

Abstract
Background:
Depression is pleiotropic and influenced by diverse genetic, environmental, and pharmacological factors. Identifying patterns of circuit activity on which many of these factors converge would be important, because studying these patterns could reveal underlying pathophysiological processes and/or novel therapies. Depression is commonly assumed to involve changes within prefrontal circuits, and dopamine D2 receptor (D2R) agonists are increasingly used as adjunctive antidepressants. Nevertheless, how D2Rs influence disease-relevant patterns of prefrontal circuit activity remains unknown.
Methods:
We used brain slice calcium imaging to measure how patterns of prefrontal activity are modulated by D2Rs, antidepressants, and manipulations that increase depression susceptibility. To validate the idea that prefrontal D2Rs might contribute to antidepressant responses, we used optogenetic and genetic manipulations to test how dopamine, D2Rs, and D2R+ neurons contribute to stress-coping behavior.
Results:
Patterns of positively correlated activity in prefrontal microcircuits are specifically enhanced by D2R stimulation as well as by two mechanistically distinct antidepressants: ketamine and fluoxetine. Conversely, this D2R-driven effect was disrupted in two etiologically distinct depression models: a genetic susceptibility model and mice that are susceptible to chronic social defeat. Phasic stimulation of dopaminergic afferents to prefrontal cortex and closed-loop stimulation of D2R+ neurons both increased effortful responses to tail suspension stress, whereas prefrontal D2R deletion reduced the duration of individual struggling episodes.
Conclusions:
Correlated prefrontal microcircuit activity represents a point of convergence for multiple depression-related manipulations. Prefrontal D2Rs enhance this activity. Through this mechanism, prefrontal D2Rs may promote network states associated with antidepressant actions and effortful responses to stress.
Keywords: Prefrontal cortex, Depression, dopamine, ketamine, calcium imaging, tail suspension test
INTRODUCTION
Prefrontal circuits play a role in human depression and depression-like states in animal models (1–6). Directly modulating prefrontal circuit activity can relieve many symptoms of clinical depression (7–9). Diverse genetic and environmental factors influence depression susceptibility (10), and a similar variety of interventions, including medications, magnetic/electrical stimulation, and exercise, alleviate depressive symptoms. Preclinical studies focusing on a single manipulation cannot easily disentangle whether observed effects on circuit physiology are important for depression or not. Moreover, poor correspondence between clinical depression and depression-like behavior in animal models has been a nearly insurmountable hurdle for identifying novel antidepressants. To overcome these challenges it is critical to discover points of convergence for disparate, clinically relevant influences on prefrontal circuit physiology. Seeking convergence is a tactical approach for filtering pleiotropic changes in neural circuits to identify those most likely to have broad clinical relevance.
The dopamine system has also increasingly been implicated in depression and its resolution. Dopamine underlies critical cognitive and emotive functions that are disrupted as core features of clinical depression, especially in the context of stress (e.g., low motivation/energy, poor concentration, altered aversive processing, anhedonia, etc.) (11–18). Furthermore, several FDA-approved medications for depression are dopamine D2 receptor (D2R) partial agonists – Aripiprazole and Brexpiprazole are approved as adjunctive treatment for major depression and Cariprazine is approved for bipolar depression; additionally, Pramipexole is commonly used off-label to treat depression (19–24). By contrast, no current medications include D1R agonism as a primary mechanism of action, although recent preclinical studies have implicated prefrontal D1Rs or D1R-expressing neurons in antidepressant-like behavioral responses in rodents (25–27).
To some extent confusion about the role of specific dopamine receptors in antidepressant responses relates to challenges associated with studying human psychiatric disorders in animal models. Rodent behavioral assays of antidepressant effects have come under increased scrutiny because of 1) general skepticism regarding ‘face validity’ for assays related to psychiatric conditions, and 2) the decades-long failure of behavioral assays to identify antidepressants with novel mechanisms (28–32). Here, we propose a different approach: leveraging a relatively simple slice calcium imaging assay to identify convergent changes in prefrontal circuit physiology focused on dopamine. We show that D2Rs, but not D1Rs, robustly enhance positively correlated activity between prefrontal neurons. Mechanistically distinct antidepressants (ketamine and fluoxetine) reproduce (and possibly occlude) the ability of D2Rs to increase correlated activity. Conversely, the presence or absence of a D2R-induced increase in correlated activity distinguishes stress resilient vs. susceptible mice, and a genetic disruption which produces a depression-like state prevents D2R activation from increasing correlated activity.
Together this shows that 1) an increase in significant positive correlations is a slice metric that consistently captures changes in circuit physiology associated with antidepressant responses and stress resilience, and 2) D2R activation promotes the same changes in circuit physiology. Notably, each of these clinically-relevant manipulations affects effortful responses to acute tail suspension stress. Therefore, to connect our slice findings with the behavioral assays that are commonly used in the field, we use cell-type specific optogenetics and prefrontal gene deletion to show that prefrontal dopamine, D2Rs, and D2R+ neurons can elicit corresponding changes in effortful stress-coping within the tail suspension test.
MATERIALS AND METHODS
See the Supplement for detailed methods.
Animals:
All experiments were conducted in accordance with procedures established by the administrative panels on laboratory animal care at the University of California, San Francisco. We used wild-type C57BL/6J mice (from Jackson Laboratories or Charles River) and the following transgenic lines: B6-CamKII::TtA (JAX: 00310), tetO-DISC1dn (JAX: 008790), TH::Cre (line FI12; www.gensat.org), D2::Cre (line ER44; www.gensat.org), Drd2loxP/loxP (JAX: 020631). Social defeat followed previously published protocols (48).
Slice GCaMP imaging and analysis:
Slice preparation and GCaMP imaging followed our previously described procedures (78). Correlations between DF/F signals and their derivatives were computed as the normalized dot product over a 20 min window, and the associated p-value was obtained using the regress function in Matlab.
Statistics:
Unless otherwise specified, nonparametric tests or ANOVA was used to assess significance.
RESULTS
Measuring network states in a slice GCaMP assay:
To examine putative neural signatures associated with antidepressant treatment and/or depression susceptibility, we targeted viral expression of GCaMP6f to deep layer prefrontal neurons and measured activity in acute prefrontal slices (Figure 1A–D). A standard approach in GCaMP imaging has been to detect large ‘events,’ presumed to reflect one or multiple action potentials. A more conservative approach avoids assumptions about the relationship between GCaMP fluorescence and underlying electrical activity, and directly analyzes fluorescence signals. One reason most studies focus on events, rather fluorescence, is that GCaMP signals obtained via single-photon imaging are contaminated by shared background fluorescence. This is problematic when examining patterns of circuit activity, rather than single cell activity, because standard pairwise correlations between signals from different cells will be dominated by autocorrelation between shared background fluorescence.
FIGURE 1. Dopamine D2 receptor stimulation increases positively correlated activity in an ex-vivo prefrontal slice assay.
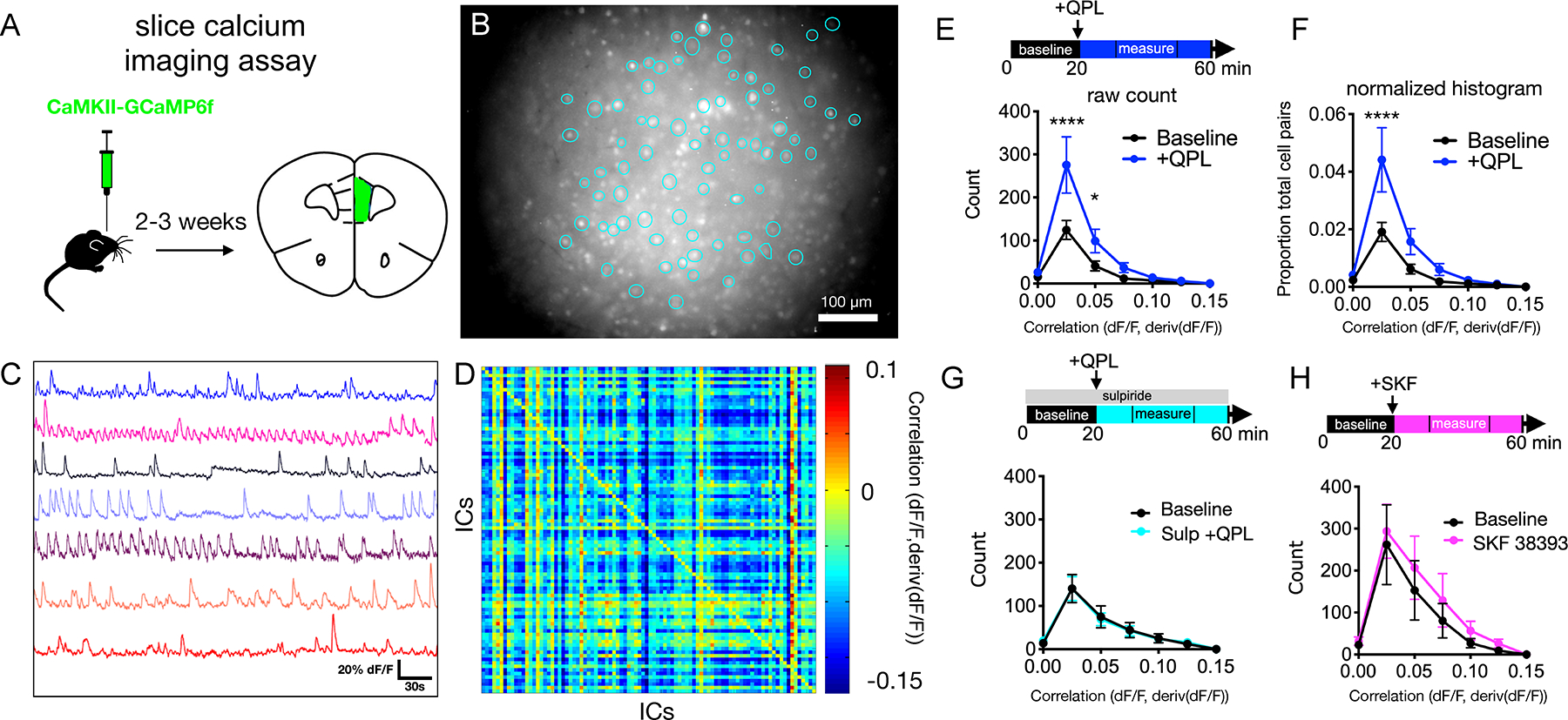
(A) Schematic of GCaMP6f injection into mPFC.
(B) Example field of view for Ca2+ imaging with regions of interest (ROIs) outlined as identified using principal and independent components analysis (PCA-ICA, putative active neurons).
(C) GCaMP6f signals from 8 example cells.
(D) Heatmap showing correlation coefficients for 85 cells in baseline condition. IC, independent component (putative neurons).
(E) Experimental design (top) and quantification of effects of quinpirole on the number of positively correlated cell pairs. baseline vs. quinpirole (n=19 slices, 9 mice) [FInteraction(6,252)=3.499, P<0.0024, FDrug(1,252)=10.89, P=0.001], 2-way ANOVA with Sidak post-hoc test.
(F) Analysis of quinpirole effects on the proportion of positively correlated cell pairs, normalized per slice. baseline vs. quinpirole (n=19 slices, 9 mice) [FInteraction(6,252)=3.404, P=0.003, FDrug(1,252)=10.63, P=0.001], 2-way ANOVA with Sidak post-hoc test.
(G) Analysis of quinpirole effects on significant positive correlations in presence of the D2R antagonist sulpiride. baseline vs. quinpirole + sulpiride (n=7 slices, 2 mice) [FInteraction(6,84)=0.031, P=0.999, FDrug(1,84)=0.0002, P=0.989], 2-way ANOVA with Sidak post-hoc test.
(H) Analysis of the effects of the D1R agonist SKF 38393 on significant positive correlations. baseline vs. SKF 38393 (n=9 slices, 4 mice) [FInteraction(6,112)=0.090, P=0.997, FDrug(1,112)=1,183, P=0.279], 2-way ANOVA with Sidak post-hoc test.
Statistics represent mean±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To overcome this, we computed correlations between fluorescence in one cell and the derivative of fluorescence from other cells. Because the integral of a signal with its derivative is zero over a closed path, this will minimize autocorrelation driven by shared background fluorescence. In previous work, we showed that when one creates simulated data composed of nominally independent signals contaminated by some amount of shared background fluorescence, correlations between signals are markedly larger in magnitude than correlations between signals and their derivatives (33). Correlations calculated from real data followed a very similar distribution, which validates this method as a means for resolving physiologically meaningful correlations. Intuitively, correlations computed this way represent the tendency for activity (i.e., the GCaMP signal) in one cell to predict increases or decreases in the activity of a second cell. The matrix of correlations between all cells represents an abstraction of network state during a given time window (Figure 1D). This relatively simple method maximizes the use of GCaMP signals to characterize network level activity, rather than simply measure changes in the activity of individual neurons.
Acute D2R stimulation and antidepressant treatment enhance positively correlated activity:
Repeated stress decreases the firing of dopaminergic neurons which project to prefrontal cortex (34) and prefrontal dopamine release (35). This has led to the hypothesis that deficient prefrontal dopamine contributes to depression (35–37). Substantial clinical evidence suggests that D2R activation can exert antidepressant effects and/or enhance the efficacy of other antidepressant interventions (19–24). Moreover, D2Rs have been implicated in the therapeutic effects of both selective serotonin reuptake inhibitors (SSRIs) and exercise (18,38–41). Therefore, we hypothesized that prefrontal D2R activation might elicit effects on neural activity similar to antidepressant agents. We found that application of the D2R agonist quinpirole (QPL, 10 μM) elicited a robust increase in the number of cell-pairs that exhibited significant positive correlations. Notably this effect was present in both sexes, but larger in males than females (Figure 1E, Figure S2). The D2R antagonist sulpiride (10 μM) blocked this effect, and the D1R agonist SKF-38393 (10 μM) did not elicit a similar increase in positive correlations (Figure 1G,H). Thus, QPL leads to an increase in significant positively correlated cell pairs that is dependent on and specific to D2Rs. To exclude non-meaningful correlations which represent the chance overlap of activity, i.e., noise, we calculated p-values for each pairwise interaction and only included those which achieved statistical significance (p<0.01). Note: we chose to quantify the total count of positively correlated cell pairs because this metric weights each cell pair equally (as opposed to the fraction of positively correlated cell pairs, which effectively give smore weight to cell pairs from slices with fewer neurons). However, to confirm that effects are not artifactually driven by potential differences in the numbers of active cells between slices, we also looked at the proportion of total cell pairs (normalized on a per slice basis). This yields the same result: quinpirole increases significant positive correlations (Figure 1F).
Next, we wondered whether the observed effects of D2Rs on correlated activity might resemble those of antidepressants. We studied two clinically effective antidepressants which have distinct mechanisms, timing and effect profiles: the SSRI fluoxetine, which is only effective after chronic treatment for weeks, and the N-methyl-D-aspartate Receptor (NMDAR) antagonist ketamine, which rapidly alleviates depression within hours, including in patients with treatment-refractory or bipolar depression (42–46). We treated mice with either chronic fluoxetine (20 mg/kg, daily injections for 3 weeks) or a single subanesthetic dose of ketamine (10 mg/kg) (Figure 2A,E). We selected 10 mg/kg of ketamine based on dose-response relationships in the tail suspension test (TST) (Figure S3). In both experiments, saline vs. antidepressant treated mice had highly similar numbers of active cells per slice (Figure 2B,F). Prefrontal slices from mice receiving either fluoxetine or ketamine both exhibited a marked increase in cell pairs with significant positive correlations at baseline (Figure 2C,G). Intriguingly, in mice treated with either antidepressant, the effects of D2R stimulation were, at least partially, occluded. There was no further increase in correlation and no difference between saline and antidepressant conditions after QPL (Figure 2D,H). This indicates that these antidepressants and QPL increase positive correlations through shared downstream mechanisms. Therefore, we hypothesized that prefrontal D2R activation might elicit antidepressant-like circuit effects, and conversely, that manipulations which increase susceptibility to depression might impair network responses to D2R activation.
FIGURE 2. In vivo antidepressant treatments increase baseline positively correlated activity in acute mPFC slices.
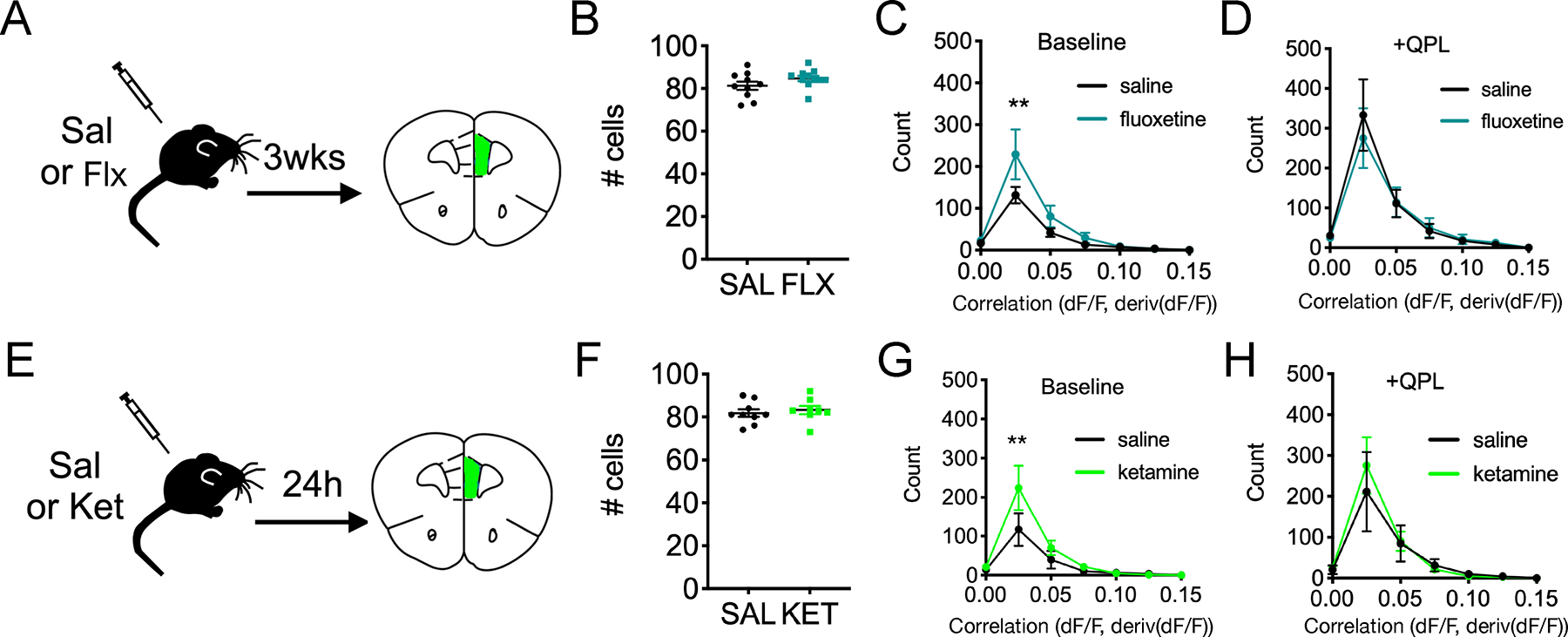
(A) Experimental design showing the timing of chronic saline vs. fluoxetine (20 mg/kg) treatments relative to slice imaging studies.
(B) Quantification of the number of active cells identified per slice for saline vs. fluoxetine.
(C-D) Quantification of positively correlated cell pairs at baseline for saline (n=10 slices, 4 mice) vs. fluoxetine (n=11 slices, 5 mice) [FInteraction(6,133)=1.64, P=0.14, FDrug(1,133)=4.68, P=0.032], 2-way ANOVA with Sidak post-hoc test and (D) quantification post-quinpirole wash-on [FInteraction(6,133)=0.215, P=0.972, FDrug(1,133)=0.110, P=0.7404], 2-way ANOVA with Sidak post-hoc test.
(E) Experimental design for the timing of acute saline vs. ketamine (10 mg/kg) treatments relative to slice imaging.
(F) Quantification of number of active cells identified per slice for saline vs. ketamine.
(G-H) Quantification of positively correlated cell pairs at baseline for saline (n=9 slices, 5 mice) vs. ketamine (n=8 slices, 5 mice) [FInteraction(6,133)=1.64, P=0.14, FDrug(1,133)=4.68, P=0.032], 2-way ANOVA with Sidak post-hoc test, and (H) post-quinpirole wash-on [FInteraction(6,105)=0.248, P=0.959, FDrug(1,105)=0.183, P=0.670], 2-way ANOVA with Sidak post-hoc test.
Statistics represent mean±SEM, **P<0.01. Sal=saline, Flx=fluoxetine, Ket=ketamine, QPL=quinpirole.
Models of depression susceptibility have deficits in D2R-driven correlated activity:
Chronic social or psychological stress is a major risk factor for depression (47). In mice, this is commonly modeled in an ethologically relevant way using chronic social defeat stress (CSDS), in which mice are repeatedly exposed to social dominance interactions from a larger, more aggressive mouse (48). Mice exposed to CSDS are classified as ‘susceptible’ or ‘resilient’ based on social avoidance when they subsequently encounter a novel mouse. All mice tend to develop stress and anxiety-like responses, but only susceptible mice (those which avoid the novel mouse) exhibit more extensive, depression-like phenotypes (48,49). Defeated mice exhibit decreased firing of VTA-to-mPFC projecting neurons and inhibiting these projections promotes susceptibility (34). We therefore hypothesized that CSDS-exposed mice might have altered prefrontal dopamine function, manifesting as changes in positive correlations either at baseline and/or after D2R stimulation.
To address this, we obtained prefrontal slices from socially-defeated mice classified as susceptible or resilient, and undefeated controls, then imaged activity before and after applying quinpirole (Figure 3A–E). Numbers of active cells per slice were similar across control, resilient and susceptible mice (Figure 3F). At baseline, slices from both resilient and susceptible mice had modestly increased positive correlations relative to undefeated controls (similar to those seen in antidepressant-treated mice) (Figure 3G). However, slices from resilient and susceptible mice exhibited strikingly different responses to D2R stimulation (Figure 3H). After quinpirole application, positive correlations in slices from resilient mice were increased several-fold compared to those from susceptible or control mice. Further, following D2R stimulation, the difference between control and susceptible mice was no longer significant, and positive correlations may have actually decreased in the susceptible condition (Figure 3G,H). The increase in baseline correlations seen in defeated mice may represent a general response to stress that may be augmented by antidepressant treatments. Conversely, only slices from resilient mice showed D2R-enhancement of positive correlations. Thus, the ability of D2Rs to increase significant positive correlations may be a circuit biomarker for resiliency. Notably, this set of control mice did not exhibit the same increase in positive correlations seen in other experiments, but this may simply reflect the fact that the CSDS protocol used mice which are considerably older than those used for all other experiments (10–11 weeks-old vs. 6–8 weeks-old, see Methods). Importantly though, the effect of quinpirole to increase significant positive correlations is restored following stress, specifically in resilient mice.
FIGURE 3. Positively correlated prefrontal activity is altered in mouse models of depression at baseline and in response to D2R stimulation.
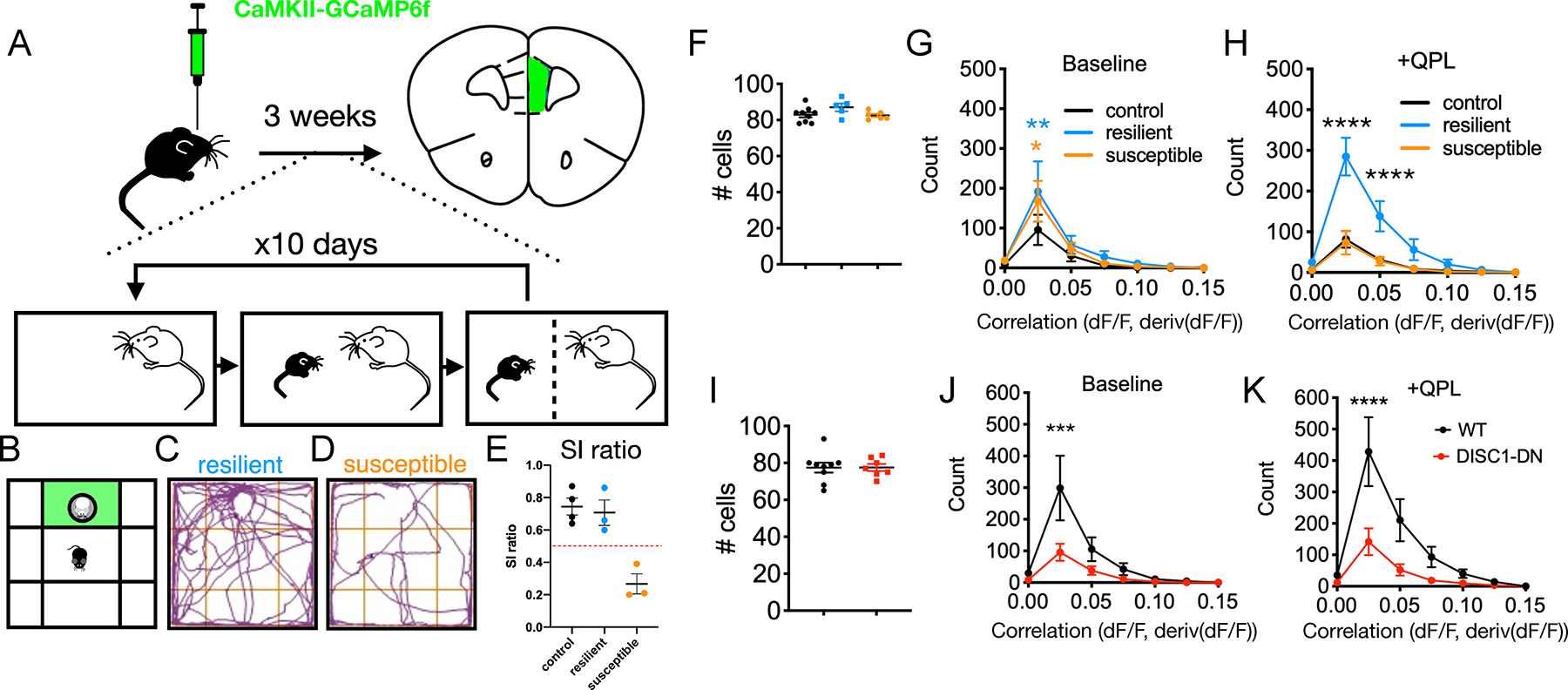
(A) Experimental design for social defeat stress protocol combined with slice calcium imaging.
(B) Social interaction assay with green interaction zone
(C) Social behavior for an example resilient mouse
(D) Social behavior for an example susceptible mouse.
(E) Social interaction ratio scoring (time in interaction zone with/without novel CD1).
(F) Quantification of number of active cells identified per slice for control, resilient and suceptible conditions. (G-H) Quantification of positively correlated cell pairs at baseline for no defeat controls (n=9 slices, 4 mice) vs. resilient (n=5 slices, 3 mice) vs. susceptible (n=6 slices, 3 mice) [FInteraction(12,119)=0.770, P=0.680, FCondition(2,119)=2.506, P=0.086], 2-way ANOVA with Sidak post-hoc test which shows significant difference for histogram bin 2 (no defeat vs. resilient, **p<0.01; no defeat vs. susceptible, *p<0.05; resilient vs. susceptible, n.s.), and (H) post-quinpirole wash-on [FInteraction(12,119)=8.247, P<0.0001, FCondition(2,119)=33.70, P<0.0001, 2-way ANOVA with Sidak post-hoc test which shows significant difference for histogram bins 2 and 3 (control vs. resilient or susceptible, ****p<0.0001; control vs. susceptible, n.s.).
(I) Quantification of number of active cells identified per slice for wildtype (WT) controls adn Disc1-DN mice.
(J-K) Quantification of positively correlated cell pairs at baseline for wildtype (n=9 slices, 4 mice) vs. Disc1-DN (n=7 slices, 3 mice) [FInteraction(6,98)=2.157, P=0.054, FGenotype(1,98)=6.605, P=0.012], 2-way ANOVA with Sidak post-hoc test, and (K) post-quinpirole wash-on [FInteraction(6,98)=3.060, P=0.009, FGenotype(1,98)=13.57, P<0.001, 2-way ANOVA with Sidak post-hoc test.
Statistics represent mean±SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
In addition to environmental influences such as stress, heritability accounts for approximately 37% of risk for developing major depressive disorder (10,47). A rare, but highly penetrant truncation in the gene Disc1 was originally associated with major mental illness, including schizophrenia, bipolar disorder, and depression, in a Scottish pedigree (50–52). Genetic studies have found evidence for an association between specific Disc1 variants and recurrent major depression, though these lack the sample sizes needed for genome-wide significance (53). Furthermore, many mouse models with disruptions in Disc1 exhibit specific changes in behavior similar to those seen in other models of depression and opposite to those elicited by antidepressant treatments (54–57). Therefore, we studied one of these to determine whether genetic factors which influence responses to stress might also exhibit altered correlated prefrontal circuit activity. Notably, we confirmed that when bred and tested in our laboratory, Disc1-DN mice had the same behavioral changes commonly associated with other mouse models of depression (Figure S3). Mice expressing a dominant negative Disc1 variant (Disc1-DN), had significantly reduced positive correlations in our slice assay, both before and after D2R activation (Figure 2H,I).
Depression is highly heterogeneous, such that individual assays and models typically capture only a portion of the complex syndrome. That being said, certain mechanisms may represent points of convergence relevant to diverse behaviors and manipulations, even if they do not capture the entire disorder. Our results suggest a simple model in which intrinsically generated, positively correlated activity between prefrontal neurons represent one such point of convergence. In this model, antidepressant manipulations and stress-resilience either directly increase intrinsically generated correlated activity between prefrontal neurons, or augment this activity in response to D2R activation. By contrast, in stress-susceptible mice and mice which exhibit behavioral changes seen in mouse models of depression, the ability of D2Rs to potentiate this correlated activity is greatly diminished. These results do not necessarily imply that all of these manipulations act directly to modulate dopamine release or D2R activation. Rather, these four manipulations (fluoxetine, ketamine, CSDS susceptibility, and Disc1 disruption) produce convergent effects when viewed at the level of network activity.
Nonetheless, to place our slice findings in the context of the existing literature and validate a potential role for prefrontal dopamine and D2Rs in behaviors that are commonly associated with mouse models of depression, we used the tail suspension test (TST). Tail suspension confronts animals with an inescapable acute stressor. Mice respond by struggling to escape (active coping) or adopting an energy conserving immobile posture (passive coping). Importantly, the TST has largely failed as a screen for discovering new antidepressants, because it is highly sensitive to compounds which recapitulate the mechanisms of action of existing antidepressants. Furthermore, aside from face validity, there is no bona fide reason to think of immobility as an analog of the depressed state. That said, all known antidepressant treatments (including both SSRIs and ketamine) increase struggling in the TST. Moreover, both Disc1 mutant mice and mice exposed to social defeat stress exhibit decreased struggling in the TST (48,57). This suggests that the disparate molecular and cellular pathways engaged by antidepressants (SSRIs and ketamine) and models of depression (Disc1-DN and social defeat) ultimately converge on common changes in neural activity that manifest within the TST, even if the original rationale for this assay (face validity) is no longer accepted. Importantly, we are not using the TST to model the sort of stress associated with CSDS or to make any direct claims about the role of positively correlated activity in vivo.
Phasic dopamine release in mPFC promotes effortful responses to acute stress:
While the relationship between the TST (or related assays) and depression may be questionable, its construct validity for coping with adversity is stronger (28–32). Mesolimbic dopamine as well as the manipulations investigated in our ex vivo assay affect TST behavior, but the role of prefrontal dopamine is less clear (34,58). VTA dopamine neurons fire either in a low frequency, sustained manner or in high frequency phasic bursts (59–62). We have previously shown that in PFC, phasic activation of VTA inputs preferentially release high concentrations of DA (63), which may elicit a D2R-driven network state associated with labile neural activity that underlies cognitive flexibility (63–65). Because D2R stimulation elicits a positively correlated state that is also associated with stress-resilience and antidepressant response (Figures 1–3), we hypothesized that phasic stimulation may enhance TST struggling. To test this, we targeted channelrhodopsin2 (ChR2) to VTA dopaminergic neurons and activated terminals in mPFC using blue laser light and fiber optics (Figure 4A–C). To model distinct tonic vs. phasic firing patterns observed in vivo, we used either a continuous 5 Hz pattern of light pulses (tonic) or arranged the same number of pulses into phasic 50 Hz bursts occurring every 5 seconds (Figure 4D). Phasic, but not tonic, stimulation increased effortful responses (i.e., struggling) specifically during light-on periods (Figure 4E,F). Neither phasic nor tonic stimulation produced nonspecific increases in locomotion in an open field (Figure 4G,H). This indicates that phasic release specifically enhances effortful responses to stress, not generic motor activity.
FIGURE 4. Phasic stimulation of dopamine terminals in mPFC increases struggling time during the tail-suspension test.
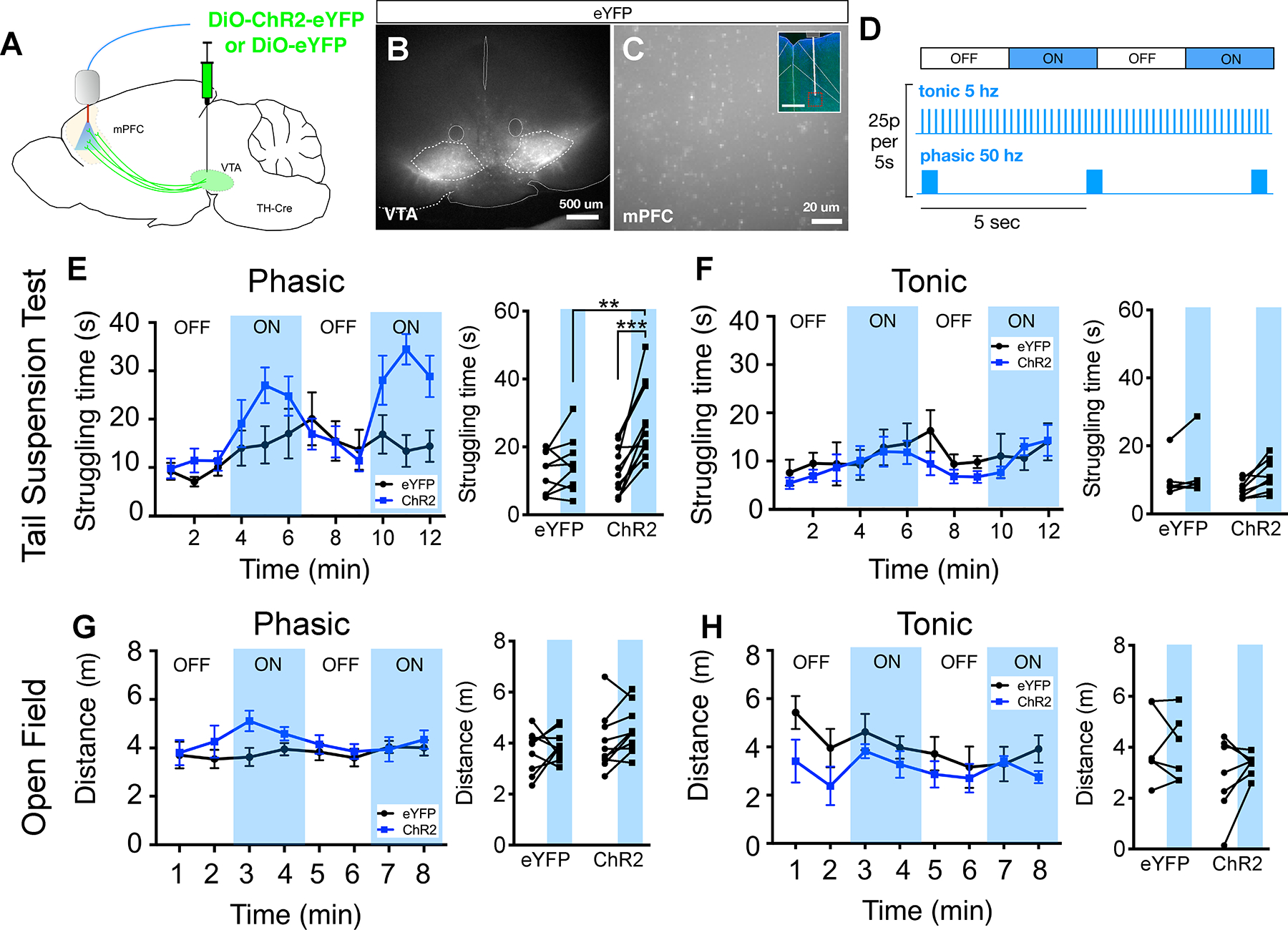
(A) Schematic for experimental design to optogenetically stimulate dopaminergic terminals in mPFC.
(B) Representative image of Cre-dependent ChR2-eYFP expression in the ventral tegmental area (VTA) of TH-Cre transgenic mice.
(C) eYFP+ axon terminals from VTA dopaminergic neurons, inset shows fiber tract targeting the mPFC with region of high magnification image (red box).
(D) Optogenetic stimulation protocols, OFF/ON blocks are 3 minutes in duration (the total duration of light pulses for phasic/tonic patterns are identical within each 5s of stimulation).
(E) Quantification of mean struggling time calculated for each minute of the tail suspension test with phasic stimulation (blue shading = light ON). Summary data for light OFF vs. ON for eYFP and ChR2 conditions on right, [FInteraction(1,36)=5.23, P=0.28, FLaser(1,36)=9.74, P=0.004, FVirus(1,36)=5.43, P=0.023], 2-way ANOVA with Sidak posthoc test.
(F) Similar plots as (E), but for tonic stimulation, [FInteraction(1,24)=0.391, P=0.538, FLaser(1,24)=1.893, P=0.182, FVirus(1,24)=0.686, P=0.416], 2-way ANOVA, Sidak posthoc test shows no significant comparisons.
(G) Quantification of mean distance traveled calculated for each minute during an open field test, with phasic stimulation summary data shown in right panel, [FInteraction(1,34)=0.179, P=0.683, FLaser(1,34)=1.511, P=0.228, FVirus(1,34)=2.604, P=0.116], 2-way ANOVA, Sidak posthoc test shows no significant comparisons.
(H) Similar plots as (G) but for tonic stimulation, [FInteraction(1,22)=0.365, P=0.552, FLaser(1,22)=0.139, P=0.713, FVirus(1,22)=3.646, P=0.069], 2-way ANOVA, Sidak posthoc test shows no significant comparisons. N=9,11,10, eYFP, ChR2 (TST), ChR2 (OF) respectively.
Statistics represent mean±SEM, **P<0.01, ***P<0.001.
Dopamine D2Rs and D2R+ prefrontal neurons modulate effortful responses to acute stress:
If, as hypothesized, phasic prefrontal dopamine increases struggling via D2Rs, then deleting D2Rs from mPFC may have the opposite effect on behavior. To test this, we injected the bilateral mPFC of floxed D2R (Drd2loxP/loxP) mice with adeno-associated virus expressing Cre recombinase or a control virus and after allowing time for expression/deletion, conducted the TST and OF tests (Figure 5B,C). Prefrontal-specific deletion of D2Rs had no significant impact on overall struggling time (Figure 5E). To analyze the fine temporal structure of TST behavior, we developed an unbiased, automated algorithm that uses tailbase velocity to assess both the number and duration of individual struggling epochs (Figure 5A,D). Mice with prefrontal-specific deletion of D2Rs exhibited a greater number of struggling episodes which were shorter in duration (Figure 5E–G). Deleting mPFC D2Rs did not result in any change in distance traveled in the OF (Figure 5H). Compensatory mechanisms may explain why the chronic deletion of D2Rs does not lead to less struggling, but these data at least raise the possibility that D2Rs have a role in maintaining effortful responses to acute stress.
FIGURE 5. D2Rs and D2R+ prefrontal neurons modulate tail suspension test behavior.
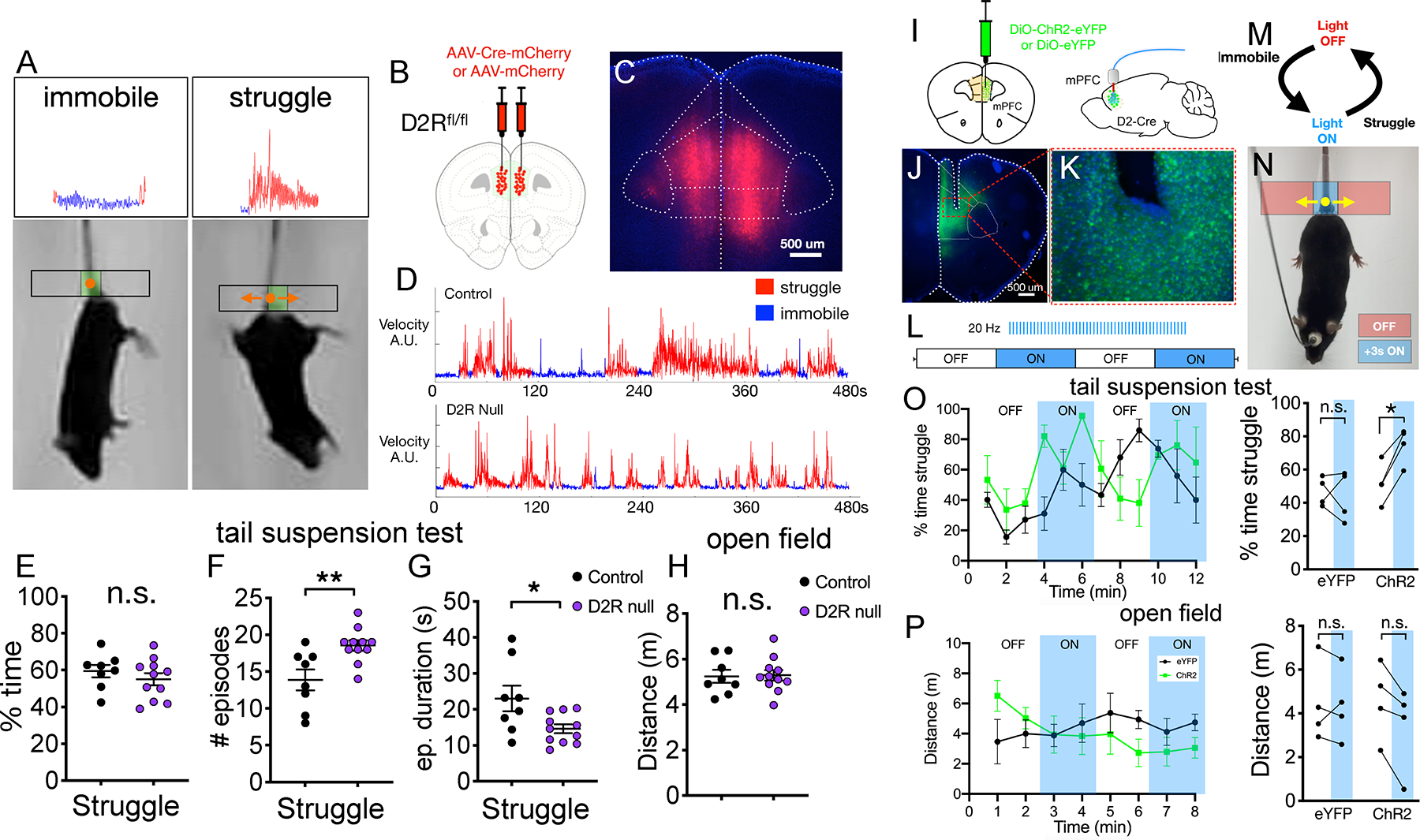
(A) Video still shots of immobile vs. struggling mice during the TST with tailbase velocity tracking and automated classification of epochs shown above.
(B) Schematic for injection of Cre-expressing vs. control virus into bilateral mPFC of Drd2flox/flox mice.
(C) Representative image showing bilateral Cre-mCherry expression limited to mPFC.
(D) Representative examples of tailbase velocity tracking for control vs. D2R deleted mice with struggling vs. immobile epoch classifications shown.
(E-G) Quantification of (E) total struggling time [unpaired t-test, t = 0.9165, df = 17], (F) number of distinct struggling episodes [unpaired t-test, t = 3.201, df = 17] and (G) mean duration of individual struggling episodes [unpaired t-test, t = 2.513, df = 17] for each mouse.
(H) Quantification of distance traveled in open field test [unpaired t-test, t = 0.114, df = 17].
(I) Experimental design for optogenetic stimulation of D2R-expressing mPFC neurons.
(J) Representative image showing virus and cannula targeting in mPFC.
(K) High magnification image of ChR2-eYFP expressing mPFC neurons.
(L-N) Block design for optogenetic stimulation (20 Hz), delivered during ON blocks via closed-loop design as in (M and N) 20 Hz stimulation triggered during immobility lasting 3 seconds, terminated at onset of struggling (detected by tailbase movement).
(O) Quantification of mean struggling time calculated per minute of tail suspension test with closed loop stimulation active during ON blocks. Summary data for light OFF vs. ON shown on right [EYFP(n=4 mice) vs. ChR2 (n=4 mice) [FInteraction(1,12)=4.594, P=0.0533, Flaser(1,12)=2.921, P=0.113, Fvirus(1,12)=8.988, P=0.011] 2-way ANOVA with Sidak post-hoc test. (P) Same plots as shown in (O), but for open field test [EYFP(n=4 mice) vs. ChR2 (n=4 mice) [FInteraction(1,12)=0.356, P=0.562, Flaser(1,12)=0.471, P=0.505, Fvirus(1,12)=0.219, P=0.648] 2-way ANOVA with Sidak post-hoc test.
Statistics represent mean±SEM, *P<0.05, **P<0.01.
As noted above, one limitation of gene knockouts is that they allow time for compensations, possibly obscuring behavioral effects. This may be addressed via more acute manipulations, e.g., optogenetics. In previous work, we showed that D2R activation enhances the excitability of prefrontal D2R+ neurons (66,67). This suggests that if dopamine promotes effortful responses by acting at prefrontal D2Rs, it may do so by driving D2R+ neuron activity. To test this, we used a viral-transgenic approach to optogenetically stimulate ChR2 expressed in D2R+ mPFC neurons (Figure 5I–K). To minimize any accommodation (e.g., due to ChR2 inactivation and/or circuit homeostasis) and stimulate neurons in a more physiologically plausible (i.e., non-continuous) manner, we used closed-loop stimulation. In this paradigm, stimulation (consisting of 4 msec light pulses delivered at 20 Hz) was triggered by immobility and terminated at the onset of struggling during light ON blocks (Figure 5L–N). This stimulation significantly increased struggling during light ON (compared to OFF) blocks in ChR2-expressing mice, but not in ChR2-negative controls (Figure 5O). Critically, similar closed-loop stimulation, delivered when mice became immobile within an open field, had no significant effect on overall locomotion (Figure 5P). Thus, directly stimulating D2R+ neurons increases struggling, whereas deleting D2Rs shortens struggling episodes (with the caveats noted above). These results are consistent with the hypothesis that prefrontal D2Rs enhance effortful coping by activating D2R+ neurons (Figure 6). More importantly though, these results implicate prefrontal dopamine, D2Rs and D2R+ neuron activity in standard behaviors of purported relevance to depression, similar to previous studies implicating D1Rs (25–27).
Fig. 6. Model.
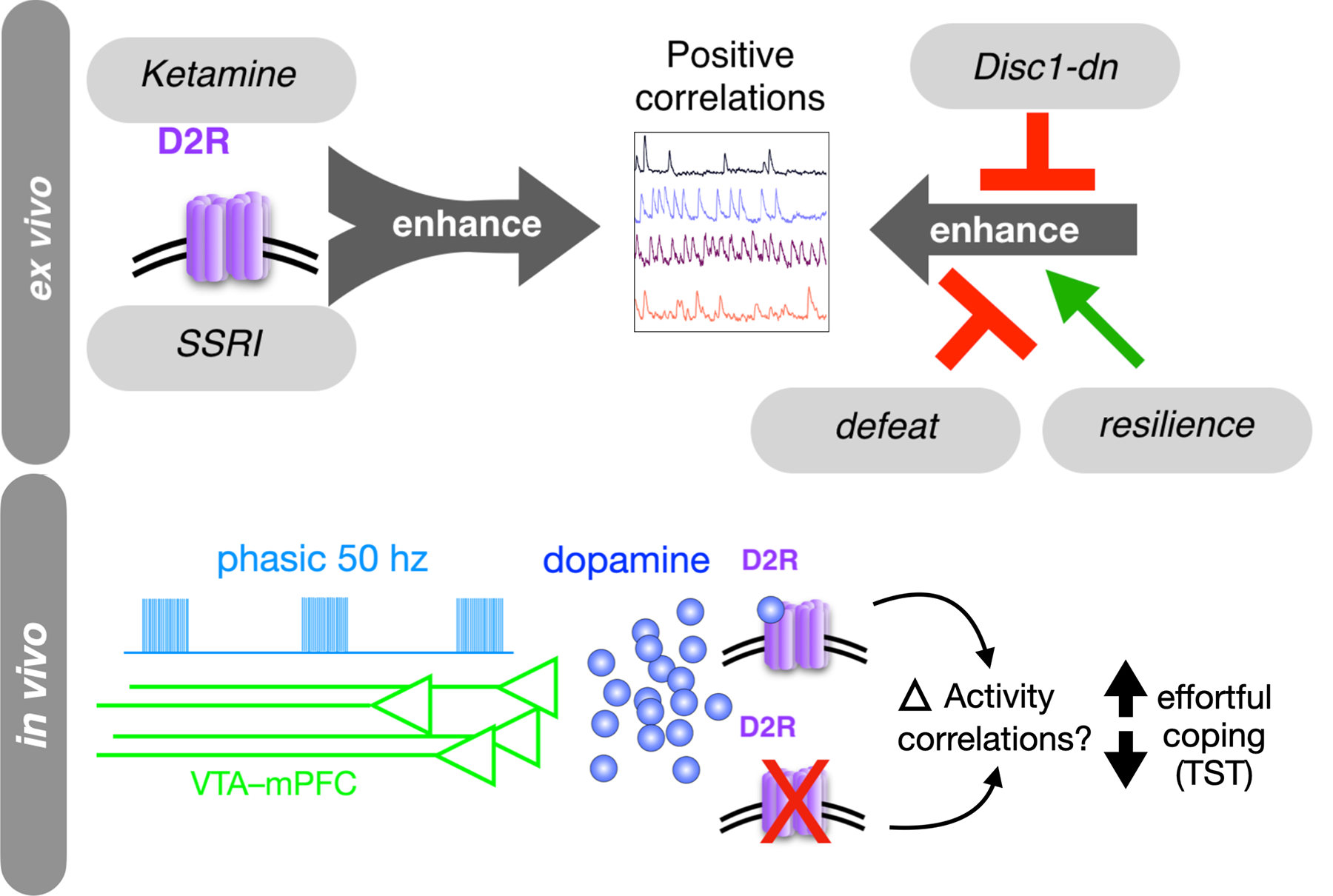
Convergent effects of D2R stimulation, antidepressants, and depression susceptibility models on positively correlated mPFC activity, ex vivo. Model also proposes that dopamine – via phasic bursting of VTA-mPFC afferents – acts via D2Rs to induce changes in effortful coping behavior in the TST. This may occur by recruiting similar prefrontal activity states in vivo.
DISCUSSION
Previous studies have largely used single preclinical models to study possible neural correlates of depression. However, a given manipulation can have pleiotropic effects, many of which may not be relevant to depression. Moreover, prior studies have relied heavily on face validity, but the presumption that behaviors such as TST, forced swim test, sucrose preference, learned helplessness, etc. are meaningful with respect to depression is questionable. Here we suggest an alternative: to develop assays that look for circuit physiology measures representing points of convergence across multiple etiologically distinct manipulations related to depression. We identify significant positive correlations between deep-layer prefrontal neurons in slice calcium imaging as one such measure. Further, we show that this metric is influenced by prefrontal D2R activation in a manner that may explain how D2R partial agonists can act synergistically with other antidepressant treatments. For context, we further explored (using genetic and optogenetic manipulations) how this mechanism (D2R activation) may influence a standard depression-related behavioral assay. Although the significance of these behavioral assays is questionable, these data validate that prefrontal D2Rs are related to behaviors that are commonly studied in the context of depression, and which are convergently affected by the other manipulations we studied. They also demonstrate a role for prefrontal D2R signaling in stress-coping, for which the TST may be more appropriate.
Multiple lines of evidence implicate prefrontal dopamine in responses to acute and chronic stress, as well as aversive processing more generally (68–70). One recent study found opposing changes and functions of VTA-NAc vs. VTA-mPFC projections (34). VTA-NAc neuron firing increased after social defeat stress, and stimulating VTA-NAc projections enhanced susceptibility. Conversely, VTA-mPFC neuron firing decreased after social defeat, and inhibiting VTA-mPFC projections enhanced susceptibility. This is consistent with our finding that stimulating VTA-mPFC projections enhances struggling, and our model that increases/decreases in D2R function and associated aspects of circuit physiology elicit antidepressant effects/increase depression susceptibility. Some data directly support a role for D2Rs in resolving depression in the context of exercise or antidepressant treatment (18,38–41). Thus, antidepressants, D2R agonists, and environmental influences (e.g., exercise, psychotherapy, bright light therapy, etc.), might work synergistically at the level of prefrontal microcircuits. Further studies will reveal whether this synergy occurs at level of dopamine/D2Rs or, alternatively, involves common downstream network processes. For example, it will be important to study the effects of manipulating D2Rs in CSDS-exposed mice to fully explicate the behavioral consequences of the effects we observed in brain slices.
Our results complement recent studies suggesting a role for D1Rs or D1R+ neurons in depression-related behaviors and antidepressant responses (25–27). Prefrontal D2Rs/D2R+ neurons have been less studied in this context, but as noted above, are clinically relevant and may contribute to resolution of depression (18,38–41). The relative balance of D1R vs. D2R activity is also important for many prefrontal-dependent functions (65,71,72), and will be important to study in the future. Our study provides evidence that prefrontal D2Rs and D2R+ neurons modulate the same behaviors (i.e., ‘antidepressant-like’ responses) as D1Rs / D1R+ neurons, possibly downstream of phasic prefrontal dopamine release (Figure 6). Future studies could investigate whether positively correlated activity is increased or decreased in vivo during specific behaviors or conditions. Regardless, slice activity patterns as described are a metric that informs us about the state of the underlying microcircuitry. The goal of this paper is really to discover measures of circuit physiology that can be used as assays to identify novel antidepressant treatments, understand mechanisms of antidepressant response, etc., rather than to identify patterns of activity associated with specific behaviors or in vivo states.
We chose to express GCaMP in (mainly) excitatory neurons to more specifically understand the effect of our manipulations on deep layer projection neurons. However, D2Rs are expressed in projection neurons and interneurons, and both are important for depression-related behavior and antidepressant-like effects (25,73–75). Rhythmic neuronal activity is important for coupling prefrontal cortex with other depression-relevant regions (76). For example, theta and low gamma synchrony in mPFC is disrupted in Disc1 mutant mice and correlates with active coping behavior in the TST (57). Because D2Rs expressed on interneurons likely play a role in synchronizing certain patterns of activity, quinpirole’s actions on interneurons may contribute to the observed changes in correlated activity. Opposing effects of stress and antidepressants on dendritic spines is a highly reproduced finding and also a potential circuit mechanism for at least some of our findings (75). Importantly, we do not necessarily intend to imply that the manipulations we studied all act directly via D2Rs expressed on either excitatory or inhibitory interneurons. Rather, different manipulations may act via different proximal mechanisms to ultimately elicit similar effects at the level of microcircuit activity. This defines an important intermediate phenotype relevant for discovering novel antidepressants.
Supplementary Material
KEY RESOURCES TABLE.
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Organism/Strain | Mouse: C57BL/6J, males/females | The Jackson Laboratory | JAX:000664 | |
| Organism/Strain | Mouse: B6-CamKII::TtA, males/females | The Jackson Laboratory | JAX: 00310 | |
| Organism/Strain | Mouse: tetO-Disc1dn, males/females | The Jackson Laboratory | JAX: 008790 | |
| Organism/Strain | Mouse: DRD2 loxP/loxP | The Jackson Laboratory | JAX: 020631 | |
| Organism/Strain | Mouse:TH::Cre +/−, males/females | www.gensat.org | line FI12 | |
| Organism/Strain | Mouse: D2::Cre +/−, males | www.gensat.org | Line ER44 | |
| Organism/Strain | Mouse: Crl:CD1(ICR), males | Charles River | strain: 022 | Retired Breeders |
| Bacterial or Viral Strain | AAV5-CaMKII-GCaMP6f | Upenn Virus Core | ||
| Bacterial or Viral Strain | AAV5-Ef1-DIO-ChR2-eYFP | Upenn Virus Core | ||
| Bacterial or Viral Strain | AAV5-Ef1-DIO-eYFP | Upenn Virus Core | ||
| Bacterial or Viral Strain | AAV5-Syn-Cre-mCherry | Upenn Virus Core | ||
| Bacterial or Viral Strain | AAV5-Syn-mCherry | Upenn Virus Core | ||
| Chemical Compound or Drug | (-)Quinpirole | Sigma-Aldrich | ||
| Chemical Compound or Drug | (±)-SKF 38393 | Sigma-Aldrich | ||
| Chemical Compound or Drug | (±)-Sulpiride | Sigma-Aldrich | ||
| Software; Algorithm | ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/index.html | |
| Software; Algorithm | ImageJ Plugin: cvMatch_Template | Tseng et al, Lab Chip, 2011 | https://sites.google.com/site/qingzongtseng/template-matching-ij-plugin | |
| Software; Algorithm | MATLAB_R2019B | Mathworks | R2019B | https://www.mathworks.com/products |
| Software; Algorithm | Prism 9 software | GraphPad Software | v9.0.0 | https://www.graphpad.com/scientific-software/prism |
Acknowledgements:
We wish to thank Dr. Laura DeNardo, Dr. Nicholas Frost and members of the Sohal lab for helpful comments and discussion related to the manuscript. A previous version of this manuscript was posted on the bioRxiv preprint server.
Funding:
This work was supported by NIH (R01 MH100292 to VSS) and NIH (K08 1K08MH116125 to SAW).
Footnotes
Competing interests: The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data and materials availability:
All code and data used in the analysis is available to academic researchers upon reasonable request.
References
- 1.Drysdale AT, Grosenick L, Downar J, Dunlop K, Mansouri F, Meng Y, et al. (2017): Resting-state connectivity biomarkers define neurophysiological subtypes of depression. Nat Med 23: 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. (2013): Reciprocal limbic-cortical function and negative mood: Converging PET findings in depression and normal sadness. Depress Sci Ment Heal 6: 245–253. [DOI] [PubMed] [Google Scholar]
- 3.Hultman R, Ulrich K, Sachs BD, Blount C, Carlson DE, Ndubuizu N, et al. (2018): Brain-wide Electrical Spatiotemporal Dynamics Encode Depression Vulnerability. Cell 173: 166–180.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hultman R, Mague SD, Li Q, Katz BM, Michel N, Lin L, et al. (2016): Dysregulation of Prefrontal Cortex-Mediated Slow-Evolving Limbic Dynamics Drives Stress-Induced Emotional Pathology. Neuron 91: 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferenczi EA, Zalocusky KA, Liston C, Grosenick L, Warden MR, Amatya D, et al. (2015): Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science (80- ) 351: aac9698–aac9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim S-Y, et al. (2012): A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492: 428–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. (2005): Deep brain stimulation for treatment-resistant depression. Neuron 45: 651–60. [DOI] [PubMed] [Google Scholar]
- 8.Gershon AA, Dannon PN, Grunhaus L (2003): Transcranial magnetic stimulation in the treatment of depression. Am J Psychiatry 160: 835–845. [DOI] [PubMed] [Google Scholar]
- 9.Wani A, Trevino K, Marnell P, Husain MM (2013): Advances in brain stimulation for depression. Ann Clin Psychiatry 25: 217–224. [PubMed] [Google Scholar]
- 10.Flint J, Kendler KS (2014): The Genetics of Major Depression. Neuron 81: 484–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang X hua, Huang J, Zhu C ying, Wang Y fei, Cheung EFC, Chan RCK, Xie G rong (2014): Motivational deficits in effort-based decision making in individuals with subsyndromal depression, first-episode and remitted depression patients. Psychiatry Res 220: 874–882. [DOI] [PubMed] [Google Scholar]
- 12.Naranjo CA, Tremblay LK, Busto UE (2001): The role of the brain reward system in depression. Prog Neuro-Psychopharmacology Biol Psychiatry 25: 781–823. [DOI] [PubMed] [Google Scholar]
- 13.Dunlop BW, Nemeroff CB (2007): The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry 64: 327–37. [DOI] [PubMed] [Google Scholar]
- 14.Post RJ, Warden MR (2018): Melancholy, anhedonia, apathy: the search for separable behaviors and neural circuits in depression. Curr Opin Neurobiol 49: 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baik JH (2020): Stress and the dopaminergic reward system. Exp Mol Med 52: 1879–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grace AA (2016): Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci 17: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szczypiński JJ, Gola M (2018): Dopamine dysregulation hypothesis: The common basis for motivational anhedonia in major depressive disorder and schizophrenia? Rev Neurosci 29: 727–744. [DOI] [PubMed] [Google Scholar]
- 18.Dailly E, Chenu F, Renard CE, Bourin M (2004): Dopamine, depression and antidepressants. Fundam Clin Pharmacol 18: 601–607. [DOI] [PubMed] [Google Scholar]
- 19.Tundo A, de Filippis R, De Crescenzo F (2019): Pramipexole in the treatment of unipolar and bipolar depression. A systematic review and meta-analysis. Acta Psychiatr Scand 140: 116–125. [DOI] [PubMed] [Google Scholar]
- 20.Mah L, Zarate CA, Nugent AC, Singh JB, Manji HK, Drevets WC (2011): Neural mechanisms of antidepressant efficacy of the dopamine receptor agonist pramipexole in treatment of bipolar depression. Int J Neuropsychopharmacol 14: 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fornaro M, Fusco A, Anastasia A, Cattaneo CI, Berardis D De (2019): Brexpiprazole for treatment-resistant major depressive disorder. 10.1080/1465656620191654457 20: 1925–1933. [DOI] [PubMed] [Google Scholar]
- 22.Berman RM, Fava M, Thase ME, Trivedi MH, Swanink R, McQuade RD, et al. (2009): Aripiprazole augmentation in major depressive disorder: A double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr 14: 197–206. [DOI] [PubMed] [Google Scholar]
- 23.Earley W, Burgess MV, Rekeda L, Dickinson R, Szatmári B, Németh G, et al. (2019): Cariprazine treatment of bipolar depression: A randomized double-blind placebo-controlled phase 3 study. Am J Psychiatry 176: 439–448. [DOI] [PubMed] [Google Scholar]
- 24.Durgam S, Earley W, Lipschitz A, Guo H, Laszlovszky I, Németh G, et al. (2016): An 8-week randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of cariprazine in patients with bipolar I depression. Am J Psychiatry 173: 271–281. [DOI] [PubMed] [Google Scholar]
- 25.Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Kozorovitskiy Y (2021): Ketamine RapidlyEnhances Glutamate-Evoked Dendritic Spinogenesis in Medial Prefrontal Cortex Through Dopaminergic Mechanisms. Biol Psychiatry 89: 1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Xiao L, Kozorovitskiy Y (2021): Attenuated dopamine signaling after aversive learning is restored by ketamine to rescue escape actions. Elife 10. 10.7554/ELIFE.64041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hare BD, Shinohara R, Liu RJ, Pothula S, DiLeone RJ, Duman RS (2019): Optogenetic stimulationof medial prefrontal cortex Drd1 neurons produces rapid and long-lasting antidepressant effects. Nat Commun 10. 10.1038/s41467-018-08168-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kara NZ, Stukalin Y, Einat H (2018): Revisiting the validity of the mouse forced swim test: Systematic review and meta-analysis of the effects of prototypic antidepressants. Neurosci Biobehav Rev 84: 1–11. [DOI] [PubMed] [Google Scholar]
- 29.Dzirasa K, Covington HE (2012): Increasing the validity of experimental models for depression. Ann N Y Acad Sci 1265: 36–45. [DOI] [PubMed] [Google Scholar]
- 30.Commons KG, Cholanians AB, Babb JA, Ehlinger DG (2017): The Rodent Forced Swim Test Measures Stress-Coping Strategy, Not Depression-like Behavior. ACS Chem Neurosci 8: 955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Kloet ER, Molendijk ML (2016): Coping with the Forced Swim Stressor: Towards Understandingan Adaptive Mechanism. Neural Plast 2016: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molendijk ML, de Kloet ER (2015, December 1): Immobility in the forced swim test is adaptive anddoes not reflect depression. Psychoneuroendocrinology, vol. 62. Elsevier Ltd, pp 389–391. [DOI] [PubMed] [Google Scholar]
- 33.Lee AT, Cunniff MM, See JZ, Wilke SA, Luongo FJ, Ellwood IT, et al. (2019): VIP Interneurons Contribute to Avoidance Behavior by Regulating Information Flow across Hippocampal-Prefrontal Networks. Neuron 102: 1223–1234.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. (2013): Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493: 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moghaddam B, Jackson M (2004): Effect of stress on prefrontal cortex function. Neurotoxicity Research, vol. 6. Springer New York LLC, pp 73–78. [DOI] [PubMed] [Google Scholar]
- 36.Ranganath A, Jacob SN (2016): Doping the Mind: Dopaminergic Modulation of Prefrontal CorticalCognition. Neuroscientist 22: 593–603. [DOI] [PubMed] [Google Scholar]
- 37.Goschke T, Bolte A (2014): Emotional modulation of control dilemmas: The role of positive affect,reward, and dopamine in cognitive stability and flexibility. Neuropsychologia 62: 403–423. [DOI] [PubMed] [Google Scholar]
- 38.Larisch R, Klimke A, Vosberg H, Löffler S, Gaebel W, Müller-Gärtner HW (1997): In vivo evidence for the involvement of dopamine-D2 receptors in striatum and anterior cingulate gyrus in major depression. Neuroimage 5: 251–260. [DOI] [PubMed] [Google Scholar]
- 39.Klimke A, Larisch R, Janz A, Vosberg H, Müller-Gärtner HW, Gaebel W (1999): Dopamine D2 receptor binding before and after treatment of major depression measured by [123I]IBZM SPECT. Psychiatry Res - Neuroimaging 90: 91–101. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt K, Nolte-Zenker B, Patzer J, Bauer M, Schmidt LG, Heinz A (2001): Psychopathological correlates of reduced dopamine receptor sensitivity in depression, schizophrenia, and opiate and alcohol dependence. Pharmacopsychiatry 34: 66–72. [DOI] [PubMed] [Google Scholar]
- 41.Chen C, Nakagawa S, Kitaichi Y, An Y, Omiya Y, Song N, et al. (2016): The role of medial prefrontal corticosterone and dopamine in the antidepressant-like effect of exercise. Psychoneuroendocrinology 69: 1–9. [DOI] [PubMed] [Google Scholar]
- 42.Monteggia LM, Zarate C (2015, February 1): Antidepressant actions of ketamine: From molecularmechanisms to clinical practice. Current Opinion in Neurobiology, vol. 30. Elsevier Ltd, pp 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH (2000): Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47: 351–354. [DOI] [PubMed] [Google Scholar]
- 44.Price RB, Nock MK, Charney DS, Mathew SJ (2009): Effects of Intravenous Ketamine on Explicit and Implicit Measures of Suicidality in Treatment-Resistant Depression. Biol Psychiatry 66: 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. (2006): A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63: 856–864. [DOI] [PubMed] [Google Scholar]
- 46.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. (2010): A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry 67: 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kendler KS, Karkowski LM, Prescott CA (1999): Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156: 837–841. [DOI] [PubMed] [Google Scholar]
- 48.Golden SA, Covington HE, Berton O, Russo SJ (2011): A standardized protocol for repeated social defeat stress in mice. Nat Protoc 6: 1183–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. (2007): Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 131: 391–404. [DOI] [PubMed] [Google Scholar]
- 50.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CAM, et al. (2000): Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet 9: 1415–1423. [DOI] [PubMed] [Google Scholar]
- 51.Hennah W, Thomson P, Peltonen L, Porteous D (2006, July 1): Beyond Schizophrenia: The role of DISC1 in major mental illness. Schizophrenia Bulletin, vol. 32. Oxford Academic, pp 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK (2008): The DISC locus in psychiatric illness. Mol Psychiatry 13: 36–64. [DOI] [PubMed] [Google Scholar]
- 53.Thomson PA, Parla JS, McRae AF, Kramer M, Ramakrishnan K, Yao J, et al. (2014): 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: Analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry 19: 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, et al. (2008): Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 13: 173–86, 115. [DOI] [PubMed] [Google Scholar]
- 55.Niwa M, Kamiya A, Murai R, Kubo K, Gruber AJ, Tomita K, et al. (2010): Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65: 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. (2007): Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A 104: 14501–14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sauer JF, Strüber M, Bartos M (2015): Impaired fast-spiking interneuron function in a genetic mouse model of depression. Elife 2015. 10.7554/eLife.04979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. (2013): Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493: 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grace AA, Floresco SB, Goto Y, Lodge DJ (2007): Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 30: 220–227. [DOI] [PubMed] [Google Scholar]
- 60.Bunney BS, Chiodo LA, Grace AA (1991): Midbrain dopamine system electrophysiological functioning: A review and new hypothesis. Synapse 9: 79–94. [DOI] [PubMed] [Google Scholar]
- 61.Grace AA, Bunney BS (1984): The control of firing pattern in nigral dopamine neurons: Burst firing. J Neurosci 4: 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grace AA, Bunney BS (1983): Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 10: 301–15. [DOI] [PubMed] [Google Scholar]
- 63.Ellwood IT, Patel T, Wadia V, Lee AT, Liptak AT, Bender KJ, Sohal VS (2017): Tonic or phasic stimulation of dopaminergic projections to prefrontal cortex causes mice to maintain or deviate from previously learned behavioral strategies. J Neurosci 37: 8315–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seamans JK, Yang CR (2004): The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74: 1–58. [DOI] [PubMed] [Google Scholar]
- 65.Trantham-Davidson H, Neely LC, Lavin A, Seamans JK (2004): Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci 24: 10652–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS (2012): Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci 32: 4959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson SE, Sohal VS (2017): Dopamine D2 receptors modulate pyramidal neurons in mouse medial prefrontal cortex through a stimulatory G-protein pathway. J Neurosci 37: 10063–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lammel S, Tye KM, Warden MR (2014): Progress in understanding mood disorders: Optogenetic dissection of neural circuits. Genes, Brain Behav 13: 38–51. [DOI] [PubMed] [Google Scholar]
- 69.Holly EN, Miczek KA (2016, January 1): Ventral tegmental area dopamine revisited: Effects of acute and repeated stress. Psychopharmacology, vol. 233. Springer Verlag, pp 163–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weele CMV, Siciliano CA, Tye KM (2019, June 15): Dopamine tunes prefrontal outputs to orchestrate aversive processing. Brain Research, vol. 1713. Elsevier B.V., pp 16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.St. Onge JR, Abhari H, Floresco SB (2011): Dissociable contributions by prefrontal D1 and D2 receptors to risk-based decision making. J Neurosci 31: 8625–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meunier CNJ, Chameau P, Fossier PM (2017): Modulation of synaptic plasticity in the cortex needs to understand all the players. Front Synaptic Neurosci 9. 10.3389/fnsyn.2017.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luscher B, Feng M, Jefferson SJ (2020): Antidepressant mechanisms of ketamine: Focus on GABAergic inhibition. Adv Pharmacol 89: 43–78. [DOI] [PubMed] [Google Scholar]
- 74.Yin YY, Wang YH, Liu WG, Yao JQ, Yuan J, Li ZH, et al. (2021): The role of the excitation:inhibition functional balance in the mPFC in the onset of antidepressants. Neuropharmacology 191. 10.1016/j.neuropharm.2021.108573 [DOI] [PubMed] [Google Scholar]
- 75.Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. (2019): Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science (80- ) 364. 10.1126/science.aat8078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mathalon DH, Sohal VS (2015): Neural oscillations and synchrony in brain dysfunction and neuropsychiatric disorders it’s about time. JAMA Psychiatry 72: 840–844. [DOI] [PubMed] [Google Scholar]
- 77.Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, et al. (2011): Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci 14: 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luongo FJ, Horn ME, Sohal VS (2016): Putative microcircuit-level substrates for attention are disrupted in mouse models of autism. Biol Psychiatry 79: 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tseng Q, Wang I, Duchemin-Pelletier E, Azioune A, Carpi N, Gao J, et al. (2011): A new micropatterning method of soft substrates reveals that different tumorigenic signals can promote or reduce cell contraction levels. Lab Chip 11: 2231–2240. [DOI] [PubMed] [Google Scholar]
- 80.Mukamel EA, Nimmerjahn A, Schnitzer MJ (2009): Automated Analysis of Cellular Signals from Large-Scale Calcium Imaging Data. Neuron 63: 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luongo FJ, Zimmerman C, Horn ME, Sohal VS (2016): Correlations between prefrontal neurons form a small world network that optimizes the generation of multineuron sequences of activity. J Neurophysiol jn.01043.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O’Connor DH, Peron SP, Huber D, Svoboda K (2010): Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron 67: 1048–1061. [DOI] [PubMed] [Google Scholar]
- 83.Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, Gould TD (2011): The tail suspension test. J Vis Exp e3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code and data used in the analysis is available to academic researchers upon reasonable request.