

Abstract
Land plant organellar genomes have extremely low rates of point mutation yet also experience high rates of recombination and genome instability. Characterizing the molecular machinery responsible for these patterns is critical for understanding the evolution of these genomes. While much progress has been made towards understanding recombination activity in land plant organellar genomes, the relationship between recombination pathways and point mutation rates remains uncertain. The organellar targeted mutS homolog MSH1 has previously been shown to suppress point mutations as well as non-allelic recombination between short repeats in Arabidopsis thaliana. We therefore implemented high-fidelity Duplex Sequencing to test if other genes that function in recombination and maintenance of genome stability also affect point mutation rates. We found small to moderate increases in the frequency of single nucleotide variants (SNVs) and indels in mitochondrial and/or plastid genomes of A. thaliana mutant lines lacking radA, recA1, or recA3. In contrast, osb2 and why2 mutants did not exhibit an increase in point mutations compared to wild type (WT) controls. In addition, we analyzed the distribution of SNVs in previously generated Duplex Sequencing data from A. thaliana organellar genomes and found unexpected strand asymmetries and large effects of flanking nucleotides on mutation rates in WT plants and msh1 mutants. Finally, using long-read Oxford Nanopore sequencing, we characterized structural variants in organellar genomes of the mutant lines and show that different short repeat sequences become recombinationally active in different mutant backgrounds. Together, these complementary sequencing approaches shed light on how recombination may impact the extraordinarily low point mutation rates in plant organellar genomes.
INTRODUCTION
Nearly all eukaryotes rely on genes encoded in endosymbiotically derived mitochondrial genomes (mtDNAs) for cellular respiration. Plants and algae additionally rely on the endosymbiotically derived plastid genome (cpDNA) for photosynthesis. In several regards, land plant organellar genome evolution is atypical compared to mtDNA evolution in other eukaryotes (Smith and Keeling 2015). For one, plant organellar genomes have low nucleotide substitution rates relative to those in plant nuclear genomes and to those of many other eukaryotic mtDNAs. The low substitution rates of plant organellar genomes extend even to synonymous sites, which likely experience very little purifying selection, suggesting that the cause of the low evolutionary rates is a low underlying point mutation rate (Wolfe et al. 1987; Drouin et al. 2008).
Compared to the small mtDNAs typical in metazoans (generally below 20 kb) and in algae and fungi (with sizes ranging from approximately 13 to 96 kb and ~20 to 235 kb, respectively), land plant mtDNAs are much larger with sequenced mtDNAs averaging 395 kb (Wu et al. 2022) and a known range extending from 70 kb to over 10 Mb (Boore 1999; Sloan et al. 2012; Skippington et al. 2017; Gualberto and Newton 2017; Sandor et al. 2018; Chen et al. 2019). Very little of this size variation stems from differences in coding capacity, as plant mtDNAs generally contain a subset of the same 41 protein-coding genes (Mower et al. 2012). Instead, the fluctuations in total mtDNA size primarily result from the acquisition and loss of noncoding DNA. Even closely related species possess very little shared noncoding sequence (Kubo and Newton 2008; Skippington et al. 2017). For example, a comparative analysis of the mtDNAs of two species within the Brassicaceae, Arabidopsis thaliana (367 kb) and Brassica napus (222 kb), revealed a mere 78 kb of shared sequence, most of which is coding (Handa 2003). Though size variation of cpDNAs is less extreme than in plant mtDNAs, variation still exists with 98.7% of sequenced land plant cpDNAs ranging from 100–200 kb in size (Xiao-Ming et al. 2017).
Plant organellar genomes also experience exceptionally high rates of structural mutation and rearrangement (Palmer and Herbon 1988). As a result, there is virtually no conservation of synteny between plant mtDNAs, as evidenced by the extensive rearrangements in alignments of mtDNAs from Col-0 and Ler ecotypes of A. thaliana (Stupar et al. 2001; Huang et al. 2005; Davila et al. 2011; Pucker et al. 2019; Zou et al. 2022). The structural instability in plant mtDNAs is partly explained by the presence of repeats of various lengths, which recombine frequently and give rise to multiple isomeric subgenomes with circular, linear and/or branched structures (Palmer and Herbon 1988; Alverson et al. 2011; Wynn and Christensen 2019). In fact, plant mtDNAs lack origins of replication, which help coordinate genome replication in many other eukaryotes, and are instead thought to replicate through break induced recombination (Gualberto and Newton 2017; Chevigny et al. 2020). Land plant cpDNAs are also recombinationally active but usually remain structurally conserved, albeit with some significant exceptions (Smith and Keeling 2015).
The seemingly disparate features of plant organellar evolution (i.e. high rates of recombination and low rates of point mutation) may be unified through a DNA repair mechanism reliant on recombination (Christensen 2014). This hypothesized mechanism hinges on the activity of the mutS homolog MSH1 (Abdelnoor et al. 2003), which is dual-targeted to mitochondria and plastids and has long been known to suppress non-allelic recombination between intermediate-sized repeats (50 to 600 bps) in the A. thaliana mtDNA (Martínez-Zapater et al. 1992; Arrieta-Montiel et al. 2009; Davila et al. 2011; Zou et al. 2022). Plant MSH1 is a chimeric fusion of a mutS gene with a GIY-YIG endonuclease domain (Abdelnoor et al. 2006) that has been proposed to introduce breaks in organellar DNA at the site of mismatches, which would then be repaired through homologous recombination (Christensen 2014, 2018; Ayala-García et al. 2018; Broz et al. 2022). Assays conducted on purified MSH1 in vitro have found that it has DNA binding and endonuclease activity with affinity for displacement loops (D-loops) (Peñafiel-Ayala et al. 2023).
We previously found support for a MSH1-mediated link between recombination and point mutations by using a high-fidelity Duplex Sequencing technique (Kennedy et al. 2014) to screen for single nucleotide variants (SNVs) and indels in msh1 mutants (Wu et al. 2020). In that study, we also included a panel of mutants lacking functional copies of other genes involved in organellar DNA replication, recombination, and/or repair, including the recombination protein RECA3, the paralogous organellar DNA polymerases POLIA and POLIB, and the glycosylases UNG, FPG, and OGG (Wu et al. 2020). Compared to wild type (WT) lines, msh1 mutants incurred SNVs at a ~10-fold increase in mtDNA and a ~100-fold increase in cpDNA, and increases in indel frequencies were even greater. In contrast, recA3 mutants showed only a small (and marginally significant) increase in mtDNA mutation, and none of the other lines in the mutant panel showed a significant increase in SNVs or indels compared to WT plants (Wu et al. 2020).
Here, we investigate additional organellar genome repair proteins (WHY2, RADA, RECA1, OSB2) known to play a role in the suppression of non-allelic recombination in the A. thaliana organellar genomes. WHY2 is a mitochondrially targeted whirly protein that binds single-stranded DNA to inhibit recombination between small repeated sequences via micro-homology mediated end joining (MMEJ) (Cappadocia et al. 2010) and is also the most abundant protein in mitochondrial nucleoids (as measured in A. thaliana cell culture; Fuchs et al. 2020). RADA is a dual-targeted DNA helicase, which has been shown to accelerate the processing of recombination intermediates and promote mtDNA stability in A. thaliana (Chevigny et al. 2022). RECA1 is a plastid-targeted protein that has been proposed to act synergistically with plastid whirly proteins to promote plastid genome integrity either by facilitating polymerase lesion bypass or by reversing stalled replication forks (Rowan et al. 2010; Zampini et al. 2015). OSB2 is a plastid-targeted single-stranded DNA binding protein that has been shown to hamper microhomology-mediated end joining in vitro (García-Medel et al. 2021). Given that we previously saw a weak signal of increased mtDNA mutation in recA3 mutants (Wu et al. 2020), we included another recA3 mutant allele in this study. In addition to these newly generated mutant lines, we also present an extended analysis of Duplex Sequencing data from Wu et al. (2020) to understand how SNVs are distributed among genomic regions, strand (template vs. non-template) of genic regions, and trinucleotide contexts. Finally, we also performed long-read Oxford Nanopore sequencing on the mutant lines, allowing us to study structural mutations and rearrangements. Collectively, these analyses provide a detailed characterization of the effects of numerous recombination-related genes on point mutations and structural variants in plant organellar genomes.
METHODS
Generation and analysis of Duplex Sequencing libraries for SNV and indel detection
We obtained seeds for A. thaliana osb2, radA, recA1, recA3, and why2 mutants from the Arabidopsis Biological Resource Center (Table S1). The generation of Duplex Sequencing data from mutants and matched WT controls (including crossing, plant growth, organelle isolation, DNA extraction, and library preparation) closely followed our previously described protocols (Wu et al. 2020). For each gene of interest, homozygous mutants were used as the paternal pollinators in crosses against WT maternal plants, which introduced ‘clean’ organellar genomes (i.e. never exposed to a mutant background) into the resulting heterozygous F1s. The presence of one WT allele in the F1 heterozygotes should be sufficient for WT-like organelle genome maintenance since the mutant alleles of the repair genes of interest are thought to act recessively (Shedge et al. 2007; Cappadocia et al. 2010; Rowan et al. 2010; Zampini et al. 2015; Wu et al. 2020; García-Medel et al. 2021; Chevigny et al. 2022). The heterozygous F1s were then allowed to self-cross and we identified three homozygous mutant and three homozygous WT F2s, which were also allowed to self-cross. Families of F3 seeds were grown together to obtain sufficient leaf tissue for organelle isolation and mutation detection via Duplex Sequencing.
The only notable differences between the methods in this study compared to Wu et al. 2020 were 1) we only isolated organelles for which the protein of interest is targeted (plastid: OSB2, RADA, and RECA1; mitochondrial: RADA, RECA3, and WHY2), whereas in Wu et al., (2020) we isolated both organelles regardless of targeting. 2) We adjusted our Duplex Sequencing library construction protocol to obtain larger inserts by ultrasonicating the DNA for only 60 seconds (three bouts of 20 seconds, with 15 second pauses between each) and size selecting libraries with a 2% gel on a BluePippin (Sage Science), using a specified target range of 400–700 bp. 3) We implemented a new approach to filter spurious variant calls resulting from nuclear insertions of mtDNA and cpDNA (NUMTs and NUPTs) by comparing putative mutations directly against the A. thaliana nuclear genome (TAIR 10.2; Berardini et al. 2015) and the new assembly of the large NUMT on chromosome 2 (Fields et al. 2022), replacing the k-mer based NUMT/NUPT filtering approach described in Wu et al. (2020).
Generation and analysis of nanopore sequencing libraries for structural variant detection
Nanopore libraries were produced from the same DNA samples that were used for Duplex Sequencing. Sequencing libraries were created following the protocol outlined in the Oxford Nanopore Technologies Rapid Barcoding Kit 96 (SQK-RBK110–96) manual (v110 Mar 24, 2021 revision) and were sequenced on MinION flow cells (FLO-MIN106) under the control of MinKNOW software v22.08.4 or 22.08.9. Multiplexed libraries from cpDNA samples were pooled and run on a single flow cell, whereas pooled mtDNA libraries were run on two flow cells. All runs were conducted for 72 hrs with a minimum read length of 200 bp. Data were processed using the Guppy Basecalling Software v6.3.4+cfaa134.
We sequenced three mutant replicates and one matched WT control for each gene of interest. Mutant lines for the cpDNA samples included msh1 (CS3246), osb2, recA1, and radA (only two radA mutants were sequenced due to a lack of DNA in mutant replicate 2), while mutant lines for the mtDNA samples included msh1 (CS3246), recA3, why2, and radA. The total sequencing yield (3.72 Gb) in our initial run of 15 cpDNA samples was an order of magnitude higher than our subsequent run with the 16 mtDNA samples (0.33 Gb). To increase mtDNA coverage we re-sequenced 12 of those mtDNA samples (all but the msh1 mutants and matched WT control) in a third run, which had a similar low yield (0.42 Gb) to the second run. In all cases, samples were run on fresh flow cells as opposed to flow cells that had been washed for a second run. Because the msh1 and radA mtDNA samples produced very little data (Table S4), we used the mtDNA contamination in the msh1 and radA cpDNA samples in downstream analyses of the nanopore data.
To calculate mitochondrial and plastid read depth, we aligned the nanopore reads to the organellar genomes with minimap2 (version 2.24; Li 2018) and tabulated depth at each position with bedtools (version 2.30.0; Quinlan and Hall 2010). We calculated the average depth in 1000-bp sliding windows tiling the organellar genomes and plotted depth as a normalized mutant:WT ratio.
The nanopore reads were analyzed with HiFiSr (https://github.com/zouyinstein/hifisr), a software tool developed to identify structural variants using BLASTn alignments of long reads in plant organellar genomes (Zou et al. 2022). Because the tool was originally developed for PacBio HiFi reads, which are more accurate than nanopore reads, we required at least two independent nanopore reads to support putative indels. In addition, we constrained our analysis to reads with only one or two BLASTn hits, disregarding the reads with three or more BLASTn hits (which may originate from reads that span two or more recombined repeats). For reads with two BLASTn hits, we compared the breakpoints of putative recombination events with the repeats in the A. thaliana organellar genomes, which are reported in Tables S10 (mtDNA) and S28 (cpDNA) by Zou et al. (2022). We calculated recombination frequencies for each repeat pair as the number of recombined reads divided by the total number of repeat-spanning reads. To compute genome-wide repeat frequencies, we restricted the analyses to repeats that showed a total of at least ten mtDNA recombination reads across all replicates. Because cpDNA recombination events were much less common, we lowered the threshold to a minimum of three recombining reads per repeat for calculating recombinaiton frequencies. All of the matched WT controls were averaged for comparisons againsnt the mutant variant frequencies because we only sequenced one WT control for each gene of interest.
RESULTS
Duplex Sequencing coverage
We generated Duplex Sequencing libraries from DNA extracted from isolated organelles to test if genes involved in recombination-suppression also impact accumulation of SNVs and short indels in A. thaliana organellar genomes. Duplex Sequencing libraries were sequenced on a NovaSeq 6000 to produce between 30.6 to 139.1 million paired-end reads (2×150 nt) per library (Table S2). Processing the Duplex Sequencing libraries to collapse Illumina reads into consensus sequences and map them to organellar genomes resulted in coverage of 94.2 to 816.3× in the mitochondrial libraries (radA, recA3, and why2) and 234.2 to 1176.6× in the plastid libraries (radA, recA1, and osb2; Table S2).
Increased SNV and indel frequency in radA, recA1, and recA3 mutants
We compared variant frequencies of each mutant to the matched WT controls (two-tailed t-test) and found significant increases in SNV and indel frequencies in the radA mutants (p-values reported in Fig. 1). We also observed significant indel and weakly significant SNV increases in the recA3 and recA1 mutants in the mtDNA and cpDNA, respectively. We analyzed our previously generated recA3 mutant from Wu et al., (2020), which represents an independent mutant allele of recA3, and similarly found significant indel and weakly significant SNV increases in mtDNA (Fig. S1). In total, we detected 204 SNVs and 123 indels in the newly generated Duplex Sequencing libraries (File S1). Dinucleotide mutations involve neighboring sites both experiencing a substitution at the same time and are increasingly being recognized as an important type of mutation (Kaplanis et al. 2019). We assessed whether these mutations increase in frequency in any of the analyzed mutant backgrounds but found no significant differences relative to WT controls (Wilcoxon signed rank test, p > 0.05, Fig. S2).
Figure 1.
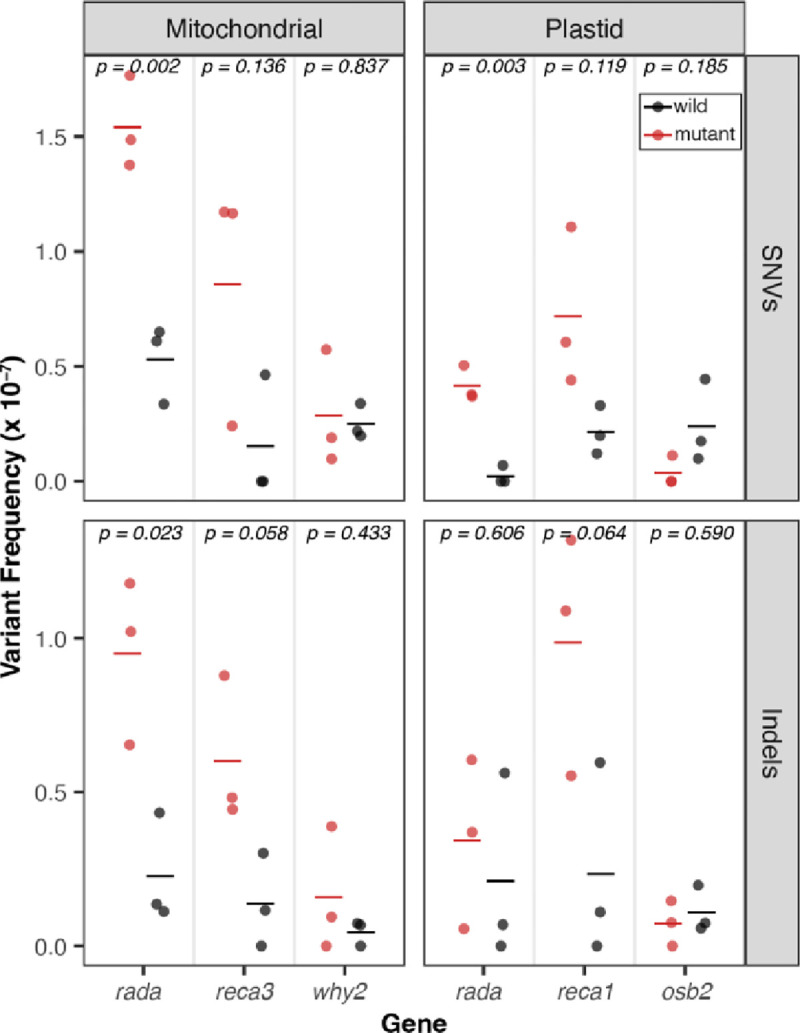
De novo point mutations measured with Duplex Sequencing. For each gene of interest (x-axis), mutant lines are plotted in red, and matched WT controls are plotted in black. The individual biological replicates are plotted as circles and group averages are plotted as dashes. Panels separate the data by genome; left column: Mitochondria and right column: Plastid, and by point mutation type; top row: SNVs and bottom row: indels. Variant frequencies (y-axis) were calculated as the total number of SNVs/total Duplex Sequencing coverage. P-values show the result of a two-tailed t-test comparing WT vs mutant mutation frequencies for each gene of interest.
Decreased frequency of CG→TA transitions in the mtDNA of newly generated WT lines
The mutant lines assayed in both this study and in Wu et al. (2020) were sequenced with matched WT controls. Surprisingly, pooled WT SNV frequencies generated in the current study were lower than the pooled WT SNV frequencies from the Wu et al. (2020) dataset (2.8×10−8 vs. 1.7×10−7, t-test, p = 8.9×10−12), driven by a decrease in CG→TA transitions (t-test, p = 2.2×10−10; Fig. 2, File S1). To understand if the decreased SNV rate in the newly generated WT libraries (Fig. 2) resulted from the changes we made to our library preparation protocol, we created a Duplex Sequencing library following our new protocol using one of the original WT DNA samples from Wu et al., (2020). This new library had an SNV rate of 1.57×10−7 which is in line with the SNV rates observed in the WT libraries from the 2020 study (Fig 2). In fact, the new SNV rate for this DNA sample was slightly higher than that of the original library (1.39×10−7). Given that the newly created libraries were all size selected on a BluePippin, which involves mixing the libraries with fluorescein labeled DNA as an internal standard for gauging DNA migration speed, we re-sequenced two stored libraries from Wu et al., (2020) with and without size selection on the BluePippin. The inclusion of the sample without size selection on the BluePippin served as a control for the sample processed on the BluePippin and also as an independent test to understand if changes in the sequencing platform could be responsible (all samples were sequenced on a NovaSeq 6000, but the chemistry of the flow cells has been updated). These re-sequenced libraries had SNV rates typical of the old WT libraries of 1.97×10−7 (size selected library) and 1.47×10−7 (not size selected). Again, these values were slightly higher than the SNV rates from the original round of sequencing (1. 36×10−7 and 1.39×10−7, respectively). Therefore, it seems highly unlikely that the decreased SNV rate in the new WT libraries is associated with the changes we made to our library preparation protocol. Instead, these appear to be genuine differences in the DNA samples, perhaps due to unknown variation in the growth conditions or DNA extraction procedures between the two batches.
Figure 2.

Comparison of the mutational spectrum of pooled WT controls from the current study (orange) vs. the WT controls from Wu et al. 2020 (blue). The two panels show the mitochondrial and plastid data and the x-axis separates substitutions type by transversions vs. transitions and further by the six types of substitutions. Individual biological replicates are plotted as circles while group averages are plotted as dashes. Only CG→TA transitions showed a significant increase in the old data set (two-tailed t-test; p=2.2×1010).
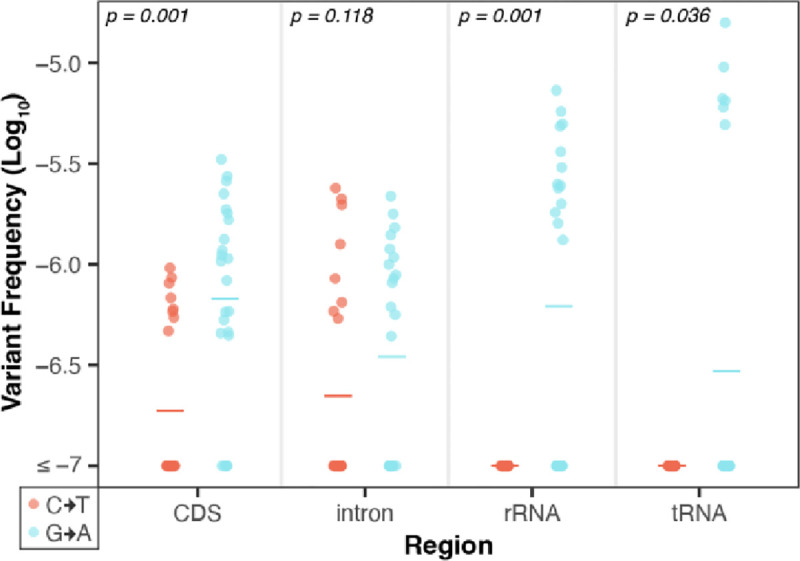
SNV frequencies are similar among different genomic regions
To gain a deeper understanding of mutational process in the organellar genomes, we next turned our attention to the distribution of SNVs, focusing primarily on the msh1 mutants and the pooled WT libraries from the Wu et al. (2020) study, given the larger number of mutations in those datasets. First, we assessed if the SNVs in msh1 mutants and pooled WT libraries from Wu et al. (2020) are evenly distributed between intergenic, protein-coding (CDS), intronic, rRNA, and tRNA regions (Fig. 3) and found no significant differences among genomic regions (Kruskal-Wallis test, p > 0.05, Table S3) except in the WT plastid comparison, which is likely not biologically meaningful, given the small number of observed WT plastid SNVs (Fig. 2). Given that the vast majority of mtDNA SNVs in the Wu et al. (2020) WT dataset are CG→TA transitions, we separately tested if this class of substitutions is evenly distributed across regions and found significant differences (Kruskal-Wallis test, p = 0.0295), driven by a decrease in tRNA genes compared to intergenic sequences (pairwise comparisons with Wilcoxon rank sum test, p=0.0013). However, tRNA genes make up a small fraction of the genome and, thus, are subject to higher sampling variance, precluding any confident conclusions about whether they actually accumulate fewer CG→TA transitions than intergenic sequence.
Figure 3.

Distribution of WT (black) and msh1 (red) SNVs (from Wu et al., 2020) across genomic region. The individual biological replicates are plotted as circles and group averages are plotted as dashes. Panels separate the data by genome; left column: Mitochondria and right column: Plastid, and by genotype with msh1 mutants on top and WT on the bottom. Note the difference in y-axis scale for msh1 mutants and WT. For each of the four panels, we performed a Kruskal-Wallis test and found no significant difference between genomic regions except the WT plastid panel (p = 0.022) where comparisons between regions are likely not biologically meaningful given the low number of WT plastid mutations. Note that for this and subsequent analyses of the msh1 Duplex Sequencing data, we pooled the two null msh1 alleles to increase statistical power.
C→T substitutions are more common on the template strand in genic regions
Next, we performed a strand asymmetry analysis to understand if the SNVs in these datasets are evenly distributed on template vs. non-template (i.e., sense or coding) strands in the CDS, intronic, rRNA, and tRNA regions of the organellar genomes. The analysis of the CG→TA transitions from the Wu et al. (2020) WT dataset revealed that G→A substitutions are significantly enriched on the non-template strand of the DNA (paired Wilcoxon signed-rank test; p < 0.05 for CDS, rRNA and tRNA genes). Conversely, C→T substitutions predominately occur on the template strand, which is read by RNA polymerases during transcription (Fig. 4). This asymmetry is most striking in rRNA and tRNA genes, where every C→T substitution occurred on the template strand (25 in rRNA and 7 in tRNA). CG→TA transitions were also asymmetrically distributed between strands in genic regions of the Wu et al. (2020) msh1 mutants (Fig. 5), though only in certain regions of the mtDNA (Fig. 5 top right panel), and not in the cpDNA (Fig. 5 bottom right panel). We also investigated strand asymmetries in the AT→GC transitions of the Wu et al. (2020) msh1 mutants and found a trend toward more C→T substitutions on the template strand of plastid genes (Fig. 5 left panels). We did not investigate strand asymmetries for the other substitution classes in WT or msh1 mutants because the small number of data points precludes meaningful comparisons between strands (see Fig. 5 of Wu et al. 2020).
Figure 4.
Strand asymmetry analysis of CG→TA transitions in the WT mtDNA Duplex Sequencing data from Wu et al. (2020). Shown are the log-transformed SNV frequencies (y-axis) of C→T (red) vs. G→A (blue) mutations on the non-template strand of all genes, separated by genomic region (x-axis). The individual biological replicates are plotted as circles and group averages are plotted as dashes. P-values show the result of paired Wilcoxon tests comparing the complementary substitution classes in each genomic region. In all but intronic regions, G→A substitutions are significantly higher on the non-template strand (conversely, C→T substitutions are significantly higher on the template strand). Strikingly, in all of the observed CG>TA transitions in the rRNA and tRNA genes the C→T substitution occurred on the template strand (i.e., all the G→A substitutions occurred on the non-template stand).
Figure 5.

Strand asymmetry analysis of CG→TA and AT→GC transitions in the msh1 Duplex Sequencing data from Wu et al. (2020). Shown are the log-transformed SNV frequencies (y-axis) of mutations on the non-template strands of all genes with complementary substitution types designated by color (see figure legends for colors of specific substitution types). The individual biological replicates are plotted as circles, and group averages are plotted as dashes. The panels divide the data by transition type, with AT→GC transitions on the left and CG→TA transitions shown on the right, and by genome, with mitochondrial data on the top and plastid data on the bottom. Transversions were not analyzed because there were relatively few observed mutations of this type in the msh1 duplex data. P-values show the result of paired t-tests comparing the complementary substitution classes in each genomic region.
CG→TA transition frequencies vary depending on trinucleotide context
To understand how surrounding nucleotides impact SNV accumulation in plant organellar genomes, we performed a trinucleotide analysis, again focusing on CG→TA transitions in WT and both transition types in msh1 mutants, due to a lack of data in other substitution classes. In the WT dataset (Wu et al. 2020), we found that CG→TA transitions are 8.4-fold more common in the mtDNA and 3.7-fold more common in the cpDNA when the C is 3′ of a pyrimidine (Fig. 6). Interestingly, this same trinucleotide context (5′ pyrimidine) is not enriched for CG→TA transitions in the msh1 mutant data. Instead CG→TA transitions are 3.0-fold more common when the C is 5′ of a G in the msh1 mutants (Fig. 7 right panels). Meanwhile AT→GC transitions are 1.8-fold more common when the A is 5′ of a C (Fig. 7 left panels). In all cases, these trinucleotide mutation frequencies are normalized by the total coverage of a given trinucleotide context so that the values are not inflated in trinucleotides that are relatively common in the mtDNA.
Figure 6.
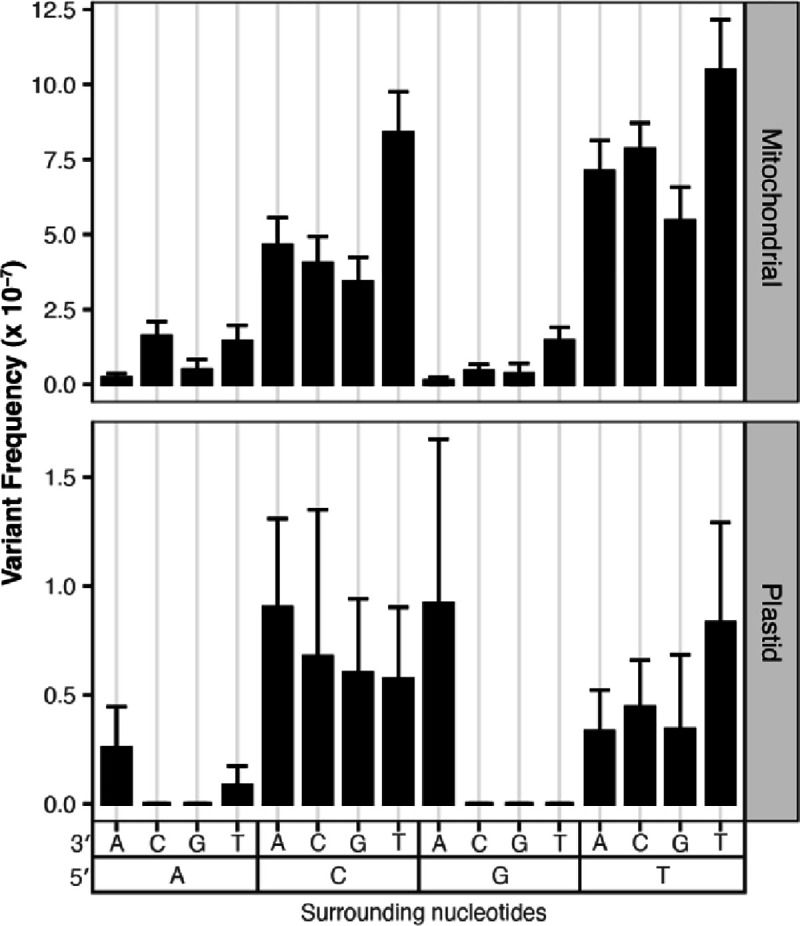
Analysis of surrounding nucleotides on C→T transition frequencies in the WT Duplex Sequencing data from Wu et al. (2020). The panels divide the data based on genome with mitochondrial data on the top and plastid data on the bottom, note the difference in the y-axis scale, as CG>TA were less frequent in the plastid. The x-axis captures the trinucleotide context with downstream nucleotides displayed next to the 3′ and upstream nucleotides display next to the 5′. The data suggest that trinucleotide contexts with upstream pyrimidines (5′ CCN 3′ and 5′ TCN 3′, where N is any nucleotide) have increased frequencies of C>T substitutions.
Figure 7.

Analysis of surrounding nucleotides on A→G and C→T transition frequencies in the msh1 Duplex Sequencing data from Wu et al. (2020). The panels divide the data based on substitution type (A→G substitutions on the left and C→T substitutions on the right) and by genome (mitochondrial data on the top and plastid data on the bottom). The x-axis captures the trinucleotide context with downstream nucleotides displayed next to the 3′ and upstream nucleotides display next to the 5′. The A→G data suggest that trinucleotide contexts with downstream Cs (5′ NAC 3′) have increased frequencies of A→G substitutions. The C→T data suggest that trinucleotide contexts with downstream Gs (5′ NCG 3′) have increased frequencies of C→T substitutions.
Chloroplast extractions produced an order of magnitude more nanopore sequencing data than mitochondrial extractions
We next generated long-read Oxford Nanopore libraries to gain a deeper understanding of how the genes in our panel impact plant organellar genome stability. Unexpectedly, the libraries produced from the mitochondrial isolations sequenced poorly compared to the plastid-derived libraries (see methods), so we investigated cross-organelle contamination (mtDNA molecules in the plastid-derived samples and cpDNA molecules in mitochondrially derived samples) to understand if poor mtDNA sequencing performance was inherit to the mtDNA or associated with differences in the organellar isolation methods. The level of mtDNA contamination in the plastid-derived nanopore libraries is similar to the level of contamination in the Duplex Sequencing libraries (Fig. S3). The average median read length of the mitochondrial derived nanopore libraries is about 2.5-fold higher than the average median read length of the plastid-derived libraries (2.48 kb vs. 1.08 kb, respectively). In the plastid derived nanopore libraries, the median lengths of the contaminating mtDNA reads tend to be slightly longer than the median lengths of native cpDNA reads (average median lengths of 1.17 kb vs 0.98 kb, respectively), though there is substantial variation between samples (Fig. S4). In the mitochondrially derived libraries, the contaminating cpDNA and native mtDNA median read lengths show more correlation (average median lengths of 2.41 kb and 2.56 kb, respectively; Fig. S4).
These analyses suggest that the difference in yields for the different nanopore runs is likely related to differences in the organellar isolation methods. One unique feature of the mitochondrial isolation protocol is the use of a DNase I treatment to remove contaminating nuclear and plastid DNA molecules (Wu et al. 2020). It is possible that this treatment results in nicking of the mtDNA that interrupts the molecules as they are threaded through the nanopore in a single-stranded fashion. Such nicking would not be expected to disrupt Duplex Sequencing library creation since the first step of making Duplex Sequencing libraries is to break DNA into small fragments via ultrasonication. However, this explanation is somewhat inconsistent with the 2.5-fold greater median read length in the mitochondrially derived nanopore libraries. Fortunately, the contaminating mtDNA derived reads in the msh1 and radA cpDNA sequenced samples provided sufficient mtDNA coverage for analyzing structural variation in the mtDNA (Table S4, Figure S3 left panel).
Repeat-mediated recombination drives distinct patterns of mtDNA instability in msh1, radA, and recA3 mutants
Given the known role of recombination-related genes in maintaining organellar genome copy number and structural stability (Arrieta-Montiel et al. 2009; Davila et al. 2011; Miller-Messmer et al. 2012; Chevigny et al. 2022; Zou et al. 2022), we analyzed the ratio of mutant coverage to WT coverage to characterize structural perturbations on a genome-wide level (Fig. 8). We see distinct variation patterns in the mtDNA coverage in msh1, radA and recA3 mutants, consistent with the expected structural effects of these genes (Fig. 8) and similar to previously documented coverage patterns (Wu et al. 2020; Chevigny et al. 2022). In contrast, the why2 coverage does not deviate from WT coverage, suggesting there is no substantial and consistent structural effect of losing why2. In recA3, the nanopore and Duplex Sequencing lines are tightly correlated, while the nanopore data tends to show greater variance in the msh1, radA, and why2 plots, perhaps because of the lower nanopore coverage in those samples (Table S5; Figs. S6 and S67). Interestingly, radA and recA3 share many major coverage peaks and valleys, suggesting genome structure is perturbed in similar ways in these mutants (Fig. 8, Figs S6 and S7). Compared to the mitochondrial samples, the cpDNA samples display much less coverage variation (Fig. S5), with a notable exception in the recA1 nanopore data. However, inspection of the coverage in the individual cpDNA replicates (Fig. S8) reveals depth irregularities in the WT control compared to the other WT samples. Regardless, the recA1 Duplex Sequencing data does not show any depth variation along the cpDNA, so the nanopore result does not appear to reflect a biological effect on cpDNA structure. One other intriguing pattern in the cpDNA plots is an apparent correlation in peaks and valleys in radA and osb2 in the Duplex Sequencing data (most notable is the shared valley at 112 kb). However, inspection of the individual recA1 mutant and matched WT control replicates (Fig. S9) reveals all samples have a dip at 112 kb and the dip is more pronounced in one or more of the osb2 and radA mutants. Given the large number of PCR cycles used to amplify the Duplex Sequencing libraries (19 cycles) the unified movement of all replicates is likely explained in part by amplification bias in AT or GC rich regions. Therefore, variation in amplification bias may result in lower coverage of AT or GC rich regions, so these patterns are likely not biological.
Figure 8.

Normalized coverage of mitochondria genomes in mutant lines of interest. Coverage of each Duplex Sequencing (red) or nanopore (blue) library was calculated in 1000-bp windows. Mutant coverage was pooled and divided by WT coverage and the resulting ratios were normalized to 1 for plotting. The total amount of sequencing data used to generate each plot is shown in the top left corner of each panel (red=Duplex Sequencing and blue=nanopore) and is included to highlight the instances where disagreement between the Duplex Sequencing and nanopore lines may be explained by increased variance in the nanopore sample due to lower mtDNA coverage. Repeats that are likely important for driving coverage variation across the mtDNA are plotted above (also see, Table 1) according to Figure 6 of Chevigny et al. 2022. Regions with altered stoichiometry and flanked by repeats are shown as colored blocks, as in Figure 6 of Chevigny et al. 2022.
We analyzed the nanopore reads for evidence of repeat-mediated recombination. To do so, we calculated recombination frequencies for each repeat pair as the count of nanopore reads that recombined at a given repeat (according the BLASTn alignments generated by HiFiSr (Zou et al. 2022)) divided by the total number of reads that mapped to the repeat. Table 1 shows the five repeats with the highest recombination frequency for each mutant genotype and the matched WT controls. Fig. 9 shows examples of how the long nanopore reads map to the mitochondrial genome following recombination at inverted (Fig. 9A) or directed repeats (Fig. 9B and C).
Table 1.
Repeat-specific recombination frequencies at the five most recombinationally active mtDNA repeats for each genotype
| Genotype | Recombined reads | Total repeat spanning reads | Recomb. freq. | Repeat name | Repeat pair coordinates | Percent ID | Length |
|---|---|---|---|---|---|---|---|
| msh1 | 49 | 178 | 0.284 | B | 41464–41999, 32196–321431 | 99.81 | 537 |
| msh1 | 51 | 199 | 0.268 | A* | 19682–20237, 34620–346763 | 99.82 | 556 |
| msh1 | 40 | 157 | 0.256 | G | 30938–31272, 27139–271061 | 99.40 | 335 |
| msh1 | 36 | 171 | 0.242 | MMJS | 134427–135193, 257452–258143 | 88.66 | 767 |
| msh1 | 48 | 203 | 0.222 | D | 6118–6569, 84540–84089 | 97.79 | 452 |
| radA | 94 | 125 | 0.692 | L* | 270775–271023, 331877–332125 | 100 | 249 |
| radA | 135 | 262 | 0.476 | A* | 19682–20237, 34620–346763 | 99.82 | 556 |
| radA | 201 | 529 | 0.4 | EE* | 65547–65673, 73611–73737 | 99.21 | 127 |
| radA | 124 | 284 | 0.357 | F* | 206095–206444, 246766–247115 | 100 | 350 |
| radA | 43 | 258 | 0.144 | X | 288315–288518, 306969–307174 | 97.57 | 206 |
| recA3 | 198 | 907 | 0.227 | L* | 270775–271023, 331877–332125 | 100 | 249 |
| recA3 | 210 | 1384 | 0.168 | EE* | 65547–65673, 73611–73737 | 99.21 | 127 |
| recA3 | 159 | 1019 | 0.149 | F* | 206095–206444, 246766–247115 | 100 | 350 |
| recA3 | 88 | 770 | 0.116 | A* | 19682–20237, 34620–346763 | 99.82 | 556 |
| recA3 | 67 | 1111 | 0.06 | I* | 30442–30722, 255122–254842 | 99.64 | 281 |
| why2 | 1 | 274 | 0.042 | unnamed | 239143–239268, 26378–263905 | 91.27 | 126 |
| why2 | 5 | 256 | 0.007 | A* | 19682–20237, 34620–346763 | 99.82 | 556 |
| why2 | 5 | 272 | 0.007 | F* | 206095–206444, 246766–247115 | 100 | 350 |
| why2 | 5 | 260 | 0.007 | L* | 270775–271023, 331877–332125 | 100 | 249 |
| why2 | 3 | 219 | 0.005 | D | 6118–6569, 84540–84089 | 97.79 | 452 |
| WT | 23 | 902 | 0.093 | A* | 19682–20237, 34620–346763 | 99.82 | 556 |
| WT | 10 | 858 | 0.057 | L* | 270775–271023, 331877–332125 | 100 | 249 |
| WT | 13 | 931 | 0.055 | B | 41464–41999, 32196–321431 | 99.81 | 537 |
| WT | 6 | 1050 | 0.041 | C | 36362–36824, 14440–143947 | 99.57 | 463 |
| WT | 11 | 933 | 0.04 | MMJS | 134427–135193, 257452–258143 | 88.66 | 767 |
Listed are the five most active repeats for each genotype, ordered by the recombination frequency within each genotype. Repeat names were sourced from Table S11 of Zou et al., 2022. For the msh1 mtDNA analysis, we relied exclusively the plastid-derived msh1 samples, and for the radA mtDNA analysis, we used a combination of the low coverage radA mitochondrial samples and the plastid radA samples (see main text). For the WT comparison, we took the average across the single matched WT libraries that were sequenced with each mutant line, including msh1and radA WT plastid samples (Table S4). The repeats that are also plotted in Figure 8 are denoted with an asterisk. Repeats which make are among the top five most active repeats in more than one genotype are bolded. Repeat-specific recombination frequencies that exceed 0.1 are shown in bold, and note that none of the WT or why2 repeat specific recombination frequencies meet this threshold.
Figure 9.

Examples of 3 nanopore reads from radA mitochondrial replicate 1 that capture repeat-mediated recombination. Nanopore reads that derive from recombination between inverted repeats map with two hits, one in the forward orientation and the other in the reverse orientation, both flanked by the sequence of a repeat, as shown in A where the 29-kb read is flanked by repeats I-1 and I-2. Recombination between direct repeats results in two hits in the same orientation with a deletion of the intervening sequence (B). The alternative product of recombination between direct repeats is the production of a small circular molecule. We identified a number of putative circular molecules or tandem duplications mediated by recombination between repeats EE-1 and EE-2, which map with two hits in the same orientation, but with a section of the end of the read mapping in front of the end of the read (C).
We calculated genome-wide recombination frequencies for the mtDNA by summing across repeats with at least 10 recombining reads (File S2). The threshold was lowered to repeats with at least three recombining reads in the cpDNA given the smaller number of recombining reads observed in the cpDNA (File S3). We found significant differences in the frequency of mtDNA rearrangements among the WT and mutant lines (one-way ANOVA, p = 1.5×10−8, Fig. 10), which were driven by increases in recombination frequency in msh1, radA and recA3 compared to WT (Tukey pairwise comparison, p = 3.0×10−7, 2.0×10−7, and 0.02, respectively). In contrast, there was no mtDNA recombination frequency difference between why2 mutants and WT samples (Tukey pairwise comparison, p = 0.99). We found that different repeats apparently become active in different mutant background as evidenced by a two-way ANOVA with a significant interaction between genotype and repeat (p < 2.0×10−16). Because our analysis focuses on reads with two or fewer BLASTn hits, we may have underestimated global recombination frequencies, especially in mutant backgrounds, as a PacBio HiFi study found that such reads with three or more BLASTn hits (which arise when reads span two or more repats that have recombined) comprise 0.34% and 8.69% of all reads in WT and msh1, respectively (Zou et al. 2022). Consistent with previous characterization of repeat mediated recombination in plant mtDNAs (Arrieta-Montiel et al. 2009; Davila et al. 2011; Miller-Messmer et al. 2012; Chevigny et al. 2022; Zou et al. 2022), we found that repeat length and percent identity are also predictive of recombination frequency through a three-way ANCOVA with repeat length and percent identity as continuous variables (p = 1.8×10−12 and 1.4×10−6, respectively) and genotype as a categorical variable (p = 2.0×10−22). There were no significant differences in repeat-mediated recombination between any of the cpDNA mutants (msh1, radA, osb2, and recA1) compared to the WT samples (one-way ANOVA, p = 0.849; Fig. 9). We identified no insertions in the HiFiSr variant calls (after requiring at least two nanopore reads to support a putative insertion) and only a single cpDNA deletion of 106 bp in msh1 mutant replicate 2, which was supported by 18 independent nanopore reads (cpDNA position 148490–148596).
Figure 10.

Frequency of repeat-mediated structural variants in the nanopore data. The individual biological replicates are plotted as circles with the size of the circle scaled by the number of repeats that are covered in the nanopore alignments. Closed circles are the libraries from mitochondrial extractions, while the open circles are libraries from the plastid extractions. In some cases, cpDNA extractions were used to harvest contaminating mtDNA-mapping reads because of low yield from direct sequencing of the mtDNA extractions. Group averages are plotted as dashes. Mutants are plotted in red, while WT samples are plotted in black. Letters represent statistically significant groupings according to Tukey pairwise comparisons on a one-way ANOVA (p<0.001). There were no differences among plastid genotypes.
DISCUSSION
Potential causes of elevated organellar mutation rates in lines with disrupted recombination machinery
By utilizing highly accurate Duplex Sequencing for point mutation detection and long-read Oxford Nanopore sequencing for structural variant detection, we have characterized the overall organellar mutational dynamics in A. thaliana lines lacking genes with roles in organellar genome recombination. The increases in point mutations we observed in radA, recA3, and recA1 are much smaller than the effects previously observed in msh1 mutants (Wu et al. 2020) where mutants experience 6.0-fold and 116.5-fold increases in SNVs (in mtDNA and cpDNA, respectively) and 86.6-fold and 790.6-fold increases in indels (in mtDNA and cpDNA, respectively). In contrast, radA mutants incurred 2.6-fold and 12.6-fold more mtDNA and cpDNA SNVs (respectively) and 5.1-fold and 3.1-fold more mtDNA and cpDNA indels (respectively) than the matched WT controls. The point mutation increases in recA3 and recA1 were even smaller than in the radA mutants. One complication with directly comparing the mutant vs. WT fold changes across the newly generated mutant lines compared to those generated in Wu et. al., (2020) is the decrease in WT mutation rates in the new genes (Fig. 2). Because of the shift in the baseline WT rates, the numbers cited above may actually underestimate the gap in effect size between msh1 and the newly analyzed genes.
The point mutation increases in msh1 mutants have clear mechanistic explanations which were first predicted based on the MSH1 mismatch recognition and GIY-YIG endonuclease domains (Christensen 2014; Wu et al. 2020). In contrast, given that RADA, RECA3 and RECA1 are all thought to function in the resolution of recombination intermediates, it is more difficult to explain the mechanisms responsible for increased point mutations in these lines. One possibility is that in the absence of one recombination pathway, recombining molecules are shuttled into an alternative, less faithful recombination pathway. For example, in mutant lines deficient in homologous recombination (HR), double-stranded breaks (DSBs) may be repaired via error prone nonhomologous end joining (NHEJ) or MMEJ, which could drive increases in indels and SNVs (Waters et al. 2014; García-Medel et al. 2019). Evidence suggests that RADA functions as the principal branch migration factor in a primary mtDNA and cpDNA homologous recombination (HR) pathway, while RECA3 may fill the same role as RADA in a partially redundant and less utilized mtDNA specific-HR pathway (Chevigny et al. 2022). Interestingly, RECA2 is thought to initiate recombination in both pathways and is essential in plants (Miller-Messmer et al. 2012; Chevigny et al. 2022). The larger SNV and indel increases in the radA mutants than in the recA3 mutants may reflect the relative utilization (and importance) of these two partially redundant HR pathways (Chevigny et al. 2022). Similarly, previous studies have documented increased NHEJ and MMEJ in cpDNA of recA1 mutants (Zampini et al. 2015), which is consistent with the significant increase in indels and marginally significant increase in SNVs reported here (Fig. 1).
Another possibility is that the rise in point mutations is an indirect effect of increased repeat-mediated recombination and its associated harm to organelle function. Increased recombination between short repeat sequences may disrupt genes, organellar genome stoichiometry, and genome organellar replication, which is recombination-dependent in plants (Shedge et al. 2007; Rowan et al. 2010; Chevigny et al. 2020). Plant organellar genomes encode proteins necessary for the electron transport chains of respiration and photosynthesis and disruption of these pathways can result in the excess production of DNA damaging reactive oxygen species (ROS; Liu et al. 2021). Although a direct link between ROS-mediated damage to DNA and mutation rates remains contentious (Kennedy et al. 2013; Itsara et al. 2014; Broz et al. 2021; Waneka et al. 2021; Sanchez-Contreras et al. 2021), ROS molecules have been shown to indirectly affect point mutation rates by impairing proofreading capabilities via damage to the metazoan mtDNA polymerase (Pol γ; Anderson et al. 2020). Impairment of organellar function is also consistent with phenotypic growth defects in radA, which include retarded development and distorted leaves with chlorotic sectors (Chevigny et al. 2022).
Potential explanations of mutational biases based on DNA strand asymmetry and flanking nucleotides
We found that SNVs in the msh1 mutants and WT plants from Wu et al., (2020) had biased distributions in terms of strand (non-template vs. template) and trinucleotide context. Such patterns are useful for understanding the underlying mechanisms driving mutation formation (Haradhvala et al. 2016; Sun et al. 2018; Moeckel et al. 2023). For example, CG→TA strand asymmetries documented in diverse metazoan mtDNAs have been proposed to result from the two DNA strands experiencing unequal time in single-stranded states during mtDNA replication, since single-stranded DNA is more vulnerable to cytosine deamination (a primary driver of CG→TA transitions) (Kennedy et al. 2013; Itsara et al. 2014; Arbeithuber et al. 2020; Waneka et al. 2021; Sanchez-Contreras et al. 2021). In mammals, C→T substitutions are ~10-fold more common than G→A substitution on the mtDNA heavy strand (H-strand), which likely spends more time in a single-stranded state as the mtDNA is copied via a strand-asynchronous replication mechanism (Kennedy et al. 2013; Arbeithuber et al. 2020). Further, the C→T substitutions form two gradients starting at the two H-strand origins of replication, consistent with the regions closest to the origin being single stranded for longer (Sanchez-Contreras et al. 2021).
The substantial CG→TA strand asymmetries we observed in the mtDNA of the Wu et al., (2020) WT libraries are unlikely to be explained by replication mechanisms given that plants mtDNAs lack discrete origins of replication or dedicated ‘leading and lagging’ strands (alternatively referred to as light and heavy strands, respectively, in some systems) and instead rely on recombination-mediated replication (Gualberto and Newton 2017; Brieba 2019; Chevigny et al. 2020). Instead, our strand asymmetry analysis focused on genic regions, motivated by well-established patterns of more C→T than G→A substitution on non-template strands which spend more time in exposed single-stranded during transcription (Haradhvala et al. 2016; Vöhringer et al. 2021; Moeckel et al. 2023). Surprisingly, we found an opposite pattern with template strands exhibiting far more C→T than G→A substitutions (Fig. 4). This effect was especially pronounced in rRNA and tRNA genes where the C→T substitutions occurred on the template strand in all 32 observed CG→TA transitions. An enrichment of C→T substitutions on template strands also occurred in the mtDNA (but not the cpDNA) of the msh1 mutants, though there was less power for detecting statistically significant effects (Fig. 5). The overabundance of A→G compared to T→C substitutions in msh1 mutant cpDNA template strands also occurs in the opposite direction of predicted effects given that the non-template strand is again expected to experience increased adenine deamination (which leads to A→G substitutions; Mugal et al. 2009; Sanchez-Contreras et al. 2021).
Enrichment of C→T and A→G substitutions on template strands is puzzling, and to our knowledge there are no other instances where this widespread transcriptional asymmetry has been reversed (Mugal et al. 2009; Moeckel et al. 2023). Reversals in strand asymmetries have been reported in metazoan mitochondrial genomes, but in these cases the asymmetries are replication based, and the reversals are proceeded by an inversion of the origin of replication, effectively switching the leading and lagging strands (Wei et al. 2010). It is notable that the WT CG→TA asymmetries are most pronounced in the rRNA and tRNA genes (Fig. 4), which are likely more highly expressed than the protein coding genes. Increases in transcription have been shown to drive genomic instability in the A. thaliana cpDNA due to the increased formation of R-loops (RNA/DNA hybrids formed by displacement of the other DNA strand), which stall replication forks and lead to DSBs (Pérez Di Giorgio et al. 2019). It is possible that increased mtDNA expression also leads to the formation of R-loops and DSBs which may then be repaired through error prone NHEJ and MMEJ. However, it is not clear how this would drive strand asymmetric mutation. Further, such a mechanism is not consistent with the relatively even distribution of SNVs across intergenic vs. transcribed regions of the genome (Fig. 3). The magnitude of the CG→TA asymmetries is decreased in the msh1 mutants (roughly 2-fold averaging across all genic sequences) compared to in the WT controls (roughly 6-fold). This shift may reflect a larger proportional contribution of mutations from simple DNA polymerase misincorporation errors (which are not expected to be strand-biased) in the absence of MSH1 activity.
The CG→TA transitions in the WT lines and both transitions in the msh1 mutants were also impacted by the identity of neighboring nucleotides (Figs. 6 and 7). Trinucleotide effects have previously been implicated to bias mutation distribution in the A. thaliana nuclear genome (Lu et al. 2021) as well as in the mtDNAs of various metazoans (Itsara et al. 2014; Arbeithuber et al. 2020; Waneka et al. 2021; Sanchez-Contreras et al. 2021). It is noteworthy that the specific trinucleotides associated with CG→TA transitions differ between WT and msh1 mutants. The 5′ YCN signature (where Y is any pyrimidine and N is any nucleotide) in the WT lines is similar to that induced by APOBEC3-mediated cytosine deamination in human cell lines (Carpenter et al. 2023), though plants lack APOBEC enzymes so the relevance of this shared pattern is unclear. Meanwhile, the 5′ NCG signature in the msh1 mutants is consistent with spontaneous water mediated cytosine deamination (Carpenter et al. 2023).
Patterns of repeat-mediated recombination differs among mutant lines
The repeat mediated mtDNA recombination activity we documented in the msh1, radA and recA3 mutants is consistent with the previously documented recombination increases of these mutant backgrounds (Shedge et al. 2007; Arrieta-Montiel et al. 2009; Rowan et al. 2010; Davila et al. 2011; Miller-Messmer et al. 2012; Zampini et al. 2015; Wu et al. 2020; Chevigny et al. 2022; Zou et al. 2022). The absence of an effect in the why2 mutants is interesting given that why2 is the most abundant protein in mitochondrial nucleoids (Fuchs et al. 2020) and plants lacking why2 display aberrant mitochondrial morphology (Golin et al. 2020; Negroni et al. 2024). On the other hand, this result is consistent with a previous study that showed why2 mutants become more recombinationally active than WT under increased genotoxic stress (ciprofloxacin treatment) but showed no recombinational difference from WT under ‘normal’ growth conditions (Cappadocia et al. 2010; Negroni et al. 2024).
Though msh1, radA and reca3 are all required for the suppression of repeat-mediated recombination in mtDNA, these proteins likely function either in independent HR pathways (radA, recA3) or in different ways (msh1). As, noted, RECA3 is thought to facilitate branch migration in an HR pathway that may be relatively minor compared to the one in which RADA functions (Chevigny et al. 2022). Previous studies of recA3/msh1 and recA3/radA double mutants have shown the double mutants are more recombinationally active than recA3 single mutants (Shedge et al. 2007), supporting the hypothesis that RECA3-mediated HR is at least partially independent of RADA-mediated HR (Miller-Messmer et al. 2012; Chevigny et al. 2022). This model is supported by the greater increase in global recombination frequency in radA compared to recA3 (Fig. 10). We might also expect different repeats to become active in recA3 compared to radA mutants. However, as seen in Table1, there is substantial overlap in the repeats with increased recombination frequencies in these mutants, though the extremely high recombination frequency at repeat L in radA is one major difference. Meanwhile, MSH1 has been proposed to suppress non-allelic recombination by recognizing and rejecting mismatches in the invading strand during heteroduplex formation (Christensen 2018; Broz et al. 2022), which could be a shared feature in both RADA and RECA3 dependent HR pathways. Supporting this idea, there is an increased number of repeats that become active in msh1 mutants compared to radA and recA3 mutants. Specifically, there are 12 repeat pairs with a recombination frequency greater than 0.1 in msh1 mutants but only four and nine repeat pairs that meet this threshold in recA3 and radA mutants, respectively (File S2).
Given that recombination is activated differently between the mutants (Fig. 8), the high degree of repeatability between replicates is fascinating (Figs. S5, S6, S7, S8). These repeatable patterns rely on consistent activation of distinct repeat pairs and/or consistent maintenance/replication of certain recombination products. Understanding why different repeats become active and how these patterns relate to the increase in point mutations reported here remains an important unanswered question in the field of plant organellar genome maintenance.
Supplementary Material
FUNDING
This work was supported by the National Institutes of Health (NIGMS R35GM148134).
DATA AVAILABILITY
The Duplex Sequencing and Oxford Nanopore reads were deposited to the NCBI Sequence Read Archive (SRA) under BioProject PRJNA1113549.
REFERENCES
- Abdelnoor R. V., Yule R., Elo A., Christensen A. C., Meyer-Gauen G., et al. , 2003. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc. Natl. Acad. Sci. U. S. A. 100: 5968–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnoor R. V., Christensen A. C., Mohammed S., Munoz-Castillo B., Moriyama H., et al. , 2006. Mitochondrial genome dynamics in plants and animals: convergent gene fusions of a MutS homologue. J. Mol. Evol. 63: 165–173. [DOI] [PubMed] [Google Scholar]
- Alverson A. J., Zhuo S., Rice D. W., Sloan D. B., and Palmer J. D., 2011. The mitochondrial genome of the legume Vigna radiata and the analysis of recombination across short mitochondrial repeats. PLoS One 6: e16404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A. P., Luo X., Russell W., and Yin Y. W., 2020. Oxidative damage diminishes mitochondrial DNA polymerase replication fidelity. Nucleic Acids Res. 48: 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeithuber B., Hester J., Cremona M. A., Stoler N., Zaidi A., et al. , 2020. Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biol. 18: e3000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta-Montiel M. P., Shedge V., Davila J., Christensen A. C., and Mackenzie S. A., 2009. Diversity of the Arabidopsis mitochondrial genome occurs via nuclear-controlled recombination activity. Genetics 183: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala-García V. M., Baruch-Torres N., García-Medel P. L., and Brieba L. G., 2018. Plant organellar DNA polymerases paralogs exhibit dissimilar nucleotide incorporation fidelity. FEBS J. 285: 4005–4018. [DOI] [PubMed] [Google Scholar]
- Berardini T. Z., Reiser L., Li D., Mezheritsky Y., Muller R., et al. , 2015. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore J. L., 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27: 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieba L. G., 2019. Structure-Function Analysis Reveals the Singularity of Plant Mitochondrial DNA Replication Components: A Mosaic and Redundant System. Plants 8. 10.3390/plants8120533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz A. K., Waneka G., Wu Z., Fernandes Gyorfy M., and Sloan D. B., 2021. Detecting de novo mitochondrial mutations in angiosperms with highly divergent evolutionary rates. Genetics 218. 10.1093/genetics/iyab039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz A. K., Keene A., Gyorfy M. F., Hodous M., Johnston I. G., et al. , 2022. Sorting of mitochondrial and plastid heteroplasmy in Arabidopsis is extremely rapid and depends on MSH1 activity. Proceedings of the National Academy of Sciences 119: e2206973119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappadocia L., Maréchal A., Parent J.-S., Lepage E., Sygusch J., et al. , 2010. Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell 22: 1849–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter M. A., Temiz N. A., Ibrahim M. A., Jarvis M. C., Brown M. R., et al. , 2023. Mutational impact of APOBEC3A and APOBEC3B in a human cell line and comparisons to breast cancer. PLoS Genet. 19: e1011043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Li Q., Fu R., Wang J., Xiong C., et al. , 2019. Characterization of the mitochondrial genome of the pathogenic fungus Scytalidium auriculariicola (Leotiomycetes) and insights into its phylogenetics. Sci. Rep. 9: 17447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevigny N., Schatz-Daas D., Lotfi F., and Gualberto J. M., 2020. DNA Repair and the Stability of the Plant Mitochondrial Genome. Int. J. Mol. Sci. 21. 10.3390/ijms21010328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevigny N., Weber-Lotfi F., Le Blevenec A., Nadiras C., Fertet A., et al. , 2022. RADA-dependent branch migration has a predominant role in plant mitochondria and its defect leads to mtDNA instability and cell cycle arrest. PLoS Genet. 18: e1010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen A. C., 2014. Genes and Junk in Plant Mitochondria—Repair Mechanisms and Selection. Genome Biol. Evol. 6: 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen A. C., 2018. Mitochondrial DNA Repair and Genome Evolution. Annual Plant Reviews online 11–32. [Google Scholar]
- Davila J. I., Arrieta-Montiel M. P., Wamboldt Y., Cao J., Hagmann J., et al. , 2011. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 9: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin G., Daoud H., and Xia J., 2008. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 49: 827–831. [DOI] [PubMed] [Google Scholar]
- Fields P. D., Waneka G., Naish M., Schatz M. C., Henderson I. R., et al. , 2022. Complete Sequence of a 641-kb Insertion of Mitochondrial DNA in the Arabidopsis thaliana Nuclear Genome. Genome Biol. Evol. 14. 10.1093/gbe/evac059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs P., Rugen N., Carrie C., Elsässer M., Finkemeier I., et al. , 2020. Single organelle function and organization as estimated from Arabidopsis mitochondrial proteomics. Plant J. 101: 420–441. [DOI] [PubMed] [Google Scholar]
- García-Medel P. L., Baruch-Torres N., Peralta-Castro A., Trasviña-Arenas C. H., Torres-Larios A., et al. , 2019. Plant organellar DNA polymerases repair double-stranded breaks by microhomology-mediated end-joining. Nucleic Acids Res. 47: 3028–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Medel P. L., Peralta-Castro A., Baruch-Torres N., Fuentes-Pascacio A., Pedroza-García J. A., et al. , 2021. Arabidopsis thaliana PrimPol is a primase and lesion bypass DNA polymerase with the biochemical characteristics to cope with DNA damage in the nucleus, mitochondria, and chloroplast. Sci. Rep. 11: 20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golin S., Negroni Y. L., Bennewitz B., Klösgen R. B., Mulisch M., et al. , 2020. WHIRLY2 plays a key role in mitochondria morphology, dynamics, and functionality in Arabidopsis thaliana. Plant Direct 4: e00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualberto J. M., and Newton K. J., 2017. Plant Mitochondrial Genomes: Dynamics and Mechanisms of Mutation. Annu. Rev. Plant Biol. 68: 225–252. [DOI] [PubMed] [Google Scholar]
- Handa H., 2003. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 31: 5907–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala N. J., Polak P., Stojanov P., Covington K. R., Shinbrot E., et al. , 2016. Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell 164: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. Y., Grünheit N., Ahmadinejad N., Timmis J. N., and Martin W., 2005. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiol. 138: 1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara L. S., Kennedy S. R., Fox E. J., Yu S., Hewitt J. J., et al. , 2014. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 10: e1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplanis J., Akawi N., Gallone G., McRae J. F., Prigmore E., et al. , 2019. Exome-wide assessment of the functional impact and pathogenicity of multinucleotide mutations. Genome Res. 29: 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S. R., Salk J. J., Schmitt M. W., and Loeb L. A., 2013. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9: e1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S. R., Schmitt M. W., Fox E. J., Kohrn B. F., Salk J. J., et al. , 2014. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 9: 2586–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T., and Newton K. J., 2008. Angiosperm mitochondrial genomes and mutations. Mitochondrion 8: 5–14. [DOI] [PubMed] [Google Scholar]
- Li H., 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhou B., Khan A., Zheng J., Dawar F. U., et al. , 2021. Reactive Oxygen Species Accumulation Strongly Allied with Genetic Male Sterility Convertible to Cytoplasmic Male Sterility in Kenaf. Int. J. Mol. Sci. 22. 10.3390/ijms22031107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., Cui J., Wang L., Teng N., Zhang S., et al. , 2021. Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures. Genome Biol. 22: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Zapater J. M., Gil P., Capel J., and Somerville C. R., 1992. Mutations at the Arabidopsis CHM locus promote rearrangements of the mitochondrial genome. Plant Cell 4: 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller-Messmer M., Kühn K., Bichara M., Le Ret M., Imbault P., et al. , 2012. RecA-dependent DNA repair results in increased heteroplasmy of the Arabidopsis mitochondrial genome. Plant Physiol. 159: 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeckel C., Zaravinos A., and Georgakopoulos-Soares I., 2023. Strand asymmetries across genomic processes. Comput. Struct. Biotechnol. J. 21: 2036–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower J. P., Sloan D. B., and Alverson A. J., 2012. Plant Mitochondrial Genome Diversity: The Genomics Revolution, pp. 123–144 in Plant Genome Diversity Volume 1: Plant Genomes, their Residents, and their Evolutionary Dynamics, edited by Wendel J. F., Greilhuber J., Dolezel J., Leitch I. J. Springer Vienna, Vienna. [Google Scholar]
- Mugal C. F., von Grünberg H.-H., and Peifer M., 2009. Transcription-induced mutational strand bias and its effect on substitution rates in human genes. Mol. Biol. Evol. 26: 131–142. [DOI] [PubMed] [Google Scholar]
- Negroni Y. L., Doro I., Tamborrino A., Luzzi I., Fortunato S., et al. , 2024. The Arabidopsis Mitochondrial Nucleoid-Associated Protein WHIRLY2 Is Required for a Proper Response to Salt Stress. Plant Cell Physiol. 65: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer J. D., and Herbon L. A., 1988. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J. Mol. Evol. 28: 87–97. [DOI] [PubMed] [Google Scholar]
- Peñafiel-Ayala A., Peralta-Castro A., Mora-Garduño J., García-Medel P., Zambrano-Pereira A. G., et al. , 2023. Plant organellar MSH1 is a displacement loop specific endonuclease. Plant Cell Physiol. 10.1093/pcp/pcad112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez Di Giorgio J. A., Lepage É., Tremblay-Belzile S., Truche S., Loubert-Hudon A., et al. , 2019. Transcription is a major driving force for plastid genome instability in Arabidopsis. PLoS One 14: e0214552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucker B., Holtgräwe D., Stadermann K. B., Frey K., Huettel B., et al. , 2019. A chromosome-level sequence assembly reveals the structure of the Arabidopsis thaliana Nd-1 genome and its gene set. PLoS One 14: e0216233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R., and Hall I. M., 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan B. A., Oldenburg D. J., and Bendich A. J., 2010. RecA maintains the integrity of chloroplast DNA molecules in Arabidopsis. J. Exp. Bot. 61: 2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Contreras M., Sweetwyne M. T., Kohrn B. F., Tsantilas K. A., Hipp M. J., et al. , 2021. A replication-linked mutational gradient drives somatic mutation accumulation and influences germline polymorphisms and genome composition in mitochondrial DNA. Nucleic Acids Res. 49: 11103–11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandor S., Zhang Y., and Xu J., 2018. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 102: 9433–9448. [DOI] [PubMed] [Google Scholar]
- Shedge V., Arrieta-Montiel M., Christensen A. C., and Mackenzie S. A., 2007. Plant mitochondrial recombination surveillance requires unusual RecA and MutS homologs. Plant Cell 19: 1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skippington E., Barkman T. J., Rice D. W., and Palmer J. D., 2017. Comparative mitogenomics indicates respiratory competence in parasitic Viscum despite loss of complex I and extreme sequence divergence, and reveals horizontal gene transfer and remarkable variation in genome size. BMC Plant Biol. 17: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan D. B., Alverson A. J., Chuckalovcak J. P., Wu M., McCauley D. E., et al. , 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10: e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. R., and Keeling P. J., 2015. Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. U. S. A. 112: 10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupar R. M., Lilly J. W., Town C. D., Cheng Z., Kaul S., et al. , 2001. Complex mtDNA constitutes an approximate 620-kb insertion on Arabidopsis thaliana chromosome 2: implication of potential sequencing errors caused by large-unit repeats. Proc. Natl. Acad. Sci. U. S. A. 98: 5099–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Li Q., Kong L., and Yu H., 2018. Multiple reversals of strand asymmetry in molluscs mitochondrial genomes, and consequences for phylogenetic inferences. Mol. Phylogenet. Evol. 118: 222–231. [DOI] [PubMed] [Google Scholar]
- Vöhringer H., Van Hoeck A., Cuppen E., and Gerstung M., 2021. Learning mutational signatures and their multidimensional genomic properties with TensorSignatures. Nat. Commun. 12: 3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waneka G., Svendsen J. M., Havird J. C., and Sloan D. B., 2021. Mitochondrial mutations in Caenorhabditis elegans show signatures of oxidative damage and an AT-bias. Genetics 219. 10.1093/genetics/iyab116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters C. A., Strande N. T., Pryor J. M., Strom C. N., Mieczkowski P., et al. , 2014. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat. Commun. 5: 4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.-J., Shi M., Chen X.-X., Sharkey M. J., van Achterberg C., et al. , 2010. New views on strand asymmetry in insect mitochondrial genomes. PLoS One 5: e12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe K. H., Li W. H., and Sharp P. M., 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U. S. A. 84: 9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Waneka G., Broz A. K., King C. R., and Sloan D. B., 2020. MSH1 is required for maintenance of the low mutation rates in plant mitochondrial and plastid genomes. Proc. Natl. Acad. Sci. U. S. A. 117: 16448–16455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.-Q., Liao X.-Z., Zhang X.-N., Tembrock L. R., and Broz A., 2022. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 60: 160–168. [Google Scholar]
- Wynn E. L., and Christensen A. C., 2019. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 9: 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao-Ming Z., Junrui W., Li F., Sha L., Hongbo P., et al. , 2017. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 7: 1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampini É., Lepage É., Tremblay-Belzile S., Truche S., and Brisson N., 2015. Organelle DNA rearrangement mapping reveals U-turn-like inversions as a major source of genomic instability in Arabidopsis and humans. Genome Res. 25: 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Zhu W., Sloan D. B., and Wu Z., 2022. Long-read sequencing characterizes mitochondrial and plastid genome variants in Arabidopsis msh1 mutants. Plant J. 112: 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Duplex Sequencing and Oxford Nanopore reads were deposited to the NCBI Sequence Read Archive (SRA) under BioProject PRJNA1113549.