

Abstract
Brain infiltration of peripheral immune cells and their interactions with brain-resident cells may contribute to Alzheimer’s disease (AD) pathology. To examine these interactions, in the present study we developed a three-dimensional human neuroimmune axis model comprising stem cell-derived neurons, astrocytes and microglia, together with peripheral immune cells. We observed an increase in the number of T cells (but not B cells) and monocytes selectively infiltrating into AD relative to control cultures. Infiltration of CD8+ T cells into AD cultures led to increased microglial activation, neuroinflammation and neurodegeneration. Using single-cell RNA-sequencing, we identified that infiltration of T cells into AD cultures led to induction of interferon-γ and neuroinflammatory pathways in glial cells. We found key roles for the C-X-C motif chemokine ligand 10 (CXCL10) and its receptor, CXCR3, in regulating T cell infiltration and neuronal damage in AD cultures. This human neuroimmune axis model is a useful tool to study the effects of peripheral immune cells in brain disease.
Adaptive immune cells have been increasingly shown to contribute to AD pathogenesis1–5. Higher numbers of T cells have been observed in brains of patients with AD6–11 and AD mouse models12–18. CD8+ T cell clonal expansion has been reported in brains of patients with AD11 and CD4+ T cells have been found in brains of patients with Lewy body dementia19. Detailed studies about how infiltrating T cells impact AD neuropathogenesis have been limited by AD transgenic mouse models, which do not fully recapitulate the complex human genetic backgrounds of AD patients or the major histocompatibility complex (MHC) context20–22. Although models derived from induced pluripotent stem cells (iPSCs) of patients with AD carry the potential to recreate complex human genetic backgrounds23, they do not fully recapitulate AD pathology—amyloid-β (Aβ) plaques, neurofibrillary tangles (NFTs) and neuroinflammation23,24. To address these limitations, we previously developed three-dimensional (3D), human, embryonic stem cell-derived, neural cell-culture models that recapitulate the full neuropathological cascade of AD25–27. In addition, we developed a triculture model of AD that recapitulates the interaction of microglia, neurons and astrocytes, leading to neuroinflammation28.
In the present study, we describe a 3D human neuroimmune axis model comprising stem cell-derived neurons, astrocytes and microglia, together with human peripheral immune cells. We used this model to investigate the molecular mechanism underlying the infiltration of peripheral immune cells into an environment of AD neuropathology. We observed a dramatic increase in the number of T cells infiltrating AD versus control cultures. Human monocytes also infiltrated more robustly into AD cultures, but this was negated by the presence of microglia. Infiltrating CD8+ T cells into AD cultures led to microglial activation and exacerbation of neuroinflammation and neurodegeneration. Finally, we found that blocking the CXCL10–CXCR3 axis robustly prevented T cell infiltration and neurodegeneration in AD cultures. Thus, our neuroimmune axis model has allowed for the successful recapitulation and dissection of the molecular mechanisms regulating infiltration of peripheral immune cells and, specifically, how CD8+ T cells contribute to AD-related neuropathogenesis.
Results
Development of a 3D human neuroimmune axis model of AD
To assess infiltration of peripheral blood mononuclear cells (PBMCs) and interaction with brain-resident cells, we developed a 3D human neuroimmune axis model using microfluidics—a peripheral immune chip (PiChip) system. The PiChip system supports a multicellular culture, including human neurons (Neu), astrocytes (ACs), iPSC-derived microglia (iMGLs) and PBMCs in a microfluidic chip. We used immortalized human neural progenitor cells (NPCs; ReN cell VM) overexpressing Aβ due to stable transfection with familial AD (FAD) mutations in APP–K670N/M671L, V717I and APPSL, and PSEN1–ΔE9 (refs. 25–27). NPCs were differentiated for 4 weeks to generate neurons and astrocytes. The iMGLs were differentiated as described previously29,30 and added to the PiChip at the end of week 4 of NPC differentiation, 1 d before addition of PBMCs (Fig. 1a). Infiltration of PBMCs was monitored using time-lapse confocal imaging before collecting cells and medium and subjecting them to biochemical assays, immunostaining and single-cell RNA-sequencing (scRNA-seq) (Fig. 1a).
Fig. 1 |. Construction and characterization of a 3D human neuroimmune axis model of AD.
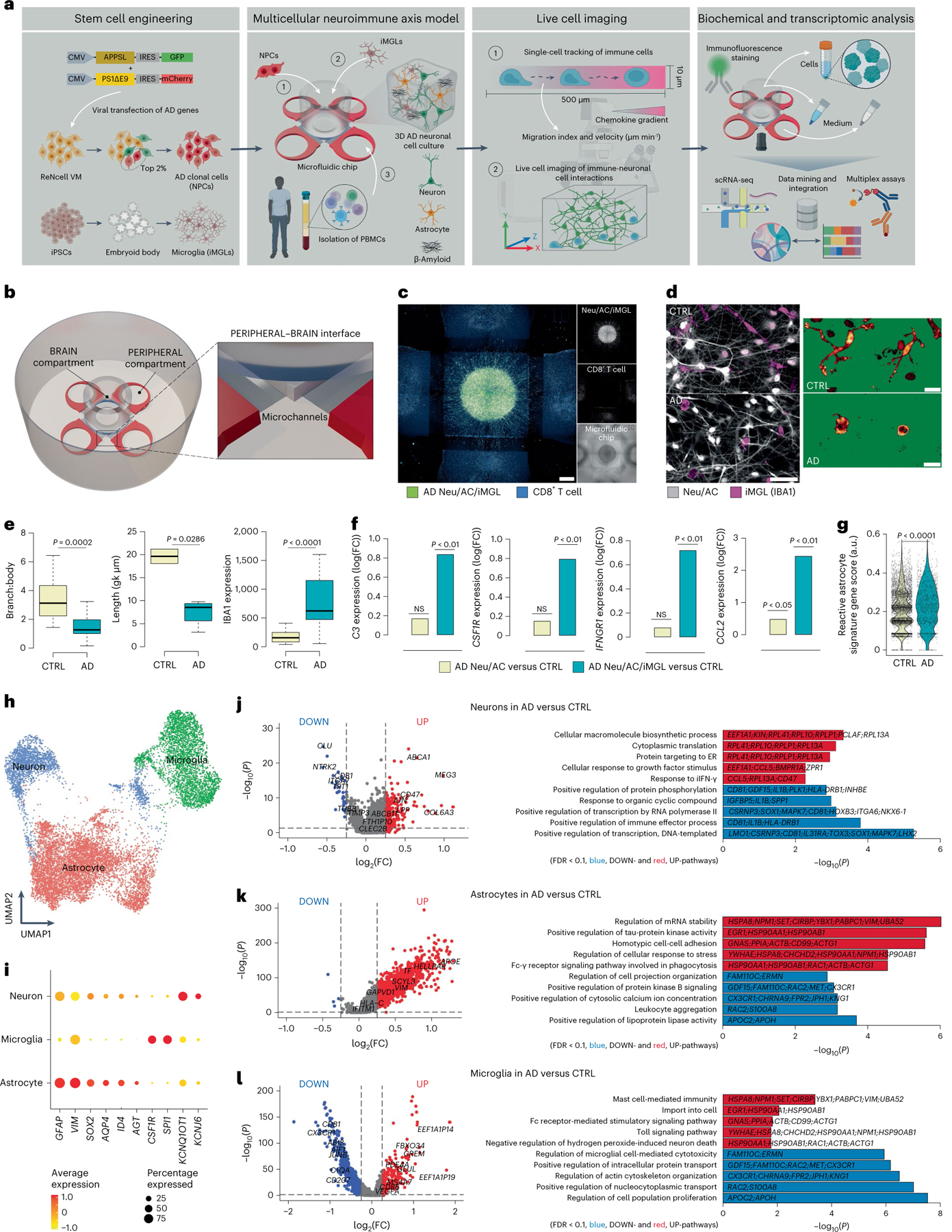
a, Schematic representing a pipeline for modeling the AD brain in a 3D human neuroimmune axis model using stem cell-derived neurons, ACs and microglia, together with peripheral immune cells in a microfluidic system (referred to as the PiChip system). b, Schematic showing the PiChip system, which contains 400 narrow migration microchannels (4 sides, 100 on each side; 10 × 10 × 500 μm3 in height, width and length) connecting the BRAIN compartment to the four PERIPHERAL compartments. c, Confocal images of 3D-differentiated AD Neu/ACs and iMGLs (green) in the BRAIN compartment and nuclei-stained CD8+ cells (blue) in the PERIPHERAL compartments. Scale bar, 500 μm. d, Representative images of microglia morphology in CTRL and AD conditions stained for IBA1 (Neu/AC, gray; iMGL, magenta—left panel). Scale bar, 50 μm (left) and 10 μm (right). e, Microglia branch:body ratio, length and IBA1 expression in CTRL and AD conditions (n = 13 independent ROIs from 5 independent experiments for CTRL and AD branch:body ratio; n = 4 independent ROIs from 4 independent experiments for CTRL and AD microglia length; n = 12 CTRL and n = 18 AD independent ROIs from 5 independent experiments for IBA1 expression). The center lines show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend to minimum and maximum values. P values are from a nonparametric, two-sided Mann–Whitney U-test. f, Whole RNA-seq analysis showing inflammatory and activated glial markers in AD cultures compared with CTRL. P values are from a quasi-likelihood edgeR:glmQLFTest method. NS, not significant. g, Highly enriched genes associated with reactive ACs in AD compared with CTRL. P values are from a nonparametric, two-sided Mann–Whitney U-test. h, UMAP visualization of the cell-type composition of the PiChip cultures by single-cell transcriptome profiles. i, Dot plot of identity genes for distinct cell types. Color scale indicates the average expression of identity genes in each cell population and dot size is proportional to the percentage of cells expressing the identity genes. a.u., arbitrary units. j–l, Volcano plots of differentially expressed genes (left) and GO of significantly upregulated and downregulated pathways (right) in neurons (j), ACs (k) and microglia (l) in AD compared with CTRL. ER, endoplasmic reticulum. P values are from Wilcoxon’s rank-sum test. Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown as red and blue dots, respectively.
To observe the infiltration of PBMCs, we engineered an open-top, multi-compartment, microfluidic system in which different cell types of the central nervous system (CNS) and PBMCs could be positioned side by side to allow live-cell imaging (Fig. 1a,b). The microfluidic system contains a BRAIN compartment with a large concentric cylinder reservoir, allowing accumulation of AD neuropathology and various soluble factors, along with four PERIPHERAL compartments supplying PBMCs (Fig. 1b and Extended Data Fig. 1a). The BRAIN and PERIPHERAL compartments are connected by 100 migration microchannels (Extended Data Fig. 1a), providing a gradient of soluble factors released by CNS cells. The 3D AD neural–glial culture (Fig. 1c and Extended Data Fig. 1b) enabled the accumulation of Aβ deposition and p-tau (Extended Data Fig. 1b–j). Glial fibrillary acidic protein-positive (GFAP+) reactive ACs were significantly higher (P = 0.0003) in AD cultures versus control (CTRL) cultures (Extended Data Fig. 1d). We observed significant increases in levels of Aβ40 (2,120-fold, P = 0.0146), Aβ42 (2,310-fold, P = 0.0033) and Aβ42:Aβ40 ratio (P = 0.0032) and a similar significant increase in p-tau (P = 0.0022) in AD versus CTRL cultures (Extended Data Fig. 1e–j).
Microglia in CTRL cultures showed widely branched filopodia (Fig. 1d,e), characteristic of homeostatic microglia31. In contrast, AD cultures contained amoeboid microglia with relatively small numbers of branches (Fig. 1d,e) and decreased length, indicative of an activated proinflammatory state31. The AD-like environment induced a significant upregulation of microglial activation markers, including ionized calcium-binding adapter molecule 1 (IBA1), versus microglia in CTRL cultures (P < 0.0001; Fig. 1e).
We next examined gliosis/inflammation-associated genes: complement C3, colony-stimulating factor-1 receptor (CSF1R), monocyte chemotactic protein-1 encoding gene (CCL2) and interferon (IFN)-γ receptor 1-associated receptor (IFNGR1). In the absence of microglia, expression of C3 and CSF1R was not significantly different in AD versus CTRL cultures (Fig. 1f). However, when microglia were introduced, C3 and CSF1R expressions significantly increased (P < 0.01) by 6.9- and 6.2-fold, respectively, in AD versus CTRL cultures (Fig. 1f). We observed a similar trend for IFNGR1, where its expression is significantly increased in AD versus CTRL when microglia were present. CCL2 expression was significantly upregulated in AD versus CTRL cultures in the presence of microglia (P < 0.01) and without (P < 0.05) (Fig. 1f). The scRNA-seq analysis revealed a substantial increase (P < 0.0001) in reactive AC-associated genes in AD versus CTRL cultures (Fig. 1g). The scRNA-seq analyses (Fig. 1h and Extended Data Fig. 1k) also identified three transcriptionally unique clusters of neurons (SYT1, KCNQ1OT1 and KCNJ6), ACs (GFAP, VIM, SOX2, AQP4, ID4 and AGT) and microglia (CSF1R and SPI1) from a total of 20,153 single cells in cultures (Extended Data Fig. 1k) and a total of 3,631 and 9,702 cells analyzed from Neu/AC and Neu/AC/iMGL cultures, respectively (Fig. 1h, i). Transcriptional analysis of differentially expressed genes identified 263 up- and 82 downregulated genes in neurons (Fig. 1j), and 1,1146 up- and 61 downregulated genes in ACs in AD versus CTRL cultures (Fig. 1k).
Pathway analysis revealed a significant increase in pathways related to endoplasmic reticulum stress, DNA damage and INF-γ in AD neurons (Fig. 1j). Gene ontology (GO) analysis revealed several significantly upregulated pathways related to messenger RNA stability, tau kinase and regulation of cellular response to stress in AD ACs versus CTRL cultures (Fig. 1k). In addition, we identified 275 up- and 4,019 downregulated genes in microglia in AD versus CTRL cultures with upregulated pathways related to Fc-receptor-mediated and toll signaling (Fig. 1l). Transcription analysis of ACs revealed 965 significantly up- and 100 downregulated genes in AD Neu/ACs/iMGLs compared with AD cultures lacking microglia (Extended Data Fig. 1l). Reactive ACs (C3-, VIM-, GFAP, and S100B-high) in the presence of microglia in AD cultures revealed significantly downregulated genes related to glutamate and γ-aminobutyric acid homeostasis (SLC1A3, SLC6A11 and CPLX1) compared with AD cultures devoid of microglia (Extended Data Fig. 1l–n), suggesting impaired homeostasis and compromised function. In addition, we observed significant upregulation (P < 0.0001) of IFN-γ- and reactive AC-associated genes in ACs of AD Neu/ACs/iMGLs compared with AD cultures without microglia (Extended Data Fig. 1o). Collectively, these data show that the PiChip system recapitulates the major pathological features of AD.
Chemotaxis of PBMCs in microfluidic systems
We next assessed the chemotaxis of T cells, monocytes and B cells in response to various chemoattractants (Supplementary Fig. 1a). We isolated PBMCs from de-identified healthy individuals and used flow cytometry to assess the purity of isolated PBMCs. Starting with whole blood, the isolated CD3+ T cell, CD14+ monocyte and CD19+ B cell purity was 96.2%, 85% and 98.3% (gated on singlets, live, CD45+), respectively (Supplementary Fig. 1b–d). We measured directional migration of PBMCs toward a panel of known chemoattractants selected for each cell type (Supplementary Fig. 1e–j). In the absence of neuronal cells, we added chemokines and PBMCs to the CHEMOKINE and PERIPHERAL compartments, respectively (Supplementary Fig. 1a). Using confocal time-lapse imaging for ~20 h, we quantified the number and velocity of PBMCs that migrated across the microchannels to the CHEMOKINE compartment (Supplementary Fig. 1e–j). CD3+ T cell chemotaxis toward CXCL12 (Supplementary Fig. 1e,f) displayed a bell-shaped, dose-dependent profile, with a maximum of 406 ± 264 recruited T cells after 15 h toward 50 nM CXCL12 (higher than 5 nM and 100 nM CXCL12) (Supplementary Fig. 1f). CD3+ T cells migrated at an average velocity of 5.5 ± 3.0 μm min−1 in response to CXCL12 (1,000 nM) (Supplementary Fig. 1f). We found no significant changes in CD3+ T cell velocity toward different concentrations of CXCL12 (Supplementary Fig. 1f).
Next, CCL2 was used to elicit chemotaxis of human monocytes (Supplementary Fig. 1g,h). Monocytes showed a classic, bell-shaped, dose-dependent chemotactic response to CCL2, with a maximum of 1,614 ± 775 recruited monocytes after 15 h at 100 nM concentration (higher than 5 nM (P = 0.0001) and 1,000 nM (P = 0.0013) CCL2). Monocytes exhibited no significant differences in the number of migrated cells toward CCL2 at 50 nM and 100 nM concentrations (Supplementary Fig. 1g,h); however, 50 nM concentration led to a higher number of migrated monocytes compared with 5 nM (P = 0.0324) CCL2 chemokine (Supplementary Fig. 1h). CCL2 induced the recruitment of monocytes with an average velocity of 1.2 ± 0.2 μm min−1 at 50 nM concentration, which was significantly higher than that observed at 100 nM (P = 0.0289) and 1,000 nM (P = 0.0055) CCL2 concentrations (Supplementary Fig. 1h).
Human B cells were tested for directional migration toward CCL19 with average recruitment of 28 ± 10 B cells at 1,000 nM CCL19 and no significant differences between varying concentrations of CCL19 (Supplementary Fig. 1i,j). B cells showed an average velocity of 1.5 ± 1.4 μm min−1 in response to 1,000 nM CCL19 (Supplementary Fig. 1j) and exhibited no significant differences in the average velocity toward varying CCL19 concentrations (Supplementary Fig. 1j). Collectively, these data reflect the robustness of the platform to generate chemokine gradients in a microfluidic system.
Selective infiltration of T cells into AD cellular models
Next, we tested whether the presence of AD neuropathology in the PiChip system affects the directional migration of PBMCs into the AD neural–glial cultures. After NPC differentiation into neurons and astrocytes for 4 weeks and the addition of microglia to the 3D cultures for 24 h as described above, PBMCs were isolated, loaded into the PERIPHERAL compartments (Fig. 2a) and monitored for infiltration using time-lapse confocal imaging for 15–20 h (Fig. 2b,c and Supplementary Video 1). We developed an unbiased/automated image analysis pipeline for high-throughput quantification of the number of infiltrating PBMCs and their corresponding velocities utilizing time-lapse confocal imaging and IMARIS software (Supplementary Fig. 2a). Each PiChip system allowed us to track PBMCs across 4 sets of 100 microchannels connecting PERIPHERAL and BRAIN compartments. The migration index was used to quantify the fold-change (FC) of immune cell infiltration into AD cultures (the BRAIN compartment) versus CTRL (Methods and Supplementary Fig. 2b,c).
Fig. 2 |. Selective infiltration of T cells into the AD cellular models and 5×FAD mice.
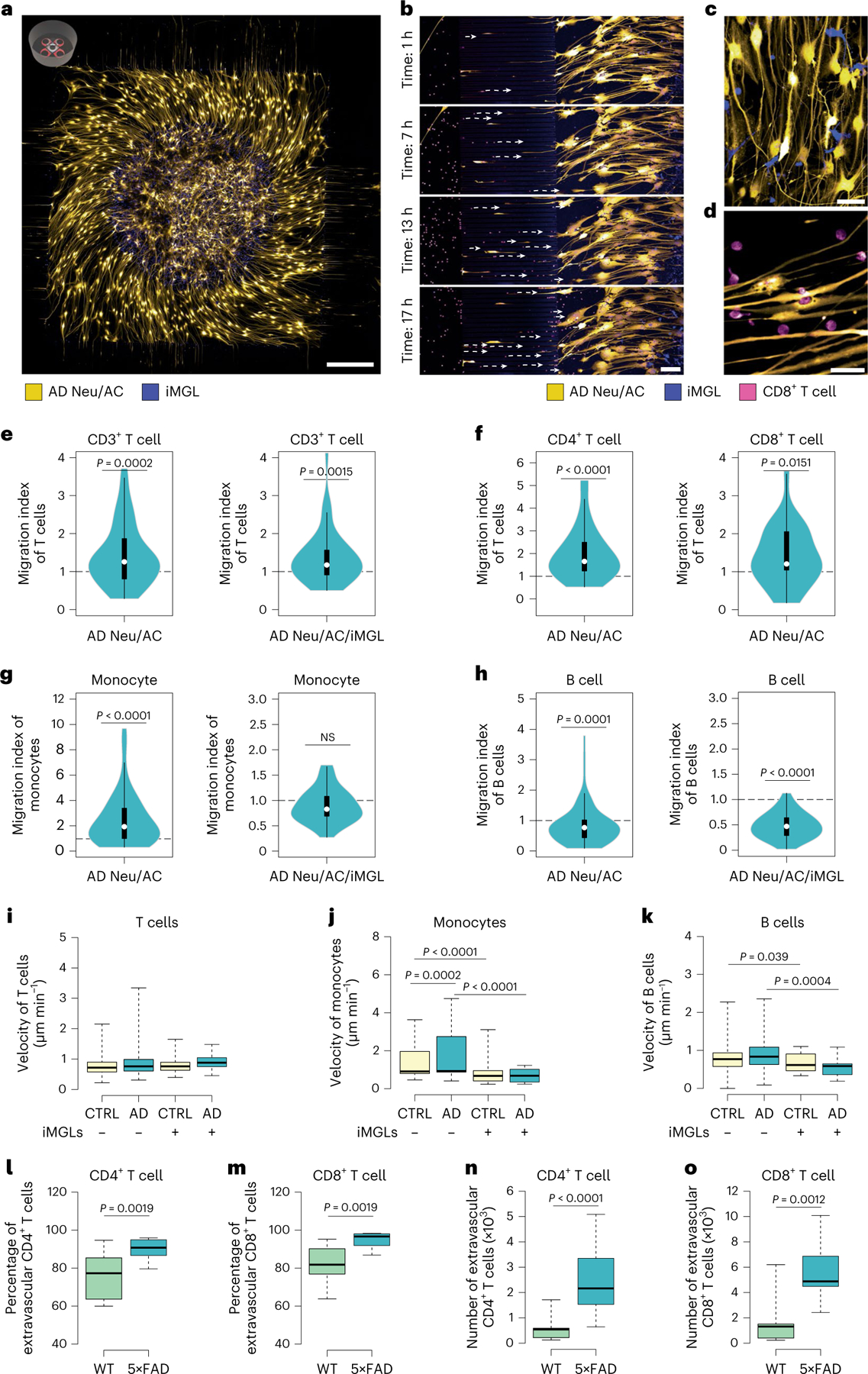
a, Representative confocal imaging of 3D-differentiated AD Neu/AC/iMGL cultures in the PiChip system (Neu/AC, yellow; iMGL, blue). Scale bar, 500 μm. b, Time-lapse confocal imaging showing migration of CD8+ T cells (nuclei stained, magenta) through microchannels over time toward the PiChip BRAIN compartment containing AD cultures (Neu/AC, yellow; iMGL, blue). Scale bar, 100 μm. c, High magnification of the 3D-differentiated AD Neu/ACs (yellow) and iMGLs (blue) in the BRAIN compartment. Scale bar, 20 μm. d, High magnification of infiltrating CD8+ T cells (nuclei stained, magenta) and AD Neu/ACs (yellow) in the BRAIN compartment. Scale bar, 20 μm. e–h, The migration index of CD3+ T cells (n = 68 biological replicates, AD Neu/AC and n = 40, AD Neu/AC/iMGL) (e), CD4+ and CD8+ T cells (n = 32 biological replicates, CD4+ and n = 26, CD8+ T cells) (f), monocytes (n = 60 biological replicates, AD Neu/AC and n = 29, AD Neu/AC/iMGL) (g) and B cells (n = 89 biological replicates, AD Neu/AC and n = 37, AD Neu/AC/iMGL) (h) in the PiChip system. P values are from two-tailed, Wilcoxon’s signed-rank tests and five or six independent experiments. i–k, Migration velocity of CD3+ T cells (n = 40–78 biological replicates) (i), monocytes (n = 28–86 biological replicates) (j) and B cells (n = 29–111 biological replicates) (k) via confined microchannels in the PiChip system. P values are from two-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test and five or six independent experiments. l–o, The percentage (l and m) and absolute number (n and o) of extravascular CD4+ and CD8+ T cells in 5×FAD and WT brains (n = 12 mice each for WT and 5×FAD for CD4+ T cells, n = 9 mice each for WT and 5×FAD for CD8+ T cells). P values are from a nonparametric, two-sided Mann–Whitney U-test. In e–h, the white circles show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend 1.5× the interquartile range (IQR) from the 25th and 75th percentiles; polygons represent density estimates of data and extend to extreme values. In i–o, The center lines show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend to minimum and maximum values.
We observed a significantly greater infiltration of CD3+ T cells into the AD Neu/AC versus CTRL cultures (1.43-fold, P = 0.0002; Fig. 2e) and found a similar trend for CD4+ and CD8+ T cells. Specifically, we observed a significantly greater infiltration of CD4+ T cells (1.66-fold, P < 0.0001; Fig. 2f) and CD8+ T cells (1.21-fold, P = 0.0151; Fig. 2f) in AD Neu/AC versus CTRL cultures. Overexpression of mCherry in the AD cells had no effect on recruitment of T cells compared with CTRL cells (Supplementary Fig. 2d). Monocytes also showed a significant increase in infiltration into the AD Neu/AC versus CTRL cultures (2.45-fold, P < 0.0001; Fig. 2g). In contrast, we observed a significant decrease in the infiltration of B cells into AD Neu/AC versus CTRL cultures (P = 0.0001; Fig. 2h).
Next, we assessed whether microglia impact the number of infiltrating peripheral immune cells. CD3+ T cell infiltration was significantly increased in AD Neu/AC/iMGL versus CTRL cultures (1.36-fold, P = 0.0015; Fig. 2e), indicating that AD pathology leads to preferential recruitment of CD3+ T cells, regardless of the presence of microglia. In contrast, the greater rate of monocyte infiltration into AD Neu/AC versus CTRL cultures was abrogated by the addition of microglia (Fig. 2g). Finally, B cells showed a noticeable decrease in infiltration rate into AD Neu/AC/iMGL cultures compared with CTRL (P < 0.0001; Fig. 2h).
We next measured the migration velocity of PBMCs during their infiltration into AD versus CTRL cultures with and without microglia. CD3+ T cells migrated slightly faster toward AD cultures, independent of microglial presence: 0.87 ± 0.05 μm min−1 and 0.81 ± 0.04 μm min−1 toward AD Neu/AC and CTRL cultures, respectively (Fig. 2i). In the presence of microglia, CD3+ T cells exhibited an average velocity of 0.91 ± 0.04 μm min−1 toward AD Neu/AC/iMGL cultures compared with 0.78 ± 0.04 μm min−1 in CTRL cultures (Fig. 2i). Monocytes showed a slightly faster velocity toward AD Neu/AC cultures at 1.7 ± 0.13 μm min−1 compared with CTRL cultures at 1.42 ± 0.1 μm min−1 (P = 0.0002; Fig. 2j). However, the presence of microglia abrogated this difference in average velocity between AD Neu/AC/iMGL (0.69 ± 0.06 μm min−1) and CTRL (0.76 ± 0.1 μm min−1) cultures (Fig. 2j). In addition, monocytes migrated faster into Neu/AC cultures than Neu/AC/iMGL cultures in both AD and CTRL conditions—2.48-fold (P < 0.0001) and 1.87-fold (P < 0.0001), respectively (Fig. 2j).
Finally, B cells exhibited similar velocity in AD (0.86 ± 0.039 μm min−1) and CTRL (0.79 ± 0.039 μm min−1; Fig. 2k) cultures without microglia, and AD (0.54 ± 0.042 μm min−1) and CTRL (0.67 ± 0.038 μm min−1) cultures with microglia present (Fig. 2k). Similar to monocytes, B cells migrated significantly faster in the absence versus the presence of microglia (1.6- and 1.2-fold into AD and CTRL cultures, respectively; Fig. 2k). Overall, the PiChip system enabled the study of infiltration characteristics of human PBMCs into 3D neural–glial cell cultures, showing that CD4+ and CD8+ T cells more robustly infiltrate AD versus CTRL cultures, regardless of the presence of microglia.
Infiltrating CD4+ and CD8+ T cells in brains of 5×FAD mice
We used intravascular CD45-FITC staining to distinguish brain-localized leukocytes from those in the bloodstream32 (Extended Data Fig. 2a). In 5×FAD mice, we observed that the percentage and number of CD45+CD11b− nonmyeloid cells were significantly increased relative to wild-type (WT) (Extended Data Fig. 2b). Further analysis demonstrated a significantly increased percentage and absolute number of extravascular CD4+ T cells (P = 0.0019 and P < 0.0001, respectively) in the brains of 5×FAD versus WT mice (Fig. 2l–o). Similarly, we found that the percentage and the absolute number of extravascular CD8+ T cells were significantly higher in the brains of 5×FAD versus WT mice (P = 0.0019 and P = 0.0012, respectively; Fig. 2l–o). The percentage of CD8+ T cells was significantly higher than CD4+ T cells (P < 0.0001) among all CD45+ extravascular nonmyeloid cells in the brains of 5×FAD mice (Extended Data Fig. 2c,d). Thus, CD4+ and CD8+ T cells infiltrate the brains of 5×FAD mice significantly more than WT, in agreement with the human PiChip system.
Elevated levels of inflammatory cytokines after infiltration of PBMCs into the AD cultures
We next assessed levels of cytokines and chemokines in the conditioned cell-culture medium of the PiChip system before and after infiltration of PBMCs (Supplementary Fig. 3a). After the infiltration of PBMCs, several inflammatory cytokines, including complement component 1q (C1q), C3, CXCL1 (GRO-α), interleukin-8 (IL-8) and CXCL10 (IP-10), were significantly elevated (P < 0.05; Supplementary Fig. 3a,b). Specifically, infiltrating CD3+ T cells led to a significant increase of C1q and C3 by 702-fold (P < 0.0001) and 193-fold (P < 0.0001), respectively, in AD cultures with CD3+ T cells versus those without (Supplementary Fig. 3b).
After infiltration of monocytes, we observed a significant elevation of C3, CXCL1, IL-8 and CXCL10: 45-fold (P = 0.0069), 59-fold (P < 0.0001), 40-fold (P < 0.0001) and 9-fold (P < 0.0001), respectively, in AD cultured with monocytes versus those without (Supplementary Fig. 3b). Although B cells infiltrated in very low numbers, they significantly elevated protein levels of C3 and CXCL10 by 80-fold (P = 0.0005) and 4.5-fold (P = 0.0001), respectively, versus cultures lacking B cells (Supplementary Fig. 3b). In summary, these data show that cytokine and chemokine levels are regulated not only by the CNS cells and AD neuropathology, but also by infiltrating PBMCs.
Dramatically increased neurodegeneration after infiltration of CD8+ T cells in the presence of microglia
Next, we assessed the effects of infiltrating T cells on AD pathogenesis and the role of microglia in this process using the PiChip system (Fig. 3a). We were able to observe infiltrating T cells in AD cultures, at which point neurons expressed strong green fluorescent protein (GFP) signals (Fig. 3b). After 72 h, we observed neurite-/axon-damaged cell bodies, which co-localized with microglia and T cells only in AD cultures (Fig. 3b and Extended Data Fig. 3a). Cleaved dystrophic neurites co-localized with microglia, indicative of physical interruption, as previously reported28 (Extended Data Fig. 3a). Time-lapse confocal imaging showed that AD neurons underwent changes in cell shape, including the presence of cleaved neurites proximal to cell bodies with shrunken soma.
Fig. 3 |. Infiltrating CD8+ T cells dramatically exacerbate neurodegeneration in the AD neuroimmune axis model via interaction with microglia.
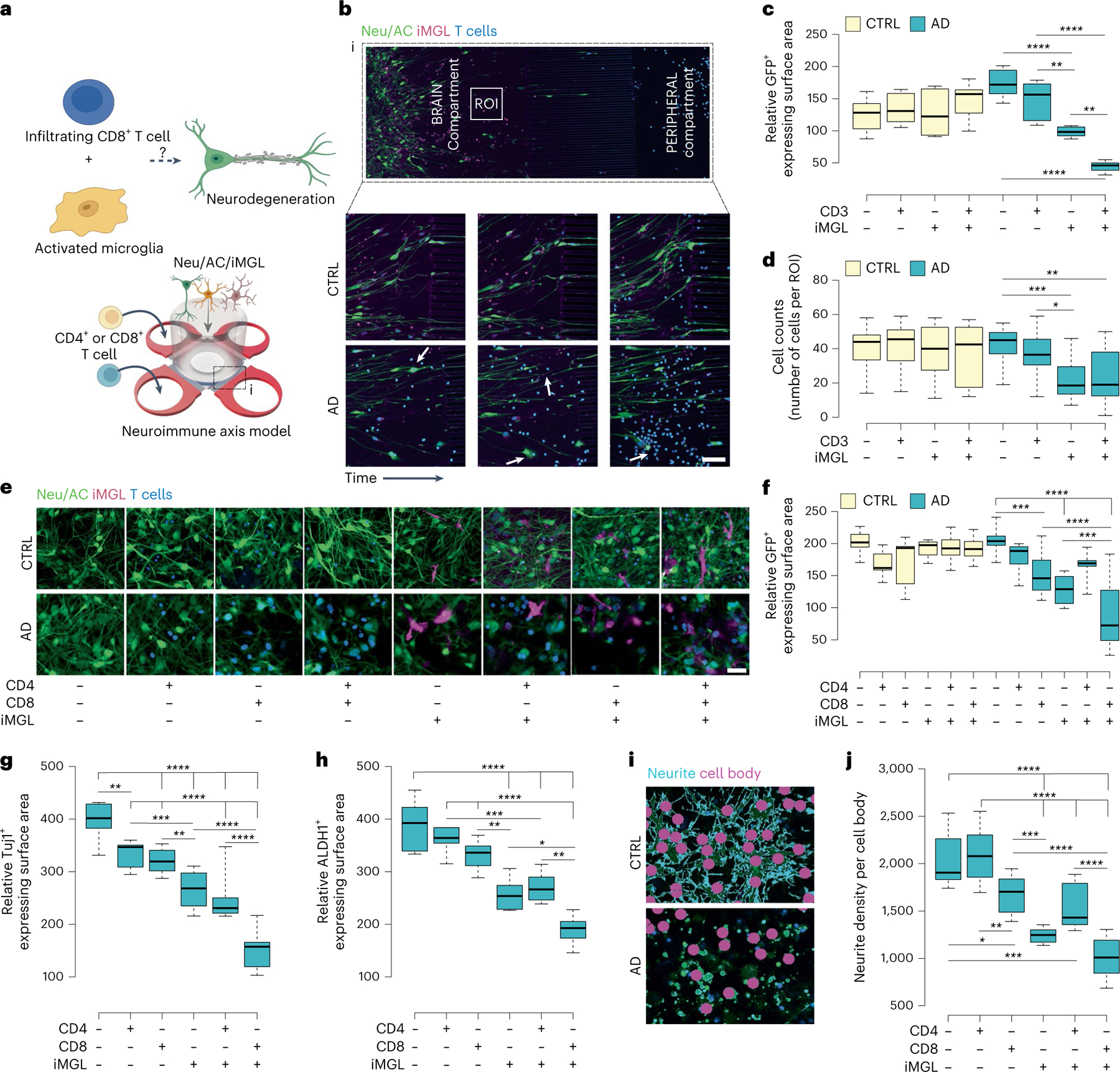
a, Schematic representing potential roles of microglia and CD8+ T cells in promoting neurodegeneration in AD. b, The 72-h time-lapse confocal imaging showing recruited T cells and cellular damage in the PiChip BRAIN compartment (AD NeuAC, green; iMGL, magenta). Scale bars, 100 μm. c,d, Quantification of cellular damage in AD and CTRL Neu/ACs ± iMGLs and T cell cultures using GFP+-expressing surface area (c) and cell body count (d) (n = 7–9 independent ROIs for GFP+ and n = 12 for cell count). P values are from two-way ANOVA with Tukey’s multiple-comparison test. e, Representative confocal imaging of cellular damage observed in the presence of CD8+ T cells and microglia in AD conditions compared with CTRL (Neu/AC, green; iMGL, magenta; CD4+/CD8+ T cells, blue) in the PiChip system. Scale bars, 25 μm. f, Quantification of cellular damage in AD Neu/ACs ± iMGLs and CTRL cultures in the presence of CD4+ or CD8+ T cells using cell surface area (GFP, green) (n = 4–15 independent ROIs). P values are from two-way ANOVA with Tukey’s multiple-comparison test. g,h, Quantification of neuronal (Tuj1+-expressing surface area) (g) and AC-damaged ALDH1+-expressing surface area (h) in the AD Neu/AC ± iMGL condition in the presence of CD4+ or CD8+ T cells (n = 10 independent ROIs for Tju1+ and n = 6 for ALDH1+). P values are from two-way ANOVA with Tukey’s multiple-comparison test. i, Representative auto-masking imaging for Neu/AC/iMGL in the presence of CD8+ T cells for quantification of the average neurite density, normalized to cell body counts (neurite, cyan; cell body, magenta) using GFP+ area. j, Quantification of the average neurite density in the AD Neu/AC ± iMGL condition in the presence of CD4+ or CD8+ T cells (n = 10–11 independent ROIs). P values are from two-way ANOVA with Tukey’s multiple-comparison test. In c, d, f, g, h and j, the center lines in the boxplots show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend to minimum and maximum values. *P < 0.0332, **P < 0.0021, ***P < 0.0002, ****P < 0.0001.
We next measured the GFP+-expressing surface area, which represents viable neurons and ACs, before and after adding T cells and/or microglia to AD Neu/AC cultures using confocal imaging. Cellular damage was significantly increased (74.5%; P < 0.0001) in AD cultures with CD3+ T cells and iMGLs compared with those without T cells/iMGLs (Fig. 3c). Based on cell numbers, the presence of infiltrating CD3+ T cells and iMGLs together in AD resulted in significant cell damage (P < 0.0021; Fig. 3d) compared with AD cultures without T cells/iMGLs. We also observed a drastic reduction in GFP+ surface area (P < 0.0001) or cell count (P < 0.0002) in AD cultures with microglia versus those without microglia (Fig. 3c,d). When we quantified the GFP+-expressing surface area of AD cultures with CD3+ T cells and iMGL compared with AD culture with only iMGLs, we observed a significant increase in cellular damage (55.1%; P < 0.0021), indicating that infiltrating CD3+ T cells partner with microglia to exacerbate cellular damage in AD cultures (Fig. 3c).
To further dissect which type of T cells was associated with neurodegeneration, we tested CD4+ and CD8+ T cells. We prepared distinct 3D cell-culture conditions in AD and CTRL cultures: (1) neuron/AC, (2) neuron/AC with CD4+ T cells, (3) neuron/AC with CD8+ T cells, (4) neuron/AC with microglia only, (5) neuron/AC with CD4+ T cells and microglia and (6) neuron/AC with CD8+ T cells and microglia (Fig. 3e). The presence of iMGL alone in AD cultures induced significant neuronal and astrocytic damage (P < 0.0001; Fig. 3f). CD8+ T cells alone (but not CD4+ T cells) in AD cultures induced significant neuronal damage (P < 0.0002; Fig. 3f). However, the greatest amount of neuronal damage was observed when CD8+ T cells and microglia were present together in AD cultures versus those without CD8+ T cells (P < 0.0002), microglia (P < 0.0001) or both (P < 0.0001) (Fig. 3f). Overall, these data indicate that the observed cellular damage in AD cultures was greatest in the presence of infiltrating CD8+ (but not CD4+) T cells and microglia (Fig. 3e, f).
We next asked whether infiltrating CD8+ T cells and microglia impact both neurons and ACs. We assessed Tuj1+ cell (a marker for differentiated neurons) surface area and found a similar decrease to previous assays. Microglia alone increased damage to Tuj1+ cells in AD cultures (32.9%; P < 0.0001) (Fig. 3g). Similarly, we observed considerable damage to Tuj1+ cells in AD when CD4+ T cells (P < 0.0021) or CD8+ T cells (P < 0.0001) were present in AD Neu/AC cultures (Fig. 3g). The greatest increase in damage on Tuj1+ areas was observed when infiltrating CD8+ T cells and microglia were both present (62%; P < 0.0001) versus AD cultures lacking T cells/microglia (Fig. 3g). We observed a similar trend in MAP2+ cell surface area as a second measure for neuronal damage. The presence of infiltrating CD8+ T cells and microglia together in AD cultures resulted in substantial cellular damage (P < 0.0001) compared with AD cultures lacking CD8+ T cells/microglia (Extended Data Fig. 3b). An ELISA assay showed that Tuj1 was significantly decreased (P = 0.0022) in AD cultures with, versus those without, T cells and microglia (Extended Data Fig. 3c).
We next utilized aldehyde dehydrogenase 1 (ALDH1), a marker for differentiated ACs. Although the presence of microglia alone in AD cultures induced significant astrocytic damage (P < 0.0001), CD8+ T cells had no impact on ALDH1+ cells in AD cultures without microglia (Fig. 3h). The greatest degree of impact on astrocytic damage was observed when infiltrating CD8+ T cells and microglia were present together in AD cultures (P < 0.0001, Fig. 3h). Thus, CD8+ T cells, alone, induced significant damage in AD Neu/AC cultures, but only at the neuronal level. Microglia, alone, induced cellular damage to neurons and ACs and, when combined with infiltrating CD8+ T cells in AD cultures, led to even greater cellular damage.
We also assessed neuritic damage using neurite-per-cell body density ratio (Fig. 3i,j). CD8+ (but not CD4+) T cell infiltration alone significantly decreased the neurite:cell body ratio in AD cultures (P < 0.0332; Fig. 3j) compared to AD cultures lacking T cells. Neurite density also decreased significantly (P < 0.0001) in AD Neu/AC/iMGL versus AD cultures lacking microglia (Fig. 3j). The most significant decrease in neurite:cell body ratio was observed when infiltrating CD8+ T cells and microglia were both present in AD cultures (P < 0.0001, Fig. 3j), consistent with the cellular damage assays. Thus, while microglia alone can induce damage at the neuronal and glial level, CD8+ T cells alone drive neuronal and/or neuritic damage (but not astrocytic damage), and microglia plus CD8+ T cells synergistically exacerbate neurodegeneration in AD cultures.
We next used time-lapse calcium imaging to discern any altered neuronal/glial activities owing to T cell infiltration. We observed a significant 1.8-fold decrease (P < 0.0001) in the frequency of calcium response (peak/time) in AD cultures in the presence of T cells and microglia compared with CTRL cutures (Extended Data Fig. 3d).
IFN-/inflammatory-associated pathways are activated in glial cells after infiltration of T cells into AD cultures
To identify cell-type-specific transcriptional changes, we performed scRNA-seq from only the PiChip BRAIN compartment with AD and CTRL cultures (Extended Data Fig. 4). Cell clusters were first assigned to five cell types: neurons (SYT1, KCNQ1OT1 and KCNJ6), ACs (GFAP, VIM, SOX2, AQP4 and ID4), microglia (CSF1R and SPI1), T cells (CD3D and CD8A) and proliferative cells (TOP2A and MKI67) (Extended Data Fig. 4a,b). The scRNA-seq data revealed several significantly up- and downregulated genes (Extended Data Fig. 4c,d) in each cell type associated with AD versus CTRL cultures. Pathways related to cytotoxicity and antigen presentation were significantly upregulated in T cells in AD Neu/AC/iMGL versus CTRL cultures (Extended Data Fig. 4e). Microglia in AD cultures with infiltrating T cells were significantly enriched with upregulated pathways involved in cell killing and antigen presentation (Extended Data Fig. 4e). IFN-associated pathways (for example, response to INF-β) were upregulated in ACs in AD cultures with microglia and T cells compared with CTRL cultures (Extended Data Fig. 4e).
We next assessed the composition of CNS cells under various conditions (Extended Data Fig. 5). Neurons and ACs were significantly decreased, whereas microglia were significantly increased in AD versus CTRL cultures (Extended Data Fig. 5c). After infiltration of T cells, we observed an increase in microglia and T cell populations and a decrease in ACs in AD Neu/AC/iMGL versus CTRL cultures (Extended Data Fig. 5d). We identified multiple transcriptionally unique clusters of microglial cells in AD cultures versus CTRL (Extended Data Fig. 5e). Disease-associated microglia (DAMs) and IFN microglia were significantly increased in AD cultures compared with CTRL (Extended Data Fig. 5f). In contrast, homeostatic microglia were significantly decreased in AD cultures versus CTRL (Extended Data Fig. 5f). This suggests that AD pathology environment together with T cell infiltration alters the composition of CNS cells.
We next carried out scRNA-seq analyses on T cells and microglia (Fig. 4). Major T cell subsets (CD4+ naive, CD4+ memory and CD8+ T cells) were annotated based on clustering of the scRNA-seq datasets (Fig. 4a,b). The percentage of CD8+ T cells was significantly increased in AD cultures with versus those without microglia (P = 0.0094; Fig. 4b,c). We also observed that, although there was an increase of total CD4+ T cells (naive and memory) in AD cultures compared with CTRL, there was no significant change in the CD4+ memory T cell population in AD versus CTRL cultures (Fig. 4a–d). In contrast, the percentage of CD4+ memory T cells was decreased in AD cultures with microglia, compared with AD cultures without microglia (Fig. 4b,d). The CD4+ naive:memory T cell ratio was 2.0 and 1.5 in the presence and absence of microglia in AD cultures, respectively.
Fig. 4 |. Compositional changes and inflammatory activity of T cells and microglia in AD conditions.
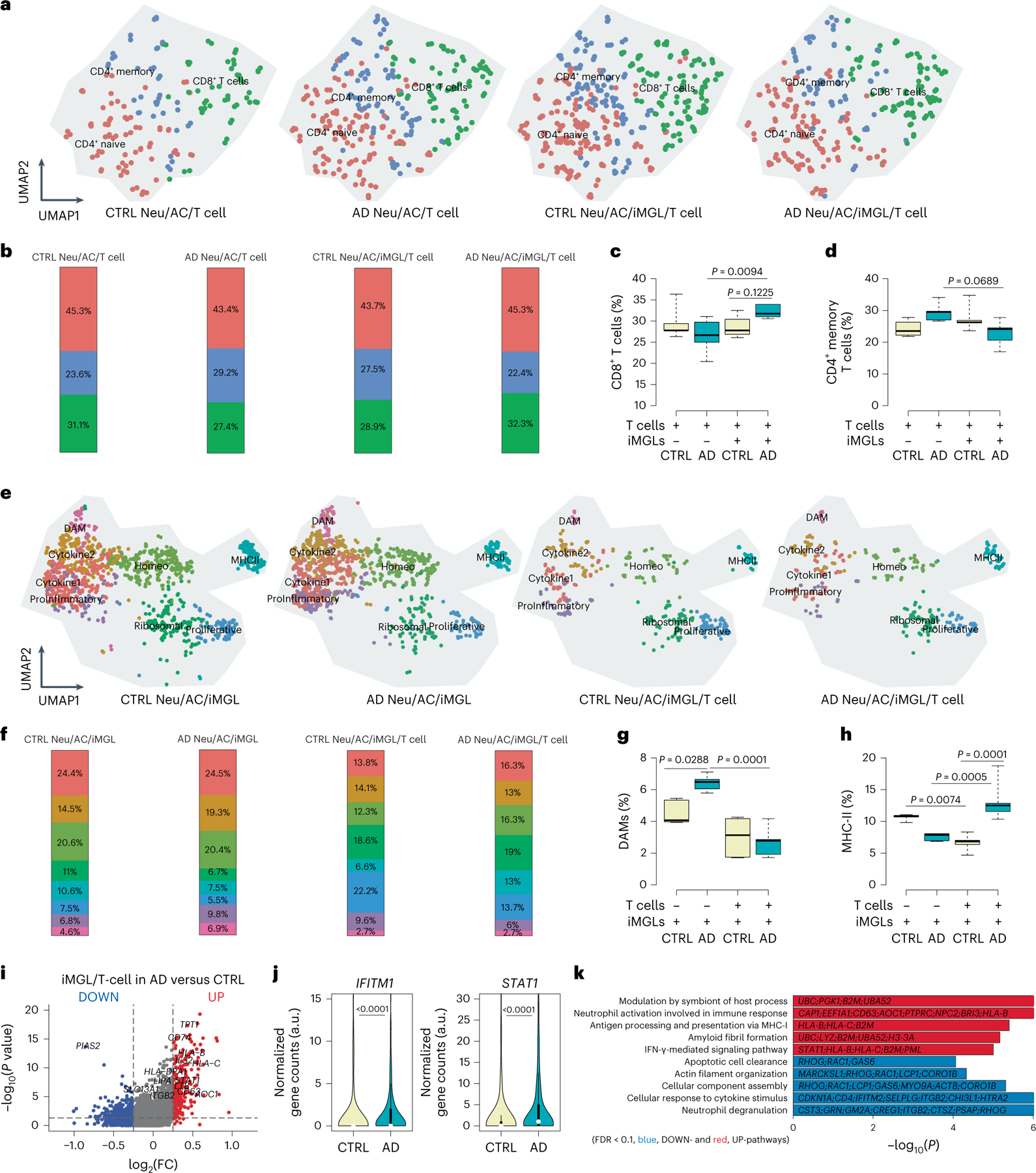
a, UMAP visualization of T cell-type composition of the PiChip cultures by single-cell transcriptome profiles. Distinct subtypes of T cells (CD4+ naive, CD4+ memory and CD8+ cytotoxic) are depicted by different colors. b, Compositional changes of CD4+ naive, CD4+ memory and CD8+ cytotoxic T cells in AD or CTRL Neu/AC ± iMGL conditions. c,d, Percentage of cytotoxic CD8+ T cells (c) and memory CD4+ T cells (d) in AD or CTRL Neu/AC ± iMGL conditions (n = 5 independent experiments for both cytotoxic CD8+ T and memory CD4+ T cells). P values are from two-way ANOVA with Tukey’s multiple-comparison test. e, UMAP visualization of T cell-type composition of the PiChip cultures by single-cell transcriptome profiles. Distinct subtypes of T cells (homeostatic, DAM, MHC-II, proinflammatory, cytokine 1, cytokine 2, proliferative and ribosomal) are depicted by different colors. f, Compositional changes of homeostatic, DAM, MHC-II, proinflammatory, cytokine 1, cytokine 2, proliferative and ribosomal microglial cells in AD or CTRL Neu/AC/iMGL ± T cells. g,h, Percentage of DAM (g) and MHC-II (h) in AD or CTRL Neu/AC/iMGL ± T cell conditions (n = 5 independent experiments for both DAM and MHC-II quantification). P values are from two-way ANOVA with Tukey’s multiple-comparison test. i, Volcano plots of differentially expressed genes in microglia in AD Neu/AC/iMGL cultures versus CTRL in the presence of infiltrating T cells. The P value is from Wilcoxon’s rank-sum test. Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown as red dots. j, Violin plots showing selected significantly enriched, IFN-associated genes (IFITM1 and STAT1) in microglia in AD cultures compared with CTRL in the presence of T cells (n = 5 independent experiments). P values are from a nonparametric, two-sided Mann–Whitney U-test. k, GO of significantly upregulated and downregulated pathways (FDR < 0.1) in microglia in AD cultures versus CTRL in the presence of T cells. P values were calculated from Fisher’s exact test and the adjusted P values are given using the Benjamini–Hochberg method. In c, d, g and h, the center lines in the boxplots show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend to minimum and maximum values. In j, the white circles show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend 1.5× the IQR from the 25th and 75th percentiles; polygons represent density estimates of data and extend to extreme values.
We next analyzed the impact of infiltrating T cells on microglial gene expression in AD versus CTRL cultures. We identified 8 transcriptionally unique clusters of microglial cells from a total of 20,153 single cells (Fig. 4e) in 4 different conditions (Fig. 4e). DAMs were significantly increased in AD cultures without T cells compared with CTRL cultures (P = 0.0288; Fig. 4f,g). Notably, the presence of T cells dramatically reduced the DAM population in AD and CTRL cultures with a significantly greater impact on AD cultures (P = 0.0001; Fig. 4f,g), suggesting that T cells can modulate the DAM population. Microglia-associated MHC-II gene expression was significantly increased in T cell-infiltrated AD cultures versus those without T cells (P = 0.0005; Fig. 4f,h) or CTRL cultures (P = 0.0001, Fig. 4f,h). Thus, microglia may act as antigen-presenting cells (APCs) to promote T cell infiltration into AD cultures.
We also identified 263 upregulated and 82 downregulated genes in microglia in AD cultures with T cells versus CTRL cultures (Fig. 4i). It is interesting that IFN-induced transmembrane protein 1 (IFITM1) and signal transducer and activator of transcription 1 (STAT1) genes were significantly enriched (P < 0.0001) in AD cultures with T cells and microglia compared with CTRL cultures (Fig. 4j), suggesting a role of IFN-associated pathways in T cell infiltration into AD cultures in the presence of microglia. GO revealed a significant increase in genes involved with the IFN-γ-mediated signaling pathway, antigen processing and presentation of exogenous peptide antigen via MHC-I, and amyloid fibril formation in AD Neu/AC/iMGL cultures with T cells (Fig. 4k). We also detected significantly downregulated genes in pathways involved with cellular response to cytokine stimulus, actin filament organization and apoptotic cell clearance in AD cultures with T cells (Fig. 4k).
Next, we assessed the effects of AD pathology in three independent transcriptomic analyses (Extended Data Fig. 6). Differentially expressed genes in microglia revealed multiple significantly upregulated pro-inflammatory cytokines and chemokines (for example, CXCL9/10/11 and IL-32 genes) in AD cultures with T cells versus those without T cells (Extended Data Fig. 6a). Cytokine- and IFN-γ-associated genes were significantly higher (P < 0.0001) in microglia when T cells were present in AD cultures (Extended Data Fig. 6b). Pathway analysis revealed upregulation of several antigen and immune-related pathways (Extended Data Fig. 6c). In contrast, pathways such as collagen-containing extracellular matrix and transition metal ion homeostasis were significantly downregulated in microglia in AD cultures with infiltrated T cells (Extended Data Fig. 6d). We performed a similar analysis on astrocytes in the presence of T cells and microglia in AD cultures and observed several significantly upregulated proinflammatory cytokines and chemokines (for example, CXCL1, CXCL10 and IL-32 genes) and IFN-associated genes (for example, IFITM1, IFIT3 and STAT1) in astrocytes in the presence of T cells and microglia in AD cultures compared with AD devoid of T cells (Extended Data Fig. 6e,f). Pathway analyses showed significant upregulation of genes involved in multiple pathways related to T cell-mediated cytotoxicity and antigen processing and presentation of peptide antigen via MHC-I (Extended Data Fig. 6g,h).
Next, we set out to identify ligand–receptor interactions between T cells and various CNS cells (Extended Data Fig. 7a). Most of the top 20 potential ligands were primarily expressed by microglia, including CCL2, IL-1β, CCL8, FGF23 and APOE (Extended Data Fig. 7a). Glia maturation factor-β (GMFB) was the most highly expressed ligand in neurons. Using NicheNet33 to predict ligand–receptor interactions (Extended Data Fig. 7b), we identified CD44, which is also associated with microglia/macrophages and serves as the receptor of T cell-associated ligands such as protein tyrosine phosphatase receptor-type C (PTPRC). We also found that high-mobility group box protein 1 (HMGB1) interacts with toll-like receptor 4 (TLR4), which is highly expressed by microglia in the CNS (Extended Data Fig. 7b). This is consistent with previous reports that HMGB1 interacts with Aβ42 and inhibits Aβ phagocytosis by microglia through TLR4 signaling34. We also predicted active target genes based on potential upstream ligand activity and receptor (Extended Data Fig. 7c). We found multiple genes with significantly altered expression, including those involved with IFN-γ- (STAT1), gliosis-(GFAP and C1Q) and inflammatory-associated expression (TNFRSF1B, CCL2, CCL4 and CCL5), as well as matrix metalloproteinase-9 (MMP9).
We next measured proinflammatory cytokine levels after infiltration of CD8+ T cells into AD cultures in the presence and absence of microglia (Extended Data Fig. 8). We observed a substantial increase in IL-15 (P < 0.0001), IL-2 (P = 0.002), tumor necrosis factor (TNF)-α (P = 0.0156) and CXCL10 (P = 0.0029) levels in AD cultures when CD8+ T cells and microglia were both present, compared with AD lacking microglia (Extended Data Fig. 8a). Granzyme B levels were also elevated (but not significantly) in the presence of CD8+ T cells and microglia in AD cultures (Extended Data Fig. 8a), consistent with the scRNA-seq data, in which the expression patterns of GZMA, GZMB, GZMH, GZMM and GZMK were altered for CD4+ and CD8+ T cells under the AD conditions (Extended Data Fig. 8b). The high expression of GZMA, GZMB and GZMH was predominantly associated with CD8+ cytotoxic T cells, in contrast to CD4+ naive T cells (Extended Data Fig. 8b). We also observed a significant increase in IFN-γ levels for CD8+ T cells together with microglia in AD compared to AD cultures without (P < 0.0001) and cultures with only microglia (P = 0.001) or CD8+ T cells (P < 0.0001) (Extended Data Fig. 8c). Collectively, these data indicate that infiltrating CD8+ T cells promote IFN signaling and probably communicate with ACs through cytokines such as IL-15 in AD cultures. The infiltrating CD8+ T cells exhibited transcriptional signatures related to elevated IFN-associated pathways, cytotoxicity, inflammatory activation and reactive gliosis in the presence of microglia under AD conditions.
Upregulation of CXCL10 in the AD neuroimmune axis model and 5×FAD brain
Next, we set out to identify secreted cytokines and chemokines that regulate infiltration of T cells into AD cultures (Supplementary Fig. 4a). We observed 2.5-fold greater production of basic fibroblast growth factor-2 in AD Neu/AC/iMGL cultures compared with CTRL (P = 0.0168; Supplementary Fig. 4b). Protein levels of several cytokines, including IL-8, IL-9 and IFN-α2, were also increased by 40-fold (P < 0.0001), 8.5-fold (P = 0.0007) and 4.8-fold (P = 0.0018), respectively, after the addition of microglia to AD cultures (Supplementary Fig. 4b). We also observed a more modest increase in IL-8 and IL-1RA by 1.1-fold (P = 0.014) and 3-fold (P = 0.029), respectively, in AD Neu/AC/iMGL cultures versus CTRL (Supplementary Fig. 4b).
Among chemokines, CXCL10 exhibited a significant increase in AD cultures compared with CTRL, both in the absence and in the presence of microglia by 5.3-fold (P = 0.0049) and 6.9-fold (P < 0.0001), respectively (Fig. 5a). We also found a highly enriched astrocytic CXCL10 in AD cultures compared with CTRL using scRNA-seq (P < 0.0001; Fig. 5b). In addition, we observed a substantial increase in CXCL10 release in the astrocytic chamber with AD Neu/ACs differentiated in the other chamber, compared with CTRL Neu/ACs (P = 0.0232) or ACs alone (P = 0.0054; Fig. 5c). Thus, AD pathology induced the release of CXCL10 from ACs.
Fig. 5 |. Upregulation of CXCL10 in the AD neuroimmune axis model and 5×FAD brain.
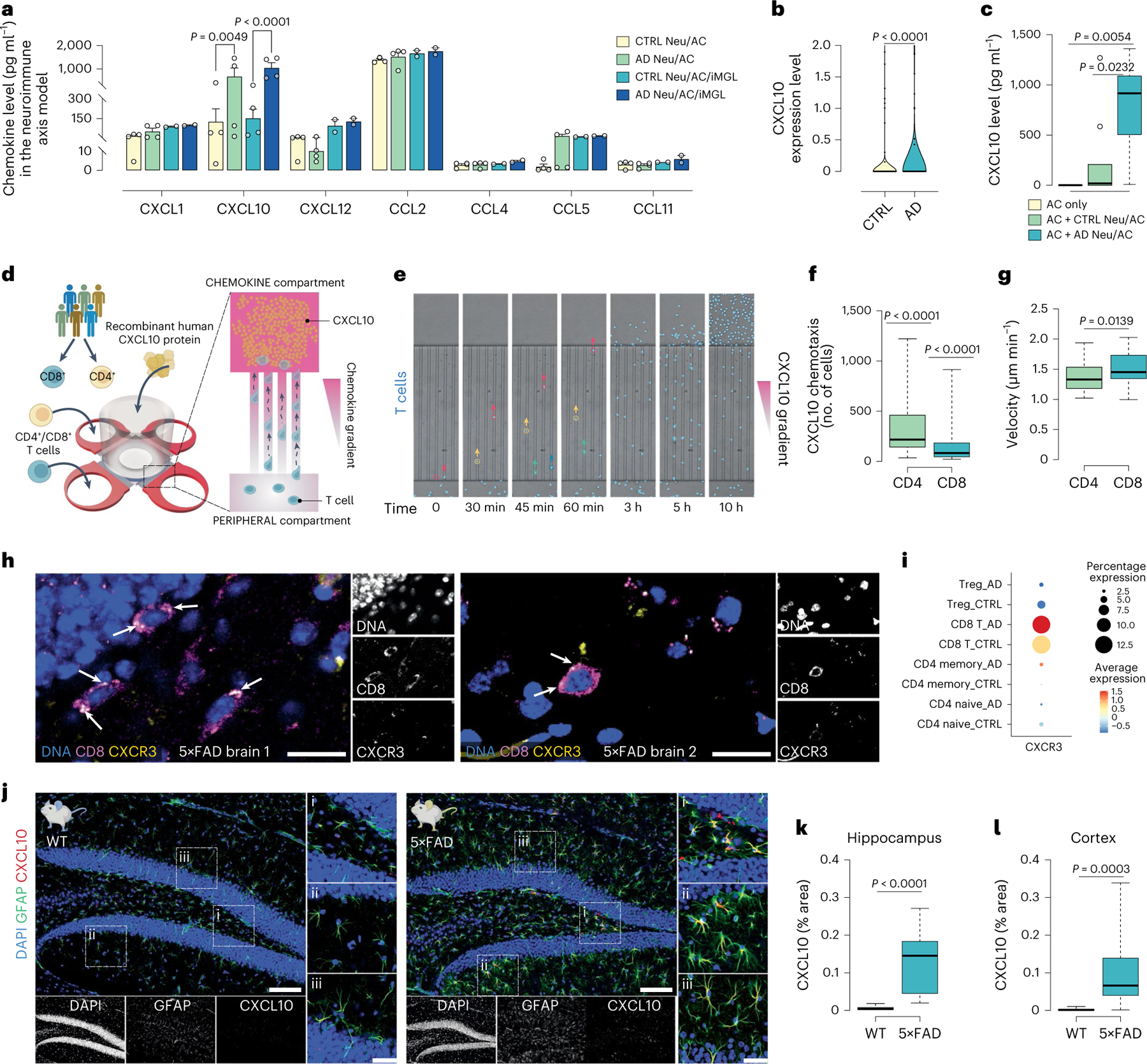
a, Quantification of chemokines in AD Neu/AC ± iMGL cultures compared with CTRL (n = 4 independent experiments all Neu/AC cultures; n = 2 for Neu/AC/iMGL cultures except CXCL10, n = 4; data are mean ± s.e.m.) P values are from two-way ANOVA with Tukey’s multiple-comparison test. b, ScRNA-seq data showing highly enriched astrocytic CXCL10 expression in AD cultures compared with CTRL (n = 5 independent experiments). P values are from a nonparametric, two-sided Mann–Whitney U-test. c, CXCL10 protein level from ACs in the presence of AD and CTRL cultures (n = 9 biological replicates except for AC only, n = 7) P values are from a nonparametric Kruskal–Wallis test with Dunn’s multiple-comparison test. d, Schematics describing an in vitro chemotaxis model where recombinant human CXCL10 was added to the CHEMOKINE compartment, whereas CD4+ and CD8+ T cells were added to the PERIPHERAL compartment. e, Time-lapse microscopy imaging showing chemotaxis of T cells (nuclei stained, blue) toward CXCL10 through confined microchannels over time. f, The number of migrated CD4+ and CD8+ T cells in response to CXCL10 (100 nM) after 17 h (n = 44 biological replicates, CD4+ and n = 47, CD8+). P values are from two-tailed, Wilcoxon’s signed-rank tests. g, Migration velocity of CD4+ and CD8+ T cells through microchannels over 17 h toward CXCL10 (100 nM) (n = 44 biological replicates, CD4+ and n = 47, CD8+). P values are from a nonparametric, two-sided Mann–Whitney U-test. h, Representative immunofluorescence staining of CD8+ T cells (magenta), co-labeled with CXCR3 (yellow) and DAPI (nuclei) in 6- to 7-month-old 5×FAD mouse brains. Scale bar, 20 μm. i, CXCR3 expression for distinct T cell types in different conditions using scRNA-seq data. The color scale (arbitrary units (a.u.)) indicates the average expression of the CXCR3 gene in each cell population and the dot size is proportional to the percentage of cells expressing the CXCR3 gene. j, Representative immunofluorescence staining of 6- to 7-month-old 5×FAD and WT mouse brains co-labeled with anti-CXCL10 (red), anti-GFAP (green) and DAPI (nuclei). Scale bar, 100 μm and 35 μm in inserts. k,l, The percentage of CXCL10 in 6- to 7-month-old 5×FAD and WT brains (n = 13, 5×FAD and n = 8, WT mice): hippocampus (k) and cortex (l). P values are from a nonparametric, two-sided Mann–Whitney U-test. In c, f, g, k and l, the boxplots show center lines as the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend 1.5× the IQR from the 25th and 75th percentiles; outliers are represented as dots.
To test whether CXCL10 modulates T cell recruitment, we first examined the migration of T cells toward purified CXCL10 in microfluidic systems without neuronal cells (Fig. 5d,e). CD3+ T cell migration toward CXCL10 exhibited linear dose-dependent behavior (Supplementary Fig. 4c). CD3+ T cells that migrated toward 1,000 nM CXCL10 were 2.63-fold and 4.6-fold higher versus 100 nM and 5 nM concentrations, respectively, and 1.8-fold higher for 100 nM versus 5 nM (Supplementary Fig. 4d). T cells migrated at an average velocity of 1.8 ± 0.8 μm min−1, with no significant differences between concentrations (Supplementary Fig. 4e).
We next tested CD4+ and CD8+ T cell propensity for CXCL10 chemotaxis (Fig. 5d–g). We observed significant migration of CD4+ and CD8+ T cells (P < 0.0001) in response to 100 nM recombinant human CXCL10, compared with medium alone (Fig. 5f): 319 ± 38 of CD4+ T cells (P < 0.0001) and 144 ± 28 of CD8+ T cells (P < 0.0001) exhibited directional migration toward CXCL10 compared with medium (Fig. 5f). CD4+ T cells had an average velocity of 1.39 ± 0.04 μm min−1 toward CXCL10. CD8+ T cells moved slightly faster than CD4+ T cells with an average velocity of 1.51 ± 0.04 μm min−1 (P = 0.0139; Fig. 5g). Thus, elevated levels of CXCL10 can induce chemotactic migration of CD4+ and CD8+ T cells into AD cultures.
CXCL10 induces T cell chemotaxis via binding to CXCR3, a G-protein-coupled seven-transmembrane-spanning receptor35. To determine whether infiltrating CD8+ T cells express CXCR3 receptor, we performed immunohistochemical analysis using antibodies against CXCR3 receptor and CD8. Most of the CXCR3 staining co-localized with CD8+ T cells in the hippocampus and cortex of 6- to 7-month-old 5×FAD mice (Fig. 5h and Supplementary Fig. 4f). This aligned with the scRNA-seq data, in which we found a substantial increase in CXCR3 expression levels in CD8+ T cells compared with other T cell subsets (Fig. 5i). We also confirmed the upregulation of CXCL10 in the parenchyma of 5×FAD and WT mice using immunohistochemical analyses (Fig. 5j). CXCL10 levels were significantly elevated in both the hippocampus (P < 0.0001) and the cortex (P = 0.0003) of 6- to 7-month-old 5×FAD mice compared with aged-matched WT mice (Fig. 5k,l). In 5×FAD mice, most of the elevated CXCL10 staining co-localized with the AC marker, GFAP (Fig. 5j).
Inhibition of CXCL10 binding to CXCR3 attenuates CD8+ T cell infiltration and neurodegeneration
Next, we asked whether blocking CXCR3 reduces T cell infiltration into the AD cultures (Fig. 6a). First, we characterized CD4+ and CD8+ T cells and CXCR3 expression in human blood samples. CD4+ T cells constituted 68%, whereas CD8+ T cells accounted for 32% of total T cells in peripheral human blood (Fig. 6b). The vast majority of CD8+ T cells (77.6 ± 2.3%) expressed CXCR3, which was significantly higher than CD4+ T cells: 39.1 ± 4.5% were CXCR3 positive (P = 0.0002; Fig. 6c).
Fig. 6 |. Blocking the binding of CXCL10 to T cell CXCR3 attenuates T cell infiltration and neurodegeneration in the neuroimmune axis model.
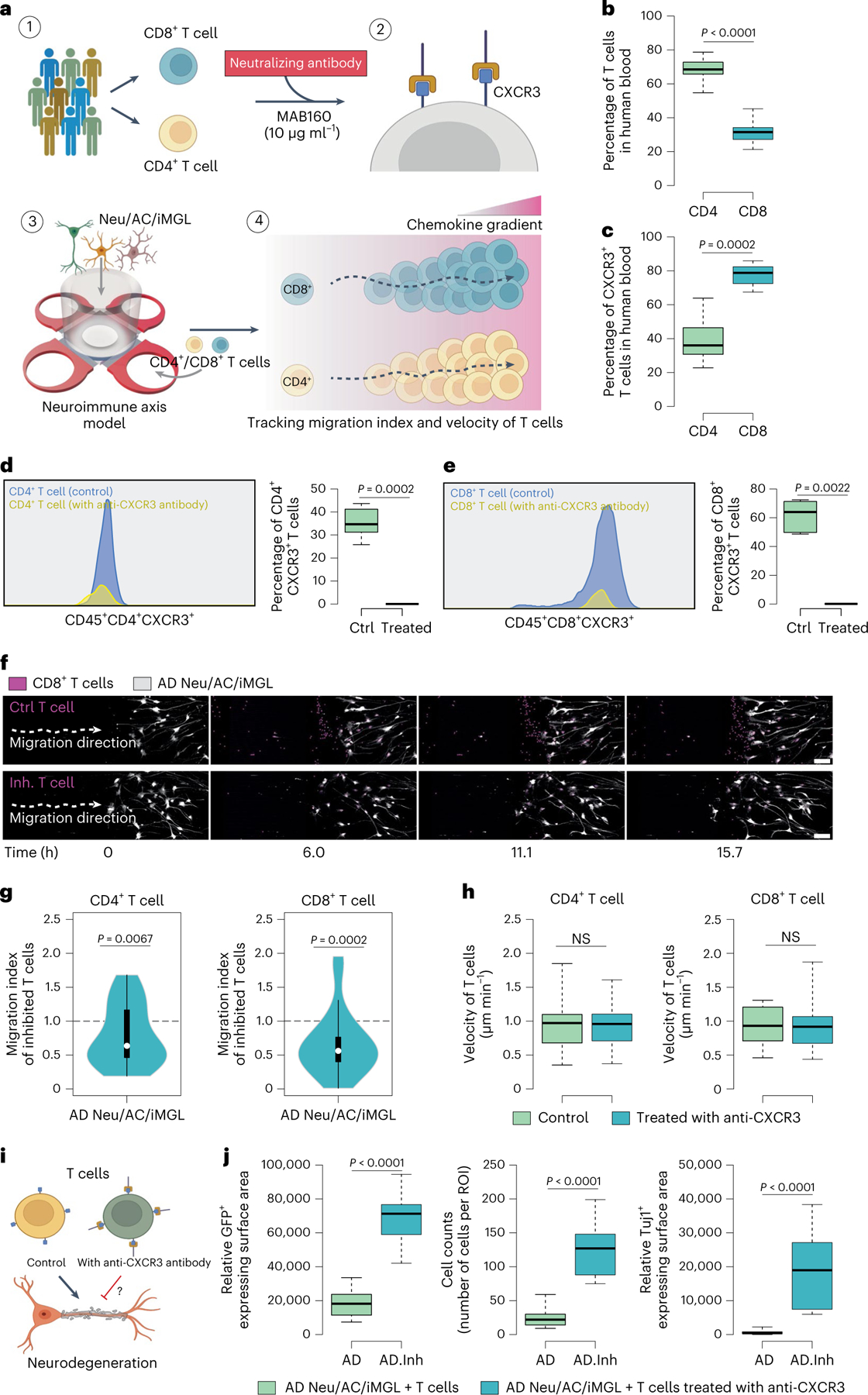
a, Schematic showing the experimental design for inhibition of the CXCL10–CXCR3 signaling axis in AD cultures using a neutralizing antibody. b,c, The percentage of CD4+ and CD8+ T cells in human blood (b) and percentage level of CXCR3 receptor expression in CD4+ and CD8+ T cells using flow cytometry (c) (n = 10 donors each for the percentage of T cells; n = 8 donors each for the percentage of CXCR3+ T cells). P values are from nonparametric, two-sided Mann–Whitney U-test. d,e, Representative histogram images of flow cytometry before and after blocking CXCR3 receptor in CD4+ (d) and CD8+ (e) T cells with a neutralizing antibody (MAB160, 10 μg ml−1). Box plots representing percentage level of CXCR3 expression in T cells (n = 8 donors for CD4+ T cells and n = 6 donors for CD8+ T cells). P values are from a nonparametric, two-sided Mann–Whitney U-test. f, Time-lapse confocal imaging showing the migratory movement of CD8+ T cells (nuclei stained, magenta) through microchannels over time before (Ctrl T cell) and after treatment with MAB160, 10 μg ml−1 of neutralizing antibody (Inh. T cell) toward the BRAIN compartment containing Neu/AC/iMGL (gray) culture. Scale bar, 100 μm. g, Quantification of migration indexinhibited (Methods) of CD4+ and CD8+ T cells with and without CXCR3-neutralizing antibody treatment (MAB160, 10 μg ml−1) in the PiChip system containing AD Neu/AC/iMGL cultures (n = 36 biological replicates each for CD4+ and CD8+ T cells). P values are from two-tailed Wilcoxon’s signed-rank tests and five or six independent experiments. h, Average migration velocity of infiltrating CD4+ and CD8+ T cells with and without MAB160 treatment (10 μg ml−1) through the microchannels over 17 h in the PiChip system (n = 33 biological replicates, CTRL and n = 30, treated for CD4+ T cells; n = 24 biological replicates, CTRL and n = 31, treated for CD8+ T cells). P values are from a nonparametric, two-sided Mann–Whitney U-test. NS, not significant. i, Schematic representing the potential effect of CXCR3-neutralizing antibody (MAB160) on cellular damage in AD cultures. j, Boxplots representing quantification of GFP+-expressing surface area, cell body count and Tuj1+-expressing surface area in AD Neu/AC/iMGL cultures before and after T cell treatment with CXCR3-neutralizing antibody (MAB160, 10 μg ml−1) (n = 13 or 14 independent ROIs for GFP+ and cell count; n = 13 for Tuj1+). P values are from a nonparametric, two-sided Mann–Whitney U-test. In b, c, d, e, h and j, the center lines in boxplots show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend to minimum and maximum values. In g, the white circles show the medians, the box limits indicate the 25th and 75th percentiles and the whiskers extend 1.5× the IQR from the 25th and 75th percentiles; polygons represent density estimates of data and extend to extreme values.
To block CXCL10 binding to CXCR3, we used either an anti-CXCR3-neutralizing antibody (MAB160) or a chemical antagonist of CXCR3 (NBI74330) and tested the effects on infiltration of T cells into AD cultures. Blocking CD3+ T cells with MAB160 led to a significant decrease in CD3+ T cell infiltration into AD cultures versus unblocked cells (P = 0.0006; Supplementary Fig. 4g), while NBI74330 showed no change in infiltration of T cells. Treating CD3+ T cells with MAB160 or NBI74330 led to no significant changes in their velocity into AD or CTRL cultures (Supplementary Fig. 4h).
Quantitative analysis of CXCR3 surface expression in T cell subsets treated with MAB160 demonstrated a significant loss of CXCR3 surface expression in both CD4+ (99.9% decrease) and CD8+ (99.8% decrease) T cells after exposure to 10 μg ml−1 of MAB160-neutralizing antibody (Fig. 6d,e). We next determined whether blocking the CXCL10–CXCR3 axis reduces the infiltration of CD4+ and/or CD8+ T cells into AD cultures. CD4+ and CD8+ T cells were isolated and immediately treated with MAB160 antibody (10 μg ml−1), loaded into the PiChip system and imaged to track their infiltration (Fig. 6f). Blocking of CD4+ T cells with MAB160 led to a 18.61% decrease (P = 0.0067) in infiltration into AD cultures versus unblocked CD4+T cells (Fig. 6g). Similarly, antibody treatment significantly decreased CD8+ T cells infiltration by 32.5% (P = 0.0002) into AD cultures versus unblocked CD8+ T cells (Fig. 6g). Treating CD4+ and CD8+ T cells with MAB160 had no significant impact on their average velocity compared with untreated T cells (Fig. 6h). Thus, CXCL10 and its T cell receptor, CXCR3, play key roles in mediating selective T cell infiltration into AD cultures versus CTRL.
We next assessed the impact of infiltrating T cells on neuronal cell death in AD cultures by blocking CXCR3 with MAB160 (Fig. 6i). MAB160 treatment significantly prevented cellular damage by 36.9-fold (P < 0.0001) and 5.2-fold (P < 0.0001) in AD cultures with T cells, respectively, as quantified by the GFP+-expressing area and cell counts (Fig. 6j). MAB160 treatments also significantly increased the number of Tuj1+ neurons by 34.6-fold (P < 0.0001) in AD cultures compared to AD cultures with infiltrating T cells without treatment (Fig. 6i). Collectively, these data strongly support a role for the CXCL10–CXCR3 axis in mediating infiltration of T cells and their pathogenic impact in AD.
Discussion
The 3D human neuroimmune axis model has allowed us to delineate the molecular events underlying the infiltration of peripheral immune cells and investigate their potential pathogenic roles in AD. We showed a significantly higher percentage of human CD4+ and CD8+ T cells infiltrating into AD neural–glial cultures independent of microglial presence. Our study supports the role of T cells in AD pathology, as suggested by the increased number of extravascular T cells in patients with AD6–11. We also demonstrated a significantly increased percentage and absolute number of extravascular CD4+ and CD8+ T cells in the brains of 5×FAD mice. Our findings agree with increased numbers of parenchymal CD8+ T cells positively correlating with advancing Braak stages of neuropathology in patients with AD10. Overall, our findings align with recent findings of increased numbers of CD8+ T cells in patients with AD and AD-like transgenic models.
We also discovered that the infiltration of human monocytes was enhanced in the presence of AD pathology compared with control cultures. This was negated by the presence of microglia. Previous studies have reported a potentially related phenomenon in which microglia depletion in mice led to increased infiltration of circulating monocytes36,37. However, the factors regulating monocyte recruitment in the CNS remain largely unknown. One proposed mechanism posits that the absence of microglia causes the release of cytokines, resulting in vascular activation and monocyte entry into the CNS36.
A recent study demonstrated increases in clonally expanded, terminally differentiated effector memory (TEMRA) CD8+ T cells in the cerebrospinal fluid (CSF) of patients with AD11, suggesting that CD8+ T cells impact neurodegeneration and cognitive impairment in patients with AD. CD8+ T cells were proximal to MAP2+ neuronal structures and NEFH+ (neurofilament heavy chain-positive) neuronal processes in APP/PS transgenic mouse brains11. Our data provide a possible underlying mechanism for CD8+ T cell-induced synaptic damage observed in previous studies11,38. We found that CD8+ T cells enhance microglial activation and exacerbate neuroinflammation and neurodegeneration in AD cultures. Specifically, we demonstrated that: (1) CD8+ T cells, alone, induce significant neuronal and/or neuritic damage (not glial) in AD cultures lacking microglia; (2) microglia, alone, can induce cellular damage at the neuronal and glial level in AD cultures; but (3) the coexistence of CD8+ T cells and microglia in AD resulted in a synergism that exacerbated neuronal and glial damage.
Our findings also emphasize the role of infiltrating T cells in triggering INF-/inflammatory-associated pathways, including IFN association (for example, IFITM1 and STAT1), antigen presentation (for example, MHC-I and MHC-II) and proinflammatory cytokines/chemokines (for example, CXCL10 and IL-32) in microglia and ACs in AD cultures. We observed a robust increase in proinflammatory cytokines (for example, IL-2, IL-15, TNF-α and IFN-γ) after infiltration of CD8+ T cells into AD cultures with microglia. Previously, it has been shown that activated CD8+ T cells secrete IFN-γ and IL-2 for astrocyte signaling and granzyme B to induce neuronal apoptosis with AC-derived IL-15 in post-traumatic injury39. Similarly, a recent study demonstrated that cytotoxic CD8+ T cells interact with microglia and oligodendrocytes through IFN signaling and contribute to white matter decay in aging40. These findings, along with our current work, underscore the critical role of IFN-γ signaling and further strengthen the link between CD8+ T cells and IFN-γ in their contribution to neurodegeneration.
Chemokines play an important role in regulating the migration and recruitment of T cells in neurodegenerative diseases, for example, AD41–43. CCL2, CCL5, CXCL8, CXCL10 and CX3CL1 are elevated in brains of patients with AD43–46. Previously, we demonstrated that CCL2 plays an important role in recruiting microglia into the AD triculture model28. In the present study, we found a significant increase in CXCL10 levels in AD cultures. CXCL10 levels have also been reported to be elevated in AD mouse models46–48. Similarly, we observed significantly elevated levels of astrocytic CXCL10 in the cortex and hippocampus of 5×FAD mice. Thus, CXCL10 is expressed in ACs and is elevated in the brains of patients with AD and mice.
Our findings also suggested a potential role for the CXCL10 receptor, CXCR3, in AD. Prior studies demonstrated the role of CXCR3 in regulating T cell trafficking and function49–52. We showed that infiltrating CD8+ T cells express the CXCR3 receptor in 5×FAD mouse brains. Similarly, a recent study has provided insight into the role of CXCR3 signaling on AD pathology in the APP/PS1 mouse model53. Other studies also showed an important role of the CXCL10–CXCR3 axis in CD8+ T cell response during chronic infection54. However, until now, the potential role of the CXCL10–CXCR3 pathway in mediating the infiltration of T cells into the AD brain has remained unknown. We showed that blocking the CXCR3 receptor using a neutralizing antibody inhibited infiltration of T cells into AD cultures and dramatically attenuated neurodegeneration. A recent longitudinal study showed that CD8+ T cells in the blood are significantly associated with brain Aβ accumulation in AD55. These findings suggest the potential for therapeutic interventions for AD based on targeting the CXCL10–CXCR3 axis to reduce CD8+ T cell infiltration into the brains of patients with AD. Another study suggested the CXCL16–CXCR6 signaling pathway may be involved in CSF T cell infiltration in the brains of those with AD56. These studies highlight the complex neuroimmune crosstalk among neurons, ACs, microglia and T cells, warranting further investigation.
In summary, we have described the creation and validation of a human neuroimmune axis model for investigating infiltration of peripheral immune cells into 3D AD neural–glial cell cultures. We observed a dramatic increase in infiltrating T cells and monocytes selectively into AD cultures. We also showed that infiltration of CD8+ T cells into AD cultures led to enhanced activation of microglia and exacerbated neuroinflammation and neurodegeneration (summarized in Fig. 7). Mechanistically, we demonstrated that the CXCL10–CXCR3 signaling axis plays a key role in regulating the infiltration of T cells into the AD cultures. Blockade of the CXCL10–CXCR3 axis prevented neuronal toxicity and offers a potential druggable therapeutic target for modulating T cells in AD patients. This neuroimmune axis model can now be employed to explore the potentially detrimental or beneficial roles of various peripheral immune cells infiltrating the brain. These studies should also guide new diagnostics and therapeutics for AD based on interventions targeting peripheral immune cell infiltration into the brain.
Fig. 7 |. Proposed mechanism for selective infiltration of CD8+ T cells into an environment of AD pathology and subsequent interaction with microglia, leading to enhanced neurodegeneration and neuroinflammation.
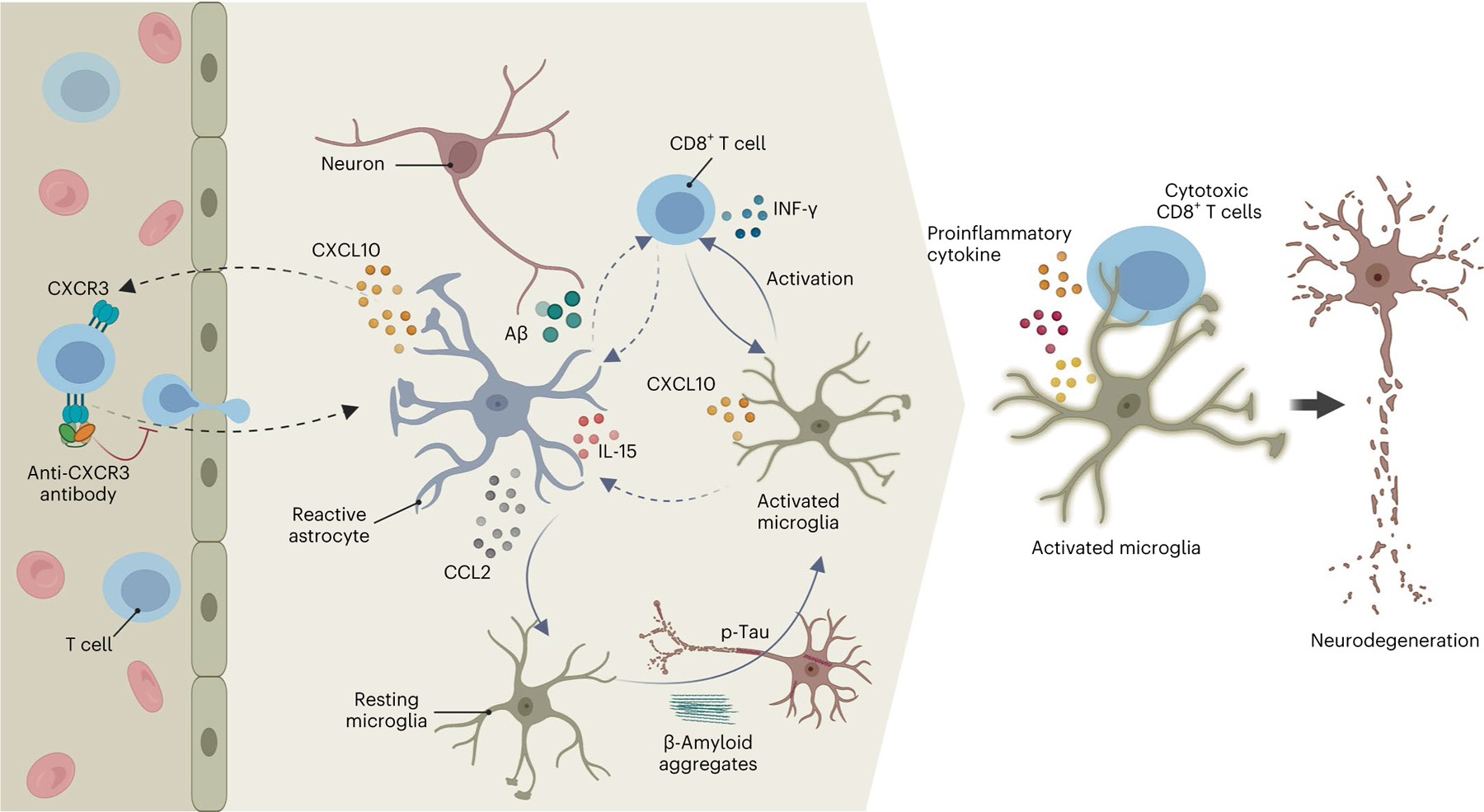
AD pathology leads to elevated levels of proinflammatory recruitment chemokines, for example, CCL2 and CXCL10. T cells selectively infiltrate an environment of AD pathology in the neuroimmune axis model after binding of CXCL10 to its T cell receptor, CXCR3. The interaction of CCL2 with CCR2 induces recruitment of microglial cells to AD pathology. Infiltrating CD8+ T cells activate IFN/inflammatory pathways in microglia and ACs, resulting in a synergistic and vicious cycle that dramatically exacerbates cellular damage and neuroinflammation in AD. Blocking the binding of CXCL10 to the CD8+ T cell CXCR3 significantly attenuates selective infiltration of CD8+ T cells into an environment of AD pathology in the AD neuroimmune axis model and prevents neurodegeneration.
Methods
NPC line, medium and reagents
An FAD cell line exhibiting substantial amyloid pathology was engineered using ReN cell VM human neural progenitor cells (Millipore Sigma, catalog no. SCC008) according to previous reports25–27. Transfected ReN VM human neural progenitor cells contained FAD-related genes encoding human amyloid precursor protein (APP) with both the K670N/M671L (Swedish) and V717I (London) mutations (APPSL), presenilin 1 (PSEN1) with the ΔE9 mutation (PSEN1(ΔE9)) with both GFP and mCherry as reporters for viral infection. The ReN cells were cultured in Matrigel-coated flasks (1:100 dilution of Matrigel:Dulbecco’s modified Eagle’s medium (DMEM)-F12 medium) using ReN cell expansion medium according to previously published protocols25–27.
iPSC-derived human microglia
The iPSC cells were acquired from the National Institute of Neurological Disorders and Stroke (NINDS) Human Cell and Data Repository (NHCDR ID—ND41865, cell line ID NN0003920 and lot no. 11335135; male, aged 64 years, no disease, ethnicity not reported, APOE3/3 and no reported TREM2 mutation). Human iPSCs were differentiated into hematopoietic progenitor cells (HPCs) and then into iMGLs following a previously published report30. Modifications of this protocol included use of 5.25 μg ml−1 of human insulin solution (Millipore Sigma, catalog no. I9278), 462 μM monothioglycerol (Millipore Sigma, catalog no. M6145-25) and 0.05% w:v bovine serum albumin (Millipore Sigma, catalog no. A9418) in the basal medium. In addition, 100 μM IDE1 (PeproTech, catalog no. 1164899) was used instead of transforming growth factor-β1 during HPC-to-iMGL differentiation.
Microfluidic chip design and fabrication
The microfluidic chip was designed using AutoCAD software and transferred on chrome photomasks. Negative photoresists SU-8 (MicroChem) were patterned using photolithography techniques on a silicon wafer. Briefly, the first (10-μm) and second (100-μm) layers were sequentially spin coated on a clean silicon wafer and patterned by ultraviolet exposure through the photomasks. The wafer was then used as a master mold for silicone elastomer polydimethylsiloxane (PDMS, Sylgard 182, Elsworth Adhesives) microfluidic devices. The PDMS array was molded by casting the liquid prepolymer composed of a mixture of 10:1 silicone elastomer and a curing agent. The mixture was cured at 75 °C overnight and then the PDMS mold was peeled off the silicon wafer. Brain and peripheral immune cell inlets were punched using a 2-mm or 2.5-mm punch (Harris Uni-Core), respectively. Individual devices were punched out of the PDMS layer using a 12-mm punch. The molded PDMS devices were bonded irreversibly after plasma treatment to 12-well glass-bottomed plates (MatTek Corporation). The cylindrical chamber was prepared by casting and curing a mixture of 10:1 silicone elastomer and a curing agent in a Petri dish overnight at 75 °C. The PDMS mold was peeled off, central inlets were punched using a 2.5-mm punch and then concentric cylinders were punched out using a 6-mm punch. The cylinder was then plasma treated and bonded irreversibly on top of the BRAIN compartment of each microfluidic device.
Neuroimmune axis model
The microfluidic devices were first plasma treated and then coated with a 1:100 dilution of Matrigel:DMEM-F12 medium, using prechilled gel-loading tips into the cell reservoirs, and incubated for at least 4 h at 37 °C. ReN cell differentiation medium was then added to the well containing the device until the medium submerged only the bottom layer of the device. Next, ReN cells (AD or CTRL) mixed with Matrigel in a 1:5 dilution of Matrigel:ReN cell differentiation medium were transferred into the central brain cell reservoir of the microfluidic chip at 5.0 × 104 cells per 5 μl using a prechilled gel-loading tip. The 3D cell culture was maintained for 4 weeks. The 3D cell cultures were given 1 ml of differentiation medium for the first week of growth, followed by 450 μl of fresh differentiation medium each week until the end of week 3 of culture. In the final week of culture, 250 μl of fresh differentiation medium was added to the 3D cell culture. Then, 1 d before adding PBMCs to the peripheral compartment, 5.0 × 103 iMGLs were added to the central brain compartment of the 3D ReN cell culture.
Isolation of human PBMCs
All donor blood specimens were acquired from healthy donors and handled according to the protocols approved by the Massachusetts General Hospital Institutional Review Board (protocol no. 2021P003192). Peripheral blood from healthy individuals aged ≥18 years was drawn in 10-ml tubes containing heparin or 5 mM EDTA (Vacutainer; Becton Dickinson) and delivered on ice within 1–3 h of collection (Research Blood Components LLC). Whole blood was processed immediately on delivery and PBMCs were carefully handled using sterile techniques. CD3+/CD4+/CD8+ T cells, monocytes and B cells were isolated directly from fresh human whole blood using EasySep Direct Human Isolation kits from STEMCELL Technologies (T cell, catalog no. 19661; CD4+ T cell, catalog no. 19662; CD8+ T cell, catalog no. 19663; monocyte, catalog no. 19669; B cell, catalog no. 19674) according to the manufacturer’s protocols. Isolated cells were stained with either Hoechst (Thermo Fisher Scientific, catalog no. 62249) or Cell Tracker Orange CMRA (Invitrogen, catalog no. C34551), washed and suspended in Iscove’s modified Dulbecco’s medium (IMDM) with 20% fetal bovine serum (FBS) (Gibco, catalog no. 1600–069) before loading into the microfluidics. Next, PERIPHERAL compartments of the matured neuroimmune axis model were loaded with 5.0 × 104 cells in 5 μl of IMDM with 20% FBS and imaged immediately.
Time-lapse imaging of PBMC migration in the neuroimmune axis model
A fully automated Nikon Eclipse Ti2 microscope was used to acquire time-lapse images of the AD or CTRL neuroimmune axis model loaded with PBMCs. A glass-bottomed plate was placed in a biochamber heated to 37 °C with 5% CO2 and confocal images of microchannels and the BRAIN compartment within each microfluidic chip were acquired every 8.5 min using a ×10 objective for at least 18 h. PBMCs were monitored using IMARIS software (Oxford Instruments, v.9.8.0).
Data processing and analysis of PBMC migration index and velocity
Migration index.
Quantification of the migration index and velocity of PBMCs was performed using an unbiased, high-throughput and automated image analysis pipeline by IMARIS software. CD3+ T cells (n = 10 donors, AD Neu/AC; n = 12, AD Neu/AC/iMGL), CD4+ (n = 8 donors), CD8+ (n = 7 donors), monocytes (n = 8 donors, AD Neu/AC; n = 9, AD Neu/AC/iMGL) and B cells (n = 12 donors, AD Neu/AC; n = 8, AD Neu/AC/iMGL) were freshly isolated as described, loaded into the peripheral compartments of the PiChip systems and imaged using automated Nikon Eclipse Ti2 microscope. Using the Spots function, the region of interest (ROI) was defined as 600 × 1,024 pixels2 and the estimated spot XY diameter of the PBMCs was 9 μm. Then, an automatic spot quality threshold and track duration were used to filter detected spots. Finally, the total number of cells migrated over time was extracted from the IMARIS software and data at 15 h post-loading were used to compute the migration index in Excel (v.16.72). Data from five or six independent PiChip systems per donor were collected and the migration index of peripheral immune cells for each donor was calculated as follows:
where migration index is considered selective infiltration into AD cultures compared with control cultures.
Velocity.
Data from independent PiChip systems for CD3+ T cells (n = 9 donors each for CTRL and AD Neu/AC; n = 10 each for CTRL and AD Neu/AC/iMGL), monocytes (n = 12 donors for CTRL Neu/AC; n = 9 for AD Neu/AC; n = 9 each for CTRL and AD Neu/AC/iMGL) and B cells (n = 14 donors each for CTRL and AD Neu/AC; n = 8 each for CTRL and AD Neu/AC/iMGL) were collected. The Spots function was also used to quantify the speed of the PBMC that migrated through the PERIPHERAL–BRAIN microchannels in microfluidic devices. A 400 × 1,024 pixel2 ROI with an estimated spot XY diameter of 9 μm was used to detect peripheral immune cells. An automatic spot quality threshold and track duration were used to filter detected spots. For tracking PBMCs, the maximum distance tracking parameter for T cells, monocytes and B cells, was 70 mm, 25 mm and 75 mm, respectively. The average velocity over 18 h was then recorded for each image. Images were excluded from analysis if ReN cell growth blocked >70% of the PERIPHERAL–BRAIN microchannels in microfluidic devices, preventing PBMC movement. Rarely, data that display large fluctuations in the number of cells in a short amount of time, which suggested inaccurate IMARIS image processing, were excluded as well.
Chemotaxis of human PBMCs
To prime the Matrigel:DMEM-F12 medium-coated microfluidic devices, 20 min before PBMCs were introduced, the BRAIN compartment was filled with 10 μl of the chemoattractant of interest (R&D Systems; monocyte chemotactic protein (MCP)-1/CCL2, catalog no. 279-MC-010; C-X-C motif chemokine ligand 10 (CXCL10), catalog no. 266-IP-010; and C-X-C motif chemokine ligand 12 (CXCL12), catalog no. 350-NS-010/CF). The wells containing microfluidic chips were then filled with IMDM with 10% FBS medium until the medium reached the top level of the cylinder without submerging the device. Isolated T cells, monocytes and B cells from whole blood were loaded by using a gel-loading tip at 5.0 × 104 cells in 10 μl of IMDM with 10% FBS for each PERIPHERAL compartment. Loaded PBMCs were recorded using time-lapse imaging on a fully automated Nikon TiE microscope with a biochamber heated to 37 °C and 5% CO2. Images were acquired every 7 min for 15–17 h, using a ×10 objective. A customized algorithm was used to map each individual migration trajectory and determine the intensity of migrated cells, as well as the cell migratory velocity. Using these data, a ‘heat map’ was generated in MATLAB (MathWorks, v.9.4 (R2018a)), which provides an analysis of the total migrated cells and their velocity in each condition.
Imaging flow cytometry
The purity of isolated human PBMCs was assessed using the ImageStream Mark II imaging flow cytometer (Amnis Corporation) equipped with a ×40 objective, 6 imaging channels and 405-, 488- and 462-nm lasers after cell isolation. Isolated cells were diluted in RPMI 1640 medium supplemented with 10 mM Hepes (Life Technologies, catalog no. 31800105) and stained (1:200 dilution) with the following fluorochrome-conjugated antibodies purchased from STEMCELL Technologies where applicable: anti-human CD45 antibody (clone HI30, catalog no. 60018), anti-human CD14 antibody (clone M5E2, catalog no. 60004), anti-human CD3 antibody (clone UCHT1, catalog no. 60011) and anti-human CD19 antibody (clone HIB19, catalog no. 60005). PBMC purity was assessed on singlet CD45+ cells to exclude debris, platelets and red blood cells. Then, 5 × 105 CD45+ events were collected for each sample and analyzed using the IDEAS software (v.6.2).
Quantitative analysis of Aβ levels
Levels of Aβ38, Aβ40 and Aβ42 in medium collected from the BRAIN compartment were simultaneously measured by a multi-array electrochemiluminescence assay kit (Meso Scale Diagnostics LLC (MSD), V-PLEX Aβ Peptide Panel 1 (6E10) kit, catalog no. K15200E-2), according to the manufacturer’s protocol and using no sample dilution. Data were collected using the MESO QuickPlex SQ 120-mm instrument and analyzed using DISCOVERY WORKBENCH (v.4.0.12).
Quantification of brain-localized T cells in 5×FAD mice
WT C57BL/6J mice were purchased from the Jackson Laboratory. The 5×FAD mice were purchased from the Jackson Laboratory (Mutant Mouse Resource and Research Center) and backcrossed on to the C57BL/6J background using a speed congenic protocol. The 5×FAD and WT mice were group housed, the specific pathogen-free facility maintained a 12 h light:dark cycle and mice could freely access food. All animal protocols were approved by the Animal Review Committee at the Massachusetts General Hospital (protocol no. 2017N000156) and were in compliance with relevant ethical regulations. The 7-month-old female WT and 5×FAD mice were anesthetized with 3–4% isoflurane and 100 μl of CD45-FITC antibody (BioLegend, clone 30-F11, catalog no. 103107). Then, 10 μg per mouse diluted in sterile phosphate-buffered saline (PBS) was injected into the retro-orbital sinus. After 5 min of circulation, mice were perfused transcardially with 25 ml of cold PBS. Brains were harvested and the olfactory bulbs and cerebellum removed. The remaining brain tissues were minced with blades and homogenized by tissue grinder in Hank’s buffered saline solution (Lonza, catalog no. BE10-527F) without addition of any enzymes. Next, 30% Percoll gradient centrifugation (1,600 r.p.m., 24 min, without breaks) was used to remove myelin and other debris. Live cells were counted by Trypan Blue. Brain cells were blocked with Fc-receptor blocking antibody (BioLegend, clone 93, catalog no. 101330, 1:200) in FACS buffer (0.5% BSA, 2 mM EDTA and 0.02% sodium azide in PBS) for 5 min at 4 °C before surface staining (30 min at 4 °C). The following anti-mouse antibodies were used for surface staining: PE anti-mouse CD45.2 (BioLegend, clone 104, catalog no. 109807, 1:200), APC/Cyanine7 anti-CD11b (BioLegend, clone M1/70, catalog no. 101225, 1:200), phycocerythrin (PE)/Cyanine7 anti-mouse CD4 (BioLegend, clone GK1.5, catalog no. 100421, 1:400) and PerCP/Cyanine5.5 anti-CD8α (BioLegend, clone 53–6.7, catalog no. 100733, 1:200). Live cells were distinguished by LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Thermo Fisher Scientific, catalog no. L34959). Flow cytometry was performed on a BD LSRFortessa X-20 flow cytometer. Flow cytometry data were analyzed using FlowJo software (Tree Star). Unpaired Student’s t-tests were performed to determine statistical significance using GraphPad Prism (v.9.4.1).
Cytokine analysis
Detection of human cytokines, chemokines, inflammatory factors, growth factors and receptors released in the 3D cell culture before and after PBMC infiltration was conducted using a high-throughput, sandwich-based, human Kiloplex quantitative proteomics array (Ray-Biotech, catalog no. QAH-CAA-X00-1). The supernatants of the 3D AD culture compartment were collected and tested against proteins. Each protein was tested in quadruplicate. A standard curve for each protein was generated using a known concentration of the protein and used for sample quantification. ‘NA’ replaced sample measurements when insufficient sample volume interfered with cytokine quantification. Fold-changes were computed as log2((AD(+MG) + PBMC)/(AD(+MG) − PBMC)) and log2((control(+MG) + PBMC)/(control (+MG) − PBMC)), where the numerator is the cytokine concentration in the presence of T cells, monocytes or B cells and the denominator is the cytokine concentration in the absence of PBMCs. If present, technical replicates were averaged before fold-change calculation. In the absence of technical replicates, single measurements were used.
Immunohistochemistry
For immunofluorescent staining of neuroimmune axis models, cells were rinsed with Dulbecco’s PBS (DPBS) twice before fixing. Cells were then incubated in a 4% paraformaldehyde solution (Electron Microscopy Sciences, catalog no. 157-4-100) overnight at 4 °C, followed by washing 3× with DPBS. Cells were permeabilized in 0.1% Triton X-100 in PBST (PBS with 0.1% Tween-20) for 15 min at room temperature and then blocked with 3% human serum albumin overnight in PBST at 4 °C. Cells were then incubated with primary antibodies for 24 h at 4 °C. The following primary antibodies (and dilutions) were used: anti-p-tau antibody (Thermo Scientific, clone AT8, catalog no. MN1020; 1:40), anti-GFAP antibody (Neuromab, clone N206A/8, catalog no. 75–240; 1:100), anti-Tuj1 antibody (Cell Signaling Technology, catalog no. 5568; 1:200), anti-IBA1 (Millipore Sigma, catalog no. MABN92, 1:100) and anti-CD11b (Novus Biologicals, catalog no. NB110-89474, 1:100). This was followed by washing the cultures with PBST 3× and incubation with species-specific secondary antibodies at 4 °C overnight.
For immunostaining of 5×FAD brain sections, harvested brain sections from 6- to 7-month-old WT and 5×FAD mice were treated with 3% w/v dextran sodium sulfate and washed 3× with Tris-buffered saline (TBS; Boston BioProducts, catalog no. BM301). Sections were then blocked for 1 h with TBS containing 0.2% Triton X-100 (Millipore Sigma, catalog no. X-100) and 5% donkey serum (abcam, catalog no. ab7475), followed by overnight incubation with primary antibodies (and dilutions), including CD8α (Cell Signaling, D4W2Z, catalog no. 98941, 1:250), CXCR3 (Santa Cruz, catalog no. sc-137140, 1:200), CXCL10 (R&D Systems, catalog no. AF-466-NA, 1:200), GFAP (Millipore Sigma, catalog no. G6171, 1:200) and 3D6 (a gift from E. Lilly, 1:1,000). Next, brain sections were washed 3× with TBS and incubated for 1 h at room temperature with secondary antibodies, including anti-mouse Cy5 ( Jackson ImmunoResearch, catalog no. 715-175-150, 1:500), anti-rabbit Cy3 ( Jackson ImmunoResearch, catalog no. 711-165-152, 1:500) and anti-rat 488 (Invitrogen, catalog no. A-21208, 1:500). Finally, brain sections were stained with Hoescht (Thermo Fisher Scientific, catalog no. 62249) for 15 min at room temperature before washing 3× with TBS and mounting them on glass slides for confocal imaging. Samples were imaged using a Nikon Eclipse Ti2 confocal microscope. The cortex and hippocampus were analyzed using the ‘Analyze Particles’ method with ImageJ (v.2.9.0) to quantify the percentage of the region of interest (ROI) covered by CXCL10+ cells. CXCR3+ T cells were not quantified due to the small number of parenchymal CD8+ T cells present in the dentate gyrus of 6- to 7-month-old 5×FAD mice57.
Quantification of neurodegeneration
Cultures with various AD and CTRL conditions, including isolated T cells (n = 4 donors for each T cell subtype) were imaged with ×20 objectives on the Nikon A1 Confocal system (Nikon), using identical settings. Quantification of GFP+ areas, which are expressed in neurons and ACs (6 imaging ROIs per well (96-well plate or PiChip)), were randomly selected. The object of the GFP+ area was identified by the threshold with automated segmentation and converted into binary images. The surface area was quantified based on the pixels and represents the total GFP+ surface area. For the quantification of cell numbers, Hoechst (Tocris, catalog no. 5117) was used and blue dots were counted with the round threshold of 0.8–1.0. The same strategy was applied for a specific immunological fluorescent stained area, including Tuj1 (abcam, catalog no. ab24629, 1:200), MAP2 (Cell Signaling Technology, catalog no. 4542, 1:200) (neuronal markers) and ALDH1L1 (EMD Millipore, catalog no. MABN495, 1:100) (AC marker).
Neurite density:cell body ratio was quantified using the GFP+ surface area as follows: (1) the whole GFP+ area in each ROI was identified (no. 1); (2) the bright spots in the GFP+ surface area were detected and counted as cell bodies (no. 2); (3) the GFP+ area except no. 2 was quantified (that is, no. 1 surface area − no. 2 surface area) (no. 4); and, last, (4) no. 3 values were divided by the number of cell bodies in step 2 (no. 2) to measure neurite density per cell body.
ScRNA-seq
To identify cell-type-specific transcriptional changes, we performed scRNA-seq from the brain compartment of AD or control cultures, 72 h post-loading peripheral immune cells into the PiChip.
ScRNA-seq via InDrop.
After generating barcoded cDNA from single cells, the droplets were broken and cDNA was processed for next-gen sequencing. Briefly, this involved purification of the cDNA library and second-strand synthesis, followed by in vitro transcription (IVT) using T7 RNA polymerase. The amplified antisense RNA was fragmented using zinc-ion-mediated cleavage and converted into a DNA library ready for Illumina sequencing by a second RT reaction and a few cycles of PCR. All samples were multiplexed and sequenced to a depth of 60,000–100,000 reads per cell. Sequencing was performed at the Bauer Core Facility of Harvard University.
Normalization and integration.
After quality control (QC), each library was individually normalized and scaled using SCTransform (https://satijalab.org/seurat/articles/sctransform_vignette.html). For all the libraries, we selected the 2,000 most variable features for downstream integration. We determined a list of variable features across libraries with SelectIntegrationFeatures (https://github.com/satijalab/seurat-data) which we used as input for integration. We next used the merge (https://github.com/satijalab/seurat-data) function of the Seurat package to correct for any potential library batch effect in our data. The integrated matrix was used for downstream analysis. In the integrated dataset, we performed principal components analysis (PCA, total number of PCs to compute and store = 20) to achieve separated cell-type clusters. We selected 1:20 dimensions for dimensionality reduction by Uniform Manifold Approximation and Projection (UMAP), which we performed with the RunUMAP() function. To minimize the batch effect across all the conditions, we performed Harmony58 and set the resolution at 1.2. The 22 clusters identified represented unique neuron/AC/microglia/T cells. The individual R objective files will be deposited with specific meta-data such as batch, resolution and condition. In neuron/AC cultures without microglia, we detected a handful of cells overlapping with the microglia cluster in Neu/AC/iMGL cultures (Extended Data Fig. 1e). In contrast to microglial cells in Neu/AC/iMGL cultures, these small number of cells in neuron/AC cultures did not fully express well-known microglial markers (for example, CSF1R and PU.1).
ScRNA-seq differential gene analysis.
Differentially expressed genes were found by applying the FindAllMarkers59,60 function for overall differential expression and FindMarkers for side-by-side comparisons, both from the Seurat R package. All the reported comparisons in the manuscript were performed with the following parameters: assay = ‘RNA’, test.use = ‘wilcox’, min.pct = 0.1, logfc.threshold = 0.01. We used Wilcoxon’s rank-sum test to calculate P values. Only genes with their adjusted P value (Padj) < 0.05 (post-hoc, Bonferroni’s correction) were considered significant. Neuronal genes and pathways are not shown because the neuronal population in the present study has been underestimated, possibly due to the damage from cell dissociation and/or in-drop sequencing.
NicheNet analysis.
To perform NicheNet ligand activity analysis33, we compiled a list of potentially active ligands and their corresponding receptors/downstream genes based on gene expression changes in the AD condition compared with controls. We utilized NicheNet to identify the top 20 active ligands based on these gene changes and then identified the ligand–receptor interactions that are significant in four different cell types (neurons, ACs, microglia and T cells). We set a cutoff value for ligands with at least one expressed receptor in each cell type, with an average log2(expression) >0.1 and false recovery rate (FDR) <0.2. Once we identified these ligand–receptor interactions, we then queried the NicheNet data sources to identify the downstream genes associated with these interactions.
Blocking CXCL10–CXCR3 axis
Flow cytometry.
T cells were immediately and carefully isolated from fresh whole blood using immunomagnetic negative selection according to the manufacturer’s protocol and sterile techniques. The isolated T cells were centrifuged at 300g for 5 min at 4 °C and the supernatant was discarded. Human CXCR3-neutralizing antibody (R&D Systems, clone: 49801, catalog no. MAB160-100) or CXCR3 chemical antagonist, NBI74330 (R&D Systems, catalog no. 4528/10), was diluted to concentrations of 10 μg ml−1, 20 μg ml−1 and 40 μg ml−1 with IMDM supplemented with 20% FBS. CD3+ T cells were resuspended at a concentration of 4 × 105 cells ml−1 in the three different concentrations of the human CXCR3 antibody or agonist and were incubated for 40 min at 37 °C and 5% CO2. The CD4+ and CD8+ T cells were incubated with 10 μg ml−1 of human CXCR3-neutralizing antibody for 40 min at 37 °C and 5% CO2. Afterwards, the volume of each conical tube containing T cells was brought up to 5 ml with cold cell-staining buffer (BioLegend, catalog no. 420201). The cells were then centrifuged at 300g for 5 min at 4 °C and the supernatant was discarded. Each cell pellet was resuspended at a concentration of 1 × 106 cells per 500 μl of cold cell-staining buffer. For CD3+ T cells, 12.5 μl (1:40 dilution) of each of the following fluorochrome-conjugated antibodies was used to stain the cells: anti-human CXCR3 PE antibody (BioLegend, catalog no. 353706), anti-human CD3 APC antibody (BioLegend, catalog no. 317318), anti-human CD45-FITC antibody (BioLegend, catalog no. 982316) and human TruStain FcX (BioLegend, catalog no. 422301). For CD4+/CD8+ T cells, the appropriate fluorochrome-conjugated antibodies were used to stain the cells: anti-human CXCR3 PE antibody (BioLegend, catalog no. 353706), anti-human CD45-FITC antibody (BioLegend, catalog no. 982316), anti-human Alexa Fluor-647 CD4+ antibody (BioLegend, clone SK3, catalog no. 344636) or anti-human Brilliant Violet CD8+ antibody (BioLegend, catalog no. 344748) and human TruStain FcX (BioLegend, catalog no. 422301). Cells were incubated with the fluorochrome-conjugated antibodies on ice in the dark for 20 min. After the incubation, the volume of each conical tube was brought up to 5 ml with cold cell-staining buffer and the cells were centrifuged at 350g for 5 min. The supernatant was aspirated off and the cell pellet was resuspended in 1 ml of cold cell-staining buffer. Cells were kept on ice, in the dark, and immediately analyzed using a BD Fortessa X-20 flow cytometer. Analysis of data was performed using FlowJo v.10.8 by first gating on singlets using forward-scatter area versus height, then on live cells based on forward versus side-scatter profiles, followed by cell subset-specific gating.
Migration analysis of inhibited T cells.
CD3+ T cells (n = 6 donors) were treated with either MAB160 or NBI74330, whereas CD4+/CD8+ cells (n = 7 donors) were incubated with MAB160 antibody, as described above. Then, 5 × 104 T cells were loaded into the peripheral compartments of the PiChip systems. Data from five or six independent PiChip systems per donor were collected and the migration index of inhibited T cells for each donor was calculated as follows:
where the migration index is considered to block T cell infiltration into AD cultures compared with untreated T cells in AD. Quantification of inhibited T cell velocity was performed as previously described. If no T cells moved through the PERIPHERAL–BRAIN microchannels of the microfluidic device during time-lapse imaging, no velocity reading was given for that image.
Chemotaxis of T cells in response to CXCL10
The bottom layer of the microfluidic device was submerged with IMDM and supplemented with 20% FBS. Recombinant human CXCL10 was loaded into the central compartment of the microfluidic device at a concentration of 100 nM. Isolated CD4+/CD8+ T cells from whole blood (n = 7–8 donors) were stained with Hoechst and added at 5 × 104 cells in 5 μl of medium to each peripheral compartment. Quantification of the T cell migration index and velocity was performed as previously described.
Quantification of cytokines after infiltration of T cells in AD
Levels of granzyme B, IL-2, IL-15 and TNF-α were simultaneously measured by a multi-array electrochemiluminescence assay kit (MSD, U-PLEX Custom Immuno-Oncology Group 1 (hu) Assay, catalog no. K151AEM-2). To quantify chemokine levels, medium was collected from CTRL and AD ReN cells that were differentiated in 1:10 Matrigel:ReN cell differentiation medium for 4 weeks in a 96-well plate, and had been co-cultured with and without microglia for 96 h and with and without CD4+ T cells or CD8+ T cells for 72 h. Then, 2.5 × 105 ReN cells were differentiated in each well and 2.5 × 104 microglia were added. Twenty-four hours after the addition of iMGL, 5,480 CD4+ or CD8+ T cells in 4 μl of human serum (Millipore Sigma, S1-100 ml) were added to the cultures. The samples were not diluted and were analyzed using the assay kit according to the manufacturer’s protocol.
CD3+ T cell migration toward ReN cells
ReN VM (Millipore Sigma, catalog no. SCC008) and ReN mCherry neural progenitor cells were cultured into the neuroimmune axis model as described above. Then, isolated human CD3+ T cells were stained with Cell Tracker Green CMFDA (Invitrogen, catalog no. C2925) and loaded at 5 × 104 cells into 5 μl of medium in to the PiChip. Images were taken at time 0 and 24 h to assess CD3+ T cell migration into the BRAIN compartment.
Quantification of microglial branches
Nikon Elements Software was used to quantify the number of branches. First, the microglial cell body was identified by bright spot detection with spherical parameter >0.9 and then associated branches were quantified using the TrackID method.
Calcium imaging
Cellular activity was monitored in AD cultures after incubation for 30 min in differentiation medium supplemented with 20 μM of Cal-590 AM (AAT Bioquest). The fluorescence intensity dynamics were measured using time-lapse imaging and two ROIs per well were used for analyses. Raw intensity values of all the active cells (>0 frequency for 100 frames) in the captured videos were extracted using ImageJ (time series analyzer v.3). The relative changes in fluorescence intensity to baseline ((Ft1 – Ft0)/Ft0) were regarded as calcium signals. For each ROI (soma and/or neurite), a transient was accepted as a signal when its amplitude was >10× the s.d. of the noise in the baseline.
Statistical analysis
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications11,19,25,28,57. We used multiple donors and at least three independent biological cell-culture replicates across experiments and, to the best of our knowledge, no donor was used more than once. For each experiment, isolated peripheral immune cells were randomly distributed in different microfluidic devices and cell-culture plates. All mice were randomly allocated into age-matched experimental groups. The data were analyzed for statistical significance using GraphPad Prism 9 and R v.4.2.1. The Shapiro–Wilk test was used to assess whether the data followed a normal distribution. Whenever the data did not follow a normal distribution, nonparametric tests were used to analyze the data. P < 0.05 was considered to be significant. The names of the statistical tests and the number of biological replicates and mice used throughout the manuscript are depicted in the figure legends. Unless otherwise noted, all data represent at least three independent experiments. Investigators were blinded during data collection and analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Characterization of the PiChip model.
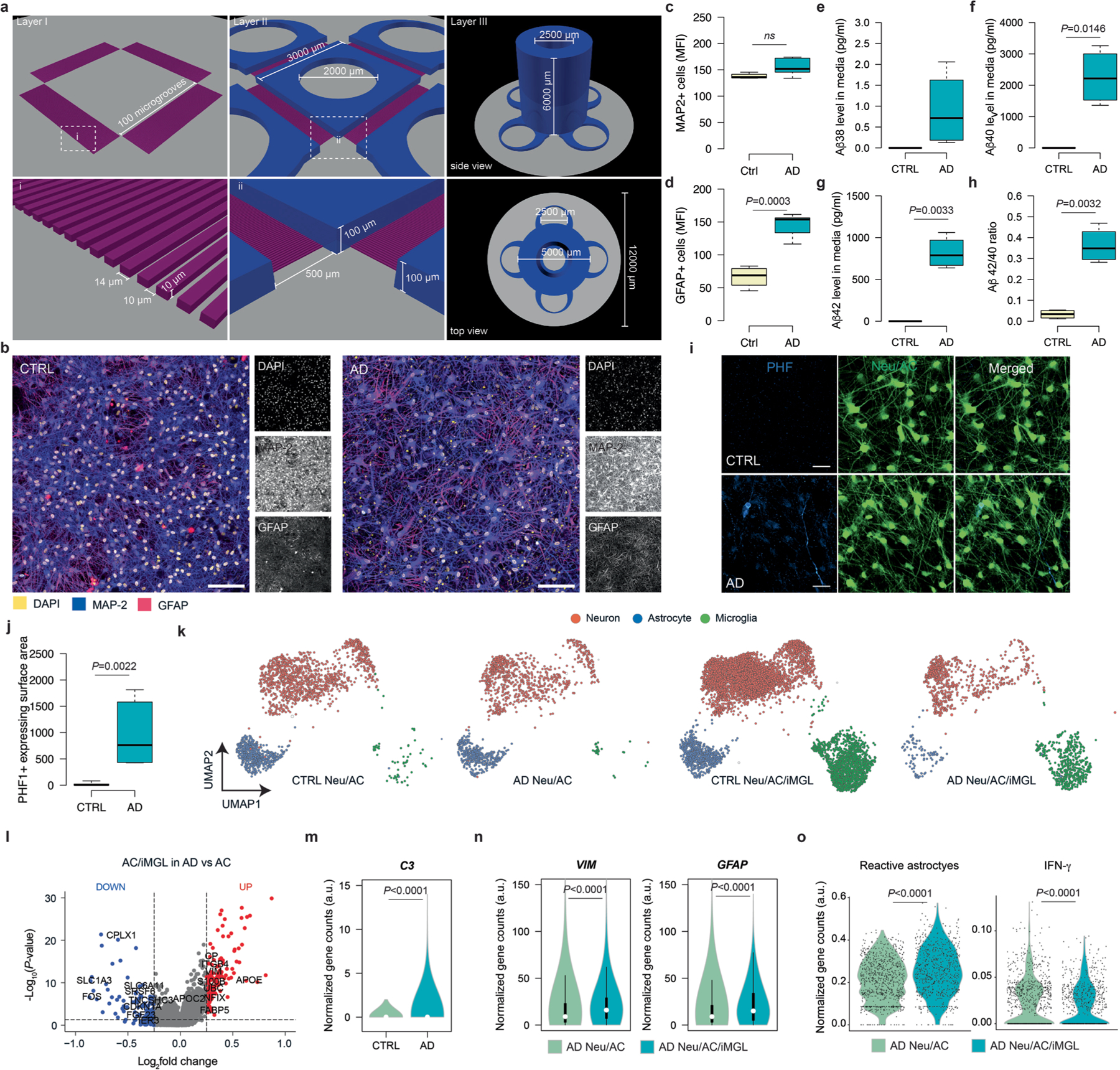
a, 3D illustration of three different layers of the microfluidic-based PiChip system. Layer I consists of an orthogonal array of 400 microgrooves of 10 μm height and width and 500 μm length. Layer II consists of BRAIN and four PERIPHERAL compartments of 100 μm height. Layer III consists of a cylindrical chamber with 6000 μm length and radius and 2500 μm open hollow. The top and bottom panels show low and high magnification of each layer, respectively. b, Representative immunofluorescence staining of 3D-differentiated neurons (MAP-2; blue) and astrocytes (GFAP; red) in AD and CTRL cultures after four weeks of differentiation. Scale bar, 100 μm. c, d, Quantification of MAP-2 (c) and GFAP+ (d) expressing surface area in AD and CTRL cultures (n = 4 independent ROIs for each MAP2+ and GFAP+ CTRL, n = 5 independent ROIs for each MAP2+ and GFAP+ AD; P-values from unpaired, two-sided, t-test). e–h, Quantification of soluble Aβ38 (e), Aβ40 (f), Aβ42 (g) and Aβ42/40 ratio (h) in AD and CTRL cultures after four weeks of differentiation (n = 4 cell cultures from two independent experiments; P-values from unpaired, two-sided, t-test with Welch’s correction). i, Representative immunofluorescence staining of p-Tau (PHF-1; blue) in neuronal cell bodies and neurites (GFP; green) in AD and CTRL cultures. Scale bar, 50 μm. j, Quantification of PHF-1+ expressing surface area in AD and CTRL cultures (n=6 ROIs from at least 5 independent experiments; P-values from nonparametric, two-sided, Mann-Whitney test). k, UMAP visualization of a total of 20153 single cells from AD and CTRL cultures by single-cell transcriptome profiles. Distinct cell types are depicted with different colors. l, Volcano plot of differentially expressed genes in astrocytes in AD Neu/AC/iMGL versus AD cultures lacking iMGL (P-values from Wilcoxon Rank Sum test). Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown in red and blue dots, respectively. m, Violin plot of highly enriched C3 gene in astrocytes in AD Neu/AC/iMGL compared to CTRL (n = 5 independent scRNA-seq data; P-values from nonparametric, two-sided, Mann-Whitney test). n, Vioin plots of selected significantly enriched genes associated with reactive astrocytes (VIM and GFAP) in AD Neu/AC/iMGL compared to AD cultures lacking iMGL (n = 5 independent single cell RNAseq data; P-values from nonparametric, two-sided, Mann-Whitney test). o, Violin plots of highly enriched genes associated with reactive astrocytes and interferon-γ in astrocytes in AD Neu/AC/iMGL compared to AD cultures lacking iMGL (n = 5 independent scRNA-seq data; P-values from nonparametric, two-sided, Mann-Whitney test). (c–h, j) In boxplots, the center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend to minimum and maximum values. (m, n) White circles show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles; polygons represent density estimates of data and extend to extreme values.
Extended Data Fig. 2 |. Increasing number of extravascular CD4+ and CD8+ T cells in 5XFAD brains.
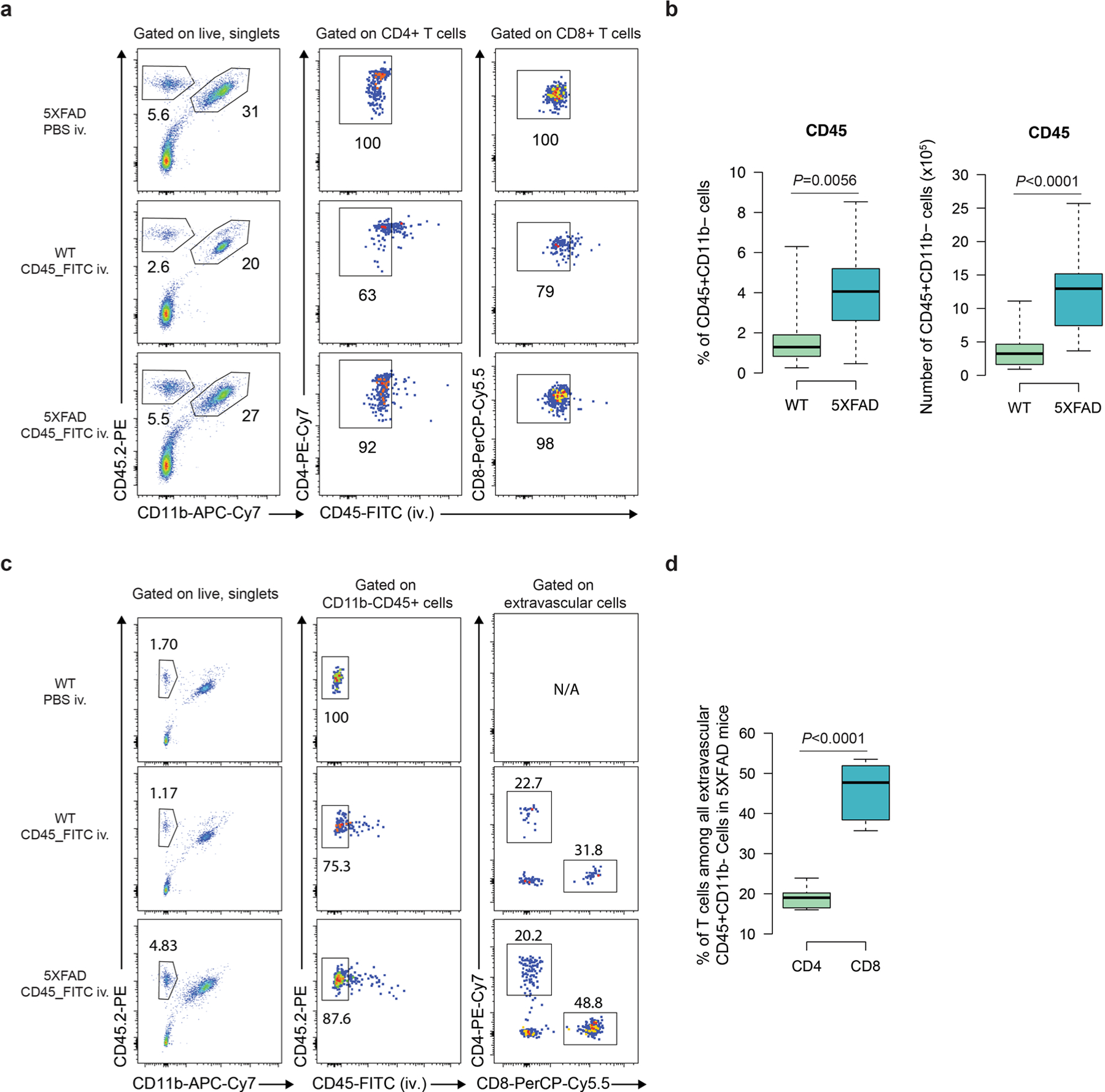
a, Representative flow cytometry plots illustrating the gating of CD45+CD11b- cells, CD45_FITC negative CD4+ and CD8+ T cells. b, Quantification of the percentages and absolute numbers of CD45+CD11b- non-myeloid cells. FITC, Fluorescein isothiocyanate (n = 12 each for WT and 5XFAD mice; P-values from nonparametric, two-sided, Mann-Whitney test). c, Representative flow cytometry plots illustrating the gating of CD4+ and CD8+ T cells among all CD45+ extravascular non-myeloid cells. d, Quantification of the percentages of CD4+ and CD8+ T cells among all CD45+ extravascular non-myeloid cells in 5XFAD mice (n = 9 WT and 5XFAD mice each; P-values from nonparametric, two-sided, Mann-Whitney test). (b, d) In boxplots, the center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend to minimum and maximum values.
Extended Data Fig. 3 |. Infiltrating T cells exacerbate neuronal damage in the presence of microglia in AD.
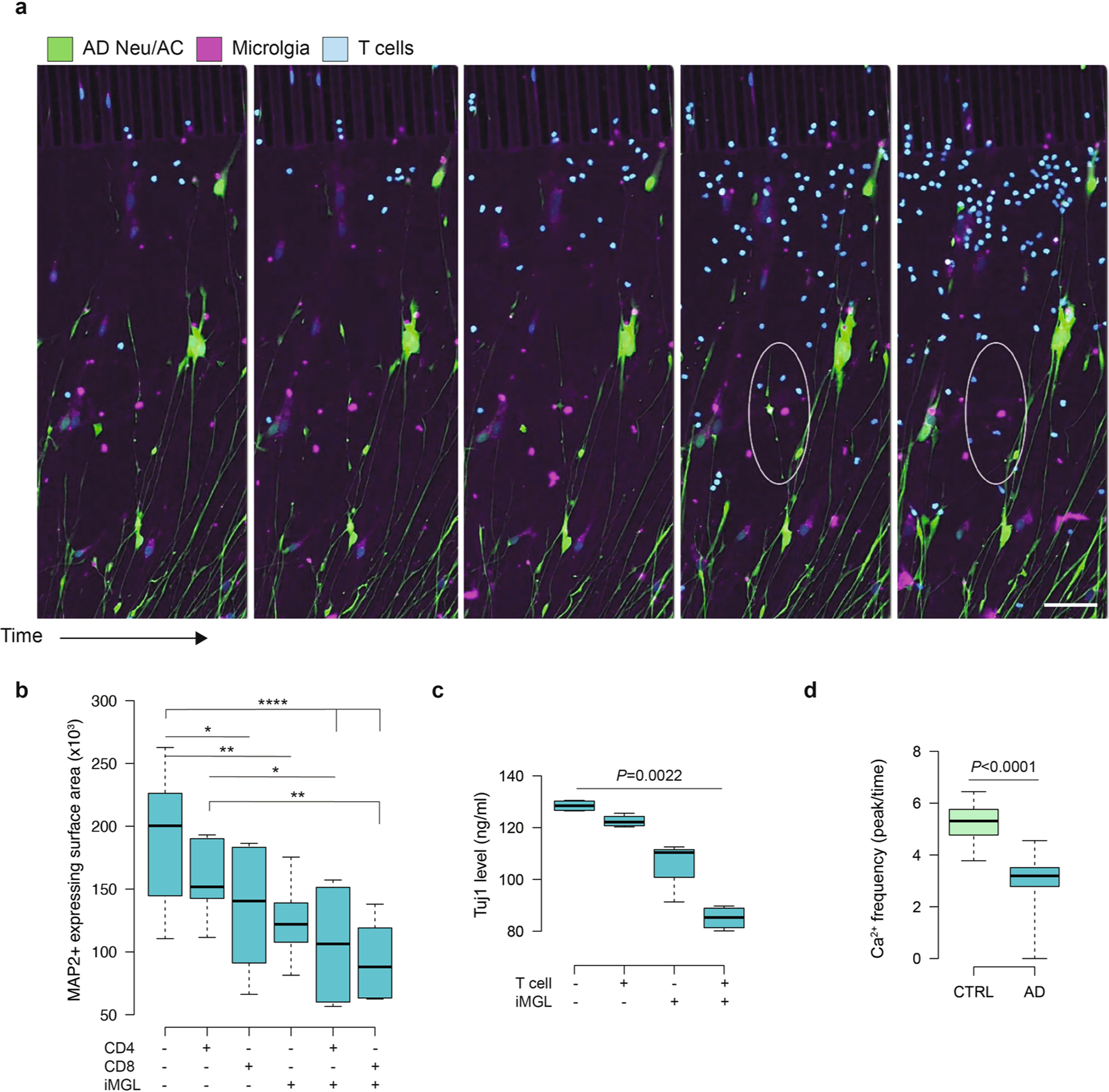
a, Time-lapse confocal imaging showing neurite cleavage in AD cultures. Scale bar, 100 μm. The circled area shows depletion of cells (GFP+ cells) in the presence of microglia and T cells in AD condition. b, Box plots represent quantification of neuronal (MAP2+ expressing surface area) damage in AD Neu/AC (+/−) iMGL conditions in the presence of CD4+ or CD8+ T cells (n = 10 independent ROIs from 4 independent experiments; P-values from two-way ANOVA with Tukey multiple comparisons tests). c, Box plots represent quantification of neuronal damage using ELISA assay for Tuj1 in AD Neu/AC (+/−) iMGL conditions in the presence of CD3+ T cells (n = 4 independent experiments; P-values from nonparametric, Kruskal-Wallis test with Dunn’s multiple comparisons test). d, Box plots represent quantification of calcium dynamics in AD cultures in the presence of T cells compared to CTRL, as monitored using Cal-520 AM, a Ca2+ indicator (n = 251 independent ROIs, CTRL and n = 222, AD from 5 independent experiments; P-values from nonparametric, two-sided, Mann-Whitney test). (b–d) In the boxplots, the center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend to minimum and maximum values.
Extended Data Fig. 4 |. Summary of scRNA-seq data, selected identity gene expression and gene ontology of significantly enriched genes.
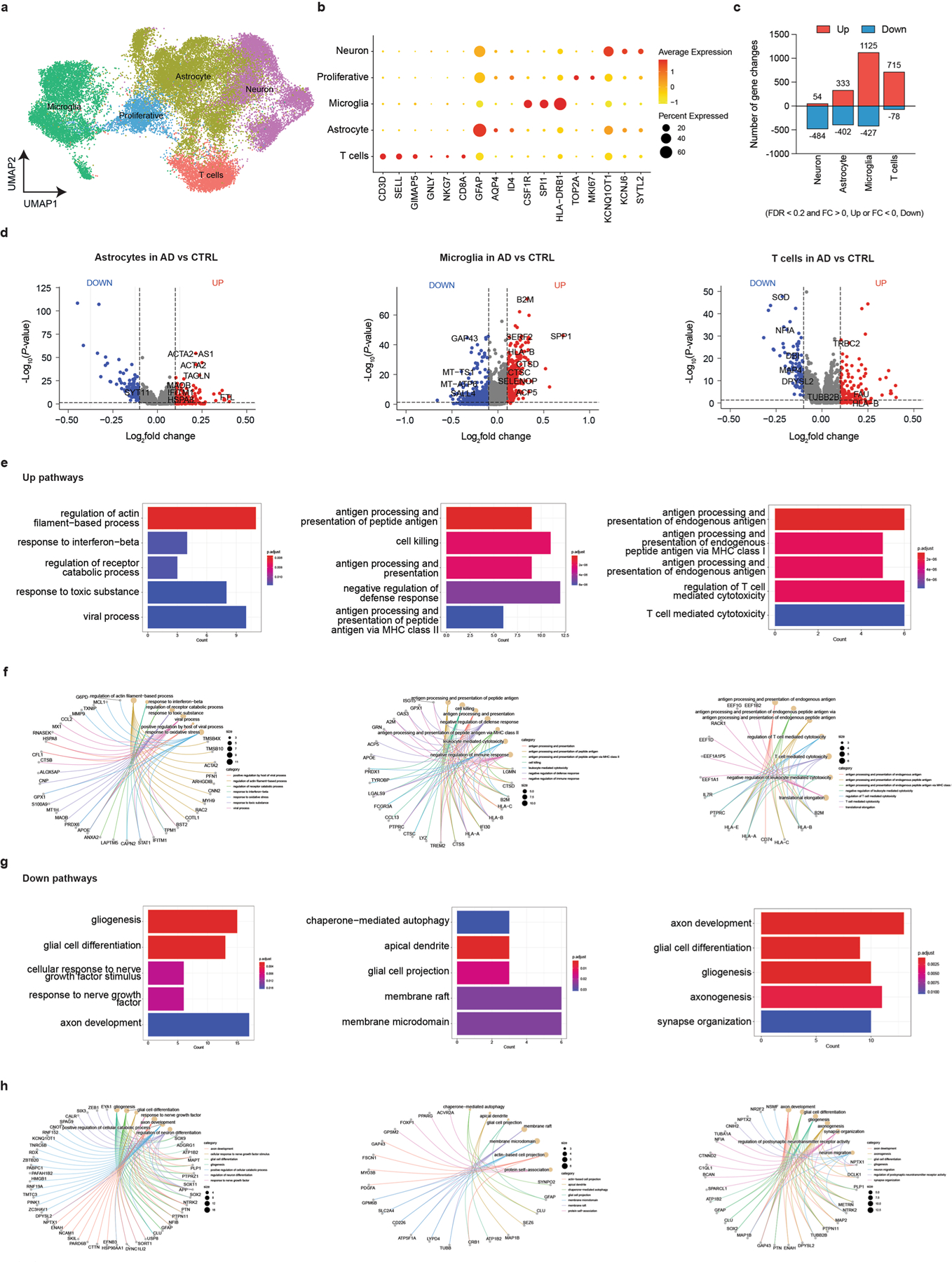
a, UMAP visualization of the cell-type composition of the PiChip cultures by single-cell transcriptome profiles. Distinct cell types are depicted with different colors. b, Dot plot of identity genes for distinct cell types. Color scale (a.u.) indicates the average of expression of identity genes in each cell population, and dot size is proportional to the percentage of cells expressing the identity genes. c, Number of significantly upregulated (FDR < 0.2 and FC > 0) and downregulated (FDR < 0.2 and FC < 0) genes across distinct cell types. Significantly upregulated genes are depicted in red, while downregulated are in blue. d, Volcano plots of differentially expressed genes in all cell types in AD cultures versus CTRL. Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown in red and blue dots, respectively (P-values from Wilcoxon Rank Sum test). e, Gene ontology of significantly upregulated pathways in all cell types in AD versus CTRL. From left to right, the order of conditions is the same as in d. Color scale indicates the adjusted P-value for significantly enriched pathways in each cell population. f, Gene ontology/network analysis of significantly upregulated genes (P < 0.05 and FC > 0.1). Top five category was visualized for associated genes and pathways. g, Gene ontology of significantly downregulated pathways in all cell types in AD versus CTRL. From left to right, the order of conditions is the same as in d. Color scale indicates the adjusted P-value for significantly enriched pathways in each cell population. h, Gene ontology/network analysis of significantly downregulated genes (P < 0.05 and FC < 0.1). Top five category was visualized for associated genes and pathways (e-h, P-values were calculated from a Fisher exact test and the adjusted p-values given by Benjamini & Hochberg method).
Extended Data Fig. 5 |. Altered proportions of neurons, astrocytes and microglia in AD cultures and upon infiltration of T cells.
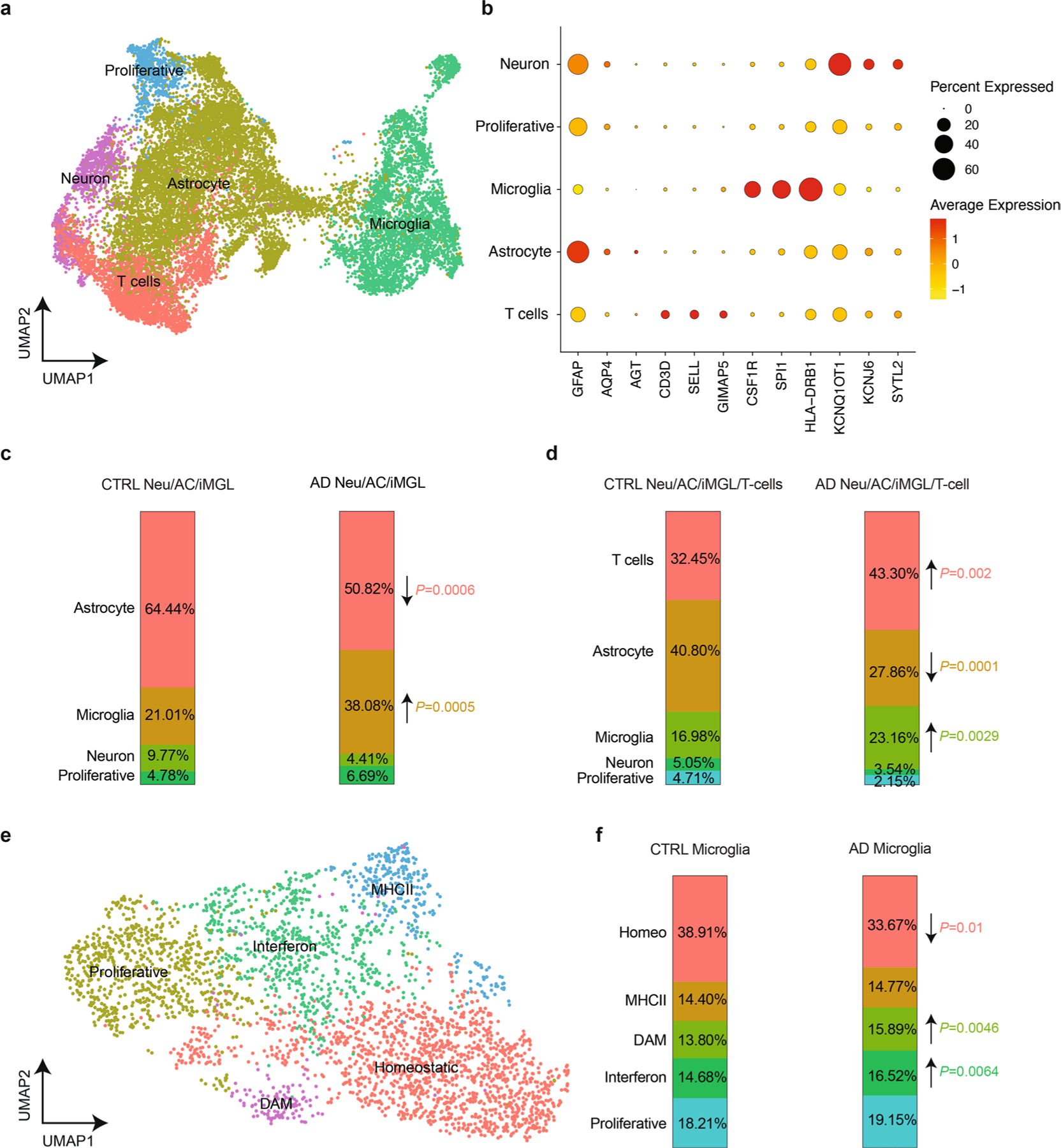
a, UMAP visualization of the cell-type composition of the PiChip cultures by single-cell transcriptome profiles. Distinct cell types are depicted with different colors. b, Dot plot of identity genes for distinct cell types. Color scale (a.u.) indicates the average of expression of identity genes in each cell population, and dot size is proportional to the percentage of cells expressing the identity genes. c, Compositional changes of neurons, astrocytes, microglia in AD Neu/AC/iMGL and CTRL cultures (P-values were calculated from the Ward test and the FDR indicates the adjusted p-values given by Benjamini & Hochberg method). d, Compositional changes of neurons, astrocytes, microglia and T cells in AD Neu/AC/iMGL (+/−) T cell and CTRL conditions (P-values were calculated from the Ward test and the FDR indicates the adjusted p-values given by Benjamini & Hochberg method). e, UMAP visualization of T cell-type composition of the PiChip cultures by single-cell transcriptome profiles. Distinct subtypes of microglia (homeostatic, DAM, MHCII, interferon and proliferative) are depicted with different colors. f, Compositional changes of homeostatic, DAM, MHCII, interferon and proliferative microglia in AD vs CTRL conditions (P-values were calculated from the Ward test and the FDR indicates the adjusted p-values given by Benjamini & Hochberg method).
Extended Data Fig. 6 |. Interferon-associated pathways are enriched in glial cells following infiltration of T cells in AD.
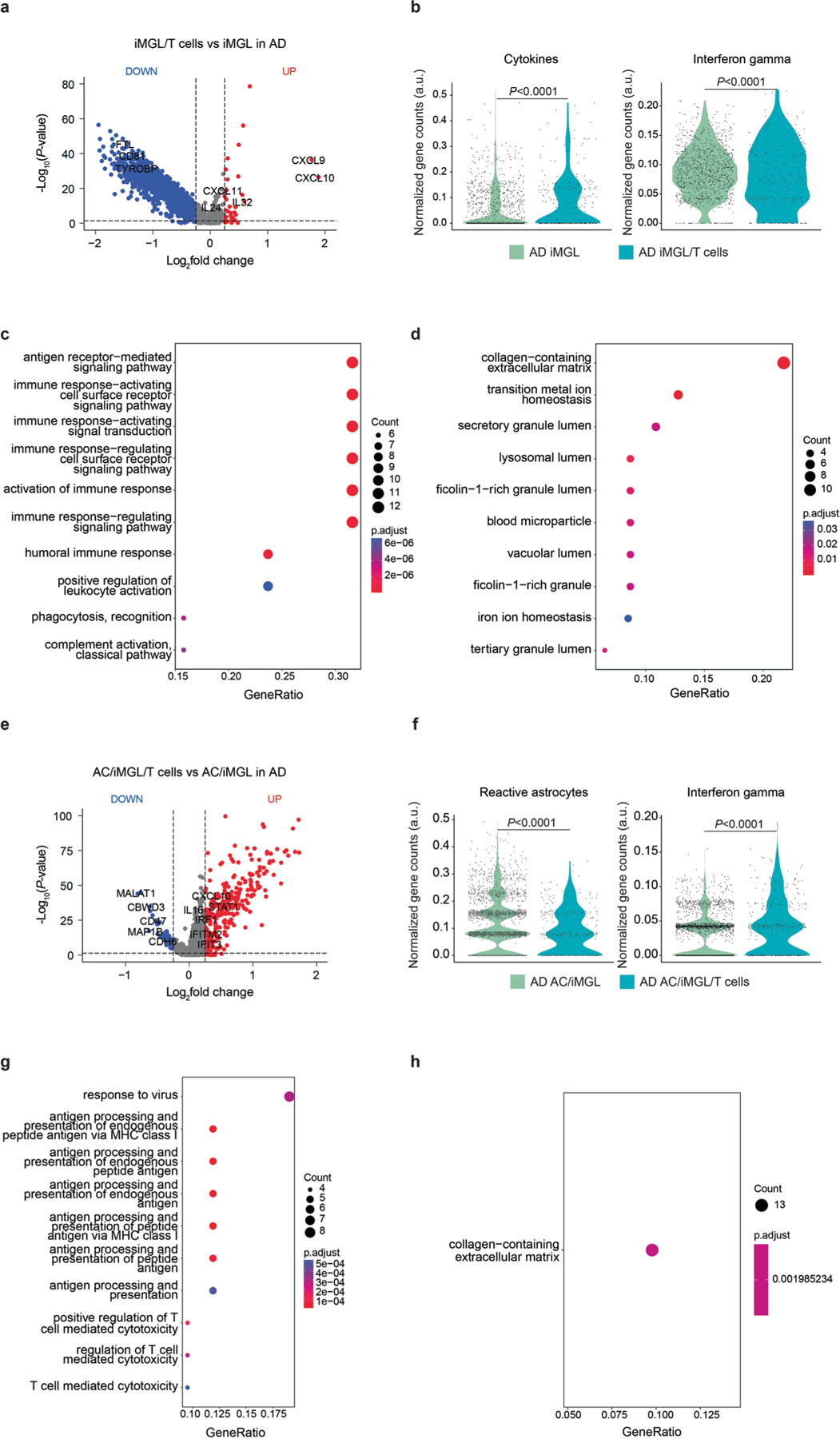
a, Volcano plot of differentially expressed genes in microglia in the presence of infiltrating T cells in AD cultures compared to AD cultures lacking T cells. Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown in red and blue dots, respectively (P-values from Wilcoxon Rank Sum test). b, Violin plots of highly enriched genes associated with cytokines (ARF1, CXCL9, CXCL10, CXCL16, CXCL8, CXCL14, CXCL5, IL24, IL32, IL17RA, IL1B, IL10, IL21R, IL18, IL6R, IL7R) and interferon-associated genes (NR1H3, CDC37, OTOP1, HCK, HPX, IRGM, IFNG, IFNGR1, IFNGR2, IRF1, JAK1, JAK2, ARG1, PARP14, PPARG, MED1, PTPN2, SP100, STAT1, TP53, TXK, NR1H2, PARP9, NLRC5, SOCS1, NMI) in microglia in the presence of infiltrating T cells in AD compared to AD cultures lacking T cells (P-values from nonparametric, two-sided, Mann-Whitney test). c, d, Gene ontology of significantly upregulated (c) and downregulated (d) pathways in microglia in the presence of infiltrating T cells in AD cultures vs AD cultures lacking T cells. Color scale indicates the adjusted P-value for significantly enriched pathways in microglia, and dot size is proportional to the count of genes (P-values were calculated from a Fisher exact test and the adjusted p-values given by Benjamini & Hochberg method). e, Volcano plot of differentially expressed genes in astrocytes in the presence of microglia and infiltrating T cells in AD compared to AD cultures lacking T cells. Significantly upregulated (P < 0.05 and FC > 0.1) and downregulated (P < 0.05 and FC < 0.1) genes are shown in red and blue dots, respectively. f, Violin plots of highly enriched genes associated with reactive astrocytes (GFAP, ALDOC, FABP7, TSPO, CRYAB, HSPB1, C3, CHI3L1, NTRK2, S100B, SOX9, STAT3) and interferon-associated genes (NR1H3, CDC37, OTOP1, HCK, HPX, IRGM, IFNG, IFNGR1, IFNGR2, IRF1, JAK1, JAK2, ARG1, PARP14, PPARG, MED1, PTPN2, SP100, STAT1, TP53, TXK, NR1H2, PARP9, NLRC5, SOCS1, NMI) in astrocytes in the presence of microglia and infiltrating T cells in AD compared to AD cultures lacking T cells (P-values from nonparametric, two-sided, Mann-Whitney test). g, h, Gene ontology of significantly upregulated (g) and downregulated (h) pathways in astrocytes in the presence of microglia and infiltrating T cells in AD compared to AD cultures lacking T cells. Color scale indicates the adjusted P-value for significantly enriched pathways in microglia, and dot size is proportional to the count of genes (P-values were calculated from a Fisher exact test and the adjusted p-values given by Benjamini & Hochberg method).
Extended Data Fig. 7 |. NicheNet analysis of upstream ligand-receptor pairs inducing the DE genes of neurons, astrocytes, and microglia upon T-cell infiltration.
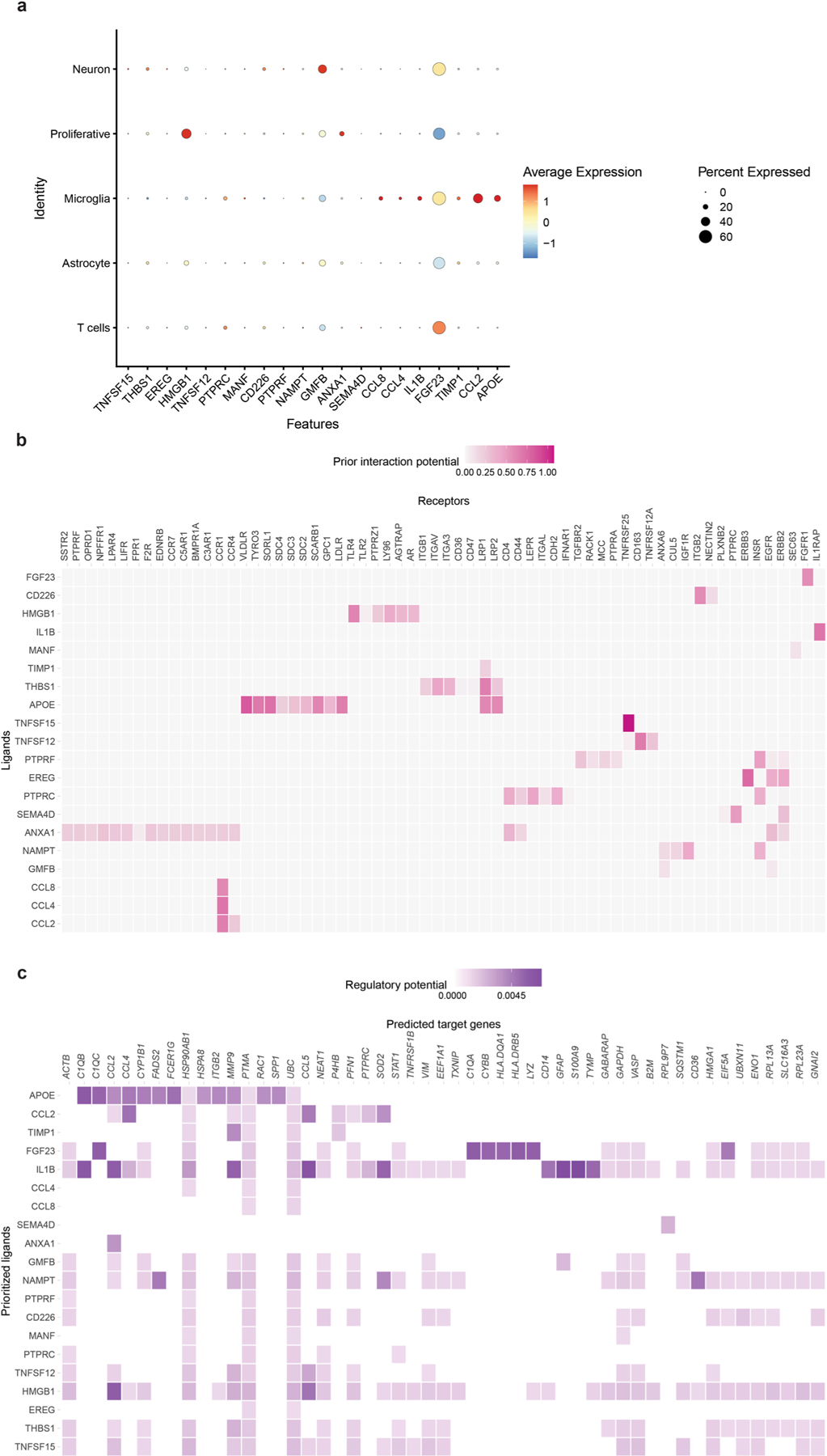
a, Dot plot of identity genes for distinct cell types. Color scale (a.u.) indicates the average expression of identity genes in each cell population, and the dot size is proportional to the percentage of cells expressing the identity genes. b, NicheNet’s ligand–target analysis represents potential upstream receptors expressed by neurons, astrocytes, microglia, and T cells associated with the top 20 potential ligands. c, Predicted target genes of the top 20 of potential ligands in AD Neu/AC/iMGL/T-cell cultures vs CTRL.
Extended Data Fig. 8 |. Increased level of pro-inflammatory cytokines in the presence of CD8+ T cells and microglia in AD cultures.
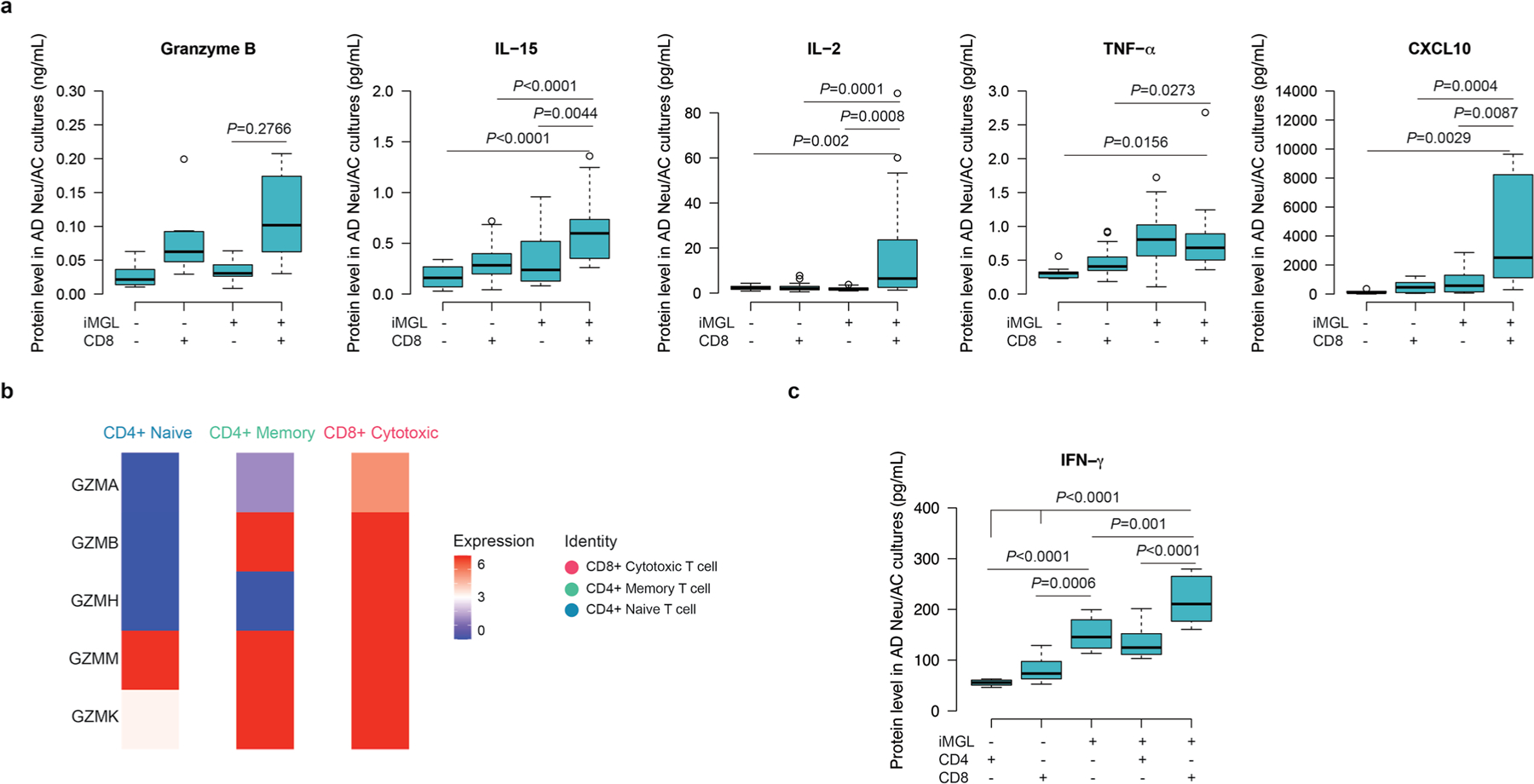
a, Quantification of granzyme B, IL-15, IL-2, TFN-α, and CXCL10 in the AD cultures (+/−) CD8+ T cells and iMGL, as measured by MSD assay (n = 6–30 biological replicates from n = 7–10 independent experiments; P-value from two-way ANOVA with Tukey multiple comparisons test). b, Enrichment of major cytotoxic genes (GZMA, GZMB, GZMH, GZMM, and GZMK) in CD4+ naïve, CD4+ memory, and CD8+ cytotoxic T cells in the AD cultures in the presence of microglia. Color scale (a.u.) indicates the expression of identity genes in each cell population. c, Quantification of INF-γ levels in the AD Neu/AC (+/−) iMGL and CD4+ or CD8+ T cells using ELISA assay (n=8 biological replicates; P-value from two-way ANOVA with Tukey multiple comparisons test). (a, c) In boxplots, the center lines show the medians; box limits indicate the 25th and 75th percentiles; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots.
Supplementary Material
Acknowledgements
This work was supported by the Cure Alzheimer’s Fund and the JPB Foundation. All microfabrication procedures of the PiChip platform were conducted at the BioMEMS Resource center. All illustrations, except the 3D microfluidic platform, were created with Biorender (https://www.biorender.com). The scRNA-seq computations in this paper were, in part, run on the FASRC Cannon cluster supported by the FAS Division of Science Research Computing Group at Harvard University.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-023-01415-3.
Code availability
The code for analysis and generating scRNA-seq figures can be found at https://github.com/jospark09/PiChip and https://zenodo.org/record/8150286.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41593-023-01415-3.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-023-01415-3.
Peer review information Nature Neuroscience thanks Julia TCW and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Data availability
The authors declare that all data supporting the findings of the present study are available within the paper, Supplementary Information files or publicly available. RNA-seq data have been deposited to the National Center for Biotechnology Information (GSE239509). The scRNA-seq was performed in five independent batches. All the relevant raw data (fastq files)/sparse matrix/R objective files are available at https://github.com/jospark09/PiChip. Unfiltered all-condition-combined list deposited; PiChip.list.ordered.Not.filtered.rds. Filtered Seurat cell type annotated R objective files deposited; PiChip.filtered_seurat.cell.type.annotated.rds. Fig. 1 and Extended Data Fig. 1e objective files deposited; PiChip.Fig.1 and extended Fig.1.rds. Microglia only re-clustered and subcluster-annotated file deposited; Mic.only.type.annotated.Fig.4.rds. T cell only re-clustered and subclustered-annotated file deposited; T.only.type.annotated.Fig.4.rds. Further information on research design is available in the Nature Research Reporting Summary linked to this article. Source data are provided with this paper.
References
- 1.Jorfi M, Maaser-Hecker A & Tanzi RE The neuroimmune axis of Alzheimer’s disease. Genome Med. 15, 6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bettcher BM, Tansey MG, Dorothée G & Heneka MT Peripheral and central immune system crosstalk in Alzheimer disease—a research prospectus. Nat. Rev. Neurol. 17, 689–701 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arlehamn CSL, Garretti F, Sulzer D & Sette A Roles for the adaptive immune system in Parkinson’s and Alzheimer’s diseases. Curr. Opin. Immunol. 59, 115–120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu K-M et al. The role of the immune system in Alzheimer’s disease. Ageing Res. Rev. 70, 101409 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Cao W & Zheng H Peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegen. 13, 51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers J, Luber-Narod J, Styren SD & Civin WH Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 9, 339–349 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Itagaki S, McGeer PL & Akiyama H Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci. Lett. 91, 259–264 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Togo T et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 124, 83–92 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Merlini M, Kirabali T, Kulic L, Nitsch RM & Ferretti MT Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener. Dis. 18, 49–56 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Unger MS et al. CD8+ T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 89, 67–86 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Gate D et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferretti MT et al. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 54, 211–225 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Browne TC et al. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 190, 2241–2251 (2013). [DOI] [PubMed] [Google Scholar]
- 14.MacPherson KP et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla AK et al. CD11a expression distinguishes infiltrating myeloid cells from plaque‐associated microglia in Alzheimer’s disease. Glia 67, 844–856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monsonego A et al. Aβ-induced meningoencephalitis is IFN-γ-dependent and is associated with T cell-dependent clearance of Aβ in a mouse model of Alzheimer’s disease. Proc. Natl Acad. Sci. USA 103, 5048–5053 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machhi J et al. CD4+ effector T cells accelerate Alzheimer’s disease in mice. J. Neuroinflamm. 18, 272 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietronigro E et al. Blockade of α4 integrins reduces leukocyte–endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 9, 12055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gate D et al. CD4+ T cells contribute to neurodegeneration in Lewy body dementia. Science 374, 868–874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wareham LK et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol. Neurodegen. 17, 23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Götz J, Bodea L-G & Goedert M Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 19, 583–598 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Scearce-Levie K, Sanchez PE & Lewcock JW Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov. 19, 1–16 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Blanchard JW, Victor MB & Tsai L-H Dissecting the complexities of Alzheimer disease with in vitro models of the human brain. Nat. Rev. Neurol. 18, 25–39 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Choi SH, Kim YH, Quinti L, Tanzi RE & Kim DY 3D culture models of Alzheimer’s disease: a road map to a ‘cure-in-a-dish’. Mol. Neurodegen. 11, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SH et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim YH et al. A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat. Protoc. 10, 985–1006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwak SS et al. Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat. Commun. 11, 1377 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abud EM et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McQuade A et al. Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol. Neurodegen. 13, 67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doorn KJ et al. Microglia in olfactory bulb of AD and PD patients. Brain Pathol. 24, 152–165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson KG et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Browaeys R, Saelens W & Saeys Y NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Takata K et al. Role of high mobility group protein-1 (HMG1) in amyloid-β homeostasis. Biochem Biophys. Res. Commun. 301, 699–703 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Smit MJ et al. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 102, 1959–1965 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Lund H et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 9, 4845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SM, Hudobenko J, McCullough LD & Chauhan A Microglia depletion increase brain injury after acute ischemic stroke in aged mice. Exp. Neurol. 336, 113530 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laurent C et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140, 184–200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu L et al. T‐cell infiltration, contribution and regulation in the central nervous system post‐traumatic injury. Cell Prolifer. 54, e13092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaya T et al. CD8+ T cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci. 25, 1446–1457 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schulz O, Hammerschmidt SI, Moschovakis GL & Förster R Chemokines and chemokine receptors in lymphoid tissue dynamics. Annu. Rev. Immunol. 34, 1–40 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Sokol CL & Luster AD The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 7, a016303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuena AR, Casolini P, Lattanzi R & Maftei D Chemokines in Alzheimer’s disease: new insights Into prokineticins, chemokine-like proteins. Front. Pharm. 10, 622 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galimberti D et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 63, 538–543 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Xia MQ, Bacskai BJ, Knowles RB, Qin SX & Hyman BT Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J. Neuroimmunol. 108, 227–235 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Wojcieszak J, Kuczyńska K & Zawilska JB Role of chemokines in the development and progression of Alzheimer’s disease. J. Mol. Neurosci. 72, 1929–1951 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaheer S et al. Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer’s disease mice. Neurochem. Res. 38, 218–225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duan R-S et al. Decreased fractalkine and increased IP-10 expression in aged brain of APPswe transgenic mice. Neurochem. Res. 33, 1085–1089 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Groom JR & Luster AD CXCR3 in T cell function. Exp. Cell Res. 317, 620–631 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurachi M et al. Chemokine receptor CXCR3 facilitates CD8+ T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 208, 1605–1620 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krummel MF, Bartumeus F & Gérard A T cell migration, search strategies and mechanisms. Nat. Rev. Immunol. 16, 193–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N & Prlic M CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat. Commun. 10, 4987 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krauthausen M et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. J. Clin. Invest. 125, 365–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ozga AJ et al. CXCL10 chemokine regulates heterogeneity of the CD8+ T cell response and viral set point during chronic infection. Immunity 55, 82–97.e8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gericke C et al. Early β‐amyloid accumulation in the brain is associated with peripheral T cell alterations. Alzheimer’s Dement. 10.1002/alz.13136 (2023). [DOI] [PubMed] [Google Scholar]
- 56.Piehl N et al. Cerebrospinal fluid immune dysregulation during healthy brain aging and cognitive impairment. Cell 185, 5028–5039 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hao Y et al. Dictionary learning for integrative, multimodal, and scalable single-cell analysis. Preprint at Nat. Biotech. 10.1038/s41587-023-01767-y (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of the present study are available within the paper, Supplementary Information files or publicly available. RNA-seq data have been deposited to the National Center for Biotechnology Information (GSE239509). The scRNA-seq was performed in five independent batches. All the relevant raw data (fastq files)/sparse matrix/R objective files are available at https://github.com/jospark09/PiChip. Unfiltered all-condition-combined list deposited; PiChip.list.ordered.Not.filtered.rds. Filtered Seurat cell type annotated R objective files deposited; PiChip.filtered_seurat.cell.type.annotated.rds. Fig. 1 and Extended Data Fig. 1e objective files deposited; PiChip.Fig.1 and extended Fig.1.rds. Microglia only re-clustered and subcluster-annotated file deposited; Mic.only.type.annotated.Fig.4.rds. T cell only re-clustered and subclustered-annotated file deposited; T.only.type.annotated.Fig.4.rds. Further information on research design is available in the Nature Research Reporting Summary linked to this article. Source data are provided with this paper.