Genes located in chromatin state TSS1 are constitutively active and enriched for nucleic acid metabolic processes. This suggests that long-living species evolved mechanisms that maintain low methylation levels in these chromatin states that would favor higher expression levels of genes essential for an organism’s survival.
The upstream regulator analysis of the EWAS of life span identified the pluripotency transcription factors OCT4, SOX2, and NANOG. Other factors, such as POLII, CTCF, RAD21, YY1, and TAF1, showed the strongest enrichment for negatively life span–related CpGs.
CONCLUSION:
The phyloepigenetic trees indicate that divergence of DNA methylation profiles closely parallels that of genetics through evolution. Our results demonstrate that DNA methylation is subjected to evolutionary pressures and selection. The publicly available data from our Mammalian Methylation Consortium are a rich source of information for different fields such as evolutionary biology, developmental biology, and aging.
Graphical Abstract
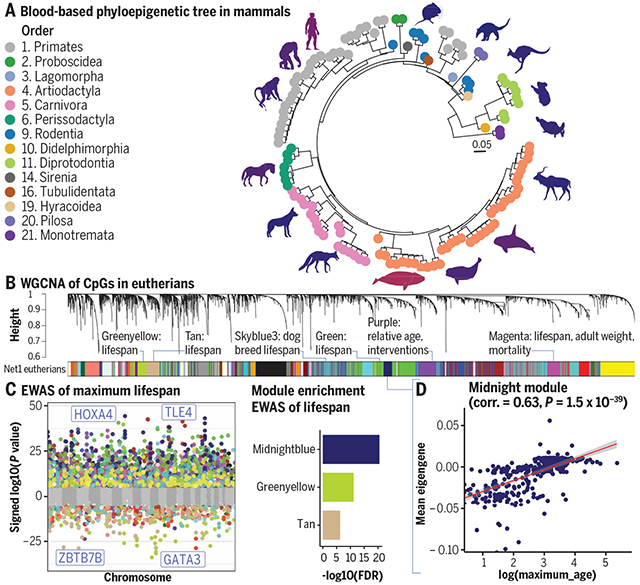
DNAm network relates to mammalian phylogeny and traits. (A) Phyloepigenetic tree from the DNAm data generated from blood samples. (B) Unsupervised WGCNA networks identified 55 comethylation modules. (C) EWAS of log-transformed maximum life span. Each dot corresponds to the methylation levels of a highly conserved CpG. Shown is the log (base 10)–transformed P value (y axis) versus the human genome coordinate Hg19 (x axis). (D) Comethylation module correlated with maximum life span of mammals. Eigengene (first principal component of scaled CpGs in the midnightblue module) versus log (base e) transformed maximum life span. Each dot corresponds to a different species.
Using DNA methylation profiles (n = 15,456) from 348 mammalian species, we constructed phyloepigenetic trees that bear marked similarities to traditional phylogenetic ones. Using unsupervised clustering across all samples, we identified 55 distinct cytosine modules, of which 30 are related to traits such as maximum life span, adult weight, age, sex, and human mortality risk. Maximum life span is associated with methylation levels in HOXL subclass homeobox genes and developmental processes and is potentially regulated by pluripotency transcription factors. The methylation state of some modules responds to perturbations such as caloric restriction, ablation of growth hormone receptors, consumption of high-fat diets, and expression of Yamanaka factors. This study reveals an intertwined evolution of the genome and epigenome that mediates the biological characteristics and traits of different mammalian species.
Comparative epigenomics is a burgeoning field that integrates epigenetic signatures with phylogenetic relationships to decipher gene-to-trait functions (1-3). Prior research has investigated the capacity of DNA methylation (DNAm) patterns in regulatory sequences to reflect evolutionary relationships among species (3, 4). A recent study compared methylation data across multiple animal species at orthologous gene promoters using a sequencing-based assay that did not specifically target conserved CpGs (4). Previous investigations faced limitations regarding the measurement platform, particularly the low sequencing depth at conserved CpGs and the sample size per species.
Our study overcomes these constraints in several ways. First, we used a measurement platform ensuring high effective sequencing depth at conserved CpGs, allowing for a more precise analysis of DNAm patterns in highly conserved DNA regions. Second, we increased the sample size per species, aiming for many samples per species. We profiled 348 species from 25 of the 26 mammalian taxonomic orders. This comprehensive dataset enables examination of phylogenetic relationships, comethylation relationships between cytosines, and their associations with maximum life span and other species characteristics.
We profiled 15,456 samples (Fig. 1A and table S1) using a methylation array platform that provides effective sequencing depth at highly conserved CpGs across mammalian species (5). This dataset is the product of the multinational Mammalian Methylation Consortium. In previous studies, we applied supervised machine learning methods to generate DNAm-based predictors of age called epigenetic clocks for numerous species (6-31).
Fig. 1. Phyloepigenetic trees parallel the mammalian evolutionary tree.

(A) The traditional phylogenetic tree from the TimeTree database (44) based on 321 (of 348) species in our study. A full description of the species in our study is reported in table S1. (B) Blood-based phyloepigenetic tree created from hierarchical clustering of DNAm data in this study (for additional analysis, see fig. S3, A and B). We formed the mean value per cytosine across samples for each species. The clustering used 1 minus the Pearson correlation (1-cor) as a pairwise dissimilarity measure and the average linkage method as intergroup dissimilarity. Phyloepigenetic trees for skin and liver can be found in fig. S2. Additional analyses, e.g., involving different choices of CpGs or intergroup dissimilarity measures, are reported in the supplementary materials (fig. S2). The colored bars reflect the branch height. (C) Scatter plot of the distances in blood phyloepigenetic (1-cor) versus the traditional evolutionary tree. (D) Scatter plots displaying the log-odds ratios of regions exhibiting phylogenetic signals relative to the TSS are presented. The phylogenetic signal is determined using Blomberg’s K statistic (32). In this analysis, CpGs were grouped into categories using sliding windows relative to the TSS. To assess enrichment, the Fisher’s exact overlap test was used, focusing on the top 500 CpGs displaying phylogenetic signals within each region. The red dots highlight the regions with the Fisher’s exact P value < 0.05. The results indicate notable enrichment (OR > 3) in certain intergenic and genic regions but not in promoters. For additional analysis, see fig. S4.
Here, we performed a large-scale cross-species unsupervised analysis of the entire dataset to reveal the relationship of DNAm with mammalian phylogeny. We show that we could construct phyloepigenetic trees that parallel traditional phylogenetic ones. We then proceed to interrogate the extent to which DNAm underpins specific biological traits by using unsupervised weighted correlation network analysis (WGCNA) to minimize the influence of bias on our observations. This approach identifies modules (clusters) of comethylated CpGs comethylation that are associated with species characteristics, including taxonomy, tissue type, sex, life span, and aging.
Results
Evolution and DNAm
We generated a dataset consisting of DNAm profiles of 15,456 DNA samples derived from 70 tissue types from 348 mammalian species using the mammalian methylation array (5). We evaluated whether methylation levels of cytosines (CpGs) in DNA sequences that are conserved across species would allow us to construct what could be called a phyloepigenetic tree. To avoid potential confounding by different tissue types, we generated tissue-specific phyloepigenetic trees (Fig. 1B and figs. S2 and S3). We defined the “congruence” between traditional phylogenetic trees and phyloepigenetic trees as the Pearson correlation coefficient between distances (branch length) based on phyloepigenetic trees and evolutionary distances in traditional phylogenetic trees. We observed high congruence (0.93) (Fig. 1C and fig. S2) for the blood-based phyloepigenetic tree (124 species) and lower congruence values for nonblood tissues (congruence = 0.58 for liver and 0.72 for skin; fig. S2). The lower congruence in liver (158 species) and skin (133 species) may have been due to potential variability in sampling between species. The varying congruence across tissue types shows that the CpG probes do not serve as genotyping proxies. The tissue dependence of congruence indicates that phyloepigenetic trees are derived based on differences in methylation levels and not sequence conservation. This point was also corroborated by three sensitivity analyses, which confirmed that the high congruence was indeed due to differences in methylation levels (see the supplementary text). In particular, the phyloepigenetic trees based on the 180 CpGs with the most significant detection P values across all 348 species still are congruent with traditional trees (fig. S2, F and G).
To identify CpGs that exhibit a pronounced phylogenetic signal in relation to methylation and phylogenetic trees, we used Blomberg’s K statistic (32). Among the top 500 CpGs showing significant phylogenetic signals (nominal Blomberg P < 0.001, selected by variance z score), we observed an enrichment in upstream intergenic regions [odds ratio (OR) = 1.4, Fisher’s exact P < 0.05; fig. S4B]. To further investigate regions with the strongest phylogenetic signal, we divided the data into groups relative to the transcriptional start sites (TSSs). This analysis also confirmed that intergenic regions exhibit significant phylogenetic signals (OR > 3, Fisher’s exact P < 0.05), whereas the promoter regions did not show such signals (Fig. 1D).
DNAm networks relate to individual and species traits
We used signed WGCNA, an unsupervised analysis (33), to cluster CpGs with similar methylation dynamics across samples into comethylation modules. We then summarized their methylation profiles as “module eigengenes.” The respective eigengenes of these modules were used to identify their potential correlations with various traits within and across mammalian species.
Our data analysis proceeded in two sequential phases. First, we developed several comethylation networks using data from 11,099 DNA samples from 174 species (discovery dataset finalized March 2021). A eutherian network [network 1 (Net1)] was formed from 14,705 conserved CpGs using this dataset (Fig. 2A). Later, we generated a second dataset of 4357 samples from 30 tissues of 240 mammalian species (174 new species and 66 that are represented in the discovery set), which were not used to define modules and were used as an independent validation set. All eutherian modules were present in the independent validation dataset according to module preservation statistics (corKME) (34), validating the presence of these modules (corKME > 0.43, P < 10−22; median corKME = 0.84) (fig. S5). These modules were designated with colors according to WGCNA convention (Fig. 2A). The smallest module (lavenderblush3) consisted of 33 CpGs and the largest (turquoise) had 1864 CpGs.
Fig. 2. DNAm network relates to species and individual characteristics in mammalian species.

(A) the WGCNA network of 14,705 conserved CpGs in eutherian species (Net1). The identified modules related to species or individual sample characteristics. Net1 modules were compared with eight additional networks (fig. S5). The modules with strong associations with species and sample characteristics are labeled below the dendrogram. Gray color indicates CpGs that are outside of modules. (B) Summary of the modules showing strong associations with species and individual sample characteristics. The plus and minus labels are the direction of association with each trait. (C) Top defined functional biological processes related to Net1 modules (for details, see fig. S9 and table S4). (D) Mammalian comethylation modules form clusters of proteins in the STRING protein-protein interaction (PPI) network. For the sake of visualization, the analysis was limited to the top 50 CpGs with the highest module membership value per module. Colors indicate mammalian Net1. The lollipop plot shows the global cluster coefficient (36) of the proteins within a module (up to 500 top CpGs) in a PPI network. Our permutation analysis matched the distribution of the original module sizes. We evaluated 1100 random permutations, i.e., 20 for each of the 55 modules. The boxplot reports the global clustering coefficient per module (y axis) versus permutation status: module resulting from a random selection of proteins (left) versus original module resulting from WGCNA (right). The modules with cluster coefficients larger than the maximum permutation cluster coefficient were considered as significant at P = 0.001. The dashed vertical line corresponds to the maximum global clustering coefficient observed in the 1100 random permutations.
To characterize the 55 modules with respect to species characteristics (e.g., maximum life span and average adult weight), module eigengenes were calculated in all samples (discovery and validation set combined, 331 eutherian species). Because information on taxonomic order, tissues, maximum life span, age, sex and adult weight of each species was available, we were able to assess whether any of the module eigengenes correlated with these traits. Of the 55 modules, 30 were found to be correlated with at least one trait (Fig. 2B, fig. S7, and table S3). Specifically, 15 modules were related to taxonomic orders such as primates, rodents, or carnivores (Fig. 2B and fig. S11). Ten modules related to tissue type (fig. S11), two to sex (fig. S11), one to age, seven to maximum life span, and four to average adult species weight. Some modules were related to multiple characteristics. In the following sections, we mainly focus on the modules that relate to mammalian maximum life span, adult weight, and age. Other modules related to taxonomic order, tissue type, and sex are described in the supplementary materials (fig. S11). We performed two analyses to ascertain whether these eutherian modules are also applicable to marsupials and monotremes. Using the discovery dataset, we first trained a network (Net2) in both eutherians and marsupials based on only 7956 probes that are mappable to both. The color bands under the hierarchical tree reveal that all the Net1 modules were also preserved in Net2 (Fig. 2A). Second, we selected CpGs in Net1 modules that were also mapped to marsupials or monotremes and confirmed that their eigengene relationships to primary traits were retained in these mammalian clades (table S3). For example, the magenta module, which is related to blood in eutherians, was also found to be so in monotremes (table S3), which confirms that the Net1 modules can indeed be applied to other mammalian clades by selecting probes that are also mapped to those clades.
A functional enrichment study, accounting for the mammalian array background, revealed that the genes neighboring to module CpGs are implicated in many biological processes including development, immune function, metabolism, reproduction, stem cell biology, stress responses, aging, and various signaling pathways (Fig. 2C and fig. S9).
Relationship with protein-protein interactions
We investigated whether the proteins encoded by cognate genes (closest to respective CpGs) within modules are known to mutually interact or predicted to do so by STRING protein-protein interaction networks, which integrate known and predicted protein associations from >14,000 organisms (35). A permutation test analysis evaluating the global cluster coefficient (36) of each module showed that 14 modules are significantly enriched (P < 0.001) for genes encoding mutually interacting proteins (Fig. 2D). Overall, these results suggest that comethylation relationships can be reflected at the protein level for a subset of modules.
Modules related to maximum life span
To adjust for potential confounders, we used four regression modeling approaches to identity modules that are associated with the log-transformed maximum life span (dependent variable): (i) a univariate regression model with a covariate that was the module eigengene (averaged per species); (ii) a phylogenetic regression model with a covariate that was again the module eigengene (averaged per species); (iii) a multivariate linear regression model that included the module eigengene, sex, tissue, and relative age as covariates; and (iv) model approach (i) applied to specific tissue types.
The marginal analysis identified four modules: magenta, black, midnightblue, and tan, that related significantly to maximum life span (the absolute value of the Pearson correlation exceeded r = 0.6, Student’s t test P < 1 × 10−33). The CpGs underlying the implicated modules exhibit the sample patterns, as can be seen from corresponding heatmaps (fig. S14C). Phylogenetic regression also identified associations of the same modules (table S3). Our fourth modeling approach, i.e., the tissue-stratified marginal analysis, indicates that the relationship of modules to maximum life span is often tissue specific. For example, the magenta and midnightblue modules relate to maximum life span in lung and liver (fig. S14A). By contrast, the black module relates to maximum life span only in skin, and the tan module exhibited a weak relationship to life span in the tissue-specific analysis.
For ease of comprehension, modules were labeled with the trait and direction of relationship by superscript plus and minus signs; for example, magenta is the Lifespan+Weight +Blood+ module). The two modules (magenta with 480 CpGs, and midnightblue with 249 CpGs) that correlated with life span in lung and liver also correlated significantly with average adult weight across all eutherian species (r = 0.47 to 0.55, P < 1 × 10−18; Fig. 3). The magenta module (Lifespan+Weight+Blood+) is enriched with developmental genes such as HOXA5, VEGFA, SOX2, and WNT11 (table S4). The midnightblue (Lifespan+Weight+) module implicates genes involved in transfer RNA metabolism (P = 2 × 10−6, e.g., URM1), lipopolysaccharides (P = 5 × 10−6, e.g., CERCAM), development (P = 10−4, e.g., the HOXL gene family), and fatty acids (P = 2 × 10−3, e.g., ACADVL). The magenta module also relates to life span and average weight of dog breeds (r = −0.30, P = 0.003; Fig. 3C). Furthermore, it is related to the hazard of human death [hazard ratio (HR) = 0.91, MetaP = 0.0016; Fig. 3D) in epidemiological cohort studies.
Fig. 3. Comethylation modules related to mammalian maximum life span, weight, human mortality, and age.

(A and B) Modules associated with log maximum life span (P < 10−20) (A) or log average species weight (P < 10−17) (B) in marginal association (correlation test with the mean module eigengene of the species). The module eigengene is defined as the first principal component of the scaled CpGs underlying a module. The species are randomly labeled by their animal number (table S1). (C) The top modules associated with median life expectancy, upper limit life expectancy, or average adult weight of 93 dog breeds, model (marginal correlation test of the mean module eigengene with target variables; for detailed breed characteristics, see table S8). R, Pearson correlation* coefficient; P, correlation test P value. (D) Forest plots of the top modules associated with mortality risk in the Framingham Heart Study Offspring Cohort (FHS), and Women’s Health Initiative (WHI) study totaling 4651 individuals (1095, 24% death). n denotes the number of deaths per total number of individuals in each study. We report the meta-analysis P value in the title of the forest plot. (E) Module that correlates significantly (P < 1 × 10−300) with relative age (defined as ratio of age/maximum life span) across mammalian species using a multivariate regression model. Covariates were tissue, sex, and species differences. Each dot corresponds to a eutherian tissue sample (n = 14,542). Dots are colored by taxonomic order as in Fig. 1. (F) Volcano plot of the rmCorr of all purple module genes in GTEx data (for additional analysis, see fig. S11).
After adjustment for phylogeny, the cyan module relates to mammalian life span phylogenetic contrast (r = 0.42, P = 4 × 10−14; fig. S13I). The Lifespan+Liver− (cyan) module consists of genes that play a role in adaptive immunity (P = 2 × 10−6), histone and protein demethylation (P = 0.0001), and metabolism (P = 0.0004) (table S4).
The multivariate model analysis included sex, tissue type, and relative age as covariates to reveal additional modules that relate to life span in different tissues. The regression analysis found two modules with opposing correlations with maximum life span: the green module (life span r = 0.42, average weight r = 0.38, P < 10−300) and the greenyellow module (life span r = −0.44, average weight r = −0.35, P < 10−300; fig. S13J). The CpGs of the Lifespan−Weight− Rodentia− (greenyellow) are located near genes that play a role in development (P = 5 × 10−13; table S4) and in RNA metabolism (P = 6 × 10−12).
Age-related consensus module in mammals
The purple module (denoted subsequently as RelativeAge+ module) exhibited the strongest positive correlation with relative age (relative age r = 0.35, P < 10−300; Fig. 3E and fig. S13).
To remove the confounding effects of species and/or tissue type, we also constructed seven consensus networks (denoted cNet3,…, cNet9; for a description, see the supplementary materials). The RelativeAge+ module was preserved in three different consensus networks (cNet3, cNet4, and cNet6; Fig. 2A), suggesting conservation in different species and tissues (scatter plot in fig. S11H). The purple RelativeAge+ module is positively enriched for CpGs in regulatory regions (e.g., promoters and 5′ untranslated regions) and depleted in intron regions (fig. S15). Functional enrichment of this module highlighted embryonic stem cell regulation, axonal fasciculation, angiogenesis, and diabetes-related pathways (table S3). The CpGs in this module are adjacent to Polycomb repressor complex 2 (PRC2, EED) targets, which are marked by H3K27me3 (table S3).
Ingenuity pathway analysis implicated POU5F1 (alias OCT4), SHH, ASCL1, SOX2, and NEUROG2 proteins as putative upstream regulators of the RelativeAge+ module. We used Genotype-Tissue Expression project (GTEx) data to determine whether the mRNA levels of any of these upstream regulators are altered with age in several human tissues. OCT4 [repeated-measures correlation (rmCorr) = 0.07, P = 2 × 10−14], which is among the four known Yamanaka factors for cellular dedifferentiation, showed a positive increase with age in several, but not all, human tissues (fig. S11F). Nine other genes (e.g., HOXD10, rmCorr = 0.16, P = 4 × 10−50; SRXN1, rmCorr = −0.14, P = 4 × 10−52) from the RelativeAge+ module also had a nominally significant rmCorr (P < 0.005) in GTEx data (Fig. 3F and fig. S11G), although opposite aging patterns could be found in select tissues. These observations highlight the relevance of genes in the RelativeAge+ module to stem cell biology and aging in human tissues.
Interventional studies in mice
We related our methylation modules to interventions that are known to modulate the life span of mice (Fig. 4, A to C). This included growth hormone receptor knockout (i.e., dwarf mice) (37) and caloric restriction (38), which extended life, and a high-fat diet, which elicited the opposite effect (12). Six modules, including the purple module (RelativeAge+) showed a significant decrease (P < 0.05) of the module eigengene in dwarf mice and after caloric restriction and, conversely, a modest increase after a high-fat diet. Although the magenta, black, midnightblue, tan, and greenyellow modules have connections to the maximum life span in mammals, they did not present a clear relationship with interventions that modify murine life span (growth hormone receptor knockout, caloric restriction, and high-fat diet). This suggests a mutual exclusivity between the modules related to the maximum mammalian life span and those affected by interventions modulating the murine life span.
Fig. 4. The effects of different pro-aging and anti-aging interventions on selected DNAm modules.

Six DNAm modules respond to life span–related intervention experiments and are associated with the life expectancy of the mouse models. By contrast, the mammalian maximum life span modules do not correspond directly to the benefits or stress triggered by the intervention in the murine samples. (A) Changes in the intervention modules in the liver parallel smaller size and longer life expectancy of growth hormone receptor mouse models (GHRKO). Sample size: GHRKO, n = 11 (n = 5 female, n = 6 male); wild type, n = 18 (n = 9 male, n = 9 female). Age range was 6 to 8 months. (B) Caloric restriction (CR) DNAm module signature predicts longer life span in this treated group (age = 18 months; sex = male; CR, n = 59; control, n = 36). (C) High-fat diet accelerates aging in five modules including the purple (RelativeAge+) module. High-fat diet, n = 133 (n = 125 females, n = 8 males); control (ad libitum feeding), n = 212 (n = 10 male, n = 202 female). Age range was 3 to 32 months. (D and E) Examining the effects of in vivo partial reprogramming on intervention modules. (D) Schematic view of the partial programming experiment in 4F mice (39). A systemic Yamanaka factors expression (Oct4, Sox2, Klf4, and Myc) was periodically induced by adding doxycycline to the drinking water for 2 days per week. Partial programming was done at three different durations. Sample size: control (C57BL/6+dox), n = 7; 1 month (1m) 4F, n = 3; 7 months (7m) 4F, n = 5; 10 months (10m) 4F. All tissues except skin, n = 3; skin, n = 2. (E) scatter plots of the linear changes of the intervention modules in the skin and kidney of mice treated with different durations (dosages) of Yamanaka factors. Intervention modules indicate a dose-dependent rejuvenation of skin and kidney by this partial programming regimen.
Transient expression of Yamanaka factors
We investigated whether a transient expression of the Yamanaka factors in the 4-factor (4F) mouse affects the module eigengenes. The experimental design is shown in Fig. 4D, with additional details reported in the original article (39). Four of six of the above-mentioned murine intervention modules showed a nominally significant dose-dependent rejuvenation in murine skin (P < 0.06), and two modules showed the same in kidney (dose refers to the duration of 4F treatment: 0, 1, 7, and 10 months of intermittent expression of 4F factors) (Fig. 4E). The purple, ivory, and lavenderblush3 modules were particularly sensitive to the 4F treatment (Pearson’s r « −0.64 in skin). In addition, the purple RelativeAge+ module’s response to the 4F treatment is consistent with bioinformatic findings that OCT4 is an upstream regulator of this module. Among the life span modules, only the black module demonstrates an increase (P = 0.007) in skin of 4F treated mice, but this was not observed in the kidney.
Epigenome-wide association analysis of maximum life span
We performed epigenome-wide association studies (EWASs) to identify individual CpGs with methylation levels that correlate with maximum life span. To reduce bias resulting from different levels of sequence conservation, our EWASs of maximum life span focused on 333 eutherian species, excluding marsupial and monotreme species. We restricted the analysis to 28,318 high-quality probes that are conserved between humans and mice.
When relating individual CpGs to log-transformed maximum life span, we used several modeling approaches (for details, see the supplementary text). Briefly, our first approach, generic modeling, applied regression analysis ignoring tissue type and age. In our second approach, we repeated the regression analysis after focusing on a given tissue type. Third, we focused on specific nonoverlapping age groups: young animals (defined as age <1.5 times the age at sexual maturity), middle-aged, and old (defined as age >3.5 times the age at sexual maturity; fig S19). Some of these regression models were further adjusted for average species weight (denoted LifespanAdjWeight).
For brevity, we will focus on linear regression models because phylogenetic regression models led to qualitatively similar conclusions (tables S13 and S14). The most significant life span–related CpGs are located in the distal intergenic region neighboring TLE4 (Pearson’s r = 0.68, P = 5.8 × 10−46; Fig. 5A and table S11) and two CpGs near the promoter region of HOXA4 (r = 0.66, P = 7.5 × 10−45; Fig. 5A, midnightblue module) and are negatively correlated with a CpG in an intron of GATA3 (r = −0.65, P =8.8 × 10−42; Fig. 5A). Many of these significant CpGs remained so after phylogenetic adjustment, such as the CpGs neighboring TLE4 and HOXA4 (P = 4.2 × 10−5 and P = 4.8 × 10−3, respectively; fig. S17 and table S11 and S12). The top 1000 life span–related CpGs (comprising 500 positively and 500 negatively life span–related CpGs) significantly overlapped (Fisher’s exact P = 5.5 × 10−134) with those found in our weight-adjusted analysis (LifespanAdjWeight).
Fig. 5. EWAS of mammalian log-transformed maximum life span.

(A) CpG-specific association with maximum life span across n = 333 eutherian species. For EWAS, the mean methylation values of each CpG (per species) were regressed on log maximum life span. The right portion of the panel reports EWAS results after adjustment for average adult weight. Genome annotation indicates human hg19. Blue dotted line indicates Bonferroni-corrected two-sided P value < 1.8 × 10−6. The point colors indicate the corresponding modules. The bar plot indicates the top enriched (hypergeometric test, eutherian probes as background) modules for the top 1000 (500 negative CpGs, nominal P < 1.1 × 10−11, FDR = 1 × 10−10; 500 negative and positive CpGs, nominal P < 1.5 × 10−21, FDR = 7.5 × 10−20) significant CpGs for different EWASs. (B) Venn diagram of the overlaps between top hits from EWAS of maximum life span and meta-analysis of age [meta-analysis results are from (7); for additional analysis, see fig. S20]. (C) Venn diagram of the overlaps between the genes adjacent to the EWAS results and top age-related mRNA changes in human tissues (P < 1 × 10−50). (D) Gene set enrichment analysis of the genes proximal to CpGs associated with mammalian maximum life span. We only report enrichment terms that are significant after adjustment for multiple comparisons (hypergeometric FDR < 0.01) and contain at least five significant genes. The top three significant terms per column (EWAS) and enrichment database are shown. (E) Ingenuity potential upstream regulator analysis (40) of the differentially methylated genes related to mammalian maximum life span. Only significant (FDR < 0.05) regulators are represented in the bar plot. (F) Venn diagram of three gene lists. Gene list 1 is the top 646 genes adjacent to 1000 life span–related CpGs (500 positive and 500 negative). Gene lists 2 and 3 are based on CpGs that are differentially methylated (nominal Wald test P < 0.005, up to 500 positive and 500 negatively related CpGs) after OSKM overexpression in murine kidney (583 genes) and skin (686 genes) (39). We observed significant overlap between the gene lists (nominal Fisher’s exact P = 9.9 × 10−30 for skin and life span; P = 4.5 × 10−25 for kidney and life span). (G) Transcriptional factor motif enrichment analysis of life span modules and life span–related CpGs. The enrichment results for LifespanAdjWeight. negative were not significant. The overlap is assessed by a hypergeometric test for the CpGs within the motifs based on the human hg19 genome.
In general, methylation of life span–related CpGs does not change with age in mammalian tissues (Fig. 5B and fig. S20). The same can be seen from EWASs of life span restricted to animals of a given age group (e.g., only very young animals; fig. S20D). The EWASs of life span in all animals (irrespective of age) is highly correlated (r > 0.7), with the analogous EWASs restricted to animals that are young, middle-aged, or old.
EWASs of life span showed good consistency with the eigengene-based analysis in the mammalian comethylation network. As expected, the following previously discussed life span–related modules were enriched with CpGs implicated by our EWAS of life span: midnightblue (hypergeometric test P = 2.2 × 10−47; 67/249 overlapped CpGs), greenyellow (hypergeometric P = 2.1 × 10−36; 70/398 overlapped CpGs), tan (hypergeometric P = 6.7 × 10−23; 52/365 overlapped CpGs), and green (hypergeometric P = 5.0 × 10−18; 104/1542 overlapped CpGs).
In total, 1006 genes had a differential methylation association with life span (union of cognate genes resulting from the marginal model analysis for life span and LifespanAdjWeight). The gene expression levels of 16 of these genes exhibited a highly significant repeated-measures correlation with chronological age (rmCorr P value < 10−50) in different human tissues (Fig. 5C). The cognate genes next to the top 500 positively life span–related CpGs play a critical role in animal organ morphogenesis [marginal model life span GREAT enrichment false discovery rate (FDR) = 3 × 10−4 and LifespanAdjWeight FDR = 3.3 × 10−7; Fig. 5D] and in increased rib number in mice (FDR = 1 × 10−21; Fig. 5D), and implicates the HOXL subclass homeobox genes (FDR = 0.004 and LifespanAdjWeight FDR = 1.3 × 10−15) in abnormal survival in mice (FDR < 4 × 10−4).
Upstream regulators of maximum life span
We used ingenuity pathway analysis (40) to identify potential upstream regulators of the genes cognate to the top 500 positively and top 500 negatively life span–related CpGs. The top-ranked candidate regulators of both gene lists included SOX2-OCT4-NANOG pluripotency factors (FDR = 5.7 × 10−4 life span negative, FDR = 5.7 × 10−4 life span positive), which play critical roles in cellular reprogramming. We performed a control analysis that ruled out potential confounding by sequence conservation (fig. S25). Upstream regulators also included several candidates related to development: sonic hedgehog (SHH), life span–negative FDR = 1.3 × 10−4; POU4F2, life span–negative FDR = 3.3 × 10−7 and ASCL1, life span–negative FDR = 1.6 × 10−3 (Fig. 5E). These findings suggest that expression of life span–related genes might be regulated to some extent by pluripotency factors. This prompted us to investigate whether expression of any of the life span–related genes identified above are altered by transient expression of pluripotency inducing factors (Yamanaka factors OSKM) in a mouse model (39). Indeed, this analysis revealed that transient expression of OSKM altered the expression of 190 of 647 life span–related genes in skin and 162 life span–related genes in the kidney (nominal Fisher’s exact P = 9.9 × 10−30 for skin and life span; P = 4.5 × 10−25 for kidney and life span; Fig. 5F and fig. S32). Genomic positions that are known to be bound by pluripotency factors in at least one human or murine cell type according to chromatin immunoprecipitation sequencing (ChIP-seq) data from the Encyclopedia of DNA Elements (ENCODE) consortium are located near CpGs that are associated with maximum species life spans: NANOG-binding sites are enriched for CpGs that are positively correlated with life span (FDR = 0.002) and to CpGs underlying the midnightblue module (FDR = 0.0006), which has high methylation levels in long-lived species (Fig. 5G). OCT4 (POU5F1) (FDR = 0.02), and cMYC (FDR = 0.003) binding sites are enriched with CpGs in the greenyellow module, which has low methylation levels in long-lived species (Fig. 5G). The ChIP-seq binding location analysis also implicates other noteworthy factors such as POLII, CTCF, RAD21, YY1, and TAF1, which show the strongest enrichment for negatively life span–related CpGs (Fig. 5G).
Given the role of CTCF in regulating the three-dimensional organization of the genome, we conducted an enrichment analysis of topologically associating domain (TAD) boundaries and loop boundaries identified in both human and mouse cell lines (fig. S26). We found that both TAD and loop boundaries demonstrated significant enrichment of negatively life span–related CpGs (FDR = 3 × 10−4) for TAD boundaries and (FDR = 6.7 × 10−4) for loop boundaries in various cell lines such as olfactory receptor cells and the human fibroblasts IMR90 and HFFc6 (fig. S26).
CpGs linked to life span in various taxonomic orders and tissues
To pinpoint CpGs associated with log maximum life span independently of phylogenetic order or tissue type, we conducted a meta-analysis of EWAS findings from 25 distinct strata comprising phylogenetic order and tissue type. Using a nonparametric meta-analysis approach (rankPvalue), we assessed the EWAS of life span (meta.lifespan) in these strata to identify CpGs unconfounded by tissue type or phylogenetic order (table S24). Our meta.lifespan results demonstrated significant overlap with the previously mentioned EWASs of life span in all eutherian species (hypergeometric P = 1 × 10−175; Fig. 6A). By contrast, none of the meta.lifespan CpGs overlapped with EWASs of age, which further supports the idea that methylation of life span–related CpGs does not change with age in mammalian tissues. The top four CpGs from the meta.lifespan analysis are depicted in Fig. 6B, showing significant positive correlations for CpGs near LOXL1 and ZSCAN29 (exons) and negative correlations for those near RAB29 (exon) and GATA3 (downstream), with log maximum life span across various taxonomic orders and tissue types. Similar to our above-mentioned results, CpGs implicated by our meta.lifespan analysis (FDR < 0.05) overlap significantly (FDR < 0.01) with genes involved in organ morphogenesis, RNA biosynthesis, increased rib number in mice, Wnt signaling (Fig. 6C), and genes altered by transient expression of pluripotency-inducing factors in mouse models (nominal Fisher’s exact P < 10−5 for skin and meta.lifespan; P < 10−11 for kidney and meta.lifespan; Fig. 6D).
Fig. 6. CpGs linked to life span in various taxonomic orders and tissues.

Using the nonparametric rankPvalue method (33), we combined 25 EWAS of life span results from various taxonomic order or tissue type strata, calculating the significance of a CpG’s consistently high (or low) rank based on the 25 EWASs of log maximum life span (meta.lifespan and underlying EWAS results can be found in table S24 and data S19). (A) The overlap of top 1000 (500 per direction) meta.lifespan CpGs with EWAS of life span in all eutherians (nominal Fisher’s exact P = 1 × 10−175). (B) Scatter plots illustrating the top meta.lifespan CpGs categorized into different tissue-phylogenetic order strata. Each panel displays only the strata that exhibit significant relationships. Each dot represents a species colored by taxonomic order. Each row corresponds to a different selection of tissue type. “bval” denotes the beta value, measuring DNAm at a CpG site, with 0 indicating no methylation and 1 indicating full methylation. (C) Gene set enrichment analysis of the genes proximal to CpGs associated with mammalian maximum life span. We only report enrichment terms that are significant after adjustment for multiple comparisons (hypergeometric FDR < 0.01) and contain at least five significant genes. The top three significant terms per column (EWAS) and enrichment database are shown. (D) Venn diagram of three gene lists. Gene list 1 (the bottom circle) is the top 407 genes adjacent to 1000 meta.lifespan CpGs (500 positive and 500 negative). Gene lists 2 and 3 (the top circles) are based on CpGs that are differentially methylated (nominal Wald test P < 0.005, up to 500 positive and 500 negatively related CpGs) after OSKM overexpression in murine kidney (583 genes) and skin (686 genes) (39).
Chromatin state analysis
Our large-scale mammalian DNAm data confirm that CpGs located in promoter regions (−2000 to 2000 bp of TSS regions) have low methylation levels (mean = 15%; Fig. 7A). By contrast, those in gene bodies and distal regions are highly methylated (mean = ~70%; Fig. 7A). CpGs having a high or low mean methylation level tend to have positive or negative correlation test Z statistics for life span, respectively (Fig. 7, A and B). We find that CpGs with low methylation levels in long-lived species are located close to the TSS of genes and near binding sites of PRC1 (P = 6.4 × 10−11; Fig. 7C) and PRC2 (P = 2 × 10−6). To test the hypothesis that long-lived species exhibit high or low methylation levels in chromosomal regions that are expected to have high or low methylation patterns, respectively, we used chromatin states that were identified and annotated based on >1000 epigenetic datasets encompassing a diverse range of human cell and tissue types (41).
Fig. 7. Chromatin state analysis and distance to the TSS for the life span–related CpGs.

(A) Illustrated plot presenting mean methylation across species (displayed on the left y axis) and EWAS of maximum life span Z statistics (shown on the right y axis), all plotted against the distances to the closest TSS (represented on the x axis). (B) Mean methylation across species (y axis) plotted against EWAS Z statistics for log maximum life span in different genomic regions (intergenic, promoter, and gene body). Additional EWAS results after adjustment for phylogenetic relationships can be found in figs. S17 to S20, and corresponding enrichment results can be found in figs. S22 to S24. Pearson correlation coefficients and P values are reported in different panels. (C and D) Chromatin annotation enrichment analysis of the top 500 negatively life span–related CpGs (C) and the top 500 positively life span–related CpGs (D). The columns in each panel correspond to EWAS results for log-transformed maximum life span across (i) all tissues combined (Lifespan.All), (ii) blood samples only (Lifespan.Blood), and (iii) skin samples only (Lifespan.Skin), (iv) meta analysis of lifespan in different tissues (meta.lifespan), and the corresponding results after adjustment for average adult weight (LifespanAdjWeight). The last column reports enrichment with respect to the RelativeAge+ module (purple). We used the same significance thresholds as in Fig. 5. Cell shading corresponds to fold enrichment between comethylation modules and each chromatin state. Numeric values correspond to the P value of such enrichments based on the hypergeometric test, and only cell values with significant P < 0.001 (equivalent to FDR < 0.02) are shown. The chromatin states are learned based on epigenetic datasets profiling chromatin mark signals in different human cell and tissue types resulting in a genome annotation shared across cell types (41). The common partially methylated domains (commonPMD), solo CpGs (WCGW), and highly methylated domain (HMD) annotations are from (42). PRC1 and PRC2 binding site: are obtained from the ChIP-seq datasets of PCR1 and PCR2 from ENCODE (45).
The negatively life span–related CpGs are enriched with a constitutive TSS chromatin state (TSS1, P = 2.5 × 10−12) and promoter flanking states (PromF4, P = 5.6 × 10−10; PromF5, P = 2.0 × 10−9; PromF2, P = 3.0 × 10−4; Fig. 7C).
The CpGs with high methylation levels in blood samples of long-lived species are enriched in gene body–associated states (notably transcribed and exon state TxEx1, P = 7.5 × 10−8 and highly transcribed state TxEx4 P = 1.7 × 10−6; Fig. 7D). A detailed description of the chromatin state enrichment for EWASs of maximum life span is provided in the supplementary text and tables S21 and S22.
A biclustering analysis between chromatin annotations and comethylation modules based on fold enrichments (Fig. 8 and tables S21 and S22) revealed that the 55 mammalian comethylation modules fall into three large groupings (referred to as meta-modules). The bar plot to the left of Fig. 8 shows different mean methylation levels of the CpGs underlying the three meta-modules: mean methylation = 0.23, 0.66, and 0.77 for meta-modules 1, 2, and 3, respectively.
Fig. 8. Mammalian methylation meta-modules based on chromatin states and external genome annotations.

The heatmap shows the enrichments between (1) mammalian comethylation modules and significant life span–related EWAS CpG groups (x axis) and (2) chromatin states or other genomic annotation (y axis). Cell shading corresponds to log-transformed fold enrichment values (observed CpG count divided by expected count). Hypergeometric tests were used to evaluate the enrichment significance in each cell. *Nominal P < 0.001 (FDR < 0.10). Only chromatin states and external genome annotations with at least one significant enrichment (FDR < 0.10) are shown. The chromatin states are based on a human-based universal chromatin annotation of human cell and tissue types (41). Other genomic annotations include the commonPMD, solo CpGs (WCGW), HMD annotations, and neither (CpGs outside these annotations) which are from (42). In addition, PRC1 and PRC2 binding sites are defined from the ChIP-seq data of PRC1 and PRC2 from ENCODE (45). The row and column hierarchical clustering trees (average linkage) are based on a dissimilarity measure (1 minus the pairwise Pearson correlation between log-transformed fold enrichment values). The left barplot indicates the mean methylation levels of the CpGs in each state for all eutherian samples in our data. We used the 14,705 eutherian CpGs as the background for enrichment of the comethylation modules. By contrast, 28,318 CpGs (high-quality probes in humans and mice) were used as a background for enrichment of significant life span–related EWAS CpG groups with chromatin states and genome annotations. Each EWAS CpG group includes up to 500 most significant CpGs per direction (positively or negatively related with life span), as detailed in the caption of Fig. 5.
Meta-module 1 contains several chromatin states that are associated with Polycomb repression, including strong polycomb-repressed state ReprPC1 and bivalent promoters (BivProm1-2). Further, meta-module 1 contains chromatin states related to TSSs (TSS1 and TSS2) and several flanking promoters (PromF2, PromF3, PromF4, and PromF5). TSS1, PromF4, and PromF5 (associated with negatively life span–related CpGs) were previously associated among universal chromatin states with the strongest enrichments for CpG islands (71 to 101 fold) (41). The color band under Fig. 8 reveals that six modules underlying meta-module 1 are sensitive to murine life span interventions. Meta-module 1 is enriched with CpGs that have low methylation levels in long-lived species (overlap with EWASs of life span, tan and greenyellow modules; Fig. 8).
Meta-module 2 can be considered as a partially methylated module (mean methylation 0.66) and is enriched with several enhancer states, late replicating domains [partially methylated domains, commonPMD (42)], and solo CpGs [WCGW (42)]. Meta-module 2 also contains the module most related with life span (midnightblue) and the human mortality risk module (magenta). These two modules overlap with the CpGs that are positively related to life span. Three out of four average weight-related modules are also located in meta-module 2.
Discussion
In this study, we present an analysis of a cross-species DNAm dataset obtained from a mammalian array platform. This platform specifically focuses on highly conserved regions of DNA, making it a valuable resource for studying methylation patterns across mammalian species (5). The successful construction of mammalian phyloepigenetic trees suggests that the divergence of DNAm profiles is closely aligned with genetic changes throughout evolution. Sensitivity assessments reveal that the observed phyloepigenetic associations are not caused by technical issues associated with our measurement platform. Instead, the phyloepigenetic signal may stem from sources such as upstream regulators, transcription factors, or DNA sequence variations in distant regions.
The conserved CpGs exhibiting the strongest phylogenetic signals are situated in intergenic regions, whereas promoter regions do not display such signals. Previous studies reported a rapid evolutionary rate of enhancers as a shared feature among mammalian genomes, but promoters demonstrate either full or partial conservation across species (2).
We found that 30 of the resulting 55 modules identified from an unsupervised machine learning method were readily associated with species traits (taxonomic order, maximum life span, and average adult weight) or individual traits (chronological age, tissue, and sex). We expect that many of the remaining 25 modules will be associated with biological characteristics about which we currently have no information. As a case in point, although the yellow module was not associated with any of our primary tested traits, it did show association with response to a murine circadian rhythm disruption study (light pollution during the night; fig. S7B). The upstream regulator analysis of the EWAS of life span identified the pluripotency transcription factors OCT4, SOX2, and NANOG. We showed that the transient overexpression of OSKM in murine tissues affects the methylation levels of CpGs near genes implicated by our EWAS of maximum life span (Fig. 5E). We speculate that the enhanced activity of the pluripotency network in long-lived species results in more efficient tissue repair and maintenance, ensuring a longer life span.
Both the EWAS and eigengene-based analyses identified methylation signatures of maximum life span presumably established at birth. Most of these were independent of aging and interventions that affect murine mortality risk. Several CpGs that are more highly methylated in long-lived species are located near HOXL subclass homeoboxes and other genes that play a role in morphogenesis and development. Some of these life span–related CpGs are located next to genes that are also implicated in our analysis of upstream regulators (e.g., ASCL1 and SMAD6).
CpGs with methylation levels that are inversely related to life span are enriched in TSS1- and promoter flanking (PromF4 and PromF5)–associated chromatin states. Genes located in chromatin state TSS1 are constitutively active and enriched for nucleic acid metabolic processes (41). This could imply that long-lived species either evolved selective mechanisms to maintain low methylation levels near TSSs or may have adaptations that promote the high expression of essential genes. This high expression may indirectly prompt more active DNA demethylation mechanisms.
Methods summary
The Mammalian Methylation Consortium generated cytosine methylation data from n = 15,456 DNA samples derived from 70 tissue types of 348 mammalian species (331 eutherians, 15 marsupials, two monotremes) using a custom-designed mammalian methylation array that targets CpGs at conserved loci in mammals (5). DNAm data were used for phyloepigenetic tree development using 1-cor dissimilarity applied to mean methylation values per species. The choice of the correlation-based dissimilarity matrix is justified in the supplementary materials and methods.
For unsupervised analysis, we formed WGCNA networks based on two sets of CpG probes in our data. The first network was generated from 14,705 conserved CpGs in 10,927 samples of 167 eutherian species. The preservation of this network was evaluated in an independent dataset comprising 3692 samples from 29 tissues of 228 mammalian species (164 new species; 64 overlapped with the training set). The second network was a subset of 7956 conserved CpGs in 11,105 samples from 167 eutherian and nine marsupial species. In addition, we developed seven consensus comethylation networks to remove the confounding effects of species and tissue type. Consensus WGCNA can be interpreted as a meta-analysis across networks in different species and tissue types (33, 43).
For the eutherian network (Net1), module eigengenes (MEs) were defined as singular vectors (corresponding to the highest singular value) from the singular value decomposition of the scaled CpGs that underlie the respective module. The eigengenes in the eutherian network (Net1) explained a range of 24 to 63% (average 43%) of the variance in the methylation data in the training set, replication set, and all data in each module (table S3). For a given module, we defined the measure of module membership (kME) as the Pearson correlation between the module eigengene and the CpGs. The association of module eigengenes was examined for different traits using individual regression models.
EWAS of life span was done in 28,318 CpGs that apply to mice and humans according to calibration and titration data (correlation with calibration exceeds 0.8) and mappability information as described in (5). Because the distribution of maximum life span and other life history traits were highly skewed, we imposed a log transformation on these phenotypes before conducting EWAS. Our tissue type–specific EWAS was conducted in tissues with enough species (N > 25) available. For our various EWAS of log-transformed maximum life span, we adopted a nominal significance threshold of 1.8 × 10−6 (0.01/28,318) based on the conservative Bonferroni adjustment. We report an FDR in our enrichment studies to adjust for multiple comparisons.
Supplementary Material
ACKNOWLEDGMENTS
This is Duke Lemur Center publication #1563.
Funding:
The majority of the funding was contributed by the Paul G. Allen Frontiers Group (S.H.) and Open Philanthropy (S.H.). Additional financial support for specific aspects of data generation was obtained from the following sources: National Geographic Society grant 8941-11 (B.L.), British Heart Foundation (FS/18/39/33684 to D.V.), Wellcome (WT202878/Z/16/Z to D.T.O.), European Research Council (788937 to D.T.O.), Cancer Research UK (20412 to D.T.O.), Max Planck Research Group Award from the Max Planck Gesellschaft (S.C.V.), UKRI Future Leaders Fellowship (MR/T021985/1 to S.C.V.), DST-NRF SARChI chair of Mammalian Behavioral Ecology and Physiology (GUN 64756 to N.C.B.), Science Foundation Ireland Future Frontiers 19/FFP/6790 (ECT), National Institute on Aging Intramural Research Program, NIH (J.A.M.), U01 AG060908 (S.H.), AG055841 (K.M.), and AG043930/AG/NIA (R.W.W.), National Institute of Health AG065403 (V.N.G.), AG047200 (V.N.G.), AG067782 (V.N.G.), and AG076607 (V.N.G.), AG047200 (V.G.), AG045795 (T.G.), AG072736 (T.G.), NIA 1R21AG078784 (K.E.S), OOD 1R21OD022988 (K.E.S), National Science Foundation IOS 2017803 (K.E.S), DEB 1854469 (K.E.S), AG047200 (A. S.), Milky Way Research Foundation (V.G.), A.H. Schultz Foundation, Department of Evolutionary Anthropology, University of Zurich (M.K.), Leverhulme Trust RPG-2019-404 and Royal Society of Edinburgh Research Reboot Grant 1107 (A. Pederson), UCLA Jonsson Comprehensive Cancer Center and Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Ablon Scholars Program (J.E.), funds provided by the College of Computer, Mathematical and Natural Sciences at the University of Maryland, College Park (G.S.W.), Taronga Conservation Society Australia (J.K.O.), S.C.V. was supported by a Max Planck Research Group Award from the Max Planck Gesellschaft and a UKRI Future Leaders Fellowship (MR/T021985/1). N.C.B. was funded by a DST-NRF SARChI chair of Mammalian Behavioral Ecology and Physiology (GUN 64756). A.N.A was funded by DSFP, King Saud University, Riyadh, Saudi Arabia. K.J.P. was supported by a postdoctoral grant from the University of Zurich. Collection of plains zebra samples was supported by the National Geographic Society to B.L. (8941-11).
Footnotes
Data and materials availability:
All data from the Mammalian Methylation Consortium are posted on the Gene Expression Omnibus website (complete dataset: GSE223748). Subsets of the data sets can also be downloaded from accession numbers, GSE174758, GSE184211, GSE184213, GSE184215, GSE184216, GSE184218, GSE184220, GSE184221, GSE184224, GSE190660, GSE190661, GSE190662, GSE190663, GSE190664, GSE174544, GSE190665, GSE174767, GSE184222, GSE184223, GSE174777, GSE174778, GSE173330, GSE164127, GSE147002, GSE147003, GSE147004). The mammalian array platform is distributed by the nonprofit Epigenetic Clock Development Foundation (https://clockfoundation.org/). The mammalian data can also be downloaded from the Clock Foundation web page: https://clockfoundation.org/MammalianMethylationConsortium. The manifest file of the mammalian array, genome annotations of the CpGs, and codes can be found on Zenodo (46).
Competing interests: S.H., A.A., and J.E. are inventors on patent/patent application number WO2020150705 held/submitted by the University of California, Los Angeles that covers the mammalian methylation array technology. S.H. and R.T.B. are founders of the nonprofit Epigenetic Clock Development Foundation, which has licensed several patents from UC Regents and distributes the mammalian methylation array.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Xiao S. et al. , Comparative epigenomic annotation of regulatory DNA. Cell 149, 1381–1392 (2012). doi: 10.1016/j.cell.2012.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villar D. et al. , Enhancer evolution across 20 mammalian species. Cell 160, 554–566 (2015). doi: 10.1016/j.cell.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qu J. et al. , Evolutionary expansion of DNA hypomethylation in the mammalian germline genome. Genome Res. 28, 145–158 (2018). doi: 10.1101/gr.225896.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klughammer J. et al. , Comparative analysis of genome-scale, base-resolution DNA methylation profiles across 580 animal species. Nat. Commun 14, 232 (2023). doi: 10.1038/s41467-022-34828-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arneson A. et al. , A mammalian methylation array for profiling methylation levels at conserved sequences. Nat. Commun 13, 783 (2022). doi: 10.1038/s41467-022-28355-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsons KM et al. , DNA methylation-based biomarkers for ageing long-lived cetaceans. Mol. Ecol. Resour 23, 1241–1256 (2023). doi: 10.1111/1755-0998.13791 [DOI] [PubMed] [Google Scholar]
- 7.Lu AT et al. , Universal DNA methylation age across mammalian tissues. bioRxiv 426733 [Preprint] (2021). doi: 10.1101/2021.01.18.426733 [DOI] [Google Scholar]
- 8.Kordowitzki P. et al. , Epigenetic clock and methylation study of oocytes from a bovine model of reproductive aging. Aging Cell 20, e13349 (2021). doi: 10.1111/acel.13349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prado NA et al. , Epigenetic clock and methylation studies in elephants. Aging Cell 20, e13414 (2021). doi: 10.1111/acel.13414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robeck TR et al. , Multi-species and multi-tissue methylation clocks for age estimation in toothed whales and dolphins. Commun. Biol 4, 642 (2021). doi: 10.1038/s42003-021-02179-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larison B. et al. , Epigenetic models developed for plains zebras predict age in domestic horses and endangered equids. Commun. Biol 4, 1412 (2021). doi: 10.1038/s42003-021-02935-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mozhui K. et al. , Genetic loci and metabolic states associated with murine epigenetic aging. eLife 11, e75244 (2022). doi: 10.7554/eLife.75244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugrue VJ et al. , Castration delays epigenetic aging and feminizes DNA methylation at androgen-regulated loci. eLife 10, e64932 (2021). doi: 10.7554/eLife.64932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robeck TR et al. , Multi-tissue methylation clocks for age and sex estimation in the common bottlenose dolphin. Front. Mar. Sci 8, 713373 (2021). doi: 10.3389/fmars.2021.713373 [DOI] [Google Scholar]
- 15.Horvath S. et al. , Methylation studies in Peromyscus: Aging, altitude adaptation, and monogamy. Geroscience 44, 447–461 (2022). doi: 10.1007/s11357-021-00472-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horvath S. et al. , Epigenetic clock and methylation studies in marsupials: Opossums, Tasmanian devils, kangaroos, and wallabies. Geroscience 44, 1825–1845 (2022). doi: 10.1007/s11357-022-00569-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath S. et al. , Epigenetic clock and methylation studies in the rhesus macaque. Geroscience 43, 2441–2453 (2021). doi: 10.1007/s11357-021-00429-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horvath S. et al. , DNA methylation age analysis of rapamycin in common marmosets. Geroscience 43, 2413–2425 (2021). doi: 10.1007/s11357-021-00438-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jasinska AJ et al. , Epigenetic clock and methylation studies in vervet monkeys. Geroscience 44, 699–717 (2021). doi: 10.1007/s11357-021-00466-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raj K. et al. , Epigenetic clock and methylation studies in cats. Geroscience 43, 2363–2378 (2021). doi: 10.1007/s11357-021-00445-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schachtschneider KM et al. , Epigenetic clock and DNA methylation analysis of porcine models of aging and obesity. Geroscience 43, 2467–2483 (2021). doi: 10.1007/s11357-021-00439-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cossette ML et al. , Epigenetics and island-mainland divergence in an insectivorous small mammal. Mol. Ecol 32, 152–166 (2023). doi: 10.1111/mec.16735 [DOI] [PubMed] [Google Scholar]
- 23.Lemaître JF et al. , DNA methylation as a tool to explore ageing in wild roe deer populations. Mol. Ecol. Resour 22, 1002–1015 (2022). doi: 10.1111/1755-0998.13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horvath S. et al. , DNA methylation clocks tick in naked mole rats but queens age more slowly than nonbreeders. Nat. Aging 2, 46–59 (2022). doi: 10.1038/s43587-021-00152-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horvath S. et al. , Pan-primate studies of age and sex. Geroscience (2023). doi: 10.1007/s11357-023-00878-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkinson GS et al. , DNA methylation predicts age and provides insight into exceptional longevity of bats. Nat. Commun 12, 1615 (2021). doi: 10.1038/s41467-021-21900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horvath S. et al. , DNA methylation aging and transcriptomic studies in horses. Nat. Commun 13, 40 (2022). doi: 10.1038/s41467-021-27754-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinho GM et al. , Hibernation slows epigenetic ageing in yellow-bellied marmots. Nat. Ecol. Evol 6, 418–426 (2022). doi: 10.1038/s41559-022-01679-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horvath S. et al. , DNA methylation clocks for dogs and humans. Proc. Natl. Acad. Sci. U.S.A 119, e2120887119 (2022). doi: 10.1073/pnas.2120887119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters KJ et al. , An epigenetic DNA methylation clock for age estimates in Indo-Pacific bottlenose dolphins (Tursiops aduncus). Evol. Appl 16, 126–133 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiavellini P. et al. , Hippocampal DNA methylation, epigenetic age and spatial memory performance in young and old rats. J. Gerontol. A Biol. Sci. Med. Sci 77, 2387–2394 (2022). doi: 10.1093/gerona/glac153 [DOI] [PubMed] [Google Scholar]
- 32.Blomberg SP, Garland T Jr.., Ives AR, Testing for phylogenetic signal in comparative data: Behavioral traits are more labile. Evolution 57, 717–745 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Langfelder P, Horvath S, WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). doi: 10.1186/1471-2105-9-559; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langfelder P, Luo R, Oldham MC, Horvath S, Is my network module preserved and reproducible? PLOS Comput. Biol 7, e1001057 (2011). doi: 10.1371/journal.pcbi.1001057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarczyk D. et al. , Correction to ‘The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets’. Nucleic Acids Res. 49, 10800 (2021). doi: 10.1093/nar/gkab835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barrat A, Barthélemy M, Pastor-Satorras R, Vespignani A, The architecture of complex weighted networks. Proc. Natl. Acad. Sci. U. S. A 101, 3747–3752 (2004). doi: 10.1073/pnas.0400087101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilcher H, Money for old mice. Nature 10.1038/news030915-13 (2003). doi: 10.1038/news030915-13 [DOI] [Google Scholar]
- 38.Acosta-Rodríguez V. et al. , Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science 376, 1192–1202 (2022). doi: 10.1126/science.abk0297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Browder KC et al. , In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging 2, 243–253 (2022). doi: 10.1038/s43587-022-00183-2 [DOI] [PubMed] [Google Scholar]
- 40.Krämer A, Green J, Pollard J Jr.., Tugendreich S, Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). doi: 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vu H, Ernst J, Universal annotation of the human genome through integration of over a thousand epigenomic datasets. Genome Biol. 23, 9 (2022). doi: 10.1186/s13059-021-02572-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou W. et al. , DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet 50, 591–602 (2018). doi: 10.1038/s41588-018-0073-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langfelder P, Horvath S, “Tutorials for the WGCNA package” (2014); https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials. [Google Scholar]
- 44.Kumar S, Stecher G, Suleski M, Hedges SB, TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol 34, 1812–1819 (2017). doi: 10.1093/molbev/msx116 [DOI] [PubMed] [Google Scholar]
- 45.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haghani A, Lu AT, Kwon SB, Arneson A, Ernst J, Horvath S, Data for: DNA methylation networks underlying mammalian traits, Zenodo; (2023). doi: 10.5281/zenodo.8180547 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.