

SUMMARY
When compared to other malignancies, the tumor microenvironment (TME) of primary and castration-resistant prostate cancer (CRPC) is relatively devoid of immune infiltrates. While androgen deprivation therapy (ADT) induces a complex immune infiltrate in localized prostate cancer, the composition of the TME in metastatic castration-sensitive prostate cancer (mCSPC), and the effects of ADT and other treatments in this context are poorly understood. Here, we perform a comprehensive single-cell RNA sequencing (scRNA-seq) profiling of metastatic sites from patients participating in a phase 2 clinical trial (NCT03951831) that evaluated standard-of-care chemo-hormonal therapy combined with anti-PD-1 immunotherapy. We perform a longitudinal, protein activity-based analysis of TME subpopulations, revealing immune subpopulations conserved across multiple metastatic sites. We also observe dynamic changes in these immune subpopulations in response to treatment and a correlation with clinical outcomes. Our study uncovers a therapy-resistant, transcriptionally distinct tumor subpopulation that expands in cell number in treatment-refractory patients.
Graphical Abstract
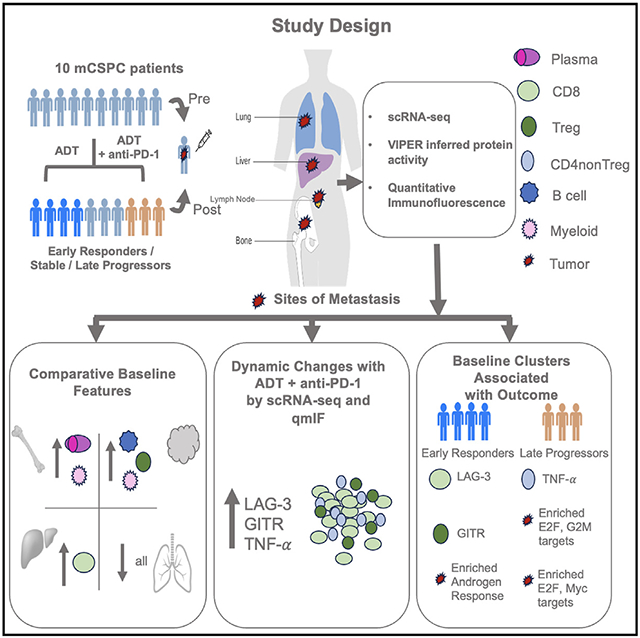
In brief
TME composition of metastatic castration-sensitive prostate cancer (mCSPC) remains elusive. Hawley et al. perform scRNA-seq analysis of metastatic sites from mCSPC patients with ADT and anti-PD-1 treatment, identify tumor and immune subpopulations conserved across multiple metastatic sites, and reveal treatment-induced dynamic changes in these subpopulations and their correlation with clinical outcomes.
INTRODUCTION
Compared to other tumor types, localized prostate cancer (PC) is characterized by an immunologically “cold” tumor microenvironment (TME), with a relative dearth of immune cells.1,2 Preclinical studies in animal models and analyses of human primary PC samples showed that tumor infiltrating immune cells are tolerogenic and immunosuppressive, as revealed by a dominant fraction of terminally differentiated cytotoxic T cells and regulatory T cells (Treg).3-5 Several studies have shown that androgen deprivation therapy (ADT), the mainstay therapy for advanced prostate cancer, induces immunogenic changes in the TME of castration-sensitive prostate cancer.6-9 Post-ADT immune infiltration may involve several mechanisms, including (a) thymic regeneration and increased production of naive T cells, (b) decreased tolerance and clonal expansion of effector T cells, and (c) stimulation of antigen-specific adaptive immune response. The latter induces a robust chemokine-mediated infiltration of functionally competent immune infiltrate into primary prostate tumors.8-19 Unfortunately, these immunogenic anti-tumor effects are not durable, as they are likely subsequently counter-balanced by concomitant increase in immunosuppressive cell subpopulations and decreases in T cell priming.13,20-22 These data provide a reasonable therapeutic rationale to combine ADT with immunotherapy, thus leveraging anti-tumor immunity while mitigating the ADT-mediated induction of suppressive mechanisms. To that end, we designed and initiated the PRIME-CUT Phase II trial (NCT03951831) treating men with metastatic castration-sensitive prostate cancer (mCSPC) with a combination of standard of care chemo-hormonal therapy and anti-PD-1 immunotherapy using the anti-PD-1 immunotherapy cemiplimab.
This trial addresses the knowledge gap that the majority of preclinical and clinical studies thus far focused on the immunogenic effects of ADT in primary prostate cancer. By contrast, the TME of metastatic, castration-sensitive prostate tumors and the effects of ADT-mediated remodeling remain poorly characterized. This knowledge gap stems from challenges associated with longitudinal tissue acquisition from metastatic castration-sensitive tumor biopsies, particularly in the pre-treatment (pre-ADT) context. In particular, whether the TME of treatment-naive metastatic sites is “cold”, i.e., similar to primary prostate cancer, is largely unknown. Indeed, it is possible that metastatic lesions might be immunologically distinct; as an analysis of PD-L1 protein expression showed that 31.6% of metastatic PC lesions had detectable PD-L1 expression, in contrast to only 7.7% of primary lesions.23 By contrast, spatial imaging and single-cell sequencing studies showed a paucity of immune infiltrates in the metastatic, castration-resistant prostate cancer (mCRPC) setting, similar to primary PC.24,25
Here, we comprehensively profiled both the tumor and non-tumor subpopulations that comprise the mCSPC TME, across distinct metastatic niches—including bone, lymph node, liver, and lung. These analyses leveraged our established pipeline for virtual inference of protein activity by enriched regulons (VIPER)26-28 to study single-cell RNA sequencing (scRNA-seq) profiles of single cells dissociated from these niches. In this approach, akin to a highly multiplexed gene reporter assay, the differential activity of each protein is quantified based on the differential expression of its lineage-specific downstream transcriptional targets. This methodology allows for full activity quantification of ~6,500 proteins in each single cell. Proteins analyzed include transcriptional regulators and signaling proteins, despite the fact that >80% of the genes are generally undetected in scRNA-seq due to the gene dropout effect. Critically, we previously showed that VIPER-based protein activity measurements in single cells compare favorably with protein-level antibody-based assays, revealing clinically relevant subpopulations that could not have been detected by gene expression analysis or fluorescence-activated cell sorting (FACS).27-29
Thus, protein activity-based cluster analysis allows for deep stratification of immune and tumor-related subpopulations, virtually eliminating gene dropout for key regulatory and signaling proteins, including critical lineage markers.27-29 Here, we applied this methodology to a series of paired, longitudinal, metastatic tumor biopsies, including both pre-treatment (baseline) and on-treatment biopsies from ten patients enrolled in the PRIME-CUT Phase 2 clinical trial. In addition to characterizing differences in TME composition across patients, we also quantified dynamic changes in the TME following ADT, both alone and in combination with anti-PD-1. In addition, we tested whether TME subpopulations at baseline correlate with early prostate-specific antigen (PSA) response. To corroborate profiling data on the immunological composition of the TME, we performed multiplex immunofluorescence staining of a subset of banked FFPE trial biospecimens29,30
RESULTS
Gene expression and protein activity clustering reveal a robust immune infiltrate in metastatic castration-sensitive prostate cancer
Since primary prostate cancer (PC) is characterized by a relative immune desert, with low representation of tumor-infiltrating immune cell subpopulations,1,2 we tested whether the tumor microenvironment (TME) of metastatic, castration-sensitive prostate cancer (mCSPC) lesions was similarly immunologically “cold”. We thus collected pre-treatment (baseline) metastatic core-needle biopsies from 8 patients (Figure 1A, Table 1), across 4 different metastatic niches (bone, lymph nodes, liver, and lung). We then generated scRNA-seq profiles, which were first used to perform standard gene expression-based cluster analysis (Figures S1 and S2). Cell lineages were inferred for each single-cell using the SingleR algorithm,30 with clustering performed using resolution-optimized Louvain.28 Standard gene expression-based clustering revealed 15 overall clusters across all metastatic sites, corresponding to 12 distinct immune cell subpopulations, as well as clusters comprising fibroblasts, endothelial, and epithelial cells (Figure S1). Inspection of the top five most differentially upregulated genes in each cluster (Figure S2) was consistent with the ascribed cellular identities assigned by SingleR. For example, granzyme M (GZMM) and natural killer granule 7 (NKG7) were differentially upregulated in CD8+ T cells, and CD37 in B cells (Figure S2). SingleR does not provide a classification of tumor vs. normal cells; as a result, tumor cells with an epithelial origin, such as prostate cancer cells, were labeled as “epithelial cells” (Figure S1). Tumor-cell identity was confirmed based on the expression of prostate-restricted tumor marker genes, such as KLK3, and by inferred copy number variation analysis using lymphoid and myeloid cells as copy-number normal reference, such that endothelial cells were also confirmed as a negative control not to have significant inferred copy number variation (Figure S3).
Figure 1. Baseline Composition of Micro-Environment by Tissue Site.

(A) Phase 2 trial design schema.
(B) Uniform manifold projection (UMAP) plot constructed from VIPER-inferred protein activity of all cells in aggregate across baseline pre-treatment patient samples. Cells are clustered by resolution-optimized Louvain algorithm with cell type inferred by SingleR.
(C) Stacked barplot of the frequency of each major cell lineage within each baseline patient sample, with each column representing a unique patient and patients grouped by metastatic site. Cell clusters from B are aggregated by shared cell type.
(D) Stacked barplot of immune vs. non-immune cell frequencies, from C.
(E) Boxplot showing distribution of frequencies for each cell cluster in B at baseline, comparing tissue sites. Also see Figure S1.
Table 1.
Clinical characteristics and cellular yield per biopsy sample
| Patient | Age | Race | Ethnicity | Baseline PSA (ng/dL) |
Biopsy Location |
Baseline (no. cells) |
ADT only (no. cells) |
ADT + anti- PD-1 (no. cells) |
Recurrence (no. cells) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 62 | White | Not Hispanic or Latino | 11.56 | Bone | 2360 | NA | 686 | NA |
| 3 | 65 | White | Not Hispanic or Latino | 156.4 | Bone | 1027 | 995 | NA | 764 |
| 5 | 82 | Black | Other Hispanic or Latino | 26.2 | LN | 4090 | 5153 | NA | NA |
| 6 | 52 | White | Dominican | 11.93 | Liver | 0 | NA | 2475 | NA |
| 7 | 60 | Other | Dominican | 146.7 | Lung | 699 | NA | 1956 | NA |
| 8 | 49 | White | Not Hispanic or Latino | 91.93 | Bone | 606 | 1784 | NA | NA |
| 10 | 76 | White | Dominican | >5000 | Bone | 521 | NA | 97 | NA |
| 12 | 65 | Black | Dominican | 95.05 | Bone | 0 | 913 | NA | NA |
| 13 | 54 | Other | Dominican | 229.9 | LN | 1212 | NA | 0 | NA |
| 14 | 63 | Black | Not indicated | 90.71 | Liver | 2161 | 0 | NA | NA |
LN, lymph node; no., number; NA, not applicable; PSA, prostate specific antigen.
Due to high gene dropout levels, scRNA-seq analyses can typically monitor ~20% of all genes in each cell.28 To address this issue, we utilized an established single cell analysis pipeline, which leverages the VIPER algorithm to measure protein activity from single-cell gene expression data. By assessing protein activity based on the expression of 100 transcriptional targets of each protein, VIPER significantly mitigates gene dropout effects, thus allowing detection of proteins whose encoding gene is completely undetected.28 Protein activity-based clustering of these scRNA-seq data revealed a much finer-grain subpopulation structure, comprising 24 distinct subpopulations, comprised mainly of immune cells but also including erythrocytes, endothelial, and three molecularly distinct transformed epithelial cell clusters (tumor cells) (Figure 1B). In particular, while gene expression-based cluster analysis yielded a single homogeneous subpopulation of monocytes and macrophages (Figures S1 and S2), activity-based analysis stratified these cells into five subpopulations, representing three distinct monocyte subtypes, as well as macrophages and neutrophils (Figures 1B and 2). There was also further refinement of T cell populations, resulting in the identification of subpopulations representing regulatory T cells (Treg) and CD8+ T cells (Figures 1B and 2). Subpopulations comprising five B cell subtypes and one plasma cell subtype were also identified using VIPER analysis (Figures 1B and 2), as compared to only one and two subtypes, respectively, by standard gene expression methods (Figures S1 and S2). Critically, VIPER analysis identified three distinct clusters of transformed epithelial cells (Figures 1B and 2), while only a single cluster was identified using gene expression analysis (Figures S1 and S2). This is critical as these clusters may have different mechanisms of drug sensitivity.
Figure 2. Top Protein Activity Cell Cluster Markers.

Heatmap of top 5 most differentially activated proteins for each cell type cluster from aggregate scRNA-seq data across all patient samples. Each row represents a protein, grouped by cluster in which they are the most active, with cluster labels on the x and y axes. Each column represents a single cell. Above the x axis cluster label there is also a treatment label indicating time point at which a given cell was profiled. Also see Figure S2.
Overall, the mean proportion of immune cells across all metastatic sites was 87% (range: 30.9% [lung] – 94.6% [lymph node]) significantly exceeding the sparse immune infiltration previously reported for primary prostate cancer.5,31 While surprising, these data are broadly consistent with prior warm-autopsy findings in advanced castrate-resistant prostate cancer,32 where CD14+/CD206+ macrophage proportions ranged from 10 to 50% in metastatic lesions of the lymph node, dura mater, liver, bone marrow, and adrenal glands. Although cryopreservation used for tissue processing has been shown to deplete epithelial cells, leading to potential methodological bias, no such bias has been previously noted from freshly dissociated tissue, as collected in this study.33 Further, since the primary goal of this study was to assess relative differences in the composition of the TME across tissue contexts and in response to treatment, albeit small numbers of samples, any artifactual depletion of non-immune cells would be expected to be consistent across samples, thus preserving the validity of subsequent inter-sample comparisons.
Protein activity analyses show distinct differences in immune cell subpopulations across metastatic sites
Prior studies showed that proportional representation of immune cell subtypes in the TME varies broadly, depending on tissue type.34,35 To compare the subtype composition of the castration-sensitive prostate cancer TME across different metastatic sites, in our relatively small dataset, we collapsed the initial 24 VIPER clusters (Figure 2) into eight lineage-specific meta-clusters, including B cells, CD4+ non-Treg (Tconv), CD8+ T cells, endothelial, erythrocyte, myeloid, Treg, and tumor cells (Figure 1C). We visualized the relative representation of these coarse-grain meta-clusters across the four metastatic niches sampled, prior to ADT or anti-PD-1 (Figures 1C and 1D). The baseline biopsy from the lung sample was comprised mostly of tumor cells, with associated CD4+ T cells, CD8+ T cells, and myeloid populations representing only 30.9% of all cells (Figure 1C). By contrast, the liver biopsies samples contained a greater proportion of immune cells, representing 77.9% of all cells, on average (Figures 1C and 1D). Immune cells comprised 94.6% of all cells in the lymph node (LN) samples (Figures 1C and 1D); perhaps unsurprisingly since LN are components of the primary immune system. Finally, and also consistent with their niche composition, bone lesions were also highly enriched in immune cells, which represented 90.5% of all cells on average (Figures 1C and 1D).
We next compared the frequency of the 24 different cellular subpopulations (Figure 2) across the four different metastatic sites sampled (bone, lymph node, liver, and lung) (Figure 1E). In bone metastases, plasma cells were highly enriched relative to other sites (p ≤ 0.05, Figure 1E). Additionally, there was an increased representation of monocyte 1 and 2 subtypes relative to other metastatic sites (Figure 1E), with increased activity of transcriptional repressors (e.g., BATF3), transcriptional activators (e.g., SH3BP2), regulators of G-protein signaling (e.g., RGS18), and serine proteases (e.g., PRTN3) (Figure 2). We also noted significant over-representation of erythrocytes in bone metastases (p ≤ 0.05, Figure 1E) with high activity of: (1) epithelial cell transforming 2 (ECT2), (2) Rho GTPase-activating protein 11A (ARHGAP11A), and (3) kinesis family member 14 (KIF14) (Figure 2); these have established roles in mitosis, cell-cycle arrest, and microtubule motor proteins, respectively.36-38 These data may represent a population of dividing erythroid progenitor cells captured incidentally during bone marrow biopsy. In lymph node samples, a robust B cell population was detected (B cell 2) (Figure 1E). There was also an increased proportion of Treg (Treg 3), with elevated activity of TNFSRF18 (GITR) in the lymph nodes as compared to other metastatic sites (p ≤ 0.05, Figure 1E). Of interest, this T regulatory population (Treg 3) showed high activity levels of ETS variant transcription factor 1 (ETV1) (p ≤ 0.05, Figure 2), a gene known to be overexpressed in prostate cancer.39,40 This finding supports recent data showing that immune cells may mimic expression of tumor marker genes.41 Liver metastases had immune infiltrations similar to bone metastases, in both overall proportion (Figure 1D) and subpopulation frequency (Figure 1E). Notably across all tissues there was a large proportion of CD8+ T cells (CD8+ T cell 1 and 2) and conventional (non Treg) CD4+ cells (CD4+ T cell 1) (Figure 1E). The CD8+ T cell 2 cluster was primarily defined by increased protein activity of lymphocyte activation gene 3 protein (LAG-3), an immune checkpoint molecule (Figure 2).42 Finally, the single lung metastasis profiled was notably the least immune-infiltrated at baseline (Figure 1D), with only 30.9% immune cells overall. Taken together, these data highlight immunological heterogeneity across sites of metastasis in castration-sensitive PC. These analyses are based on 40,270 high-quality cells across 11 patients.
Combination treatment of ADT andanti-PD-1 results in a significant expansion of CD8+ T cells across several metastatic sites
To understand the immunologic effects of ADT on the TME of the four different metastatic sites, either alone or in combination with anti-PD-1 (cemiplimab), we compared the pre-vs. post-treatment frequency of each subpopulation across the four metastatic sites (Figure 3). As mentioned previously, all patients on trial were required to have a baseline metastatic biopsy, as well as an on-treatment biopsy, with patients randomized to one of two time points for the on-treatment biopsy, at either four weeks after ADT (degarelix) or after ADT plus two cycles of anti-PD1. Overall, sufficient patients with bone and lymph node metastases were enrolled to enable collection of biopsy samples at baseline and at both on-treatment time points. Additionally, we were also able to obtain a tumor progression biopsy from a patient with late tumor recurrence in the bone, after 11 months on treatment. The liver and lung metastatic biopsy samples shown here were collected at baseline and after ADT with two cycles of anti-PD-1 (Figures 3E-3H). No samples of liver and lung metastases were collected after ADT alone given the randomization procedures based on patients’ order of enrollment.
Figure 3. Treatment with ADT+anti-PD-1 Induces Dramatic Changes in the Tumor Micro-Environment.

(A) UMAP plot of all cells from patients with metastatic bone lesions, split by treatment time point (Baseline, ADT-only, ADT+anti-PD-1, and post-treatment recurrence) and labeled by cell cluster.
(B) Stacked barplot showing the relative frequency of each major cell lineage by treatment time point for patients with metastatic Bone lesions, with each column representing aggregate of all samples profiled at a specific treatment time point.
(C) UMAP plot, as in A, for patients with metastatic lymph node lesions.
(D) Stacked barplot, as in B, for patients with metastatic lymph node lesions.
(E) UMAP plot, as in A, for patients with metastatic liver lesions.
(F) Stacked barplot, as in B, for patients with metastatic liver lesions.
(G) UMAP plot, as in A, for patients with metastatic lung lesions.
(H) Stacked barplot, as in B, for patients with metastatic lung lesions.
(I) Boxplot showing distribution of frequencies for each cell cluster, comparing frequencies across treatment time points including baseline, ADT-only, and ADT+anti-PD-1. Also see Figure S4.
We next leveraged pre- and on-treatment scRNA-seq profiles to determine the effects of ADT or ADT plus anti-PD-1-on the castrate-sensitive prostate cancer TME. Using the coarser meta-cluster representation mentioned previously (B cells, CD4+ non-Tregs, Tregs, CD8+ T cells, myeloid cells, endothelial cells, erythrocytes, and tumor cells), we quantified treatment-mediated changes at each metastatic site (Figures 3B, 3D, 3F, and 3H). In bone metastases, ADT induced increased representation of myeloid cells (p = 2.8e-118 by Fisher’s exact test), consistent with pre-clinical data13 (Figures 3A and 3B). In bone lesions, ADT was also associated with a decrease in CD4+ Tconv as well as in tumor cells (p = 0.01 and 1.1e-15) (Figures 3A and 3B). By contrast, the ADT and anti-PD-1 combination reduced myeloid cell representation (p = 3.6e-10) while concomitantly increasing CD8+ T cells (p = 3.1e-115) (Figures 3A and 3B). In the lone progression biopsy obtained from a bone lesion, the relative subpopulation frequencies resembled that of the baseline samples, albeit with a greater proportion of tumor cells (Figures 3A and 3B). Finally, in lymph node-derived samples, ADT induced an expansion of CD4+ non-Treg (p = 5.3e-36) (Figures 3C and 3D) These findings are consistent with the notion that treatment-induced immunologic changes vary based on the metastatic niche, although confirmation in additional datasets of pre- and on-treatment metastatic biopsies in hormone-sensitive prostate cancer are warranted. As external validation of these trends, we leveraged the only previously existing transcriptional dataset profiling metastatic castrate-sensitive tumors, to our knowledge, which includes bulk RNA sequencing of 17 hormone-naive and 21 short-term castrated samples, in bone only.43 We applied the CIBERSORTx algorithm to infer relative abundance of each cell type in these bulk data (Figure S4), such that ADT was found to significantly increase myeloid infiltrate (p = 0.028) from pre-treatment mean abundance of 7.01% to post-treatment mean of 10.89%, with a non-significant trend toward decrease in CD non-Treg (p = 0.37) and in tumor cells (p = 0.14).
Few cells were recovered from lymph nodes after combination treatment with ADT and anti-PD-1. However, the recovered cells showed a greater proportion of Treg (p = 0.002) and myeloid cells (p = 1.4e-8), with virtually no representation of CD8+ T cells or B cells (Figures 3C and 3D). Surprisingly, in both the bone and lymph node samples, there was a relative increase in tumor cell composition following combination therapy with ADT and anti-PD-1, as compared to baseline and ADT-only samples (Figures 3A-3D). This was surprising, and contrasts with observations from the viscera (liver and lungs), where combination therapy was associated with a substantial reduction in overall tumor cell composition (p = 4.1e-119 in liver, p = 4.0e-222 in lungs) (Figures 3E-3H). Additionally, the myeloid compartment expansion observed in bone-derived samples (after ADT) and in lymph node-derived samples (after combination therapy) (Figures 3A-3D) was not detected in the viscera (liver and lungs) (Figures 3E-3H. In contrast, and similar to bone samples, combination therapy induced significant increase in CD8+ T cells in liver and lung-derived samples (p = 1.6e-29 and 7.5e-66, respectively), which was not observed with ADT alone. Taken together, these data show that anti-PD-1 increased CD8+ T cell infiltration into metastatic sites, when used in combination with ADT to an extent that was not observed with ADT alone.
We next focused on the finer-grain subpopulations detected by inferred protein activity analysis (Figure 2). Specifically, we assessed which subpopulations showed a treatment-related relative increase or decrease (Figure 3I). After ADT, the fraction of Cluster 2 CD8+ T cells decreased relative to baseline (p = 0.034 by Mann-Whitney U test), while all monocyte subtypes increased, across all metastatic sites (p = 0.036) (Figure 3I). In contrast, following combination therapy, we observed significant expansion of Cluster 1 CD4+ T cells (characterized by high TNF-α activity; Figure 2), Cluster 2 CD8+ T cells (characterized by high LAG-3 activity; Figure 2), and Cluster 3 Treg cells (characterized by high TNFRSF18 activity; Figure 2), across all metastatic sites (p = 0.033, 0.026, and 0.008, respectively) (Figure 3I). These three immune cell subpopulations represented the bulk of tumor-infiltrating immune cells whose composition is increased by anti-PD-1 therapy and are broadly consistent with reported effects of PD-1 blockade on the T cell compartment.44
Immune infiltration correlates with quantitative multiplex immunofluorescence analysis (QmIF)
To confirm the significant degree of immune infiltration observed by scRNA-seq profiling at the protein level, we performed quantitative multiplex immunofluorescence (qmIF) staining on banked FFPE tissue from a subset of six patient samples, including two baseline pre-treatment samples, two ADT-only samples, and two ADT+anti-PD-1 samples. Of these, paired pre-treatment baseline and on-treatment specimen were profiled for the same patient for one set of samples. The quantitative immunofluorescence panel used included markers designed to profile tumor cells (panCK), T cells (CD4+ or CD8+), Treg (CD8-CTLA-4+CD4+), myeloid cells (CD68/163+), and LAG-3+ CD8+ T cells. Representative slide sections highlighting differences in T cell infiltrate are shown in Figure 4A.
Figure 4. Immune Infiltration at Baseline and in Response to Treatment is Recapitulated by Immunofluorescence Analysis.

(A) Representative immunofluorescence staining images of samples with low T cell infiltration (top – a baseline lymph node slide section) and high T cell infiltration (bottom – an on-treatment liver slide section). Images on the left show PanCK tumor cell staining and images on the right additionally overlay CD4+ and CD8+ staining intensity.
(B) Comparison of cumulative T cell frequencies in paired specimens as determined by scRNA-seq (y axis) versus quantitative immunofluorescence (x axis), where pre-treatment samples are colored in red at lower T cell frequency and on-treatment samples are colored in blue at higher frequency. Linear regression line is plotted, with correlation value as shown and p value = 0.031.
(C) Comparison of cumulative tumor cell frequencies in paired specimens as determined by scRNA-seq (y axis) versus quantitative immunofluorescence (x axis), where pre-treatment samples are colored in red at higher frequency and on-treatment samples are colored in blue. Linear regression line is plotted, with correlation value as shown and p value = 0.026.
(D) Comparison of treatment-induced fold-change for a representative patient where baseline and on-treatment tissue was profiled by both scRNA-seq and quantitative immunofluorescence. Fold-change in each major cell population profiled by quantitative immunofluorescence is shown in blue, and fold-change by scRNA-seq is shown in orange. Fold-changes are broadly concordant across all cell populations between the two modalities, with statistically significant correlation across cell types (p value = 0.00442).
Importantly, the overall degree of T cell infiltration determined by scRNA-seq was significantly correlated with the degree of T cell infiltration by quantitative immunofluorescence (R2 = 0.7304, p = 0.031), such that T cell cumulative frequency ranged from 5 to 65% of total cell count by qmIF, versus 20–80% by scRNA-seq (Figure 4B). These data support the notion that the significant immune infiltrates determined by scRNA-seq were not an artifact of tissue dissociation. Further, immunofluorescence analysis confirmed the treatment-induced increase in immune infiltrate described previously (Figure 4B), as well as a treatment-induced reduction in tumor cell count (Figure 4C), with tumor cell frequencies similarly well-correlated between the two orthogonal tissue profiling modalities (R2 = 0.7454, p = 0.026).
Although immunofluorescence analysis is limited in the number of cell subtypes which can be simultaneously profiled, the treatment-induced fold-change in each cell population profiled by qmIF was consistent with scRNA-seq for paired analysis of baseline and on-treatment tissue in the same patient (Figure 4D). Specifically, combination therapy was shown to induce an increase in T cells (overall as well as for CD4+, CD8+, and LAG-3+ CD8+ T cells) (Figure 4B), as well as an overall decrease in tumor cells (Figure 4C), Treg, and myeloid cells. Directionality of fold-change was consistent between qmIF and scRNA-seq across all cell types, and fold-change values are significantly correlated (R2 = 0.9102, p = 0.004) (Figure 4D). Taken together, these qmIF data provide key protein-level support for the scRNA-seq data.
Baseline immune cell population frequencies correlate with treatment response
We next correlated baseline subpopulation composition with treatment response, with the goal of determining whether the presence of specific pre-treatment subpopulations was associated with PSA response or progression after therapy. To that end, we empirically categorized patients into three treatment response groups (early PSA response, stable disease, or late progressors) based on PSA log10 fold-change (Figure 5A). In this classification, early responders showed a PSA decrease to below 1% of the pre-treatment value, indicating robust response, while late progressors initially responded to therapy, with marked PSA decrease, but showed a PSA increase by week 28. We then compared the frequencies of each of the 24 subpopulations in “early responders” vs. “late progressors” (Figure 5B). To mitigate potential confounding factors, patients with stable disease were excluded from these analyses.
Figure 5. Differences in Baseline Immune Composition Associate with Differences in Treatment Response.

(A) Spider-plot of log10(Fold-Change) from baseline in prostate-specific antigen (PSA) over time with treatment, for each patient, such that four patients, labeled in blue, exhibited rapid and dramatic decrease to below 1% of initial PSA and were identified as early responders to treatment, and two patients, labeled in orange, initially responded to treatment with a rapid increase in PSA observed after on-treatment week 28. These were considered late progressors on-treatment. The remaining patients, in gray, generally trended toward a decreasing PSA, though not as rapidly as the early responders.
(B) Boxplot showing distribution of frequencies at Baseline for each cell cluster, comparing frequencies in early responders vs. late progressors, such that clusters with significant difference at baseline (p < 0.05 by Student’s t test) included CD4+ T cell 1, CD8+ T cell 2, Treg 3, and Epithelial 2.
At baseline, increased Cluster 2 LAG-3+ CD8+T cell representation was significantly associated with early PSA response (p = 0.006, Figure 5B). Interestingly, this represents the same population of LAG-3+ CD8+ T cells whose representation was expanded following anti-PD-1 therapy (Figure 3I). The subpopulation of TNFRSF18+ Treg (Cluster 3 Tregs) also trended with an early PSA response, though the association was not statistically significant (p = 0.13, Figure 5B). Conversely, over-representation of Cluster 1 CD4+ T cells was significantly associated with late PSA progression (p = 0.024, Figure 5B). Notably, one of the most differentially active proteins in this CD4+ subtype was tumor necrosis family (TNF-α, Figure 2), a multifunction proinflammatory cytokine implicated in tumor progression45-47 as discussed in the following text.
Transformed cells show transcriptional heterogeneity across metastatic sites
An initial analysis of tumor cells by inferred protein activity-based clustering resulted in three “epithelial” subtypes (EPI1, EPI2, and EPI3) (Figure 1B). Copy number alteration (CNA) inference and expression of the KLK3 prostate tumor marker gene confirmed that all three subtypes represent malignantly transformed epithelial cells (Figure S3). Following identification of transformed epithelial cells across pre-treatment samples from all metastatic sites, we found that all three epithelial subtypes were represented in bone, lymph node, and lung-derived samples (Figure 6). To further understand heterogeneity in the tumor cell compartment, we performed a more stringent cluster analysis by excluding all non-transformed cells. This analysis yielded eight molecularly distinct, co-existing tumor cell subclusters (REF-EPI1–REF-EPI8) (Figure 6A) that were further analyzed to assess enrichment of cancer hallmarks and cancer-related pathways in differentially active proteins (Figure S5). We found that REF-EPI1 was the subtype most enriched in androgen response proteins, REF-EPI2 and REF-EPI3 were defined by upregulation of E2F and MYC targets, and G2M checkpoint proteins, REF-EPI4 and REF-EPI5 were defined by upregulation of TNF-α signaling and interferon response proteins, REF-EPI6 was identified by heme metabolism, REF-EPI7 by unfolded protein response and androgen response, and finally REF-EPI8 by activation of the reactive oxygen species pathway (Figure S5).
Figure 6. Sub-Clustering Reveals Heterogeneity of Tumor Cells by Tissue Site.

(A) UMAP plot showing sub-clustering by resolution-optimized Louvain algorithm of only tumor cells (Epithelial 1, Epithelial 2, and Epithelial 3 from Figure 1B). Plot shows aggregate of all 2,550 tumor cells across all patients at all time-points.
(B) Stacked barplot of tumor cluster frequency by treatment time point in patients with metastatic bone tumors.
(C) Stacked barplot of tumor cluster frequency by treatment time point in patients with metastatic liver tumors.
(D) Stacked barplot of tumor cluster frequency by treatment time point in patients with metastatic lymph node tumors.
(E) Stacked barplot of tumor cluster frequency by treatment time point in patients with metastatic lung tumors.
(F) Boxplot showing distribution of frequencies at Baseline for each tumor subcluster, comparing frequencies in early responders vs. late progressors, such that the only cluster with significant difference at baseline (p < 0.05 by Student’s t test) was REF-EPI2, with higher baseline frequency in late progressors. Also see Figures S3 and S5.
The relative frequency of each tumor cell subcluster varied across metastatic sites at baseline, after ADT, after combination therapy, and at recurrence (Figures 6B-6E) in our small cohort of patients. At baseline, there was wide variability in the composition of tumor subclusters across metastatic sites (Figures 6B-6E). Bone and lymph node samples were the most heterogeneous, with representation of nearly all tumor cell subclusters (Figures 6B and 6D). In contrast, liver and lung-derived samples were less heterogeneous (Figures 6C and 6E). In particular, the lung sample was comprised nearly entirely of cells from the REF-EPI1 and REF-EPI2 subtypes (Figure 6E). These data are consistent with reports that patients with pulmonary-only prostate cancer metastases may have an overall favorable prognosis.48 Post-treatment, tumor subcluster representation was differentially affected across distinct metastatic sites (Figures 6B-6E). In bone samples, there was an increase in REF-EPI6 cells following ADT (p = 4.4e-14, Figure 6B). With the addition of anti-PD-1 REF-EPI6 cells became almost undetectable (p = 0.02, Figure 6B), while REF-EPI5 cells increased to comprise 65% of tumor cells (p = 9.9e-27, Figure 6B). However, in the recurrent bone sample, REF-EPI6 cells comprised nearly 75% of all tumor cells, while REF-EPI5 were reduced to only 1% of all tumor cells (Figure 6B), suggesting that REF-EPI6 may play an important role in treatment resistance.
In lymph node pre-treatment samples, the REF-EPI1 subcluster accounted for nearly 50% of all tumor cells; after ADT, their representation increased to ~65% (Figure 6D). Similar to bone-derived samples, following ADT, the fractional representation of tumor cell subtypes in lymph node samples changed slightly, while preserving the original heterogeneity (Figure 6D). However, addition of anti-PD-1 led to the emergence of REF-EPI2 as a predominant subcluster, comprising 89% of cells (p = 9.4e-7, Figure 6D). Liver and lung samples also maintained heterogeneity but did not show the emergence of a predominant tumor cell subcluster after combination therapy (Figures 6C and 6E). We were unable to assess the effects of ADT alone on liver or lung metastases as no patients with those lesions were randomized to the ADT only biopsy time point.
We next determined whether the presence of individual tumor cell subclusters at baseline was associated with differential PSA response (early responders vs. late progressors) (Figure 6F). Indeed, REF-EPI1, associated with an androgen response signature, was significantly overrepresented in baseline samples from “early responders” (p = 0.05) as compared to “late progressors” (Figure 6F). In contrast, REF-EPI2 and REF-EPI3 were overrepresented in baseline samples from “late progressors”, who ultimately progressed on treatment (p = 0.0008 and 0.08, respectively) (Figure 6F).
Interestingly, two proteins (TMPRSS2 and NKX3.1), which are regulated by the androgen receptor (AR) were among the most differentially active in REF-EPI1.49-51 Taken together, these data are consistent with the notion that activation of AR-regulated proteins in subclusters representing the majority of tumor cells is associated with an early treatment response. Notably, REF-EPI2 is characterized by aberrant activity of KIF14 (most-upregulated protein), which has previously been described as a candidate oncogene correlating with poor prognosis in prostate cancer.38
Validation of association between tumor cell subclusters and outcome
To assess the correlation of the tumor cell subclusters identified previously with outcome, we tested for enrichment of each subcluster within larger cohorts of bulk RNA-sequencing data. For these analyses, we defined the set of differentially active proteins in each subcluster as a unique protein activity signature. With these signatures and our protein activity inference algorithm, we next assessed the prognostic significance of each tumor cell subcluster across treatments in the TCGA dataset. By Cox regression on patient-by-patient normalized enrichment scores (Figure 7A), enrichment of tumor cell subcluster REF-EPI2 was significantly associated with a shorter recurrence-free survival (hazard ratio 1.37, p = 0.002). The leading-edge genes in the REF-EPI2 protein activity signature differentially enriched in patients with recurrence compared to non-recurrence are shown in Figure 7B and include KIF14 as well as TOP2A, both significant markers of REF-EPI2. Log rank testing of enrichment scores binarized to “high” vs. “low” showed significant association of both REF-EPI2 and REF-EPI3 protein activity profiles with shorter recurrence-free survival (p = 0.0087 and 0.022, respectively) (Figures 7C and 7D), and significant association of REF-EPI1 and REF-EPI7 protein activity profiles with longer recurrence-free survival (p = 0.0062 and 0.042, respectively) (Figures 7E and 7F). No other subcluster specific signature was statistically significantly associated with survival. In two smaller datasets that profiled metastatic CRPC (East Coast Stand Up to Cancer, West Coast Stand Up to Cancer),52,53 trends were observed toward association of REF-EPI2 and REF-EPI3 with decreased overall survival (Figures S6 and S7). Specifically, REF-EPI3 was significantly associated with worse overall survival in the East Coast SU2C dataset (p = 0.045) (Figure S7B), while REF-EPI2 did not achieve statistical significance. Additional correlations were not possible, since neither dataset includes recurrence-free survival or PSA response in the clinical metadata. Taken together, these results are consistent with the concept that overrepresentation of REF-EPI1 cells at baseline correlates with an improved treatment response while overrepresentation of REF-EPI2 and REF-EPI3 at baseline is associated with less favorable treatment response.
Figure 7. Tumor Single-Cell Subcluster Signatures Associate with Differential Outcomes in TCGA.

(A) Forest plot of Cox regression hazard ratios testing association in TCGA of patient-by-patient normalized enrichment score for each tumor subcluster gene set with recurrence-free survival. REF-EPI2 gene set enrichment is significantly associated with worse survival outcomes (p = 0.002).
(B) Heatmap of leading-edge gene set from REF-EPI2 comparing all recurrent vs. non-recurrent patients in TCGA.
(C) Kaplan-Meier curve testing association of binarized REF-EPI2 gene set enrichment (greater than 0 = high, less than 0 = low) with recurrence-free survival in TCGA, such that REF-EPI2 enrichment significantly associates with worse recurrence-free survival.
(D) Kaplan-Meier curve testing association of binarized REF-EPI3 gene set enrichment (greater than 0 = high, less than 0 = low) with recurrence-free survival in TCGA, such that REF-EPI3 enrichment significantly associates with worse recurrence-free survival.
(E) Kaplan-Meier curve testing association of binarized REF-EPI1 gene set enrichment (greater than 0 = high, less than 0 = low) with recurrence-free survival in TCGA, such that cluster 0 enrichment significantly associates with improved recurrence-free survival.
(F) Kaplan-Meier curve testing association of binarized REF-EPI7 gene set enrichment (greater than 0 = high, less than 0 = low) with recurrence-free survival in TCGA, such that REF-EPI7 enrichment significantly associates with improved recurrence-free survival, up to 2800 days. Kaplan-Meier curves are not shown for the remaining clusters as log rank p values for these were not statistically significant (p > 0.05). Also see Figures S6 and S7.
DISCUSSION
Studies on primary prostate cancer and metastatic, castration-resistant disease using high-throughput transcriptomic sequencing,25,41,54-56 showed that the TME is relatively immune-depleted. Here, we used scRNA-seq profiles to comprehensively characterize the TME of metastatic, castration-sensitive prostate cancer (mCSPC), across a variety of tissue types. Using longitudinal samples from 10 patients over a treatment course with ADT and anti-PD-1, we profiled the baseline TME and tumor cells, longitudinal changes induced by treatment, and correlated baseline features with PSA response. These studies leveraged our expertise in inferred protein-activity computational methods to increase resolution of the immune cell subpopulations and tumor cell subclusters as compared to conventional gene expression and transcriptomic methods. In particular, inferred protein activity enabled stratification of tumor cell subclusters and associations with post-treatment outcome, whereas tumor cells did not subcluster by standard gene expression methods at all due to excessive transcriptional background noise.
Profiling transcriptomes from a cumulative 40,270 single-cells, our study uncovered a previously undescribed and rich immune infiltrate in untreated mCSPC samples, albeit in a small cohort of 11 patients. Here, baseline bone, lymph node, and liver samples were similarly immune-infiltrated while the lung metastasis was relatively immune-depleted (Figures 1C and 1D) and distinctly different; the latter should be considered within the context of a small sample size. These data add to the notion that pulmonary-tropic and non-pulmonary metastatic mCSPC may be biologically and/or immunologically fundamentally distinct.48 Regarding the metastatic castration-sensitive tumor cells (transformed epithelial cells), our data adds to the literature by demonstrating significant intra- and inter-patient tumor cell heterogeneity.57 However, these results expand our knowledge through the identification of 8 distinct tumor cell subclusters and reporting their differential representation in patients as a function of response to treatment and in pre vs. on-treatment samples (Figure 6A) (Figures 6B-6E). These subclusters were not apparent using standard gene expression profiling (Figures S1 and S2) and could only be detected by VIPER-based inferred protein activity analysis. In that regard, we showed phenotypic changes in tumor cell types induced by treatment, correlated baseline tumor cell phenotypes with clinical response, and defined pathways enriched longitudinally and upon recurrent, progressive disease.
Limitations of the study
These findings are somewhat limited by the total number of patient samples per tissue type and analysis of a single metastatic site per patient. However, even this relatively small sample size was sufficient to arrive at statistical significance based on over or under representation of specific clusters in the mCSPC TME. In addition, longitudinal analyses of prostate cancer tumor metastases at the single cell level, over a course of treatment in a carefully conducted clinical trial, have not been previously reported or even collected to our knowledge. Other thus far published single-cell datasets in prostate cancer have notable differences from ours, specifically (1) that prior single-cell metastasis data are in the castration-resistant setting and (2) prior datasets reflect substantial heterogeneity with respect to treatment length and regimen at time of biopsy.58 Indeed, to our knowledge previous data in the metastatic castrate-sensitive setting are limited to bulk RNA sequencing in a cohort of 17 hormone-naive and 21 short-term castrate samples.43 A second limitation is that our findings are potentially biased toward more aggressive biology given that only patients with radiographically apparent disease at the time of on-treatment biopsy were able to safely undergo the procedure. As such, we are unable to comment regarding the on-treatment changes in the TME and tumor cell profiles of participants who rapidly responded to therapy. The time points for on-treatment biopsies were fixed due to the design of the clinical trial and are not based on tumor kinetics - although that may be an option in future studies. Here, we focused on cell types that were represented across all tissue types to avoid analyzing subpopulations that may be less relevant to tumor-immune crosstalk given their presence in a specific metastatic niche. This approach highlights broad changes in transcriptional program across tissue types in lieu of a deep dive on tissue-specific features.
The observation that treatment induces changes in both the TME cellular composition and transcriptional program of tumor cells, i.e., lineage plasticity, is consistent with other studies in prostate cancer.59-61 However, the TME immune subpopulation changes we observed after administration of ADT in mCSPC were in contrast to those described in primary prostate cancer. In primary prostate cancer, an immune infiltrate rich in T cells invades the TME after ADT administration,6,13 whereas in mCSPC biopsies we observed a decrease in CD4+ and CD8+ T cells after ADT. Of note, the combination of ADT and anti-PD-1 was effective at recruiting CD8+ effector T cells. It is possible that these observed differences between primary and metastatic CSPC are due to the baseline TME composition, i.e., an “immune desert” vs. “immune replete,” respectively, and additional immunomodulatory factors already present in the milieu.
Importantly, in our study, we observed significant increases in TNF-α + CD4+ non-T reg T cells (CD4+ 1), LAG-3+ CD8+ T cells (CD8+ 2), and GITR+ T regs (Treg 3) after combination therapy (Figure 3I). These data highlight the notion that combination therapy with ADT and anti-PD-1 therapy in men with castration-sensitive prostate cancer may be an immunologically active combination, even in bone metastasis. This is also in contrast to the metastatic castration resistant state, where two large Phase III randomized controlled trials comparing next generation hormonal therapy (NHT) alone to the combination of NHT + anti-PD(L)-1 were negative.62,63
Upregulation of lymphocyte activation gene-3 (LAG-3, CD223) is of interest; this a CD4+ homolog that binds to MHC class II,42 and which remains expressed on CD8+ T cells in the context of chronic antigen exposure; thus mediating T cell exhaustion.64,65 Dual-inhibition of PD-1 and LAG-3 was recently shown to improve outcomes in patients with melanoma in a large phase 3 clinical trial.66 In prostate cancer models that are resistant to single-agent PD-1, dual blockade of PD-1 and LA-G3 improved vaccine efficacy providing evidence that combination immune checkpoint therapy may be critical to improving clinical response outcomes.67 Taken together, these data suggest that LAG-3 may be a potential adjunct to combination immune checkpoint therapy in prostate cancer. Another potential target, glucocorticoid-induced TNFR-related (GITR) protein is an immune checkpoint receptor that belongs to tumor necrosis factor receptor superfamily (TNFRSF). GITR is preferentially expressed on CD8+ and Treg cells and agonistic antibodies have been shown to potentiate the former and reduce functionality of the latter.68-70 Although several preclinical and early phase studies showed that anti-GITR agonist antibodies are safe, clinical results have been modest thus far.71,72 Trials of dual immune checkpoint blockade targeting GITR and PD-1 have shown slight advantage over single-agent anti-GITR agonists antibodies,73,74 although data in mCSPC is limited.
Tumor necrosis factor alpha (TNF-α), an innate cytokine that can signal either as a membrane-bound protein and or a soluble ligand, was initially implicated as an anti-tumor cytokine but has since been strongly associated with tumor progression.45,46,75 In prostate cancer, our group previously found that elevated TNF-α levels were associated with PSA progression in men with biochemically recurrent prostate cancer, consistent with studies in metastatic prostate cancer.76-78 Related mechanistic data showed that TNF-α promotes prostate cancer dissemination from metastatic lymph nodes through activation of the CCL21/CCR7 axis79 and that inhibiting TNF-α, and its downstream pro-survival signaling molecules such as NF-κB and Bcl-2, may be a therapeutic approach for androgen-independent prostate cancer.80 Taken together, the novel data presented here showing significant upregulation of TNF-α in CD4+ T cells after ADT and anti-PD-1 (Figures 2 and S5), suggest that inhibiting TNF-α, either concurrently or sequentially, with ADT and immune checkpoint therapy in CSPC may be a rational treatment combination.
Due to the general tropism of metastatic prostate cancer to bone, tissue-specific changes occurring in the TME of bone samples were of particular interest. Notably, following ADT alone, we observed a relative increase of myeloid cells (Figures 3A and 3B) and a noticeable decrease in CD4+ non-Treg (Tconv) as well as tumor cells. As discussed earlier, the observed expansion of CD8+ T cells was most pronounced in the bone TME after combination therapy compared to other tissue types (Figure 3B). Although these findings should be confirmed in subsequent carefully controlled clinical trial datasets, these data highlight the notion that future precision medicine approaches for prostate cancer will likely need to incorporate data on tissue location.
As previously noted, the association of baseline immune subpopulations with PSA response showed that TNF-α + CD4+ T cells (CD4+ 1) were statistically significantly associated with late PSA progression, and also that LAG-3+ CD8+ T cells (CD8+ 2) and GITR+ T reg cells (T reg 3) were both statistically significantly associated with early PSA response (Figure 5B). In the case of Treg this is somewhat paradoxical as these cells are classically considered to be an immune suppressive subpopulation. However, a recent analysis of Treg using high-dimensional flow cytometry from an NPK-C1 transplantable prostate tumor model revealed significant phenotypic diversity within Tregs, including a Treg subpopulation enriched in regressing tumors.81 Thus, our data are consistent with the preclinical observation that “favorable” Tregs may be present in the TME at various states of treatment. Taken together, these data demonstrate the capacity of single-cell and high-dimensional data to provide more granularity on immune subpopulations and may even challenge the definitions of classical “immunosuppressive” or “immune effector cells.”
We observed both expected and unexpected findings upon association of baseline tumor subclusters with PSA response. REF-EPI1, which has high protein activity levels of the androgen receptor (AR) regulated proteins (TMPRSS2 and NKX3.1), was unsurprisingly associated with early PSA response (Figure 6F). Using the “hallmarks of cancer” pathway enrichment analysis,82 we further confirmed that REF-EPI1 was enriched with androgen response genes (Figure S5). We also found positive association between this gene set and clinical outcomes in the TCGA dataset using GSEA (Figure 7E). In contrast, REF-EPI2 and REF-EPI3, defined by upregulation of E2F targets, Myc targets, and G2M checkpoint (Figure S5), were associated with late on-treatment PSA progression (Figure 6F). These associations were validated in a similar manner (Figures 7C, 7D, and S7). Interestingly, we did not observe tumor REF-EPI4, the subcluster enriched with TNF-α signaling genes (Figure S5), to be associated with worse outcomes (Figure 6F). This association is opposite to what we observed in the TNF-α + CD4+ T cells (CD4+ 1), which was associated with decreased clinical outcomes (Figure 5B). Taken together, these observations highlight the notion that the specific cell of protein activity and/or expression (CD4+ T cells vs. tumor cells) reflects the underlying immunologic processes at play.
Building on this rich longitudinal transcriptomic dataset, we propose that the ultimate treatment paradigm for men with mCSPC requires a multi-pronged and adaptive combination regimen to elicit complete and durable responses. Similar to terminology used in the HIV field, we propose the term “Highly Active Anti-Tumor Therapy” (or HAATT), to describe a potential regimen with the following properties: (1) potent upfront tumor cell killing perhaps directed at known resistant tumor cell clones, (2) activation of CD8+ effector T cell function via combination immune checkpoint therapy (anti-PD-1 with anti-LAG-3, anti-GITR agonist antibodies), (3) depletion or blocking of regulatory T cells, and (4) targeting immunosuppressive or tumor-permissive molecules in the TME (i.e., cytokines, chemokine receptors, metabolomic pathways, or transcription factors). In summary, the data presented here highlight the importance of deeply profiling the underlying tumor biology and immunology using advanced systems biology methods in an effort to drive future clinical trial design.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Charles G. Drake, MD, PhD (cgd2139@columbia.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All original code and scRNA-seq data generated by this study have been deposited at Mendeley and are publicly available as of the date of publication at https://doi.org/10.17632/5nnw8xrh5m.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Clinical cohort
The PRIME-CUT study (modulating the PRostate cancer Immune MicroEnvironment with ChemoimmUnoTherapy for metastatic prostate cancer), is an open-label, single-arm, phase 2, single institution trial (NCT03951831) conducted at Columbia University Irving Medical Center (New York, NY). The study was approved by the IRB at Columbia University Irving Medical Center. The trial examined the clinical activity of phased administration of ADT, anti-PD-1, and docetaxel in men with newly diagnosed metastatic, castration-sensitive prostate cancer. The study was approved by the institutional review board at Columbia University and all participants provided written consent. Key eligibility criteria included a diagnosis of metastatic, castration-sensitive prostate cancer with a non-castrate testosterone level (> 150 ng/dL). Recruitment was restricted to patients with metastatic lesions amenable to biopsy. Patients with bone metastases were allowed. Treatment consisted of ADT (degarelix 240 mg subcutaneously (SC) for one dose, followed by leuprolide 22.5 mg SC every 12 weeks) followed by anti-PD-1 antibody (cemiplimab-rwlc 350 mg IV every 3 weeks) beginning four weeks after ADT initiation. With the intent of immune priming, a two-cycle lead-in of anti-PD-1 therapy was administered prior to standard of care docetaxel (75 mg/m2 every 3 weeks for six cycles). Participants received ADT and anti-PD-1 until study completion (52 weeks) or until a lack of clinical benefit or intolerable side effects. The primary clinical activity endpoint was the rate of undetectable PSA at 6 months after chemotherapy initiation (37 weeks on-study) as compared to the historical rate following ADT plus chemotherapy from the phase III CHAARTED trial.86 Secondary endpoints included time to progression to CRPC and rate of radiographic response at study completion. To ensure patient safety within the limitations of a small phase 2 study, toxicity was monitored using a Bayesian method which provided continuous monitoring boundaries for termination of the trial if the toxicity rate was unacceptable.87
Sample collection
From May 2019 through December 2020, twelve patients were enrolled. At the time of enrollment, all patients were scheduled for a pretreatment, interventional radiology-guided baseline biopsy of the most accessible metastasis. For on-treatment biopsies, subjects were randomized to one of two time points, either 4 weeks or 10 weeks on-study. Subsequent biopsies at the time of disease progression were optional. All on-treatment biopsies were obtained from the same anatomical location as the baseline biopsy. Patients randomized to the week 4 time point had been treated with four weeks of ADT (degarelix) alone at the time of second biopsy. Patients randomized to the week 10 time point were treated with 10 weeks of ADT (4 weeks of degarelix and 6 weeks of leuprolide) as well as two cycles (6 weeks) of anti-PD-1 at biopsy. We subsequently refer to these time points as ‘ADT only’ and ‘ADT+anti-PD-1,’ respectively. Given the phased administration of ADT and anti-PD-1, these data allowed us to compare the effects of ADT monotherapy versus ADT+anti-PD-1 on the transcriptional program of immune cell and tumor cell populations in the TME across a variety of tissues.
Of the twelve enrolled patients, two patients’ samples were excluded from further analyses due to either an insufficient number of viable cells for loading onto the 10X Genomics instrument or a lack of tumor cells identified in the biopsy sample using copy number inference (see STAR Methods below). We thus report on ten patients’ samples from bone, lymph node, liver, and lung metastases. We recovered an adequate number of cells in both baseline and on-treatment biopsy samples from six of the ten patients (four patients with bone metastases, one patient with lymph node metastases, and one patient with lung metastases). In the four remaining patients, only one of the two samples (baseline or on-treatment) per patient yielded adequate cells for sequencing and analysis (one baseline lymph node sample, one baseline liver sample, and one on-treatment bone sample [ADT only], and one on-treatment [ADT+anti-PD-1] liver sample.
METHOD DETAILS
Tissue dissociation
Fresh biopsy material was minced to 2-4 mm sized pieces with micro-scissors. For bone metastases, minced tissue was resuspended and examined with light microscopy. If dissociated to a single-cell suspension, the entire sample was passed through a 70-um filter for downstream processing. For bone metastases not found to be dissociated. and for all other sites, tissue was digested with 2.5 mL of tumor dissociation medium (L-15 medium with 1g/L glucose, 5% FBS, 15 mM HEPES, 800 U/ml collagenase IV, and 0.1 mg/ml DNase I) with gentle agitation in a glass vial in a 37°C water bath for 30 min. Dissociated cells were passed through a 70-um filter and centrifuged at 300 g for 5 min at 4°C. If the resulting pellet showed signs of red blood cells (RBC), cells were incubated with ACK lysis buffer and centrifuged a second time at 300 g for 5 min at 4°C. Cells were then resuspended in L-15 medium with 1g/L glucose, 5% FBS, and 15 mM HEPES and counted with Trypan Blue for viability. An aliquot of 10,000-20,000 cells per sample was used for subsequent scRNA-seq analysis.
Single-cell RNA sequencing and data processing
Single-cell RNA sequencing (scRNA-seq) was performed using the 10x Genomics (Pleasanton, CA) Chromium Single Cell 5’ Library & Gel Bead Kit at the Columbia University Human Immune Monitoring Core (HIMC). Manufacturers’ protocols were followed for the preparation of gene expression libraries and sequencing on the Illumina (San Diego, CA) NovaSeq 6000 Sequencing System. Sequencing reads (base call files) were converted to FASTQ files using the 10x Genomics data processing pipeline “cellranger mkfastq”, followed by “cellranger count” for cell calling, gene mapping to the pre-built human reference set of 30,727 genes (10x Genomics), and gene counting. Cell-gene count matrix data were processed using the publicly available Seurat package to filter for cells with less than 10% mitochondrial RNA content, more than 1,500 UMI counts, and fewer than 15,000 UMI counts, followed by the Seurat SCTransform command to perform a regularized negative binomial regression based on the 3000 most variable genes.
Due to separate harvesting a processing of each specimen as fresh surgical tissue, without anchor integration, significant patient-level batch effect was observed, with cells clustering by patient rather than cell type. Thus, after filtering low-quality cells and scaling each sample separately with SCTransform, all samples were combined for subsequent analyses by the Seurat AnchorIntegration algorithm. To ensure statistically valid clustering, the combined matrix was clustered using the Louvain Algorithm at a range of resolution values from 0 to 1 in intervals of 0.01, with optimal resolution selected to maximize mean silhouette score, as previously described in.28 Gene Expression data were projected into their first 50 principal components using the RunPCA function in Seurat, and further reduced into a 2-dimensional visualization space using the RunUMAP function with method umap-learn and Pearson correlation as the distance metric between cells. Differential Gene Expression between clusters was computed by the MAST hurdle model for single-cell gene expression modeling, as implemented in the Seurat FindAllMarkers command, with log fold change threshold of 0.5 and minimum fractional expression threshold of 0.25, indicating that the resulting gene markers for each cluster were restricted to those with log fold change greater than 0 and non-zero expression in at least 25% of the cells in the cluster.
Semi-supervised cell type calling
For each single cell gene expression sample, cell-by-cell identification of cell types was performed using the SingleR package30 and the preloaded Blueprint-ENCODE reference, which includes normalized expression values for 259 bulk RNA-seq samples generated by Blueprint and ENCODE from 43 distinct cell types representing pure populations of stroma and immune cells.88,89 The SingleR algorithm computes the correlation between each individual cell and each of the 259 reference samples, and then assigns both a label of the cell type with highest average correlation to the individual cell and a p-value computed by Wilcox test of correlation to that cell type as compared to all other cell types. Cell-by-cell SingleR labels were restricted to those with p < 0.05, and unsupervised clusters are labelled as a particular cell type based on the most-represented SingleR cell type label within that cluster. Since tumor cells are not represented within the Blueprint-ENCODE reference, tumor cells are assigned as ‘epithelial’. The tumor cell identity of these cells was confirmed by expression of KLK3, a prostate cancer marker gene, as well as by inference of copy number variations using the InferCNV algorithm84 with all lymphoid and myeloid cell clusters specified as a copy-number-normal reference.
Regulatory network and protein activity inference
Protein activity was quantified from single-cell gene expression profiles according to the pipeline previously described90 and subsequently used for analysis of single-cell ccRCC samples.28 From the combined and batch-corrected dataset of all patients, metaCells were independently assembled from each distinct single cell subpopulation, as identified by gene expression clustering. Specifically, metaCells were generated by summing SCTransform-corrected template counts for the 10 nearest neighbors of randomly selected cells, based on Pearson correlation distance in gene expression space. 200 cells were randomly sampled from each cluster to generate metaCells and the latter were then used to generate cluster-specific regulatory networks using the ARACNe algorithm.27-29 ARACNe was run with 100 bootstrap iterations using 1,785 transcription factors (genes annotated in gene ontology molecular function database as GO:0003700, “transcription factor activity”, or as GO:0003677, “DNA binding” and GO:0030528, “transcription regulator activity”, or as GO:0003677 and GO:0045449, “regulation of transcription”), 668 transcriptional cofactors (a manually curated list, not overlapping with the transcription factor list, built upon genes annotated as GO:0003712, “transcription cofactor activity”, or GO:0030528 or GO:0045449), 3,455 signaling genes (annotated in GO biological process database as GO:0007165, “signal transduction” and in GO cellular component database as GO:0005622, “intracellular” or GO:0005886, “plasma membrane”), and 3,620 surface markers (annotated as GO:0005886 or as GO:0009986, “cell surface”). ARACNe was used to analyze each of these gene sets independently to ensure that the data processing inequality (DPI) was not affected by different baseline mutual information for different protein classes. Moreover, we did not use ARACNe to infer the regulatory targets of proteins with no known signaling or transcriptional activity, for which the effect on downstream transcriptional targets may be difficult to interpret. Parameters were set to zero DPI tolerance and Mutual Information (MI) p-value threshold of p = 10−8, computed by shuffling the original dataset as a null model.
Protein activity was inferred by VIPER applying all ARACNe networks to the integrated and scaled gene expression signature of the combined dataset across all patients. The resulting protein activity matrix was loaded into a Seurat Object, then projected into its first 50 principal components using the RunPCA function, and further reduced into a 2-dimensional visualization space using RunUMAP function with method umap-learn and Pearson correlation as the distance metric between cells. Clustering on protein activity was performed exactly as above for clustering on gene expression, with optimal Louvain resolution selected to maximize silhouette score as an objective metric of robust cluster separation. Differential protein activity between clusters identified by resolution-optimized Louvain was computed using Student’s t-test, and the top proteins for each cluster were ranked by p-value.
Association of TME subpopulations with tissue site and PSA response
Cluster frequencies were computed as a percentage of patient cells within each cluster divided by the total number of patient cells. For example—in a sample of 1000 total cells with 200 T-cells, the frequency of T-cells would be computed as 200/1000=0.2. Subsequent comparisons were made between cluster frequencies in different tissue sites, treatment time-points, and at baseline between patients who responded or did not respond to treatment, as assessed by change in PSA over time. Specifically, for each set of comparisons the combined clustering across all patients was used to define clusters, and relative abundance of each cluster in each sample was computed. Cluster frequencies were compared by Mann-Whitney U-test. A separate sub-clustering of tumor cells only was also performed by Louvain algorithm with silhouette score optimization as above, with relative abundance representing percentage of each tumor cell subcluster among total tumor cell count per sample.
Differential protein activity was computed for each tumor cell subcluster by Student’s t-test, and pathway enrichment within each cluster was assessed by the Enrichr browser tool.91 Cluster frequencies were compared with respect to tissue site, treatment timepoint, and subsequent response to treatment, such that early response to treatment was defined by reduction to less than 1% of initial PSA within 10 weeks of treatment.
Tumor cell subcluster association with outcome in external datasets
For each tumor cell subpopulation, a protein signature was defined based on the proteins that were significantly differentially activated in that cluster. Relative enrichment of these protein signatures was assessed in three independent, external, prostate cancer bulk RNA-seq datasets, (TCGA, East Coast SU2C, West Coast SU2C)52,53,92; for these analyses the bulk RNA-seq dataset was first internally scaled by z-score, then VIPER protein activity inference was performed using the single-cell ARACNe networks, and finally enrichment of each tumor subcluster signature was determined in each bulk RNA-seq sample using Gene Set Enrichment Analysis (GSEA),82 where genes were ranked from the highest to lowest activity of their encoded protein. The resulting normalized enrichment scores were tested for correlation with recurrence-free-survival in TCGA or with overall survival time in SU2C by Cox regression. Since enrichment of tumor cell cluster 1 was found to be significantly associated with a shorter recurrence-free-survival in TCGA, the genes in the leading-edge, and their encoded proteins, were further identified by GSEA analysis of all proteins ranked by differential activity in TCGA samples with and without recurrence. Finally, patient-by-patient enrichment scores were binarized to less than zero = “low” and greater than zero = “high” and assessed for effect on survival by log-rank test and Kaplan-Meier curve.
Quantitative immunofluorescence analysis
Quantitative immunofluorescence (QmiF) was performed as previously described.93 Briefly, FFPE sections were dewaxed and stained for seven relevant proteins (key resources table). Slides were cured at room temperature, then whole slide images were acquired on the Vectra Polaris Quantitative Pathology Imaging System (Akoya Biosciences, Marlborough, MA). The entire tissue was selected for imaging using Phenochart and multispectral image tiles were acquired using the Polaris. Images were spectrally unmixed using Phenoptics inForm software and exported as multi-image TIF files.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CTLA-4 | BioSB | Cat#BSB2884; RRID: AB_2762365 |
| Rabbit monoclonal anti-PD-L1 | Cell Signaling Technology | Cat#13684; RRID: AB_2687655 |
| Mouse monoclonal anti-CD8 | Dako | Cat# M7103; RRID: AB_2075537 |
| Rabbit monoclonal anti-CD163 | BioSB | Cat#BSB3276 |
| Mouse monoclonal anti-CD68 | Dako | Cat#M0876; RRID: AB_2074844 |
| Rabbit monoclonal anti-LAG-3 | BioSB | Cat#BSB3366 |
| Mouse monoclonal ant-PanCK | Dako | Cat#M3515; RRID: AB_2132885 |
| Rabbit monoclonal anti-CD4 | Cell Marque | Cat#104R-15; RRID: AB_1516770 |
| LAG-3 | BioSB | Cat# BSB3366; RRID: AB-3073657 |
| CD163 | Cell Marque | Cat# AC-0316; RRID: AB_3073656 |
| Biological samples | ||
| Untreated metastatic castration-sensitive needle core biopsies: Fresh Samples | Columbia University Irving Medical Center | N/A |
| On-treatment (ADT or ADT + a-PD-1) metastatic castration-sensitive needle core biopsies: Fresh Samples | Columbia University Irving Medical Center | N/A |
| Untreated metastatic castration-sensitive needle core biopsies: FFPE tissue | Columbia University Irving Medical Center | N/A |
| On-treatment (ADT or ADT + a-PD-1) metastatic castration-sensitive needle core biopsies: FFPE tissue | Columbia University Irving Medical Center | N/a |
| Critical commercial assays | ||
| 10X Genomics Chromium Single Cell 5’ Library & Gel Bead Kit | 10X Genomics | N/A |
| Illumina NovaSeq 6000 Sequencing System | Illumina | N/A |
| Opal-7-color multiplex IHC kit | Akoya Biosciences | SKU NEL811001KT |
| Deposited data | ||
| Raw and Analyzed data used to generate analyses shown in this Manuscript: scRNASeq, bulkRNASeq, qmIF | This manuscript | Available on Mendeley Data https://doi.org/10.17632/5nnw8xrh5m.1 |
| Software and algorithms | ||
| HALO Image Analysis Software | Indica Labs | http://www.indicalab.com/halo/ |
| Seurat v3 | Stuart et al., 201983 | https://cran.r-project.org/web/packages/Seurat/index.html |
| SingleR | Aran et al., 201930 | https://bioconductor.org/packages/release/bioc/html/SingleR.html |
| InferCNV | (Tickle et al., 2019)84 | https://github.com/broadinstitute/infercnv |
| ARACNe | Lachmann et al., 201685 | https://github.com/califano-lab/ARACNe-AP |
| VIPER | Alvarez et al., 201627 | http://bioconductor.org/packages/release/bioc/html/viper.html |
| R Code Used to Generate Figures and perform analyses shown in this manuscript | This manuscript | Available on Mendeley Data https://doi.org/10.17632/5nnw8xrh5m.1 |
Images were analyzed with HALO image analysis software (Indica Labs, Cooales, NM) using the Highplex FL module, v4.1.3. Cellular analysis was performed by first identifying cells based on nuclear recognition (DAPI stain), then measuring fluorescence intensity of the estimated cytoplasmic areas of each cell. A mean intensity threshold above background was used to determine positivity for each fluorochrome within the cytoplasm, thereby, defining cells as either positive or negative for each marker. The positive cell data was then used to define co-localized populations.
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantitative and statistical analyses were performed using the R programming environment and packages as described above. Differential gene expression was assessed at the single-cell level by the MAST single-cell statistical framework as implemented in Seurat v3, and differential VIPER activity was assessed by t-test, each with Bonferroni multiple-testing correction. Comparisons of cell frequencies were performed by non-parametric Wilcox rank-sum test, and survival analyses were performed by log-rank test and cox regression. In all cases, statistical significance was assigned for p values ≤ 0.05.
ADDITIONAL RESOURCES
The phase 2 clinical trial is registered on clinicaltrials.gov and registration number is NCT03951831.
Supplementary Material
Highlights.
Protein-inferred activity clustering shows complex immune cells in CSPC metastases
ADT and aPD-1 treatment induces robust infiltrates of CD8+, CD4+ T cells, and Tregs
T cell infiltration determined by scRNA-seq correlated with T cells shown by qmIF
Baseline tumor and immune clusters associated with clinical outcomes
ACKNOWLEDGMENTS
This work was supported by grants from the Conquer Cancer Foundation (2019YIA to J.E.H.), the Prostate Cancer Foundation (2020YIA to J.E.H.) and the National Institutes of Health (T32CA203703 to J.E.H., UL1TR001873 to J.E.H., F30CA260765-01 to A.Z.O.), as well as by an NCI Outstanding Investigator Award (R35 CA197745), the NIH Shared Instrumentation Grants (S10 OD012351 and S10 OD021764), and U54CA209997 all to AC. We also acknowledge the P30CA013696 Cancer Center grant for the support of the Cancer Biostatistics Shared Resource and the JP Sulzberger Columbia Genome Center at the Herbert Irving Comprehensive Cancer Center at Columbia University. This research was additionally supported by the Experimental Histopathology Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704). Regeneron Pharmaceuticals provided the funding for the clinical trial.
INCLUSION AND DIVERSITY
We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2023.10.006.
DECLARATION OF INTERESTS
Dr. Hawley has served as a paid consultant to Seagen, Daiichi Sankyo, and ImmunityBio and has received sponsored research funding to her institution from Astra Zeneca, Bristol Meyers Squibb, Crescendo Biologics, Macrogenics, and Vaccitech. Dr. Drake is a co-inventor on patents licensed from JHU to BMS and Janssen and is currently an employee of Janssen Research. Dr. Califano isfounder, equity holder, and consultant of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth Inc. Dr. Lowy is an employee and stockholder of Regeneron Pharmaceuticals.
REFERENCES
- 1.Stultz J, and Fong L (2021). How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 24, 697–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bilusic M, Madan RA, and Gulley JL (2017). Immunotherapy of Prostate Cancer: Facts and Hopes. Clin. Cancer Res 23, 6764–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, Battistini L, Iafrate M, Prayer-Galetti T, Pagano F, and Viola A (2005). Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med 201, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sfanos KS, Bruno TC, Meeker AK, De Marzo AM, Isaacs WB, and Drake CG (2009). Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate 69, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vargas Roditi LD, Jacobs A, Rueschoff JH, Bankhead P, Chevrier S, Jackson HW, Hermanns T, Fankhauser CD, Poyet C, Chun F, et al. (2021). Single-Cell proteomics defines the cellular heterogeneity of localized prostate cancer. Preprint at bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obradovic AZ, Dallos MC, Zahurak ML, Partin AW, Schaeffer EM, Ross AE, Allaf ME, Nirschl TR, Liu D, Chapman CG, et al. (2020). T-cell infiltration and adaptive treg resistance in response to androgen deprivation with or without vaccination in localized prostate cancer. Clin. Cancer Res 26, 3182–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drake CG, Doody ADH, Mihalyo MA, Huang CT, Kelleher E, Ravi S, Hipkiss EL, Flies DB, Kennedy EP, Long M, et al. (2005). Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell 7, 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gannon PO, Poisson AO, Delvoye N, Lapointe R, Mes-Masson AM, and Saad F (2009). Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 348, 9–17. [DOI] [PubMed] [Google Scholar]
- 9.Mercader M, Bodner BK, Moser MT, Kwon PS, Park ES, Manecke RG, Ellis TM, Wojcik EM, Yang D, Flanigan RC, et al. (2001). T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc. Natl. Acad. Sci. USA 98, 14565–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutherland JS, Goldberg GL, Hammett MV, Uldrich AP, Berzins SP, Heng TS, Blazar BR, Millar JL, Malin MA, Chidgey AP, and Boyd RL (2005). Activation of thymic regeneration in mice and humans following androgen blockade. J. Immunol 175, 2741–2753. [DOI] [PubMed] [Google Scholar]
- 11.Drake CG, Doody ADH, Mihalyo MA, Huang CT, Kelleher E, Ravi S, Hipkiss EL, Flies DB, Kennedy EP, Long M, et al. (2005). Androgen ablation mitigates tolerance to a prostate/prostate cancer-restricted antigen. Cancer Cell 7, 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morse MD, and McNeel DG (2010). Prostate cancer patients on androgen deprivation therapy develop persistent changes in adaptive immune responses. Hum. Immunol 71, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen YC, Ghasemzadeh A, Kochel CM, Nirschl TR, Francica BJ, Lopez-Bujanda ZA, Carrera Haro MA, Tam A, Anders RA, Selby MJ, et al. (2018). Combining intratumoral Treg depletion with androgen deprivation therapy (ADT): preclinical activity in the Myc-CaP model. Prostate Cancer Prostatic Dis. 21, 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greenstein BD, Fitzpatrick FT, Adcock IM, Kendall MD, and Wheeler MJ (1986). Reappearance of the thymus in old rats after orchidectomy: inhibition of regeneration by testosterone. J. Endocrinol 110, 417–422. [DOI] [PubMed] [Google Scholar]
- 15.Greenstein BD, Fitzpatrick FT, Kendall MD, and Wheeler MJ (1987). Regeneration of the thymus in old male rats treated with a stable analogue of LHRH. J. Endocrinol 112, 345–350. [DOI] [PubMed] [Google Scholar]
- 16.Roden AC, Moser MT, Tri SD, Mercader M, Kuntz SM, Dong H, Hurwitz AA, McKean DJ, Celis E, Leibovich BC, et al. (2004). Augmentation of T cell levels and responses induced by androgen deprivation. J. Immunol 173, 6098–6108. [DOI] [PubMed] [Google Scholar]
- 17.Kissick HT, Sanda MG, Dunn LK, Pellegrini KL, On ST, Noel JK, and Arredouani MS (2014). Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc. Natl. Acad. Sci. USA 111, 9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson CA, Mrose SA, and Thomas DW (1995). Enhanced production of B lymphocytes after castration. Blood 85, 1535–1539. [PubMed] [Google Scholar]
- 19.Windmill KF, and Lee VW (1998). Effects of castration on the lymphocytes of the thymus, spleen and lymph nodes. Tissue Cell 30, 104–111. [DOI] [PubMed] [Google Scholar]
- 20.Sorrentino C, Musiani P, Pompa P, Cipollone G, and Di Carlo E (2011). Androgen deprivation boosts prostatic infiltration of cytotoxic and regulatory T lymphocytes and has no effect on disease-free survival in prostate cancer patients. Clin. Cancer Res 17, 1571–1581. [DOI] [PubMed] [Google Scholar]
- 21.Pu Y, Xu M, Liang Y, Yang K, Guo Y, Yang X, and Fu YX (2016). Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med 8, 333ra47. [DOI] [PubMed] [Google Scholar]
- 22.Tang S, Moore ML, Grayson JM, and Dubey P (2012). Increased CD8+ T-cell function following castration and immunization is countered by parallel expansion of regulatory T cells. Cancer Res. 72, 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haffner MC, Guner G, Taheri D, Netto GJ, Palsgrove DN, Zheng Q, Guedes LB, Kim K, Tsai H, Esopi DM, et al. (2018). Comprehensive Evaluation of Programmed Death-Ligand 1 Expression in Primary and Metastatic Prostate Cancer. Am. J. Pathol 188, 1478–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, Gulati R, Plymate SR, Zhou Z, Birditt B, et al. (2021). Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun 12, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He MX, Cuoco MS, Crowdis J, Bosma-Moody A, Zhang Z, Bi K, Kanodia A, Su MJ, Ku SY, Garcia MM, et al. (2021). Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat. Med 27, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding H, Douglass EF, Sonabend AM, Mela A, Bose S, Gonzalez C, Canoll PD, Sims PA, Alvarez MJ, and Califano A (2018). Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun 9, 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obradovic A, Chowdhury N, Haake SM, Ager C, Wang V, Vlahos L, Guo XV, Aggen DH, Rathmell WK, Jonasch E, et al. (2021). Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988–3005.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet 50, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox JJ, Navarro HI, Hashimoto T, Garcia AJ, and Goldstein AS (2019). Mass cytometry reveals species-specific differences and a new level of complexity for immune cells in the prostate. Am. J. Clin. Exp. Urol 7, 281–296. [PMC free article] [PubMed] [Google Scholar]
- 32.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, and Pienta KJ (2009). CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem 284, 34342–34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denisenko E, Guo BB, Jones M, Hou R, De Kock L, Lassmann T, Clément O, Poppe D, Clément O, Lister R, and Forrest ARR (2020). Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH, and Slaney CY (2018). Tissue-dependent tumor microenvironments and their impact on immunotherapy responses. Front. Immunol 9, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 36.Chen M, Pan H, Sun L, Shi P, Zhang Y, Li L, Huang Y, Chen J, Jiang P, Fang X, et al. (2020). Structure and regulation of human epithelial cell transforming 2 protein. Proc. Natl. Acad. Sci. USA 117, 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawson CD, Fan C, Mitin N, Baker NM, George SD, Graham DM, Perou CM, Burridge K, Der CJ, and Rossman KL (2016). Rho GTPase Transcriptome Analysis Reveals Oncogenic Roles for Rho GTPase-activating Proteins in Basal-like Breast Cancers. Cancer Res. 76, 3826–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Yuan Y, Liang P, Zhang Z, Guo X, Xia L, Zhao Y, Shu S, Sun S, Ying Y, and Cheng Y (2017). Overexpression of a novel candidate oncogene KIF14 correlates with tumor progression and poor prognosis in prostate cancer. Oncotarget 8, 45459–45469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clark JP, and Cooper CS (2009). ETS genefusions in prostate cancer. Nat. Rev. Urol 6, 429–439. [DOI] [PubMed] [Google Scholar]
- 40.Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, Chen Y, Nau M, Ravindranath L, Chen Y, Dobi A, et al. (2005). Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene 24, 3847–3852. [DOI] [PubMed] [Google Scholar]
- 41.Chen S, Zhu G, Yang Y, Wang F, Xiao YT, Zhang N, Bian X, Zhu Y, Yu Y, Liu F, et al. (2021). Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol 23, 87–98. [DOI] [PubMed] [Google Scholar]
- 42.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, and Hercend T (1990). LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med 171, 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thysell E, Köhn L, Semenas J, Järemo H, Freyhult E, Lundholm M, Thellenberg Karlsson C, Damber JE, Widmark A, et al. (2022). Clinical and biological relevance of the transcriptomic-based prostate cancer metastasis subtypes MetA-C. Mol. Oncol 16, 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LaFleur MW, Muroyama Y, Drake CG, and Sharpe AH (2018). Inhibitors of the PD-1 Pathway in Tumor Therapy. J. Immunol 200, 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balkwill F (2006). TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 25, 409–416. [DOI] [PubMed] [Google Scholar]
- 46.Montfort A, Colacios C, Levade T, Andrieu-Abadie N, Meyer N, and Ségui B (2019). The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol 10, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, Belsue V, de Andrea C, Rodriguez-Ruiz ME, Perez-Gracia JL, Marquez-Rodas I, et al. (2019). Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569, 428–432. [DOI] [PubMed] [Google Scholar]
- 48.Shenderov E, Isaacsson Velho P, Awan AH, Wang H, Mirkheshti N, Lotan TL, Carducci MA, Pardoll DM, Eisenberger MA, and Antonarakis ES (2019). Genomic and clinical characterization of pulmonary-only metastatic prostate cancer: A unique molecular subtype. Prostate 79, 1572–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, Curtis RT, Shell BK, Bostwick DG, Tindall DJ, et al. (1997). A Novel Human Prostate-Specific, Androgen-Regulated Homeobox Gene (NKX3.1) That Maps to 8p21, a Region Frequently Deleted in Prostate Cancer. Genomics 43, 69–77. [DOI] [PubMed] [Google Scholar]
- 50.Clinckemalie L, Spans L, Dubois V, Laurent M, Helsen C, Joniau S, and Claessens F (2013). Androgen Regulation of the TMPRSS2 Gene and the Effect of a SNP in an Androgen Response Element. Mol. Endocrinol 27, 2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648. [DOI] [PubMed] [Google Scholar]
- 52.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et al. (2018). Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 174, 758–769.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dong B, Miao J, Wang Y, Luo W, Ji Z, Lai H, Zhang M, Cheng X, Wang J, Fang Y, et al. (2020). Single-cell analysis supports a luminal-neuroendocrine transdifferentiation in human prostate cancer. Commun. Biol 3, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karthaus WR, Hofree M, Choi D, Linton EL, Turkekul M, Bejnood A, Carver B, Gopalan A, Abida W, Laudone V, et al. (2020). Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 368, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song H, Weinstein HNW, Allegakoen P, Wadsworth MH, Xie J, Yang H, Feng FY, Carroll PR, Wang B, Cooperberg MR, et al. (2020). Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. Preprint at bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haffner MC, Zwart W, Roudier MP, True LD, Nelson WG, Epstein JI, De Marzo AM, Nelson PS, and Yegnasubramanian S (2021). Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol 18, 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kfoury Y, Baryawno N, Severe N, Mei S, Gustafsson K, Hirz T, Brouse T, Scadden EW, Igolkina AA, Kokkaliaris K, et al. (2021). Human prostate cancer bone metastases have an actionable immunosuppressive microenvironment. Cancer Cell 39, 1464–1478.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 32, 474–489.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, Nguyen HM, Yang YC, Da Costa RMG, Kaipainen A, et al. (2019). Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Invest 129, 4492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powles T, Yuen KC, Gillessen S, Kadel EE, Rathkopf D, Matsubara N, Drake CG, Fizazi K, Piulats JM, Wysocki PJ, et al. (2022). Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: a randomized phase 3 trial. Nat. Med 28, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.(2023). Merck Announces KEYNOTE-991 Trial Evaluating KEYTRUDA® (pembrolizumab) Plus Enzalutamide and Androgen Deprivation Therapy in Patients With Metastatic Hormone-Sensitive Prostate Cancer to Stop for Futility -. Merck.com. https://www.merck.com/news/merck-announces-keynote-991-trial-evaluating-keytruda-pembrolizumab-plus-enzalutamide-and-androgen-deprivation-therapy-in-patients-with-metastatic-hormone-sensitive-prostate-cancer-to-stop-for/. [DOI] [PubMed] [Google Scholar]
- 64.Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen H-R, Pyle KJ, Hipkiss E, Vignali DAA, Pardoll DM, and Drake CG (2009). Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T Cells. J. Immunol 182, 6659–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grosso JF, and Drake CG (2007). Current immunotherapeutic strategies in prostate cancer. Surg. Oncol. Clin 16, 861–871. [DOI] [PubMed] [Google Scholar]
- 66.Lipson EJ, Tawbi HA-H, Schadendorf D, Ascierto PA, Matamala L, Gutiérrez EC, Rutkowski P, Gogas H, Lao CD, Janoski de Menezes J, et al. (2021). Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). J. Clin. Oncol 39, 9503. [Google Scholar]
- 67.Zahm CD, Moseman JE, Delmastro LE, and G Mcneel D (2021). PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy. OncoImmunology 10, 1912892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fares CM, Allen EMV, Drake CG, Allison JP, and Hu-Lieskovan S (2019). Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book, 147–164. [DOI] [PubMed] [Google Scholar]
- 69.Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, Yang X, Zhong H, Newman W, Qi J, et al. (2019). Rational design of anti-GITR-based combination immunotherapy. Nat. Med 25, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vence L, Bucktrout SL, Fernandez Curbelo I, Blando J, Smith BM, Mahne AE, Lin JC, Park T, Pascua E, Sai T, et al. (2019). Characterization and Comparison of GITR Expression in Solid Tumors. Clin. Cancer Res 25, 6501–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Paz-Ares L, Meniawy T, Rottey S, Moreno V, Gazzah A, et al. (2020). Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor–Related Protein Agonist Alone or in Combination With Nivolumab for Patients With Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 6, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tran B, Carvajal RD, Marabelle A, Patel SP, LoRusso PM, Rasmussen E, Juan G, Upreti VV, Beers C, Ngarmchamnanrith G, and Schöffski P (2018). Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor–related protein agonist AMG 228 in patients with advanced solid tumors. J. Immunother. Cancer 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Killock D. (2019). GITR agonism — combination is key. Nat. Rev. Clin. Oncol 16, 402. [DOI] [PubMed] [Google Scholar]
- 74.Geva R, Voskoboynik M, Dobrenkov K, Mayawala K, Gwo J, Wnek R, Chartash E, and Long GV (2020). First-in-human phase 1 study of MK-1248, an anti-glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors. Cancer 126, 4926–4935. [DOI] [PubMed] [Google Scholar]
- 75.Balkwill F. (2009).Tumour necrosis factor and cancer. Nat. Rev. Cancer 9, 361–371. [DOI] [PubMed] [Google Scholar]
- 76.Hawley JE, Pan S, Figg WD, Lopez-Bujanda ZA, Strope JD, Aggen DH, Dallos MC, Lim EA, Stein MN, Hu J, and Drake CG (2020). Association between immunosuppressive cytokines and PSA progression in biochemically recurrent prostate cancer treated with intermittent hormonal therapy. Prostate 80, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharma J, Gray KP, Harshman LC, Evan C, Nakabayashi M, Fichorova R, Rider J, Mucci L, Kantoff PW, and Sweeney CJ (2014). Elevated IL-8, TNF-alpha, and MCP-1 in men with metastatic prostate cancer starting androgen-deprivation therapy (ADT) are associated with shorter time to castration-resistance and overall survival. Prostate 74, 820–828. [DOI] [PubMed] [Google Scholar]
- 78.Michalaki V, Syrigos K, Charles P, and Waxman J (2004). Serum levels of IL-6 and TNF-α correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 90, 2312–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maolake A, Izumi K, Natsagdorj A, Iwamoto H, Kadomoto S, Makino T, Naito R, Shigehara K, Kadono Y, Hiratsuka K, et al. (2018). Tumor necrosis factor-α induces prostate cancer cell migration in lymphatic metastasis through CCR7 upregulation. Cancer Sci. 109, 1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Srinivasan S, Kumar R, Koduru S, Chandramouli A, and Damodaran C (2010). Inhibiting TNF-mediated signaling: A novel therapeutic paradigm for androgen independent prostate cancer. Apoptosis 15, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ager CR, Obradovic AZ, Arriaga JM, Chaimowitz MG, Califano A, Abate-Shen C, and Drake CG (2021). Longitudinal immune profiling reveals unique myeloid and T-cell phenotypes associated with spontaneous immunoediting in a prostate tumor model. Cancer Immunol. Res 9, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tickle T, Tirosh I, Georgescu C, Brown M, Haas B (2019). inferCNV of the Trinity CTAT Project.. Klarman Cell Observatory, Broad Institute of MIT and Harvard, Cambridge, MA, USA. https://github.com/broadinstitute/inferCNV.Infer Copy Number Variation from Single-Cell RNA-Seq Data 2018. https://bioconductor.org/packages/release/bioc/html/infercnv.html. [Google Scholar]
- 85.Lachmann A, Giorgi Federico M., Lopez G, Califano A (2016). ARACNeAP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32: 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sweeney CJ, Chen Y-H, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong Y-N, Hahn N, Kohli M, Cooney MM, et al. (2015). Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med 373, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thall PF, Simon RM, and Estey EH (1996). New statistical strategy for monitoring safety and efficacy in single-arm clinical trials. J. Clin. Oncol 14, 296–303. [DOI] [PubMed] [Google Scholar]
- 88.Martens JHA, and Stunnenberg HG (2013). BLUEPRINT: Mapping human blood cell epigenomes. Haematologica 98, 1487–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.ENCODE Project Consortium, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Obradovic A, Vlahos L, Laise P, Worley J, Tan X, Wang A, and Califano A (2021). PISCES: A pipeline for the Systematic, Protein Activity-based Analysis of Single Cell RNA Sequencing Data. Preprint at bioRxiv. [Google Scholar]
- 91.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cancer Genome Atlas Research Network, Ahn J, Akbani R, Ally A, Amin S, Andry CD, Annala M, Aprikian A, Armenia J, Arora A, et al. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veatch JR, Lee SM, Shasha C, Singhi N, Szeto JL, Moshiri AS, Kim TS, Smythe K, Kong P, Fitzgibbon M, et al. (2022). Neoantigen-specific CD4+ T cells in human melanoma have diverse differentiation states and correlate with CD8+ T cell, macrophage, and B cell function. Cancer Cell 40, 393–409.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original code and scRNA-seq data generated by this study have been deposited at Mendeley and are publicly available as of the date of publication at https://doi.org/10.17632/5nnw8xrh5m.1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.