

Abstract
Background:
Heart failure involves metabolic alterations including increased glycolysis despite unchanged or decreased glucose oxidation. The mitochondrial pyruvate carrier (MPC) regulates pyruvate entry into the mitochondrial matrix, and cardiac deletion of the MPC in mice causes heart failure. How MPC deletion results in heart failure is unknown.
Methods:
We performed targeted metabolomics and isotope tracing in wildtype (fl/fl) and cardiac-specific Mpc2−/− (CS-Mpc2−/−) hearts after in vivo injection of U-13C-glucose. Cardiac glycogen was assessed biochemically and by transmission electron microscopy. Cardiac uptake of 2-deoxyglucose was measured and western blotting performed to analyze insulin signaling and enzymatic regulators of glycogen synthesis and degradation. Isotope tracing and glycogen analysis was also performed in hearts from mice fed either low-fat diet or a ketogenic diet previously shown to reverse the CS-Mpc2−/− heart failure. Cardiac glycogen was also assessed in mice infused with angiotensin-II that were fed low-fat or ketogenic diet.
Results:
Failing CS-Mpc2−/− hearts contained normal levels of ATP and phosphocreatine, yet these hearts displayed increased enrichment from U-13C-glucose and increased glycolytic metabolite pool sizes. 13C enrichment and pool size was also increased for the glycogen intermediate UDP-glucose, as well as increased enrichment of the glycogen pool. Glycogen levels were increased ~6-fold in the failing CS-Mpc2−/− hearts, and glycogen granules were easily detected by electron microscopy. This increased glycogen synthesis occurred despite enhanced inhibitory phosphorylation of glycogen synthase and reduced expression of glycogenin-1. In young, non-failing CS-Mpc2−/− hearts, increased glycolytic 13C enrichment occurred, but glycogen levels remained low and unchanged compared to fl/fl hearts. Feeding a ketogenic diet to CS-Mpc2−/− mice reversed the heart failure and normalized the cardiac glycogen and glycolytic metabolite accumulation. Cardiac glycogen levels were also elevated in mice infused with angiotensin-II, and both the cardiac hypertrophy and glycogen levels were improved by ketogenic diet.
Conclusions:
Our results indicate that loss of MPC in the heart causes glycogen accumulation and heart failure, while a ketogenic diet can reverse both the glycogen accumulation and heart failure. We conclude that maintaining mitochondrial pyruvate import and metabolism is critical for the heart, unless cardiac pyruvate metabolism is reduced by consumption of a ketogenic diet.
Keywords: heart failure, mitochondria, pyruvate, ketogenic diet
Introduction
Currently, 6.7 million adults in the United States suffer from heart failure, which has risen from 6.0 million in just two years1. Those diagnosed with heart failure have a 5-year mortality rate of over 50%, which has remained unchanged in recent years despite existing treatment options1. Heart failure can arise from a multitude of causes, including ischemia/infarction, cardiac hypertrophy, chronic hypertension, or valvular diseases1, 2. A better understanding of the development and pathophysiology of heart failure is needed to generate improved treatment options.
Since the heart lacks the ability to store ample amounts of metabolites or high energy phosphates, a continuous supply of nutrients is required to fuel its constant contractile work. Mitochondrial oxidative metabolism plays a large role in the production of high energy phosphates in the adult heart. The preferred fuel source of the heart are fatty acids, with fatty acid oxidation (FAO) accounting for ~70% of the adenosine triphosphate (ATP) produced3, 4. Yet the healthy heart is metabolically flexible and can adapt to use alternative fuel sources to meet its energy demands, including the use of glucose, lactate, ketone bodies, and amino acids3, 4. Glycolysis accounts for ~5% of ATP produced in the healthy heart3, 4. However, during hypertrophy and heart failure metabolic changes occur, including significantly reduced FAO, increased glycolysis, but relatively decreased glucose oxidation3–6. This mismatch between increased glycolysis, yet unchanged or decreased glucose oxidation has been suggested to be important for stimulating cardiac hypertrophy and failure7–9.
The mitochondrial pyruvate carrier (MPC) is responsible for transporting pyruvate, the end product of glycolysis or oxidized lactate, into the mitochondrial matrix10, 11. The mammalian MPC is a heterodimeric complex composed of MPC1 and MPC2 proteins, both of which are necessary for MPC complex formation and pyruvate transport10, 11. Once pyruvate has been transported into the mitochondria, it can be converted into acetyl-CoA and used in the citric acid cycle10, 11. Recent studies by us and others revealed that cardiac MPC gene and protein expression is significantly reduced in humans with heart failure12–15 and that deletion of MPC14, 12, 14 or MPC212, 13 in mice leads to spontaneous heart failure with age. However, it is still not understood how reduction or deletion of MPC expression contributes to heart failure.
In the present study, we suggest that this heart failure is associated with accumulation of glycogen in the MPC-deficient heart. Even with dilated cardiomyopathy, cardiac-specific MPC2−/− (CS-Mpc2−/−) hearts contain normal levels of ATP and phosphocreatine compared to wildtype littermates, suggesting that lack of pyruvate oxidation does not simply result in “energetic stress”. Glucose uptake is increased, and glycolytic metabolites accumulate in both young, non-failing and failing CS-Mpc2−/− hearts. However, glycogen accumulation is only observed in failing CS-Mpc2−/− hearts and the heart failure and glycogen accumulation can be reversed by providing a highfat, low-carbohydrate ketogenic diet (KD). Lastly, we also observe increased glycogen in hearts from angiotensin-II infused mice, a hypertrophic stimulus that reduces Mpc expression12, and both the cardiac glycogen and hypertrophy in this model are also normalized by ketogenic diet feeding.
Methods
Animals
All animal procedures were performed in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at Saint Louis University. Generation of mice with Mpc2 gene flanked by loxP sites, as well as cardiac-specific deletion of Mpc2 by crossing to Mlc2v-Cre knock-in mice (Jackson Laboratory Strain #029465) have been described previously13, 16. All mice used in these experiments were from a C57BL6/J background. All experiments were performed with a mixture of male and female littermate mice. Mice were housed in a climate-controlled specific-pathogen free facility maintained at 22–24°C and 40–60% humidity in ventilated cages with a 12-hour light/dark cycle. Ad libitum access to drinking water was provided by automatic cage waterers. Food was also provided ad libitum. All mice were grouphoused, up to five mice per cage. Based on our previous study13, the experiments in young, non-failing CS-Mpc2−/− mice were performed at 6 weeks of age and experiments in CS-Mpc2−/− mice with heart failure were performed at 16–30 weeks of age. For ketogenic diet (KD) experiments, fl/fl and CS-Mpc2−/− mice were initially fed chow and aged to 16 weeks to develop heart failure in CS-Mpc2−/− mice, then switched to either low-fat (LF) control diet or KD for a duration of 3 weeks. The LF diet (F1515, BioServ) composition is 68.9%kcal carbohydrate, 19.0%kcal protein, and 12.1%kcal fat. Ketogenic diet (F3666, BioServ) composition is 1.8%kcal carbohydrate, 4.7%kcal protein, and 93.5%kcal fat. Diets were randomly assigned for whole cages based on genotypes contained within to result in roughly even numbers for each group. Unless otherwise stated, at study culmination mice were fasted for 2–3 hours, and euthanized by CO2 asphyxiation. Blood was collected via cannulation of the inferior vena cava and placed into EDTA-treated tubes on ice. Tissues were then excised, weighed, and snap frozen in liquid nitrogen. Plasma was collected by centrifuging blood tubes at 8,000g for 8 minutes at 4°C and freezing the plasma supernatant in liquid nitrogen. Plasma and tissues were stored at −80°C until analyzed.
AngII infusion was performed in 9-week-old wildtype C57BL6/J male mice purchased from the Jackson Laboratory (Strain #000664). Subcutaneous osmotic pumps (#1004, Alzet) with either AngII (1.44mg/kg/day, Sigma Aldrich) or saline vehicle were implanted for an infusion duration of three weeks similarly to as described previously17. Mice were anesthetized by inhaled 2–3% isoflurane in 100% oxygen via a nose cone, and the upper back area shaved with electric clippers and scrubbed with 7.5% betadine antiseptic. A ~1cm incision was made in the prepped area of the mouse’s back, and forceps inserted to create a subcutaneous pocket. After inserting the pump, the wound was sealed with 2–3 surgical staples. Mice were treated with 1 mg/kg subcutaneous buprenorphine-SR and several drops of lidocaine were applied to wound edges for pain management. Mice were allowed to arouse while on a heat pad and placed back into their home cage. Mice were observed daily, and surgical staples were removed within 7–10 days. Mice were switched to LF or KD 3 days prior to pump implant. Diet was assigned randomly for each cage, and then vehicle versus AngII pumps were assigned for mice for each diet to result in roughly even numbers per group. Mice were euthanized by CO2 and plasma/tissues harvested as described above.
RNA sequencing and analysis
Hearts used for RNA sequencing were excised from 6 fl/fl and 6 CS-Mpc2−/− 30-week-old chow-fed mice. RNA was isolated from ~25 mg of frozen left ventricular tissue using RNA-STAT (Tel-Test, Inc) and purified using the PureLink RNA Mini Kit with DNase treatment (Invitrogen). RNA was then pooled between 2 different fl/fl and CS-Mpc2−/− hearts, resulting in n=3 samples per group. RNA concentration and quality were determined by 2100 Bioanalyzer RNA 6000 Nano Kit (Agilent) and depleted of rRNA using RiboMinus Eukaryote Kit v2 (Ambion). DNA libraries were constructed using Ion Total RNA-seq Kit v2 (Thermo Fisher Scientific) and sequenced on an Ion Torrent Proton DNA sequencer (Thermo Fisher Scientific). Raw data were aligned to the mm10 genome. Gene counts for each sample were normalized by read depth, low expression genes filtered from the data, and t-test performed for statistical comparison. Lists of upregulated and downregulated differentially expressed genes (p < 0.05) between fl/fl and CS-Mpc2−/− hearts were subjected to pathway-level overrepresentation analysis using IMPaLA18, which included Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis, among others. Heat map of differentially expressed genes was generated with Shiny heatmap19. RNA sequencing files were uploaded to NCBI GEO Series Number GSE268694.
Gene expression analysis
Relative gene expression was determined by reverse transcription quantitative PCR (RT-qPCR). Total RNA was extracted from snap frozen tissues using RNA-STAT (Tel-Test, Inc). [RNA] was assessed by Nanodrop 2000c (Thermo Fisher Scientific). Then 1 μg of sample was reverse transcribed into complementary DNA by Superscript VILO (Thermo Fisher Scientific) using a 2720 Thermal cycler (Applied Biosystems). Relative quantification of target gene expression was measured in duplicate using Power SYBR Green (Thermo Fisher Scientific) with a QuantStudio 3 PCR System (Thermo Fisher Scientific). Target gene threshold cycle (Ct) values were normalized to reference gene (Rplp0) Ct values by the 2−ΔΔCt method. Oligonucleotide primer sequences used in 5’ to 3’ were: Rplp0 FWD: GCAGACAACGTGGGCTCCAAGCGAA, Rplp0 REV: GGTCCTCCTTGGTGAACACGAAGCCC, Mpc1 FWD: GACTTTCGCCCTCTGTTGCTA, Mpc1 REV: GAGGTTGTACCTTGTAGGCAAAT, Mpc2 FWD: CCGCCGCGATGGCAGCTG, and Mpc2 REV: GCTAGTCCAGCACACACCAATCC.
Western blotting and protein expression analysis
Protein extracts were prepared by homogenizing ~30 mg of frozen ventricular tissue in lysis buffer (15mM NaCl, 25 nM Tri Base, 1 mM EDTA, 0.2% NP-40, 10% glycerol) supplemented with 1X cOmplete protease inhibitor cocktail (Millipore Sigma) and a phosphatase inhibitor cocktail (1 mM Na3VO4, 1 mM NaF, and 1 mM PMSF) by using 2–3 zirconia beads (Biospec products) and homogenization for 1 min with a Mini Beadbeater (Biospec products). Protein concentrations were measured using MicroBCA Assay kit (Thermo Fisher Scientific) and absorbance measured with a Synergy HTX multi-mode plate reader (Biotek). Then 50 μg of protein lysate was electrophoresed on criterion TGX 4–15% precast gel (Bio-Rad Laboratories) and transferred onto PVDF membrane (Millipore). Membranes were blocked with 5% bovine serum albumin (BSA) (#BAH65 Equitech-Bio, Inc) in Tris-buffered saline with Tween-20 (TBST) for at least 1 hour.
Primary antibodies were used at 1:1000 dilution in 5% BSA-TBST, with gentle rocking at 4°C overnight. AKT (9272), phosphorylated AKT (serine 473) (4060), MPC1 (14462), MPC2 (46141), glycogen synthase (3886), and phosphorylated glycogen synthase (serine 640) (47043) were from Cell Signaling. α-tubulin (T6199) was from Sigma. PYGM (ab231963) was from abcam. Glycogenin-1 (sc-271109) was from Santa Cruz Biotechnology. After primary antibody incubation, membranes were washed 3X for 10 minutes in TBST and probed with IRDye secondary antibodies at 1:10,000 (Li-Cor Biosciences, 926–32213 or 926–68072) in 5% BSA-TBST for 1 hour, then washed 3X 10 min. Membranes were imaged on an Odyssey CLx imaging system and analyzed with Image Studio Lite software (Li-Cor Biosciences).
Targeted metabolomics and 13C enrichment from glucose
Mice were fasted for 2 hours then injected intraperitoneally (i.p.) with 1g/kg uniformly labelled U-13C-glucose (CLM-1396, Cambridge Isotope Labs) in saline. 30 min later the mice were anesthetized with 1–2% isoflurane (Covetrus) in 100% oxygen via an induction chamber then transferred to a nose cone. A thoracotomy was rapidly performed and the beating heart snap-frozen using a metal clamp chilled in liquid nitrogen. Hearts were stored at −80°C until processed. The heart tissue was pulverized with a mortar and pestle chilled in a liquid nitrogen bath. To every 1 mg of pulverized tissue, 40 μl of 2:2:1 acetonitrile:methanol:water was added. The samples were then briefly vortexed, placed in a liquid nitrogen bath for 1 minute, and then sonicated for 10 minutes at 25°C. This was repeated 3X. Samples were stored at −20°C overnight. The following day, the samples were centrifuged at 14,000g for 10 min at 4°C, and supernatants added to glass liquid chromatography vials with autosampler inserts and stored at −80°C until analyzed.
Ultra-high-performance liquid chromatography (UHPLC)-MS/MS was performed with a Vanquish Flex UHPLC system interfaced with an Orbitrap ID-X Tribid Mass spectrometer (Thermo Fisher Scientific) similar to as previously described20. Hydrophilic interaction liquid chromatography (HILIC) separation was performed using a HILICON iHILIC-(P) Classic column (Tvistevagen, Umea, Sweden) with the following specifications: 100 × 2.1 mm, 5 μm. Mobile-phase solvents were composed of A = 20 mM ammonium bicarbonate, 0.1% ammonium hydroxide and 2.5 μM medronic acid in water:acetonitrile (95:5) and B = acetonitrile:water (95:5). The column compartment was 45°C for all experiments. The following linear gradient was applied at a flow rate of 250 μL min −1: 0–1 min: 90% B, 1–12 min: 90–35% B, 12–12.5 min: 35–25% B, 12.5–14.5 min: 25% B. The column was re-equilibrated with 20 column volumes of 90% B. The injection volume was 4 μL for all experiments.
Data were collected with the following settings: spray voltage, 3.5kC (positive) / −2.8 kV (negative); sheath gas, 50; auxiliary gas, 10; sweep gas, 1; ion transfer tube temperature, 300°C; vaporizer temperature, 200°C; mass range, 67–1,000 Da, resolution, 120,000 (MS1), 30,000 (MS/MS); maximum injection time, 100 ms; isolation window, 1.6 Da. LC/MS data were processed and analyzed with the open-source Skyline software21. Natural abundance correction of 13C for tracer experiments was performed with AccuCor22. All studies contained 3 quality control samples and a blank sample, and only metabolites with sufficient abundance and fractional enrichment over the limit of detection were included in analysis. These methods were used to analyze the pool size and 13C enrichment of glucose and glycolytic metabolites, organic acids and amino acids, and the pool sizes of nucleotide phosphates and phosphocreatine and creatine. Data are presented as the % enrichment of the specified 13C isotopologue, or as the normalized peak area for each metabolite’s pool size.
Transmission electron microscopy
The papillary muscle from hearts (under 1 mm3) was excised and fixed in 2.5% glutaraldehyde in 0.1M sodium cacodylate, pH 7.4 overnight. Samples were then post-fixed with 1% osmium tetroxide for 1 h and then serial dehydration with alcohol (50%, 75%, 90%, and 100%) was performed and then embedded in Epon 12. Ultrathin sections were then post-stained with uranyl acetate and lead citrate. Samples were imaged on a JEOL 1400 Plus transmission electron microscope equipped with an AMT camera.
Cardiac glycogen assay
Cardiac glycogen concentrations were measured similarly to as preformed previously13. Briefly, 15–30 mg of heart tissue were boiled in 30% KOH at 100°C for 30 min. Then, 2% Na2SO4 and 100% EtOH were added, and the tubes were vortexed. Samples were boiled again to help aid in the precipitation of glycogen and then tubes were centrifuged at 16,000g for 5 min and supernatant aspirated. The pellet was dissolved in 200 μL H2O and 800 μL of 100% ethanol was added. The samples were then boiled for 5 minutes, and recentrifuged at 16,000g for 5 min. This wash step was repeated 3X. The final pellets were suspended in 200 μL of 0.3 mg/ml amyloglucosidase (Roche) in 0.2 M sodium acetate (pH 4.8). Serial dilutions of 10 mg/ml oyster glycogen (Sigma) were prepared as standards. Samples and standards were then incubated in a 40°C water bath for 3 hours. Samples and standards were then diluted 1:1 with H2O, and the samples were added to a 96-well plate. Glucose assay buffer (0.3 M triethanolamine, 4 mM MgCl2, 2 mM ATP, 0.9 mM NADP+, and 5 μg/mL glucose-6-phosphate dehydrogenase (Roche)) was added to each well, and the absorbance was measured at 340 nm with a Synergy HTX multi-mode plate reader (Biotek). Then 1 μg hexokinase (Roche) was added to each well and the plate was incubated at room temperature in the dark for 30 minutes, and absorbance measured again at 340 nm. The glycogen concentration was calculated from the difference in absorbance readings plotted in relation to the oyster glycogen standards.
Glycogen 13C enrichment and pool size measurement was performed similarly as described previously23. Briefly, glycogen from cardiac tissue of mice injected with U-13C-glucose as described above was precipitated and digested by 0.3 mg/mL amyloglucosidase in 0.2 M sodium acetate as described for the biochemical glycogen assay. Following the 3 hour digestion with amyloglucosidase, the samples were added to a 2:2:1 acetonitrile:methanol:water mixture maintaining a 1:9 ratio of sample: extraction solvent. The samples were vortexed for 1 min then stored at −20°C for 1 hour. Then the samples were centrifuged at 14,000g for 10 min at 4°C. The supernatant was then transferred to glass LC vials for UHPLC-MS/MS analysis as described above.
Myocardial glucose uptake assay
Glucose uptake was measured similarly to methods used for skeletal muscle described previously24, 25. Mice were anesthetized with 50 mg/kg sodium pentobarbital i.p., the heart removed, and two ~2–4 mg left ventricular muscle strips collected from each heart. Individual cardiac strips were placed into a well of a 24-well plate containing Krebs-Henseleit Buffer recovery solution with non-glucose fuel sources (4.5 mM lactate, 0.5 mM pyruvate, and 200 μM BSA-palmitate), 34.8 mM mannitol, and 0.1% radioimmunoassay-grade BSA at 35°C. Incubation buffers were pre-gassed with 95% O2:5% CO2, and each well of the 24-well plate had its own gas supply. After 60 min in recovery buffer, samples were moved to another well containing the same solution ± 30 μU/mL (submaximal) insulin for 30 min. Samples were then moved to another well containing glucose transport medium ± 30 μU/mL insulin. Glucose transport assay medium was the same as recovery medium but also contained 1 mM 2-deoxyglucose (2-DG), 2 μCi/mL 3H-2-DG, and 0.3 mCi/mL 14C-mannitol at 30°C. After 10 min in the glucose transport assay medium, samples were clamp frozen. Samples were homogenized in ice-cold buffer containing protease and phosphatase inhibitors (50 mM HEPES, pH 7.4, 2 mM Na3VO4, 150 mM NaF,10 μg/mL leupeptin, 10 μg/mL aprotinin, 0.5 μg/mL pepstatin and 1 mM phenylmethylsulfonyl fluoride) and centrifuged at 4°C for 10 min at 13,500g. Supernatant protein was assessed by the Bradford method, and aliquots were mixed with scintillation fluid (Ultima Gold XR, Perkin Elmer, Boston, MA, USA) before counting of disintegrations per minute (DPM, TriCarb 3110TR, Perkin Elmer). The DPM of 14C mannitol was used to measure extracellular volume and correct for DPM of 3H-2-DG in the extracellular fluid.
Insulin signaling assessment
Insulin signaling was evaluated as described previously26. Briefly, mice were fasted for 3 hours, then injected i.p. with 5mU/g insulin or saline vehicle. 10 minutes after injection, mice were euthanized by CO2 and the hearts collected and snap frozen in liquid nitrogen and stored at −80°C. Western blotting was then performed for phosphorylated and total AKT as described above.
Plasma ketone body assay
Plasma ketone concentrations were measured using a β-hydroxybutyrate colorimetric assay kit (Cayman Chemical). Samples were incubated in the dark for 25 min and absorbance measured at 445 nm with a Synergy HTX multi-mode plate reader (Biotek) and plotted against a standard curve for quantification.
Mitochondrial isolation and respirometry
Mitochondria were isolated from freshly excised hearts by ten passes of a glass-on-glass Dounce homogenizer on ice in 4 mL of buffer containing 250 mM sucrose, 10 mM Tris, and 1 mM EDTA, pH 7.4. Homogenates were centrifuged at 1,000g × 5 min at 4°C, and the supernatant transferred to a new tube and centrifuged at 10,000g × 10 min at 4°C. This mitochondria-enriched pellet was washed with fresh homogenization buffer and recentrifuged at 10,000g × 10 min at 4°C twice. The mitochondrial pellet was then solubilized in ~200 μL of Mir05 respiration buffer consisting of 0.5 mM EGTA, 3 mM MgCl2, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1g/L fatty-acid free BSA, pH 7.1. Mitochondrial protein concentration was measured by MicroBCA (Thermo Fisher Scientific).
50 μg of mitochondria were added to each chamber of an Oxygraph O2k FluoRespirometer (Oroboros Instruments), and respiration was stimulated with 5 mM pyruvate, 2 mM malate, and then coupled with 2.5 mM ADP+Mg2+. Lastly, 5 μM UK-5099 was added to the chambers to inhibit the MPC and pyruvate respiration.
Respiration was also performed on saponin-permeabilized left-ventricular muscle fibers using the Oroboros O2k similar to as previously performed13. Respiration was again stimulated with 5 mM pyruvate, 2 mM malate, and then coupled with 2.5 mM ADP+Mg2+, and inhibited with 5 μM UK-5099. Respiration data was recorded and analyzed with DatLab version 7.4.0.1 software, and is presented as oxygen consumption rate normalized to mitochondrial mass or tissue mass added to the chamber.
Data availability
Data supporting the findings of this study are available from the corresponding author upon reasonable request. The RNA sequencing data generated in this study have been deposited in NCBI GEO Series Number GSE268694.
Statistical analysis
All data are presented as dot plots with mean±S.D. All data were graphed and statistically analyzed with GraphPad Prism version 10.0.2. An unpaired, two-tailed Student’s t-test with Welch correction was used for comparison of two groups. Experiments with more than two groups were analyzed using a two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test. A P value of less than 0.05 was considered statistically significant.
Results
Heart failure in CS-Mpc2−/− mice is not due to energetic stress
We and others have previously described spontaneous heart failure development in MPC-deficient murine hearts4, 12–14, but what causes these hearts to fail is not understood. In attempt to uncover insights we performed bulk RNA sequencing on CS-Mpc2−/− hearts with dilated cardiomyopathy and fl/fl littermates (Figure 1A–C). This analysis uncovered 956 significantly differentially expressed genes in the failing hearts compared to control, with ribosome and protein synthesis pathways upregulated and multiple pathways related to fatty acid metabolism, pyruvate metabolism, and amino acid catabolism downregulated (Figure 1C). The downregulation of fatty acid metabolism is a well-known metabolic alteration in failing hearts2, including in these CS-Mpc2−/− hearts13. Despite transcriptional downregulation of fatty acid, pyruvate, and amino acid metabolic pathways, surprisingly CS-Mpc2−/− hearts maintained normal levels of the high energy phosphates ATP and phosphocreatine as well as normal phosphocreatine/creatine and phosphocreatine/ATP ratios (Figure 1D–J). Although AMP levels were slightly elevated and ADP levels trended to be elevated, the lack of a decrease in ATP or phosphocreatine suggests normal energetic status of these failing CS-Mpc2−/− hearts. Additionally, there were no significant differences in nicotinamide nucleotides NAD+, NADH, NADP+, or NADP in failing CS-Mpc2−/− hearts (Figure 1K–N), suggesting that redox control of metabolic pathways is also normal. Altogether, these results suggest that despite multiple decreased oxidative pathways, the heart failure in CS-Mpc2−/− mice is not caused by overt energetic stress.
Figure 1. Heart Failure in CS-Mpc2−/− mice is not due to energetic stress.

A, Heart weight/tibia length (HW/TL) in CS-Mpc2−/− and fl/fl littermates, n=6. B, Representative gross images of excised fl/fl and CS-Mpc2−/− hearts. Scale bar = 5 mm. C, Heatmap of differentially expressed genes identified by RNA sequencing of hearts from fl/fl and CS-Mpc2−/− littermates, n=3. Most prominently dysregulated KEGG pathways upregulated and downregulated are listed. D through N, mass spectrometry normalized peak area pool sizes of adenosine triphosphate (ATP) (D), adenosine diphosphate (ADP) (E), adenosine monophosphate (AMP) (F), phosphocreatine (G), creatine (H), phosphocreatine/creatine (PCr/Cr) ratio (I), PCr/ATP ratio (J) oxidized nicotinamide adenine dinucleotide (NAD+) (K), reduced nicotinamide adenine dinucleotide (NADH) (L), oxidized nicotinamide adenine dinucleotide phosphate (NADP+) (M), and reduced nicotinamide adenine dinucleotide phosphate (NADPH) (N) in hearts of fl/fl and CS-Mpc2−/− littermates that had been injected i.p. with U-13Cglucose, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Failing CS-Mpc2−/− hearts display increased glycolysis
As it has been well described that heart failure is associated with increased glucose utilization, often with unchanged or decreased glucose oxidation27, we hypothesized that MPC deletion would mimic these metabolic changes. To evaluate glucose metabolism, we injected mice i.p. with U-13C-glucose, excised the hearts 30 min post injection, and assessed metabolite pool sizes and 13C isotopologues by UHPLC-MS/MS. We first focused on fully 13C labeled glycolytic metabolites (Figure 2A) and their pool sizes (Figure S1). While glucose enrichment and pool size were unaltered, glucose-6-phosphate (G6P) enrichment showed a trend to be increased and all other downstream glycolytic metabolites displayed significantly greater 13C enrichment and pool size in CS-Mpc2−/− hearts (Figure 2B–G and Figure S1A–H). Additionally, CS-Mpc2−/− hearts displayed increased enrichment and pool sizes of both lactate and alanine (Figure 2H–I and Figure S1I–J), indicative of decreased pyruvate carbon entry into the TCA cycle. Altogether, these results suggest increased glycolytic metabolism in failing CS-Mpc2−/− hearts.
Figure 2. Failing CS-Mpc2−/− hearts display increased glycolysis and accumulate glycogen.

A, Schematic of U-13C-glucose labeling of glycolytic metabolites and glycogen, created with BioRender. B through I, 13C % enrichment of glucose (B), glucose-6-phosphate (G6P) (C), fructose bisphosphate (FBP) (D), dihydroxyacetone phosphate (DHAP) (E), phosphoenolpyruvate (PEP) (F), pyruvate (G), lactate (H), and alanine (I) in hearts from fl/fl and CS-Mpc2−/− mice after injection with U-13C-glucose, n=11–12. J, Cardiac glycogen levels measured biochemically in fl/fl and CS-Mpc2−/− littermates, n=16. K, Representative transmission electron microscopy images at 5,000X magnification of the cardiac papillary muscle of fl/fl and CS-Mpc2−/−, white arrowhead denotes glycogen granules. Scale bars = 1 μm. L and M, 13C % enrichment (L) and pool size (M) of uridine diphosphate (UDP)-glucose, n=11–12. N and O, 13C % enrichment of the glucose within glycogen for fully labeled M6 (N), and fully unlabeled M0 (O), n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Surprisingly, CS-Mpc2−/− hearts do not display significantly decreased enrichment of TCA cycle metabolites (Figure S2A–L). Likewise, pool sizes of TCA cycle metabolites were also unaltered in CS-Mpc2−/− hearts apart from α-ketoglutarate being significantly decreased (Figure S2A–L). This normal TCA cycle enrichment from U-13C-glucose contrasts studies of MPC deletion or inhibition in other cell types, such as hepatocytes16, when provided U-13C-glucose or -pyruvate. However, while one previous study identified decreased TCA cycle enrichment with MPC1 deletion in isolated cardiomyocytes14, another previous study observed normal or even significantly increased enrichment of the TCA cycle in MPC1−/− hearts perfused with 13C-glucose4. While these puzzling 13C enrichment results do not support it, it is likely that MPC deletion reduces cardiac pyruvate oxidation, as both isolated mitochondria and permeabilized cardiac muscle fibers display significantly reduced oxygen consumption when provided with pyruvate/malate as substrates (Figure S3)13. Additionally, while mitochondria from fl/fl hearts are sensitive to the MPC inhibitor UK-5099 which decreases pyruvate respiration, there is little-to-no effect of UK-5099 in CS-Mpc2−/− mitochondria or muscle fibers (Figure S3). Potential explanations for these surprising 13C enrichment results are discussed in more detail below.
CS-Mpc2−/− hearts do not increase glucose enrichment of the pentose phosphate pathway, but do increase flux into glycerol-3-phosphate
Several recent studies have suggested the importance of glucose flux through the pentose phosphate pathway to fuel hypertrophied or failing hearts with amino acids, RNA, and nucleotides7, 9. Interestingly, in failing CS-Mpc2−/− hearts we observed no significant increases in 13C enrichment of 6-phosphogluconate, ribose-5-phosphate, sedoheptulose-7-phosphate, nor serine or glycine which can be end products of the pentose phosphate pathway (Figure S4A–F). As described above, we also identified no changes in NADP+ or NADPH (Figure 1M–N), also indicative of unaltered flux through the pentose phosphate pathway. Several pentose phosphate pathway metabolites displayed increased pool sizes in CS-Mpc2−/− hearts (Figure S4B–F), however the unaltered enrichment from U-13C glucose suggests that CS-Mpc2−/− hearts do not significantly increase glucose incorporation into the pentose phosphate pathway. Lastly, we observed increased 13C enrichment and pool size of glycerol-3-phosphate (Figure S1A,K–L), suggesting the failing CS-Mpc2−/− hearts increasingly shuttle glucose carbons into glycerophospholipid synthesis.
Failing CS-Mpc2−/− hearts accumulate glycogen
Another potential fate of glucose is storage as glycogen, and indeed we13 and others4 previously observed increased glycogen content in MPC-deficient hearts, without expanding upon this finding. Here we confirmed again increased glycogen concentrations in failing CS-Mpc2−/− hearts measured biochemically (Figure 2J) as well as by detection of abundant glycogen granules on transmission electron microscopy images of failing CS-Mpc2−/− hearts (Figure 2K). In hearts from U-13C-glucose injected mice, we observed significantly elevated 13C enrichment and pool size of UDP-glucose (Figure 2L–M), indicating increased flow of carbon into this glycogen intermediate. When glucose was liberated from the cardiac glycogen, fl/fl hearts displayed essentially zero 13C enrichment, while all but one of the CS-Mpc2−/− hearts displayed detectable enrichment (Figure 2N). Conversely, the percentage of completely unlabeled glucose liberated from glycogen was significantly decreased in CS-Mpc2−/− hearts (Figure 2O). Taken together, these results suggest that loss of MPC in the heart results in increased glycogen synthesis and accumulation of glycogen.
Glucose uptake is increased, and glycogen synthase should be inhibited in CS-Mpc2−/− hearts
While the 13C-glucose enrichment studies suggest increased glycolysis in CS-Mpc2−/− hearts, the bolus of glucose injected makes it difficult to determine if cardiac glucose uptake is altered, and we observed similar labeling of glucose in fl/fl and CS-Mpc2−/− hearts in those studies (Figure 2B). To more appropriately assess glucose uptake, myocardial muscle fibers were incubated with 3H-2-deoxyglucose in the absence or presence of insulin. Basal glucose uptake in CS-Mpc2−/− myocardial fibers was nearly double that of fl/fl myocardial fibers and remained significantly higher with mild insulin-stimulation (Figure 3A). Since heart failure is often associated with cardiac insulin-resistance28, we assessed insulin signaling by blotting for phosphorylated and total AKT after i.p. insulin injection, which was not significantly altered in CS-Mpc2−/− mice (Figure 3B). Lastly, we also investigated the regulatory enzymes for glycogen synthesis and degradation. Interestingly, in the failing CS-Mpc2−/− hearts (Figures 3C–E), we observed significantly elevated phosphorylation of glycogen synthase at serine 640 (Figure 3C,F), which is a strong inhibitory phosphorylation site29. Additionally, expression of glycogenin-1, which acts as a primer for glycogen synthase by initializing the glucosyl chain30, was decreased in failing CS-Mpc2−/− hearts (Figure 3C,G). We did not observe any difference in glycogen phosphorylase expression, therefore cannot suggest whether glycogenolysis, or the breakdown of glycogen, is altered in CS-Mpc2−/− hearts. Altogether, these results suggest that cardiac MPC loss increases glucose uptake and glycogen synthesis, despite the heart’s attempt to inhibit glycogen synthesis by phosphorylation of glycogen synthase and decreased glycogenin-1 expression.
Figure 3. Glucose uptake is increased, and glycogen synthesis should be inhibited in CS-Mpc2−/− hearts.

A, Glucose uptake assessed by 2-deoxyglucose in the present or absence of insulin in cardiac muscle fibers from fl/fl and CS-Mpc2−/− littermates, n=9–12. B, Western blot images of phosphorylated (serine-473) and total protein kinase B (AKT) and the ratio of quantified densitometry for pAKT/AKT from hearts of fl/fl and CS-Mpc2−/− mice injected with either saline vehicle or 5mU/g insulin. C through G, Western blot images of phosphorylated glycogen synthase (pGS) serine-640, total glycogen synthase (GS), muscle glycogen phosphorylase (PYGM), glycogenin-1, mitochondrial pyruvate carrier (MPC) 1, MPC2, and α-tubulin (C), and the normalized densitometry quantification for MPC1/tubulin (D), MPC2/tubulin (E), pGS/total GS (F), and glycogenin-1/tubulin (G) in hearts of fl/fl and CS-Mpc2−/− littermates, n=4. Data are presented as the mean±SD. Data in A were evaluated by two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test. Data in B were evaluated by unpaired, two-tailed Student’s t-test with Welch correction due to only n=2 in saline groups. Data in D-G were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Non-failing CS-Mpc2−/− hearts from young mice display increased glycolysis, but no glycogen accumulation
As increased glucose uptake and glycolysis is known to occur in failing hearts, we investigated whether these metabolic changes were due to the presence of heart failure in CS-Mpc2−/− hearts, or a direct consequence of MPC deletion. To assess this, we repeated the U-13C-glucose enrichment experiment in 6-week-old fl/fl and CS-Mpc2−/− mice before the onset of cardiac remodeling and dysfunction13. The 13C enrichment of all glycolytic metabolites was increased in these young, non-failing CS-Mpc2−/− hearts (Figure 4A–I). The pool size of glucose in these hearts was increased, but the pool size of all glycolytic metabolites was unaltered in the non-failing CS-Mpc2−/− hearts (Figure S5A–J). However, neither UDP-glucose enrichment nor pool size were significantly altered in the non-failing CS-Mpc2−/− hearts (Figure 4J–K), suggesting no increase in glycogen synthesis. Indeed, glycogen concentrations were normal in these young, non-failing CS-Mpc2−/− hearts (Figure 4L) and unlike in the failing hearts, we failed to detect glycogen granules on electron micrographs (Figure 4M). The increased glycolysis in these non-failing CS-Mpc2−/− hearts did not appear to increase pentose phosphate pathway flux as ribose-5-phosphate, and the enrichment of serine and glycine and their pool sizes remained normal (Figure S5K–O). However, enrichment into glycerol-3-phosphate was increased, while pool size remained unchanged (Figure S5P,Q), suggesting increased flow of glucose carbons into glycerophospholipid synthesis. As within the failing CS-Mpc2−/− hearts, these nonfailing CS-Mpc2−/− hearts also displayed normal enrichment and pool size of TCA cycle intermediates after 13C-glucose injection (Figure S6). Cumulatively, these data suggest that cardiac MPC deletion on its own increases glycolytic metabolism, but only with the development of heart failure does glycogen accumulate.
Figure 4. Non-failing CS-Mpc2−/− hearts from young mice display increased glycolysis, but no glycogen accumulation.

A through K, 13C % enrichment of glucose (A), gluocse-6-phosphate (G6P) (B), fructose bisphosphate (FBP) (C), dihydroxyacetone phosphate (DHAP) (D), 2/3-phosphoglycerate (PG) (E), phosphoenolpyruvate (PEP) (F), pyruvate (G), lactate (H), alanine (I), and uridine diphosphate (UDP)-glucose (J), and the UDP-glucose normalized pool size (K) in hearts of young non-failing CS-Mpc2−/− and fl/fl littermates injected i.p. with U-13C-glucose, n=8–10. L, Heart glycogen content measured biochemically in young non-failing CS-Mpc2−/− and fl/fl littermates, n=6–7. M, Representative transmission electron microscopy images at 15,000X magnification of the cardiac papillary muscle in young fl/fl and CS-Mpc2−/− littermates. Scale bars = 1 μm. Data are presented as the mean±SD, and were analyzed by unpaired, two-tailed Student’s t-test with Welch correction.
Ketogenic diet normalizes the CS-Mpc2−/− heart failure and glycogen accumulation
Previous reports by us and others showed that feeding a ketogenic diet could either prevent or reverse the heart failure in MPC-deficient hearts4, 13. Here we again normalized the CS-Mpc2−/− hearts within 3 weeks of ketogenic diet feeding (Figure 5A). This was associated with complete normalization of the glycogen content of the hearts measured both biochemically and by electron microscopy (Figure 5B,C). Phosphorylation of glycogen synthase serine-640, which was dramatically increased in the failing CS-Mpc2−/− hearts from low fat (LF)-fed mice, was also normalized by KD-feeding (Figure 5D). To evaluate glucose metabolism, we once again injected mice with U-13C-glucose and measured metabolite enrichment and pool sizes by mass spectrometry. Interestingly, glucose enrichment was significantly increased in both fl/fl and CS-Mpc2−/− mice fed a KD compared to mice fed LF diet (Figure 5E), suggesting that these hearts that have been chronically subjected to low blood glucose and insulin from KD feeding13 have the ability to enhance glucose uptake when provided with a bolus. This led to increased glycolytic enrichment down to elevated enrichment of pyruvate, lactate, and alanine in hearts from KD-fed mice (Figure S7A–C). However, 13C enrichment of TCA cycle intermediates were very low in hearts of KD-fed mice, suggesting that ketogenic diet strongly downregulates glucose carbon incorporation into the TCA cycle and oxidation (Figure S7D–L). The failing CS-Mpc2−/− hearts from LF-fed mice once again displayed increased enrichment of the glucose within glycogen, however the increased U-13C-glucose uptake with KD-feeding also generally increased 13C-enrichment of glycogen (Figure 5F). However, the metabolite pool sizes of glucose-6-phosphate, pyruvate, and the glycogen precursor UDP-glucose, paint the clear picture of accumulation in the failing CS-Mpc2−/− heart, and normalization with KD-feeding (Figure 5G–I). Lastly, in confirmation of our biochemical assay and electron micrographs, glucose within the glycogen pool measured by mass spectrometry was significantly elevated in CS-Mpc2−/− hearts from LF-fed mice and normalized by KD-feeding (Figure 5J). All together, these data suggest that KD rescues the heart failure in CS-Mpc2−/− mice likely by decreasing the need for pyruvate entry into the TCA cycle and normalizing glycolytic metabolism and glycogen stores.
Figure 5. Ketogenic diet normalizes the CS-Mpc2−/− heart failure and glycogen accumulation.

A, Heart weight to tibia length ratio (HW/TL) of fl/fl and CS-Mpc2−/− mice fed either low fat (LF) or ketogenic diet (KD), n=4–7. B, Heart glycogen content measured biochemically in fl/fl and CS-Mpc2−/− mice fed either a LF diet or KD, n=4–7. C, Representative transmission electron microscopy images at 5,000X magnification of the cardiac papillary muscle of fl/fl and CS-Mpc2−/− mice fed LF or KD. Arrowheads denote glycogen granules. Scale bars = 1 μm. D, Western blot images of phosphorylated glycogen synthase (serine 640) and total glycogen synthase from hearts of fl/fl or CS-Mpc2−/− mice fed either LF or KD, n=2. E through J, 13C % enrichment of glucose (E) and the glucose within glycogen (F), and the normalized pool size of glucose-6-phosphate (G6P) (G), pyruvate (H), uridine diphosphate (UDP)-glucose (I), and glycogen (J) in fl/fl and CS-Mpc2−/− mice that were fed LF or KD and injected i.p. with U-13C-glucose, n=6–7. Data are presented as the mean±SD, and were analyzed by two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test.
AngII-induced cardiac hypertrophy increases glycogen and is improved by ketogenic diet
To evaluate if increased cardiac glycogen was characteristic of non-genetic cardiomyopathy, we subjected mice to AngII infusion via subcutaneous osmotic pump in combination with LF or KD-feeding. As expected, KD-feeding significantly lowered blood glucose concentrations and increased plasma β-hydroxybutyrate levels in both saline and AngII infused mice (Figure 6A–B). AngII infused mice fed LF displayed significant hypertrophy as shown by increase in HW/TL, which was significantly reduced by KD-feeding (Figure 6C). Cardiac glycogen more than doubled in AngII infused mice fed LF diet, and again cardiac glycogen levels normalized by KD (Figure 6D). AngII was previously shown to reduce cardiac MPC1 and MPC2 expression12, and we confirmed that both Mpc1 and Mpc2 expression were significantly decreased by AngII in LF-fed mice (Figure 6E–F). Interestingly, ketogenic diet significantly reduced cardiac Mpc expression compared to LF (Figure 6E–F), potentially contributing to the significant decrease of 13C-glucose enrichment of the TCA cycle we observed previously with ketogenic diet (Figure S7). Like in the failing CS-Mpc2−/− hearts, there was a significant increase in phosphorylation of glycogen synthase in the hearts of AngII infused mice fed LF diet (Figure 6G–H), suggesting a cellular attempt to inhibit glycogen synthesis. This phosphorylation of glycogen synthase was also slightly increased in hearts from KD-fed mice (Figure 6G–H), likely due to the low dietary carbohydrate and decreased glycemia (Figure 6A). All together, these data suggest that AngII-induced cardiac hypertrophy decreases Mpc expression and increases glycogen stores; however, this can be normalized through KD-feeding which also improves the cardiac hypertrophy.
Figure 6. AngII-induced cardiac hypertrophy increases glycogen and is improved by ketogenic diet.
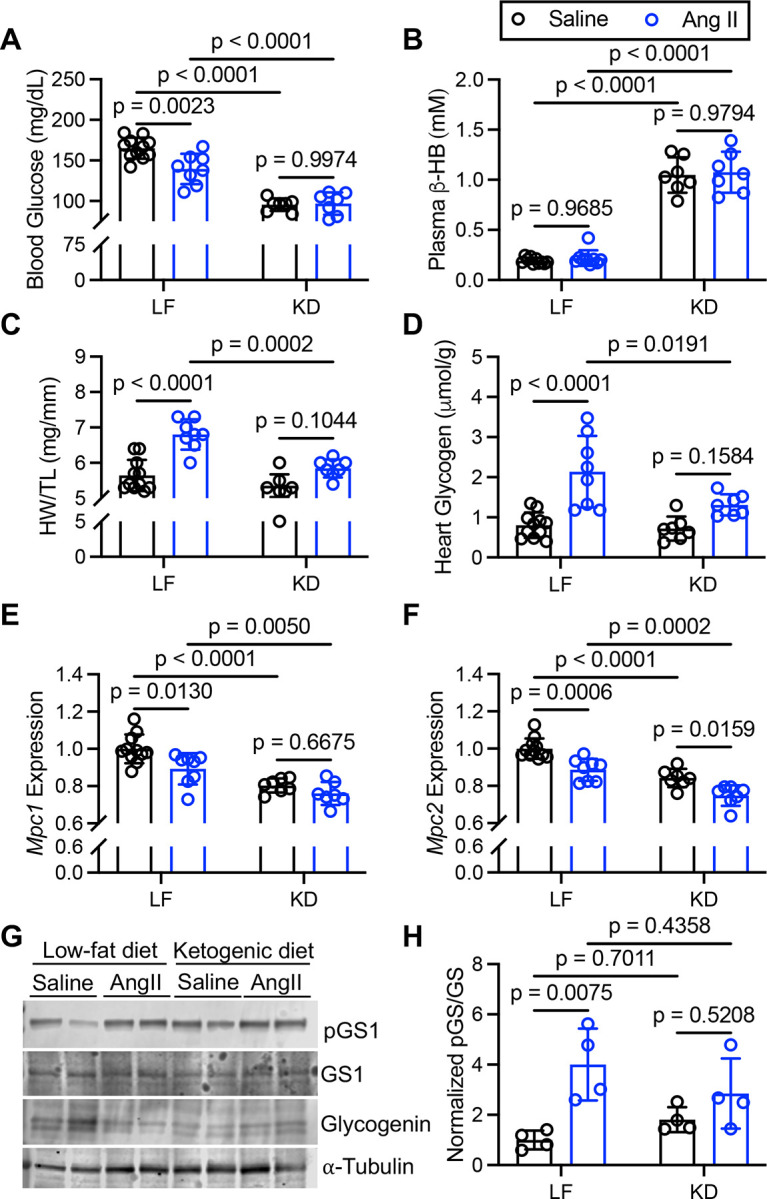
A and B, Blood glucose (A) and plasma β-hydroxybutyrate (β-HB) concentrations (B) of saline vehicle or AngII-infused mice fed low fat (LF) diet or ketogenic diet (KD), n=7–10. C, Heart weight to tibia length ratio (HW/TL) of saline vehicle and AngII infused mice fed LF or KD, n=7–10. D, Cardiac glycogen measured biochemically from mice infused with saline or AngII fed either LF or KD, n=7–10. E and F, Normalized gene expression for Mpc1 (E) and Mpc2 (F) in hearts from mice infused with saline or AngII fed LF or KD, n=7–10. G and H, representative western blot images of phosphorylated glycogen synthase (pGS1), total glycogen synthase (GS1), glycogenin-1, and α-tubulin (G), and the normalized densitometry quantified for pGS/GS (H) from hearts of mice infused with saline or AngII fed LF or KD, n=4. Data are presented as the mean±SD, and were analyzed by two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test.
Discussion
This work describes that decreases in MPC expression, or genetic deletion of the MPC, increases cardiac glucose utilization, glycogen accumulation, and heart failure. Altered cardiac metabolism is a hallmark of heart failure, generally consisting of decreased mitochondrial oxidative capacity, particularly for fatty acids which the normal heart prefers2. It is generally believed that this decrease in mitochondrial oxidation creates an “energy starved” heart that then causes an increase in myocardial glucose uptake and glycolysis to attempt to fuel the heart27. Our current study suggests this may not represent the full story beyond these metabolic changes. In hearts deficient for the mitochondrial pyruvate carrier, which limits the ability of pyruvate from glucose or lactate to enter the mitochondria for oxidation, profound dilated cardiomyopathy develops despite normal levels of high energy phosphates ATP and phosphocreatine. ATP was also previously found to be normal in MPC1−/− hearts that had begun to fail12. Although this indicates a lack of “energetic stress” or the “energy starved failing heart”, glucose uptake and glycolysis is increased in these CS-Mpc2−/− hearts. Previous studies suggest that increased glucose utilization is important for fueling the hypertrophied or failing heart, predominantly by increasing nucleotide and amino acid anabolism through the pentose phosphate pathway7, 9. Here, we suggest that accumulation of glycogen is contributing to the heart failure in CS-Mpc2−/− hearts and in AngII-induced hypertrophy.
The heart has very little capacity to store nutrients or high energy phosphates31, making constant tissue perfusion critical. This includes very little storage of glucose as glycogen, with the mouse heart typically containing 1–5 μmol glucosyl units per gram, compared to 40–80 μmol/g in skeletal muscles and 500–800 μmol/g in the liver in the fed condition32. Of course, skeletal muscle glycogen is important during exhaustive exercise, and liver glycogen stores are critical for maintaining glycemia during fasting, and these physiologic stressors can deplete the skeletal muscle and liver glycogen pools. Interestingly, heart glycogen levels remain normal or are even elevated in the fasted state13, 33–35, with this elevation likely due to the hearts switching to even more predominant fat oxidation in the fasted state inhibiting glycolysis more than glucose uptake36. High cardiac workloads increase glycogenolysis and may decrease cardiac glycogen concentrations37, 38, however these perfused heart studies showed that perfusion with ample substrates (glucose or lactate) abrogated the depletion of glycogen. Thus, it is not well understood if cardiac glycogen levels are decreased or maintained under higher workloads in vivo. Exhaustive exercise has been shown to decrease cardiac glycogen content by 25%, which was rapidly repleted39. Overall, these previous studies suggest that while cardiac glycogen levels are low, the consistent flux of glucose in and out of the glycogen pool and relative maintenance of glycogen concentrations indicates an important role.
Another interesting point from these previous studies is that glucose released from cardiac glycogen is preferentially oxidized instead of converted to lactate37, 40. This would have significant effects in MPC-deficient hearts that cannot efficiently oxidize this glycogen-derived pyruvate. This inability to oxidize glucose from glycogen may partially explain the accumulation of glycogen in these MPC-deficient hearts, in addition to the increased glucose uptake and incorporation of 13C-glucose into glycogen we observed. That said, a limitation of our current study was the inability to assess rates of glycogenolysis. A puzzling finding from this study, and a previous study4, is that enrichment of the TCA cycle from 13C-glucose is not decreased in MPC−/− hearts (Figures S2 and S6), even though we observe significantly reduced respiration with pyruvate as substrate in isolated mitochondria and permeabilized muscle fibers from CS-Mpc2−/− hearts (Figure S3). There are several potential explanations for this maintained TCA cycle 13C enrichment. In this whole-heart method, 13C enrichment of the TCA cycle would be maintained in the non-cardiomyocyte cells in the heart which would not have Mpc2 deleted by Mlc2vCre. This explanation is supported in that decreased TCA cycle enrichment was observed in isolated cardiomyocytes from MPC1−/− hearts14. Another possibility is that there could be a metabolic bypass of the MPC, such as alanine transamination and alanine mitochondrial transport as we have previously identified in hepatocytes16, or cytosolic and mitochondrial malic enzymes. This sort of bypass may be occurring to a degree, as we do observe much greater reduction of pyruvate respiration in isolated mitochondria (devoid of cytosol) than we do when using permeabilized cardiac tissue (Figure S3). Lastly, while we mostly think of these organic acids as TCA cycle metabolites, it is clear many of these can be released from mitochondria and found in the cytosol and other organelles41. Therefore, non-mitochondrial pools of these metabolites may somehow be labeled by 13C-glucose in the CS-Mpc2−/− hearts. Overall, with the dramatic reduction in mitochondrial pyruvate respiration and the pronounced pathologic phenotype of the hearts, we infer that pyruvate oxidation is reduced despite normal 13C enrichment of TCA cycle metabolites in CS-Mpc2−/− hearts.
Perhaps due to its low abundance, cardiac glycogen has not been well studied in the context of heart failure. The one exception is the association between glycogen accumulation and hypertrophic cardiomyopathy that occurs in multiple forms of glycogen storage diseases (GSD). For example, GSD type-II, or Pompe disease, is caused by mutation in the enzyme acid α-glucosdiase, which catalyzes the lysosomal degradation of glycogen42. This results in cardiac glycogen accumulation, up to 200-fold in Pompe disease mouse models, and development of hypertrophic cardiomyopathy43. Thus, these GSDs clearly support the connection between increased cardiac glycogen and heart failure.
This current work also describes the normalization of cardiac glycogen and rescuing of heart failure by a ketogenic diet in both the CS-Mpc2−/− model, and in AngII-induced hypertrophy. We hypothesize that ketogenic diet is improving these hearts by decreasing glycemia and cardiac glucose metabolism, ultimately reducing the need for mitochondrial pyruvate metabolism and the MPC as shown by very low 13C-glucose enrichment of the TCA cycle in hearts from KD-fed mice (Figure S7). Significantly reduced glucose oxidation, even with insulin stimulation, was recently observed in hearts from mice fed a KD44. One interesting point to this current study however is that hearts from KD-fed mice appeared to have increased uptake of the bolus 13C-glucose (Figure 5E). This is somewhat contrary to the KD-induced insulin resistance recently described44, and argues that the hearts of KD-fed mice can readily perform glucose utilization when challenged with a bolus. Ultimately, this chronic decrease in glycemia and cardiac glucose metabolism from ketogenic diet feeding allows the glycogen levels to return to normal. Thus, our findings not only provide new insights into how reduced mitochondrial pyruvate metabolism contributes to heart failure, but also provides important implications for the development of strategies to reduce cardiac glucose utilization and glycogen levels. One could speculate this decrease in glycogen may also be a mechanism for the profound cardioprotective effects of sodium-glucose cotransporter-2 (SGLT2) inhibitors, as recently postulated that SGLT2 inhibitors improve heart failure by reducing intermediates of glucose and lipid metabolism45. However, cardiac glycogen content with SGLT2 inhibitor treatment requires further investigation.
Supplementary Material
Supplemental Figure 1. Glycolytic pool sizes in failing MPC−/− hearts. A, Schematic pathway of glycolysis and accessory glucose pathways. B through L, Normalized pool size of glucose (B), glucose-6-phosphate (G6P) (C), fructose bisphosphate (FBP) (D), dihydroxyacetone phosphate (DHAP) (E), 2/3-phosphoglycerate (PG) (F), phosphoenolpyruvate (PEP) (G), pyruvate (H), lactate (I), alanine (J), and the 13C % enrichment of glycerol 3-phosphate (Glycerol 3P) (K) and glycerol 3P pool (L) of failing CS-Mpc2−/− hearts and fl/fl littermates injected with U-13C-glucose, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 2. TCA cycle enrichment and pool sizes in failing CS-Mpc2−/− hearts. A through L, the 13C % enrichment and pool sizes of citrate (A-B), aconitate (C-D), α-ketoglutarate (KG) (E-F), succinate (G-H), fumarate (I-J), and malate (K-L) in fl/fl and failing CS-Mpc2−/− hearts of mice injected i.p. with U-13C-glucose, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 3. MPC deletion reduces pyruvate oxidation. A and B, Oxygen consumption rates (OCR) measured from isolated cardiac mitochondria (A) and permeabilized cardiac muscle fibers (B) from hearts of fl/fl and CS-Mpc2−/− littermates stimulated with pyruvate/malate (P/M), P/M plus adenosine diphosphate (ADP), and P/M, ADP, and the MPC inhibitor UK-5099 (5 μM), n=4–5. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 4. No major changes in the pentose phosphate pathway in failing MPC−/− hearts. A, Schematic of 13C enrichment of the pentose phosphate pathway from U-13C-glucose. Metabolites in grey were not measured. B through F, 13C % enrichment and normalized pool size of 6-phosphogluconate (PG) (B), ribose-5-phosphate (R5P) (C), sedoheptulose 7-phosphate (Sedo7P) (D), serine (E), and glycine (F), in hearts from fl/fl and failing CS-Mpc2−/− mice injected with U-13C-gluocse, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 5. Glycolytic pool sizes in young, non-failing MPC−/− hearts. A, Schematic of glycolysis and accessory glucose pathways. B through Q, Normalized pool sizes of glucose (B), glucose-6-phosphate (G6P) (C), fructose bisphosphate (FBP) (D), dihydroxyacetone phosphate (DHAP) (E), 2/3 phosphoglycerate (PG) (F), phosphoenolpyruvate (PEP) (G), pyruvate (H), lactate (I), alanine (J), ribose-5-phosphate (R5P) (K), and the 13C enrichment and pool size of serine (L-M), glycine (N-O), and glycerol 3-phosphate (glycerol 3P) (P-Q) in young non-failing CS-Mpc2−/− and fl/fl littermates injected with U-13C-glucose, n=8–10. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 6. TCA cycle enrichment and pool sizes in young, non-failing CS-Mpc2−/− hearts. A through L, 13C % enrichment and normalized pool sizes of citrate (A-B), aconitate (C-D), α-ketoglutarate (αKG) (E-F), succinate (G-H), fumarate (I-J), and malate (K-L), in hearts of young, nonfailing CS-Mpc2−/− and fl/fl littermates injected with U-13C-glucose, n=8–10. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 7. Ketogenic diet decreases TCA cycle enrichment from glucose. A through L, 13C % enrichment of pyruvate (A), lactate (B), alanine (C), citrate (D), aconitate (E), α-ketoglutarate (αKG) (F), glutamate (G), glutamine (H), succinate (I), fumarate (J), malate (K), and aspartate (L) in hearts from fl/fl and CS-Mpc2−/− fed either low-fat (LF) or ketogenic diet (KD) and injected with U-13C-glucose, n=6–7. Data are presented as the mean±SD. Data were evaluated by two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test.
Novelty and Significance.
What Is Known?
Heart failure involves metabolic changes such as increased glycolysis despite unchanged or increased glucose oxidation.
This metabolic alteration is due at least in part by decreased expression of the mitochondrial pyruvate carrier (MPC) proteins identified in animal models and failing human hearts.
Deletion of either MPC protein in the murine heart leads to development of dilated cardiomyopathy.
What New Information Does This Article Contribute?
What drives heart failure with loss of the MPC is not understood, and these studies suggest that the defective pyruvate oxidation does not simply cause “energetic stress” and the “energy starved failing heart” as the failing CS-Mpc2−/− hearts display normal ATP and phosphocreatine levels.
Loss of the MPC instead increases glycolytic metabolism and causes accumulation of glycogen in the failing heart, and both the glycogen and the heart failure can be normalized by decreasing cardiac glucose metabolism by feeding a ketogenic diet.
Cardiac hypertrophy from angiotensin-II infusion increases heart glycogen levels and both the glycogen and hypertrophy are also improved by ketogenic diet in this model.
It has become clear that metabolic changes that occur in heart failure are not just adaptive alterations but can be maladaptive drivers of cardiac remodeling and dysfunction. Hypertrophied and failing hearts often increase glycolytic metabolism, generally thought to be a response to decreased mitochondrial oxidative metabolism. We find that deletion of the MPC increases glycolytic metabolism despite normal levels of high energy phosphates ATP and phosphocreatine. This increased glycolytic metabolism causes glycogen to accumulate in the failing heart. However, interventions to decrease glucose metabolism, such as feeding a ketogenic diet, were able to normalize the cardiac glycogen levels and the heart failure. This study reveals that maintaining mitochondrial pyruvate metabolism may be a strategy to improve heart failure by decreasing glycolytic metabolism and the accumulation of glycogen. Conversely, interventions to decrease cardiac glucose delivery and metabolism such as the ketogenic diet likely improves heart failure by preventing the accumulation of glycolytic intermediates and glycogen.
Sources of Funding
R.C.W. is supported by a SLU Biochemistry and Molecular Biology Doisy Scholar Fellowship. This work was supported by NIH R00-HL136658 to K.S.M and R35 ES028365 to G.J.P.
Nonstandard Abbreviations and Acronyms
- 2-DG
2-deoxyglucose
- αKG
alpha-ketoglutarate
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- AngII
angiotensin-II
- Cr
creatine
- DHAP
dihydroxyacetone phosphate
- FAO
fatty acid oxidation
- FBP
fructose bisphosphate
- G6P
glucose-6-phosphate
- GSD
glycogen storage disease
- KD
ketogenic diet
- KEGG
Kyoto encyclopedia of genes and genomes
- LF
low-fat
- MPC
mitochondrial pyruvate carrier
- NAD+ and NADH
oxidized and reduced nicotinamide adenine dinucleotide
- NADP+ and NADPH
oxidized and reduced nicotinamide adenine dinucleotide phosphate
- PCr
phosphocreatine
- PEP
phosphoenolpyruvate
- P/M
pyruvate/malate
- R5P
ribose-5-phosphate
- RT-qPCR
reverse transcription quantitative PCR, Sedo7P, sedoheptulose 7-phosphate
- UDP
uridine diphosphate
- UHPLC
ultra-high performance liquid chromatography
Footnotes
Disclosures
G.J.P. has a research collaboration agreement with Thermo Fisher Scientific and is a scientific advisor for Cambridge Isotope Laboratories. All other authors declare no conflicts of interest.
References
- 1.Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK, Buxton AE, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Fugar S, Generoso G, Heard DG, Hiremath S, Ho JE, Kalani R, Kazi DS, Ko D, Levine DA, Liu J, Ma J, Magnani JW, Michos ED, Mussolino ME, Navaneethan SD, Parikh NI, Poudel R, Rezk-Hanna M, Roth GA, Shah NS, St-Onge MP, Thacker EL, Virani SS, Voeks JH, Wang NY, Wong ND, Wong SS, Yaffe K, Martin SS, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation. 2023;147:e93–e621. [DOI] [PubMed] [Google Scholar]
- 2.Lopaschuk GD, Karwi QG, Tian R, Wende AR and Abel ED. Cardiac Energy Metabolism in Heart Failure. Circ Res. 2021;128:1487–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Jong KA and Lopaschuk GD. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can J Cardiol. 2017;33:860–871. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, Rauckhorst AJ, Tayyari F, Pewa AD, Gray LR, Teesch LM, Puchalska P, Funari TR, McGlauflin R, Zimmerman K, Kutschke WJ, Cassier T, Hitchcock S, Lin K, Kato KM, Stueve JL, Haff L, Weiss RM, Cox JE, Ruger J, Taylor EB, Crawford PA, Lewandowski ED, Des Rosiers C and Abel ED. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat Metab. 2020;2:1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahma MK, Ha CM, Pepin ME, Mia S, Sun Z, Chatham JC, Habegger KM, Abel ED, Paterson AJ, Young ME and Wende AR. Increased Glucose Availability Agenuates Myocardial Ketone Body Utilization. J Am Heart Assoc. 2020;9:e013039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, Lopaschuk GD and Oudit GY. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological aherload. Cardiovasc Res. 2013;97:676–85. [DOI] [PubMed] [Google Scholar]
- 7.Karlstaedt A, Khanna R, Thangam M and Taegtmeyer H. Glucose 6-Phosphate Accumulates via Phosphoglucose Isomerase Inhibition in Heart Muscle. Circ Res. 2020;126:60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davogusgo GE, Salazar RL, Vasquez HG, Karlstaedt A, Dillon WP, Guthrie PH, Martin JR, Vitrac H, De La Guardia G, Vela D, Ribas-Latre A, Baumgartner C, Eckel-Mahan K and Taegtmeyer H. Metabolic remodeling precedes mTORC1-mediated cardiac hypertrophy. J Mol Cell Cardiol. 2021;158:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rigerhoff J, Young S, Villet O, Shao D, Neto FC, Begcher LF, Hsu YA, Kolwicz SC Jr., Rahery D and Tian R. Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis. Circ Res. 2020;126:182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS and Ruger J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER and Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–6. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, Crespo-Leiro MG, Vieites MG, Bianchi K, Morales V, Domenech N and Eaton P. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab. 2020;2:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW and Finck BN. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab. 2020;2:1232–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cluntun AA, Badolia R, Leglova S, Parnell KM, Shankar TS, Diakos NA, Olson KA, Taleb I, Tatum SM, Berg JA, Cunningham CN, Van Ry T, Bog AJ, Krokidi AT, Fogarty S, Skedros S, Swiatek WI, Yu X, Luo B, Merx S, Navankasagusas S, Cox JE, Ducker GS, Holland WL, McKellar SH, Ruger J and Drakos SG. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021;33:629–648 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Vazquez P, Fernandez-Caggiano M, Barge-Caballero E, Barge-Caballero G, Couto-Mallon D, Grille-Cancela Z, Blanco-Canosa P, Paniagua-Martin MJ, Enriquez-Vazquez D, Vazquez-Rodriguez JM, Domenech N and Crespo-Leiro MG. Reduced mitochondrial pyruvate carrier expression in hearts with heart failure and reduced ejection fraction patients: ischemic vs. non-ischemic origin. Front Cardiovasc Med. 2024;11:1349417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC and Finck BN. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015;22:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang T, Song C, Ralph DL, Andrews P, Sparks MA, Koller BH, McDonough AA and Coffman TM. Cell-Specific Actions of the Prostaglandin E-Prostanoid Receptor 4 Agenuating Hypertension: A Dominant Role for Kidney Epithelial Cells Compared With Macrophages. J Am Heart Assoc. 2022;11:e026581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamburov A, Cavill R, Ebbels TM, Herwig R and Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27:2917–8. [DOI] [PubMed] [Google Scholar]
- 19.Khomtchouk BB, Hennessy JR and Wahlestedt C. shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS One. 2017;12:e0176334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferguson D, Eichler SJ, Yiew NKH, Colca JR, Cho K, Pak GJ, Shew TM, Lutkewige AJ, Mukherjee S, McCommis KS, Niemi NM and Finck BN. Mitochondrial pyruvate carrier inhibition initiates metabolic crosstalk to stimulate branched chain amino acid catabolism. Mol Metab. 2023;70:101694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams KJ, Prag B, Bose N, Dubois LG, St John-Williams L, Perrog KM, Ky K, Kapahi P, Sharma V, MacCoss MJ, Moseley MA, Colton CA, MacLean BX, Schilling B, Thompson JW and Alzheimer’s Disease Metabolomics C. Skyline for Small Molecules: A Unifying Sohware Package for Quantitative Metabolomics. J Proteome Res. 2020;19:1447–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su X, Lu W and Rabinowitz JD. Metabolite Spectral Accuracy on Orbitraps. Anal Chem. 2017;89:5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ullman JC, Mellem KT, Xi Y, Ramanan V, Merrig H, Choy R, Gujral T, Young LEA, Blake K, Tep S, Homburger JR, O’Regan A, Ganesh S, Wong P, Sagerfield TF, Lin B, Situ E, Yu C, Espanol B, Sarwaikar R, Fastman N, Tzitzilonis C, Lee P, Reiton D, Morton V, Santiago P, Won W, Powers H, Cummings BB, Hoek M, Graham RR, Chandriani SJ, Bainer R, DePaoli-Roach AA, Roach PJ, Hurley TD, Sun RC, Gentry MS, Sinz C, Dick RA, Noonberg SB, Beake DT, Morgans DJ Jr. and Green EM. Small-molecule inhibition of glycogen synthase 1 for the treatment of Pompe disease and other glycogen storage disorders. Sci Transl Med. 2024;16:eadf1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher JS, Nolte LA, Kawanaka K, Han DH, Jones TE and Holloszy JO. Glucose transport rate and glycogen synthase activity both limit skeletal muscle glycogen accumulation. Am J Physiol Endocrinol Metab. 2002;282:E1214–21. [DOI] [PubMed] [Google Scholar]
- 25.Ching JK, Spears LD, Armon JL, Renth AL, Andrisse S, Collins RLt and Fisher JS. Impaired insulin-stimulated glucose transport in ATM-deficient mouse skeletal muscle. Appl Physiol Nutr Metab. 2013;38:589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamm DR, Pyles KD, Sharpe MC, Healy LN, Colca JR and McCommis KS. Novel insulin sensitizer MSDC-0602K improves insulinemia and fagy liver disease in mice, alone and in combination with liraglutide. J Biol Chem. 2021;296:100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran DH and Wang ZV. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J Am Heart Assoc. 2019;8:e012673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riehle C and Abel ED. Insulin Signaling and Heart Failure. Circ Res. 2016;118:1151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y and Roach PJ. Inactivation of rabbit muscle glycogen synthase by glycogen synthase kinase-3. Dominant role of the phosphorylation of Ser-640 (site-3a). J Biol Chem. 1993;268:23876–80. [PubMed] [Google Scholar]
- 30.Pitcher J, Smythe C and Cohen P. Glycogenin is the priming glucosyltransferase required for the initiation of glycogen biogenesis in rabbit skeletal muscle. Eur J Biochem. 1988;176:391–5. [DOI] [PubMed] [Google Scholar]
- 31.Doenst T, Nguyen TD and Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pederson BA, Cope CR, Schroeder JM, Smith MW, Irimia JM, Thurberg BL, DePaoli-Roach AA and Roach PJ. Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J Biol Chem. 2005;280:17260–5. [DOI] [PubMed] [Google Scholar]
- 33.Evans G. The glycogen content of the rat heart. J Physiol. 1934;82:468–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichelt ME, Mellor KM, Curl CL, Stapleton D and Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol. 2013;65:67–75. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Nguyen VT and Taegtmeyer H. Feeding and fasting determine postischemic glucose utilization in isolated working rat hearts. Am J Physiol. 1991;260:H542–8. [DOI] [PubMed] [Google Scholar]
- 36.Depre C, Vanoverschelde JL and Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–88. [DOI] [PubMed] [Google Scholar]
- 37.Goodwin GW, Taylor CS and Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998;273:29530–9. [DOI] [PubMed] [Google Scholar]
- 38.McConville P, Lakaga EG and Spencer RG. Greater glycogen utilization during 1- than 2-adrenergic receptor stimulation in the isolated perfused rat heart. Am J Physiol Endocrinol Metab. 2007;293:E1828–35. [DOI] [PubMed] [Google Scholar]
- 39.Conlee RK and Tipton CM. Cardiac glycogen repletion aher exercise: influence of synthase and glucose 6-phosphate. J Appl Physiol Respir Environ Exerc Physiol. 1977;42:240–4. [DOI] [PubMed] [Google Scholar]
- 40.Goodwin GW, Ahmad F and Taegtmeyer H. Preferential oxidation of glycogen in isolated working rat heart. J Clin Invest. 1996;97:1409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Reyes I and Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puzzo F, Colella P, Biferi MG, Bali D, Paulk NK, Vidal P, Collaud F, Simon-Sola M, Charles S, Hardet R, Leborgne C, Meliani A, Cohen-Tannoudji M, Astord S, Gjata B, Sellier P, van Wigenberghe L, Vignaud A, Boisgerault F, Barkats M, Laforet P, Kay MA, Koeberl DD, Ronzik G and Mingozzi F. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang JY, Kan SH, Sandfeld EK, Dalton ND, Rangel AD, Chan Y, Davis-Turak J, Neumann J and Wang RY. CRISPR-Cas9 generated Pompe knock-in murine model exhibits early-onset hypertrophic cardiomyopathy and skeletal muscle weakness. Sci Rep. 2020;10:10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho KL, Karwi Q, Wang F, Wagg C, Zhang L, Panidarapu S, Chen B, Pherwani S, Greenwell AA, Oudit G, Ussher JR and Lopaschuk GD. The ketogenic diet does not improve cardiac function and blunts glucose oxidation in ischemic heart failure. Cardiovasc Res. 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Packer M. SGLT2 inhibitors: role in protective reprogramming of cardiac nutrient transport and metabolism. Nat Rev Cardiol. 2023;20:443–462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Glycolytic pool sizes in failing MPC−/− hearts. A, Schematic pathway of glycolysis and accessory glucose pathways. B through L, Normalized pool size of glucose (B), glucose-6-phosphate (G6P) (C), fructose bisphosphate (FBP) (D), dihydroxyacetone phosphate (DHAP) (E), 2/3-phosphoglycerate (PG) (F), phosphoenolpyruvate (PEP) (G), pyruvate (H), lactate (I), alanine (J), and the 13C % enrichment of glycerol 3-phosphate (Glycerol 3P) (K) and glycerol 3P pool (L) of failing CS-Mpc2−/− hearts and fl/fl littermates injected with U-13C-glucose, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 2. TCA cycle enrichment and pool sizes in failing CS-Mpc2−/− hearts. A through L, the 13C % enrichment and pool sizes of citrate (A-B), aconitate (C-D), α-ketoglutarate (KG) (E-F), succinate (G-H), fumarate (I-J), and malate (K-L) in fl/fl and failing CS-Mpc2−/− hearts of mice injected i.p. with U-13C-glucose, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 3. MPC deletion reduces pyruvate oxidation. A and B, Oxygen consumption rates (OCR) measured from isolated cardiac mitochondria (A) and permeabilized cardiac muscle fibers (B) from hearts of fl/fl and CS-Mpc2−/− littermates stimulated with pyruvate/malate (P/M), P/M plus adenosine diphosphate (ADP), and P/M, ADP, and the MPC inhibitor UK-5099 (5 μM), n=4–5. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 4. No major changes in the pentose phosphate pathway in failing MPC−/− hearts. A, Schematic of 13C enrichment of the pentose phosphate pathway from U-13C-glucose. Metabolites in grey were not measured. B through F, 13C % enrichment and normalized pool size of 6-phosphogluconate (PG) (B), ribose-5-phosphate (R5P) (C), sedoheptulose 7-phosphate (Sedo7P) (D), serine (E), and glycine (F), in hearts from fl/fl and failing CS-Mpc2−/− mice injected with U-13C-gluocse, n=11–12. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 5. Glycolytic pool sizes in young, non-failing MPC−/− hearts. A, Schematic of glycolysis and accessory glucose pathways. B through Q, Normalized pool sizes of glucose (B), glucose-6-phosphate (G6P) (C), fructose bisphosphate (FBP) (D), dihydroxyacetone phosphate (DHAP) (E), 2/3 phosphoglycerate (PG) (F), phosphoenolpyruvate (PEP) (G), pyruvate (H), lactate (I), alanine (J), ribose-5-phosphate (R5P) (K), and the 13C enrichment and pool size of serine (L-M), glycine (N-O), and glycerol 3-phosphate (glycerol 3P) (P-Q) in young non-failing CS-Mpc2−/− and fl/fl littermates injected with U-13C-glucose, n=8–10. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 6. TCA cycle enrichment and pool sizes in young, non-failing CS-Mpc2−/− hearts. A through L, 13C % enrichment and normalized pool sizes of citrate (A-B), aconitate (C-D), α-ketoglutarate (αKG) (E-F), succinate (G-H), fumarate (I-J), and malate (K-L), in hearts of young, nonfailing CS-Mpc2−/− and fl/fl littermates injected with U-13C-glucose, n=8–10. Data are presented as the mean±SD. Data were evaluated by unpaired, two-tailed Student’s t-test with Welch correction.
Supplemental Figure 7. Ketogenic diet decreases TCA cycle enrichment from glucose. A through L, 13C % enrichment of pyruvate (A), lactate (B), alanine (C), citrate (D), aconitate (E), α-ketoglutarate (αKG) (F), glutamate (G), glutamine (H), succinate (I), fumarate (J), malate (K), and aspartate (L) in hearts from fl/fl and CS-Mpc2−/− fed either low-fat (LF) or ketogenic diet (KD) and injected with U-13C-glucose, n=6–7. Data are presented as the mean±SD. Data were evaluated by two-way analysis of variance (ANOVA) with Tukey post-hoc multiple-comparisons test.
Data Availability Statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request. The RNA sequencing data generated in this study have been deposited in NCBI GEO Series Number GSE268694.